

Abstract
Chronic pain is a common cause of disability worldwide and remains a global health and socio‐economic challenge. Current analgesics are either ineffective in a significant proportion of patients with chronic pain or associated with significant adverse side effects. The PPARs, a family of nuclear hormone transcription factors, have emerged as important modulators of pain in preclinical studies and therefore a potential therapeutic target for the treatment of pain. Modulation of nociceptive processing by PPARs is likely to involve both transcription‐dependent and transcription‐independent mechanisms. This review presents a comprehensive overview of preclinical studies investigating the contribution of PPAR signalling to nociceptive processing in animal models of inflammatory and neuropathic pain. We examine current evidence from anatomical, molecular and pharmacological studies demonstrating a role for PPARs in pain control. We also discuss the limited evidence available from relevant clinical studies and identify areas that warrant further research.
Linked Articles
This article is part of a themed section on 8th European Workshop on Cannabinoid Research. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.10/issuetoc
Abbreviations
- ACC
anterior cingulate cortex
- AEA
anandamide
- CCI
chronic constriction injury
- CFA
complete Freund's adjuvant
- DRG
dorsal root ganglion
- EMSA
electrophoretic mobility shift assay
- FAAH
fatty acid amide hydrolase
- iNOS
inducible NOS
- OEA
N‐oleoylethanolamide
- PAG
periaqueductal grey
- lPAG
lateral PAG
- VlPAG
ventrolateral PAG
- PEA
N‐palmitoylethanolamide
- PPRE
peroxisome proliferator response element
- RXR
retinoid X receptor
- RVM
rostroventromedial medulla
- SD
Sprague–Dawley
- VTA
ventral tegmental area
- WKY
Wistar‐Kyoto
Peroxisome proliferator‐activated receptors
The PPARs are ligand‐dependent transcription factors that belong to the nuclear hormone superfamily of receptors. Three major isoforms have been identified: PPARα, cloned from mouse liver (Issemann and Green, 1990), PPARβ/δ and PPARγ, both cloned from Xenopus (Dreyer et al., 1992). These three isoforms share a common structure, typified by the presence of a highly conserved DNA binding domain, with two zinc finger motifs, that recognize the peroxisome proliferator response element (PPRE) in the promoter regions of target genes (Desvergne and Wahli, 1999). They also contain two transcription activation domains; ligand independent AF‐1 in the N‐terminal domain (Delerive et al., 2002) and the AF‐2 in the C‐terminal domain, which is ligand‐dependent and has a large ligand binding domain. This large ligand binding domain makes it possible for PPARs to interact with a wide array of synthetic and natural lipid ligands.
PPARs exist as heterodimers with the retinoid X receptor (RXR), bound to co‐repressor proteins in the inactive state. Upon ligand activation, the co‐repressors dissociate from the PPAR/RXR complex, allowing for the recruitment of co‐activators. The activated PPAR/RXR‐co‐activator complex subsequently binds to specific DNA sequences or PPRE, resulting in the transcriptional activation of target genes (Green et al., 1992, Tugwood et al., 1992). Genes regulated by PPARs via this PPRE‐dependent mechanism are mainly involved in lipid and lipoprotein metabolism (Tugwood et al., 1992). Alternate mechanisms of action that do not involve PPRE binding have been reported, especially for PPARα, which has known anti‐inflammatory effects (Delerive et al., 2001). These latter mechanisms involve inhibition of NF‐κB and AP‐1 inflammatory signalling and the consequent trans‐repression of pro‐inflammatory genes such as inducible NOS (iNOS), COX‐2 and TNF‐α (Crisafulli and Cuzzocrea, 2009, Cuzzocrea et al., 2008, Delerive et al., 2000). These anti‐inflammatory consequences of PPARα activation are fundamental to the role of this receptor in modulating both inflammatory and neuropathic pain as discussed later.
PPARs are widely distributed in mammalian tissues, including the peripheral and CNSs (Braissant et al., 1996, Moreno et al., 2004) (Table 1), and are activated by a variety of endogenous compounds derived from saturated and unsaturated fatty acid, of which N‐palmitoylethanolamide (PEA) (LoVerme et al., 2005) and N‐oleoylethanolamide (OEA) (Fu et al., 2003) remain the best characterized to date. The ubiquitous distribution of PPARs also reflects their roles in many physiological processes, including an emerging role as key modulators in nociceptive processing. In particular, and as reviewed below, anatomical, molecular and pharmacological studies suggest that the PPAR signalling system may be a viable therapeutic target for the treatment of chronic pain and its comorbidity with stress‐related psychiatric disorders.
Table 1.
Expression of mRNA or protein for PPAR isoforms within neuroanatomical loci involved in pain
| Neuroanatomical locus | PPARα | PPARβ/δ | PPARγ | |
|---|---|---|---|---|
| Peripheral | Dorsal root ganglion | ✔ b , c | ? | ? |
| C fibres | ? | ? | ? | |
| Aδ fibres | ? | ? | ? | |
| Aβ fibres | ? | ? | ? | |
| Spinal | Spinal cord | ✔ * d | ✔d | ✔ a , d , g |
| Supraspinal | Frontal cortex | ✔d | ✔d | ✔d |
| Pre‐frontal cortex (PFC) | ✔e | ✔e | ✔e | |
| Hippocampus | ✔d | ✔d | ✔d | |
| Thalamus | ✔d | ✔d | ✔d | |
| Hypothalamus | ✘ d | ✔d | ✔d | |
| Basal ganglia | ✔d , e | ✔d , e | ✔d , e | |
| Amygdala | ✔e | ✔e | ✔e | |
| PAG | ✔f | ? | ✔f | |
| Rostroventral medulla (RVM) | ? | ? | ? | |
| Ventral tegmental area (VTA) | ✔e | ✔e | ✔e |
Expression of PPARs in key components of the pain pathway
A role for PPAR signalling in pain processing is suggested by studies demonstrating the presence of the different PPAR isoforms at key peripheral, spinal and supraspinal sites involved in pain processing (Table 1; Figure 1). Within the periphery, PPARα protein is expressed in the dorsal root ganglion (DRG) (LoVerme et al., 2006), although the relative distribution within DRG sub‐nuclei remains unreported. Unlike PPARα, to our knowledge, the expression of PPARβ or PPARγ in the DRG remains unexplored. Despite the paucity of data on the distribution pattern of PPARα on nociceptive primary afferents (Aδ‐fibres and C‐fibres), the reported analgesic effects of PPARα agonists, administered locally or peripherally, in animal models of inflammatory and neuropathic pain (see Table 2 for comprehensive list) suggest a modulatory influence on peripheral nociceptive afferents such that PPARα activation in the DRG results in the suppression or silencing of nociceptive afferent fibre firing. However, the validation of this hypothesis requires further characterization of PPARα in DRG nuclei using double‐labelling immunohistochemistry or in situ hybridization techniques.
Figure 1.
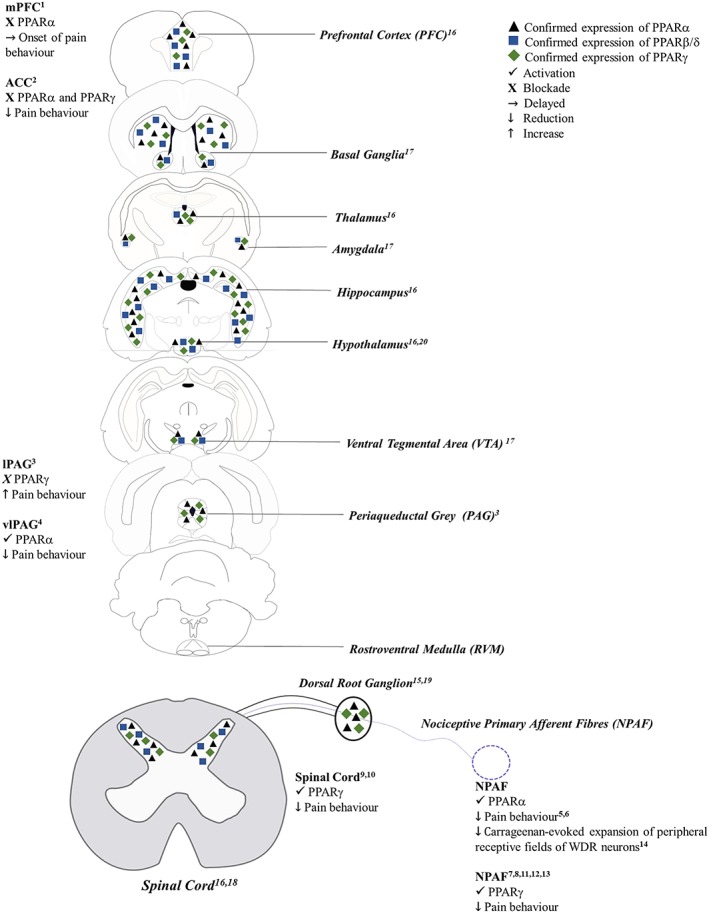
Anatomical localization of PPAR isoforms in key components of the pain pathway and their role in pain modulation. (1) Okine et al. (2014); (2) Okine et al. (2017); (3) Okine et al. (2016); (4) De Novellis et al. (2012); (5) LoVerme et al. (2006); (6) Russo et al. (2007); (7) Hasegawa‐Moriyama et al. (2013); (8) Mansouri et al. (2017a, b); (9) Churi et al. (2008); (10) Griggs et al. (2015); (11) Saito et al. (2016); (12) Hasegawa‐Moriyama et al. (2012); (13) Takahashi et al. (2011); (14) Sagar et al. (2008); (15) D'Agostino et al. (2009); (16) Moreno et al. (2004); (17) Warden et al. (2016); (18) Churi et al. (2008); (19) Maeda et al. (2008); (20) Chakravarthy et al. (2007).
Table 2.
