

Abstract
Aims: Quantitative imaging of glutathione (GSH) with high spatial and temporal resolution is essential for studying the roles of GSH in redox biology. To study the long-standing question of compartmentalization of GSH, especially its distribution between the nucleus and cytosol, an organelle-targeted quantitative probe is needed.
Results: We developed a reversible reaction-based ratiometric fluorescent probe—HaloRT—that can quantitatively measure GSH dynamics with subcellular resolution in real time. Using HaloRT, we quantitatively measured the GSH concentrations in the nucleus and cytosol of HeLa cells and primary hepatocytes under different treatment conditions and found no appreciable concentration gradients between these two organelles.
Innovation and Conclusion: We developed the first reversible ratiometric GSH probe that can be universally targeted to any organelle of interest. Taking advantage of this new tool, we provided definitive evidence showing that GSH concentrations are not significantly different between the nucleus and cytosol, challenging the view of nuclear compartmentalization of GSH.
Keywords: glutathione, organelle-targeted probe, HaloTag, small-molecule fluorescent probe, compartmentalization of glutathione, live-cell imaging
Innovation.
Currently, there is no glutathione (GSH) probe available that can target any organelle of interest. Our HaloRT probe is the first quantitative GSH probe that can be used to measure GSH levels in different organelles independent of the probe concentration. A long-standing debate in the field is whether the nucleus has higher GSH over the cytosol. Our observation showed that GSH concentrations are not significantly different in these two organelles, providing the first direct evidence challenging the view of nuclear compartmentalization of GSH.
Introduction
Glutathione (GSH), the most abundant nonprotein small peptide in eukaryotic cells, regulates the intracellular redox homeostasis with its oxidized disulfide partner, oxidized glutathione. Imbalanced redox states have been shown to lead to various abnormal conditions such as diseases and drug resistance (17, 20). Understanding the intracellular concentration and distribution of GSH provides the most direct evidence to a localized cellular redox state and is the key step to study GSH-related redox biology. Just as pH is essential to all cellular events, GSH level is also crucial to a considerable number of biochemical processes inside cells. Importantly, the activity of GSH is tightly linked to its spatial and temporal dynamics, which, however, remains poorly characterized due to lack of effective methods for quantifying subcellular GSH levels in real time.
Our group pioneered the use of reversible reaction-based ratiometric fluorescent probes for measuring GSH concentrations in live cells. We developed the first fluorescent probe ThiolQuant Green that can quantitatively measure intracellular GSH levels in live cells (10). Building on this work, we developed the second-generation GSH probe RealThiol (RT), which allows monitoring dynamic changes of GSH levels in real time (9). The groups of Urano and Yoon also independently reported two probes that can follow GSH dynamics (11, 18). Recently, we developed a mitochondria-targeted GSH probe, MitoRT (5), taking advantage of the high binding affinity of a triphenylphosphonium moiety toward the highly negatively charged mitochondrial membrane (16) and revealed the specificity of mitochondrial GSH dynamics. Unfortunately, this small-molecule-based subcellular targeting strategy is not suitable to all different organelles.
To develop a universal strategy that can be easily and broadly applied to any organelle of interest, we designed a HaloTag-enabled GSH probe, named HaloRT (Fig. 1a). In this study, using different signaling peptide sequences, we are able to specifically locate HaloRT in the nucleus, cytosol, mitochondria, and the endoplasmic reticulum (ER), and measure organelle-specific GSH concentrations. To our surprise, we discovered that there is no appreciable GSH concentration gradient between the nucleus and cytosol in HeLa cells and in primary hepatocytes under several different treatment conditions, strongly challenging the existing view of a nuclear pool of GSH (7).
FIG. 1.

Characterization of HaloRT probe. (a) Chemical structure of HaloRT ligand and the modular design diagram. (b) Reversible reaction between HaloRT and GSH. (c) Deconvoluted, normalized UV-vis absorption spectra (unshaded) and fluorescent spectra (shaded) of HaloRT (green, λex = 488 nm) and HaloRT-GSH conjugate (blue, λex = 405 nm). (d) Reaction kinetics of HaloRT and GSH measured on a stopped-flow rapid mixing device with a fluorescence detector. Forward and backward reactions were monitored at λex = 405 nm, λem = 480 nm, which correspond to the formation and dissociation of HaloRT-GSH conjugate. (e) Calibration curve determined by mixing a series of known concentrations of GSH with HaloRT, followed by imaging on a coverslip using confocal microscopy. The calculated Kd′ is 23.7 mM, and the R2 is 0.991 in the physiologically relevant concentration range of GSH. (f) Selectivity of HaloRT probe in solution phase. Relative fluorescent ratio between blue and green channels was measured following mixing HaloRT probe with GSH (10 mM), H2O2 (100 μM), tBuOOH (100 μM), Na2SO3 (100 μM), lipoic acid (100 μM), homocysteine (100 μM), hydrogen sulfide (100 μM), and cysteine (100 μM). (g) Gel permeation chromatography trace of standard HaloRT-GSH conjugate and lysate from cyto-HaloRT-stained HeLa cells. Fluorescent signal (λex = 405 nm, λem = 480 nm) was used for detecting HaloRT-GSH conjugate. Calculation of the area under the curve revealed that 87% of the signal can be attributed to HaloRT-GSH, indicating high selectivity toward GSH over protein thiols inside cells. FL, fluorescence; GSH, glutathione; H2O2, hydrogen peroxide; RT, RealThiol probe. Color images are available online.
