

Abstract
Herein we report a dearomative syn-1,4-diamination protocol using simple nonactivated arenes and amines. This one-pot method utilizes arene–arenophile para-cycloadducts, formed via visible-lightmediated [4+2]-photocycloaddition that undergoes formal allylic substitution with amine nucleophiles under Pdcatalysis. The products are obtained with exclusive syn-1,4-selectivity; the method permits enantioselective desymmetrization of naphthalene, as well as elaborations of amine-containing drug molecules. Furthermore, the resulting unsaturated products are amenable to numerous options for diversification. Overall, this novel dearomative functionalization strategy offers rapid and straightforward access to complex building blocks, which are difficult to prepare otherwise, from simple arenes.
Dearomatization represents one of the most prominent and effective complexity-generating strategies,1 as it directly converts aromatic building blocks into functionalized, high-value added compounds.2 In addition to the venerable Birch reduction3 and dearomative oxidation of phenols,4 the field has witnessed numerous developments in recent years, mainly in the area of stoichiometric transition-metal-mediated dearomatizations5 and catalytic dearomative elaborations of phenols and heterocycles.6 However, such transformations involving nonactivated arenes are widely underdeveloped and catalytic methods that result in concomitant introduction of functionality are particularly scarce.7
Recently, we have reported several dearomative functionalization methods using small organic molecules called arenophiles,8 such as N-methyl-1,2,4-triazoline-3,5-dione (MTAD, 1), which can undergo visible-light-mediated para-cycloaddition with simple arenes (Figure 1a).9 The resulting arene–arenophile bicycles of type I provide ample opportunities for subsequent in situ catalytic functionalizations, as demonstrated with Pd- and Ni-catalyzed dearomative carboaminations.10 These transformations enable the direct introduction of multiple functionalities onto the arene, displaying a high degree of atom, step, and redox economy11 compared to the traditional approaches needed for preparation of such products. Consequently, we have been interested in extending the scope of these catalytic processes, anticipating that the arenophile-mediated dearomative functionalization would also be feasible beyond carbon nucleophiles. Specifically, we postulated that the intermediate allylpalladium species (II) would be electrophilic enough to react with other nucleophiles,12 such as neutral amines, to provide syn-1,4-aminofunctionalized unsaturated products.13
Figure 1.

(a) Pd-catalyzed dearomative syn-1,4-diamination (this work). (b) Examples of biologically active compounds that feature a syn-1,4-cyclohexanediamine motif.
Syn-1,4-cyclohexanediamines are important structural motifs that exist in many natural products and biologically active compounds, as exemplified by aminoglycoside antibiotic astromicin (2),14 VLA-4 antagonist tetrahydrobenzoquinoline 3,15 or fungicidal carboxamide 416 (Figure 1b). Despite their abundance, preparation of decorated syn-1,4-cyclohexanediamines is not straightforward; thus, a more general and efficient strategy is needed for the synthesis of these compounds.17 Herein, we disclose a conceptually different approach to syn-1,4-cyclohexanediamine derivatives based on the dearomative 1,4-diamination of arenes. This process involves arenophile-mediated photochemical para-cycloaddition and subsequent palladium-catalyzed ring-opening of the resulting cycloadducts with amines (Figure 1a). A range of simple arenes and amines provided products with exclusive syn-1,4-selectivity, and high enantioselectivity was achieved in the case of naphthalene. The dearomatized products contain multiple handles amenable to further derivatizations and functional group interconversions, providing rapid access to a diverse set of highly functionalized molecules. Finally, this dearomatization process was used for structural elaboration of memantine, a drug that is used to treat Alzheimer’s disease.