Summary of pharmacological studies investigating the role of PPARs in animal models of pain
| Type of pain | Model | Drug | Dose | Route of administration | Animal | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Inflammatory | Carrageenan | PEA and GW7647 (PPARα agonist) | 2 μL per mouse (i.c.v) 0.3 μL (spinal) | i.c.v and spinal | Swiss mice | Reduced hyperalgesia in mice via inhibition of pro‐inflammatory signalling in the carrageenan model of inflammatory pain. The results were mimicked by the PPARα agonist (GW7647). | D'Agostino et al. (2009) |
| Inflammatory | Subcutaneous carrageenan | PEA | 200, 400 and 800 μg·mL−1 | Subcutaneous | Wistar rats | Treatment with PEA reduced allodynia evaluated by Von Frey. | De Filippis et al. (2011) |
| Inflammatory | Carrageenan | PFOA (PPARα agonist) and rosiglitazone (PPARγ agonist) | 100 mg·kg−1 | Systemic | Sprague–Dawley rats | Pretreatment with either drug inhibited carrageenan‐induced oedema in a dose‐dependent manner and also reduced carrageenan‐induced hyperalgesia. | Taylor et al. (2002) |
| Inflammatory | Carrageenan | Rosiglitazone and 15d‐PGJ2 (PPARγ agonist) | 0–5 to 50 mg (rosiglitazone) and 50–200 mg (15d‐PGJ2) | i.c.v | Sprague–Dawley rats | i.c.v. administration of the drugs dose‐dependently reduced behavioural withdrawal responses to noxious heat. The administration of antagonists (BADGE and GW9662) reversed the anti‐hyperalgesic effects. | Morgenweck et al. (2009) |
| Inflammatory | Carrageenan and collagen‐induced arthritis | Adelmidrol (PEA analogue) | 10 mg·kg−1 | Intraperitoneal | Sprague–Dawley rats | The administration of adelmidrol produced a significant inhibition in the development of carrageenan‐induced and collagen‐induced thermal and mechanical allodynia. This anti‐allodynic effect was reversed by GW9662 (PPARγ antagonist). | Impellizzeri et al. (2016) |
| Inflammatory | Carrageenan | URB597 (FAAH inhibitor) | 25 and 100 μg | Intraplantar | Sprague–Dawley rats | The administration of URB597 attenuated the hyperalgesia induced by carrageenan. GW6471 (PPARα antagonist) reversed this effect. | Jhaveri et al. (2008) |
| Inflammatory | Carrageenan | GW0742 and ATRA(PPARβ/δ agonist) | 0.1 mg·kg−1 (GW0742) and 5 mg·kg−1 (ATRA) | Intraperitoneal | Wistar rats | The administration of both drugs has reduced mechanical and thermal hyperalgesia induced by carrageenan. The co‐treatment with a PPARβ/δ antagonist (GSK0660) has blocked the effects of the drugs. | Gill et al. (2013) |
| Inflammatory | Formalin | GW7647 (PPARα agonist) | 0.1–10 μg/10 μL | Intraplantar | Swiss mice | GW7647 inhibited phase I and phase II pain behaviour in the formalin‐induced inflammatory pain model at the highest dose. | Russo et al. (2007) |
| Inflammatory | Formalin and carrageenan | PEA | 0.01–50 μg/200 μL | Intraperitoneal | Swiss mice | Reduction in both early and late phases of formalin‐induced nociception by PEA at 5 and 50 mg per paw. PPARα knockout animals failed to respond to PEA compared to wild‐type animals. The injection of carrageenan resulted in a significant reduction of mechanical and thermal threshold values. Both hyperalgesic parameters were strongly reduced by PEA (50 mg). | Sasso et al. (2012) |
| Inflammatory | Formalin | GW7647 (PPARα agonist) and GW6471 (PPARα antagonist) | 10 μg (GW7647); 10 μg (GW6471) | Intra‐mPFC | Sprague–Dawley rats | Intra‐mPFC administration of GW6471, but not GW7647, resulted in delayed onset of the early second phase of formalin‐evoked nociceptive behaviour. Formalin‐evoked nociceptive behaviour was associated with significant reductions in mPFC levels of endogenous PPARα ligands (PEA and OEA). | Okine et al. (2014) |
| Inflammatory | Formalin | GW6471 (PPARα antagonist) and GW9662 (PPARγ antagonist) | 3 nmol–5 μL (GW6471); 36 nmol–5 μL (GW9662) | Intra‐ACC | Sprague–Dawley rats | Both antagonists significantly reduced formalin‐evoked nociceptive behaviour, suggesting facilitatory/permissive roles for these receptors in the ACC in inflammatory pain. | Okine et al. (2017) |
| Inflammatory | Formalin | GW9662 (PPARγ antagonist) | 14.4 nmol/0.2 μL | Intra‐lPAG | Sprague–Dawley rats and Wistar‐Kyoto rats | Pharmacological blockade of PPARγ in the lPAG enhanced formalin‐evoked nociceptive behaviour in WKY, but not SD, rats. | Okine et al. (2016) |
| Inflammatory | Formalin and carrageenan | Fenofibrate and pioglitazone | 100 or 300 mg·kg−1 (fenofibrate; acute and chronic – 7 days); 25, 50 or 100 mg·kg−1 (acute pioglitazone) |
Per os (p.o.) – fenofibrate Intraperitoneal – pioglitazone |
Swiss mice and Wistar rats | Chronic and acute administration of fenofibrate and acute administration of pioglitazone did not inhibit nociceptive responses of mice in the hot plate or in the first phase of the formalin test. The chronic treatment with fenofibrate and acute administration of pioglitazone (same doses) attenuated the second phase of the formalin‐induced nociceptive response. The prolonged treatment with fenofibrate also attenuated the initial phase of the carrageenan‐induced nociceptive behaviour in rats. | Oliveira et al. (2007) |
| Inflammatory | Complete Freund's adjuvant (CFA)‐induced inflammation | Rosiglitazone | 0.3, 3 or 30 μg | Intraplantar | C57BL6 mice | Hyperalgesia to mechanical stimuli was dose‐dependently attenuated on days 5 and 7 after the procedure in mice that received rosiglitazone, which was reversed to the level of vehicle‐injected mice by coadministration of GW9662. In contrast to the effects of rosiglitazone on mechanical stimuli, rosiglitazone had little effect on withdrawal latency to heat stimuli. | Hasegawa‐Moriyama et al. (2013) |
| Inflammatory | Formalin | GW9662 and pioglitazone | 2 mg·kg−1 (GW9662) and 10, 20, 30 or 50 mg·kg−1 (pioglitazone) | Intraperitoneal | Wistar rats | Pioglitazone at doses 30 and 50 mg·kg−1 significantly inhibited the flinching behaviour in phase 1 and, at dose 30 mg·kg−1, in phase 2. GW9662 had no effect in nociceptive behaviour per se, but it attenuated antinociceptive effects of the combined treatment of simvastatin and pioglitazone. | Mansouri et al. (2017a, b) |
| Inflammatory | Formalin | GW9662 and pioglitazone | 2 mg·kg−1 (GW9662) and 10, 20, 30 or 50 mg·kg−1 (pioglitazone) | Intraperitoneal and intraplantar | Wistar rats | Both routes of pioglitazone administration produced antinociception in both phases of formalin‐induced pain. Antinociception caused by i.p. and i.pl. pioglitazone was blocked by GW‐9662 at doses 2 mg·kg−1 (i.p.) and 3 μg per paw (i.pl.). | Mansouri et al. (2017a, b) |
| Inflammatory | Complete Freund's adjuvant (CFA)‐induced | – | – | – | Wistar rats | PPARα was rapidly activated in lumbar spinal cord after CFA intraplantar injection. | Benani et al. (2004) |
| Inflammatory | Formalin, carrageenan and writhing test | Oleanoic acid (OA) | 10, 20 and 40 mg·kg−1 | Oral | Swiss mice | OA treatment inhibits acetic acid‐induced abdominal writhing in mice. OA alone did not produce a significant effect on the first phase of the formalin test but reduced the number of paw licks in the second phase of the formalin test. | Vasconcelos et al. (2006) |
| Inflammatory | Formalin | PEA and GW6471 |
0.2 and 2 mg·kg−1 (GW6471 i.p.), 0.2 or 1 g/5 μL per mouse (GW6471 i.t.), 1 μg/20 μL per mouse (GW6471 i.pl.) 1 or 3 mg·kg−1 (PEA i.p.) |
Intraperitoneal (GW6471 was also administered intraplantar and intrathecal) | ICR mice and −/−α7 mice (C57BL/6 background) | GW6471 blocks and PEA potentiates the antinociceptive effects of α7 nAChR full agonist. PEA and GW6471 alone do not affect formalin‐evoked nociceptive responses. | Donvito et al. (2017) |
| Inflammatory | Formalin – Tempomandibular joint (TMJ) | 15d‐PGJ2 and GW9662 |
0.3, 1 or 3 ng/15 μL per TMJ (GW9662) 100 ng/15 μL per TMJ (15d‐PGJ2) |
Intra‐TMJ | Wistar rats | Treatment with 15d‐PGJ2 attenuated formalin‐evoked nociceptive behaviour in the TMJ. This effect was blocked by GW9662 (PPARγ antagonist). | Pena‐dos‐Santos et al. (2009) |
| Inflammatory | Carrageenan | URB597, GW6471 and WY14643 (PPARα agonist) |
25 μg in 50 μL (URB597) 30 μg in 50 μL (GW6471) 100 μg in 50 μL (WY14643) |
Intraplantar | Sprague–Dawley rats | GW6471 completely abolished the inhibitory effects of URB597 on the carrageenan‐evoked expansion of receptive fields (8 g) and WY14643 significantly attenuated carrageenan‐evoked expansion of peripheral receptive fields of WDR neurons. | Sagar et al. (2008) |
| Inflammatory | Carrageenan and formalin – Tempomandibular joint (TMJ) | 15d‐PGJ2 and GW9662 | 30–300 ng per paw (15d‐PGJ2) | Intraplantar or intra‐TMJ | Wistar rats | 15d‐PGJ2 inhibits the mechanical hypernociception induced carrageenan in the hind paw and formalin in the TMJ. These effects were blocked by GW9662. | Napimoga et al. (2007) |
| Inflammatory | Carrageenan | PEA and siRNA for PPARα | 1.0 μg·h−1 | i.c.v. | Sprague–Dawley rats | Lower PPARα expression was observed in the spinal cord of high fat‐fed rats. PEA significantly attenuated thermal and mechanical hyperalgesia in HF‐fed rats. Intrathecal administration of PPARα siRNA completely abolished the effects of i.c.v. PEA on pain sensitivity. | Wang et al. (2014) |
| Inflammatory | Carrageenean (diet‐induced obesity) | siRNA for PPARα | 10 μL | Intrathecal | Sprague–Dawley rats | Knockdown of spinal PPARα eradicated the beneficial effects of ursolic acid on thermal hyperalgesia and paw oedema, and reversed the spinal cord inflammatory response. | Zhang et al. (2016) |
