

Abstract
Scavenger receptor CD36 is a multifunctional membrane protein that promotes thrombosis in conditions of oxidative stress such as metabolic disorders including dyslipidemia, diabetes mellitus, and chronic inflammation. In these conditions, specific reactive oxidant species are generated that are context and cell dependent. In the vasculature, CD36 signaling in smooth muscle cells and endothelial cells promote generation of reactive oxygen species, genetic downregulation of antioxidant genes, and impaired smooth muscle and endothelial function. In hematopoietic cells, CD36 signaling enhances platelet dysfunction thus decreasing the threshold for platelet activation and accelerating arterial thrombosis, whereas in macrophages, CD36 promotes lipid-laden foam cell formation and atherosclerosis. These clinically significant processes are mediated through complex redox regulated signaling mechanisms that include Src-family kinases, MAP kinases and other downstream effectors. We provide an overview of CD36 signaling in vascular redox stress highlighting the role on oxidant generation in vascular and hematopoietic cells, but with special emphasis on platelets and dyslipidemia.
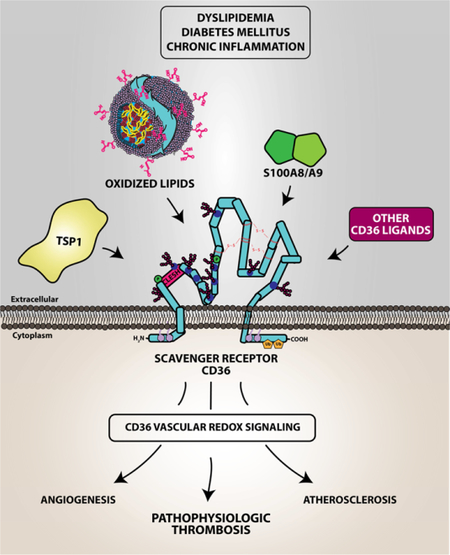
Graphical Abstract
Introduction
Arterial thrombosis is the major cause of death and disabilities in conditions associated with vascular redox stress including cardiovascular disease (1), chronic inflammation (2), and diabetes mellitis (3,4).Thrombosis in these contexts is mediated by activation of platelets, which are cell fragments derived from megakaryocytes in the bone marrow and are essential to maintain hemostasis (5). Inappropriate platelet activation induced by vascular redox stress promotes risk for vascular occlusion and its clinical complications.
Dyslipidemia refers to a group of metabolic disorders characterized by elevated levels of lipoprotein and/or triglycerides and is a risk factor for enhanced platelet activation in cardiovascular diseases (6). Cholesterol carrying molecules, such as low-density lipoprotein (LDL) particles, are susceptible to oxidation during the inflammatory processes of atherosclerotic plaque formation within the vessel wall. Over the course of time, LDL particles become oxidized. Signature oxidation sites have been described on the all components of the particle, including protein, cholesterol, and phospholipids (all shown in (Figure 1A). The oxidants and mechanisms of altering LDL physiology in vivo has been a topic of debate and has been excellently reviewed (7). Oxidation of fatty acids with unsaturated carbon-carbon bonds (generally in the sn-2 position of the glycerol phospholipid) by reactive oxygen species promotes beta scission, release of the aldehyde products, and generates a truncated carbon chain whereby incorporation of diatomic oxygen renders the lipids hydrophilic. These lipids, which were characterized as predominantly oxidized phosphatidylcholine, are termed oxPCcd36 due to exhibiting high affinity binding to the scavenger receptor CD36. They are characterized by having a carboxylic acid or aldehyde in the terminal position, a double bond in the beta carbon, and a ketone or alcohol in the gamma position (see oxPCCD36 motif in Figure 1A). If the carbon backbone is of sufficient length, the hydrophilic nature of the incorporated oxygen in oxPCCD36 results in translocation of the lipid in the lipoprotein particles to the exterior face of the lipoprotein, forming so-called “lipid whiskers”(8) (oxLDL of Figure 1A). This translocation event is likely thermodynamically driven and enzyme-independent. Exposure of oxPCCD36 as lipid whiskers promotes its selective recognition by the pattern recognition receptor CD36 (8–10). These lipid species are among a large group of endogenous danger-associated molecular patterns (DAMPs) formed in the setting of tissue injury, cell damage, inflammation and redox stress, and are a major risk factor for myocardial infarction or stroke.
Figure 1. A) Oxidized lipids/cholesterol are risk factors for clinically significant thrombotic events in cardiovascular disease.


Low density lipoprotein particles, which are cholesterol carrying molecules, become oxidized during the inflammatory and oxidative processes of atherosclerotic plaque formation. The major targets of redox active species are the protein, cholesterol, and lipid components of the particle. Phospholipid oxidation at the sn-2 position by these species promotes truncation of the lipid and subsequent incorporation of oxygen. The specific oxidized lipid motif, known as oxPCCD36, is shown, which is predominantly oxidized phosphatidylcholine species with a carboxylic acid or aldehyde in the terminal position, a double bond in the beta carbon, and a ketone or alcohol in the gamma position. These oxidized lipids translocate to the aqueous plasma milieu forming so-called lipid “whiskers” that are recognized selectively by scavenger receptor CD36 present abundantly on platelets. Cholesterol oxidation yields keto- and alcohol-derivatives of the cholesterol. Modification of the protein component of the lipoprotein is amino acid-dependent (e.g. tyrosine nitration, aldehyde adduction on lysines). B.) Scavenger receptor CD36 topology and structure. The depiction of CD36 on the membrane is shown with various post-translational modifications. CD36 is palmitoylated on the two short N- and C-terminal tails with ubiquitination sites in the C-terminal tail. CD36 has two membrane-spanning domains that are involved in protein-protein interaction with itself to form dimers and with other receptors clustered in membrane lipid microdomains, such as toll-like receptors (55,104), sodium-potassium ATPase (57), tetraspanin CD9 (54), and others (11). The extracellular domain of CD36 is heavily glycosylated, which functions to traffic the protein to the correct membrane compartment. Residues 92–120 are known as the CLESH domain, and are essential for binding to the type 1 thrombospondin repeat domain (TSR) found in anti-angiogenic proteins of the thrombospondin family. Furthermore, a hydrophobic pocket represents a potential domain for fatty acid and oxidized lipid interaction. CD36 has two potential phosphorylation sites on the extracellular domain (Thr92 and Ser237). Three disulfide bonds within the extracellular domain are essential to maintain the structure of the protein. Entrance 1 in the figure represents a hydrophobic pocket for interaction with various ligands. Entrance 2 in the figure represents a hypothesized channel for fatty acid transportation as suggested by structural modeling with the LIMP-II member of the scavenger receptor family. The mouse anti-CD36 monoclonal antibody [clone FA6–152], which is predominantly used as an inhibitor, was shown to bind to amino acids 155–183 and inhibits the ligand binding capacity. However, the detailed mechanism of inhibiting CD36 signal transduction by this antibody is not clear and is potentially due to masking the receptor-ligand binding site or changing the conformation of the protein.
