

Abstract
The ribosome exit tunnel is an important structure involved in the regulation of translation and other essential functions such as protein folding. By comparing 20 recently obtained cryo-EM and X-ray crystallography structures of the ribosome from all three domains of life, we here characterize the key similarities and differences of the tunnel across species. We first show that a hierarchical clustering of tunnel shapes closely reflects the species phylogeny. Then, by analyzing the ribosomal RNAs and proteins, we explain the observed geometric variations and show direct association between the conservations of the geometry, structure and sequence. We find that the tunnel is more conserved in the upper part close to the polypeptide transferase center, while in the lower part, it is substantially narrower in eukaryotes than in bacteria. Furthermore, we provide evidence for the existence of a second constriction site in eukaryotic exit tunnels. Overall, these results have several evolutionary and functional implications, which explain certain differences between eukaryotes and prokaryotes in their translation mechanisms. In particular, they suggest that major co-translational functions of bacterial tunnels were externalized in eukaryotes, while reducing the tunnel size provided some other advantages, such as facilitating the nascent chain elongation and enabling antibiotic resistance.
INTRODUCTION
Ribosomes are the key actors of mRNA translation, a fundamental process underlying all forms of life. While decoding the mRNA nucleotides into their associated polypeptide sequence, ribosomes regulate the dynamics of translation and other central co-translational processes such as the translocation to cell membranes and protein folding (1–3). These processes rely on the structural properties of the ribosome, through interactions with different elements such as binding factors, tRNAs or the nascent polypeptide chain. For example, specific amino acid sequence motifs in certain nascent chains can stall the ribosome and subsequently arrest translation in an antibiotic-dependent manner (4–6). This phenomenon is caused by interactions between the ribosome and the nascent polypeptide chain itself: prior to leaving the ribosome, nascent polypeptides first pass through a structure called the ribosome exit tunnel, spanning from the peptidyl-transferase center (PTC)—where amino acids are polymerized onto the growing nascent chain—to the surface of the ribosome. As the tunnel can accommodate up to ∼40 amino acids (7), its geometry and biophysical properties potentially impact translation dynamics (8–11), as well as co-translational folding events (12–14).
The important role of the ribosome tunnel hence suggests that some of its key elements should be well conserved across species. On the other hand, the selectivity of arrest sequences to specific species (5,7) or differences of translational and co-translational mechanisms between eukarya and bacteria (e.g. initiation and termination; nascent chain quality control and folding; interacting chaperones and so on) (15–18) suggest that important variations of the exit tunnel structure likely exist, with most extreme examples having been observed in mitochondria (19). As such variations have potentially important consequences on the regulation of translation or antibiotic resistance (9,20), it is thus crucial to identify and catalog these differences, and more generally understand the evolution of the ribosome exit tunnel. While the ribosome has been extensively used in the past to elucidate phylogenetic relationships via sequence analysis (21), several studies have more recently shed light on the relation between the evolution of the ribosome and its function. Specifically, the availability of high-resolution 3D structures of the ribosome from X-ray crystallography and cryo-electron microscopy (cryo-EM) has been combined with sequence information to reveal that the evolution of ribosomal RNA (rRNA) has been locally constrained at the beginning of the tunnel around the PTC, ribosomal intersubunit bridges or tRNA contact regions (22–24). Over the past few years, an increasing number of new ribosome structures has been obtained at ∼3 Å resolution, enabling full atomic models of ribosomes from all domains of life to be obtained. Hence, it is now possible to extend our understanding of the relation between the biophysical structure of the entire exit tunnel and its evolution across many different species, thereby unraveling local specificities of the tunnel function.
In the present work, we provide a quantitative analysis of the ribosome exit tunnel structure across a diverse set of species, by compiling and comparing 20 recently obtained ribosome structures from all three domains of life (bacteria, archae and eukarya). Upon extracting the coordinates of the tunnels, we investigate the relation between the geometry of the tunnel and the evolution of the ribosomal structure and its constituent sequences. To achieve this, we introduce and apply a suite of computational methods to study the geometric properties of the tunnel, the local structure of the ribosome near the tunnel. and the conservation of rRNA and ribosomal protein sequences. Our comparative approach hence reveals the extent of conservation and variation along the tunnel across species. We discuss the implications of these results on the evolution of the ribosome, the transit of the nascent polypeptide chain and various major co-translational processes, which explain some important differences observed between prokaryotic and eukaryotic modes of translation.
MATERIALS AND METHODS
Ribosome structures
Cryo-EM reconstructions and X-ray crystallography structures of ribosomes were downloaded from the protein data bank https://www.rcsb.org/. References and details are given in Table 1. The fitting of the model’s residues to the original map was evaluated for each structure using coot (25) density-fit-score function (2Fo-Fc maps were used for X-Ray data). We checked that modifications of the ribosome structure due to binding or presence of other molecules did not substantially affect the exit tunnel structure and geometry.
Table 1.
Ribosomes structures used in our study
| Species/Organelles | Resolution | Reference | PDB codename |
|---|---|---|---|
| Chloroplast (Spinacia) | 3.8 Å (EM) | Ahmed et al. (2017) (82) | 5X8T |
| Mitochondria (H. Sapiens) | 3.1 Å (EM) | Amunts et al. (2015) (83) | 3J9M |
| B. subtilis (b) | 3.8 Å (EM) | Beckert et al. (2017) (84) | 5NJT |
| E. coli (b) | 2.9 Å (EM) | Fischer et al. (2015) (85) | 5AFI |
| D. radiodurans (b) | 3.4 Å (X-ray) | Krupkin et al. (2016) (86) | 5JVG |
| L. lactis (b) | 5.6 Å (EM) | Franken et al. (2017) (87) | 5MYJ |
| M. smegmatis (b) | 3.2 Å (EM) | Hentschel et al. (2017) (88) | 5O60 |
| M. tuberculosis (b) | 3.4 Å (EM) | Yang et al. (2017) (89) | 5V7Q |
| S. aureus (b) | 3.4 Å (X-ray) | Matzov et al. (2017) (90) | 5NRG |
| T. thermophilus (b) | 2.5 Å (X-ray) | Polinakov et al. (2015) (91) | 4Y4P |
| H. marismortui (a) | 2.4 Å (X-ray) | Gabdulkhakov et al. (2013) (92) | 4V9F |
| P. furiosus (a) | 6.6 Å (EM) | Armache et al. (2012) (93) | 4V6U |
| H. sapiens (e) | 2.9 Å (EM) | Natchiar et al. (2017) (94) | 6EK0 |
| L. donovani (e) | 2.9 Å (EM) | Zhang et al. (2016) (95) | 5T2A |
| P. falciparum (e) | 3.2 Å (EM) | Wong et al. (2014) (96) | 3J79 |
| S. cerevisiae (e) | 3.9 Å (EM) | Schmidt et al. (2016) (97) | 5GAK |
| T. aestivum (e) | 5.5 Å (EM) | Gogala et al. (2014) (98) | 4V7E |
| T. cruzi (e) | 2.5 Å (EM) | Liu et al. (2016) (99) | 5T5H |
| T. gondii (e) | 3.2 Å (EM) | Li et al. (2017) (34) | 5XXB |
| T. vaginalis (e) | 3.4 Å (EM) | Li et al. (2017) (34) | 5XY3 |
| E. coli (b) | 3.9 Å (EM) | Arenz et al. (2014) (100) | 3J7Z |
| T. thermophilus (b) | 2.8 Å (X-ray) | Osterman et al. (2017) (101) | 5VP2 |
| H. sapiens (e) | 3.6 Å (EM) | Khatter et al. (2015) (102) | 4UG0 |
First column contains the species, with the domain they belong to (b: bacteria, a: archaea, e: eukarya). The last three structures are replicates for H. sapiens, E. coli and T. thermophilus, used to assess the robustness of our results (see Supplementery Data).