| Neuropathic and inflammatory | Chronic constriction injury of sciatic nerve (CCI), Freund's adjuvant and carrageenan | PEA and GW7647 (PPARα agonist) | PEA (50 mg) and GW7647 (50 mg); PEA (20 mg·kg−1) s.c. | Intraplantar and subcutaneous | Swiss mice | PEA reduced formalin‐induced pain at i.pl. doses; GW7647 and PEA reduced hyperalgesic responses in the CCI model of neuropathic pain; acute administration of GW7647 and PEA reduced hyperalgesic responses in the complete Freund's adjuvant and carrageenan models of inflammatory pain. | LoVerme et al. (2006) |
| Inflammatory and neuropathic | Chronic constriction injury (CCI) and acetic acid‐, magnesium sulphate‐ and kaolin‐evoked writhing and formalin | GW9662 and PEA |
PEA (20 mg·kg−1 per i.p.) GW9662 (2 mg·kg−1) |
Intraperitoneal | Swiss CD1 mice and PPARα null type mice | PEA had antihyperalgesic effect on mechanical and thermal stimulus. Single intraperitoneal administration of GW9662 produced a reversion of analgesic effect of both compounds tested (butyrate and FBA). | Russo et al. (2016) |
| Inflammatory and visceral | Formalin and writhing |
OEA WY14643 |
0.5, 1, 5, 10 and 20 mg·kg−1 (OEA) 5 and 20 mg·kg−1 (WY14643) |
Intraperitoneal | CD1 mice (wild type and PPARα null) | Treatment with OEA decreased the writhing response induced by acetic acid and formalin‐evoked nociceptive behaviour (both phases) in both wild and KO mice. WY14643 did not affect the early phase of the formalin test whereas it slightly decreased the late phase. | Suardíaz et al. (2007) |
| Neuropathic | Chronic constriction injury of sciatic nerve (CCI) | PEA | 10 mg·kg−1 | Intraperitoneal | Murine | Administration of PEA reduced thermal hyperalgesia and mechanical allodynia; these effects were mediated by PPARγ. | Costa et al. (2008) |
| Neuropathic | Chronic constriction injury of sciatic nerve (CCI) | PEA | 10 mg·kg−1 | Intraperitoneal for 7 days | C57BL/6J mice | Administration of PEA reduced thermal hyperalgesia and mechanical allodynia. | Bettoni et al. (2013) |
| Neuropathic | Spared nerve injury | 15d‐PGJ2 and rosiglitazone (PPARγ agonists) | 25, 50, 100 and 200 μg (15d‐PGJ2) amd 25, 50 and 100 μg (rosiglitazone) | Intrathecal | Sprague–Dawley rats | Treatments with the drugs (dose of 100 μg) decreased mechanical and cold hypersensitivity. Concomitant treatment with PPARγ antagonist (BADGE) reversed these effects. | Churi et al. (2008) |
| Neuropathic | Spared nerve injury | Pioglitazone (PPARγ agonist) | 1, 3 and 10 mg·kg−1·day−1 during 7 days (i.p.) 0, 0.3, 3.0, 30.0 mg·kg−1 daily during 7 days (included in the diet) | Intraperitoneal and oral | Sprague–Dawley RATS | Treatment with pioglitazone had anti‐allodynic and anti‐hyperalgesic effects, reversed by the PPARγ antagonist (GW9662). | Morgenweck et al. (2013) |
| Neuropathic | Spared nerve injury | Pioglitazone (PPARγ agonist) | 2 and 10 mg·kg−1 (i.p.) and 0–300 μg (i.t.) | Intraperitoneal and intrathecal | Sprague–Dawley rats | Treatment rapidly reduced hyperalgesia induced by SNI; administration of GW9662 (PPARγ antagonist) reversed the effects. | Griggs et al. (2015) |
| Neuropathic | Diabetic‐induced hyperalgesia | Pioglitazone (PPARγ agonist) | 30 mg·kg−1·day−1 | Oral (diet) | ZL and ZDF rats | Treatment reduced mechanical and thermal (hot and cold) hyperalgesia in diabetic rats. | Griggs et al. (2016) |
| Neuropathic | Silk suture thread of the sciatic nerve | Pioglitazone (PPARγ agonist) | 1–25 mg·kg−1 | Oral (diet) | ICR mice | Treatment attenuated tactile allodynia. | Maeda et al. (2008) |
| Neuropathic | Partial sciatic nerve ligation | Rosiglitazone (PPARγ agonist) | 3 and 10 mg·kg−1 | Intraperitoneal and local | C57BL6 mice | Systemic rosiglitazone treatment early in the course of progressive inflammation ameliorated tactile allodynia. | Takahashi et al. (2011) |
| Neuropathic | Diabetic‐induced hyperalgesia | PEA | 30 mg·kg−1 | Intraperitoneal | Mice | Treatment with PEA relieves mechanical allodynia. | Donvito et al. (2015) |
| Neuropathic | Paclitaxel‐induced allodynia | PEA and GW6471 (PPARα antagonist) | 30 mg·kg−1 (PEA) and 2 mg·kg−1 (GW6471) | Intraperitoneal, intraplantar, intrathecal and i.c.v. | ICR mice | PEA treatment had anti‐allodynic effects and the treatment with GW6471 (PPARα antagonist) reversed these effects. | Donvito et al. (2016) |
| Neuropathic | Chronic constriction injury of sciatic nerve (CCI) | PEA | 30 mg·kg−1·day−1 | Subcutaneous (daily) | Wild‐type and PPARα−/− (KO) C57BL6 mice | On day 14, PEA prevented pain threshold alterations in Randall–Selitto and Dynamic Plantar Aesthesiometer tests. In PPAR‐α null mice PEA treatment failed to induce pain relief. | Di Cesare Mannelli et al. (2013) |
| Neuropathic | Spared nerve injury | PEA and OEA | 10 mg·kg−1·day−1 during 15 days (OEA and PEA) and 6 nmol per mouse (PEA and OEA) | Intraperitoneal and intra‐mPFC | CD‐1 mice | Repeated PEA and OEA treatments significantly increased both the thermal and mechanical thresholds in SNI mice. PEA microinjection decreased mechanical threshold with maximum effect at 75 min post‐drug. OEA microinjections immediately and transiently reduced mechanical allodynia that lasted up to 30 min post‐injection. | Guida et al. (2015) |
| Neuropathic | Oxaliplatin‐induced neuropathy | PEA | 30 mg·kg−1 (acute and chronic administration) | Intraperitoneal | Sprague–Dawley rats | Single administration of PEA was able to reduce oxaliplatin‐dependent pain induced by mechanical and thermal stimuli. The repeated treatment with PEA prevented lowering of pain threshold as well as increased pain on suprathreshold stimulation. | Di Cesare Mannelli et al. (2015) |
| Neuropathic | Spinal cord injury | Pioglitazone | 0.5, 1.5 or 3.0 mg·kg−1 (pioglitazone) and 2 mg·kg−1 (GW9662) | Intraperitoneal | Sprague–Dawley rats | Pioglitazone treatment significantly increased thermal threshold in spinal cord injured rats compared to the vehicle group. The administration of pioglitazone + GW9662 (PPARγ antagonist) or GW9662 alone did not result in significant differences to post‐SCI surgery rats treated with vehicle. | Park et al. (2007) |
| Neuropathic | Diabetes‐induced neuropathic pain | Pioglitazone | 10 mg·kg−1 | Oral (chronic administration – 28 days) | Sprague–Dawley rats | The chronic administration of pioglitazone did not attenuate the hyperalgesia induced by the high fat diet/streptozotocin (HFD/STZ) model of diabetes. | Byrne et al. (2015) |
| Neuropathic | Sciatic nerve ligation (SNL) | Pioglitazone | 4.5 and 9.0 mg·mg−1 | Intraperitoneal | Wistar rats | Both doses of pioglitazone attenuated hyperalgesia in the hot plate test and the cold allodynia effect of rats submitted to SNL. | Garg et al. (2017) |
| Neuropathic | Partial sciatic nerve ligation (PSL) | Pioglitazone | 1, 5 or 25 mg·kg−1 | Per os (p.o.) during 5 days | ICR mice | Pioglitazone reduced the tactile allodynia at all doses. However, pioglitazone did not affect nociceptive responses in sham mice. | Iwai et al. (2008) |
| Neuropathic | Tibial and sural nerve transection (TSNT) | Rosiglitazone | 2.5, 5 or 10 mg·kg−1 | Per os (p.o.) daily for 28 days | Wistar rats | Administration of rosiglitazone (at 5 and 10 mg·kg−1) reduced the mechanical and cold hyperalgesia induced by TSNT without affecting heat hyperalgesia. | Jain et al. (2009) |
| Neuropathic | Lumbar 5 spinal nerve transection | Pioglitazone | 2.5, 5 or 10 mg·kg−1 | Per os (p.o.) daily for 14 days | Sprague–Dawley rats | Pioglitazone (5 and 10 mg·kg−1) attenuated mechanical hyperalgesia produced by lumbar 5 spinal nerve transection | Jia et al. (2010) |
| Neuropathic | Trigeminal inflammatory compression (TIC) | Pioglitazone, GW0742 (PPAR β/δ agonist), bezafibrate, fenofibrate, GW9662 | 100, 300 or 600 mg·kg−1 (pioglitazone); 1 or 6 mg·kg−1 (GW0742); 100 mg·kg−1 (bezafibrate); 200 mg·kg−1 (fenofibrate); 30 mg·kg−1 (GW9662) |
Oral – pioglitazone 600 mg·kg−1 and bezafibrate 100 mg·kg−1
Intraperitoneal – all the others |
C57BL/6 mice | Systemic administration of pioglitazone attenuates whisker pad mechanical allodynia at doses of 300 and 600 mg·kg−1. Administration of GW9662 prior to pioglitazone (300 mg·kg−1) blocked the analgesic effect of pioglitazone. GW0742 (6 mg·kg−1) partially attenuated mechanical allodynia in mice with TIC injury compared to vehicle treated mice. | Lyons et al. (2017, 2018) |
| Neuropathic | Chronic constriction injury (CCI) | Pioglitazone | 20 mg·kg−1·day−1 | Oral – daily for 14 days | Sprague–Dawley rats | Pioglitazone attenuated the CCI‐induced mechanical and thermal hyperalgesia. | Murad and Ayuob (2015) |
| Neuropathic | Spinal nerve ligation (SNL) | Pioglitazone | 5, 10 or 20 mg·kg−1 | Intraperitoneal – daily for 28 days | Sprague–Dawley rats | Higher doses of pioglitazone attenuated the SNL‐induced mechanical allodynia, cold allodynia, mechanical hyperalgesia and thermal hyperalgesia. | Pottabathini et al. (2016) |
| Neuropathic | Oxaliplatin‐induced neuropathic pain | 15 Thiazolidinones (TZDs) and GW9662 |
40 mg·kg−1 (TZDs) 4 mg·kg−1 (GW9662) |
Intraperitoneal | C57BL/6 mice | Except for compound 14, all TZDs showed antinociceptive properties; these TZDs attenuated oxaliplatin‐induced mechanical hyperalgesia. This effect was prevented by GW9662 (PPARγ antagonist). | Moreira et al. (2017) |
| Neuropathic | Sciatic nerve ligation | GW6471 | 5, 10 or 20 mg·kg−1 | Intraperitoneal | C57BL/6 wild‐type and PPARα null lineages | Higher sensitivity to thermal and mechanical non‐noxious and noxious stimuli, and cold and mechanical allodynia and heat hyperalgesia was observed in mice lacking PPARα. Writhing after acetic acid were also enhanced in mutant mice. The blockade of PPARα did not alter nociceptive behaviour. | Ruiz‐Medina et al. (2012) |
