

Abstract
Enantioselective total syntheses of the anticancer isocarbostyril alkaloids (+)-7-deoxypancratistatin, (+)-pancratistatin, (+)-lycoricidine, and (+)-narciclasine are described. Our strategy for accessing this unique class of natural products is based on the development of a Nicatalyzed dearomative trans-1,2-carboamination of benzene. The effectiveness of this dearomatization approach is notable, as only two additional olefin functionalizations are needed to construct the fully decorated aminocyclitol cores of these alkaloids. Installation of the lactam ring has been achieved through several pathways and a direct interconversion between natural products was established via a late-stage C-7 cupration. Using this synthetic blueprint, we were able to produce natural products on a gram scale and provide tailored analogs with improved activity, solubility, and metabolic stability.
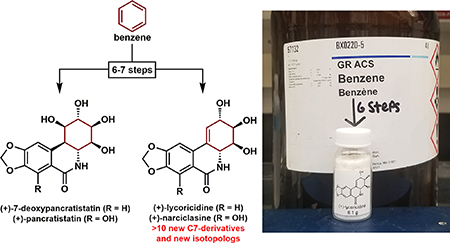
Graphical Abstract
INTRODUCTION
Plants belonging to the Amaryllidaceae family have long been known for their medicinal properties; their importance was recognized by the Ancient Greeks, as crude plant extracts were prescribed by Hippocrates and his School of Medicine as remedies against various illnesses, including tumors.1 Isolation studies revealed numerous compounds associated with the significant anticancer effects of these plants, including the isocarbostyril-type alkaloids (+)-7-deoxypancratistatin (1),2 (+)-pancratistatin (2),3 (+)-lycoricidine (3),4 and (+)-narciclasine (4)5 (Figure 1). These compounds exhibited significant growth-inhibitory potencies against several human cancer cell lines and showed unique cytotoxicity patterns that do not correlate with any known anticancer agents.6 For example, experiments examining the cytotoxic profile of these compounds revealed noticeably reduced death in noncancerous cells relative to cancer cells, suggesting that their development as chemotherapeutics could result in fewer adverse side-effects.7 Moreover, narciclasine (4) exhibited considerable activity in in vivo tumor models, including highly invasive human glioblastomas and apoptosis-resistant brain metastases,8 albeit with toxicity also being reported in certain cases.9 In addition to their potent anticancer activity, pancratistatin (2) and 7-deoxypancratistatin (1) also showed significant antiviral activity, such as in in vivo models for Japanese encephalitis,10 and narciclasine (4) has been found to attenuate diet-induced obesity11 and to possess anti-inflammatory properties.12
Figure 1.

Structures of isocarbostyril alkaloids (+)-7-deoxypancratistatin (1), (+)-pancratistatin (2), (+)-lycoricidine (3), and (+)-narciclasine (4).
Despite these encouraging biological properties, the precise biomolecular mechanisms of action have not been fully elucidated. Pancratistatin is postulated to induce apoptosis through the intrinsic pathway, as evidenced by an increase in caspase-9 and caspase-3 activity, exposure of phosphatidyl serine, and destabilization of mitochondrial membrane potential.13 Interestingly, the cytotoxic activity of narciclasine has been attributed to the extrinsic caspase-8 apoptotic pathway via the activation of the Fas and death receptor 4 (DR4) death inducing signaling complex (DISC).7d Furthermore, narciclasine has exhibited activity in cytostatic pathways that complement its cytotoxic ones. It has been shown to block peptide synthesis through direct inhibition of the A-site of the 60S ribosome.14 These findings were further corroborated by cocrystallization of narciclasine (4) in the A-site of the 60S ribosome.15 Additionally, 4 binds the translation elongation factor eEF1A, thereby impeding this protein’s secondary function of actin bundle formation and disrupting polysome organization and impairing cytokinesis.8b Likewise, similar activities are observed through the activation of GTPase RhoA in glioblastoma cells, causing the formation of F-actin stress fibers that disrupt cytokinesis.8a
These promising biological attributes sparked significant interest for large-scale production of isocarbostyrils 1–4 to enable preclinical evaluations. To this end, the highest yielding isolation of (+)-pancratistatin (2), from 100 kg of Hymenocallis litoralis grown in the Hawaiian wilderness, yielded 15 g (150 mg/kg, 0.015% yield) of 2.16 To secure more sustainable access, a biotechnological approach was developed involving a plant tissue culture cloning of the same plant species. Unfortunately, cultivation of these plants in fields and greenhouses in Arizona delivered only 9–24 mg/kg (0.0009–0.0024%) of pure material.17 On the other hand, various isolation protocols for narciclasine have been reported in the literature, yielding 30–140 mg/kg of natural product from wet Narcissus plant bulbs.18
Due to the challenging isolation from natural sources, the isocarbostyrils 1–4 have been exceptionally attractive targets for chemical synthesis, with nearly 50 distinct strategies reported to date.19–22 Despite many elegant approaches, the discovery of a sustainable route with practical access to these natural products has remained elusive, as nearly all biological evaluations of 1–4 have been conducted using isolated natural material. Nevertheless, these impressive synthetic endeavors enabled basic SAR studies that identified the core pharmacophore and provided more potent and selective analogs that could not be accessed through direct modification of the natural products.6,13a
Considering the lack of scalable approaches, we initially became interested in the synthesis of (+)-pancratistatins (1 and 2).23 However, it was apparent at the outset of this work that the newly developed methodology, which was needed to streamline this task, would also create opportunities to explore the synthesis of (+)-lycoricidine, (+)-narciclasine, and tailored analogs thereof. Herein, we describe our synthetic approaches to isocarbostyrils 1–4, and several designed analogs with improved physiochemical properties and metabolic stability. This work ultimately resulted in an interesting methodological development and led to an efficient and scalable synthesis of these intriguing alkaloids.
RESULTS AND DISCUSSION
Olefin-like Functionalization of Benzene
The ultimate synthetic challenge posed by isocarbostyrils 1–4 is the construction of the densely decorated aminocyclitol cores containing six or four contiguous stereocenters. We postulated that these motifs could be traced back to benzene using distinct alkene difunctionalization reactions, which would ideally set all the required functionality of pancratistatins (1 and 2), as well as lycoricidine (3) and narciclasine (4) in a stereoselective manner (Figure 2). Based on their substitution pattern, the pancratistatins could be derived from aminotetraols 5 or 6. These hexafunctionalized cyclohexanes could be traced back to the corresponding dienes 7 and 8 by applying two different dihydroxylations. Finally, we hypothesized that dienes of this type could be obtained from benzene (9) through dearomative trans-1,2-carboamination with N-methyl-1,2,4-triazoline-3,5-dione (MTAD, 12) and aryl Grignard reagents 10 or 11. Using similar yet distinct disconnections, lycoricidine (3) and narciclasine (4) could originate from the corresponding functionalized lactam precursors 13 and 14, which in turn, could be obtained from dienes 7 and 8.
Figure 2.

Retrosynthetic analysis of isocarbostyril alkaloids 1–4 from benzene (9) using an olefin-functionalization approach.
Based on the above retrosynthetic analysis, benzene (9) could be considered as a surrogate for the hypothetical 1,3,5-cyclohexatriene; thus, three olefin-type difunctionalization reactions would enable the key retrosynthetic disconnections and provide natural products 1–4 in a rapid and controlled fashion. However, due to the inherent resonance stabilization of benzene, these and related olefin-like dearomative transformations are practically nonexistent in synthetic organic chemistry. Only certain stoichiometric reactions of transitionmetal complexes24 and microbial arene oxidation25 can affect olefin-like dearomative functionalizations; however, such processes are not suitable for the desymmetrization of benzene.
Dearomative trans-1,2-Carboamination
At the onset of our studies, it was clear that the invention of a novel dearomatization process was crucial to the success of our synthetic plan. Since our laboratory has been involved in the development of dearomative functionalizations based on visible-light-promoted para-cycloaddition with arenophiles,26 we postulated that the application of this chemistry in the presence of a transition metal catalyst and an aryl nucleophile could result in the desired trans-1,2-carboamination (Figure 3a). Particularly, we were keen to explore if the intermediate arenophile MTAD-benzene cycloadduct 15 could serve as a viable substrate for oxidative addition with low-valent transition metals, as it possesses an electron-deficient bis-allylic bridgehead urazole. We envisioned that a diene of type 15 could serve as a π-ligand, coordinating to the metal center and facilitating oxidative addition in an anti-fashion to the urazole moiety (15 → I). This step would lead to cyclohexadienyl intermediate II, which could undergo transmetalation with an organometallic reagent to form η5-species III. Finally, reductive elimination (III → IV) and diene decomplexation would yield product 16 and regenerate the metal catalyst.
Figure 3.

(a) Concept and mechanistic rationale for dearomative trans-1,2-carboamination strategy. (b) General reactivity of cyclohexadienyliron complexes with nucleophiles. (c) Ligand scope for Ni-catalyzed dearomative trans-1,2-carboamination.