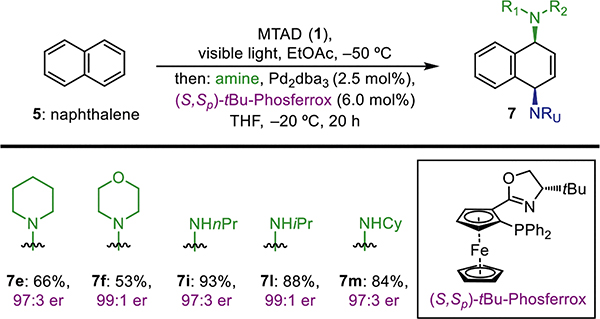
Our preliminary investigations commenced with exposure of a cold solution of naphthalene (5) and MTAD (1) to visible light, followed by the addition of amine and Pd catalysts in THF and subsequent warming of the reaction mixture to 0 °C (Table 1). Thus, using CH2Cl2 as the solvent and Pd2(dba)3/PPh3 as the catalysts, product 7a was obtained in 52% yield and as a single constitutional and diastereoisomer (entry 1). Importantly, this initial result demonstrated the feasibility of catalytic dearomative syn-1,4-diamination. Next, we turned our attention toward the evaluation of reaction parameters (see Supporting Information for full details). Probing the steric and electronic properties of monodentate phosphines, exemplified by PPhCy2, P(o-MeC6H4)3, and P(p-MeOC6H4)3 (entries 2–4), as well as using bidentate dppf (entry 5), typically used in allylic substitution reactions, did not increase the yield of the desired product. Use of an alternative Pd source (entries 6 and 7) revealed that Pd(PPh3)4 gave a slight increase in efficiency. However, in all cases a significant amount of unreacted MTAD-naphthalene cycloadduct was observed after analyzing the crude reaction mixtures. In order to improve conversion, we kept the temperature of the ring-opening step at −20 °C and used longer reaction times, which proved highly beneficial for product formation (entries 8–10).18 Finally, using this procedure with EtOAc as the solvent provided the highest yield of product (62%, entry 10).
Table 1.
Optimization of Reaction Conditionsa
 | |||||
|---|---|---|---|---|---|
| entry | [Pd] source (mol %) | solvent | solvent temp (°C) | time (h) | yield (%)b |
| 1 | Pd2(dba)3/PPh3 (2.5/6) | CH2Cl2 | −50 to 0 | 5 | 52 |
| 2 | Pd2(dba)3/PPhCy2 (2.5/6) | CH2Cl2 | −50 to 0 | 5 | 22 |
| 3 | Pd2(dba)3/P(o- MeOC6H4)3 (2.5/6) |
CH2Cl2 | −50 to 0 | 5 | 5 |
| 4 | Pd2(dba)3/P(p- MeOC6H4)3 (2.5/6) |
CH2Cl2 | −50 to 0 | 5 | 44 |
| 5 | Pd2(dba)3/dppf (2.5/6) | CH2Cl2 | −50 to 0 | 5 | 36 |
| 6 | [Pd(allyl)Cl]2/PPh3 (2.5/6) | CH2Cl2 | −50 to 0 | 5 | 51 |
| 7 | Pd(PPh3)4 (5) | CH2Cl2 | −50 to 0 | 5 | 57 |
| 8 | Pd(PPh3)4 (5) | CH2Cl2 | −20 | 20 | 70 |
| 9 | Pd(PPh3)4 (5) | EtCN | −20 | 20 | 70 |
| 10 | Pd(PPh3)4 (5) | EtOAc | −20 | 20 | 72 (62) |
Standard reaction conditions: MTAD (1, 0.5 mmol, 1.0 equiv), naphthalene (5, 1.0 mmol, 2.0 equiv), solvent (0.1 M), visible light, −50 °C, 12 h; then addition of BnNHMe (1.0 mmol, 2.0 equiv) and [Pd] catalyst in THF.
Determined by 1H NMR integration relative to the internal standard. Isolated yield shown in parenthesis.