| Neuropathic | Oxaliplatin‐induced neuropathic pain | Rosiglitazone | 3 and 10 mg·kg−1 | Per os (p.o.) daily for 20 days | Sprague–Dawley rats | Rosiglitazone attenuated hyperalgesia and allodynia resulted by oxaliplatin neuropathy. | Zanardelli et al. (2014) |
| Neuropathic | Trigeminal nerve injury (TNI) | Pioglitazone | 100 mg·kg−1 | Intraperitoneally – daily for 7 days | C57BL/6 mice | DCS (NMDA agonist) and pioglitazone combination n attenuated orofacial neuropathic pain and anxiety related behaviours. The treatment with pioglitazone alone did not alter nociceptive behaviour. | Lyons et al. (2017, 2018) |
| Neuropathic | Peripheral nerve injury – L5 Spinal nerve transection | Pioglitazone and GW9662 |
10 mg·kg−1 (pioglitazone) 2 mg·kg−1 (GW9662) |
Intraperitoneal | Sprague–Dawley rats | Pioglitazone improved the mechanical hyperalgesia in operated rats. This effect was reversed by GW9662. | Jia et al. (2013) |
| Neuropathic | Chronic constriction injury (CCI) | Pioglitazone and GW9662 |
10 mg·kg−1 (pioglitazone) 2 mg·kg−1 (GW9662) |
Intraperitoneal | Sprague‐Dawley rats | Treatment with pioglitazone attenuated CCI‐induced hyperalgesia. This effect was reverrsed by GW9662 administration. | Jia et al. (2016) |
| Neuropathic and Visceral | Acetic acid induced visceral pain, sciatic nerve injury (SNI) | MK886 (PPARα antagonist) | 2 mg·kg−1 | Intraperitoneal |
ICR mice, Kunming mice and C57BL/6J mice and PPAR‐α knockout mice (−/−) |
F96 (selective NAAA inhibitor) had an overall antinociceptive effect in the different models and tests carried out in the study. This effect was widely blocked by PPARα antagonist MK886 and by genetic disruption of PPAR‐α. | Yang et al. (2015) |
| Visceral | Diarrhoea‐predominant irritable bowel syndrome (D‐IBS) | Pioglitazone and GW9662 | 2 mg·kg−1 (pioglitazone); 3 mg·kg−1 (GW9662) | Intraperitoneal on days 7, 9 and 11 | Wistar rats | Pioglitazone reduced visceral hypersensitivity and defecation frequency and increased nociceptive thresholds. | Paragomi et al. (2014) |
| Other | – | PEA | 3 and 6 nmol | Intra‐vlPAG | Wistar rats | Reduced thermo‐nociceptive threshold, as well as on/off cell activity in the rostro‐ventromedial medulla (RVM). | De Novellis et al. (2012) |
| Other | – | Pioglitazone and GW9662 (PPARγ antagonist) |
10 or 30 mg·kg−1 (pioglitazone) 2.5 or 5 mg·kg−1 (GW9662) |
Oral (via gavage) – Daily (concomitant with morphine administration or for the prior 2 days before testing) |
C57 mice | Treatment with pioglitazone attenuated the development of morphine tolerance. Pioglitazone administration in mice that were not chronically treated with morphine does not have an effect in nociception. Pretreatment with GW9662 reversed the effects of pioglitazone in morphine‐treated rats. GW9662 alone does not have an effect in nociceptive responses. The development of tolerance for morphine is more pronounced in PPARγ knockout mice. | De Guglielmo et al. (2014) |
| Other | – | Pioglitazone and GW9662 |
20 or 40 mg·kg−1 (pioglitazone) 2 mg·kg−1 (GW9662) |
Per os (p.o.) (pioglitazone) – daily for 17 days concomitant with morphine treatment Subcutaneous (GW9662) |
Wistar rats | Treatment with pioglitazone attenuated the development of morphine tolerance. GW‐9662 administration 30 min before pioglitazone antagonized the mentioned pioglitazone‐induced effects. | Ghavimi et al. (2015) |
| Other | – | Pioglitazone and GW9662 |
5, 10, 20 or 40 mg·kg−1 (pioglitazone) 2 mg·kg−1 (GW9662) |
Per os (p.o.) (pioglitazone) – daily for 17 days concomitant with morphine treatment Subcutaneous (GW9662) – daily before pioglitazone administration |
Wistar Rats | The highest dose of pioglitazone per se did not alter the pain threshold in tail‐flick test. Treatment with pioglitazone attenuated the development of morphine tolerance and GW‐9662 administration 30 min before pioglitazone antagonized the pioglitazone‐induced effects. | Ghavimi et al. (2014) |
| Other | Incisional pain | Rosiglitazone | 25 μg | Intraplantar (in loci) | BKS.Cg − +Leprdb/+Leprdb/Jcl Mice | Rosiglitazone alleviates mechanical hyperalgesia resulted by the incision. | Saito et al. (2016) |
| Other | Incisional pain | Rosiglitazone | 0.5 mg·mL−1 | Local (intraplantar) | C57BL/6 mice | Local administration of rosiglitazone immediately after the procedure ameliorates thermal and mechanical hyperalgesia. | Hasegawa‐Moriyama et al. (2012) |
The Table was constructed based on searches within Pubmed central (https://www.ncbi.nlm.nih.gov/pmc/) and Google scholar (https://scholar.google.co.uk/) using combinations of one or more of the following search terms: PPARs, PPARα, PPARβ, PPARγ, PEA, OEA, synthetic PPAR ligands, pharmacological effects, CNS, brain, spinal cord, dorsal root ganglion, animal models and pain.
PPARα protein expression in the spinal cord has also been demonstrated in earlier studies (Benani et al., 2004, Okine et al., 2015). The functional relevance of PPARα signalling in the spinal cord to nociceptive processing is demonstrated at least in part by the reported increases in PPARα activation or protein expression in animal models of chronic inflammatory and neuropathic pain states. For example, using electrophoretic mobility shift assays (EMSA), Benani and colleagues were able to demonstrate a rapid increase in activation of the PPARα protein isoform in the rat spinal cord after injection of complete Freund's adjuvant into the hind paw (Benani et al., 2004). Moreover, increased PPARα protein expression in the ipsilateral spinal cord was observed in the rat spinal nerve ligation model of neuropathic pain (Okine et al., 2015). Furthermore, down‐regulation of PPARα protein expression in the spinal cord contributed to augmented peripheral inflammation and inflammatory hyperalgesia in diet‐induced obese rats (Wang et al., 2014). While the pathophysiological relevance of PPARα activation or changes in PPARα protein expression in the spinal cord during hyperalgesia requires further investigation, these findings provide evidence for PPARα, as a potentially important player in spinal pain processing. In addition to PPARα, PPARγ is also expressed in the spinal cord (Table 1). Increased PPARγ protein expression in the spinal trigeminal caudalis 3 weeks after trigeminal nerve injury in mice has been reported to play a significant role in trigeminal nociceptive transmission, as demonstrated by the attenuation of whisker pad mechanical allodynia (Lyons et al., 2017), and identifies PPARγ as a potential therapeutic target for orofacial neuropathic pain.
In comparison, there is a paucity of data on the expression or activation of PPARβ/δ in the spinal cord in inflammatory or neuropathic pain states. In this regard, further characterization of PPARβ/δ is essential to establish whether changes in expression or endogenous activation of the receptor occur in pain states, and the extent to which they contribute to spinal pain processing.
As shown in Table 1, all three PPAR isoforms are widely expressed supraspinally, in key brain regions involved in pain processing. The expression of PPARs at key relay sites such as the thalamus and the midbrain periaqueductal grey (PAG) may reflect a role for PPAR signalling in modulating the activity of ascending and descending pain pathways. Furthermore, the presence of PPARs within cortical regions and the amygdala suggests potential involvement of PPAR signalling in modulating cognitive or affective components of pain. While there is currently no direct evidence in support of this hypothesis, this view is however consistent with the role of both the cortex and amygdala as key brain regions involved in the modulation of the cognitive‐affective components of pain (see Fuchs et al., 2014; Neugebauer, 2015).
Evidence from pharmacological or genetic manipulation studies for a role of PPARs in pain
Pharmacological or genetic manipulation of PPARα and PPARγ using selective agonists, antagonists or gene knockout approaches specifically targeting these receptors within the pain pathway has been shown to alter nociceptive processing, demonstrated by changes in neuronal activity or behavioural responses in animal models of inflammatory and neuropathic pain (Figure 1). Both PPARα and PPARγ regulate the release of pro‐inflammatory mediators associated with tissue or nerve injury through the inhibition of pro‐inflammatory signalling pathways such as NF‐κB activation (Cuzzocrea et al., 2008, Delerive et al., 2000) and suppression of downstream pro‐inflammatory molecules including COX‐2 and iNOS (D'Agostino et al., 2009), two key players in the development of chronic pain states. The majority of pharmacological studies to date demonstrate antinociceptive effects of both endogenous and synthetic agonists of PPARα and PPARγ in animal models of inflammatory and neuropathic pain (for a comprehensive summary of the published studies to date see, Table 2). It is pertinent to note that a significant proportion of preclinical studies investigating the role of endogenous PPAR ligands in nociceptive processing in the periphery and the CNS have mainly focused on the effects of PEA, with relatively little attention given to the role of OEA. One possible reason that may account for this apparent bias is the reported activation of TRPV1 pronociceptive non‐selective cation channels (the vanilloid receptors), by OEA (Ahern, 2003). Thus, it is possible that any PPAR‐mediated analgesic effects of OEA are likely to be nullified by its TRPV1‐mediated pronociceptive effects, as demonstrated in an animal model of neuropathic pain (Guida et al., 2015).