Extraction of the ribosome tunnel geometry
To extract the tunnel coordinates, we used a tunnel search algorithm developed by Sehnal et al. (26). The tunnel search was initiated at the PTC. To locate the PTC, we aligned the sequences of 23S and 28S rRNAs and selected the nucleotide aligned with U4452 in human. The tunnel search algorithm was applied after editing the structure to contain atoms located <80 Å from the constriction site. For the human ribosome, the constriction site was obtained by computing the center of mass of amino acids G70, R71, in uL4 and H133 in uL22. The same procedure was applied for the other species after alignment of proteins uL4 and uL22. We manually picked the ribosome exit tunnel by looking at its shape, length and position relative to uL4 and uL22 (as the search procedure also found artifactual tunnels, e.g. oriented from the PTC toward the small subunit, or artificially created by truncating the ribosome structure before applying the tunnel search algorithm). Each structure in our comparative study contained only a single tunnel path, with no exception. Coordinates were extracted using Pymol and Python custom scripts. The origin of the tunnel was set at a distance of 5 Å from the nucleotide used to initiate the tunnel search algorithm to remove sensitive regions generated by the tunnel search algorithm close to the initial point. Downstream analysis for computing global and local geometric features of the tunnel was done using Matlab and Python (more details in Supplementary Data). An alternative method, which provides a more detailed visualization, is described and analyzed in Supplementary Data.
Distance metric for pairwise comparison and clustering of radius plots
For two tunnels T1 and T2 parametrized by 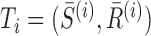 , where i = 1, 2,
, where i = 1, 2, 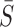 is an arc length parametrization of the tunnel centerline 3D coordinates and
is an arc length parametrization of the tunnel centerline 3D coordinates and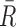 is the associated radius (in ångström), we introduced the following distance metric D(T1, T2) given by
is the associated radius (in ångström), we introduced the following distance metric D(T1, T2) given by
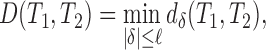 |
(1) |
where ℓ is the maximum shift length and
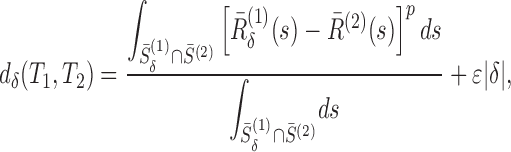 |
(2) |
where 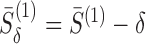 and
and 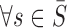 ,
, 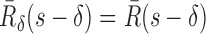 . In other words, we looked for the best alignment of the two tunnels such that it minimizes the average Lp difference between the two aligned radius plots, with a negative penalty for the size of the alignment shift (which is also limited to be less than ℓ). We then take this measurement as the distance between the two tunnels. For p = 2 and considering ϵ as a dimensionless parameter, the units in Eq. (2) are consistent and the distance D is in Å. In practice, we took ℓ = 20 Å, ϵ = 0.01 and p = 2 (for more details on how sensitive our results are with these parameters, see Supplementary Data), and evaluated the integrals by computing their Riemann sum. After computing the pairwise distance matrix associated with our dataset, we constructed the associated phylogenetic tree using the Unweighted Pair Group Method Average (UPGMA) algorithm.
. In other words, we looked for the best alignment of the two tunnels such that it minimizes the average Lp difference between the two aligned radius plots, with a negative penalty for the size of the alignment shift (which is also limited to be less than ℓ). We then take this measurement as the distance between the two tunnels. For p = 2 and considering ϵ as a dimensionless parameter, the units in Eq. (2) are consistent and the distance D is in Å. In practice, we took ℓ = 20 Å, ϵ = 0.01 and p = 2 (for more details on how sensitive our results are with these parameters, see Supplementary Data), and evaluated the integrals by computing their Riemann sum. After computing the pairwise distance matrix associated with our dataset, we constructed the associated phylogenetic tree using the Unweighted Pair Group Method Average (UPGMA) algorithm.
Sequence alignment and conservation of ribosomal proteins
Ribosomal proteins are named according to the system set by Ban et al. (27). Sequences were aligned using MAFFT (28) with default parameters and visualized in Jalview (29). Conservation scores were computed using Jensen-Shannon divergence and software provided by Capra and Singh (30), using the default parameters.
Conserved motifs in ribosomal RNA
For each of the three domains (bacteria, archaea and eukarya), conserved motif sequences were obtained from Doris et al. (24), with their positions along the rRNA sequence computed using exact string matching. For each nucleotide, the distance from the tunnel (or PTC) was computed using MATLAB.
Charge score
To quantify the enhancement and conservation of positive charges among the ribosomal proteins in our study, we introduce the following measure: For a given multiple sequence alignment (MSA) M containing N sequences, let 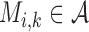 denote the symbol in row i and column k of M, where
denote the symbol in row i and column k of M, where 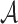 is the set of 20 amino acids plus the gap symbol. Then, for each element x of
is the set of 20 amino acids plus the gap symbol. Then, for each element x of 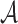 , we assign a charge c(x) = +1 for lysine and arginine; −1 for aspartic acid and glutamic acid; and 0 for all other amino acids and the gap symbol. For each column k of the MSA, we define the charge score S(k), as
, we assign a charge c(x) = +1 for lysine and arginine; −1 for aspartic acid and glutamic acid; and 0 for all other amino acids and the gap symbol. For each column k of the MSA, we define the charge score S(k), as
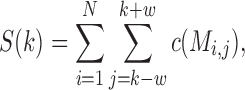 |
where the window size w is set in our analysis to be 2. Charge score analysis was done using Matlab custom scripts.
Visualization tools
Structures were visualized using Pymol. Maps of ribosomal RNA secondary structures, distance of the rRNA nucleotides to the tunnel (or PTC) and conserved motifs were visualized in RiboVision (31).
Data availability
The tunnel coordinates, summary statistics, pairwise distance matrix, and ribosomal RNA and protein conservation scores are provided in Supplementary Data. The tunnels can be visualized and downloaded from the ChannelsDB online server (32).
RESULTS
Extraction of the ribosome exit tunnel structure
We compiled and analyzed publicly available ribosome structures obtained from cryo-EM and X-ray crystallography (see ‘Materials and methods’ section and Table 1). These recent structures (9 published in 2017) accounted for 20 different organisms, including 2 archaea, 2 organelles, 8 bacteria and 8 eukaryotes. Most of these structures (16 out of 20) came from cryo-EM maps, with an average resolution of 3.74 Å; the X-ray structures had an average resolution of 2.92 Å. Replicates of Escherichia coli, Thermus thermophilus and Homo sapiens ribosome structures were also included in our analysis to examine the robustness of our results (see Supplementary Data). For each structure, we applied a procedure described in ‘Materials and methods’ section and illustrated in Figure 1 to extract the ribosome exit tunnel geometry, encoded as a set of coordinates describing the trajectory of the centerline and the tunnel radius at each point of the centerline. Upon obtaining these data, we checked the quality of each tunnel reconstruction by comparing the density-fitting score (see ‘Materials and methods’ section) between residues located close to the tunnel and the rest of the structure. All the structures in our dataset showed an increase of the density-fitting score for tunnel residues, suggesting the good quality of maps in the tunnel region.
Figure 1.

Extraction of the ribosome exit tunnel coordinates. For a given structure of the ribosome large subunit (LSU) (from Schmidt et al. (97)), we first locate the PTC and then apply a tunnel search algorithm (26) to reconstruct the geometry of the exit tunnel (more details in ‘Materials and methods’ section). The inset in the right panel shows the exit tunnel with the PTC and the constriction site that separates the tunnel between its upper and lower parts.
Analysis of global features indicates larger bacterial tunnels and more variation in the lower part
To compare the geometry of the ribosome exit tunnel across different species, we first examined global geometric features, such as the length, tunnel-wide average radius and volume (see Supplementary Data for specific values). The range of values for both the tunnel length (88.8 ± 6.0 Å) and that for average radius (5.4 ± 0.4 Å) were consistent with previous observations (7,33). Upon ordering the species by their exit tunnel volume (see Figure 2), we found a perfect separation between bacteria and eukaryotes, with archaea in between. Specifically, bacterial tunnels (including the ones from organelles) are larger than eukaryotic ones, with mean (3.85 ± 0.37) × 104 Å3 for bacteria compared to (2.78 ± 0.13) × 104 Å3 for eukaryotes. We similarly analyzed the length and average radius to see if the volume variation could be mainly explained by one of these two variables. We obtained a less clear separation among the three domains of life, but still observed a similar trend for the length (91.6 ± 3.6 Å for bacteria and 83.3 ± 2.9 Å for eukarya) and average radius (5.7 ± 0.3 Å for bacteria and 5.1 ± 0.1 Å for eukarya), suggesting that both contribute to the observed difference in volume.
Figure 2.

Volume, length and average radius of the ribosome exit tunnel across different species. Horizontal bar plots represent the ordered volume (left), length (middle) and average radius (right) of the tunnels from our dataset. The volume and length are decomposed into two subparts, separated by the constriction site (see ‘Materials and methods’ section). Species are specified and colored by their respective domains (plus organelles, in yellow): bacteria (red), archaea (black), eukarya (blue).