Image (B) was originally printed in Yang, X. et al. (2017) Nature Review It was modified, and reprinted with permission from Springer Nature
Scavenger Receptor CD36
Cluster of Differentiation 36 (CD36) is a multifunctional pattern recognition membrane receptor that is highly expressed on vascular and hematopoietic cells. CD36 belongs to the class B scavenger receptor family (11) and was initially named platelet Glycoprotein IV because it is the fourth major glycoprotein band seen on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of platelet lysates (12). Multiple vascular cells and hematopoietic cells express this protein, including vascular smooth muscle cells and endothelial cells, innate immune cells such as macrophages, and platelets (9,13,14). The human CD36 gene is located on chromosome 7q11.2 and has 15 exons (15) encoding a 471 amino acid protein of about 50 kilodalton molecular weight (16). Like most membrane proteins, glycosylation is required for proper trafficking to the membrane; heavy N-glycosylation increases the molecular weight of the protein to 88 kilodalton (17). CD36 localizes in cholesterol-rich membrane microdomains and has been used as an immune-marker for caveolae in microvascular endothelial cells.
In humans, the blood group polymorphism called Naka-was identified on platelet CD36 (18). Up to 5–10% of Japanese carry the Naka-negative phenotype; they do not express detectable levels of platelet CD36 (18,19). Two types of Naka negative phenotype have been described: Type I is the result of being homozygous or compound heterozygous for null mutations at both alleles and is associated with absence of CD36 in all cells. Type II is a platelet-specific deficiency (20,21) and its genetic basis is not well understood. The frequency of CD36 null alleles in Asian and African populations is very high, (7–10%) and the selective pressure has not been identified (22). Furthermore, platelet CD36 expression levels vary considerably in humans. Quantitative flow cytometry analysis showed an average copy number of 17,000 per cell (23) and genetic studies revealed specific polymorphisms in the CD36 gene that are associated with platelet surface expression levels and with responsiveness to its model ligand, oxLDL (23). Importantly, some of these single nucleotide polymorphisms have been associated with risk for myocardial infarction (23,24).
CD36 Structure and Function
CD36 has at least 3 well characterized cellular functions. The first to be described was as a membrane receptor for the matricellular protein thrombospondin-1 (TSP1). On microvascular endothelial cells interaction of TSP1 with CD36 mediates a potent anti-angiogenic effect (25). CD36 also facilitates translocation of free fatty acids from the extracellular milieu into the cytoplasm. This function is important in adipocyte and muscle cell physiology and in some published papers CD36 is referred to as fatty acid translocate (FAT)(26). Our lab has been interested in the function of CD36 as a scavenger receptor, that is a receptor that recognizes and internalizes specific exogenous and endogenous danger signals. These include microbial surface liposaccharides(27), malaria parasitized erythrocytes (28,29), advanced glycated proteins (30), and modified low density lipoproteins (LDL), such as oxidized LDL (oxLDL)(9). The latter has been shown by our group and others to be critically important in the pathogenesis of atherosclerosis by mediating macrophage foam cell formation in the vessel wall and trapping of foam cell in atheromatous plaque. CD36 also recognizes HDL and other lipoproteins; however, the functional relevance of these recognition is not well understood.
A high resolution molecular structure for CD36 has not been solved, but homology modeling based on published extracellular structure of the related protein Lysosomal Integral Membrane Protein II (LIMP-II) indicates distinct hydrophobic pockets potentially for lipid recognition and fatty acid trafficking; these are shown in Entrance 1 and Entrance 2 of Figure 1B (31). Topological analysis showed that CD36 has two short N- and C-terminal intracellular tails, two transmembrane domains, and a very large extracellular domain that is heavily glycosylated. There are three disulfide bonds in the extracellular domain required to maintain its structure (11). CD36 is palmitoylated on both N- and C-terminal tails to anchor the protein to the membrane bilayer. Additionally, CD36 was shown to be regulated by ubiquitination on the C-terminal tail. However, the functional impact of ubiquitination on CD36 is not very well understood but was proposed to regulate CD36 expression that is context dependent. Smith et al. showed in Chinese Hamster Ovary cells that CD36 was ubiquitinated on Lys48 and Lys63. Exogenous fatty acids promote ubiquitination whereas insulin-treatment in insulin-expressing cells lead to decreased ubiquitination (32). In addition, CD36 has a consensus protein kinase C (PKC) phosphorylation site in its extracellular domain at Thr92(33). This site can be phosphorylated in vitro by PKC, and when phosphorylated blocks thrombospondin 1 binding (33) and modulates recognition of Plasmodium falciparum-infected erythrocytes (34). An additional putative extracellular phosphorylation site on Ser237 by PKA was proposed to regulate fatty acid binding and uptake (35). The in vivo relevance of these post-translational modifications, however, has not been convincingly demonstrated.
The molecular mechanisms by which CD36 recognizes its diverse ligands are an active area of research. CD36 recognizes many ligands in addition to oxPCCD36, including free fatty acids (26), TSP1 (25) and other proteins containing the so-called thrombospondin type 1 repeat (TSR) domain (36), cell-derived microparticles (37), advanced glycation end products (AGE) (30), a calcium binding S100A protein family member known as myeloid-related protein 14 (38), high density lipoprotein (HDL) (39), and microbial products such as staphylococcal lipoteichoic acid (40). Ligand recognition sites on the extracellular domain of CD36 are not fully characterized; however, the binding site for TSR was mapped to an approximate 30 amino acid (aa) residue region on the extracellular surface of the protein called the CLESH (CD36, LIMP-2, Emp sequence homology) domain extending from aa90 (25). This binding site was shown to be important for the anti-angiogenic potential of CD36 and is modulated by electrostatic and steric properties, including perhaps phosphorylation of Thr92 (33). The CLESH domain is lined with positively charged amino acid residues and interacts with negatively charged residues of TSR. The motif on CD36 that recognizes oxidized lipids is still unclear; however, mutational analyses showed that two lysine residues (K164 and K166) located on the extracellular domain are required for oxPCCD36 binding, potentially by electrostatic interaction with the negatively charged oxygen molecules of oxidized lipids (41).
In seminal work by Podrez et al, platelet CD36 was shown to couple inflammation and dyslipidemia to a prothrombotic phenotype (9). Using the apoE null strain of mice, a widely used model to study dyslipidemia with pathophysiology similar to that observed in human (42), they showed that dyslipidemia augments thrombosis induced by vascular injury in small and large arteries, and that this phenotype was rescued by genetically deleting CD36. Furthermore, the absence of CD36 in mice did not impact normal platelet activation by “classic” physiologic activators, such as adenosine diphosphate (ADP), collagen or thrombin. These studies were replicated using human platelets, which showed that the absence of CD36, its inhibition by specific monoclonal antibodies or with the competing lipid ligand sulfosuccinimidyl oleate did not impact normal platelet physiology but prevented platelet activation by oxidized lipids. Subsequent work by our lab and others extended these observations to show that pathological conditions associated with redox stress, such as diabetes (30) and chronic inflammation (37) also generated CD36 ligands, including advanced glycation end products and cell-derived microparticles that promoted platelet activation and arterial thrombosis. These data suggest that CD36 signaling acts as a platelet “rheostat” to reduce the threshold for platelet activation and thus promote a prothrombotic phenotype. CD36 signaling pathways thus represent potential therapeutic targets. Although the initial studies by Podrez et al were published more than 10 years ago, the pathophysiology of platelet CD36 is still under intense investigation with significant efforts put forth to understand the signaling mechanisms driving platelet activation.