Results and Discussion
To achieve subcellular specificity for GSH probes, we adopted the widely used HaloTag technology for organelle-specific protein tagging (12). Derived from our GSH RT probe, we substituted the dicarboxylic acid moiety with the HaloTag protein substrate elongated with a four-carbon linker (HaloRT ligand, Fig. 1a). Then, by expressing organelle-specific HaloTag proteins in live cells, the HaloRT ligand can be recruited to different organelles and used for quantitative real-time imaging of GSH at subcellular resolution.
After successfully synthesizing the HaloRT ligand, we first verified that HaloRT, the conjugate between the HaloRT ligand and recombinant HaloTag protein, exhibits similar GSH sensing abilities as RT. Similar to RT that has absorption maxima at 478 and 406 nm before and after reacting with GSH, respectively, HaloRT exhibits a bright orange color, an absorption maximum at 480 nm, and shows fluorescence maximum at 565 nm on excitation at 488 nm. Importantly, addition of GSH shifts the absorption and fluorescence peaks to 390 and 488 nm (excited at 405 nm), respectively (Fig. 1b, c).
The kinetics between HaloRT and GSH is also very similar to that of RT, which typically equilibrates with physiologically relevant concentration of GSH within minutes. We measured the reaction using a stopped-flow rapid mixing device at room temperature and monitored the formation and disappearance of HaloRT-GSH by fluorescence (λex = 405 nm, λem = 480 nm). We observed that both forward and reverse reactions equilibrate in about 200 s (Fig. 1d). We also tested the reversibility of the sensing reaction by sequentially adding GSH and an irreversible thiol scavenger, N-methylmaleimide (NMM), to the HaloRT ligand solution. As expected, the fluorescent signals from both blue and green channels as well as the ratio can reversibly reflect the corresponding GSH concentrations (Supplementary Fig. S1).
To measure the dissociation equilibrium constant (Kd) of the reversible sensing reaction, we mixed a series of known concentrations of GSH (from 0.39 to 50 mM) with HaloRT (∼10 μM) and measured the fluorescence in two channels (λex1 = 405 nm, λem1 = 480 nm; λex2 = 488 nm, λem2 = 565 nm). The Kd for the sensing reaction is 121 mM, 32-fold higher than that of RT.
The low reactivity of the protein-conjugated RT may be due to the steric hindrance of the protein or the hydrophobic HaloTag protein surface shifts the Kd of thiol-Michael addition reactions. It should be noted that the apparent dissociation equilibrium constant (Kd′), which is dependent on instrument settings, is more relevant in determining the dynamic range of the probe (please refer to the supporting information in reference 11 for detailed explanation). By adjusting the gains of both emission channels, Kd′ values are 24.8 and 23.7 mM measured by a plate reader and a confocal microscope used in this study. In both cases, the calibration curve confers a superb linear relationship with R2 above 0.99 within the physiologically relevant GSH concentration range (1.25–15 mM, Fig. 1e and Supplementary Fig. S2).
The high Kd of HaloRT results in a very small percentage of HaloRT-GSH formed under physiological conditions. Compared with RT, we increased the gain for the 405 nm excitation channel to achieve sensitive detection of the subtle changes of HaloRT-GSH level. It should be noted that similar to RT, lower pH will alter the kinetics and thermodynamics of reaction between HaloRT and GSH. Thus, lysosomal GSH measurement using HaloRT may require recharacterization of the probe.
Next, we determined if the probe is selective toward GSH in cells. As shown in Figure 1f and Supplementary Figure S3, both HaloRT probe and HaloRT ligand only respond to physiologically relevant concentration of GSH but not to other biothiols or reactive oxygen species. Similar to the RT probe, we suggest validating measured GSH concentrations with other methods such as liquid chromatography-mass spectrometry to ensure that the GSH level is at least 10-fold higher than that of cysteine. In cases that GSH is lower than 1 mM and cysteine is still at its physiologically relevant concentration of 100 μM, the accuracy of GSH measurements based on HaloRT may be compromised.
We also applied the gel permeation chromatography (GPC) method with fluorescence detection that we developed for assessing the selectivity of RT in a complicated intracellular environment (9). The lysate of HaloRT-stained cells was washed using a denature buffer at pH 4.5 to prevent reverse reactions, and the fluorescence signal of HaloRT-GSH (λex = 405 nm, λem = 480 nm) was used for detection. Compared with the HaloRT-GSH, standard underwent the same processing, we did not observe significant molecular-weight changes (Fig. 1g), suggesting that the HaloRT probe selectively reacts with small-molecule thiols but not protein thiols in cells. Together, based on the fluorescence spectra, kinetic and thermodynamic constants, and the selectivity profile, we conclude that HaloRT is suitable to quantitatively monitor GSH level changes under cellular conditions in real time.
To express the HaloTag protein in different organelles, we fused it with organelle targeting sequences, Halo-nuclear localization sequence (NLS) with a nuclear localization sequence, Halo-MapKK with a cytosolic localization sequence from MapK kinase (MapKK) (4), Halo-KDEL with an ER retention sequence, and Halo-MTS with a mitochondria targeting sequence. HeLa cells were transiently transfected with Halo-NLS, Halo-MapKK, Halo-KDEL, or Halo-MTS, incubated with the HaloRT ligand at 37°C for 30 min and then imaged using a confocal microscope. We confirmed that Halo-NLS, Halo-MapKK, Halo-KDEL, and Halo-MTS specifically target the HaloRT ligand to the nucleus (nuc-HaloRT), cytosol (cyto-HaloRT), endoplasmic reticulum (ER-HaloRT), and mitochondria (Mito-HaloRT), respectively (Fig. 2a). Thus, our strategy is effective to target the GSH probe to different cellular organelles.
FIG. 2.