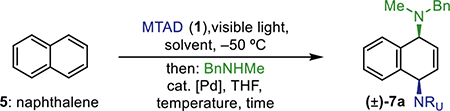
With optimized conditions in hand (Table 1, entry 10), we examined the amine scope for this protocol using naphthalene (5) and benzene (6, Table 2). Aside from methylbenzylamine (7a), other acyclic secondary amines proved to be viable nucleophiles, as exemplified with dimethylamine (7b) and diethylamine (7c), which gave products with similar yields. Moreover, cyclic secondary amines, such as pyrrolidine, piperidine, morpholine, and N-methylpiperazine, were good substrates for this transformation as well (7d–7h). In addition to naphthalene (5), benzene (6) also showed the desired reactivity, delivering products 8a–8d with a representative set of linear (8a and 8b) and cyclic (8c and 8d) secondary amines. Next, we explored the dearomative diamination process with primary amines as substrates and observed significant erosion in yields using Pd(PPh3)4 as the catalyst. Gratifyingly, after performing an additional screen, we found that changing the catalyst to Pd2(dba)3/dppf (2.5/6.0 mol %) greatly improved efficiency for these substrates. Thus, the reaction of naphthalene (5) with a range of aliphatic amines, such as linear propyl-, pentyl-, and benzylamine (7i–7k), or branched isopropyl-, cyclohexyl-, and tert-butylamine (7l–7n), all gave products in good yields. Notably, this dearomative difunctionalization is mild enough to tolerate a variety of functionality as demonstrated with products derived from amines incorporating alkene (7o), silyl-protected alcohol (7p), and ester groups (7h and 7q). We also tested the scalability of this transformation by conducting dearomative difunctionalization of naphthalene with propylamine on a gram scale; accordingly, we obtained 7i in 74% yield on an 8.8 mmol scale. Finally, benzene (6) reacted successfully with primary amines, albeit slightly lower yields of products 8e–8h were obtained compared to naphthalene. Throughout these experiments, disubstituted products are formed as single diastereo- and constitutional isomers (see Table 2 for representative X-ray structures of 7a, 8d, 7i, and 8f).
Table 2.
Amine Scope of the Dearomative syn-1,4-Diamination of Naphthalene (5) and Benzene (6)a
 |