CD36 REDOX SIGNALING
Ligand-mediated CD36 signaling characteristically promotes generation of intracellular reactive oxygen species that are context-and cell type-dependent. The mechanisms generating reactive oxygen species vary among vascular and hematopoietic cells. This section highlights key publications regarding oxidant signaling by CD36 in vascular smooth muscle cells, endothelial cells, monocytes/macrophages, and platelets.
CD36 redox signaling in vascular smooth muscle and endothelial cells.
The vasculature is a complex and dynamic environment fundamental to maintain homeostasis. Vascular smooth muscle cells, for example, are essential for maintaining vascular tone and blood flow. In dyslipidemia, smooth muscle cell dysfunction promotes atherosclerotic plaque progression increasing the risk for plaque rupture and thrombosis (13). CD36 is expressed in vascular smooth muscle cells and was shown to modulate vascular function and arterial thrombosis. In particular, CD36 signaling downregulates key antioxidant factors, such as the redox-sensitive nuclear factor Nrf2, when vascular smooth muscle cells are stimulated with oxidized lipids. Nrf2 downregulation is via its phosphorylation by Src family kinase member Fyn (see Figure 2A) (13). Although not directly shown in vascular smooth muscle cells, Fyn was shown to be recruited to and associate with CD36 upon DAMP recognition in endothelial cells and platelets (43–45). Nrf2 promotes transcription of the peroxide detoxifying enzyme peroxiredoxin 2 (Prdx2). The downregulation of Nrf2 leads to the accumulation of reactive oxygen species, since Prdx2 is consequentially downregulated. However, the downregulation of Nrf2 in the CD36 signaling pathway is not exclusive. Activators of Nrf2, such as oxLDL and 4-hydroxy-2-nonenal, promote CD36 expression in murine macrophages (46), the macrophage-like cell line RAW264.7 (47), and the pre-adipocyte cell line 3T3-L1 (47). This mechanism could be related to an increase demand in CD36 expression to uptake its ligands and is exemplified further by Nrf2-mediating CD36 expression to support of Plasmodium phagocytosis (48,49). Nrf2 is intricately linked to the NLRP3 inflammasome pathway through multiple signaling points, including the activation of NF-kB and regulating reactive oxygen species. This link is highlighted by Sheedy and colleagues on the role for CD36 to promote priming of monocytes/macrophages through the NLRP3 inflammasome and NF-kB activation, cholesterol uptake, and cholesterol crystal formation (50). Although specific sources of reactive oxygen species generated by CD36 signaling are yet to be defined in smooth muscle cells, pharmacologic studies suggest that reactive oxygen species are likely generated from both mitochondrial and non-mitochondrial sources, such as NADPH oxidase (51). Specific sources of ROS in vascular smooth muscle cell CD36 signaling requires further investigation.
Figure 2. CD36 redox signaling in vascular smooth muscle cells, microvascular endothelial cells, and monocytes/macrophages.


(A) In vascular smooth muscle cells, CD36 signaling was shown to downregulate the antioxidant transcription factor Nrf2. Nrf2 is normally a cytosolic protein held in an inactive state by binding to an inhibitory partner known as KEAP or Inhibitor of Nrf2 (INrf2). Upon oxidative stress the two proteins dissociate and Nrf2 translocates to the nucleus where it binds to specific DNA sequences known as antioxidant response elements (ARE) to promote transcription of multiple antioxidant genes, including peroxiredoxin 2 (Prdx2) and heme oxygenase-1 (HO-1). CD36 signaling via the Src family kinase Fyn, leads to phosphorylation of nuclear Nrf2, nuclear export, and degradation, thereby preventing upregulation of antioxidants and thus promoting reactive oxygen species accumulation and pro-atherogenic cellular activity. (B) In microvascular endothelial cells (MVEC), vascular endothelial growth factor receptor 2 (VEGFR2) recognizes its ligand VEGF and becomes phosphorylated. VEGFR2 than transduces signals to mediate cellular migration, proliferation, and tube formation important for angiogenesis. However, CD36 through recognition of thrombospondin-1 and related proteins promotes activation of Fyn, p38 MAP kinase, and apoptosis initiating caspases, which overall limits endothelial cell migration and tube formation. CD36 also forms functional complexes with VEGFR2, promoting a spleen tyrosine kinase (Syk)-dependent phosphorylation and activation of the Src-homology Protein Tyrosine Phosphatase 1 (SHP-1), which dephosphorylates VEGFR2 to limit MVEC function. (C) In macrophages, CD36 was shown to form a complex with multiple proteins in membrane microdomains, including CD9, Toll-Like Receptors, Fc Receptor γ, and Na-K ATPase. Upon oxLDL binding, CD36 promotes direct recruitment and activation of Src family kinases (particularly Lyn via its association with Na-K ATPase) for signal transduction. This signal transduction promotes the phosphorylation of focal adhesion kinases for cytoskeletal rearrangement, loss of cellular locomotion, and macrophage trapping. In addition, this signal transduction increases the generation of reactive oxygen species from NADPH oxidase. These species modify the protein tyrosine phosphatase SHP2 inactivating the enzyme, which regulates Src kinase-mediated signaling. oxLDL recognition by CD36 also promotes Vav family guanine nucleotide exchange factors that promote Rac activity to modulate non-muscle myosin II and regulation of cellular locomotion. Vavs also connect CD36 to an oxLDL-uptake mechanism that is dependent on dynamin. In relation to oxLDL uptake, MAP kinase activation (JNK1/2) was shown to be essential for CD36-mediated macrophage foam cell formation. The mechanistic connection between CD36, Toll-like receptors, and Na-K ATPase were shown to be linked to activation of pro-inflammatory pathways, including the NF-kB pathway and activation of the NLRP3 inflammasome, that promotes atherosclerosis. The CD36 and Toll-like receptor signaling pathway was linked to cholesterol crystal deposition and foam cell formation.
Image (A) was originally printed in Li W. et al. (2010) Journal of Clinical Investigations by the American Society of Clinical Investigations. It was modified and reprinted with permission.
Image (B) was originally published in Chu L.Y. et al. (2013) Blood by the American Society of Hematology. It was modified and reprinted with permission.
Image (C) image was originally printed in Park Y.M. (2014) Experimental and Molecular Medicine by Springer Nature. It was modified and reprinted.