The pharmacological effects of PEA involve both transcription‐dependent and transcription‐independent or non‐genomic mechanisms. While the former account primarily for the anti‐inflammatory effects associated with PPAR activation, the non‐genomic mechanisms are thought to underlie the rapid antinociceptive effects of not only PEA but also other synthetic PPAR agonists in animal models of inflammatory and neuropathic pain (Churi et al., 2008, LoVerme et al., 2006; Donvito et al., 2015). It is, however, important to note that the non‐genomic mechanisms mediating the effects of PEA are not independent of PPAR expression or activation. Indeed, evidence from studies with PPAR knockout mice suggests that the modulation of medium and large Ca2+ channels associated with the rapid antinociceptive effects of PEA and other synthetic PPARα agonists on inflammatory pain behaviour in mice are contingent upon PPARα receptor expression in the DRG (LoVerme et al., 2006). Given that these rapid antinociceptive effects are incompatible with the duration of longer term transcription‐dependent mechanisms, the modulation of Ca2+ channels in this instance may be a by‐product of protein–protein interactions induced by changes in PPAR protein conformation following the binding of agonist to the receptor. Further studies aimed at identifying the precise mechanisms involved could potentially lead to the identification of novel therapeutic targets for the treatment of pain.
The non‐genomic effects of PEA may also involve the indirect activation of other receptors, such as cannabinoid receptors, mediated by anandamide (AEA) through the so‐called ‘entourage hypothesis’ (LoVerme et al., 2005). In this context, competition by PEA for fatty acid amide hydrolase (FAAH)‐mediated hydrolysis, is thought to provide a ‘sparing effect’ on AEA hydrolysis by FAAH, resulting in enhanced signalling at endocannabinoid targets, in particular CB1 or CB2 receptors, to produce analgesia (Tuo et al., 2017). In keeping with this, we have recently demonstrated a role for CB1 receptors in the antinociceptive effects of PEA injected directly into the anterior cingulate cortex (ACC) in the rat formalin test (Okine et al., 2016). Moreover, given the preferential binding of AEA over PEA to PPARγ (Bouaboula et al., 2005), it is possible that entourage‐mediated signalling involving AEA is likely to underpin the PPARγ‐mediated antinociceptive effects of PEA (Costa et al., 2008). Indeed, AEA binds to and activates PPARα in addition to PPARγ (O'Sullivan and Kendall, 2010). Here, it is important to note that these findings have implications not only for the interpretation of data demonstrating the analgesic effects of endogenous PPAR ligands such as PEA and OEA or inhibitors of FAAH which enhance levels of endogenous PPAR agonists but also the design of such experiments given the possible involvement of other receptor systems other than PPARs. In this regard, the inclusion of appropriate robust control experiments involving PPAR antagonists or gene knockout should always be considered when examining a role for PPARs as mediators of the pharmacological effects of endogenous PPAR ligands.
The analgesic effects of PPAR agonists may also be mediated via modulation of cellular organelles. For example, a combination drug therapy of the synthetic PPARγ agonist pioglitazone with D‐cycloserine attenuates chronic orofacial neuropathic pain and associated anxiety by improving mitochondrial function following trigeminal nerve injury (Lyons et al., 2018). With the molecular mechanisms continuing to be elucidated, these findings widen the scope and increase the appeal of PPAR agonists as therapeutic agents for treating pain. Furthermore, given the involvement of both genomic and non‐genomic mechanisms in mediating the effects of PPAR agonists, further studies aimed at determining which mechanisms are predominant in different types of pain will be important for the optimization of the analgesic effects of PPAR agonists.
It is however important to note that while the weight of evidence is in favour of antinociceptive effects of PPARα or PPARγ activation at multiple sites within the pain pathway, recent findings from our group also reveal a pain permissive or facilitatory role for PPAR signalling in discrete brain regions such as the ACC (Okine et al., 2017, Okine et al., 2014). Intra‐ACC injection of GW6471 (selective PPARα antagonist) or GW9662 (selective PPARγ antagonist) significantly suppressed the onset of formalin‐evoked nociceptive behaviour in rats (Okine et al., 2017). Such permissive or facilitatory roles of endogenous PPAR activation within the ACC may allow the animal to perceive pain and take the necessary actions to escape from, or avoid injury.
The specific role of PPARβ/δ activation in pain processing remains largely unknown, despite molecular evidence demonstrating the presence of the receptor at key sites within the pain pathway such as the spinal cord, thalamus and PAG (Table 1; Figure 1). However, in a previous study, administration of GW0742, a selective PPARβ/δ receptor agonist (0.1 mg·kg−1 per i.p. for 4 days) significantly decreased mechanical and thermal hyperalgesia in adult male Wistar rats, induced by carrageenan injection into the hind paw, compared with vehicle‐treated controls. These effects were reversed in the presence of the selective PPAR β/δ antagonist GSK0660 (0.3 mg·kg−1 per i.p. for 4 days) (Gill et al., 2013). These findings demonstrate the potential of PPAR β/δ agonists as therapeutic agents for the treatment pain. Further preclinical studies are however needed to understand fully the extent to which PPARβ/δ‐mediated signalling modulates nociceptive transmission within the CNS.
Evidence from clinical trials
Over the last couple of decades, the analgesic effects of PEA, an endogenous PPAR agonist, or its derivatives, have been demonstrated in several clinical trials in different pain conditions. In a recent comprehensive review of 21 clinical trials, Gabrielsson et al., reported that oral or sublingual treatment with PEA or micronized PEA (PEA‐μm; reduced crystal particles of PEA that enhance the dissolution and reduce the absorption variability) reduced pain intensity in patients with neuropathic and inflammatory joint pain phenotypes (Gabrielsson et al., 2016). Significantly, these treatments were not associated with any marked side effects. Similar reports of the analgesic effects of PEA in clinical trials have been discussed in another comprehensive review by Hesselink and Hekker (2012). These studies report that administration of PEA (doses ranging from 300 to 600 mg·day−1; mostly orally administered as tablets) is effective against a range of pain conditions including neuropathic pain, low back pain and postoperative pain.
In contrast, Andresen and colleagues report that a 12‐week treatment with PEA‐μm did not alleviate pain in patients with spinal cord injury‐induced neuropathic pain, compared to placebo‐treated patients (Andresen et al., 2016). The authors however point out that the limited knowledge on PEA‐μm pharmacokinetics, including information on diffusion to the CSF, make it difficult to draw more specific conclusions. It is also possible that the heterogeneity in the population of spinal cord injury pain phenotypes could have affected the outcome of this study. These clinical effects of PEA however, while suggestive of a role for PPAR signalling, do not necessarily rule out the involvement of other receptor systems, given the multiple signalling pathways mediating the pharmacological effects of PEA as demonstrated in preclinical studies and discussed elsewhere in this review. In this regard, the use of synthetic PPAR agonists in clinical trials may be more beneficial and informative. In keeping with this line of argument, a more defined role for PPAR signalling in modulating human pain conditions was demonstrated in a study by Smith and colleagues, who reported a reduction in occurrence of myalgia, a muscoskeletal pain disorder, in men receiving clofibrate, an approved PPARα agonist used clinically for the treatment of dyslipidemia (Smith et al., 1970). However, subsequent attempts at replicating these early promising results using other fibrates to alleviate muscular pain have not been successful (Biga et al., 2005). Nevertheless, these drugs were found to be effective in attenuating pain associated with rheumatoid arthritis and osteoarthritic pain (van Eekeren et al., 2013). These findings indicate that synthetic PPARα agonists can have analgesic effects in specific types of pain.
Synthetic agonists of PPARγ are currently used clinically as insulin sensitizers in the treatment of non‐insulin dependent (Type 2) diabetes. However, despite preclinical evidence demonstrating their analgesic effects in a variety of animal models of inflammatory and neuropathic pain (see Table 2), to our knowledge, there are currently no published clinical studies investigating their effects on pain in human subjects or patients. Similarly, there is a paucity of clinical studies investigating the effects of synthetic PPARβ agonists on pain.
A potential role for PPAR signalling in interactions between pain and negative affect
The close relationship between stress (and stress‐related disorders such as anxiety and depression) and chronic pain is now widely recognized (Jennings et al., 2014, Olango and Finn, 2014). Although the role of PPAR signalling in the modulation of stress‐pain interactions remains largely unexplored, the abundant expression of PPARs in key brain regions involved in the modulation of the cognitive and affective components of pain such as the amygdala and PAG (Table 1 and Figure 1), and the availability of endogenous PPAR ligands in these brain structures, supports a potential role for the PPAR signalling system in stress–pain interactions and as a potential therapeutic target for the treatment of comorbid chronic pain and affective disorders. This view is also consistent with our recent demonstrations of enhanced second phase formalin‐evoked nociceptive behaviour following selective blockade of PPARγ in the lateral PAG in Wistar‐Kyoto (WKY) but not Sprague–Dawley (SD) rats (Okine et al., 2017). The WKY rat strain is stress‐hyperresponsive and exhibits a hyperalgesic phenotype to nociceptive stimuli compared with SD rats and is considered a suitable genetic model for studying stress–pain interactions (Burke et al., 2010, Rea et al., 2014, O′Mahony et al., 2013). While the specific contribution of PPARγ signalling to the stress hyperresponsive phenotype of WKY rats is currently not clear, the differential effects of pharmacological modulation of PPARγ in the lateral PAG on formalin‐evoked nociceptive behaviour in SD and WKY rats suggests an important role for this receptor in a genetic background that is prone to stress and hypersensitivity to nociceptive stimuli. These findings also suggest that PPARγ‐mediated signalling in the lateral PAG may represent a potential therapeutic target for future development of effective therapies for treating comorbid chronic pain and stress‐related disorders such as anxiety and depression. The therapeutic potential of PPARγ for treatment of pain and mood disorder comorbidity is also supported by evidence that pioglitazone attenuates chronic constriction injury (CCI)‐induced depression‐related behaviour in the forced swim test in rats (Garg et al., 2017)), reduces anxiety‐like behaviour in a mouse model of chronic orofacial neuropathic pain (Lyons et al., 2018) and augments both the anti‐depressant and the antinociceptive effects of fluoxetine in the rat CCI model of neuropathic pain (Murad and Ayuob, 2015). Additional studies on the therapeutic potential of PPAR agonists (including those for PPARα and PPARβ/δ) for treatment of the affective/emotional component of chronic pain are warranted.
Future perspectives
Synthetic PPARα and PPARγ agonists such as clofibrate and rosiglitazone, respectively, are currently available clinically for the treatment of medical conditions such as dyslipidemia and non‐insulin dependent (Type 2) diabetes, the latter of which is often associated with altered sensory processing in the extremities. The current clinical use of these drugs provides an opportunity for clinical trials to explore and evaluate their potential as novel analgesics for the treatment of intractable chronic pain conditions such as diabetic neuropathy.