Though catalysis involving η5-species had not been previously reported, our studies were inspired by the wealth of chemistry employing stoichiometric reactions of η5-complexes. Early work from Birch, and later findings from Pearson, Davies, Green, and Mingos, showcased highly regioand stereoselective outcomes in cationic cyclohexadienylmetal complexes with nucleophiles (Figure 3b).27 For example, the most widely studied iron η5-intermediate 19, prepared from the corresponding 1,3-diene via complexation (17 → 18) and subsequent C–H abstraction (18 → 19), reacts with nucleophiles with exclusive 1,2-site-selectivity (19 → 20), due to the greater positive charge localized on the termini of the η5-system.27d Encouraged by these precedents, we expected that symmetrical intermediate III should follow a similar mechanistic course to diene IV through an inner-sphere pathway, resulting in syn-delivery of a nucleophile relative to the metal center. Moreover, since the η5-intermediate III is symmetrical, a reductive elimination step could enable enantiodiscrimination through differentiation of the enantiotopic termini of the cyclohexadienyl system. Thus, the desired product 16 could be formed in an enantioselective fashion by using a suitable chiral ligand bound to the metal center.
To probe the above-described reactivity of the arenophilebenzene cycloadduct with transition metals, we performed a series of prospecting investigations with aryl nucleophiles in combination with transition metals complexes. Catalysts based on Co, Ir, Rh, Cu, and Ni were primarily investigated as these metals have exceptionally rich repertoires of nucleophilic additions to their complexes containing allyl and dienyl ligands. Gratifyingly, by using the combination of [Ni(cod)2] and phosphine ligands with organomagnesium bromide 10, we were able to observe the desired product (Figure 3c). While monodentate phosphines and NHC-based ligands proved to be ineffective, most bisphosphines we tested furnished product 21, with dppf delivering the highest yield in this series. Specifically, we identified that conducting the MTAD-benzene cycloaddition reaction in dichloromethane, followed by the addition of a Ni-catalyst ([Ni(cod)2]/dppf = 10/20 mol %) and aryl Grignard reagent 10 delivered the desired dearomatized product 21 in 74% yield as a single diastereoand constitutional isomer (for X-ray of 21, see Figure 3c). Although these experiments established the viability of a diastereoselective process, they did not address the feasibility of rendering the process enantioselective. Accordingly, we performed a comprehensive evaluation of chiral P,P- and P,N-bidentate ligands, and discovered that the PHOX-type ligand (R,Rp)-iPr-Phosferrox afforded the desired product 21 in 75% yield and with high enantioselectivity (98:2 er).
Initial Approaches to (+)-Pancratistatins
With the first vicinal stereocenters in place, the stage was set for the introduction of the remaining four hydroxy substituents in a stereoselective manner to complete the pancratistatin core (Figure 4a). The initial plan involved formation of the trans-diol through hydrolytic opening of an epoxide, followed by Upjohn cis-dihydroxylation. However, early experiments with diene 21 and mCPBA or NBS/H2O gave mixtures of products, likely due to the undesired directing effects of the urazole hydrazyl group (pKa = 5.8 in water). Therefore, methylation of the urazole’s nitrogen proved crucial for stereo- and chemoselective diene functionalization. This effect is likely due to a more rigid conformation, where the methyl group shields the bottom face of the diene. Such a conformation was supported by NOESY experiments revealing through-space correlation between the methyl group and several diene protons (see inset at the bottom of Figure 4). The methyl group was conveniently introduced (21 → 7) by simply adding Me2SO4 at the end of dearomatization sequence. Importantly, using a decreased catalyst loading ([Ni]/ligand = 5/10 mol %), this one-pot process allowed us to routinely prepare decagram batches of diene 7.
Figure 4.

First generation approaches to (+)-7-deoxypancratistatin (1). (a) Synthesis of aminotetraol 5. (b) Conversion of aminotetraol 5 to (+)-7deoxypancratistatin (1). Reagents and conditions: 1. benzene (9), MTAD (12), CH2Cl2, visible light, −78 °C; then [Ni(cod)2] (5 mol %), (R,Rp)-iPr-Phosferrox (10 mol %), Grignard reagent 10, CH2Cl2, THF, −78 to +25 °C; then Me2SO4, K2CO3, 65% (98:2 er); 2. mCPBA, NaHCO3, CH2Cl2, 25 °C; 3. NaOBz, H2O, 100 °C, 63% over two steps; 4. NMO, OsO4 (5 mol %), tBuOH, H2O, 25 °C, 91%; 5. LiAlH4, THF, reflux; then Rochelle salt; then Raney-Co, H2 (1 atm), 60 °C, 60%; 6. NBS, THF, H2O, 25 °C, 79%; 7. OsO4 (5 mol %), NMO, citric acid, acetone, H2O, tBuOH (1:1:2), 25 °C; 8. NaOBz, H2O, 100 °C, 42% over two steps; 9. LiAlH4, THF, reflux; then Rochelle salt; then Raney-Co, H2 (1 atm), 60 °C, 69%; 10. HMTA, TFA, AcOH, 90 °C 95%; 11. NaClO2, NaH2PO4, 2-methyl-2-butene, THF, H2O, 25 °C, 57%; 12. Boc2O, Et3N, 1,4-dioxane, H2O, 25 °C; then Ac2O, Et3N, DMAP, CH2Cl2, 25 °C, 76%; 13. Ph3P(O), Tf2O, BF3·Et2O, CH2Cl2, 0 °C; then NaOMe, MeOH, 25 °C, 75%.
With a robust sequence that allowed for the preparation of sufficient amounts of key diene 7, we turned our attention to the next two olefin difunctionalization steps. Due to the electron-withdrawing effect of the urazole nitrogen, it was expected that the alkene distal to this moiety should react preferentially with electrophilic reagents. Indeed, installation of the trans-diol by means of a two-step sequence involving epoxidation with mCPBA and subsequent epoxide hydrolysis (NaOBz, H2O, 100 °C) proceeded smoothly, delivering product 22 as a single diastereo- and constitutional isomer (for X-ray of 22, see bottom of Figure 4). The last alkene transformation needed for establishing the hexasubstituted aminocyclitol core was Upjohn dihydroxylation,28 which provided tetraol 23 in 91% yield.
The final sequence required to complete the synthesis of (+)-7-deoxypancratistatin (1) was the deprotection of urazole 23 to free amine 5 and its conversion to the corresponding lactam. Exploring known conditions to effect hydrolysis and N–N bond cleavage, such as heating in highly acidic or basic solutions followed by hydrogenolysis, led to complete decomposition of the starting material. Gratifyingly, we observed promising reactivity with hydride-based reducing agents. For example, exposure of urazole 23 to LiAlH4 gave cyclic hydrazine hemiaminal 24 (Figure 4, bottom); however, this compound readily underwent oxidation under an ambient atmosphere, complicating its isolation and reproducibility. Therefore, we developed a one-pot procedure that directly reduced this sensitive intermediate to amine 5 by carefully quenching the LiAlH4 reduction with Rochelle’s salt, followed by immediate addition of Raney-Co and exposure of the reaction mixture to a hydrogen atmosphere.29 Using this protocol, we consistently obtained free amine 5 in 60% yield on a multigram scale.
Additionally, we were able to secure amine 5 from diene 7 by an alternative pathway. Thus, subjecting diene 7 to NBS and H2O gave bromohydrin 25 in 79% yield (for X-ray of 25, see bottom of Figure 4). This intermediate underwent Upjohn dihydroxylation, and the resulting dibromotriol was subsequently exposed to weakly basic aqueous NaOBz to provide bromotetraol 26 through concomitant epoxide formation and hydrolysis. Similar, highly chemoselective hydrolytic opening of the corresponding intermediate epoxide diol was demonstrated by Hudlický during his approach to pancratistatins.20b Finally, the above-described sequential reduction with LiAlH4 and Raney-Co furnished amine 5 through urazole fragmentation and protodehalogenation.
The ultimate objective, the construction of the lactam ring and completion of (+)-7-deoxypancratistatin (1), was initially achieved through Duff formylation (5 → 27) followed by Pinnick oxidation (27 → 1).30 While formylation efficiently delivered the desired aldimine 27, separation of this product from HMTA and its byproducts proved challenging, and oxidation to 1 continuously gave inconsistent results. Therefore, an alternative pathway was sought that could still rely on the readily available hexafunctionalized precursor 5. Accordingly, we explored the Bischler–Napieralski reaction, as this key lactam forming strategy was successfully used by Banwell and others in similar molecular settings.31 The corresponding isocyanate precursor 28 was readily prepared by sequential protection of amine with Boc and alcohols with acetates. The key cyclization was successfully accomplished with Hendrickson’s reagent (triphenylphosphonium anhydride trifluoromethanesulfonate), delivering acetylated (+)-7-deoxypancratistatin that was converted to natural product 1 upon treatment with K2CO3 in MeOH.32
With the synthesis of (+)-7-deoxypancratistatin (1) completed, we turned our attention to (+)-pancratistatin (2), expecting that the lessons learned from the synthesis of 1 could be translated toward preparation of its congener as well (Figure 5). Indeed, enantioselective dearomative trans-1,2-carboamination of benzene (9) with more elaborate aryl Grignard reagent 11 furnished the desired diene 8 in 66% yield and 97:3 er. The subsequent epoxidation of this compound with mCPBA, consistently provided low yields, likely due to an increased steric hindrance introduced by an additional methoxy group on the arene moiety. Gratifyingly, in situ generated dimethyldioxirane33 provided the desired allylic epoxide that was hydrolyzed to trans-diol 29 using aqueous NaOBz. The remaining steps, Upjohn dihydroxylation (29 → 30), urazole fragmentation (30 → 6), carbamate preparation (6 → 31), and Bischler–Napieralski reaction (31 → 32) proceeded smoothly, furnishing protected pancratistatin (32) in 87% yield. It is important to note that application of Hendrickson’s reagent for this cyclization proved to be more efficient and selective (10:1 r.r) compared to most of the previously reported cyclizations in similar systems.31 Finally, global deprotection of 32 with BBr3 followed by NaOMe provided (+)-pancratistatin (2) in 50% yield over two steps.