CD36 is also expressed on microvascular endothelial cells (MVEC) and plays an essential role in endothelial cell function. Specifically, CD36 binds to the TSR domain in several anti-angiogenic proteins including thrombospondin-1 and thrombospondin-2 (25). Recognition of TSR by CD36 promotes MVEC apoptotic signaling, enhances dephosphorylation of pro-angiogenic Vascular Endothelial Growth Factor Receptor (VEGFR), and decreases endothelial cell migration and formation of tube-like structures (Figure 2B) (52). CD36 on MVECs also recognizes anionic phosphatidylserine on the surface of extracellular vesicles, leading to inhibition of cell migration (53). This is mediated by a pathway requiring Src family kinase Fyn, activation of NADPH oxidase, and generation of reactive oxygen species. Genetic deletion of CD36, pharmacologic inhibition of Src family kinases or NADPH oxidase, or scavenging superoxide radical anion by the superoxide dismutase mimetic MnTMPyP restored endothelial cell migration in cells treated with extracellular vesicles. These data suggest that reactive oxygen species generated by CD36 modulate MVEC migration in specific conditions where circulating extracellular vesicles are abundantly present, such as in chronic inflammation and cardiovascular disease.
CD36 redox signaling in macrophages.
In dyslipidemia, cholesterol in low-density lipoprotein particles accumulates in the subendothelial space in the vascular wall and triggers sterile inflammation. Blood monocytes migrate into the vascular wall, differentiate to macrophages, and promote lipid clearance. However, cholesterol overload and lipoprotein particle oxidation augments lipid-laden macrophage “foam” cell formation, trapping of the macrophages in the neointima, and promotes cell death. These processes, which are in part CD36-dependent as demonstrated with genetic, pharmacologic and immunologic approaches, promote inflammation and atherosclerotic plaque progression.
While CD36 functions as a direct ligand-dependent signal transducer, it colocalizes in membrane microdomains with other cellular receptors, including tetraspanins (54), Toll-like receptors 2/4/6 (55,56) and Na/K ATPase (57); interaction with these receptors modulates cellular responses (Figure 2C). For example, in the presence of oxLDL, Na/K ATPase is recruited to the CD36 signaling complex and participates in signaling via its constitutively bound Src family tyrosine kinase Lyn (57). We found that macrophages from mice heterozygous for deficiency of the alpha subunit of Na/K ATPase had diminished CD36-dependent oxLDL uptake and foam cell formation. These mice also displayed decreased atherosclerotic plaque progression when bred into the dyslipidemic apoE null background. CD36 expression on the membrane surface in macrophages was shown in elegant imaging studies by Jaqaman et al. to be modulated by cytoskeletal dynamics which promoted signal transduction (58). It is possible that other co-receptors for CD36 exists within membrane microdomains that are yet to be identified.
Of relevance to redox signaling, macrophage CD36 promotes a signaling pathway that generates reactive oxygen species (Figure 2C) (59). While much remains to be learned about downstream consequences of macrophage ROS generation, we showed that these species modify critical cysteine residues of the protein tyrosine phosphatase SHP2 leading to disruption of actin cytoskeletal rearrangements and macrophage migration (59). We also showed that CD36 signaling leads to loss of macrophage cellular polarity due to dysregulation of Rac and non-muscle myosin II (60), key determinants of cellular symmetry and lamellipodia formation. The specific redox species modifying cytoskeletal function in this setting is not clear and were not investigated, but since inhibition of NADPH oxidase or scavenging reactive oxygen species by N-acetyl cysteine restored phosphorylation of focal adhesion kinase, we speculate that hydrogen peroxide is the major reactive oxygen species generated.
Our lab also showed that macrophage CD36 promotes atherosclerosis through Vav family guanine nucleotide exchange factors (61,62). Vavs are highly regulated by Src-mediated tyrosine phosphorylation and act as central signaling hubs to several signaling pathways (63). Indeed, in the macrophage system CD36-mediated Vav activation requires Src family kinases in addition to phospholipase Cγ2 (61,62). Mechanistically, Vav activation promotes calcium mobilization and nucleotide exchange of the GTPase Dynamin 2, which is required for oxLDL uptake and foam cell formation as shown in Figure 2C. Importantly, tissue extracts taken from aortas of hyperlipidemic mice showed that Vavs1 and 3 were activated in a CD36-dependent manner.
Kotla et al showed CD36 gene expression was mechanistically regulated by reactive oxygen species in the macrophage system (64). These species generate cholesterol crystals, which drive CD36 expression through a pathway dependent on the signaling effector Bruton tyrosine kinase (BTK) to phosphorylate acyl transferase p300. P300 then acetylate the transcription factor STAT1. These signaling events subsequently promote association of STAT1 with transcriptional co-activator Peroxisome Proliferator Activating Receptor Gamma (PPARγ) to drive CD36 expression. These studies link reactive oxygen species generation in the macrophage system to genetic transcription of the protein. We and others report that oxidized lipid stimulation of macrophages also upregulates CD36 expression by a PPARγ-mediated transcriptional pathway (65).
Oxysterols, which are oxidized cholesterols generated enzymatically or non-enzymatically, are pro-atherogenic in monocytes/macrophages through mechanisms that are partially dependent on scavenger receptors. 7-ketocholesterol, in particular, was shown to promote CD36 gene expression in macrophages and subsequent oxidized LDL uptake/foam cell formation (66). Furthermore, oxysterols was shown to promote CD36 expression on cells of the macrophage lineage(67). This pathway was shown to be dependent on PKC and MAP kinase ERK1/2 based on RNAi or pharmacologic inhibition (67,68).
Platelet CD36 signaling in dyslipidemia.
In platelets, CD36 interacts with multiple signaling pathways. A role for CD36 in platelet signal transduction was initially suggested by studies in which CD36 was found to co-immuno-precipitate with members of the Src family kinases Fyn, Lyn, and Yes (44). Subsequent work from our lab demonstrated that CD36 recruits and activates Fyn and Lyn, with Fyn being the predominant Src family kinase in platelets in response to oxLDL and other ligands (43). These then further promote activation of non-receptor tyrosine kinase Syk (69,70), the Rho/Rho-associated protein kinase ROCK (69), Vav 1/3(71), generation of reactive oxygen species(72,73), and activation of MAP kinases (61,71). The multiple pathways activated by CD36 suggest that further details are needed to pinpoint specific signaling nodes mediating enhanced platelet activation, aggregation, and ultimately thrombus formation.
NADPH oxidase is a major source of oxidant generation by platelet CD36
Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase (NOX) is a membrane multi-subunit complex that generates superoxide radical anion by transferring electrons from the electron carrier NADPH to diatomic oxygen. NOX expression in platelets is not surprising given the evolutionary link between platelets and phagocytes and their developmental origin from a common hematopoietic progenitor cell. NOX1 and NOX2 are reported as the major complexes to generate reactive oxygen species in platelets (74–76). Earlier studies by Violi and colleagues suggested that reactive oxygen species generated from NOX promote platelet activation (77–80) implicated NOX in platelet function. Studies of individuals with chronic granulomatous disease, an x-linked genetic deficiency in the cybb1 allele, or with deficiencies in any member of the NOX protein complex, showed decreased platelet activation (77,80–82). Delaney and colleagues further showed that murine models deficient in NOX1 or NOX2 had impaired platelet response to the classic physiologic agonists thrombin and collagen (74), consistent with studies showing that thrombin and collagen are potent platelet activators that generate reactive oxygen species (83). These studies suggest a functional role for reactive oxygen species and NOX in promoting platelet activation and thrombosis.