It is however important to note that the development of analgesics targeting the PPAR signalling system must also take into account their potential adverse side effects (Table 3), given the important physiological roles of the PPAR signalling system. For example, the inhibitory effects of PEA on gastrointestinal motility (Capasso et al., 2001) or on contraction of human aortic smooth muscle, associated with PPARα activation (Staels et al., 1998,) may limit the therapeutic efficacy of PPAR‐targeting analgesics. Nonetheless, evidence from clinical studies using PEA so far suggests that these side effects are unlikely to be of major concern over a short duration of administration of PPAR agonist and that the therapeutic benefits may outweigh the side effects. Moreover, the demonstration of cardioprotective and anti‐tumour effects (Wright et al., 2014) of some synthetic PPAR agonists, but not others, suggests that these reported adverse side effects may be agonist‐dependent rather than PPAR target‐dependent. Further studies are however required to determine the long‐term effects of persistent pharmacological modulation of PPAR signalling in chronic pain states.
Table 3.
Summary of adverse effects associated with PPAR activation
|
Cardiovascular effects Tumorigenic effects Others peripheral effects |
• Increased risk of myocardial infarctiona
,
b
,
c
• Increased risk of bladder cancer in patients on long‐term therapy with synthetic PPAR agonistsc , d • Haemangioma and renal pelvic tumoursd • PPAR agonist induced peripheral oedema formation via fluid retentione , f • Increased liver toxicityg • Inhibition of gastrointestinal motilityh • Inhibition of human aortic smooth‐muscle contraction associated with PPARα activationi |
The role of PPAR signalling in the development or modulation of other chronic pain conditions, such as osteoarthritis, cancer pain and migraine requires further study, as does the interaction of PPAR signalling with other well‐characterized endogenous pain control systems and currently prescribed analgesics. On this latter point, the PPARγ agonist pioglitazone has been shown to attenuate tolerance to morphine in a rat model of inflammatory pain (Ghavimi et al., 2015, Ghavimi et al., 2014) and in the mouse tail immersion test (de Guglielmo et al., 2014). Similar potential synergistic antinociceptive interactions with the cannabinoid (Russo et al., 2007) and TRPV1 (Ambrosino et al., 2014) signalling systems have been reported. In respect of the latter study, the evidence suggests that the potential antinociceptive effects of this synergistic interaction are likely to be facilitated by PPARα‐dependent activation of TRPV1 channels and subsequent desensitization of the receptor. Elsewhere, a synergistic antinociceptive interaction between PEA and the opiate drug, tramadol, has been demonstrated in the mouse formalin test (Déciga‐Campos et al., 2015). The antinociceptive mechanisms of the PEA and tramadol combination involved the opioid receptor, TRPV1 and PPARα. Significantly, the sedative effects of the combination of PEA and tramadol were minimal compared with those observed with individual treatments. Collectively, these findings make a compelling case for an increased understanding of PPAR signalling and its crosstalk with other analgesic targets. Such knowledge could lead to the development of novel PPAR signalling based‐analgesic strategies.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was funded by grants from Science Foundation Ireland (10/IN.1/B2976), the Irish Research Council, CNPq ‐ Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil (207530/2014‐9) and the National University of Ireland Galway.
Okine, B. N. , Gaspar, J. C. , and Finn, D. P. (2019) PPARs and pain. British Journal of Pharmacology, 176: 1421–1442. 10.1111/bph.14339.
References
- Ahern GP (2003). Activation of TRPV1 by the satiety factor oleoylethanolamide. J Biol Chem 278: 30429–30434. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosino P, Soldovieri MV, De Maria M, Russo C, Taglialatela M (2014). Functional and biochemical interaction between PPARα receptors and TRPV1 channels: potential role in PPARα agonists‐mediated analgesia. Pharmacol Res 87: 113–122. [DOI] [PubMed] [Google Scholar]
- Andresen SR, Bing J, Hansen RM, Biering‐Sorensen F, Johannesen IL, Hagen EM et al (2016). Ultramicronized palmitoylethanolamide in spinal cord injury neuropathic pain: a randomized, double‐blind, placebo‐controlled trial. Pain 157: 2097–2103. [DOI] [PubMed] [Google Scholar]
- Aoki T (2007). Current status of carcinogenicity assessment of peroxisome proliferator‐activated receptor agonists by the US FDA and a mode‐of‐action approach to the carcinogenic potential. J Toxicol Pathol 20: 197–202. [Google Scholar]
- Bełtowski J, Rachańczyk J, Włodarczyk M (2013). Thiazolidinedione‐induced fluid retention: recent insights into the molecular mechanisms. PPAR Res 2013: 628628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benani A, Heurtaux T, Netter P, Minn A (2004). Activation of peroxisome proliferator‐activated receptor alpha in rat spinal cord after peripheral noxious stimulation. Neurosci Lett 369: 59–63. [DOI] [PubMed] [Google Scholar]
- Bettoni I, Comelli F, Colombo A, Bonfanti P, Costa B (2013). Non‐neuronal cell modulation relieves neuropathic pain: efficacy of the endogenous lipid palmitoylethanolamide. CNS Neurol Disord Drug Targets 12: 34–44. [DOI] [PubMed] [Google Scholar]
- Biga J, Vigreux P, Lapeyre‐Mestre M, Montastruc JL (2005). Muscular pain and hypolipidaemic drugs: a cross‐sectional study in cardiological outpatient practice. Therapie 60: 295–297. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Hilairet S, Marchand J, Fajas L, Fur GL, Casellas P (2005). Anandamide induced PPARγ transcriptional activation and 3T3‐L1 preadipocyte differentiation. Eur J Pharmacol 517: 174–181. [DOI] [PubMed] [Google Scholar]
- Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W (1996). Differential expression of peroxisome proliferator‐activated receptors (PPARs): tissue distribution of PPAR‐alpha, ‐beta, and ‐gamma in the adult rat. Endocrinology 137: 354–366. [DOI] [PubMed] [Google Scholar]
- Burke NN, Hayes E, Calpin P, Kerr DM, Moriarty O, Finn DP et al (2010). Enhanced nociceptive responding in two rat models of depression is associated with alterations in monoamine levels in discrete brain regions. Neuroscience 171: 1300–1313. [DOI] [PubMed] [Google Scholar]
- Byrne FM, Cheetham S, Vickers S, Chapman V (2015). Characterisation of pain responses in the high fat diet/streptozotocin model of diabetes and the analgesic effects of antidiabetic treatments. J Diabetes Res 2015: 752481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso R, Izzo AA, Fezza F, Pinto A, Capasso F, Mascolo N et al (2001). Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. Br J Pharmacol 134: 945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy MV, Zhu Y, López M, Yin L, Wozniak DF, Coleman T et al (2007). Brain fatty acid synthase activates PPARα to maintain energy homeostasis. J Clin Invest 117: 2539–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churi SB, Abdel‐Aleem OS, Tumber KK, Scuderi‐Porter H, Taylor BK (2008). Intrathecal rosiglitazone acts at peroxisome proliferator‐activated receptor‐gamma to rapidly inhibit neuropathic pain in rats. J Pain 9: 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G (2008). The endogenous fatty acid amide, palmitoylethanolamide, has anti‐allodynic and anti‐hyperalgesic effects in a murine model of neuropathic pain: involvement of CB(1), TRPV1 and PPARγ receptors and neurotrophic factors. Pain 139: 541–550. [DOI] [PubMed] [Google Scholar]
- Crisafulli C, Cuzzocrea S (2009). The role of endogenous and exogenous ligands for the peroxisome proliferator‐activated receptor alpha (PPAR‐alpha) in the regulation of inflammation in macrophages. Shock 32: 62–73. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Bruscoli S, Mazzon E, Crisafulli C, Donato V, Di Paola R et al (2008). Peroxisome proliferator‐activated receptor‐α contributes to the anti‐inflammatory activity of glucocorticoids. Mol Pharmacol 73: 323–337. [DOI] [PubMed] [Google Scholar]
- D'Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E et al (2009). Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF‐κB nuclear signalling in dorsal root ganglia. Eur J Pharmacol 613: 54–59. [DOI] [PubMed] [Google Scholar]
- Déciga‐Campos M, Ramírez‐Marín PM, López‐Muñoz FJ (2015). Synergistic antinociceptive interaction between palmitoylethanolamide and tramadol in the mouse formalin test. Eur J Pharmacol. 765: 68–74. [DOI] [PubMed] [Google Scholar]
- De Filippis D, Luongo L, Cipriano M, Palazzo E, Cinelli MP, De Novellis V et al (2011). Palmitoylethanolamide reduces granuloma‐induced hyperalgesia by modulation of mast cell activation in rats. Mol Pain 7: 3–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guglielmo G, Kallupi M, Scuppa G, Stopponi S, Demopulos G, Gaitanaris G et al (2014). Analgesic tolerance to morphine is regulated by PPARγ. Br J Pharmacol 171: 5407–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Novellis V, Luongo L, Guida F, Cristino L, Palazzo E, Russo R et al (2012). Effects of intra‐ventrolateral periaqueductal grey palmitoylethanolamide on thermoceptive threshold and rostral ventromedial medulla cell activity. Eur J Pharmacol 676: 41–50. [DOI] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Vanden Berghe W, Fruchart JC, Haegeman G, Staels B (2002). DNA binding‐independent induction of IκBα gene transcription by PPARα. Mol Endocrinol 16: 1029–1039. [DOI] [PubMed] [Google Scholar]
- Delerive P, Fruchart JC, Staels B (2001). Peroxisome proliferator‐activated receptors in inflammation control. J Endocrinol 169: 453–459. [DOI] [PubMed] [Google Scholar]
- Delerive P, Gervois P, Fruchart JC, Staels B (2000). Induction of IκBα expression as a mechanism contributing to the anti‐inflammatory activities of peroxisome proliferator‐activated receptor‐α activators. J Biol Chem 275: 36703–36707. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Wahli W (1999). Peroxisome proliferator‐activated receptors: nuclear control of metabolism. Endocr Rev 20: 649–688. [DOI] [PubMed] [Google Scholar]