Figure 5.

First generation approach to (+)-pancratistatin (2). Reagents and conditions: 1. benzene (9), MTAD (12), CH2Cl2, visible light, −78 °C; then [Ni(cod)2] (5 mol %), (R,Rp)-iPr-Phosferrox (10 mol %), Grignard reagent 11, CH2Cl2, THF, −78 to +25 °C; then Me2SO4, K2CO3, 66% (97:3 er); 2. Oxone, NaHCO3, acetone, H2O, 25 °C; 3. NaOBz, H2O, 100 °C, 55% over two steps (18% of 8 recovered); 4. NMO, OsO4 (5 mol %), tBuOH, H2O, 25 °C, 91%; 5. LiAlH4, THF, reflux; then Rochelle salt; then Raney-Co, H2 (1 atm), 60 °C, 65%; 6. Boc2O, Et3N, 1,4-dioxane, H2O, 25 °C; then Ac2O, Et3N, DMAP, CH2Cl2, 25 °C, 72%; 7. Ph3P(O), Tf2O, BF3·Et2O, CH2Cl2, 25 °C, 87% (10:1 r.r.); 8. BBr3, CH2Cl2, 25 °C; 9. NaOMe, MeOH, 25 °C, 50% over two steps.
Streamlined Synthesis of (+)-Pancratistatins
The above-described synthetic campaigns resulted in concise preparation of both (+)-pancratistatins in seven and nine steps, respectively. Though this accomplishment represents the shortest enantioselective approach to the pancratistatins to date, we felt there was still room for improvement, mainly to increase atom, step, and redox economy, all of which are desired for the practical synthesis of such compounds.34 Specifically, installation of the trans-diol and lactam formation required intermediary steps, such as protecting group manipulations and discrete oxidation level alterations. Moreover, as in all previous synthetic endeavors, 1 and 2 each required individual de novo total synthesis from the corresponding C-7 substituted aromatic building blocks, which provided an additional roadblock in synthetic development. Finally, the precursor for functionalized Grignard reagent 11 required three synthetic operations from o-vanillin, whereas the precursor for Grignard 10 was commercially available.
With these key challenges in mind, we set forth to further streamline our synthesis of the pancratistatins and to provide a direct synthetic connection between 1 and 2 (Figure 6). During initial studies of the epoxidation of diene 7, we frequently observed small amounts of the desired trans-diol 22 during the epoxidation step; therefore, we explored the viability of a one-pot, trans-dihydroxylation protocol. Indeed, this approach proved to be successful when the epoxidation reaction was conducted in the presence of pTsOH and a large excess of water.35 In addition, the use of hexafluoroisopropanol (HFIP) as a solvent was essential36 to obtain diol product 22 in 74% yield. This represented a marked increase in yield (63 → 74%) as well as removed a chromatographic purification from the early sequence, improving the preparation of diol 22. At this point, the remaining transformations toward aminotetraol 5 remained the same, as they were already scalable and reproducible.
Figure 6.

Streamlined synthesis of pancratistatins 1 and 2. Reagents and conditions: 1. benzene (9), MTAD (12), CH2Cl2, visible light, −78 °C; then [Ni(cod)2] (5 mol %), (R,Rp)-iPr-Phosferrox (10 mol %), Grignard reagent 10, CH2Cl2, THF, −78 to +25 °C; then Me2SO4, K2CO3, 65% (98:2 er); 2. mCPBA, pTsOH, CH2Cl2, HFIP, H2O, 50 °C, 74%; 3. NMO, OsO4 (5 mol %), tBuOH, H2O, 25 °C, 91%; 4. LiAlH4, THF, reflux; then Rochelle salt; then Raney-Co, H2 (1 atm), 60 °C, 60%; 5. Br2, AcOH, 25 °C; 6. NaCo(CO)4 (30 mol %), nBu4NBr, CO (1 atm), NaHCO3, H2O, 1,4-dioxane, 365 nm light, 60 °C, 72% over two steps; 7. HMDS, I2 (1 mol %), MeCN, 80 °C; then solvent removal and (TMP)2Cu(CN)Li2, THF, − 78 °C → 0 °C; then tBuOOH, THF, − 78 °C; acidic workup, 62%.
We next wanted to improve the overall efficiency of the lactam formation as our previously described approach required five protecting groups for the installation of a single carbonyl group. However, we knew that the introduction of carbonyl functionality in the presence of four free alcohols would prove challenging with respect to achieving the desired chemoselectivity. To this end, we were able to install a bromine substituent at the desired position (8:1 r.r.) on the electron-rich arene ring by exposing aminotetraol 5 to bromine under acidic conditions. The installed halogen provided a handle for the exploration of carbonyl insertion through carbonylative coupling chemistry. Accordingly, we began investigating various benchmark Pd-catalyzed carbonylative coupling procedures;37 however, we unfortunately observed only complex mixtures of oxidized intermediates and precipitation of Pd black. This was somewhat expected, as palladium is well-known to oxidize similar substrates38 and, to the best of our knowledge, carbonylative couplings involving aryl bromides in the presence of free primary and secondary alcohols has yet to be developed.
We then turned our focus to explore catalytic carbonylation based on other metals that are known to tolerate free alcohols. Along these lines, we tested a set of conditions reported by Caubere and co-workers that employed dicobalt octacarbonyl under highly basic aqueous conditions (5 M NaOH) and UV irradiation.39 According to the proposed mechanism,40 such transformations proceed through photoinduced electron- or charge-transfer complexes between aryl halides and [Co(CO)4]−, resulting in the formation of aroylcobalt carbonyl complexes of type I-1, which should readily collapse to lactam product 1 with concurrent regeneration of catalyst [Co(CO)4]−. Indeed, using Caubere’s protocol we were encouraged to find that (+)-7-deoxypancratistatin (1) could be observed in low yield, with rest of the mass balance being recovered starting material and an amino acid, likely resulting from subsequent base-induced hydrolysis. This suspicion was further validated by the fact that longer reaction times led to lower yields and larger amounts of the amino acid, which proved challenging to convert back to 1. Therefore, it became apparent that the highly basic reaction environment was causing the lower yields, and development of more neutral reaction conditions was needed. The main role of NaOH in the original report was to convert Co2(CO)8 to active NaCo(CO)4, and to serve as a base that sequestered HBr. Thus, we prepared pure NaCo(CO)4, and utilized it in the reaction alongside NaHCO3 as a mild HBr scavenger, resulting in formation of (+)-7-deoxypancratistatin (1) in 72% overall yield from aminotetraol 5. For ease of operation, the bromination and carbonylative coupling were performed in a single reaction vessel, with only a solvent exchange as an intermediary step.
The final synthetic challenge left was to establish a direct connection between the pancratistatins (1 → 2), which would completely remove the need for the use of tailored Grignard reagent 11 as well as de novo synthesis of 2. In addition to providing this link, such a C-7 functionalization could also enable facile synthesis of analogs at this position. Many methods for this formal oxidative transformation were investigated, including direct sp2 C–H oxidation,41 indirect sp2 C–H borylation/oxidation,42 and various directed ortho metalation/oxidation procedures.43 Undeterred, we eventually found that treating 1 with hexamethyldisilazane (HMDS) in the presence of catalytic amounts of iodine allowed for in situ generation of tetrasilylated 7-deoxypancratistatin 33,44 which could immediately be subjected to a cupration/oxidation sequence. This formal sp2 C–H oxidation was accomplished using directed cupration with (TMP)2Cu(CN)Li2 and subsequent arylcuprate I-2 oxidation with tBuOOH,45 conditions recently reported by Uchiyama and co-workers that proved to be robust enough for our complex system, affording (+)-pancratistatin (2) in 62% yield after acidic workup. Of note is the use of HMDS/I2 for global silylation of (+)-7-deoxypancratistatin prior to the deprotonative cupration, as other common silyl transfer agents, such as TMSCl, TMSCN, TMSN3, N,O-bis(trimethylsilyl)acetamide (BSA), N-methyl-N-trimethylsilylacetamide (MSA), N-methyl-N-trimethylsilyltrifluoroacetamide (MSTFA), and N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) left stoichiometric impurities that could not be removed without chromatographic purification. Though purification of 33 was possible, the rapid hydrolysis of the silyl groups on silica led to issues in reproducibility. On the other hand, as the only byproduct from the silylation with HMDS is NH3, all volatiles and excess of the reagent could be removed simply through azeotropic distillation with toluene, leaving 33 in sufficient purity for the next operation. Thus, the described synthesis delivers (+)-7-deoxypancratistatin (1) and (+)-pancratistatin (2) in six and seven operations and in 19% and 12% overall yield. It is important to note that using this streamlined synthetic sequence, we have prepared several grams of both pancratistatins to date, showcasing the scalability of the abovedescribed approach.