In 2014, Magwenzi and colleagues showed that NOX2 was the major source of reactive oxygen species generated by platelet CD36 signaling (73), an unprecedented finding given reactive oxygen species from this source is generally thought to be linked to signaling by strong platelet agonists, such as thrombin and collagen. They showed that pharmacologic or genetic interruption of NOX2 rescued the CD36-mediated arterial thrombosis phenotype observed in dyslipidemic conditions. This is mediated by direct recognition of oxLDL, activation of Src kinases, subsequent phosphorylation and activation of Syk, and activation of phospholipase C gamma 2 (PLCγ2). PLCγ2 thus links CD36 signaling to the activation of Protein Kinase C, which is a serine/threonine kinase that coordinates with TRAF4 and Src family kinase member Lyn to promote phosphorylation of the p47 phox subunit of NOX2 (76). This phosphorylation event couples assembly of the NOX complex to generate reactive oxygen species. Inhibiting CD36 on human platelets by a monoclonal blocking antibody or by using CD36 deficient murine platelets prevented the generation of reactive oxygen species (72,73,84). Direct evidence of PLCγ2 and NOX2 in diet-induced prothrombotic phenotype was shown by using gp91phox null mice or mice lacking the PLCg2 isoform in the ferric chloride-induced arterial thrombosis model (84). Although these studies are provocative, direct evidence of specific reactive oxygen species generated and their functional output was not provided.
In a more analytical approach, we measured levels of superoxide radical anion generated by platelet NOX2 in the CD36 signaling cascade using a sensitive and quantitative high-performance liquid chromatography (HPLC) assay with fluorescence detection (72). This assay relies on quantifying fluorescence of the superoxide-specific oxidation product of hydroethidium, 2-hydroxyethidine, and fitting quantified data to a synthetic 2-hydroxyethidine standard curve. Mechanistically, hydroethidium undergoes non-specific one electron oxidation to hydroethidine radical cation. It is the hydroethidine radical cation that is oxidized by superoxide radical anion to generate 2-hydroxyethidine (85). However, hydroethidine radical cation is nonspecifically oxidized to ethidium or to form dimers, which may titrate the radical intermediate away from superoxide radical anion. In addition, the fluorescence emission maxima of 2-hydroxyethidine (586 nm) significantly overlaps with ethidium (603 nm) (86) which warrants separation methods to distinguish the two hydroethidium oxidized products. HPLC was chosen because it is a reliable method to separate and quantify 2-hydroxyethidine from ethidium and is sensitivity and suitable for superoxide anion detection in platelets, which has been challenging due to the available tools and lack of robust reactive oxygen species generated.
Using this analytical approach, we showed that oxidized lipids promoted accumulation of superoxide radical anion in a time-and concentration-dependent manner (72). This was blunted by inhibiting CD36 with a CD36 monoclonal antibody or by inhibiting NOX with the small molecule inhibitor VAS2870. We then showed that preventing NOX activation with this agent inhibited platelet activation by oxidized lipids. Hydrogen peroxide was detected by HPLC using boronate-based probes. Stimulating platelets with oxidized LDL promoted hydrogen peroxide generation. Furthermore, we used polyethyleneglycol-conjugated catalase, which is an enzyme that specifically degrades hydrogen peroxide to water, and showed that oxidized lipid-induced platelet aggregation was inhibited. This was not observed when the enzyme was denatured by boiling. These data showed that specific reactive oxygen species are functionally important for platelet aggregation in the setting of dyslipidemia.
Activation of redox sensitive MAP kinases by CD36 promotes platelet activation, maladaptive caspase activity, and pathophysiologic thrombosis.
Platelet signaling pathways activated by reactive oxygen species are incompletely defined; however, platelets do express a number of redox-sensitive effectors that could activate upon treatment with the reactive oxygen species hydrogen peroxide, including MAP kinase family members Jnk and ERK5 (72), Rac (87), and the ribosomal S6 family kinase p70S6k (87).
MAP kinases are a family of serine/threonine kinases that are important for cell survival, proliferation, differentiation, and apoptosis (88). There are three major families of classical MAP kinases: extracellular-regulated kinases (ERK), p38, and c-Jun n-terminal kinase (Jnk), all of which are activated through a three-tier kinase signaling cascade that results in phosphorylation and activation of the target MAP kinases on a signature T-X-Y motif. Of the canonical map kinase family members, CD36 signal transduction in the platelet system promotes activation of Jnk2 (43) and ERK5 (72). Both MAP kinases are sensitive to redox stress and are activated by bolus addition of exogenous hydrogen peroxide or by signaling pathways generating intracellular reactive oxygen species (87,89).
Although direct activation of Jnk by reactive oxygen species is unclear in platelets, Jnk is necessary for platelet activation and accelerated arterial thrombosis by CD36 (43,72,87,90). Blocking Jnk with a pharmacologic inhibitor, SP600125, prevented platelet activation by oxLDL (43), and platelets from high fat diet-fed hyperlipidemic mice showed basal phosphorylation of Jnk compared to chow diet-fed animals. Direct activation of Jnk was seen in a growing thrombus by immunohistochemistry in wild type mice but not CD36 null animals. Jnk is also known to promote platelet activation by classic physiologic activators, such as by the protease-activated receptors or the collagen receptor glycoprotein VI (GPVI) (91), which are physiologic activators known to generate reactive oxygen species in platelets.
Platelet ERK5 was shown by Cameron and colleagues to be differentially activated by physiologic agonists that generate reactive oxygen species. In addition, direct activation of platelet ERK5 was observed after exposing the cells to hydrogen peroxide. Using a murine myocardial infarction model, they showed that in tissue ischemia where reactive oxygen species are abundantly generated, platelet ERK5 activation promotes infarct expansion (87). The mechanism of ERK5 activation by reactive oxygen species is not clear but was proposed by Abe et al to be by a Src-dependent pathway (92). ERK5 phosphorylation and activation in the setting of myocardial infarction regulates protein expression of p70s6k, the small GTPase family member Rac, and matrix metalloproteinase 9. Expression of these proteins was not static during tissue infarction and was agonist-dependent. This was the first study suggesting that ERK5 is present and functional in platelets as a redox sensor in conditions of greatly elevated reactive oxygen species generation.
Since we found that specific reactive oxygen species were generated by CD36 signaling events and that oxidative stress and dyslipidemia are coupled processes, we hypothesized a potential role for ERK5 in CD36 prothrombotic signaling pathways (72). ERK5 was rapidly activated in a sustained manner when platelets were treated with oxLDL; this was not observed with the control unoxidized native LDL. ERK5 activation was also elevated in basal conditions in platelets isolated from apoE null mice fed a high fat diet (Figure 3A). ERK5 activation is not exclusive to oxLDL, since we showed that non-oxidized CD36 ligands such as MRP14 and TSR peptide also activated the kinase in platelets, suggesting a receptor-dependent mechanism that is independent of lipid hydroperoxides in oxLDL particles. Studies with pharmacologic inhibitors, monoclonal antibodies and cells from mice with specific genetic deletions showed that ERK5 activation by oxLDL in platelets was dependent on CD36, Src family kinases, NADPH oxidase, and hydrogen peroxide (Figure 3B). Platelet stimulation by oxLDL also promotes increased expression of Rac (Figure 3C) which is consistent with other studies showing that ERK5 was necessary for H2O2 induced Rac upregulation in platelets (87).