Standard reaction conditions for naphthalene (5): MTAD (1, 0.5 mmol, 1.0 equiv), naphthalene (5, 1.0 mmol, 2.0 equiv), EtOAc (0.1 M), visible light, −50 °C, 12 h; then addition of amine (1.0 mmol, 2.0 equiv) and [Pd] catalyst in THF, −20 °C, 20 h. Reaction conditions for benzene (6): MTAD (1, 1.0 mmol, 1.0 equiv), benzene (6, 10 mmol, 10 equiv), CH2Cl2 (0.2 M), visible light, −78 °C, 12 h; then addition of amine (2.0 mmol, 2.0 equiv) and [Pd] catalyst in THF, −20 °C, 20 h. Reported yields are of isolated products.
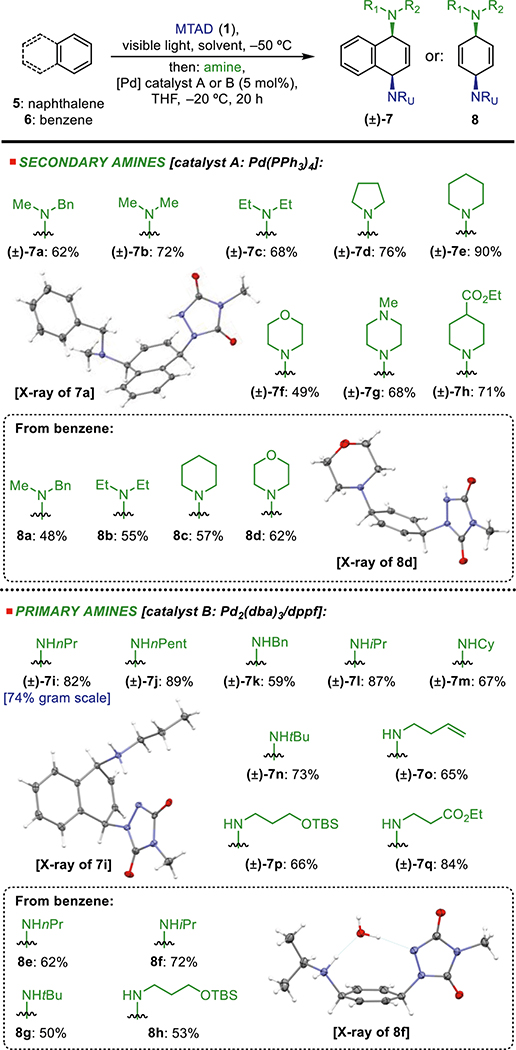
We next investigated the scope of arenes using propylamine as an amine source (Table 3). While benzene worked well (Table 2, insets), substituted mononuclear analogs proved to be unproductive substrates for this reaction. On the other hand, polynuclear arenes delivered desired products 10a–10d. In the case of 10b–10e, mixtures of constitutional isomers were observed, resulting from the lack of regioselectivity in opening the nonsymmetrical arene–arenophile cycloadducts.19 Additionally, polynuclear heteroarenes were also amenable to dearomative syn-1,4-diaminofunctionalization, providing products 10e–10h. Compared to arene-derived products 10a–10d, these heteroarene-based compounds were obtained with noticeably higher selectivities. In all cases, dearomative cycloaddition with polynuclear arenes proceeded in a highly site-selective manner; functionalization was observed only at the terminal, nonsubstituted ring.
Table 3.
Arene Scope of the Dearomative syn-1,4-Diaminationaa
 |
Standard reaction conditions: MTAD (1, 1.0 mmol, 1.0 equiv), arene (9, 2.0 mmol, 2.0 equiv), EtOAc (0.1 M), visible light, −50 °C, 12 h; then addition of nPrNH2 (2.0 mmol, 2.0 equiv) and [Pd] catalyst (5 mol %) in THF, −50 to 0 °C, 5 h. Reported yields are of isolated products, with ratios of constitutional isomers (in parentheses) determined by 1H NMR of the crude reaction mixtures.
[Pd] catalyst in THF, −20 °C, 20 h.
CH2Cl2 was used instead of EtOAc.
10 mol % of [Pd] catalyst was used.
Cycloaddition was run at 0.05 M concentration.
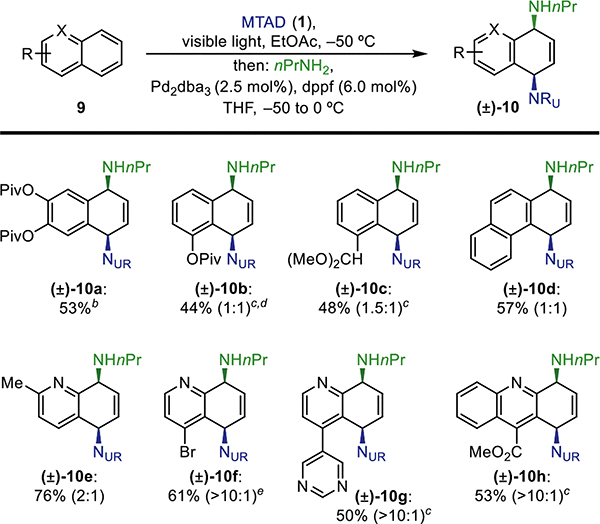
We then focused on providing an enantioselective variant of this transformation for the arene–arenophile cycloadducts that are amenable to desymmetrization (Table 4). Accordingly, we screened chiral ligands that could enable asymmetric diamination of naphthalene, and observed high enantioselectivities with Pd2(dba)3 and (S,Sp)-tBu-Phosferrox (2.5/6.0 mol %). Using this protocol, a representative collection of amine–naphthalene adducts were obtained from secondary (7e and 7f) and primary amines (7i, 7l, and 7m) with selectivities ranging from 97:3 to 99:1 er.
Table 4.