- Di Cesare Mannelli L, D'Agostino G, Pacini A, Russo R, Zanardelli M, Ghelardini C et al (2013). Palmitoylethanolamide is a disease‐modifying agent in peripheral neuropathy: pain relief and neuroprotection share a PPAR‐alpha‐mediated mechanism. Mediators Inflamm 2013: 328797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cesare Mannelli L, Pacini A, Corti F, Boccella S, Luongo L, Esposito E et al (2015). Antineuropathic profile of N‐palmitoylethanolamine in a rat model of oxaliplatin‐induced neurotoxicity. PLoS ONE 10: e0128080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donvito G, Bettoni I, Comelli F, Colombo A, Costa B (2015). Palmitoylethanolamide relieves pain and preserves pancreatic islet cells in a murine model of diabetes. CNS Neurol Disord Drug Targets 14: 452–462. [DOI] [PubMed] [Google Scholar]
- Donvito G, Wilkerson JL, Damaj MI, Lichtman AH (2016). Palmitoylethanolamide reverses paclitaxel‐induced allodynia in mice. J Pharmacol Exp Ther 359: 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donvito G, Bagdas D, Toma W, Rahimpour E, Jackson A, Meade JA et al (2017). The interaction between alpha 7 nicotinic acetylcholine receptor and nuclear peroxisome proliferator‐activated receptor‐α represents a new antinociceptive signaling pathway in mice. Exp Neurol 295: 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W (1992). Control of the peroxisomal beta‐oxidation pathway by a novel family of nuclear hormone receptors. Cell 68: 879–887. [DOI] [PubMed] [Google Scholar]
- Floyd JS, Barbehenn E, Lurie P, Wolfe SM (2009). Case series of liver failure associated with rosiglitazone and pioglitazone. Pharmacoepidemiol Drug Saf 18: 1238–1243. [DOI] [PubMed] [Google Scholar]
- Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F et al (2003). Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR‐[alpha]. Nature 425: 90–93. [DOI] [PubMed] [Google Scholar]
- Friedland SN, Leong A, Filion KB, Genest J, Lega IC, Mottillo S et al (2012). The cardiovascular effects of peroxisome proliferator‐activated receptor agonists. Am J Med 125: 126–133. [DOI] [PubMed] [Google Scholar]
- Fuchs PN, Peng YB, Boyette‐Davis JA, Uhelski ML (2014). The anterior cingulate cortex and pain processing. Front Integr Neurosci 8: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielsson L, Mattsson S, Fowler CJ (2016). Palmitoylethanolamide for the treatment of pain: pharmacokinetics, safety and efficacy. Br J Clin Pharmacol 82: 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Deshmukh Vishwajit R, Prasoon P (2017). Possible modulation of PPAR‐γ cascade against depression caused by neuropathic pain in rats. J Basic Clin Physiol Pharmacol 28: 593–600. [DOI] [PubMed] [Google Scholar]
- Ghavimi H, Charkhpour M, Ghasemi S, Mesgari M, Hamishehkar H, Hassanzadeh K et al (2015). Pioglitazone prevents morphine antinociceptive tolerance via ameliorating neuroinflammation in rat cerebral cortex. Pharmacol Rep 67: 78–84. [DOI] [PubMed] [Google Scholar]
- Ghavimi H, Hassanzadeh K, Maleki‐Dizaji N, Azarfardian A, Ghasami S, Zolali E et al (2014). Pioglitazone prevents morphine antinociception tolerance and withdrawal symptoms in rats. Naunyn Schmiedebergs Arch Pharmacol 387: 811–821. [DOI] [PubMed] [Google Scholar]
- Gill N, Bijjem K, Sharma P (2013). Anti‐inflammatory and anti‐hyperalgesic effect of all‐trans retinoic acid in carrageenan‐induced paw edema in Wistar rats: involvement of peroxisome proliferator‐activated receptor‐β/δ receptors. Indian J Pharmacol 45: 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S, Tugwood JD, Issemann I (1992). The molecular mechanism of peroxisome proliferator action: a model for species differences and mechanistic risk assessment. Toxicol Lett, 64–65 Spec No : 131–139. [DOI] [PubMed] [Google Scholar]
- Griggs RB, Donahue RR, Adkins BG, Anderson KL, Thibault O, Taylor BK (2016). Pioglitazone inhibits the development of hyperalgesia and sensitization of spinal nociresponsive neurons in type 2 diabetes. J Pain 17: 359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griggs RB, Donahue RR, Morgenweck J, Grace PM, Sutton A, Watkins LR et al (2015). Pioglitazone rapidly reduces neuropathic pain through astrocyte and nongenomic PPARγ mechanisms. Pain 156: 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guida F, Luongo L, Marmo F, Romano R, Iannotta M, Napolitano F et al (2015). Palmitoylethanolamide reduces pain‐related behaviors and restores glutamatergic synapses homeostasis in the medial prefrontal cortex of neuropathic mice. Mol Brain 8: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa‐Moriyama M, Kurimoto T, Nakama M, Godai K, Kojima M, Kuwaki T et al (2013). Peroxisome proliferator‐activated receptor‐gamma agonist rosiglitazone attenuates inflammatory pain through the induction of heme oxygenase‐1 in macrophages. Pain 154: 1402–1412. [DOI] [PubMed] [Google Scholar]
- Hasegawa‐Moriyama M, Ohnou T, Godai K, Kurimoto T, Nakama M, Kanmura Y (2012). Peroxisome proliferator‐activated receptor‐gamma agonist rosiglitazone attenuates postincisional pain by regulating macrophage polarization. Biochem Biophys Res Commun 426: 76–82. [DOI] [PubMed] [Google Scholar]
- Hesselink JMK, Hekker TAM (2012). Therapeutic utility of palmitoylethanolamide in the treatment of neuropathic pain associated with various pathological conditions: a case series. J Pain Res 5: 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impellizzeri D, Di Paola R, Cordaro M, Gugliandolo E, Casili G, Morittu VM et al (2016). Adelmidrol, a palmitoylethanolamide analogue, as a new pharmacological treatment for the management of acute and chronic inflammation. Biochem Pharmacol 119: 27–41. [DOI] [PubMed] [Google Scholar]
- Issemann I, Green S (1990). Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347: 645–650. [DOI] [PubMed] [Google Scholar]
- Iwai S, Maeda T, Kiguchi N, Kobayashi Y, Fukazawa Y, Ozaki M et al (2008). Pioglitazone attenuates tactile allodynia and microglial activation in mice with peripheral nerve injury. Drug Discov Ther 2: 353–356. [PubMed] [Google Scholar]
- Jain V, Jaggi AS, Singh N (2009). Ameliorative potential of rosiglitazone in tibial and sural nerve transection‐induced painful neuropathy in rats. Pharmacol Res 59: 385–392. [DOI] [PubMed] [Google Scholar]
- Jennings EM, Okine BN, Roche M, Finn DP (2014). Stress‐induced hyperalgesia. Prog Neurobiol 121: 1–18. [DOI] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Robinson I, Garle MJ, Patel A, Sun Y et al (2008). Inhibition of fatty acid amide hydrolase and cyclooxygenase‐2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator‐activated receptor‐alpha in a model of inflammatory pain. Neuropharmacology 55: 85–93. [DOI] [PubMed] [Google Scholar]
- Jia HB, Wang XM, Qiu LL, Liu XY, Shen JC, Ji Q et al (2013). Spinal neuroimmune activation inhibited by repeated administration of pioglitazone in rats after L5 spinal nerve transection. Neurosci Lett 543: 130–135. [DOI] [PubMed] [Google Scholar]
- Jia H, Xu S, Liu Q, Liu J, Xu J, Li W et al (2016). Effect of pioglitazone on neuropathic pain and spinal expression of TLR‐4 and cytokines. Exp Ther Med 12: 2644–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Zhu S, Ji Q, Hui K, Duan M, Xu J et al (2010). Repeated administration of pioglitazone attenuates development of hyperalgesia in a rat model of neuropathic pain. Exp Clin Psychopharmacol 18: 359–365. [DOI] [PubMed] [Google Scholar]
- Loke YK, Kwok CS, Singh S (2011). Comparative cardiovascular effects of thiazolidinediones: systematic review and meta‐analysis of observational studies. BMJ 342: d1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D (2005). The search for the palmitoylethanolamide receptor. Life Sci 77: 1685–1698. [DOI] [PubMed] [Google Scholar]
- LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace‐Raso G et al (2006). Rapid broad‐spectrum analgesia through activation of peroxisome proliferator‐activated receptor‐α. J Pharmacol Exp Ther 319: 1051–1061. [DOI] [PubMed] [Google Scholar]
- Lyons DN, Zhang L, Danaher RJ, Miller CS, Westlund KN (2017). PPARγ agonists attenuate trigeminal neuropathic pain. Clin J Pain 33: 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DN, Zhang L, Pandya JD, Danaher RJ, Ma F, Miller CS et al (2018). Combination drug therapy of pioglitazone and D‐cycloserine attenuates chronic orofacial neuropathic pain and anxiety by improving mitochondrial function following trigeminal nerve injury. Clin J Pain 34: 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Kiguchi N, Kobayashi Y, Ozaki M, Kishioka S (2008). Pioglitazone attenuates tactile allodynia and thermal hyperalgesia in mice subjected to peripheral nerve injury. J Pharmacol Sci 108: 341–347. [DOI] [PubMed] [Google Scholar]
- Mansouri MT, Naghizadeh B, Ghorbanzadeh B, Alboghobeish S (2017a). Systemic and local anti‐nociceptive effects of simvastatin in the rat formalin assay: role of peroxisome proliferator‐activated receptor γ and nitric oxide. J Neurosci Res 95: 1776–1785. [DOI] [PubMed] [Google Scholar]
- Mansouri MT, Naghizadeh B, Ghorbanzadeh B, Rajabi H, Pashmforoush M (2017b). Pharmacological evidence for systemic and peripheral antinociceptive activities of pioglitazone in the rat formalin test: role of PPARγ and nitric oxide. Eur J Pharmacol 805: 84–92. [DOI] [PubMed] [Google Scholar]
- Moreira DRM, Santos DS, Espírito Santo RFD, Santos FED, de Oliveira Filho GB, Leite ACL et al (2017). Structural improvement of new thiazolidinones compounds with antinociceptive activity in experimental chemotherapy‐induced painful neuropathy. Chem Biol Drug Des 90: 297–307. [DOI] [PubMed] [Google Scholar]
- Moreno S, Farioli‐Vecchioli S, Ceru MP (2004). Immunolocalization of peroxisome proliferator‐activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 123: 131–145. [DOI] [PubMed] [Google Scholar]