Figure 3. Redox-sensitive platelet ERK5 activation in mice is CD36-dependent and targets Rac.

In (A), 8–10 weeks old atherogenic apoE null mice were fed a “Western” high fat or control diet for 2 weeks. Platelets were isolated, purified, and normalized to 300,000/µL followed by immunoblotting for phospho-ERK5 to indicate its activity. Total ERK5 was used as a loading control. Samples from 5 control dieted and 5 high fat diet-fed animals are shown and quantified as the densitometric ratio of phospho-ERK5 to total ERK5. In (B), 250,000/µL wild type C57Bl/6 or cd36 null mice platelets were stimulated with 50 µg/mL oxLDL over time. Levels of ERK5 activation were determined by blotting for phospho-ERK5 and comparing it to total ERK5. CD36 expression was shown to confirm CD36-deficiency, and beta actin was blotted as a loading control. In (C), 300,000/µL platelets were isolated from healthy human donors and stimulated with 50 µg/mL oxLDL or 500 µM H2O2 over time. Expression of the downstream GTPase Rac was determined by western blot. Beta actin is a loading control and levels of Rac1 expression was determined by quantifying the densitometric ratio of Rac1 to beta actin. Data represented as standard error of the mean. p<0.05 was considered statistically significant.
A functional role for ERK5 activation by CD36 in vivo was determined using two different murine models of arterial thrombosis induced by injury to the vasculature. All the commonly used models of arterial thrombosis rely on exposing flowing blood to thrombogenic surfaces (e.g. uncovering the extracellular matrix by endothelial damage) allowing for platelets and fibrin to accumulate (93). Thrombosis can be observed in real time using a doppler transonic flow indicator (94) or by sophisticated imaging systems to detect platelets using fluorescently-tagged monoclonal antibodies or fluorophores such as mepacrine, which are taken up by platelets and concentrated in intracellular granules. Similarly, fibrin can be detected using a fluorescently-tagged anti-fibrin monoclonal antibody that does not recognize fibrinogen (95). These labels are injected intravenously and allowed to circulate before inducing thrombosis.
Topical application of chemical oxidants, such as ferric chloride (FeCl3), onto the adventitial wall of large arteries or small arterioles (94), or localized light-induced activation of the photo-activated oxidant Rose Bengal, are widely used models of in vivo thrombosis in mice (93). Although the specific mechanisms underlying FeCl3-mediated vascular injury are still being elucidated, the potent oxidant causes endothelial cell death, aggregation of cells, and protein accumulation (96). In this model of thrombosis, CD36 deficiency in mice does not impact the time it takes to vessel occlusion in the carotid and mesenteric arteries compared to wild type controls (9,13,94), suggesting that CD36 does not impact normal platelet physiology. However, when mice are fed a high fat/high cholesterol diet in the setting of apoE deletion, severe hyperlipidemia ensues and time to occlusion of the vessels decreases significantly (increased thrombosis) (9). CD36 deficiency in this model rescued the decreased time to occlusion of the vessel in dyslipidemia back to levels seen in chow-dieted control animals. Importantly, we generated atherogenic apoE null chimeric mice by bone marrow transplantation using donor mice deficient in platelet ERK5. We found that in the FeCl3-injury model, high fat diet-fed apoE null chimeric mice expressing ERK5 displayed accelerated time to vessel occlusion, which was not observed when ERK5 was absent in platelets (72).
A second model of arterial thrombosis was employed that we believe is amenable to study redox-regulating signaling pathway without artifactual generation of reactive oxygen species that could confound results. This model relies on autologous insertion of a segment of a small artery from the gut into the lumen of the large carotid artery. Flowing blood is subsequently exposed to the collagen-rich adventitial tissue of the epigastric artery segment creating a thrombogenic surface to promote platelet adhesion and aggregation. This model was developed by Dr. Brian Cooley and while technically challenging is highly reproducible (97). With this model we found that high fat diet-fed apoE null chimeric mice expressing platelet ERK5 showed rapid and enhanced accumulation of platelets and formed thrombi that subsequently embolizes over time. Control diet-fed, normolipidemic animals showed significantly lower levels of platelet accumulation, suggesting that the model is sensitive to diet-induced platelet hyper-reactivity. Importantly, when ERK5 was deleted in platelets, arterial thrombosis in high fat-diet fed apoE null chimeric mice phenocopied the control-dieted animals, suggesting a functional role for ERK5 in promoting thrombosis in dyslipidemia. To our knowledge, this is the most rigorous approach to studying redox regulated signaling pathways in thrombosis in mice. The redox-regulated prothrombotic pathway by CD36 and ERK5 is highlighted in Figure 4A showing that using multiple approaches to interrupt the signaling pathway prevented platelet activation, aggregation, and accumulation in dyslipidemic conditions.
Figure 4. Platelet CD36 redox signaling promotes phosphatidylserine externalization.



CD36 signal transduction in platelets is via a redox pathway that promotes pathophysiologic thrombosis. In (A), CD36 signaling activates Src family kinases, predominantly Fyn and Lyn, that promotes downstream generation of specific reactive oxygen species, superoxide radical anion and hydrogen peroxide, from NADPH oxidase. Superoxide radical and hydrogen peroxide then activate redox-sensitive signaling pathways, including the MAP kinase ERK5. ERK5 links platelet CD36 to a prothrombotic phenotype. Interruption of this pathway by either pharmacologically, by blocking antibodies, or through genetic deficiency of key components prevented platelet activation and subsequent thrombosis in dyslipidemia. In (B), under pathophysiologic conditions, redox-dependent events initiated by CD36 enhances phosphatidylserine externalization predominantly through apoptotic-like caspase signaling. This ERK5 dependent pathway cross-talks with the collagen receptor GPVI pathway to enhance phosphatidylserine exposure, subsequent fibrin formation, and promote pathological arterial thrombosis. In (C), the physiologic pathways of phosphatidylserine externalization and integrin activation are described to the left, where physiologic agonists activate their receptors and triggers receptor-dependent signaling pathways. In one phenotype, signaling by physiologic agonists promote activation of integrin aIIbb3 that leads to platelet aggregation and subsequent thrombosis. In another phenotype, potent physiologic agonists activate the peptidylprolyl isomerase cyclophilin D to sensitize the formation of the mitochondrial permeability transition pore that enhances procoagulant phosphatidylserine externalization. This mechanism promotes assembly of the prothrombinase complex to promote thrombin generation and subsequent fibrin deposition to support thrombosis. An alternative physiologic pathway to phosphatidylserine externalization was described, in which aged platelets promote phosphatidylserine externalization through apoptotic-like signaling. Phosphatidylserine exposure in this setting enhances platelet clearance.