Pd-Catalyzed Enantioselective Dearomative syn-1,4-Diaminationaa
 |
Standard reaction conditions: MTAD (1, 0.5 mmol, 1.0 equiv), naphthalene (5, 1.0 mmol, 2.0 equiv), EtOAc (0.1 M), visible light, −50 °C, 12 h; then addition of amine (1.0 mmol, 2.0 equiv) and Pd2(dba)3 (2.5 mol %) and (S,Sp)-tBu-Phosferrox (6.0 mol %) in THF, −20 °C, 20 h. Reported yields are of isolated products.
The dearomative elaboration described herein can serve as an entry point for rapid molecular diversification and structural elaboration of amine-containing drugs (Figure 2). For example, representative product 7i encompasses several handles for further functionalization (Figure 2a). Accordingly, the corresponding unsaturated amines 11 and 12, saturated amine 13, aminoketone 14, and differentially substituted diamine 15 were obtained from 7i in one to three steps.20 We were also interested in probing this dearomative diamination protocol as a tool for diversification of medicinally relevant amines (Figure 2b). Thus, memantine (16), an FDA-approved drug used for the treatment of dementia associated with Alzheimer’s disease, was further elaborated with naphthalene. Using our standard conditions (see Table 2), dearomatized product 7r was obtained in 66% yield. Moreover, in one to three steps, this intermediate was further diversified to alkene 17, saturated ketone 18, and diaminodiol 19, showcasing the diverse functionalization opportunities this chemistry provides.
Figure 2.

(a) Diversification of product 7i. (b) Elaboration of anti-Alzheimer drug memantine (16). Reagents and conditions: (a) Li, NH3, 60%; (b) (i) H2, Rh/Al2O3 (cat.), 83%; (ii) HCl, 48%; (c) (i) H2, Rh/Al2O3 (cat.), 83%; (ii) Li, NH3, 71%; (d) (i) H2, Rh/Al2O3 (cat.), 83%; (ii) tBuOCl, 50%; (e) (i) Boc2O; then NaOMe 79%; (ii) PhCOCH2Br, K2CO3, 81%; (iii) KOH, 64%; (f) Li, NH3, 64%; (g) H2, Rh/Al2O3 (cat.), 57%; (ii) tBuOCl, 80%; (h) (i) PhCOCH2Br, K2CO3, 71%; (ii) OsO4 (cat.), NMO, 80%; (iii) KOH, 58%.
In summary, we have reported a dearomative diamination strategy. This process involves visible-light-mediated para-cycloaddition of arenes with an arenophile and subsequent Pd-catalyzed ring-opening of the resulting cycloadducts with amines as nucleophiles. A variety of amines and arenes provided products with exclusive syn-1,4-selectivity, and high enantioselectivity was observed for the desymmetrization of naphthalene. The corresponding dearomatized products offered unique access to functionalized small molecules, as they contained unsaturation and the arenophile motif, which could be used for further manipulations. The synthetic value of this method has also been demonstrated by rapid and selective elaboration of memantine, an anti-Alzheimer drug, into new analogs. Finally, from a practical perspective, it is noteworthy that this dearomatization protocol could be conducted on a gram scale without significant loss of efficiency. Further studies regarding scope and utility, as well as the development of related transformations and applications of this method, are ongoing and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work was provided by the University of Illinois, the National Science Foundation (CAREER Award No. CHE-1654110), the NIH/National Institute of General Medical Sciences (R01 GM122891), and the donors of the American Chemical Society Petroleum Research Fund (PRF#57175-DNI1). D.S. is an Alfred P. Sloan Fellow. W.C.W. is a Springborn and NSF Graduate Fellow (GRFP). M.O. thanks the Honjo International Scholarship Foundation. Solvias AG is acknowledged for a generous gift of chiral ligands. We also thank Dr. D. Olson and Dr. L. Zhu for NMR spectroscopic assistance, Dr. D. L. Gray and Dr. T. Woods for X-ray crystallographic analysis assistance, and F. Sun for mass spectrometric assistance.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b13030.