Synthesis of (+)-Narciclasine
With the practical synthesis of pancratistatins completed, we turned our attention toward (+)-lycoricidine (3) and (+)-narciclasine (4). As shown in our retrosynthetic analysis (Figure 2), we surmised that compounds 13 and 14 could be viable intermediates to reach the unsaturated aminocyclitol core of 3 and 4 through a base-promoted epoxide isomerization and concurrent arylmetal attack into the urazole ring, resulting in carbonyl transfer and assembly of the lactam. We were particularly interested in exploring such intermediates because they could be readily traced back to dienes 7 and 8, using olefin functionalization chemistry. In addition to our previous success in handling these compounds, we also recently improved the dearomative trans-1,2-carboamination strategy,46 which permitted the preparation of these intermediates on a multidecagram scale without the use of a glovebox. Although we initially used 5 mol % of [Ni(cod)2] as a precatalyst for the synthesis of the pancratistatins (Figures 4–6), we further optimized this protocol to permit the application of air-stable [Ni(acac)2] in much lower loadings ([Ni]/ligand = 1.5/2.0 mol %).
At the onset of our studies toward (+)-narciclasine (4), we commenced by targeting key intermediate 14 (Figure 7). Thus, diene 8, which was prepared in 66% yield and 97:3 er using the new protocol, was exposed to an excess of NBS in THF/H2O, affording dibromide 34 as a single diastereo- and constitutional isomer in 85% yield. Exposure of this compound to Upjohn dihydroxylation conditions with a basic work up (K2CO3), followed by acetonide protection of the intermediate epoxy diol, gave key bromoepoxide 14 in 77% yield over two steps. One pot dihydroxylation/bromohydrin closure was developed for practical reasons, as the dibromotriol proved challenging to extract and purify. This compound, which is prepared from benzene in four steps, contains all the necessary atoms that are present in narciclasine (4), as well as strategically placed functional handles for conversion to the natural product. Specifically, the stereochemical relationship between the epoxide and the benzylic hydrogen is appropriate for a formal syn-epoxide elimination,47 which would provide the desired allylic alcohol. Moreover, the aryl bromide moiety serves as a precursor to an arylmetal species that could add into the nearby urazole carbonyl group and form the desired lactam. To our delight, we discovered that the slow addition of tBuLi to a cold solution of 14 could achieve both epoxide isomerization and benzamide formation, likely through the intermediate I-3, providing compound 35 in 65% yield. Furthermore, although 14 contains two neighboring hydrogens, both in syn-quasi axial position (for an X-ray of similar compound 13, see Figure 8, bottom) only product 35 was observed, resulting from elimination of the more acidic benzylic proton.48
Figure 7.

Synthesis of (+)-narciclasine (4). Reagents and conditions: 1. benzene (9), MTAD (12), CH2Cl2, visible light, −78 °C; then [Ni(acac)2] (1.5 mol %), (R,Rp)-iPr-Phosferrox (2.0 mol %), Grignard reagent 11, CH2Cl2, THF, −78 to +25 °C; then Me2SO4, K2CO3, 66% (97:3 er); 2. NBS, H2O, THF, 25 °C, 85%; 3. OsO4 (5 mol %), NMO, citric acid, acetone, H2O, tBuOH, 25 °C; then K2CO3, 25 °C; 4. 2,2-dimethoxypropane, pTsOH (10 mol %), CH2Cl2, 25 °C, 77% over two steps; 5. tBuLi, THF, −78 °C, 65%; 6. SmI2, MeOH, 0 °C; then 40 °C; then HCl, 0 °C, 89%.
Figure 8.

Synthesis of (+)-lycoricidine (3) and (+)-narciclasine (4). Reagents and conditions: 1. benzene (9), MTAD (12), CH2Cl2, visible light, −78 °C; then [Ni(acac)2] (1.5 mol %), (R,Rp)-iPr-Phosferrox (2.0 mol %), Grignard reagent 10, CH2Cl2, THF, −78 to +25 °C; then Me2SO4, K2CO3, 65% (97:3 er); 2. NBS, H2O, THF, 25 °C, 79%; 3. OsO4 (5 mol %), NMO, citric acid, acetone, H2O, tBuOH, 25 °C; then K2CO3, 25 °C; 4. 2,2-dimethoxypropane, pTsOH (10 mol %), CH2Cl2, 25 °C, 78% over two steps; 5. tBuLi, THF, −78 °C, 70%; 6. SmI2, MeOH, 0 °C, then HCl, 0 °C, 94%; 7. HMDS, TFA (1.0 mol %), MeCN, 25 °C; then solvent removal and (TMP)2Cu(CN)Li2, THF, −78 → 0 °C; then tBuOOH, THF, −78 °C; acidic workup, 57% (14% of 3 recovered); 8. NaI, nBu4NPF6, SmI2 (10 mol %), DMF, 25 °C, Mg anode, Pt cathode, 5.0 mA, 45% (52% of 36 recovered).
With the phenanthridone skeleton completed, the final task en route to narciclasine (4) was reductive N–N bond cleavage of acylsemicarbazide 35 and global deprotection. This was achieved by slow addition of freshly prepared SmI2, followed by mild heating and subsequent acidic workup, to deliver (+)-narciclasine (4) in 89% yield. Heating this reaction to 40 °C proved crucial to achieve full Sm(III)-mediated deprotection of the aryl methoxy group,21k and the acidic workup was favored over other common procedures, which gave heterogeneous mixtures that were tedious to separate and extract. Thus, using this six step protocol with 25% overall yield, we were able to obtain more than 600 mg of (+)-narciclasine (4) in a single pass from benzene (9).
Scalable Route to (+)-Lycoricidine and (+)-Narciclasine
With the successful application of the late-stage hydroxylation at C-7 in the case of pancratistatins (1 → 2, see Figure 6), we wondered if the same chemical connection could also be feasible between lycoricidine (3) and narciclasine (4). The major benefits of such a direct conversion would be (1) application of readily available Grignard 10 instead of noncommercial reagent 11; (2) avoidance of individual de novo total synthesis of 3 and 4; and (3) rapid preparation of C-7 analogs to fully explore this position of the pharmacophore.
By employing a glovebox-free procedure and operationally simple photoreactor, we prepared dearomatized product 7 on more than 100 mmol scale (>25 g) in 65% yield and 97:3 er after methylation with Me2SO4. Following a similar sequence to the one described above, diene 7 was subjected to two equivalents of NBS in THF and H2O to produce bromohydrin 25 in 79% yield. Subsequent Upjohn dihydroxylation, base-mediated epoxide formation, and diol protection, furnished epoxy acetonide 13 in 78% overall yield, setting the stage for the key epoxide isomerization/lactam formation cascade (see bottom of Figure 8 for an X-ray of 13). Thus, dropwise addition of tBuLi to a cold solution of 13 provided intermediate 36 in 70% yield on >20 g scale. Interestingly, only 2.35 equiv of tBuLi were needed to achieve full conversion on large scale, as addition of further equivalents only led to decomposition of the product. Treatment of lactam 36 with SmI2, followed by acidic work up delivered (+)-lycoricidine (3) in 94% yield, producing slightly over 8 g of this natural product in a single pass. Moreover, we obtained single crystals suitable for X-ray crystallographic analysis (Figure 8, bottom).49 To avoid stoichiometric use of SmI2, we have also explored reductive N–N bond cleavage employing catalytic amounts of SmI2 with an electrochemical method recently reported by the Ackermann group (Figure 8 inset).50 While the scalability of this reaction has yet to be tested, preliminary results suggest that a more economical approach could be feasible, as product 37 was obtained in 45% yield (52% of 36 recovered).
Finally, with ample amounts of (+)-lycoricidine (3) in hand, we set out to examine C-7 functionalization using the deprotonative cupration/oxidation conditions previously developed to convert (+)-7-deoxypancratistatin (1) to (+)-pancratistatin (2).51 Our initial attempts employed lycoricidine acetonide 37 and required catalytic amounts of TFA to mediate the silylation, as I2 led to the formation of byproducts. After numerous conditions were screened with lycoricidine acetonide 37, only minimal (<10%) conversion to the desired product was observed. However, we found that direct conversion of lycoricidine (3) to silylated lycoricidine, followed by addition of (TMP)2Cu(CN)Li2, subsequent in situ oxidation of arylcuprate species with tert-butyl hydroperoxide, and acidic workup delivered (+)-narciclasine (4). Importantly, we were able to run this oxidation on a 3 g scale, obtaining a 57% yield and isolating >1.8 g of (+)-narciclasine (4). Over the course of this study, we conveniently prepared >20 g of 3 and >5 g of 4 in total, demonstrating the scalability of the approach described herein.
Synthesis and Biological Evaluation of C-7 Analogs
With gram amounts of (+)-lycoricidine (3) now readily available, as well as an established late-stage cupration procedure, we turned our attention to the preparation of the corresponding C-7 analogs (Figure 9). Previous synthetic efforts have provided basic structure–activity correlations, revealing the importance of certain functionalities and their stereochemical orientation, mainly on the aminocyclitol core.8b However, the influence of C-7 substitution has not been significantly investigated.8a This comes as no surprise, as each C-7 derivative would previously require a multistep synthesis, making the preparation of a library of C-7 analogs very time-consuming. By using Uchiyama’s cupration-based strategy, alkyl- and amine-based functionalities were selectively introduced at position 7 in a single operation from 3 (Figure 9a). For example, exposure of lycoricidine-derived cuprate intermediate I4 to alkyl electrophiles or O-benzoyl hydroxylamines provided a range of C- (38–42, 44, and 46) and N-substituted analogs (47–51), respectively.52 Simple saponification of esters 42 and 44 provided carboxylic acids 43 and 45. This C-7 diversification could be also applied to the pancratistatin series, as demonstrated with the preparation of 7-aminopancratistatin (52, Figure 9b). However, amine analogs 47, 48, and 52 could not be made directly due to purification issues and consequently had to be synthesized from the corresponding allyl substituted O-benzoyl hydroxylamine, followed by allyl deprotection (for example, see Figure 9b).