In an unexpected finding, platelet CD36 signaling through ERK5 was also shown to promote maladaptive caspase activity and subsequent loss of membrane asymmetry with externalization of procoagulant phosphatidylserine (Figure 4B) (98). This signaling pathway is dependent on activation of CD36, Src kinases, and generation of hydrogen peroxide to activate ERK5. The CD36-Src kinase-ERK5 pathway also sensitizes platelets to phosphatidylserine externalization by the collagen receptor GPVI, but not by the physiologic agonists thrombin or ADP (98). Phosphatidylserine externalization promotes surface assembly of the prothrombinase and tenase coagulation enzyme complexes to localize and augment thrombin generation from prothrombin. Thrombin selectively cleaves fibrinogen causing rapid polymerization to form a fibrin clot. Interestingly, phosphatidylserine externalization by oxLDL alone was not sufficient to promote fibrin formation ex vivo; however, fibrin formation was accelerated when the cells were first sensitized with oxLDL before activation with the GPVI agonist convulxin. These data are not surprising given the crosstalk between CD36 and the GPVI signaling pathway that promote phosphatidylserine externalization. Pharmacologic or antibody-mediated inhibition of CD36, ERK5, or caspases prevented the accelerated fibrin formation by oxidized LDL (98). Furthermore, in diet-induced dyslipidemia, genetic deletion of CD36 or platelet ERK5 in mice rescued enhanced fibrin and platelet accumulation in vivo.
Two pathways for cell surface phosphatidylserine exposure have been described in the literature; one mediated by calcium-dependent cyclophilin D-sensitized mitochondrial permeability transition pore formation (Figure 4C; left) and the other by activation of pro-apoptotic caspases (Figure 4C; right). In platelets, the cyclophilin D pathway was felt to be the sole mechanism for agonist-mediated pro-coagulant phosphatidylserine externalization and to require activation by so-called strong platelet activators, such as collagen and thrombin (99,100); in fact, the combination of both agonists provides the most robust response. The caspase pathway exists in platelets, but was felt to play a role only in clearance of aged platelets, not in procoagulant activity (101,102). Our studies showing that CD36 signaling activates the caspase pathway to support fibrin formation in vitro and in vivo independently of the cyclophilin D pathway is the first reported evidence to our knowledge that the caspase pathway has a role beyond platelet clearance and that CD36 signaling through ERK5 can link the two procoagulant pathways.
Platelet CD36 signaling desensitizes the nitric oxide/cGMP inhibitory pathway.
Magwenzi et al. showed that ROS generated by CD36 inhibited a natural platelet inhibitory pathway mediated by nitric oxide/cGMP signaling (73). They showed that ROS generated by CD36/NADPH oxidase modulate protein kinase G (PKG) thereby preventing downstream activation of the PKG substrate VASP. VASP is a central hub for inhibitory signaling in platelets and is phosphorylated by PKG at serine 239. This phosphorylation event inhibits cytoskeletal rearrangement and prevents platelet integrin activation (103). OxLDL-mediated desensitization was prevented when platelets were incubated with a CD36 blocking monoclonal antibody or inhibitors to Src family kinases, PLCγ2, PKC, and NADPH oxidase. The specific redox active species modulating PKG activity were not identified and requires further investigation. Data from this study suggests that redox-signaling events by CD36 negatively impacts platelet inhibitory pathways in addition to the well-described effect on promoting platelet activation pathways.
Key knowledge gaps and conclusions
Although much has been learned about CD36 signaling in the vasculature since its initial descriptions as a pro-thrombotic, pro-atherosclerotic and anti-angiogenic receptor, answers to several key questions remain elusive. Based on the findings of redox-regulated pathways activated by CD36 signaling, we propose that oxidant stress is a chronic and sustained process in dyslipidemia and other chronic inflammatory states to promote thrombosis via CD36-mediated platelet activation and fibrin formation. Redox signaling by CD36 is multifaceted, but likely involves other sensitive pathways activated or inhibited. Could it be possible that platelet CD36 signaling requires generation of other oxidant species including reactive nitrogen, carbon, sulfur, and chloro- and bromo-species? To date, the tools available to study redox signaling are limited and advances in detection methods could elucidate a role for these species in a temporal and spatial manner during thrombosis in vivo using sophisticated imaging systems. In addition, mechanisms linking reactive oxygen species to ERK5 activation are unclear. Furthermore, understanding the mechanistic initiation of apoptotic-like signaling by reactive oxygen species and ERK5 is central to elucidating a pathway novel to what is thought of as a pro-survival, -proliferation, and -differentiation pathway for MAP kinases. This mechanism could involve coordinated activation of specific caspase members in order to promote thrombosis. We also believe that identifying other sources of reactive oxygen species are crucial to determining additional redox sensitive pathways. This supposition is based on evidence from other cellular systems that show spatial and temporal modulation of signaling pathways by redox active species from specific sources (e.g. mitochondrial versus membrane bound). In some cases, signaling effectors proximal to the microenvironment of a source of oxidant could be influence by reactive species more than effectors distal from the source. Furthermore, longer half-life reactive species could diffuse further to influence signaling events essential for platelet activation.
Overall, understanding how CD36 acts as a “rheostat” for increased platelet sensitivity by studying its signaling pathways could identify potential targets for therapeutic intervention not just in dyslipidemia but in a wide variety of pathophysiologic conditions, including diabetes mellitus and chronic inflammation.
HIGHLIGHTS.
Scavenger Receptor CD36 is a multifunctional membrane protein.
CD36 signaling characteristically generates reactive oxygen species.
CD36 signaling promotes arterial thrombosis, atherosclerosis, and inhibits angiogenesis.
Platelet CD36 promotes thrombosis by activating redox-sensitive signaling pathways.
Studying CD36 signaling pathways could identify potential targets for therapeutic intervention in dyslipidemia, diabetes mellitus, and chronic inflammation.