We then carried out a more refined analysis by partitioning the tunnel into two subparts separated by the ‘constriction site’ (see Figure 1), a conserved central region constricted by the uL4 and uL22 protein loops, which we located at position 34.1 ± 4.8 Å from the start of the tunnel (Figure 2). We studied how the volume in the upper (from start to constriction site) and lower (from constriction site to exit) parts respectively correlated with the volume of the entire tunnel, and found a strong correlation for the lower part (Pearson correlation r2 = 0.9649, P-value P < 10−4), while no significant correlation was observed for the upper part (r2 = 0.085), suggesting that the upper part of the tunnel is geometrically quite conserved, and most of the variation across species comes from the lower part. The total length also showed a better correlation with the lower part length (r2 = 0.72, P < 10−3, while r2 = 0.32, P < 10−3 for the upper part), and the average radius for the lower part (5.8 ± 0.6 Å) was larger than that of the upper part (4.7 ± 0.3 Å).
Hierarchical clustering of tunnel radius variation plots reflects the species phylogeny
As we found domain-specific variation of the tunnel geometry that is more amplified in the lower part of the tunnel, we aimed to quantify this pattern more precisely. We first examined the 3D coordinates of the points describing the tunnel centerline (see Supplementary Data). Upon fitting the points to a straight line, we found that 96.7 ± 1.0% of their variation could be explained by the fit. We therefore simply parametrized the centerline of the tunnel by its arc length and studied the associated radii (see ‘Materials and methods’ section), leading to a radius variation plot for each species (Figure 3A).
Figure 3.

Clustering of species obtained from pairwise comparison of the tunnel geometry. (A) For all our structures, we plot the tunnel radius as a function of the distance across the tunnel. These plots are used to compare the tunnel geometries. (B) Clustering obtained after applying our tunnel geometric distance metric to our dataset (for a definition of the metric and more details, see ‘Materials and methods’ section). The first main branch encompasses the bacterial ribosomes, highlighted in red, while the second contains eukarya (blue) and archaea (black) (scale bar: 0.2 Å). For the full clustering and phylogenetic trees obtained from 16S/18S rRNA sequences, see Supplementary Figure S2. (C) We divide for each couple of species their common domain after alignment in 4 quarters (see ‘Materials and methods’ section) and use the same metric to compute the distance in each of the subregions. The bar plots represent for each quarter the average and std of the geometric distance for subset of pairs made of 2 prokaryotes (red), 2 eukaryotes (blue), and 1 prokaryote and 1 eukaryote (violet).
To compare these plots, we introduced a distance function (see ‘Materials and methods’ section) and evaluated it for each pair of species. The resulting pairwise distance matrix was then used to cluster the species, yielding the hierarchical tree shown in Figure 3B. As in the global feature analysis, we obtained a clear separation of domains, with bacteria clustering separately from archaea and eukaryotes. Among eukaryotes, T. vaginalis is rather special in that it clustered with archaea (P. furiosus and H. marismortui), which then grouped together with a cluster of trypanosomes (L. donovani and T. cruzi); separated from these, the remaining eukaryotes formed a larger cluster.
The separations of T. vaginalis and trypanosomes from the other eukaryotes are also consistent with the evolutionary relationships obtained from 16S-like rRNA sequences (from the ribosome small subunit) (34). We confirmed this result by carrying out a phylogenetic analysis for the species in our dataset (see Supplementary Figure S2a). At a finer resolution, our hierarchical clustering from tunnel geometry comparison (see Supplementary Figure S2b) starts to deviate from the phylogenetic tree from sequence analysis, suggesting that smaller differences of the tunnel geometry are more difficult to relate evolutionarily. By including ribosomes from mitochondria and chloroplast (Supplementary Figure S2c), we also found that they clustered with prokaryotes, which is consistent with their bacterial origin (35). To assess the robustness of these results, we used replicate structures from same species and studied the sensitivity of the clustering to the parameters of the geometric distance (see Supplementary Figure S1 and Supplementary Table S1). Overall, this did not lead to significant changes, demonstrating the robustness of our results to replicate structures and the choice of the comparison metric.
Relative contributions of different tunnel subregions to the intra- and inter-domain variation in tunnel geometry
To understand why we obtained a clear separation between the prokaryotic and eukaryotic tunnels, we more closely studied the variation of tunnel geometry across the two domains of life. Distinguishing intra- and inter-domain pairs (respectively accounting for pairs of species from the same domain, and pairs with one from each), we found that while intra-domain distances are on average similar for bacteria and eukaryotes (Supplementary Figure S3), inter-domain distances are noticeably larger (with an increase of 40% for the average).
As the distance that we introduced integrates the geometric variation along the tunnel, we sought to examine which part of the tunnel contributes most significantly to the intra- and inter-domain distances. To this end, we divided up the tunnel for each pair of species into four quarters (the first and fourth respectively corresponding to the start and the exit parts) and applied the same metric as before to compute the geometric distance in each of these subregions (Figure 3C). While the inter-domain distance was on average always larger than the intra-domain one in every subregion, we found that across these regions, the inter-domain distance was substantially larger for the last two quarters (each carrying on average 30% of the total variation) compared to the first two (∼20% each). A similar trend was observed among eukaryotes (with 33% and 26% of the total variation respectively carried by the third and last quarters). In stark contrast, in bacteria the third quarter was the one carrying the least variation (21% compared to 24%, 26%, and 29% for the first, second and fourth subregions, respectively).
Existence of a second constriction site in eukaryotes and the role of ribosomal protein uL4
To understand the local geometric variation of the exit tunnel observed across different species, we sought to determine how the ribosomal structure can explain the aforementioned clustering pattern of bacterial and eukaryotic tunnels. In the region associated with most inter-domain variation (see Figure 3C), we first looked at the constriction site, where proteins uL4 and uL22 meet. Interestingly, we found that the structure of uL4 at the tunnel generally differs in eukarya and bacteria, due to an extension of the uL4 loop in eukarya that yields a second constriction site. Such an extension is also present but less prominent in archaea (see Supplementary Figure S4a). We illustrate this structural difference in Figure 4A for E. coli and H. sapiens. The presence of a second constriction site in H. sapiens exit tunnel is clearly illustrated in Figure 4B, which shows that the second trough of the radius plot is even lower than the first trough for H. sapiens, while the opposite is true for E. coli.
Figure 4.

The presence of a second constriction site in eukarya explains the geometric difference observed between bacterial and eukaryotic tunnels. (A) We show the constriction site region in E. coli (left) obtained from Fischer et al. (85), and H. Sapiens (right) obtained from Natchiar et al. (94). The structure is surrounded by ribosomal proteins uL4 and uL22. An extended arm in H. sapiens uL4 produces a second constriction site (see also Supplementary Figure S4). (B) The plots of the tunnel radius as a function of the tunnel distance shows a first trough associated with the constriction site (around position 30), common to E. coli and H. sapiens. A second trough appears around position 50. (C) We compare the distance between the troughs for bacteria (left box plot) and eukarya (right box plot) in our dataset. The interquartile range is indicated by the box, the median by a line inside, and upper and lower adjacent values by whiskers. (D) In left, we provide the same comparison as in (C) for the tunnel radii associated with the first and second troughs. In right, we compare the radius of the first and second troughs for each species of our dataset. The second trough radius is larger than the first one for all archaea (black dots) and bacteria (red dots). In contrast, this is only the case in eukarya for trypanosome species L. donovani and T. cruzi.
To confirm that such a difference persists between other bacteria and eukarya, we analyzed in Figure 4C and D the relative positions and the tunnel radii associated with the first and second troughs of the radius plot for each species in our entire dataset. We found the distance between the troughs to be on average larger in bacteria (23.6 ± 2.7 Å) than in eukarya (19.1 ± 2.3 Å), suggesting structural changes in the constriction site region. While at the first trough (which corresponds to the universally shared constriction site) bacteria and eukarya have similar radii (4.2 ± 0.6 Å for bacteria and 4.2 ± 0.4 Å for eukarya), the radius at the second trough is significantly larger in bacteria (5.0 ± 0.5 Å) than in eukarya (3.9 ± 0.6 Å). Furthermore, direct comparison between the first and second troughs for each species shows that the second trough radius is larger than the first one for all archaea and bacteria (Figure 4D). In contrast, we observed the opposite in eukarya, except for L. donovani and T. cruzi (also explaining the clustering of these two with archaea in Figure 3B). Therefore, the second constriction site in eukarya is in general narrower than the first one.