Representative confocal images of HaloRT-stained HeLa cells. (a) Nuclear-, cytosolic-, ER-, and mitochondria-specific HaloTag expressing HeLa cells were stained with HaloRT and imaged with a confocal microscopy at blue and green channels sequentially. Ratio maps were calculated by dividing the blue channel by the green channel at each corresponding pixel. Scale bars are 10 μm. (b) Nuclear-specific HaloTag expressing HeLa cells were stained with HaloRT and quantitatively measured using a confocal microscopy. Position 1 illustrated a nontransfected cell, position 2 illustrated a high-expression transfected cell, and position 3 illustrated a low-expression transfected cell. The quantification results are presented in Table 1, showing the probe concentration independent ratiometric readouts. ER, endoplasmic reticulum. Color images are available online.
It should be noted that our transient transfection protocol does not yield 100% transfection efficiency and the protein expression levels of the transfected cells may vary. The nontransfected cells can serve as an internal control showing that the HaloRT ligand will only stain transfected cells. As shown in Figure 2b, the cell at location 1 was completely nontransfected and the fluorescence signal was close to the background (Table 1). In contrast, the transfected cells at locations 2 and 3 showed significantly higher fluorescence signals than the background in both channels, indicating a successful transfection and staining. Due to the inhomogeneous protein expression levels, the absolute intensities from locations 2 and 3 varied by ∼3-fold. While this may pose a great challenge for intensity-based quantification, it does not affect the final ratiometric readout of HaloRT (Table 1). The advantageous ratiometric quantification by HaloRT probe is clearly independent of the absolute probe concentration.
Table 1.
Quantification Results of Figure 2b
| Area | Fluorescent intensity | Ratio (blue/green) | |
|---|---|---|---|
| Blue channel | Green channel | ||
| Background | 125 ± 60 | 51 ± 12 | NA |
| 1 | 118 ± 95 | 69 ± 36 | NA |
| 2 | 3067 ± 375 | 3464 ± 325 | 0.893 ± 0.109 |
| 3 | 949 ± 210 | 1070 ± 136 | 0.913 ± 0.230 |
It is generally accepted that GSH is compartmentalized in different organelles and regulated separately there, but whether there is a nuclear pool of GSH remains a long-standing debate (2, 3, 15). Bellomo et al. were the first to report a nuclear pool of GSH in primary hepatocytes using monochlorobimane (MCB) as a GSH probe (2). However, this study was challenged by Briviba et al. based on their observation that the MCB-GSH conjugate microinjected into the cytosol can preferentially accumulate in the nucleus, suggesting that MCB could react with cytosolic GSH (cGSH) first and the conjugate is further enriched in the nucleus (3).
Although some other studies favor the view of nuclear pool of GSH especially at early phases of cell proliferation, the fluorescent probes used suffer the same potential artifact as MCB (7, 13). It should be noted that 5-chloromethylfluorescein diacetate (CMFDA) was used as a GSH probe in several studies (7, 13). However, the fluorescence changes of CMFDA are due to fluorescein formation through esterase-mediated hydrolysis and independent of GSH conjugation. Therefore, it is questionable to use CMFDA as a GSH probe. Moreover, most previously studies using small-molecule GSH probes could not rule out the possibility that the irreversibly formed probe-GSH conjugate could diffuse through nuclear pore complex.
In our system, the readouts of HaloRT are independent of probe concentrations due to the ratiometric nature. The Halo-NLS and Halo-MapKK are first transfected into the cells and expressed into their targeted organelles before seeing the HaloRT ligand. In addition, we have confirmed that nuc-HaloRT and cyto-HaloRT have high spatial specificity. Thus, we have applied our system to reexamine the nuclear GSH (nGSH) postulate.
We discovered no appreciable concentration gradient of GSH between the nucleus and cytosol. Despite the absolute fluorescence signals from the nucleus and cytosol being slightly different due to the variation of transfection efficiencies, the ratios of fluorescence signals remain statistically the same in these two locations. This ratiometric feature of our GSH probes is a unique advantage to avoid the nonspecific influence from different transfection efficiencies and probe availability (Fig. 3a, b).
FIG. 3.

Organelle-specific HaloRT probes show no nuclear pool of GSH. (a) Representative images of HaloRT ligand-stained HeLa cells cotransfected with both nuclear-specific and cytosolic-specific HaloTag proteins. Scale bar is 10 μm. (b) Quantitative analysis of GSH concentrations in the nucleus and cytosol from untreated HeLa cells. A total of 394 and 420 cells from >25 images from >5 individual experiments were statistically analyzed for nGSH and cGSH groups, respectively. (c) Time-lapsed ratio maps from HeLa cells transiently treated with 500 μM of H2O2 at time 0. Scale bar is 10 μm. (d) Quantitative analysis of GSH concentrations from transient H2O2-treated HeLa cells. A total of 15 and 18 cells from the representative experiment group, shown in (c), were statistically analyzed for nGSH and cGSH groups, respectively. (e) Quantitative analysis of GSH concentration from HeLa cells treated with vehicle, 50 μM of BSO for 24 hours, 500 μM of BSO for 24 hours, 10 μM of TM for 4 hours, 10 μM of NEL for 4 hours, and 100 μM of DEM for 2 hours and then allowed to recover for 22 hours, respectively (from left to right). A total of 394, 188, 229, 178, 172, and 184 cells from >25 images from >5 individual experiments were statistically analyzed for nGSH group at each corresponding treatment condition, respectively; and a total of 420, 219, 262, 234, 236, and 198 cells from >25 images from >5 individual experiments were statistically analyzed for cGSH group at each corresponding treatment condition, respectively. (f) Quantitative analysis of GSH concentrations from HeLa cells cultured in crowded region and in sparse region. A total of 122 and 65 cells from >15 images from >3 individual experiments were statistically analyzed for nGSH group in the two regions, respectively; and a total of 119 and 96 cells from >15 images from >3 individual experiments were statistically analyzed for cGSH group in the two regions, respectively. (g) Quantitative analysis of GSH concentrations from untreated primary hepatocytes. A total of 38 and 46 cells from >15 images from >3 individual experiments were statistically analyzed for nGSH and cGSH groups, respectively. Standard unpaired t-test was used for all statistical analyses. All the error bars represent standard deviations. BSO, buthionine sulfoximine; cGSH, cytosolic GSH; DEM, diethyl maleate; NEL, nelfinavir; nGSH, nuclear GSH; NLS, nuclear localization sequence; TM, tunicamycin. Color images are available online.