Figure 9.

(a) Late-stage preparation and anticancer activity of C-7 analogs of lycoricidine (3). Cell viability was assessed after 72 h using the Alamar Blue assay, n ≥ 3, SEM for each measurement is in the Supporting Information. Doxorubicin (Dox) was used as a reference. (b) Preparation and activity of 7-aminopancratistatin 52. (c) Role of C-7 amine substituent on solubility. High throughput equilibrium solubility using miniaturized shake flask approach was used. Error is SEM, n ≥ 3. (d) Evaluation of metabolic stability of narciclasine (3) and its deuterated isotopologs (4-d5 and 4-d2). Stability was assessed in mouse liver microsomes. Compounds were incubated with microsomes for 3 h, and the percentage remaining was quantified relative to t0 using an internal standard. Error is SEM, n ≥ 2.
With a small library of analogs in hand (38–52), their anticancer activities, as well as those of the natural products (1–4), were measured using human lung and colon cancer cell lines (A549 and HCT116). As expected, the C-7 substituent plays an important role in the activity of these compounds. Alkyl substituents drastically reduced the activity as C-analogs 38–45 displayed weaker potency than lycoricidine (3), with activities ranging from 29 to >100 μM. Even derivatives containing hydrogen-bond donor or acceptor groups, such as homologue 41, differing from narciclasine by an additional CH2 group, proved to be less potent. Interestingly, 7cyanolycoricidine (46) showed increased activity when compared to lycoricidine (3) in HCT116 cells. Furthermore, introduction of an amino group improved activity over lycoricidine, as exemplified with 7-aminolycoricidine (47) which possesses an IC50 of 0.39 μM in HCT116 cells. However, other N-analogs containing alkylated amines, such as methylamine (48), acetamide (49), pyrrolidine (50), and morpholine (51) did not show significant cytotoxicity, pointing at the importance of an –XH type of motif (X = O, NH) for enhanced activity.
Finally, we also evaluated the solubility of the most active amino analogs (Figure 9c). Natural isocarbostyril alkaloids 1–4 are known to be highly insoluble; however, 7-aminolycoricidine (47) and 7-aminopancratistatin (52) showed 11and 6-fold increased solubility when compared to their natural counterparts. The improved aqueous solubility and comparable activity make these new C-7 amino analogs attractive targets for further diversification and study.
Metabolic Stability
Though numerous in vitro and several in vivo evaluations of isocarbostyril alkaloids gave promising results, there are no reports describing metabolism of these compounds, despite the fact that the results obtained from such studies would help with the planning or interpretation of clinical and toxicological studies. Over the years, many procedures have emerged for studying metabolic stability and the identification of metabolites, including deuterium labeling.53 By taking advantage of the kinetic isotope effect (KIE) one can increase the metabolic stability of a compound by incorporating deuterium at the potentially metabolically compromised site.54
One of the unique advantages of the arenophile-based approach is the ability to provide selective access to tailored stable isotopologs, as most of the starting aromatic compounds are readily available in their deuterated forms (Figure 9d). Using our previous synthetic strategy, substitution of benzene (9) for benzene-d6 (9-d6) led to narciclasine analog 4-d5, which has each proton on the cyclitol core replaced with a deuterium. Likewise, employing selectively labeled Grignard precursor 11-d2, which can be easily prepared using CD2Cl2, we have been able to synthesize narciclasine isotopolog 4-d2, which has deuterium incorporated into the methylene bridge. With these differentially deuterated compounds in hand, we tested their activity and metabolic stability using a microsome assay. While both labeled compounds 4-d5 and 4-d2 have equipotent activity, they showed noticeably (~30%) greater metabolic stability when compared to narciclasine (4), suggesting that both the methylene bridge and the cyclitol core are susceptible to metabolic degradation.
CONCLUSION
The Amaryllidaceae isocarbostyril alkaloids have been inspiring the synthetic community for many decades, serving as benchmark molecules to showcase many creative approaches toward their unique molecular architectures and stereochemical complexity. The syntheses of isocarbostyril alkaloids (+)-7-deoxypancratistatin (1), (+)-pancratistatin (2), lycoricidine (3), and narciclasine (4) described herein utilize a new methodology and strategy, developed specifically for their highly decorated aminocyclitol core. The key asymmetric dearomative trans-1,2-carboamination of benzene (9) provided facile access to these natural products through diene intermediate 7, which served as a divergence point for the synthesis of 1–4. Moreover, late-stage C-7 cupration of (+)-7-deoxypancratistatin (1) and lycoricidine (3) provided direct synthetic connection to (+)-pancratistatin (2) and narciclasine (4) in a practical, single operation.
Our streamlined route to the pancratistatins featured three olefin-like difunctionalizations of benzene and a late stage carbonylative coupling reaction that gave (+)-7-deoxypancratistatin (1) in six steps and 19% overall yield. One pot amidedirected deprotonative cupration and subsequent arylcuprate oxidation allowed for the installation of the C-7 hydroxyl group present in (+)-pancratistatin (2). The route to lycoricidine (3) featured a base-promoted epoxide isomerization/lactam formation cascade reaction followed by SmI2 mediated N–N reductive cleavage, delivering the natural product in six steps and 26% overall yield. Utilizing this strategy, we have synthesized several grams of each natural product to date, showcasing the scalability of our approach. Furthermore, large scale access to these natural products, coupled with an enabling directed cupration, resulted in the synthesis of a small library of C-7 analogs. Of these, 7-aminolycoricidine (47) showed enhanced activity over its natural counterpart, as well as significantly improved aqueous solubility. The brevity of the described approach and the availability of deuterated starting arenes also prompted the synthesis of differentially deuterated narciclasine isotopologs 4-d5 and 4-d2, both of which had improved metabolic stability when compared to nonlabeled natural product 4. We anticipate that the concise and scalable syntheses, as well as new avenues for derivatization reported in this article, will provide a new practical means of supplying these medicinally important compounds and further invigorate their biological investigations.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work was provided by the University of Illinois, the NIH/National Institute of General Medical Sciences (GM122891) and the ACS Petroleum Research Fund (57175-DNI1). D.S. is an Alfred P. Sloan Fellow. L.W.H. and R.L.S acknowledge the NIH-NIGMS CBI Training Grant (T32-GM070421). L.W.H. thanks the NSF for a Graduate Fellowship (GRFP). W. R. Grace and Co. is acknowledged for a gift of Raney-Co and Solvias AG for a generous donation of chiral ligands. We also thank Dr. D. Olson and Dr. L. Zhu for NMR spectroscopic assistance, Dr. D. L. Gray for X-ray crystallographic analysis assistance, and F. Sun for mass spectrometric assistance. Finally, the authors dedicate this paper to Professor Scott E. Denmark on the occasion of his 65th birthday.
Footnotes
The authors declare the following competing financial interest(s): The University of Illinois has filed a provisional patent on this work.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b12123.
Experimental procedures, as well as spectroscopic and analytical data for all new compounds (PDF)
Crystallographic data for C14H13NO6, H2O (CIF)
Crystallographic data for C17H19N3O6 (CIF)
Crystallographic data for C20H22BrN3O7 (CIF)
Crystallographic data for C17H17Br2N3O5 (CIF)
REFERENCES
- (1).Hartwell JL Plants used against cancer. A survey. Lloydia 1967, 30, 379. [PubMed] [Google Scholar]
- (2).Ghosal S; Singh SK; Kumar Y; Srivastava RS Isocarbostyril alkaloids from Haemanthus kalbreyeri. Phytochemistry 1989, 28, 611. [Google Scholar]
- (3).Pettit GR; Gaddamidi V; Cragg GM Antineoplastic Agents, 105. Zephyranthes grandiflora. J. Nat. Prod 1984, 47, 1018. [DOI] [PubMed] [Google Scholar]
- (4).Okamoto T; Torii Y; Isogai YO Lycoricidinol and lycoricidine, new plant-growth regulators in the bulbs of Lycoris radiata herb. Chem. Pharm. Bull 1968, 16, 1860. [DOI] [PubMed] [Google Scholar]
- (5).Ceriotti G Narciclasine: An Antimitotic Substance from Narcissus Bulbs. Nature 1967, 213, 595. [DOI] [PubMed] [Google Scholar]
- (6).For recent reviews, see:Ingrassia L; Lefranc F; Mathieu V; Darro F; Kiss R Amaryllidaceae Isocarbostyril Alkaloids and Their Derivatives as Promising Antitumor Agents. Transl. Oncol 2008, 1, 1.Kornienko A; Evidente A Chemistry, Biology, and Medicinal Potential of Narciclasine and its Congeners. Chem. Rev 2008, 108, 1982.Fürst R Narciclasine - an Amaryllidaceae Alkaloid with Potent Antitumor and Anti-Inflammatory Properties. Planta Med. 2016, 82, 1389.He MM; Qu CR; Gao OD; Hu XM; Hong XC Biological and pharmacological activities of amaryllidaceae alkaloids. RSC Adv. 2015, 5, 16562.Ghavre M; Froese J; Pour M; Hudlicky T Synthesis of Amaryllidaceae Constituents and Unnatural Derivatives. Angew. Chem., Int. Ed 2016, 55, 5642.