To explain the discrepancy observed for L. donovani and T. cruzi, we looked at the ribosomal structure in the constriction site region. Aside from ribosomal proteins uL4 and uL22, the tunnel is surrounded there by rRNA. In trypanosomes like L. donovani and T. cruzi, the large subunit (LSU) rRNA breaks from the standard 28S rRNA into six smaller chains (36,37). The two main chains precisely meet in the constriction site region (see Supplementary Figure S4b), suggesting that less constraint is locally applied to the ribosomal proteins and tunnel structure, potentially increasing the size of the second constriction site. To summarize, we concluded that an important part of the geometrical differences and clustering observed between the ribosome exit tunnels comes from the structure at the constriction site. More precisely, there exists a second constriction site specific to eukarya, which narrows the tunnel after the first, universally shared constriction site.
Replacement of uL23 by eL39 ribosomal protein in eukarya affects the tunnel geometry
In addition to the structure of the constriction site region, we further examined the structure of the lower part of the tunnel, where we also detected some important geometric variations. In bacteria, the tunnel in this region is mainly surrounded by rRNA and the protein uL23. In eukarya and archaea, uL23 is also present, but the segment covering the tunnel region is replaced by the protein eL39 (33,38,39). Upon close examination and comparison of these structures, we found that eL39 not only does cover the region originally occupied by uL23 in bacteria, but also extends to the tunnel exit, as illustrated in Figure 5A. More precisely (Figure 5B), the coverage distance of uL23 in bacterial ribosomes is 19.0 ± 2.8 Å, compared to 31.6 ± 2.3 Å for eL39 in eukarya. Such a difference affects the tunnel geometry (Figure 5B), as the tunnel radius in the corresponding regions is significantly larger for bacteria (6.0 ± 0.4 Å) than for eukarya (4.6 ± 0.4 Å ). As a result, the exit region of the tunnel is wider in bacteria than in eukarya, with an average radius difference of ∼1 Å in the last 30 Å of the tunnel, hence contributing to the clustering of the radius plots that we previously obtained.
Figure 5.

The replacement of uL23 by eL39 in eukarya affects the tunnel geometry. (A) The structures of the lower part of the tunnel in Escherichia coli (left) and Homo sapiens (right) show the replacement of ribosomal protein uL23 by eL39 in H. sapiens, which also covers a larger portion of the tunnel. (B) Upper plot shows a comparison of the distance covered by uL23 and eL39 in bacteria (right box plot) and eukarya (left box plot). Lower plot shows the same comparison for the average radius.
Association between rRNA sequence conservation and the exit tunnel
After finding evidence that the geometric variation of the tunnel across different species can be explained by rRNA and protein structural variations that have emerged through evolution, we sought to study how the tunnel and its geometry directly relate to the evolution of the ribosome at the sequence level. First, we investigated the conservation of rRNAs, focusing on the main chain (23S for bacteria, and 28S for archaea and eukarya) that constitutes the ribosome LSU. Comparative analysis of rRNA sequences has been extensively used in the past to elucidate phylogenetic relationships (40,41), and has recently (24) led to the identification of stretches of evolutionarily conserved sequences, the so-called ‘conserved nucleotide elements’ (CNE). In particular, it has been suggested that a large part of the CNEs may have a function in nascent polypeptide transit through the tunnel (24). To verify this, we compared the presence of CNEs with their distance to the tunnel (see Figure 6A and Supplementary Figure S5).
Figure 6.

Association between geometric and sequence conservations of ribosomal rRNA. (A) A map of the secondary structure of the 23S rRNA in E. coli, colored by the distance from tunnel (see also Supplementary Figure S5). (B) For a given species and distance d, we look at all the rRNA nucleotides located within distance d from the tunnel, and we compute the frequency of conserved elements (24). We plot this frequency as a function of d for all the species of our dataset (see also Supplementary Figure S6). (C) We study the local conservation of rRNA nucleotides along the tunnel: Upon dividing the tunnel into regions of 15 Å along the centerline, we consider for each region all the rRNA nucleotides that are the closest and located within 25 Å, and we compute the associated number of conserved, domain-specific and universally conserved elements. We show here the resulting plots for E. coli (up) and H. sapiens (down) (for other species, see Supplementary Figure S6).
Upon computing for each species the frequency of CNEs at a given distance d from the tunnel, we observed in Figure 6B a global decrease in the frequency of CNEs as d increases, which means that nucleotides located farther from the tunnel are less conserved. For much divergent rRNAs such as the ones from organelles or T. vaginalis (34), the low number of CNEs yields no association with the distance to the tunnel. For bacteria, archaea and eukaryotic trypanosomes, we found that the frequency of CNEs within 25 Å from the tunnel is 21 ± 5%, which is more than the double the frequency of CNEs located between 25 and 50 Å (10 ± 2%) and also that located farther than 50 Å from the tunnel (8 ± 2%). In the remaining eukaryotes, we found a much larger frequency of CNEs within 25 Å from the tunnel (45 ± 12%), with a sharp decrease for the region between 25 and 50 Å (18 ± 5%) and the region beyond 50 Å (10 ± 3%). Distinguishing universal and domain-specific CNEs (24), we found a similar trend for both, with a larger contribution from domain-specific CNEs (Supplementary Figure S6a). Overall, these results confirm the association between the tunnel and the conservation of surrounding rRNA nucleotides.
To determine whether such conservation is homogeneous or specific to some local parts of the tunnel, we computed for each species the local frequency of CNEs and compared the level of conservation across the tunnel, as shown in Figure 6C and Supplementary Figure S6b. Overall, we found that the sequence conservation along the tunnel is heterogeneous and strongly enhanced toward the upper part of the tunnel, as more than 80% of the CNEs were associated with the first 40 Å of the tunnel (while all the nucleotides positioned along the first 40 Å of the tunnel represent only 48% of all nucleotides). In this region, we also observed a stronger conservation closer to the PTC, as 43% of the CNEs were located <10 Å from the tunnel start position. These results are in agreement with the comparative geometric study of the tunnels, and consistent with the results obtained by Mears et al. (22) (who used a similar approach, but with different ribosome structures and measure of conservation), also showing more conservation in the upper part region.
Conservation of ribosomal protein sequence and positive charges at the exit tunnel
At last, we studied the relation between the geometric and sequence conservations at the tunnel for ribosomal proteins. To do so, we aligned across species the main proteins located close to the tunnel, namely uL4, uL22 and uL23 for bacteria and eL39 for eukarya (see ‘Materials and methods’ section, Figure 7A and Supplementary Figure S7a). For uL4, because of multiple insertions that prevent a good alignment of all sequences (like the one leading to the aforementioned second constriction site), we separately aligned uL4 for bacterial and eukaryotic sequences. Upon computing a conservation score (30) for each alignment, we found peaks of conservation in regions located in proximity with the ribosomal tunnel, suggesting that association between the sequence conservation and the exit tunnel also occurs for proteins (Figure 7B and Supplementary Figure S7b). Upon closer examination of the consensus motif sequences at the tunnel, we found a large fraction (∼30%) of positively charged amino acids (arginine R and lysine K). Interestingly, these positively charged amino acids sometimes co-occur at the same position of an alignment, e.g. position 159 of the uL22 alignment (with 8 arginines and 8 lysines, see Figure 7A), position 69 of bacterial uL4 alignment and position 34 of eL39 alignment (Supplementary Figure S7a). This suggests that not only the sequence but also the charge properties are important to maintain the integrity of the tunnel.
Figure 7.

Conservation of sequence and positive charge of ribosomal protein uL22 at the tunnel. (A) We show the multiple sequence alignment of ribosomal protein uL22 close to the tunnel. Highlighted residues are the ones located within 10 Å from the tunnel. (B) We plot the sequence and charge conservation scores (see ‘Materials and methods’ section) along the sequence alignment. Continuous lines represent the signal averaged over a window of five sites. Larger highlighted region is the same as in (A). We also highlight a subregion of residues close to the tunnel, with a peak in charge or sequence conservation. (C) The associated structure of uL22 in H. sapiens, where residues in green and red correspond to the ones highlighted in (B). In particular, the region of high charge and sequence conservation is also in direct contact with the constriction sites. For the other ribosomal proteins associated with the tunnel, see Supplementary Figure S7.