Next, to investigate whether the nGSH can be better maintained compared with cGSH, we treated Halo-NLS and Halo-MapKK cotransfected HeLa cells with different compounds and measured the corresponding nGSH and cGSH under these conditions. In a time-lapsed experiment, we treated cells with a bolus of 500 μM of hydrogen peroxide (H2O2) to induce oxidative stress and imaged for 5 min consecutively. As shown in Figure 3c, both nGSH and cGSH levels decrease synchronously, but there is no significant GSH gradient between the nucleus and cytosol throughout this period. Further quantification demonstrates that nGSH and cGSH levels are not different from each other before and after H2O2 treatments (Fig. 3d).
We confirmed the quantification results with the same experiment performed using RT as a GSH probe (Supplementary Fig. S4). We also applied a non-GSH responsive HaloTag substrate, JF549, as our control and its fluorescence signal did not change on the same H2O2 treatment (Supplementary Fig. S5), indicating that the fluorescence signal changes of HaloRT are due to H2O2-induced GSH level changes.
We also challenged cells with other stressors and examined their effects on nGSH and cGSH. Buthionine sulfoximine (BSO) is a specific GSH synthase inhibitor that decreases intracellular GSH in a concentration-dependent manner. When we treated HeLa cells with 50 or 500 μM of BSO for 48 hours, we found that both nGSH and cGSH decrease as BSO concentration increases (Fig. 3e and Supplementary Fig. S6), but their levels are statistically the same.
ER is a critical cellular compartment for protein folding, modification, and quality control, which is linked to the oxidizing balance of GSH. We thus have treated cells with tunicamycin (TM) and nelfinavir (NEL) to induce ER stress (14a) and test whether these treatments would affect nGSH and/or cGSH levels. We found that after 4 hours of treatments, nGSH and cGSH are decreased to similar levels (Fig. 3e), suggesting that ER stress-induced oxidative insults affect both the nucleus and cytosol. Together, these results show that shifting the oxidative balance toward oxidation reduces nGSH and cGSH levels at the same time and to the same degree, not supporting an independent pool of GSH in the nucleus.
On the contrary, we examined whether an increase in GSH would affect the concentration gradient between nGSH and cGSH. To this end, we stimulated cells with 100 μM of diethyl maleate (DEM) transiently for 2 hours and then allowed the cells to recover for 24 hours. We found that this treatment greatly increases GSH levels in the cell, but there is no difference between the nGSH and cGSH increases (Fig. 3e and Supplementary Fig. S6).
Densely cultured cells are known to have higher GSH levels compared with sparsely cultured counterparts (8, 14). We thus generated a cell culture condition where crowded and sparse regions coexist within the same culture dish and show two- to threefolds of GSH concentration difference (Supplementary Fig. S7). Under this physiological condition without exogenous stimulants, we did not observe an appreciable difference between nGSH and cGSH (Fig. 3f).
Furthermore, we also examined nGSH and cGSH levels in primary hepatocytes transfected with Halo-NLS and Halo-MapKK and stained with the HaloRT ligand. Consistent with our observation in cell line studies, there is no appreciable difference between nGSH and cGSH in these primary cells despite a lower transfection efficiency, compared with HeLa cells, being observed (Fig. 3g and Supplementary Fig. S8). Therefore, we did not observe a significant concentration difference between nGSH and cGSH with increased GSH levels in HeLa cells and under physiological conditions in primary hepatocytes.
In conclusion, we have developed an organelle-specific GSH probe, HaloRT, which can be universally applied to determine GSH levels in any organelle of interest with targeted HaloTag protein expression.
This HaloRT system enables us to quantify GSH concentrations with subcellular resolution in real time. Taking advantage of this new system, we revisited the nGSH postulate. Our measurements do not reveal any appreciable GSH concentration gradients between the cytosol and nucleus and thus do not support an independent pool of nGSH. Considering the nuclear pore complex has an inner diameter of ∼9–12 nm and typically allows free diffusion of molecules less than 60 kDa (1, 19), the small tripeptide GSH should freely diffuse through the nuclear pores. In organelles with enclosed membrane structures, GSH can be regulated separately, such as in the mitochondria, as demonstrated previously (5). In addition, our findings are consistent with a previous study using redox fluorescent proteins to measure GSH redox potentials in these organelles (5).
Although we did not observe any nuclear pool of GSH under our experimental conditions, we cannot rule out the possibility that the nuclear pores can be closed to restrict the free diffusion of GSH under other experimental conditions, thus creating a GSH concentration gradient. In addition, although demonstrated only for the nucleus, cytosol, ER, and mitochondria, with the HaloRT probe in hand, this system can be potentially applied to any organelle and will be a valuable addition to the redox biology toolbox.