- Morgenweck J, Abdel‐Aleem OS, Mcnamara KC, Donahue RR, Badr MZ, Taylor BK (2009). Activation of peroxisome proliferator‐activated receptor gamma in brain inhibits inflammatory pain, dorsal horn expression of Fos, and local edema. Neuropharmacology 58: 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenweck J, Griggs RB, Donahue RR, Zadina JE, Taylor BK (2013). PPARγ activation blocks development and reduces established neuropathic pain in rats. Neuropharmacology 70: 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murad H, Ayuob N (2015). Co‐administration of pioglitazone improves fluoxetine's antinociceptive, neuroprotective, and antidepressant effects in chronic constriction injury in rats. Pain Physician 18: 609–620. [PubMed] [Google Scholar]
- Napimoga MH, Souza GR, Cunha TM, Ferrari LF, Clemente‐Napimoga JT, Parada CA et al (2007). 15d‐Prostaglandin J2 inhibits inflammatory hypernociception: involvement of peripheral opioid receptor. J Pharmacol Exp Ther 324: 313–321. [DOI] [PubMed] [Google Scholar]
- Neugebauer V (2015). 15. Amygdala pain mechanisms. Handb Exp Pharmacol 227: 261–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Wolski K (2007). Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine 356: 2457–2471. [DOI] [PubMed] [Google Scholar]
- O'Mahony SM, Clarke G, Mckernan DP, Bravo JA, Dinan TG, Cryan JF (2013). Differential visceral nociceptive, behavioural and neurochemical responses to an immune challenge in the stress‐sensitive Wistar‐Kyoto rat strain. Behav Brain Res 253: 310–317. [DOI] [PubMed] [Google Scholar]
- Okine BN, Madasu MK, Mcgowan F, Prendergast C, Gaspar JC, Harhen B et al (2016). N‐palmitoylethanolamide in the anterior cingulate cortex attenuates inflammatory pain behaviour indirectly via a CB1 receptor‐mediated mechanism. Pain 157: 2687–2696. [DOI] [PubMed] [Google Scholar]
- Okine BN, Gaspar JC, Madasu MK, Olango WM, Harhen B, Roche M et al (2017). Characterisation of peroxisome proliferator‐activated receptor signalling in the midbrain periaqueductal grey of rats genetically prone to heightened stress, negative affect and hyperalgesia. Brain Res 1657: 185–192. [DOI] [PubMed] [Google Scholar]
- Okine BN, Rea K, Olango WM, Price J, Herdman S, Madasu MK et al (2014). A role for PPARα in the medial prefrontal cortex in formalin‐evoked nociceptive responding in rats. Br J Pharmacol 171: 1462–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okine BN, Spicer C, Millns P, Bennett A, Chapman V (2015). Systemic administration of WY‐14643, a selective synthetic agonist of peroxisome proliferator activator receptor‐alpha, alters spinal neuronal firing in a rodent model of neuropathic pain. Scand J Pain 9: 42–48. [DOI] [PubMed] [Google Scholar]
- Olango WM, Finn DP (2014). Neurobiology of stress‐induced hyperalgesia. Curr Top Behav Neurosci 20: 251–280. [DOI] [PubMed] [Google Scholar]
- Oliveira AC, Bertollo CM, Rocha LT, Nascimento EB Jr, Costa KA, Coelho MM (2007). Antinociceptive and antiedematogenic activities of fenofibrate, an agonist of PPARalpha, and pioglitazone, an agonist of PPARgamma. Eur J Pharmacol 561: 194–201. [DOI] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA (2010). Cannabinoid activation of peroxisome proliferator‐activated receptors: potential for modulation of inflammatory disease. Immunobiology 215: 611–616. [DOI] [PubMed] [Google Scholar]
- Paragomi P, Rahimian R, Kazemi MH, Gharedaghi MH, Khalifeh‐Soltani A, Azary S et al (2014). Antinociceptive and antidiarrheal effects of pioglitazone in a rat model of diarrhoea‐predominant irritable bowel syndrome: role of nitric oxide. Clin Exp Pharmacol Physiol 41: 118–126. [DOI] [PubMed] [Google Scholar]
- Park SW, Yi JH, Miranpuri G, Satriotomo I, Bowen K, Resnick DK et al (2007). Thiazolidinedione class of peroxisome proliferator‐activated receptor gamma agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J Pharmacol Exp Ther 320: 1002–1012. [DOI] [PubMed] [Google Scholar]
- Pena‐dos‐Santos DR, Severino FP, Pereira SAL, Rodrigues DBR, Cunha FQ, Vieira SM et al (2009). Activation of peripheral κ/δ opioid receptors mediates 15‐deoxy‐Δ12,14‐prostaglandin J2 induced‐antinociception in rat temporomandibular joint. Neuroscience 163: 1211–1219. [DOI] [PubMed] [Google Scholar]
- Pottabathini R, Kumar A, Bhatnagar A, Garg S, Ekavali E (2016). Ameliorative potential of pioglitazone and ceftriaxone alone and in combination in rat model of neuropathic pain: targeting PPARÎ3 and GLT‐1 pathways. Pharmacol Rep 68: 85–94. [DOI] [PubMed] [Google Scholar]
- Rea K, Olango WM, Okine BN, Madasu MK, Mcguire IC, Coyle K et al (2014). Impaired endocannabinoid signalling in the rostral ventromedial medulla underpins genotype‐dependent hyper‐responsivity to noxious stimuli. Pain 155: 69–79. [DOI] [PubMed] [Google Scholar]
- Ruiz‐Medina, J. , Flores, J.A. , Tasset, I. , Tunez, I. , Valverde, O. , AND Fernandez‐Espejo, E. (2012). Alteration of neuropathic and visceral pain in female C57BL/6J mice lacking the PPAR‐α gene. Psychopharmacology (Berl) 222: 477–488. [DOI] [PubMed] [Google Scholar]
- Russo R, Caro CD, Avagliano C, Cristiano C, La Rana G, Mattace Raso G et al (2016). Sodium butyrate and its synthetic amide derivative modulate nociceptive behaviors in mice. Pharmacol Res 103: 279–291. [DOI] [PubMed] [Google Scholar]
- Russo R, LoVerme J, La Rana G, D'Agostino G, Sasso O, Calignano A et al (2007). Synergistic antinociception by the cannabinoid receptor agonist anandamide and the PPAR‐alpha receptor agonist GW7647. Eur J Pharmacol 566: 117–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Kendall DA, Chapman V (2008). Inhibition of fatty acid amide hydrolase produces PPAR‐alpha‐mediated analgesia in a rat model of inflammatory pain. Br J Pharmacol 155: 1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hasegawa‐Moriyama M, Kurimoto T, Yamada T, Inada E, Kanmura Y (2016). Resolution of inflammation by resolvin D1 is essential for peroxisome proliferator‐activated receptor‐γ‐mediated analgesia during postincisional pain development in type 2 diabetes. Anesthesiology 123: 1420–1434. [DOI] [PubMed] [Google Scholar]
- Sasso O, Russo R, Vitiello S, Raso GM, D'Agostino G, Iacono A et al (2012). Implication of allopregnanolone in the antinociceptive effect of N‐palmitoylethanolamide in acute or persistent pain. Pain 153: 33–41. [DOI] [PubMed] [Google Scholar]
- Smith AF, Macfie WG, Oliver MF (1970). Clofibrate, serum enzymes, and muscle pain. Br Med J 2: 86–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP et al (1998). Activation of human aortic smooth‐muscle cells is inhibited by PPARα but not by PPARγ activators. Nature 393: 790–793. [DOI] [PubMed] [Google Scholar]
- Suardíaz M, Estivill‐Torrús G, Goicoechea C, Bilbao A, Rodríguez De Fonseca F (2007). Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain 133: 99–110. [DOI] [PubMed] [Google Scholar]
- Sunder Mudaliar MF, Chang AR, Henry RR (2003). Thiazolidinediones, peripheral edema, and type 2 diabetes: incidence, pathophysiology, and clinical implications. Endocrine Practice 9: 406–416. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Hasegawa‐Moriyama M, Sakurai T, Inada E (2011). The macrophage‐mediated effects of the peroxisome proliferator‐activated receptor‐gamma agonist rosiglitazone attenuate tactile allodynia in the early phase of neuropathic pain development. Anesth Analg 113: 398–404. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Dadia N, Yang CB, Krishnan S, Badr M (2002). Peroxisome proliferator‐activated receptor agonists inhibit inflammatory edema and hyperalgesia. Inflammation 26: 121–127. [DOI] [PubMed] [Google Scholar]
- Tugwood JD, Issemann I, Anderson RG, Bundell KR, Mcpheat WL, Green S (1992). The mouse peroxisome proliferator activated receptor recognizes a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J 11: 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo W, Leleu‐Chavain N, Spencer J, Sansook S, Millet R, Chavatte P (2017). Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N‐acylethanolamine acid amidase inhibitors. J Med Chem 60: 4–46. [DOI] [PubMed] [Google Scholar]
- van Eekeren ICM, Clockaerts S, Bastiaansen‐Jenniskens YM, Lubberts E, Verhaar JAN, Van Osch GJVM et al (2013). Fibrates as therapy for osteoarthritis and rheumatoid arthritis? A systematic review. Ther Adv Musculoskelet Dis 5: 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos MAL, Royo VA, Ferreira DS, Crotti AEM, e Silva MLA, Carvalho JCT et al (2006). In vivo analgesic and anti‐inflammatory activities of ursolic acid and oleanoic acid from Miconia albicans (melastomataceae). Z Naturforsch C 61:477–482. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang Q, Zhao L, Li D, Fu Z, Liang L (2014). Down‐regulation of PPARα in the spinal cord contributes to augmented peripheral inflammation and inflammatory hyperalgesia in diet‐induced obese rats. Neuroscience 278: 165–178. [DOI] [PubMed] [Google Scholar]
- Warden A, Truitt J, Merriman M, Ponomareva O, Jameson K, Ferguson LB et al (2016). Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep 6: 27618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MB, Bortolini M, Tadayyon M, Bopst M (2014). Minireview: challenges and opportunities in development of PPAR agonists. Mol Endocrinol 28: 1756–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li L, Chen L, Li Y, Chen H, Li Y et al (2015). Potential analgesic effects of a novel N‐acylethanolamine acid amidase inhibitor F96 through PPAR‐α. Sci Rep 5: 13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanardelli M, Micheli L, Cinci L, Failli P, Ghelardini C, Di Cesare Mannelli L (2014). Oxaliplatin neurotoxicity involves peroxisome alterations. PPARγ agonism as preventive pharmacological approach. PLoS One 9: e102758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Song, C. , Li, H. , Hou, J. , AND Li, D. (2016). Ursolic acid prevents augmented peripheral inflammation and inflammatory hyperalgesia in high‐fat diet‐induced obese rats by restoring downregulated spinal PPARα. Mol Med Rep 13: 5309–5316. [DOI] [PubMed] [Google Scholar]