- (7).(a) Pettit GR; Pettit GR III; Backhaus RA; Boyd MR; Meerow AW Antineoplastic Agents, 256. Cell Growth Inhibitory Isocarbostyrils from Hymenocallis. J. Nat. Prod 1993, 56, 1682. [DOI] [PubMed] [Google Scholar]; (b) Griffin C; Hamm C; McNulty J; Pandey S Pancratistatin induces apoptosis in clinical leukemia samples with minimal effect on non-cancerous peripheral blood mononuclear cells. Cancer Cell Int. 2010, 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ingrassia L; Lefranc F; Dewelle J; Pottier L; Mathieu V; Spiegl-Kreinecker S; Sauvage S; El Yazidi M; Dehoux M; Berger W; Van Quaquebeke E; Kiss R Structure–Activity Relationship Analysis of Novel Derivatives of Narciclasine (an Amaryllidaceae Isocarbostyril Derivative) as Potential Anticancer Agents. J. Med. Chem 2009, 52, 1100. [DOI] [PubMed] [Google Scholar]; (d) Dumont P; Ingrassia L; Rouzeau S; Ribaucour F; Thomas S; Roland I; Darro F; Lefranc F; Kiss R The Amaryllidaceae isocarbostyril narciclasine induces apoptosis by activation of the death receptor and/or mitochondrial pathways in cancer cells but not in normal fibroblasts. Neoplasia 2007, 9, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pettit GR; Gaddamidi V; Herald DL; Singh SB; Cragg GM; Schmidt JM; Boettner FE; Williams M; Sagawa Y Antineoplastic Agents, 120. Pancratium Littorale. J. Nat. Prod 1986, 49, 995. [DOI] [PubMed] [Google Scholar]; (f) Griffin C; Karnik A; McNulty J; Pandey S Pancratistatin Selectively Targets Cancer Cell Mitochondria and Reduces Growth of Human Colon Tumor Xenografts. Mol. Cancer Ther 2011, 10, 57. [DOI] [PubMed] [Google Scholar]
- (8).Lefranc F; Sauvage S; Van Goietsenoven G; Mégalizzi V; Lamoral-Theys D; Debeir O; Spiegl-Kreinecker S; Berger W; Mathieu V; Decaestecker C; Kiss R Narciclasine, a plant growth modulator, activates Rho and stress fibers in glioblastoma cells. Mol. Cancer Ther 2009, 8, 1739.Van Goietsenoven G; Hutton J; Becker JP; Lallemand B; Robert F; Lefranc F; Pirker C; Vandenbussche G; Van Antwerpen P; Evidente A; Berger W; Prévost M; Pelletier J; Kiss R; Kinzy TG; Kornienko A; Mathieu V Targeting of eEF1A with Amaryllidaceae isocarbostyrils as a strategy to combat melanomas. FASEB J. 2010, 24, 4575.(c) ref 7c.(d) ref 7.
- (9).Van Goietsenoven G; Mathieu V; Lefranc F; Kornienko A; Evidente A; Kiss R Narciclasine as well as other Amaryllidaceae isocarbostyrils are promising GTP-ase targeting agents against brain cancers. Med. Res. Rev 2013, 33, 439.(b) ref 7e.
- (10).Gabrielsen B; Monath TP; Huggins JW; Kefauver DF; Pettit GR; Groszek G; Hollingshead M; Kirsi JJ; Shannon WM; Schubert EM; DaRe J; Ugarkar B; Ussery MA; Phelan MJ Antiviral (RNA) Activity of Selected Amaryllidaceae Isoquinoline Constituents and Synthesis of Related Substances. J. Nat. Prod 1992, 55, 1569. [DOI] [PubMed] [Google Scholar]
- (11).Julien SG; Kim SY; Brunmeir R; Sinnakannu JR; Ge X; Li H; Ma W; Yaligar J; Kn BP; Velan SS; Roder PV; Zhang Q; Sim CK; Wu J; Garcia-Miralles M; Pouladi MA; Xie W; McFarlane C; Han W; Xu F Narciclasine attenuates dietinduced obesity by promoting oxidative metabolism in skeletal muscle. PLoS Biol. 2017, 15, No. e1002597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mikami M; Kitahara M; Kitano M; Ariki Y; Mimaki Y; Sashida Y; Yamazaki M; Yui S Suppressive activity of lycoricidinol (narciclasine) against cytotoxicity of neutrophil-derived calprotectin, and its suppressive effect on rat adjuvant arthritis model. Biol. Pharm. Bull 1999, 22, 674. [DOI] [PubMed] [Google Scholar]; (b) Fuchs S; Hsieh LT; Saarberg W; Erdelmeier CA; Wichelhaus TA; Schaefer L; Koch E; Fürst R Haemanthus coccineus extract and its main bioactive component narciclasine display profound anti-inflammatory activities in vitro and in vivo. J. Cell. Mol. Med 2015, 19, 1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Ma D; Pignanelli C; Tarade D; Gilbert T; Noel M; Mansour F; Adams S; Dowhayko A; Stokes K; Vshyvenko S; Hudlicky T; McNulty J; Pandey S Cancer Cell Mitochondria Targeting by Pancratistatin Analogs is Dependent on Functional Complex II and III. Sci. Rep 2017, 7, 42957. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kekre N; Griffin C; McNulty J; Pandey S Pancratistatin causes early activation of caspase-3 and the flipping of phosphatidyl serine followed by rapid apoptosis specifically in human lymphoma cells. Cancer Chemother. Pharmacol 2005, 56, 29. [DOI] [PubMed] [Google Scholar]
- (14).Carrasco L; Fresno M; Vazquez D Location Of Resistance To The Alkaloid Narciclasie In The 60S Ribosomal Subunit. FEBS Lett. 1975, 52, 236. [DOI] [PubMed] [Google Scholar]
- (15).Garreau de Loubresse N; Prokhorova I; Holtkamp W; Rodnina MV; Yusupova G; Yusupov M Structural basis for the inhibition of the eukaryotic ribosome. Nature 2014, 513, 517. [DOI] [PubMed] [Google Scholar]
- (16).Pettit GR; Pettit GR III; Backhaus RA; Boettner FE Antineoplastic Agents, 294. Variations in the Formation of Pancratistatin and Related Isocarbostyrils in Hymenocallis littoralis. J. Nat. Prod 1995, 58, 37. [DOI] [PubMed] [Google Scholar]
- (17).Backhaus RA; Pettit GR III; Huang D-S; Pettit GR; Groszek G; Odgers JC; Ho J; Meerow A Biosynthesis of the Antineoplastic Pancratistatin Following Tissue Culture of Hymenocallis Littoralis (Amaryllidaceae). Acta Hortic. 1992, 306, 364. [Google Scholar]
- (18).(a) For an overview of several isolation methods, see: ref 6b.For the most recent optimized procedure, see:Borra S; Lapinskaite R; Kempthorne C; Liscombe D; McNulty J; Hudlicky T Isolation, Synthesis, and Semisynthesis of Amaryllidaceae Constituents from Narcissus and Galanthus sp.: De Novo Total Synthesis of 2-epi-Narciclasine. J. Nat. Prod 2018, 81, 1451.
- (19).For 7-deoxypancratistatin syntheses, see:Paulsen H; Stubbe M Chirale synthese von (+)-lycoricidin. Tetrahedron Lett. 1982, 23, 3171.Keck GE; McHardy SF; Murry JA Total Synthesis of (+)-7-Deoxypancratistatin: A Radical Cyclization Approach. J. Am. Chem. Soc 1995, 117, 7289.Tian X; Maurya R; Königsberger K; Hudlicky T Asymmetric Total Synthesis of (+)-7-Deoxypancratistatin. Synlett 1995, 1995, 1125.Hudlicky T; Tian X; Konigsberger K; Maurya R; Rouden J; Fan B Toluene Dioxygenase-Mediated cis-Dihydroxylation of Aromatics in Enantioselective Synthesis. Asymmetric Total Syntheses of Pancratistatin and 7-Deoxypancratistatin, Promising Antitumor Agents. J. Am. Chem. Soc 1996, 118, 10752.Chida N; Jitsuoka M; Yamamoto Y; Ohtsuka M; Ogawa S Total Synthesis of (+)-7-Deoxypancratistatin and (+)-7-Deoxy-trans-dihydronarciclasine. Heterocycles 1996, 43, 1385.Keck GE; Wager TT; McHardy SF A SecondGeneration Radical-Based Synthesis of (+)-7-Deoxypancratistatin. J. Org. Chem 1998, 63, 9164.Akgun H; Hudlicky T Total syntheses of ert-conduramine A and ent-7-deoxypancratistatin. Tetrahedron Lett. 1999, 40, 3081.Acena JL; Arjona O; Leon MA; Plumet J Total Synthesis of (+)-7-Deoxypancratistatiń from Furan. Org. Lett 2000, 2, 3683.Hakansson AE; Palmelund A; Holm H; Madsen R Synthesis of 7-Deoxypancratistatin from Carbohydrates by the Use of Olefin Metathesis. Chem. - Eur. J 2006, 12, 3243.Zhang H; Padwa A Application of a stereospecific RhCl(PPh3)3 decarbonylation reaction for the total synthesis of 7-(±)-deoxypancratistatin. Tetrahedron Lett. 2006, 47, 3905.Nieto-García O; Lago-Santome H; Cagide-Fagín F; Ortiz-Lara JC; Alonso R A formal [3 + 3]-annulation-based approach to pancratistatins: total synthesis of (±)-7-deoxy-pancratistatin and its 2-epi and 2,4-diepi analogues. Org. Biomol. Chem 2012, 10, 825.Cai S-L; Yuan B-H; Jiang Y-X; Lin G-Q; Sun X-W Asymmetric cinnamylation of N-tert-butanesulfinyl imines with cinnamyl acetates: total syntheses of (+)-lycoricidine and (+)-7-deoxypancratistatin. Chem. Commun 2017, 53, 3520.