Therefore, we introduced a measure of positive charge conservation (see ‘Materials and methods’ section) that specifically accounts for the local presence of positively charged amino acids in an alignment. Upon computing the charge score for our sequence alignments, we indeed found some significant correlation between the sequence conservation and charge conservation scores for bacterial proteins with Pearson’s correlation r2 = 0.70, 0.51 and 0.47 (P-values < 10−4) for bacterial alignments of uL4, uL23 and uL22, respectively (see Supplementary Data). We also found that overall, regions with the highest presence of positive charges are located in the tunnel region (see Figure 7B and Supplementary Figure S7b). In uL22, the residues in direct contact with the tunnel (Figure 7B and C) are highly conserved, with the highest charge conservation score. Larger charge and sequence conservations were also found in uL4, with more conservation at the first constriction site compared to the second one in eukarya (Supplementary Figure S7b). In the lower part of the tunnel, we detected a strong peak of both charge and sequence conservation in bacterial uL23. Interestingly, while we did not observe in eukarya a conservation signal as strong in the whole region of the tunnel covered by eL39, the region where eL39 overlaps with uL23 in bacteria also exhibits large charge and sequence conservation (Supplementary Figure S7b and c), suggesting persistence of the local charge properties in this region although the ribosome structure differs between bacteria and eukarya. To assess the global impact of charged residues in ribosomal proteins, we computed their induced electrostatic Coulomb potential along with the tunnel centerline (Supplementary Figure S8). We found that the concentration of positively charged residues yielded a peak in the electrostatic potential near the second trough of the tunnel radius plot (Figure 4). This peak was more pronounced in eukaryotes, due to the presence of additional charges in the extended arm of uL4 (shown in Figure 4A). Overall, we concluded that ribosomal proteins are more locally conserved at the proximity of the tunnel, with a bias toward positively charged amino acids, suggesting that these amino acids and the electrostatic environment are important for the tunnel function (see ‘Discussion’ section).
DISCUSSION
In this study, we carried out a comparative analysis of the ribosome exit tunnel geometry that is most comprehensive to date. Recent advances in cryo-EM (42,43) allowed us to observe the ribosome at a resolution fine enough to detect significant differences across species. While the eukaryotic 80S ribosome is ∼50% greater in mass and rRNA lengths compared to the prokaryotic 70S ribosome (23,44), we found the bacterial exit tunnel to be larger (Figure 2), in both length and average radius. Such differences mostly come from the lower part of the tunnel. We classified the tunnels using their radius variation plots and interestingly found that the resulting hierarchical clustering closely reflects the species phylogeny (Figure 3). We then found evidence of two notable evolutionary changes that affect the tunnel structure and explain our clustering result. The first is the insertion of an extended loop sequence in eukaryotic ribosomal protein uL4 (Figure 4), which leads to a second constriction site that is narrower than the first one (except in trypanosomes). The second is the replacement of the uL23 loop in the lower part of the tunnel in bacteria by the small protein eL39 in archaea and eukarya (Figure 5). While the existence of the first constriction site and the overlapping positions of eukaryotic eL39 and prokaryotic uL23 proteins have been previously established (33,38,45), our comparative analysis shows that these evolutionary changes have led to more important modifications that significantly affect the tunnel geometry and hence the path of the nascent chain through the tunnel.
Comparison with other methods of tunnel extraction
To extract the geometry of the tunnel, we used an algorithm developed by Sehnal et al. (26,46), which utilizes Voronoi diagrams to search for the shortest path from a starting point to the molecular outer surface. Over the past years, several other computational methods have been developed to detect empty spaces in biomacromolecules (for a recent survey, see Krone et al. (47)), and have been applied to the ribosome exit tunnel (48–51). While some of these methods may provide a more detailed visualization of the tunnel, they are less adapted for comparative analysis. To assess the robustness of our results, we developed an alternative method to extract and quantify the tunnel volume for a more detailed representation of the exit tunnel (methods are detailed in the Supplementary Data). We found a high correlation with the volume that we originally computed (see Supplementary Figure S9), yielding no changes in the order obtained in Figure 2. While the results presented in this study are thus robust with respect to the computational method used, it would be interesting to study and interpret finer variations of the tunnel by developing improved tools that can extract and integrate more detailed geometric and structural information from the exit tunnel.
Implications on the evolution of the ribosome
Since both ribosomal RNA and protein sequences located near the exit tunnel are more conserved, there is a strong relationship between the evolution of ribosome components and the variation in the geometry of the exit tunnel (24). The high geometric conservation of the upper part of the tunnel coincides with the high sequence conservation of the associated rRNAs and ribosomal proteins, suggesting that it is an important part of the ribosome core. The observed contrast in conservation between the upper and lower parts of the tunnel is in agreement with previously proposed models of the evolution of the rRNA structure, suggesting that the exit tunnel continuously got more elaborate and extended in nearly all phases of ribosomal evolution (23). More precisely, the model by Petrov et al. distinguishes an early phase of creation of a short tunnel, well conserved from prokaryotes to eukaryotes, which was subsequently completed by a phase of tunnel extension and expansion of the LSU. These two phases are also preceded by the maturation of the PTC, commonly considered as the oldest ribozyme (23,52,53) and which also appeared in our analysis as the most universally conserved region (24,54). Complementing Petrov et al.’s study, we here provided further details on the involvement of specific ribosomal proteins during the later phase of ribosome evolution that led to distinct modes of translation.
While there is a clear association between the geometry of the exit tunnel and the evolution of its components, some other biophysical properties of the tunnel may also be influencing the evolution of the ribosome. We indeed found a significant contribution of positively charged amino acids (see Figure 7 and Supplementary Figure S7). While it has been proposed that this general enrichment may be needed to create electrostatic interaction during ribosome assembly (55,56), our analysis suggests that near the tunnel, it could be tied to maintain an appropriate electrostatic environment inside the tunnel (see below).
Impact on the nascent polypeptide chain transit
The geometric and structural variation of the exit tunnel may have several functional and biophysical implications, as the nascent chain transit can be affected. On the one hand, a too narrow tunnel might alter the elongation rate during protein synthesis (9–11). On the other hand, a too large tunnel radius could lead to an increase of conformational sampling (12–14) and hence misfolding events. Our results indicate that before reaching the constriction site in the upper tunnel, the nascent chain propagates in an environment that seems relatively well conserved, with an increase of the tunnel radius followed by a decrease near the constriction site. We previously found (8) that this radial increase creates a strong ‘entropic’ barrier for a diffusing particle inside the tunnel, which can be compensated by the electrostatic potential if the particle is positively charged. More generally, it was also experimentally shown that electrostatics in the ribosomal tunnel modulate chain elongation rates (8,57) and can even induce ribosome stalling (58).
Once the N-terminus exits from the tunnel, the elongation of the nascent chain may be affected by additional factors, such as the co-translational processes that generate pulling forces (59). For example, the translocon, involved in the translocation of membrane proteins, can relieve elongation arrest due to the SecM sequence, thereby enabling translation restart (60). In eukaryotes, a similar mechanism has been suggested for the chaperone Hsp70 (61). More generally, proteins that start to fold co-translationally while still in contact with the ribosome, exert some pulling force sufficient to weaken or even abolish stalling (62,63). As the confined geometry of the tunnel can actually play a role in stabilizing α-helices (64), thereby improving the nascent chain stiffness (59)), it would be interesting to test whether the second narrower constriction site in eukaryotes facilitates the pulling and elongation process.
Functional implications of the tunnel variation
We detected more variation in the lower part of the tunnel, and showed that the structural modifications associated with ribosomal proteins eL39 and uL23 led to a decrease in the eukaryotic tunnel size. These modifications can explain some of the previously observed differences in translation modes. While past studies have shown that the tunnel can be large enough to accommodate a substantial degree of structure that affects co-translational protein folding (in the so-called ‘folding vestibule’ located near the exit port) (65–70), our results suggest that eukaryotic tunnels are less favorable to such folding inside the ribosome.