Materials and Methods
Chemical synthesis of HaloRT ligand
All the chemicals were purchased from Sigma-Aldrich or Alfa Aesar unless otherwise specified. 3-Carbaldehyde-7-azetidinylcoumarin was purchased from Ascendex Scientific, LLC. Tetrahydrofuran was distilled over sodium benzophenone ketyl, and CH2Cl2 was distilled over phosphorus pentoxide. All the other solvents and reagents were used as received without further purification.
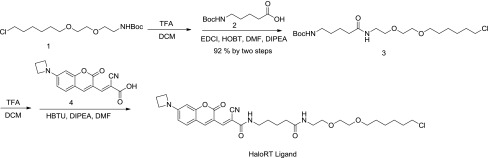
Tert-butyl (2-(2-((6-chlorohexyl)oxy)ethoxy)ethyl)carbamate (1, 65 mg, 0.2 mmol) (refer to Supplementary Fig. S9 for 1H-NMR) was dissolved in dichloromethane (DCM) (2 mL) and treated with trifluoroacetic acid (TFA) (1 mL) at room temperature for 2 hours. The reaction mixture was concentrated, dried, and used in the next step without further purification. The residue was dissolved in dimethylformamide (DMF) (2 mL). 5-((tert-butoxycarbonyl)amino)pentanoic acid (2, 43 mg, 0.2 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (38 mg, 0.3 mmol), hydroxybenzotriazole (41 mg, 0.3 mmol), and N,N-diisopropylethylamine (DIPEA) (0.18 mL, 1.0 mmol) were subsequently added at room temperature. After stirring overnight, the solvent was removed under vacuum. The residue was purified by silica gel column chromatography to give tert-butyl (5-((2-(2-((6-chlorohexyl)oxy)eth-oxy)ethyl)amino)-5-oxopentyl)carbamate as a colorless oil (3, 78 mg, yield 92% by two steps).
1H NMR (400 MHz, CDCl3): δ 6.12 (s, 1H), 4.72 (s, 1H), 3.62–3.52 (m, 8H), 3.47–3.43 (m, 4H), 3.12–3.09 (m, 2H), 2.20 (t, J = 7.4 Hz, 2H), 1.81–1.74 (m, 2H), 1.71–1.58 (m, 4H), 1.55–1.34 (m, 15H) (Supplementary Fig. S10). 13C NMR (100 MHz, CDCl3) δ 172.72, 156.05, 79.05, 71.25, 70.25, 70.02, 69.83, 45.01, 39.98, 39.13, 35.98, 32.51, 29.53, 29.45, 28.43, 26.67, 25.41, 22.69 (Supplementary Fig. S11).
Tert-butyl (5-((2-(2-((6-chlorohexyl)oxy)ethoxy)ethyl)amino)-5-oxopentyl)carbamate (3, 21 mg, 0.05 mmol) was dissolved in DCM (2 mL) and treated with TFA (1 mL) at room temperature for 2 hours. The reaction mixture was concentrated, dried, and used in the next step without further purification. The residue was dissolved in DMF (1 mL). 3-(7-(azetidin-1-yl)-2-oxo-2H-chro-men-3-yl)-2-cyano-acrylic acid (4, 15 mg, 0.05 mmol), N,N,N',N'-tetramethyl-O-1H-benzotriazol-1-yl)uronium hexafluorophosphate (38 mg, 0.1 mmol), and DIPEA (0.044 mL, 0.25 mmol) were added subsequently at room temperature. After stirring overnight, the solvent was removed under vacuum. The residue was purified by silica gel column chromatography to give 5-(3-(7-(azetidin-1-yl)-2-oxo-2H-chromen-3-yl)-2-cyanoacrylamido)-N-(2-(2-((6-chlorohexyl)oxy)ethoxy)ethyl)pentanamide as a red solid (HaloRT Ligand, 15 mg, yield 50%).
1H NMR (400 MHz, CDCl3) δ 8.80 (s, 0.5H), 8.64 (s, 0.5H), 8.47 (s, 0.5H), 7.65 (s, 0.5H), 7.34 (t, J = 7.9 Hz, 1H), 6.78 (t, J = 4.4 Hz, 0.5H), 6.53 (t, J = 4.8 Hz, 0.5H), 6.31–6.15 (m, 2H), 6.08 (d, J = 9.7 Hz, 1H), 4.14–4.03 (m, 4H), 3.62–3.47 (m, 8H), 3.46–3.35 (m, 6H), 2.52–2.43 (m, 2H), 2.22 (q, J = 7.0 Hz, 2H), 1.78–1.58 (m, 8H), 1.49–1.34 (m, 4H) (Supplementary Fig. S12). 13C NMR (100 MHz, CDCl3) δ 172.86, 172.78, 161.42, 161.11, 160.63, 157.40, 155.01, 154.90, 148.28, 145.69, 143.92, 131.71, 131.52, 118.80, 117.61, 111.95, 111.78, 109.19, 109.04, 108.71, 108.58, 106.00, 101.68, 95.65, 95.55, 77.24, 71.27, 70.24, 70.21, 70.01, 69.99, 69.81, 69.79, 51.33, 45.05, 39.92, 39.73, 39.22, 35.75, 32.52, 32.51, 29.43, 28.86, 28.63, 26.68, 25.40, 25.39, 22.63, 22.57, 16.15 (mixture of cis and trans isomers) (Supplementary Fig. S13). ESI m/z = 601.3 [M+H]+.
Synthesis of HaloRT probe
HaloRT ligand (400 μM, 200 μL) was incubated with purified His-HaloTag protein (Promega, G4491, 6.5 mg/mL, 50 μL) for 1 hour at room temperature in phosphate-buffered saline (PBS). The reaction mixture was then concentrated with a 10k cutoff viva-spin 500 centrifugation tube and washed twice with fresh PBS. The resulting bright orange supernatant was then used as HaloRT probe for further testing. The HaloRT probe was validated by GPC to ensure that no free HaloRT ligand remained in solution.