- (20).For pancratistatin syntheses, see:Danishefsky S; Lee JY Total synthesis of (±)-pancratistatin. J. Am. Chem. Soc 1989, 111, 4829.Tian X; Hudlicky T; Koenigsberger K First Total Synthesis of (+)-Pancratistatin: An Unusual Set of Problems. J. Am. Chem. Soc 1995, 117, 3643.Trost BM; Pulley SR Asymmetric Total Synthesis of (+)-Pancratistatin. J. Am. Chem. Soc 1995, 117, 10143.(d) ref 28.Magnus P; Sebhat IK Synthesis of the Antitumor Alkaloid (+)-Pancratistatin Using the β-Azidonation Reaction via a Prochiral 4-Arylcyclohexanone Derivative. J. Am. Chem. Soc 1998, 120, 5341.Rigby JH; Maharoof USM; Mateo ME Studies on the Narciclasine Alkaloids: Total Synthesis of (+)-Narciclasine and (+)-Pancratistatin. J. Am. Chem. Soc 2000, 122, 6624.Kim S; Ko H; Kim E; Kim D Stereocontrolled Total Synthesis of Pancratistatin. Org. Lett 2002, 4, 1343.Li M; Wu A; Zhou P A concise synthesis of (+)-pancratistatin using pinitol as a chiral building block. Tetrahedron Lett. 2006, 47, 3707.Dam JH; Madsen R Convergent Synthesis of Pancratistatin from Piperonal and Xylose. Eur. J. Org. Chem 2009, 2009, 4666.Jung Y-G; Kang H-U; Cho H-K; Cho C-G β-Silyl Styrene As a Dienophile in the Cycloaddition with 3,5-Dibromo-2-pyrone for the Total Synthesis of (±)-Pancratistatin. Org. Lett 2011, 13, 5890.Cagide-Fagín F; Nieto-García O; Lago-Santome H; Alonso R Enantioselective Synthesis of Protected Nitrocyclohexitols with Five Stereocenters. Total Synthesis of (+)-Pancratistatin. J. Org. Chem 2012, 77, 11377.Akai S; Kojima M; Yamauchi S; Kohji T; Nakamura Y; Sato K.-i. A Concise Total Synthesis of (+)-Pancratistatin from D-Glucose Featuring the Henry Reaction. Asian J. Org. Chem 2013, 2, 299.Potter TJ; Ellman JA Total Synthesis of (+)-Pancratistatin by the Rh(III)-Catalyzed Addition of a Densely Functionalized Benzamide to a Sugar-Derived Nitroalkene. Org. Lett 2017, 19, 2985.
- (21).For previous syntheses of lycoricidine, see:Ohta S; Kimoto S Total synthesis of (±)-lycoricidine. Tetrahedron Lett. 1975, 16, 2279.(b) ref 19a.Paulsen H; Stubbe M Cyclit-Reaktionen, VIII. Synthese von enantiomerenreinem (+)-Lycoricidin aus D-Glucose. Liebigs Ann. Chem 1983, 1983, 535.Ugarkar BG; Dare J; Schubert EM Improved Synthesis of Lycoricidine Triacetate. Synthesis 1987, 1987, 715.Chida N; Ohtsuka M; Ogawa S Stereoselective total synthesis of (+)-lycoricidine. Tetrahedron Lett. 1991, 32, 4525.Chida N; Ohtsuka M; Ogawa S Total synthesis of (+)-lycoricidine and its 2-epimer from D-glucose. J. Org. Chem 1993, 58, 4441.Hudlicky T; Olivo HF A short synthesis of (+)-lycoricidine. J. Am. Chem. Soc 1992, 114, 9694.Hudlicky T; Olivo H; McKibben B Microbial Oxidation of Aromatics in Enantiocontrolled Synthesis. 3. Design of Amino Cyclitols (exo-Nitrogenous) and Total Synthesis of (+)-Lycoricidine via Acylnitrosyl Cycloaddition to Polarized 1-Halo-1,3-cyclohexadienes. J. Am. Chem. Soc 1994, 116, 5108.Martin SF; Tso HH Synthetic Studies on the Narciclasine Alkaloids. A Synthesis of (±)-Lycoricidine. Heterocycles 1993, 35, 85.Keck GE; Wager TT Total Synthesis of ent-Lycoricidine via a Thiyl Radical Addition–Cyclization Sequence. J. Org. Chem 1996, 61, 8366.Keck GE; Wager TT; Rodriguez JFD Total Syntheses of (−)-Lycoricidine, (+)-Lycoricidine, and (+)-Narciclasine via 6-exo Cyclizations of Substituted Vinyl Radicals with Oxime Ethers. J. Am. Chem. Soc 1999, 121, 5176.Elango S; Yan T-H A short synthesis of (+)-lycoricidine. Tetrahedron 2002, 58, 7335.Padwa A; Zhang H Synthesis of Some Members of the Hydroxylated Phenanthridone Subclass of the Amaryllidaceae Alkaloid Family. J. Org. Chem 2007, 72, 2570.Matveenko M; Kokas OJ; Banwell MG; Willis AC Chemoenzymatic Approaches to Lycorine-Type Amaryllidaceae Alkaloids: Total Syntheses of ent-Lycoricidine, 3-epient-Lycoricidine, and 4-Deoxy-3-epi-ent-lycoricidine. Org. Lett 2007, 9, 3683.Yadav JS; Satheesh G; Murthy CVSR Synthesis of (+)-Lycoricidine by the Application of Oxidative and Regioselective Ring-Opening of Aziridines. Org. Lett 2010, 12, 2544.(p) ref 19l.Southgate EH; Holycross DR; Sarlah D Total Synthesis of Lycoricidine and Narciclasine by Chemical Dearomatization of Bromobenzene. Angew. Chem., Int. Ed 2017, 56, 15049.
- (22).For previous syntheses of narciclasine, see:Rigby JH; Mateo ME Total Synthesis of (+)-Narciclasine. J. Am. Chem. Soc 1997, 119, 12655.Gonzalez D; Martinot T; Hudlicky T A short chemoenzymatic synthesis of (+)-narciclasine. Tetrahedron Lett. 1999, 40, 3077.(c) ref 30.Hudlicky T; Rinner U; Gonzalez D; Akgun H; Schilling S; Siengalewicz P; Martinot TA; Pettit GR Total Synthesis and Biological Evaluation of Amaryllidaceae Alkaloids: Narciclasine, ent-7-Deoxypancratistatin, Regioisomer of 7-Deoxypancratistatin, 10b-epi-Deoxypancratistatin, and Truncated Derivatives. J. Org. Chem 2002, 67, 8726.Elango S; Yan T A Short Synthesis of (+)-Narciclasine via a Strategy Derived from Stereocontrolled Epoxide Formation and SnCl4-Catalyzed AreneEpoxide Coupling. J. Org. Chem 2002, 67, 6954.Matveenko M; Banwell MG; Willis AC A chemoenzymatic total synthesis of entnarciclasine. Tetrahedron 2008, 64, 4817.(h) See also refs 20f and 21q.
- (23).Hernandez LW; Pospech J; Klöckner U; Bingham TW; Sarlah D Synthesis of (+)-Pancratistatins via Catalytic Desymmetrization of Benzene. J. Am. Chem. Soc 2017, 139, 15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).For selected reviews, see:Liebov BK; Harman WD Group 6 Dihapto-Coordinate Dearomatization Agents for Organic Synthesis. Chem. Rev 2017, 117, 13721.Keane JM; Harman WD A New Generation of π-Basic Dearomatization Agents. Organometallics 2005, 24, 1786.Pape AR; Kaliappan KP; Kündig EP Transition-Metal-Mediated Dearomatization Reactions. Chem. Rev 2000, 100, 2917.
- (25).For recent review, see:Boyd DR; Bugg TDH Arene cisdihydrodiol formation: from biology to application. Org. Biomol. Chem 2006, 4, 181.
- (26).Southgate EH; Pospech J; Fu J; Holycross DR; Sarlah D Dearomative dihydroxylation with arenophiles. Nat. Chem 2016, 8, 922.For early reports, see:.Hamrock SJ; Sheridan RS Para photoaddition of N-methyltriazolinedione to benzene. Synthesis of energy-rich azo compounds comprising benzene + nitrogen. J. Am. Chem. Soc 1989, 111, 9247.Kjell DP; Sheridan RS Photochemical cycloaddition of N-methyltriazolinedione to naphthalene. J. Am. Chem. Soc 1984, 106, 5368.