Such a difference could actually reflect the differences in complexity and division of labor between the eukaryotic and prokaryotic chaperone networks (18,65). In prokaryotes, both co-translational folding and denaturation of proteins during stress are ensured by an overlapping set of chaperones that primarily relies on the bacterial trigger factor (71). In contrast, distinct specialized networks of downstream chaperones evolved in eukaryotes to separately carry out these processes (72). While the prokaryotic tunnel can assist the chaperone network by pre-folding the nascent chain inside the tunnel (73), the Prefoldin family of proteins (74) plays such a role instead in eukarya and archaea. Another direct implication of the replacement of uL23 by eL39 has been observed for the translocation of membrane proteins, which is mediated in both eukarya and bacteria by the Signal Recognition Particle (SRP), a ribonucleoprotein that recognizes the signal peptide emerging from the ribosome (7). In bacteria, the SRP can be recruited prior to the emergence of the peptide from the tunnel (75), via uL23 that both recognizes the nascent sequence inside the tunnel and interacts with the SRP at the outer surface of the ribosome. In eukaryotes, however, forward signaling does not occur and it has been shown that eukaryote-specific signaling mechanisms, involving the structure of eL39 at the exit port, can compensate for the lacking function that the tunnel can play in promoting the SRP binding (76).
While the reduction of the tunnel size in eukarya seems to reflect the externalization of major co-translational functions, the increase of tunnel confinement may also provide some other advantages. Besides the aforementioned facilitation of nascent chain elongation, the reduction of the tunnel radius at the exit port by eL39 and the addition of a second constriction site also contribute to restricting the access to the tunnel and the PTC from external threats. For example, as the insertion of amino acids in the loop of uL4 in E. coli confers resistance to larger size macrolides (77,78), it has been suggested that the narrower size of the constriction site in eukarya can block the access of these antibiotics to the targeted PTC; we actually found a second eukaryote-specific constriction site, located below the universal constriction site, to be responsible for this narrower access. Similarly, mutants lacking eL39 are more vulnerable against ribosome targeting antibiotics (79), in addition to an increased translation error rate and cold sensitive phenotype. Molecular dynamic simulations have predicted interaction between the 28S rRNA tetraloop and eL39, potentially leading to even more obstruction of the tunnel, suggesting that eL39 acts as an energy barrier to ensure protein quality control and protect the ribosome from deleterious external agents (39).
Future directions
While our study highlights important variations of the exit tunnel, a better understanding of the dynamics of the nascent polypeptide chain inside the tunnel is needed to assess the impact of these variations on translation. Recent long-time-scale, coarse-grained simulations of the nascent chain have modeled the tunnel as a simple cylinder (80). It would be interesting to include in such a study the geometric variation induced by constricted regions, which differ between prokaryotes and eukaryotes. Furthermore, since our work shows how positively charged residues are maintained and concentrated in these regions, it would be interesting to study the impact of the electrostatic potential for different configurations of charged polypeptides inside the tunnel. We note that computing the electrostatic potential inside the tunnel is challenging, however, because of the complexity of the structure, the spatial resolution to achieve, and the heterogeneous solvent properties inside the tunnel (81).
Based on the present study and the aforementioned involvement of the tunnel structure in various co-translational processes, it would also be interesting to thoroughly investigate the inter-domain differences in the chaperone machinery engaging the ribosome exit tunnel, especially near the exit port. For example, the class of chaperones involved in guiding de novo protein folding includes multiple domain-specific actors (71), the diversity of which may be the result of different strategies developed in conjunction with the tunnel structure. Elucidating the evolutionary basis of these processes might also require the development of more refined tools to extract and analyze the tunnel geometry and its properties. In particular, it would be interesting to build a stochastic model describing the evolution of the tunnel geometry, to decipher the larger variation detected in bacteria and to determine if it is mainly due to the difference in evolutionary time scale or whether natural selection played a role. More generally, the methods presented here could be extended and applied to more structures in the future, and as a result provide useful insights into many of the essential biological processes in which the ribosome takes part.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [R01-GM065050, R01-GM094402]; Packard Fellowship; Chan Zuckerberg Biohub (to Y.S.S.). Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Nyathi Y., Wilkinson B.M., Pool M.R.. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim. Biophys. Acta. 2013; 1833:2392–2402. [DOI] [PubMed] [Google Scholar]
- 2. Thommen M., Holtkamp W., Rodnina M.V.. Co-translational protein folding: progress and methods. Curr. Opin. Struct. Biol. 2017; 42:83–89. [DOI] [PubMed] [Google Scholar]
- 3. Rodnina M.V., Wintermeyer W.. Protein elongation, co-translational folding and targeting. J. Mol. Biol. 2016; 428:2165–2185. [DOI] [PubMed] [Google Scholar]
- 4. Vazquez-Laslop N., Thum C., Mankin A.S.. Molecular mechanism of drug-dependent ribosome stalling. Mol. Cell. 2008; 30:190–202. [DOI] [PubMed] [Google Scholar]
- 5. Davis A.R., Gohara D.W., Yap M.-N.F.. Sequence selectivity of macrolide-induced translational attenuation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:15379–15384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tenson T., Ehrenberg M.. Regulatory nascent peptides in the ribosomal tunnel. Cell. 2002; 108:591–594. [DOI] [PubMed] [Google Scholar]
- 7. Ito K. Regulatory Nascent Polypeptides. 2014; Tokyo: Springer. [Google Scholar]
- 8. Dao Duc K., Song Y.S.. The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation. PLoS Genet. 2018; 14:e1007166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choi J., Grosely R., Prabhakar A., Lapointe C., Wang J., Puglisi J.. How messenger RNA and nascent chain sequences regulate translation elongation. Ann. Rev. Biochem. 2018; 87:421–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilson D.N., Beckmann R.. The ribosomal tunnel as a functional environment for nascent polypeptide folding and translational stalling. Curr. Opin. Struct. Biol. 2011; 21:274–282. [DOI] [PubMed] [Google Scholar]
- 11. Kannan K., Vázquez-Laslop N., Mankin A.S.. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 2012; 151:508–520. [DOI] [PubMed] [Google Scholar]
- 12. Cabrita L.D., Dobson C.M., Christodoulou J.. Protein folding on the ribosome. Curr. Opin. Struct. Biol. 2010; 20:33–45. [DOI] [PubMed] [Google Scholar]
- 13. Fedyukina D.V., Cavagnero S.. Protein folding at the exit tunnel. Ann. Rev. Biophys. 2011; 40:337–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen C., Wang E., Liu P., Xiao Y.. Simulation study of the role of the ribosomal exit tunnel on protein folding. Phys. Rev.E. 2013; 87:022701. [DOI] [PubMed] [Google Scholar]
- 15. Kisselev L. Polypeptide release factors in prokaryotes and eukaryotes: same function, different structure. Structure. 2002; 10:8–9. [DOI] [PubMed] [Google Scholar]
- 16. Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999; 234:187–208. [DOI] [PubMed] [Google Scholar]
- 17. Ryazanov A., Ovchinnikov L., Spirin A.. Development of structural organization of protein-synthesizing machinery from prokaryotes to eukaryotes. Biosystems. 1987; 20:275–288. [DOI] [PubMed] [Google Scholar]
- 18. Pechmann S., Willmund F., Frydman J.. The ribosome as a hub for protein quality control. Mol. Cell. 2013; 49:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amunts A., Brown A., Bai X.-c., Llácer J.L., Hussain T., Emsley P., Long F., Murshudov G., Scheres S.H., Ramakrishnan V.. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014; 343:1485–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kannan K., Mankin A.S.. Macrolide antibiotics in the ribosome exit tunnel: species-specific binding and action. Ann. N.Y. Acad. Sci. 2011; 1241:33–47. [DOI] [PubMed] [Google Scholar]
- 21. Olsen G.J., Woese C.R.. Ribosomal RNA: a key to phylogeny. FASEB J. 1993; 7:113–123. [DOI] [PubMed] [Google Scholar]
- 22. Mears J.A., Cannone J.J., Stagg S.M., Gutell R.R., Agrawal R.K., Harvey S.C.. Modeling a minimal ribosome based on comparative sequence analysis. J. Mol. Biol. 2002; 321:215–234. [DOI] [PubMed] [Google Scholar]