UV-Vis and fluorescence spectra of HaloRT probe
HaloRT probe was diluted with PBS to 10 μM and mixed with an equal volume of PBS or GSH (700 mM). The resulting mixture was then measured on a BioTek H1 plate reader for absorption and fluorescence at corresponding wavelengths using a Take3 Trio microvolume plate. A deconvolution of the overlapping spectra was performed to yield the individual spectrum.
Kinetics of HaloRT probe reacting with GSH
The reaction kinetics was measured on a stopped-flow rapid mixing device with a fluorescence detector. The formation and dissociation of HaloRT-GSH conjugate were monitored at λex = 405 nm and λem = 480 nm. The forward reaction was measured by mixing an equal volume of 10 μM HaloRT probe with 10 mM of GSH and the reverse reaction was measured by diluting a pre-equilibrated 10 μM HaloRT probe and 10 mM GSH mixture with an equal volume of PBS.
Reversibility of HaloRT ligand reacting with GSH
HaloRT ligand was diluted with PBS to 10 μM and monitored using a fluorimeter at λex = 405 nm, λem = 480 nm and λex = 488 nm, λem = 565 nm. To 1 mL of the HaloRT ligand solution was added 50 μL of 100 mM GSH solution. After the reaction equilibrated (∼300 s), 50 μL of 100 mM NMM solution was added to the mixture. GSH concentration reflected by the ratio was confirmed with standard GSH solution with >99% accuracy.
Equilibrium constant of the reaction between HaloRT probe and GSH
HaloRT probe was diluted with PBS to 10 μM and mixed with an equal volume of a series of known concentrations of GSH (0.39, 0.78, 1.56, 3.13, 6.25, 12.5, 25, 50, 100 mM) and equilibrated for 5 min at room temperature. The resulting mixture was then measured on a plate reader at two fluorescent channels: λex = 405 nm, λem = 480 nm and λex = 488 nm, λem = 565 nm. The ratio between the two channels was used for calibration of equilibrium constant.
Similarly, the mixture of HaloRT probe and GSH was prepared and imaged using a confocal microscope. Before imaging, the mixture was mixed with 15 μm polystyrene beads for focusing purpose. A typical sample can be prepared by sealing 5 μL of solution with a coverslip.
Selectivity of HaloRT ligand and HaloRT probe in solution
HaloRT ligand and HaloRT probe were diluted with PBS to 10 μM and mixed with equal volumes of PBS, GSH (20 mM), H2O2 (200 μM), tBuOOH (200 μM), Na2SO3 (200 μM), lipoic acid (200 μM), homocysteine (200 μM), hydrogen sulfide (200 μM), and cysteine (200 μM), respectively. The resulting mixture was then measured on plate reader at two fluorescent channels: λex = 405 nm, λem = 480 nm and λex = 488 nm, λem = 565 nm. The ratio between the two channels was used for calculation.
Intracellular selectivity of HaloRT probe measured with GPC-fluorescence
To acquire enough amount of material for analysis, we choose to overexpress HaloTag protein in the cytosol, which has the highest overall quantity of protein. HeLa cells (from a confluent 15 mm dish) transiently transfected with cytosolic-specific HaloTag protein were stained with 5 μM of HaloRT ligand at 37°C for 30 min. The cells were washed twice with cold PBS to remove excessive small-molecule probes and then lysed using 1 mL of 10% trichloroacetic acid on ice. The lysate was then centrifuged at 15,000 g at 4°C for 15 min.
The supernatant was ready for GPC-fluorescence (FL) measurement as the small-molecule mixture. The resulting precipitate was then redissolved using 0.1 M pH 4.5 citric acid-buffered 5% SDS at 58°C for 5 min. Supernatant was then collected after quickly centrifuging at 15,000 g at room temperature for 1 min. This second supernatant was ready for GPC-FL measurement as the redissolved protein mixture.
The GPC-FL measurement was performed using 0.1 M pH 4.5 citric acid-buffered 0.1% SDS solution with a cationic column. Flow rate was set at 1.5 mL/min and the temperature was set at 45°C. A standard HaloRT-GSH sample, a small-molecule mixture from cell lysate, and a redissolved protein mixture from the same cell lysate were measured on GPC-FL with a fluorescent detector set at λex = 405 nm, λem = 480 nm for detection of HaloRT-GSH conjugate. The small-molecule mixture resulted in no fluorescent signal, and the HaloRT-GSH standard and the redissolved protein mixture showed similar retention time on their fluorescent signal. Area under the curve after normalization was used for quantification purpose. Analysis revealed that 87% of the protein mixture signal comes from HaloRT-GSH compared with the standard.
General cell culture
The HeLa cell line used in this study was purchased from American Type Culture Collection (ATCC) and grown in DMEM (11965; Gibco) supplemented with 10% FBS and 1% 1003 penicillin/streptomycin (Gibco). Cells were cultured under a controlled atmosphere (37°C, 5% CO2). Glass-bottomed dishes were used for cell culture due to confocal scanning requirements.