Experimental procedures and spectral data for all new compounds (PDF)
X-ray crystallographic data for (±)-7a (CIF)
X-ray crystallographic data for (±)-7i (CIF)
X-ray crystallographic data for (±)-8d (CIF)
X-ray crystallographic data for (±)-8f (CIF)
REFERENCES
- (1).For recent review on application of dearomatizations, see:Roche SP; Porco JA Dearomatization Strategies in the Synthesis of Complex Natural Products. Angew. Chem., Int. Ed 2011, 50, 4068.Pape AR; Kaliappan KP; ndig EP Transition-Metal-Mediated Dearomatization Reactions. Chem. Rev 2000, 100, 2917.For recent reviews on catalytic asymmetric dearomatizations, see:Zhuo C-X; Zhang W; You S-L Catalytic Asymmetric Dearomatization Reactions. Angew. Chem., Int. Ed 2012, 51, 12662.Zheng C; You S-L Catalytic Asymmetric Dearomatization by Transition-Metal Catalysis: A Method for Transformations of Aromatic Compounds. Chem. 2016, 1, 830.
- (2).For selected recent examples involving dearomatizations of arenes, see:Wiesenfeldt MP; Nairoukh Z; Li W; Glorius F Hydrogenation of fluoroarenes: Direct access to all-cis-(multi)-fluorinated cycloalkanes. Science 2017, 357, 908.James MJ; Schwarz JL; Strieth-Kalthoff F; Wibbeling B; Glorius F Dearomative Cascade Photocatalysis: Divergent Synthesis through Catalyst Selective Energy Transfer. J. Am. Chem. Soc 2018, 140, 8624.Farndon JJ; Ma X; Bower JF Transition Metal Free C–N Bond Forming Dearomatizations and Aryl C–H Aminations by in Situ Release of a Hydroxylamine-Based Aminating Agent. J. Am. Chem. Soc 2017, 139, 14005.Good SN; Sharpe RJ; Johnson JS Highly Functionalized Tricyclic Oxazinanones via Pairwise Oxidative Dearomatization and N-Hydroxycarbamate Dehydrogenation: Molecular Diversity Inspired by Tetrodotoxin. J. Am. Chem. Soc 2017, 139, 12422.Wilson KB; Myers JT; Nedzbala HS; Combee LA; Sabat M; Harman WD Sequential Tandem Addition to a Tungsten–Trifluorotoluene Complex: A Versatile Method for the Preparation of Highly Functionalized Trifluoromethylated Cyclohexenes. J. Am. Chem. Soc 2017, 139, 11401.Nakayama H; Harada S; Kono M; Nemoto T Chemoselective Asymmetric Intramolecular Dearomatization of Phenols with α-Diazoacetamides Catalyzed by Silver Phosphate. J. Am. Chem. Soc 2017, 139, 10188.Zheng J; Wang S-B; Zheng C; You S-L Asymmetric Dearomatization of Naphthols via a Rh-Catalyzed C(sp2)–H Functionalization/Annulation Reaction. J. Am. Chem. Soc 2015, 137, 4880.García-Fortanet J; Kessler F; Buchwald SL Palladium-Catalyzed Asymmetric Dearomatization of Naphthalene Derivatives. J. Am. Chem. Soc 2009, 131, 6676.Zhuo C-X; You S-L Palladium-Catalyzed Intermolecular Asymmetric Allylic Dearomatization Reaction of Naphthol Derivatives. Angew. Chem., Int. Ed 2013, 52, 10056.Phipps RJ; Toste FD Chiral Anion Phase-Transfer Catalysis Applied to the Direct Enantioselective Fluorinative Dearomatization of Phenols. J. Am. Chem. Soc 2013, 135, 1268.Oguma T; Katsuki T Iron-Catalyzed Dioxygen-Driven C–C Bond Formation: Oxidative Dearomatization of 2-Naphthols with Construction of a Chiral Quaternary Stereocenter. J. Am. Chem. Soc 2012, 134, 20017.