- (27).(a) Birch AJ; Haas M Removal of OMe from tricarbonyl-1or –2-methoxycyclohexa-1,–3-dieneiron complexes: a novel preparation of tricarbonyl-π-cyclohexadienyliron salts. Tetrahedron Lett. 1968, 9, 3705. [Google Scholar]; (b) Birch AJ; Chamberlain KB; Thompson DJ Organometallic complexes in synthesis. Part VI. Some oxidative cyclisations of tricarbonylcyclohexadieneiron complexes. J. Chem. Soc., Perkin Trans. 1 1973, 1, 1900. [Google Scholar]; (c) Davies SG; Green MLH; Mingos DMP Nucleophilic addition to organotransition metal cations containing unsaturated hydrocarbon ligands: A survey and interpretation. Tetrahedron 1978, 34, 3047. [Google Scholar]; (d) Pearson AJ Tricarbonyl(diene)iron complexes: synthetically useful properties. Acc. Chem. Res 1980, 13, 463. [Google Scholar]
- (28).VanRheenen V; Kelly RC; Cha DY An improved catalytic OsO4 oxidation of olefins to –1,2-glycols using tertiary amine oxides as the oxidant. Tetrahedron Lett. 1976, 17, 1973. [Google Scholar]
- (29).Raney-Co was used as a safer alternative to Raney-Ni. For recent review, see:Banwell MG; Jones MT; Reekie TA; Schwartz BD; Tan SH; White LV RANEY® cobalt – an underutilised reagent for the selective cleavage of C–X and N–O bonds. Org. Biomol. Chem 2014, 12, 7433.
- (30).Bal BS; Childers WE; Pinnick HW Oxidation of α,β-un saturated aldehydes. Tetrahedron 1981, 37, 2091.For oxidation of imines to amides, see:Mohamed MA; Yamada K.-i.; Tomioka K Accessing the amide functionality by the mild and lowcost oxidation of imine. Tetrahedron Lett. 2009, 50, 3436.
- (31).Banwell MG; Cowden CJ; Gable RW Lycoricidine and pancratistatin analogues from cyclopentadiene. J. Chem. Soc., Perkin Trans. 1 1994, 1, 3515.(b) See also refs 19c, 20e, 20g, 20l and 22b.
- (32).Cho H; Lee JO; Hwang S; Seo JH; Kim S Hendrickson-Reagent-Mediated Conversion of N-Boc Carbamates to Isocyanates: Applications for the Synthesis of 3,4-Dihydroisoquinolin-1-ones and Ureas. Asian J. Org. Chem 2016, 5, 287. [Google Scholar]
- (33).(a) Curci R; Fiorentino M; Troisi L; Edwards JO; Pater RH Epoxidation of alkenes by dioxirane intermediates generated in the reaction of potassium caroate with ketones. J. Org. Chem 1980, 45, 4758. [Google Scholar]; (b) Adam W; Hadjiarapoglou LP; Smerz A Dioxirane Epoxidation of α,β-Unsaturated Ketones. Chem. Ber 1991, 124, 227. [Google Scholar]; (c) Kurihara M; Ito S; Tsutsumi N; Miyata N Stereoselective epoxidation with dioxiranes generated from ketones. Tetrahedron Lett. 1994, 35, 1577. [Google Scholar]
- (34).(a) Trost BM The atom economy–a search for synthetic efficiency. Science 1991, 254, 1471. [DOI] [PubMed] [Google Scholar]; (b) Newhouse T; Baran PS; Hoffmann RW The economies of synthesis. Chem. Soc. Rev 2009, 38, 3010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Burns NZ; Baran PS; Hoffmann RW Redox Economy in Organic Synthesis. Angew. Chem., Int. Ed 2009, 48, 2854. [DOI] [PubMed] [Google Scholar]
- (35).For a review on the site-selective opening of vinylepoxides, see:Olofsson B; Somfai P In Aziridines and Epoxides in Organic Synthesis; Yudin AK, Ed.; Wiley-VCH: Weinheim, Germany, 2006; p 315.
- (36).For the beneficial role of HFIP in epoxidation and epoxide opening reactions, see:Berkessel A; Adrio JA Dramatic Acceleration of Olefin Epoxidation in Fluorinated Alcohols: Activation of Hydrogen Peroxide by Multiple H-Bond Networks. J. Am. Chem. Soc 2006, 128, 13412.Byers JA; Jamison TF Entropic factors provide unusual reactivity and selectivity in epoxideopening reactions promoted by water. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 16724.
- (37).For recent review, see:Wu X-F; Neumann H; Beller M Palladium-catalyzed carbonylative coupling reactions between Ar–X and carbon nucleophiles. Chem. Soc. Rev 2011, 40, 4986.
- (38).For selected early examples, see:Blackburn TF; Schwartz J Homogeneous catalytic oxidation of secondary alcohols to ketones by molecular oxygen under mild conditions. J. Chem. Soc., Chem. Commun 1977, 157, 157.Peterson KP; Larock RC Palladium-Catalyzed Oxidation of Primary and Secondary Allylic and Benzylic Alcohols. J. Org. Chem 1998, 63, 3185.Nishimura T; Onoue T; Ohe K; Uemura S Palladium(II)-Catalyzed Oxidation of Alcohols to Aldehydes and Ketones by Molecular Oxygen. J. Org. Chem 1999, 64, 6750.
- (39).Brunet J-J; Sidot C; Caubere P Sunlamp-irradiated phase-transfer catalysis. 1. Cobalt carbonyl catalyzed SRN1 carbonylations of aryl and vinyl halides. J. Org. Chem 1983, 48, 1166. [Google Scholar]
- (40).Rossi RA; Pierini AB; Penenory AB Nucleophilic Substitution Reactions by Electron Transfer. Chem. Rev 2003, 103, 71. [DOI] [PubMed] [Google Scholar]
- (41).For selected examples, see:Desai LV; Malik HA; Sanford MS Oxone as an Inexpensive, Safe, and Environmentally Benign Oxidant for C–H Bond Oxygenation. Org. Lett 2006, 8, 1141.Chen X-Y; Ozturk S; Sorensen EJ Pd-Catalyzed Ortho C–H Hydroxylation of Benzaldehydes Using a Transient Directing Group. Org. Lett 2017, 19, 6280.Liu W; Ackermann L Ortho- and Para-Selective Ruthenium-Catalyzed C(sp2)–H Oxygenations of Phenol Derivatives. Org. Lett 2013, 15, 3484.
- (42).For example, see:Ishiyama T; Takagi J; Ishida K; Miyaura N; Anastasi NR; Hartwig JF Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc 2002, 124, 390.Ros A; Estepa B; Lopez-Rodriguez R; Alvarez E; Fernandez R; Lassaletta JM Use of Hemilabile N,N Ligands in Nitrogen-Directed Iridium-Catalyzed Borylations of Arenes. Angew. Chem., Int. Ed 2011, 50, 11724.Ishiyama T; Isou H; Kikuchi T; Miyaura N Ortho-C–H borylation of benzoate esters with bis(pinacolato)diboron catalyzed by iridium–phosphine complexes. Chem. Commun 2010, 46, 159.
- (43).For reviews, see:Snieckus V Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics. Chem. Rev 1990, 90, 879.Chevallier F; Mongin F; Takita R; Uchiyama M In Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds; Mortier J, Eds.; John Wiley & Sons: Hoboken, NJ, 2015; p 777.
- (44).Karimi B; Golshani B Mild and Highly Efficient Method for the Silylation of Alcohols Using Hexamethyldisilazane Catalyzed by Iodine under Nearly Neutral Reaction Conditions. J. Org. Chem 2000, 65, 7228. [DOI] [PubMed] [Google Scholar]
- (45).Tezuka N; Shimojo K; Hirano K; Komagawa S; Yoshida K; Wang C; Miyamoto K; Saito T; Takita R; Uchiyama M Direct Hydroxylation and Amination of Arenes via Deprotonative Cupration. J. Am. Chem. Soc 2016, 138, 9166. [DOI] [PubMed] [Google Scholar]
- (46).Hernandez LW; Klöckner U; Pospech J; Hauss L; Sarlah D Nickel-Catalyzed Dearomative trans-1,2-Carboamination. J. Am. Chem. Soc 2018, 140, 4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).For a comprehensive review on base-promoted isomerization of epoxides, see:Crandall JK; Apparu M Base-Promoted Isomerizations of Epoxides. Org. React 1983, 29, 345.
- (48).For elimination involving similar β-phenyl substituted epoxides, see:Thummel RP; Rickborn B Base-induced rearrangement of epoxides. V. Phenyl-substituted epoxides. J. Org. Chem 1972, 37, 3919.
- (49).Though lycoricidine has been isolated and synthesized numerous times, its X-ray crystallographic structure has not been documented in CCDC.
- (50).Mei R; Sauermann N; Oliveira JCA; Ackermann L Electroremovable Traceless Hydrazides for Cobalt-Catalyzed Electro-Oxidative C–H/N–H Activation with Internal Alkynes. J. Am. Chem. Soc 2018, 140, 7913. [DOI] [PubMed] [Google Scholar]
- (51).For our previous application of Uchiyama’s reagent in synthesis of (±)-narciclasine, see ref 21q.
- (52).(a) Beak P; Kokko BJ A modification of the Sheverdina-Kocheshkov amination: the use of methoxyamine-methyl lithium as a convenient synthetic equivalent for NH2+. J. Org. Chem 1982, 47, 2822. [Google Scholar]; (b) Berman AM; Johnson JS Copper-Catalyzed Electrophilic Amination of Functionalized Diarylzinc Reagents. J. Org. Chem 2005, 70, 364. [DOI] [PubMed] [Google Scholar]
- (53).Prakash C; Shaffer CL; Nedderman A Analytical strategies for identifying drug metabolites. Mass Spectrom. Rev 2007, 26, 340. [DOI] [PubMed] [Google Scholar]
- (54).Harbeson SL; Tung RD Chapter 24 - Deuterium in Drug Discovery and Development. Annu. Rep. Med. Chem 2011, 46, 403. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.