Organelle-specific quantitative imaging of GSH using HaloRT probe
Halo-NLS, Halo-MapKK, Halo-KDEL, and Halo-MTS plasmids were constructed by integrating organelle-specific signal sequences into the Halo cDNA sequence. The following signal sequences are used for plasmid constructs: 5′-CCAAAAAAGAAGAGAAAGGTAGAAGACCCC-3′ for nuclear specific, 5′-TTGCAGAAGAAGCTGGAGGAGCTAGAGCTTGAT-3′ for cytoplasm specific, 5′-AAAGATGAGCTATAA-3′ (KDEL sequence) for ER specific, and 5′-ATGGCCTCCACTCGTGTCCTCGCCTCTCGCCTGGCCTCCCAGATGGCTGCTTCCGCCAAGGTTGCCCGCCCTGCTGTCCGCGTTGCTCAGGTCAGCAAGCGCACCATCCAGACTGGCTCCCCCCTCCAGACCCTCAAGCGCACCCAGATGGCCACCATGAGCGCCCTGCTGATCCTGGCTCTCGTGGGCGCTGCCGTGGCC-3′ for mitochondria specific.
Whole piece of DNA fragment, including signal sequence and Halo cDNA sequence, was synthesized by GenScript and then inserted into pCDNA 3.1 (+) vector through KpnI (5′end) and NotI (3′end) sites.
HeLa cells (4 × 105) were seeded on a 35-mm glass-bottomed culture dish and incubated for 24 hours. The cells were then transfected with one or two plasmids using Lipofectamine 3000 transfection agent and incubated for an additional 36 hours. Before imaging, cells were stained with 1 μM HaloRT ligand in culture medium at 37°C for 30 min and then washed with fresh medium twice, each for 10 min. Imaging was performed using a Zeiss LSM 780 confocal microscope with a temperature control chamber. Green channel signal was acquired with λex = 488 nm, λem = 499–615 nm; blue channel signal was acquired with λex = 405 nm, λem = 418–495 nm.
The usage of transient transfection for all our experiments inevitably leads to nonhomogeneous transfection efficiency. Some cells are not transfected, while other cells are overexpressing the transfected plasmids. This inhomogeneous transfection resulted in inhomogeneous fluorescent signal from cell to cell. Theoretically, ratiometric imaging will not be affected by the differences in absolute fluorescent signal, but the instrument dynamic range is only accurate within limited range. For quantitation purpose, we have to compromise and use the settings that we are able to take the calibration curve with. Thus, for quantitative analysis, we only picked cells with S/N > 5 signals from both channels and do not have saturating pixels. In addition, because we could hardly define the boundary of nucleus from the ratio map in Halo-NLS and Halo-MapKK cotransfected cells, we manually labeled the nucleus and cytosol by comparing with the images from other channels.
Treatment conditions for HeLa cells
Transient treatment with H2O2 was performed at the site of imaging. Stained, transfected HeLa cells were treated with a bolus of 500 μM of H2O2 (100 μL 5 mM H2O2 in PBS diluted to 1 mL medium) in the imaging chamber and imaged at 37°C continuously for 5 min with 20 s per scan. This condition was designed for testing rapid GSH consumption after a bolus of external oxidative insult. The same H2O2 treatment was also applied to transfected cells stained with both HaloRT and JF549.
BSO (50 and 500 μM) was added to HeLa cells at seeding, and the cells were incubated for 24 hours before transfection of cells. Cells were imaged 48 hours after transfection. BSO is a GSH synthase inhibitor. This condition was designed for decreasing overall GSH concentrations in a controlled, dose-dependent manner.
DEM (100 μM) was added to HeLa cells 10 hours after transfection. Cells were incubated with DEM for 2 hours and then washed with fresh culture medium. Then, the cells were incubated for an additional 22 hours in fresh culture medium before imaging. This condition was designed for increasing GSH concentration.
ER stressors (10 μM of TM and 10 μM of NEL) were added to cells 4 hours before imaging. This condition was designed to induce ER stress, which will transiently decrease GSH concentration.
HeLa cells were seeded on a confined spot on a culture dish without stirring, which created a crowded region. The peripheral of the crowded cells was sparse regions. This condition was designed for creating high- and low-concentration GSH in cells on the same plate but at different regions.
Primary hepatocyte imaging
Mice were euthanized with isoflurane and 25G needle was inserted into the inferior vena cava. After their portal vein is cut, peristaltic pump was used to perfuse the liver. First, Earle's balanced salt solution (EBSS) (14155063; Invitrogen) containing 5 mM EGTA was used. Following EBSS perfusion, Hank's balanced salt solution (24020117; Invitrogen) containing 100 U/mL collagenase and trypsin inhibitor was used. The liver was then harvested and massaged to obtain dissociated cells in hepatocyte wash medium (17704024; Invitrogen).
After cells went through mesh, the cells were sublayered onto Percoll (P4937; Sigma). Only viable cells at the bottom layer were washed twice with wash medium, and then, the cells were plated onto dishes in Wiliams E medium (12551; Invitrogen) containing insulin/transferrin/selenium supplementation. Then, the cells were kept in DMEM overnight. Transfection, staining, and confocal imaging were performed following the same procedure for HeLa cells.
Data availability
All relevant data are available from the authors.
Supplementary Material
Acknowledgments
The research was supported, in part, by the National Institutes of Health (R01-GM115622, R01-CA207701, R21-CA213535, and RF1-AG062257 to J.W., R01-AG045183, R01-AT009050, R21-EB022302, DP1-DK113644, and RF1-AG062257 to M.C.W., R01-GM120033 to M.M-S.), the Welch Foundation (Q-1912 to M.C.W.), Cancer Prevention and Research Institute of Texas (RP130573 to M.M.S.), the IDDRC Microscopy Core (P30HD024064 and 1U54 HD083092 Intellectual and Developmental Disabilities Research Grant from the Eunice Kennedy Shriver National Institute of Child Health and Human Development), the Optical Imaging and Vital Microscopy Core, and the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (AI036211, CA125123, and RR024574), and the expert assistance of Joel M. Sederstrom. We also thank Dr. Lavis for providing the JF549 HaloTag substrate and the Department of Pharmacology and Chemical Biology at Baylor College of Medicine for providing partial support for the NMR.