- (3).For a comprehensive review, see:Rabideau PW; Marcinow Z The Birch Reduction of Aromatic Compounds. Org. React 1992, 42, 1.
- (4).For a recent review, see:Pouységu L; Deffieux D; Quideau S Hypervalent Iodine-Mediated Phenol Dearomatization in Natural Product Synthesis. Tetrahedron 2010, 66, 2235.
- (5).For an overview of this area, see: Liebov BK; Harman WD Group 6 Dihapto-Coordinate Dearomatization Agents for Organic Synthesis. Chem. Rev 2017, 117, 13721.Keane JM; Harman WD A New Generation of π-Basic Dearomatization Agents. Organometallics 2005, 24, 1786. See also ref 1b.
- (6).For recent reviews, see:Ding Q; Zhou X; Fan Recent Advances in Dearomatization of Heteroaromatic Compounds. Org. Biomol. Chem 2014, 12, 4807. See also ref 1d.
- (7).Wertjes WC; Southgate EH; Sarlah D Recent Advances in Chemical Dearomatization of Nonactivated Arenes. Chem. Soc. Rev 2018, 47, 7996. [DOI] [PubMed] [Google Scholar]
- (8).Southgate EH; Pospech J; Fu J; Holycross DR; Sarlah D Dearomative Dihydroxylation with Arenophiles. Nat. Chem 2016, 8, 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Hamrock SJ; Sheridan RS Para Photoaddition of N-Methyltriazolinedione to Benzene. Synthesis of Energy-rich Azo Compounds Comprising Benzene + Nitrogen. J. Am. Chem. Soc 1989, 111, 9247. [Google Scholar]; (b) Kjell DP; Sheridan RS Photochemical Cycloaddition of N-Methyltriazolinedione to Naphthalene. J. Am. Chem. Soc 1984, 106, 5368. [Google Scholar]
- (10).(a) Okumura M; Shved AS; Sarlah D Palladium-Catalyzed Dearomative syn-1,4-Carboamination. J. Am. Chem. Soc 2017, 139, 17787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hernandez LW; Klöckner, U.; Pospech, J.; Hauss, L.; Sarlah, D. Nickel-Catalyzed Dearomative trans-1,2-Carboamination. J. Am. Chem. Soc 2018, 140, 4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Trost BM The Atom Economy-a Search for Synthetic Efficiency. Science 1991, 254, 1471. [DOI] [PubMed] [Google Scholar]; (b) Newhouse T; Baran PS; Hoffmann RW The Economies of Synthesis. Chem. Soc. Rev 2009, 38, 3010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Burns NZ; Baran PS; Hoffmann RW Redox Economy in Organic Synthesis. Angew. Chem., Int. Ed 2009, 48, 2854. [DOI] [PubMed] [Google Scholar]
- (12).For reviews on Pd-catalyzed allylic substitution, see:Trost BM; Van Vranken DL Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev 1996, 96, 395.Trost BM; Crawley ML Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev 2003, 103, 2921.Lu Z; Ma S Metal-Catalyzed Enantioselective Allylation in Asymmetric Synthesis. Angew. Chem., Int. Ed 2008, 47, 258.Weaver JD; Recio A III; Grenning AJ; Tunge JA Transition Metal-Catalyzed Decarboxylative Allylation and Benzylation Reactions. Chem. Rev 2011, 111, 1846.