- 23. Petrov A.S., Bernier C.R., Hsiao C., Norris A.M., Kovacs N.A., Waterbury C.C., Stepanov V.G., Harvey S.C., Fox G.E., Wartell R.M. et al.. Evolution of the ribosome at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:10251–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doris S.M., Smith D.R., Beamesderfer J.N., Raphael B.J., Nathanson J.A., Gerbi S.A.. Universal and domain-specific sequences in 23S–28S ribosomal RNA identified by computational phylogenetics. RNA. 2015; 21:1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of coot. Acta Crystallogr. Sec. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sehnal D., Vařeková R.S., Berka K., Pravda L., Navrátilová V., Banáš P., Ionescu C.-M., Otyepka M., Koča J.. MOLE 2.0: advanced approach for analysis of biomacromolecular channels. J. Cheminform. 2013; 5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ban N., Beckmann R., Cate J.H., Dinman J.D., Dragon F., Ellis S.R., Lafontaine D.L., Lindahl L., Liljas A., Lipton J.M. et al.. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014; 24:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katoh K., Standley D.M.. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J.. Jalview Version 2 multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009; 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Capra J.A., Singh M.. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007; 23:1875–1882. [DOI] [PubMed] [Google Scholar]
- 31. Bernier C.R., Petrov A.S., Waterbury C.C., Jett J., Li F., Freil L.E., Xiong X., Wang L., Migliozzi B.L., Hershkovits E. et al.. RiboVision suite for visualization and analysis of ribosomes. Faraday Discuss. 2014; 169:195–207. [DOI] [PubMed] [Google Scholar]
- 32. Pravda L., Sehnal D. Svobodová, Vařeková R., Navrátilová V., Toušek D., Berka K., Otyepka M., Koča J.. ChannelsDB: database of biomacromolecular tunnels and pores. Nucleic Acids Res. 2017; 46:D399–D405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilson D.N., Cate J.H.D.. The structure and function of the eukaryotic ribosome. Cold Spring Harbor Perspect. Biol. 2012; 4:a011536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Z., Guo Q., Zheng L., Ji Y., Xie Y.-T., Lai D.-H., Lun Z.-R., Suo X., Gao N.. Cryo-EM structures of the 80S ribosomes from human parasites Trichomonas vaginalis and Toxoplasma gondii. Cell Res. 2017; 27:1275–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gray M.W., Burger G., Lang B.F.. The origin and early evolution of mitochondria. Genome Biol. 2001; 2:reviews1018.1–reviews1018.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. White T.C., Rudenko G., Borst P.. Three small RNAs within the 10 kb trypanosome rRNA transcription unit are analogous to domain VII of other eukaryotic 28S rRNAs. Nucleic Acids Res. 1986; 14:9471–9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao H., Ayub M.J., Levin M.J., Frank J.. The structure of the 80S ribosome from Trypanosoma cruzi reveals unique rRNA components. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:10206–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harms J., Schluenzen F., Zarivach R., Bashan A., Gat S., Agmon I., Bartels H., Franceschi F., Yonath A.. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001; 107:679–688. [DOI] [PubMed] [Google Scholar]
- 39. Petrone P.M., Snow C.D., Lucent D., Pande V.S.. Side-chain recognition and gating in the ribosome exit tunnel. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:16549–16554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yilmaz P., Parfrey L.W., Yarza P., Gerken J., Pruesse E., Quast C., Schweer T., Peplies J., Ludwig W., Glöckner F.O.. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2013; 42:D643–D648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yarza P., Ludwig W., Euzéby J., Amann R., Schleifer K.-H., Glöckner F.O., Rosselló-Móra R.. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analyses. Syst. Appl. Microbiol. 2010; 33:291–299. [DOI] [PubMed] [Google Scholar]
- 42. Frank J. Advances in the field of single-particle cryo-electron microscopy over the last decade. Nat. Protoc. 2017; 12:209–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Razi A., Britton R.A., Ortega J.. The impact of recent improvements in cryo-electron microscopy technology on the understanding of bacterial ribosome assembly. Nucleic Acids Res. 2016; 45:1027–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berg J.M., Tymoczko J.L., Stryer L.. Freeman WH. Biochemistry. 2002; NY. [Google Scholar]
- 45. Nissen P., Hansen J., Ban N., Moore P.B., Steitz T.A.. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000; 289:920–930. [DOI] [PubMed] [Google Scholar]
- 46. Pravda L., Sehnal D., Berka K., Navrátilová V., Toušěk D., Bazgier V., Vařeková R.S., Otyepka M., Koča J.. Channelsdb and moleonline-database and tool for analysis of biomacromolecular tunnels and pores. Biophys. J. 2018; 114:342a–343a. [Google Scholar]
- 47. Krone M., Kozlíková B., Lindow N., Baaden M., Baum D., Parulek J., Hege H.-C., Viola I.. Visual analysis of biomolecular cavities: state of the art. Computer Graphics Forum. 2016; 35:527–551. [Google Scholar]
- 48. Voss N.R., Gerstein M.. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010; 38:W555–W562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coleman R.G., Sharp K.A.. Finding and characterizing tunnels in macromolecules with application to ion channels and pores. Biophys. J. 2009; 96:632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ho B.K., Gruswitz F.. HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 2008; 8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chovancova E., Pavelka A., Benes P., Strnad O., Brezovsky J., Kozlikova B., Gora A., Sustr V., Klvana M., Medek P. et al.. CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012; 8:e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fox G.E. Origin and evolution of the ribosome. Cold Spring Harbor Perspect. Biol. 2010; 2:a003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Krupkin M., Matzov D., Tang H., Metz M., Kalaora R., Belousoff M.J., Zimmerman E., Bashan A., Yonath A.. A vestige of a prebiotic bonding machine is functioning within the contemporary ribosome. Philos. Trans. R. Soc. B: Biol. Sci. 2011; 366:2972–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Agmon I., Bashan A., Yonath A.. On ribosome conservation and evolution. Israel J. Ecol. Evol. 2006; 52:359–374. [Google Scholar]
- 55. Wyant G.A., Abu-Remaileh M., Frenkel E.M., Laqtom N.N., Dharamdasani V., Lewis C.A., Chan S.H., Heinze I., Ori A., Sabatini D.M.. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science. 2018; 360:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lott B.B., Wang Y., Nakazato T.. A comparative study of ribosomal proteins: linkage between amino acid distribution and ribosomal assembly. BMC Biophys. 2013; 6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lu J., Deutsch C.. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J. Mol. Biol. 2008; 384:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Javed A., Christodoulou J., Cabrita L.D., Orlova E.V.. The ribosome and its role in protein folding: looking through a magnifying glass. Acta Crystallogr. Sect. D: Struct. Biol. 2017; 73:509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ismail N., Hedman R., Schiller N. Von, Heijne G.. A biphasic pulling force acts on transmembrane helices during translocon-mediated membrane integration. Nat. Struct. Mol. Biol. 2012; 19:1018–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Butkus M.E., Prundeanu L.B., Oliver D.B.. Translocon “pulling” of nascent SecM controls the duration of its translational pause and secretion-responsive secA regulation. J. Bacteriol. 2003; 185:6719–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu B., Han Y., Qian S.-B.. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol. Cell. 2013; 49:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goldman D.H., Kaiser C.M., Milin A., Righini M., Tinoco I., Bustamante C.. Mechanical force releases nascent chain–mediated ribosome arrest in vitro and in vivo. Science. 2015; 348:457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fritch B., Kosolapov A., Hudson P., Nissley D.A., Woodcock H.L., Deutsch C., O’Brien E.P.. Origins of the mechanochemical coupling of peptide bond formation to protein synthesis. J. Am. Chem. Soc. 2018; 140:5077–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ziv G., Haran G., Thirumalai D.. Ribosome exit tunnel can entropically stabilize α-helices. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:18956–18961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kramer G., Boehringer D., Ban N., Bukau B.. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol. 2009; 16:589–597. [DOI] [PubMed] [Google Scholar]
- 66. Lu J., Deutsch C.. Folding zones inside the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 2005; 12:1123–1129. [DOI] [PubMed] [Google Scholar]