Abbreviations Used
- BSO
buthionine sulfoximine
- cGSH
cytosolic glutathione
- CMFDA
5-chloromethylfluorescein diacetate
- DCM
dichloromethane
- DEM
diethyl maleate
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- EBSS
Earle's balanced salt solution
- ER
endoplasmic reticulum
- FL
fluorescence
- GPC
gel permeation chromatography
- GSH
glutathione
- H2O2
hydrogen peroxide
- MapKK
MapK kinase
- MCB
monochlorobimane
- NEL
nelfinavir
- nGSH
nuclear glutathione
- NLS
nuclear localization sequence
- NMM
N-methylmaleimide
- PBS
phosphate-buffered saline
- RT
RealThiol probe
- TFA
trifluoroacetic acid
- TM
tunicamycin
Authors' Contributions
X.J., J.C., X.C., M.M-S., T.P., D.M., M.C.W., and J.W. designed the experiments; X.J., J.C., C.Z., S.C., Y.Z., M.Z., and X.S. performed the experiments; X.J., M.C.W., and J.W. performed data analysis and wrote the article.
Author Disclosure Statement
X.J., J.C., and J.W. are coinventors of a patent application related to this work.
References
- 1. Beck M, Lučić V, Förster F, Baumeister W, and Medalia O. Snapshots of nuclear pore complexes in action captured by cryo-electron tomography. Nature 449: 611–615, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Bellomo G, Vairetti M, Stivala L, Mirabelli F, Richelmi P, and Orrenius S. Demonstration of nuclear compartmentalization of glutathione in hepatocytes. Proc Natl Acad Sci U S A 89: 4412–4416, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Briviba K, Fraser G, Sies H, and Ketterer B. Distribution of the monochlorobimane-glutathione conjugate between nucleus and cytosol in isolated hepatocytes. Biochem J 294: 631–633, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, and Goodman RH. Biosensor reveals multiple sources for mitochondrial NAD+. Science 352: 1474–1477, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen J, Jiang X, Zhang C, MacKenzie KR, Stossi F, Palzkill T, Wang MC, and Wang J. Reversible reaction-based fluorescent probe for real-time imaging of glutathione dynamics in mitochondria. ACS Sens 2: 1257–1261, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.This reference has been deleted
- 7. García-Giménez JL, Markovic J, Dasí F, Queval G, Schnaubelt D, Foyer CH, and Pallardó FV. Nuclear glutathione. Biochim Biophys Acta 1830: 3304–3316, 2013 [DOI] [PubMed] [Google Scholar]
- 8. Gutscher M, Pauleau A-L, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, and Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods 5: 553–559, 2008 [DOI] [PubMed] [Google Scholar]
- 9. Jiang X, Chen J, Bajić A, Zhang C, Song X, Carroll SL, Cai Z-L, Tang M, Xue M, Cheng N, Schaaf CP, Li F, MacKenzie KR, Ferreon ACM, Xia F, Wang MC, Maletić-Savatić M, and Wang J. Quantitative real-time imaging of glutathione. Nat Commun 8: 16087, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang X, Yu Y, Chen J, Zhao M, Chen H, Song X, Matzuk AJ, Carroll SL, Tan X, Sizovs A, Cheng N, Wang MC, and Wang J. Quantitative imaging of glutathione in live cells using a reversible reaction-based ratiometric fluorescent probe. ACS Chem Biol 10: 864–874, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Z, Zhou X, Miao Y, Hu Y, Kwon N, Wu X, and Yoon J. A reversible fluorescent probe for real-time quantitative monitoring of cellular glutathione. Angew Chem Int Ed Engl 56: 5812–5816, 2017 [DOI] [PubMed] [Google Scholar]
- 12. Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, and Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3: 373–382, 2008 [DOI] [PubMed] [Google Scholar]
- 13. Markovic J, Borrás C, Ortega A, Sastre J, Viña J, and Pallardó FV. Glutathione is recruited into the nucleus in early phases of cell proliferation. J Biol Chem 282: 20416–20424, 2007 [DOI] [PubMed] [Google Scholar]
- 14. Pani G, Colavitti R, Bedogni B, Anzevino R, Borrello S, and Galeotti T. A redox signaling mechanism for density-dependent inhibition of cell growth. J Biol Chem 275: 38891–38899, 2000 [DOI] [PubMed] [Google Scholar]
- 14a. Peñaranda Fajardo NM, Meijer C, and Kruyt FAE. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem Pharmacol 118: 1–8, 2016 [DOI] [PubMed] [Google Scholar]
- 15. Smith CV, Jones DP, Guenthner TM, Lash LH, and Lauterburg BH. Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicol Appl Pharmacol 140: 1–12, 1996 [DOI] [PubMed] [Google Scholar]
- 16. Smith RA, Porteous CM, Gane AM, and Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A 100: 5407–5412, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Townsend DM, Tew KD, and Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother 57: 145–155, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Umezawa K, Yoshida M, Kamiya M, Yamasoba T, and Urano Y. Rational design of reversible fluorescent probes for live-cell imaging and quantification of fast glutathione dynamics. Nat Chem 9: 279–286, 2017 [DOI] [PubMed] [Google Scholar]
- 19. Wang R. and Brattain MG. The maximal size of protein to diffuse through the nuclear pore is larger than 60 kDa. FEBS Lett 581: 3164–3170, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waxman DJ. Glutathione S-transferases: role in alkylating agent resistance and possible target for modulation chemotherapy—a review. Cancer Res 50: 6449–6454, 1990 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the authors.