- (13).Alternatively, formal dearomative aminofunctionalizations with complementary 1,2-selectivity could be achieved using transitionmetal-catalyzed ring-opening of strained benzyne-derived azabenzonorbornadienes, as pioneered by Lautens. For selected reviews, see:Lautens M; Fagnou K; Hiebert S Transition Metal-Catalyzed Enantioselective Ring-Opening Reactions of Oxabicyclic Alkenes. Acc. Chem. Res 2003, 36, 48.Rayabarapu DK; Cheng C-H New Catalytic Reactions of Oxa- and Azabicyclic Alkenes. Acc. Chem. Res 2007, 40, 971.Woo S; Keay BA An Improved Synthesis of Ethyl 2-(dicyanomethylene)propanoate. Synthesis 1996, 1996, 669.Chiu P; Lautens M Using Ring-Opening Reactions of Oxabicyclic Compounds as a Strategy in Organic Synthesis. Top. Curr. Chem 1997, 190, 1.For selected examples of formal dearomative 1,2-diamination, see: Cho Y; Zunic V; Senboku H; Olsen M; Lautens M Rhodium-Catalyzed Ring-Opening Reactions of N-Boc-Azabenzonorbornadienes with Amine Nucleophiles. J. Am. Chem. Soc 2006, 128, 6837.Cho Y-H; Fayol A; Lautens M Enantioselective Synthesis of Chiral 1,2-Diamines by the Catalytic Ring Opening of Azabenzonorbornadienes: Application in the Preparation of New Chiral Ligands. Tetrahedron: Asymmetry 2006, 17, 416.Lautens M; Fagnou K; Zunic V An Expedient Enantioselective Route to Diaminotetralins: Application in the Preparation of Analgesic Compounds. Org. Lett 2002, 4, 3465.
- (14).Nara T; Yamamoto M; Kawamoto I; Takayama K; Okachi R; Takasawa S; Sato T; Sato S Fortimicins A and B, new aminoglycoside antibiotics. I. Producing organism, fermentation a biological properties of fortimicins. J. Antibiot 1977, 30, 533. [DOI] [PubMed] [Google Scholar]
- (15).Ho W-B; Broka C Synthesis of a Peptidomimetic Tricyclic Tetrahydrobenzo[ij]quinoline as a VLA-4 Antagonist. J. Org. Chem 2000, 65, 6743. [DOI] [PubMed] [Google Scholar]
- (16).Bruhn JA; Pasteris RJ Preparation of Carboxamide Derivative Fungicides for Synergistic Fungicidal Mixtures. Patent WO 2008091594, 2008. [Google Scholar]
- (17).For selected preparations of 1,4-diamines, see:Akermark B; Bäckvall J-E; Löwenborg A; Zetterberg K Palladium(II)-promoted 1,4-Diamination of 1,3-Dienes. Stereochemistry of Amination of a π-Allylpalladium Complex. J. Organomet. Chem 1979, 166, C33.Bäckvall J-E Palladium in Some Selectivë Oxidation Reactions. Acc. Chem. Res 1983, 16, 335.Lishchynskyi A; Muñiz K An Approach to the Regioselective Diamination of Conjugated Di- and Trienes. Chem. - Eur. J 2012, 18, 2212.Martínez C; Martínez L; Kirsch J; Escudero-Adán EC; Martin E; Muñiz, K. Copper-Mediated 1,4-Diamination of 1,3-Butadienes. Eur. J. Org. Chem 2014, 2014 (10), 2017.For a previous dearomative approach to saturated syn-1,4-diamines, see: Okumura M; Nakamata Huynh SM; Pospech J; Sarlah D Arenophile-mediated Dearomative Reduction. Angew. Chem., Int. Ed 2016, 55, 15910.
- (18).Full conversion of the MTAD-naphthalene cycloadduct was observed under these conditions. The lower isolation yield was mainly due to the instability of the product 7a.
- (19).All constitutional isomers were produced with exclusive syn-1,4-selectivity, and were readily separable by flash chromatography. See the Supporting Information for details.
- (20).The urazole moiety was converted to the corresponding amine through N-alkylation of urazole with α-bromoacetophenone and subsequent carbanion-assisted cleavage of the N–N-bond, as described previously:Adam W; Pastor A; Wirth T Org. Lett 2000, 2, 1295..In the case of 7i, the basic amine had to be protected with a Boc group before alkylation. However, in the case of 7r, this protection was not required due to the significant steric protection by the adamantane scaffold.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.