- 67. O’Brien E.P., Christodoulou J., Vendruscolo M., Dobson C.M.. New scenarios of protein folding can occur on the ribosome. J. Am. Chem. Soc. 2011; 133:513–526. [DOI] [PubMed] [Google Scholar]
- 68. Nilsson O.B., Hedman R., Marino J., Wickles S., Bischoff L., Johansson M., Müller-Lucks A., Trovato F., Puglisi J.D., O’Brien E.P. et al.. Cotranslational protein folding inside the ribosome exit tunnel. Cell Rep. 2015; 12:1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marino J., Heijne G., Beckmann R.. Small protein domains fold inside the ribosome exit tunnel. FEBS Lett. 2016; 590:655–660. [DOI] [PubMed] [Google Scholar]
- 70. Kudva R., Tian P., Pardo-Avila F., Carroni M., Best R.B., Bernstein H.D., von Heijne G.. The shape of the bacterial ribosome exit tunnel affects cotranslational protein folding. eLife. 2018; 7:e36326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Preissler S., Deuerling E.. Ribosome-associated chaperones as key players in proteostasis. Trends Biochem. Sci. 2012; 37:274–283. [DOI] [PubMed] [Google Scholar]
- 72. Albanese V., Yam A. Y.-W., Baughman J., Parnot C., Frydman J.. Systems analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell. 2006; 124:75–88. [DOI] [PubMed] [Google Scholar]
- 73. Kudlicki W., Coffman A., Kramer G., Hardesty B.. Ribosomes and ribosomal RNA as chaperones for folding of proteins. Fold. Design. 1997; 2:101–108. [DOI] [PubMed] [Google Scholar]
- 74. Vainberg I.E., Lewis S.A., Rommelaere H., Ampe C., Vandekerckhove J., Klein H.L., Cowan N.J.. Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell. 1998; 93:863–873. [DOI] [PubMed] [Google Scholar]
- 75. Bornemann T., Jöckel J., Rodnina M.V., Wintermeyer W.. Signal sequence–independent membrane targeting of ribosomes containing short nascent peptides within the exit tunnel. Nat. Struct. Mol. Biol. 2008; 15:494–499. [DOI] [PubMed] [Google Scholar]
- 76. Berndt U., Oellerer S., Zhang Y., Johnson A.E., Rospert S.. A signal-anchor sequence stimulates signal recognition particle binding to ribosomes from inside the exit tunnel. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zaman S., Fitzpatrick M., Lindahl L., Zengel J.. Novel mutations in ribosomal proteins L4 and L22 that confer erythromycin resistance in Escherichia coli. Mol. Microbiol. 2007; 66:1039–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tu D., Blaha G., Moore P.B., Steitz T.A.. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell. 2005; 121:257–270. [DOI] [PubMed] [Google Scholar]
- 79. Dresios J., Derkatch I.L., Liebman S.W., Synetos D.. Yeast ribosomal protein L24 affects the kinetics of protein synthesis and ribosomal protein L39 improves translational accuracy, while mutants lacking both remain viable. Biochemistry. 2000; 39:7236–7244. [DOI] [PubMed] [Google Scholar]
- 80. Bui P.T., Hoang T.X.. Protein escape at the ribosomal exit tunnel: Effects of native interactions, tunnel length, and macromolecular crowding. Journal Chem. Phys. 2018; 149:045102. [DOI] [PubMed] [Google Scholar]
- 81. Lucent D., Snow C.D., Aitken C.E., Pande V.S.. Non-bulk-like solvent behavior in the ribosome exit tunnel. PLoS Comput. Biol. 2010; 6:e1000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ahmed T., Shi J., Bhushan S.. Unique localization of the plastid-specific ribosomal proteins in the chloroplast ribosome small subunit provides mechanistic insights into the chloroplastic translation. Nucleic Acids Res. 2017; 45:8581–8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Amunts A., Brown A., Toots J., Scheres S.H., Ramakrishnan V.. The structure of the human mitochondrial ribosome. Science. 2015; 348:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Beckert B., Abdelshahid M., Schäfer H., Steinchen W., Arenz S., Berninghausen O., Beckmann R., Bange G., Turgay K., Wilson D.N.. Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. EMBO J. 2017; 36:2061–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fischer N., Neumann P., Konevega A.L., Bock L.V., Ficner R., Rodnina M.V., Stark H.. Structure of the E. coli ribosome–EF-Tu complex at< 3 Å resolution by C s-corrected cryo-EM. Nature. 2015; 520:567–570. [DOI] [PubMed] [Google Scholar]
- 86. Krupkin M., Wekselman I., Matzov D., Eyal Z., Posner Y.D., Rozenberg H., Zimmerman E., Bashan A., Yonath A.. Avilamycin and evernimicin induce structural changes in rProteins uL16 and CTC that enhance the inhibition of A-site tRNA binding. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E6796–E6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Franken L.E., Oostergetel G.T., Pijning T., Puri P., Arkhipova V., Boekema E.J., Poolman B., Guskov A.. A general mechanism of ribosome dimerization revealed by single-particle cryo-electron microscopy. Nat. Commun. 2017; 8:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hentschel J., Burnside C., Mignot I., Leibundgut M., Boehringer D., Ban N.. The complete structure of the mycobacterium smegmatis 70S ribosome. Cell Rep. 2017; 20:149–160. [DOI] [PubMed] [Google Scholar]
- 89. Yang K., Chang J.-Y., Cui Z., Li X., Meng R., Duan L., Thongchol J., Jakana J., Huwe C.M., Sacchettini J.C. et al.. Structural insights into species-specific features of the ribosome from the human pathogen Mycobacterium tuberculosis. Nucleic Acids Res. 2017; 45:10884–10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Matzov D., Eyal Z., Benhamou R.I., Shalev-Benami M., Halfon Y., Krupkin M., Zimmerman E., Rozenberg H., Bashan A., Fridman M. et al.. Structural insights of lincosamides targeting the ribosome of Staphylococcus aureus. Nucleic Acids Res. 2017; 45:10284–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Polikanov Y.S., Melnikov S.V., Söll D., Steitz T.A.. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol. 2015; 22:342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gabdulkhakov A., Nikonov S., Garber M.. Revisiting the Haloarcula marismortui 50S ribosomal subunit model. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013; 69:997–1004. [DOI] [PubMed] [Google Scholar]
- 93. Armache J.-P., Anger A.M., Márquez V., Franckenberg S., Fröhlich T., Villa E., Berninghausen O., Thomm M., Arnold G.J., Beckmann R. et al.. Promiscuous behaviour of archaeal ribosomal proteins: implications for eukaryotic ribosome evolution. Nucleic Acids Res. 2012; 41:1284–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Natchiar S.K., Myasnikov A.G., Kratzat H., Hazemann I., Klaholz B.P.. Visualization of chemical modifications in the human 80S ribosome structure. Nature. 2017; 551:472–477. [DOI] [PubMed] [Google Scholar]
- 95. Zhang X., Lai M., Chang W., Yu I., Ding K., Mrazek J., Ng H.L., Yang O.O., Maslov D.A., Zhou Z.H.. Structures and stabilization of kinetoplastid-specific split rRNAs revealed by comparing leishmanial and human ribosomes. Nat. Commun. 2016; 7:13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wong W., Bai X.-c., Brown A., Fernandez I.S., Hanssen E., Condron M., Tan Y.H., Baum J., Scheres S.H.. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. Elife. 2014; 3:e03080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schmidt C., Becker T., Heuer A., Braunger K., Shanmuganathan V., Pech M., Berninghausen O., Wilson D.N., Beckmann R.. Structure of the hypusinylated eukaryotic translation factor eIF-5A bound to the ribosome. Nucleic Acids Res. 2016; 44:1944–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gogala M., Becker T., Beatrix B., Armache J.-P., Barrio-Garcia C., Berninghausen O., Beckmann R.. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature. 2014; 506:107–110. [DOI] [PubMed] [Google Scholar]
- 99. Liu Z., Gutierrez-Vargas C., Wei J., Grassucci R.A., Ramesh M., Espina N., Sun M., Tutuncuoglu B., Madison-Antenucci S., Woolford J.L. et al.. Structure and assembly model for the Trypanosoma cruzi 60S ribosomal subunit. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12174–12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Arenz S., Meydan S., Starosta A.L., Berninghausen O., Beckmann R., Vázquez-Laslop N., Wilson D.N.. Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol. Cell. 2014; 56:446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Osterman I.A., Khabibullina N.F., Komarova E.S., Kasatsky P., Kartsev V.G., Bogdanov A.A., Dontsova O.A., Konevega A.L., Sergiev P.V., Polikanov Y.S.. Madumycin II inhibits peptide bond formation by forcing the peptidyl transferase center into an inactive state. Nucleic Acids Res. 2017; 45:7507–7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Khatter H., Myasnikov A.G., Natchiar S.K., Klaholz B.P.. Structure of the human 80S ribosome. Nature. 2015; 520:640–645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The tunnel coordinates, summary statistics, pairwise distance matrix, and ribosomal RNA and protein conservation scores are provided in Supplementary Data. The tunnels can be visualized and downloaded from the ChannelsDB online server (32).