ACKNOWLEDGEMENT
This work was supported by NIH NHLBI grants R01 HL111614 (RLS), T32 HL134643 (MY), R01 HL111614-S (MY), and the Cardiovascular Center’s A. O. Smith Fellowship Scholars Program (MY).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Siti HN, Kamisah Y, Kamsiah J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul Pharmacol 2015;71:40–56. [DOI] [PubMed] [Google Scholar]
- 2.Kim YW, West XZ, Byzova TV. Inflammation and oxidative stress in angiogenesis and vascular disease. J Mol Med (Berl) 2013;91(3):323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rochette L, Zeller M, Cottin Y, Vergely C. Diabetes, oxidative stress and therapeutic strategies. Biochim Biophys Acta 2014;1840(9):2709–2729. [DOI] [PubMed] [Google Scholar]
- 4.Asmat U, Abad K, Ismail K. Diabetes mellitus and oxidative stress-A concise review. Saudi Pharm J 2016;24(5):547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clemetson KJ. Platelets and primary haemostasis. Thromb Res 2012;129(3):220–224. [DOI] [PubMed] [Google Scholar]
- 6.Obermayer G, Afonyushkin T, Binder CJ. Oxidized low-density lipoprotein in inflammation-driven thrombosis. J Thromb Haemost 2018;16(3):418–428. [DOI] [PubMed] [Google Scholar]
- 7.Parthasarathy S, Raghavamenon A, Garelnabi MO, Santanam N. Oxidized low-density lipoprotein. Methods Mol Biol 2010;610:403–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenberg ME, Li XM, Gugiu BG, et al. The lipid whisker model of the structure of oxidized cell membranes. J Biol Chem 2008;283(4):2385–2396. [DOI] [PubMed] [Google Scholar]
- 9.Podrez EA, Byzova TV, Febbraio M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nature Medicine 2007;13(9):1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Podrez EA, Poliakov E, Shen Z, et al. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem 2002;277(41):38503–38516. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, Okamura DM, Lu X, et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat Rev Nephrol 2017;13(12):769–781. [DOI] [PubMed] [Google Scholar]
- 12.Clemetson KJ, Pfueller SL, Luscher EF, Jenkins CS. Isolation of the membrane glycoproteins of human blood platelets by lectin affinity chromatography. Biochim Biophys Acta 1977;464(3):493–508. [DOI] [PubMed] [Google Scholar]
- 13.Li W, Febbraio M, Reddy SP, Yu DY, Yamamoto M, Silverstein RL. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J Clin Invest 2010;120(11):3996–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med 2014;46:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Ruiz E, Armesilla AL, Sanchez-Madrid F, Vega MA. Gene encoding the collagen type I and thrombospondin receptor CD36 is located on chromosome 7q11.2. Genomics 1993;17(3):759–761. [DOI] [PubMed] [Google Scholar]
- 16.Armesilla AL, Vega MA. Structural organization of the gene for human CD36 glycoprotein. J Biol Chem 1994;269(29):18985–18991. [PubMed] [Google Scholar]
- 17.Hoosdally SJ, Andress EJ, Wooding C, Martin CA, Linton KJ. The Human Scavenger Receptor CD36: glycosylation status and its role in trafficking and function. J Biol Chem 2009;284(24):16277–16288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamoto N, Ikeda H, Tandon NN, et al. A platelet membrane glycoprotein (GP) deficiency in healthy blood donors: Naka-platelets lack detectable GPIV (CD36). Blood 1990;76(9):1698–1703. [PubMed] [Google Scholar]
- 19.Curtis BR, Aster RH. Incidence of the Nak(a)-negative platelet phenotype in African Americans is similar to that of Asians. Transfusion 1996;36(4):331–334. [DOI] [PubMed] [Google Scholar]
- 20.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 2001;108(6):785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto N, Akamatsu N, Sakuraba H, Yamazaki H, Tanoue K. Platelet glycoprotein IV (CD36) deficiency is associated with the absence (type I) or the presence (type II) of glycoprotein IV on monocytes. Blood 1994;83(2):392–397. [PubMed] [Google Scholar]
- 22.Hirano K, Kuwasako T, Nakagawa-Toyama Y, Janabi M, Yamashita S, Matsuzawa Y. Pathophysiology of human genetic CD36 deficiency. Trends Cardiovasc Med 2003;13(4):136–141. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh A, Murugesan G, Chen K, et al. Platelet CD36 surface expression levels affect functional responses to oxidized LDL and are associated with inheritance of specific genetic polymorphisms. Blood 2011;117(23):6355–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love-Gregory L, Sherva R, Schappe T, et al. Common CD36 SNPs reduce protein expression and may contribute to a protective atherogenic profile. Hum Mol Genet 2011;20(1):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klenotic PA, Page RC, Li W, Amick J, Misra S, Silverstein RL. Molecular basis of antiangiogenic thrombospondin-1 type 1 repeat domain interactions with CD36. Arterioscler Thromb Vasc Biol 2013;33(7):1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coburn CT, Knapp FF Jr., Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem 2000;275(42):32523–32529. [DOI] [PubMed] [Google Scholar]
- 27.Hoebe K, Georgel P, Rutschmann S, et al. CD36 is a sensor of diacylglycerides. Nature 2005;433(7025):523–527. [DOI] [PubMed] [Google Scholar]
- 28.Cabrera A, Neculai D, Kain KC. CD36 and malaria: friends or foes? A decade of data provides some answers. Trends Parasitol 2014;30(9):436–444. [DOI] [PubMed] [Google Scholar]
- 29.Smith TG, Serghides L, Patel SN, Febbraio M, Silverstein RL, Kain KC. CD36-mediated nonopsonic phagocytosis of erythrocytes infected with stage I and IIA gametocytes of Plasmodium falciparum. Infect Immun 2003;71(1):393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu W, Li W, Silverstein RL. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012;119(25):6136–6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neculai D, Schwake M, Ravichandran M, et al. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 2013;504(7478):172–176. [DOI] [PubMed] [Google Scholar]
- 32.Smith J, Su X, El-Maghrabi R, Stahl PD, Abumrad NA. Opposite regulation of CD36 ubiquitination by fatty acids and insulin: effects on fatty acid uptake. J Biol Chem 2008;283(20):13578–13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu LY, Silverstein RL. CD36 ectodomain phosphorylation blocks thrombospondin-1 binding: structure-function relationships and regulation by protein kinase C. Arterioscler Thromb Vasc Biol 2012;32(3):760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho M, Hoang HL, Lee KM, et al. Ectophosphorylation of CD36 regulates cytoadherence of Plasmodium falciparum to microvascular endothelium under flow conditions. Infect Immun 2005;73(12):8179–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guthmann F, Maehl P, Preiss J, Kolleck I, Rustow B. Ectoprotein kinase-mediated phosphorylation of FAT/CD36 regulates palmitate uptake by human platelets. Cell Mol Life Sci 2002;59(11):1999–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asch AS, Barnwell J, Silverstein RL, Nachman RL. Isolation of the thrombospondin membrane receptor. J Clin Invest 1987;79(4):1054–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh A, Li W, Febbraio M, et al. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J Clin Invest 2008;118(5):1934–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Fang C, Gao H, et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J Clin Invest 2014;124(5):2160–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvo D, Gomez-Coronado D, Suarez Y, Lasuncion MA, Vega MA. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J Lipid Res 1998;39(4):777–788. [PubMed] [Google Scholar]
- 40.Nilsen NJ, Deininger S, Nonstad U, et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol 2008;84(1):280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kar NS, Ashraf MZ, Valiyaveettil M, Podrez EA. Mapping and characterization of the binding site for specific oxidized phospholipids and oxidized low density lipoprotein of scavenger receptor CD36. J Biol Chem 2008;283(13):8765–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Getz GS, Reardon CA. ApoE knockout and knockin mice: the history of their contribution to the understanding of atherogenesis. J Lipid Res 2016;57(5):758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen K, Febbraio M, Li W, Silverstein RL. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res 2008;102(12):1512–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang MM, Bolen JB, Barnwell JW, Shattil SJ, Brugge JS. Membrane glycoprotein IV (CD36) is physically associated with the Fyn, Lyn, and Yes protein-tyrosine kinases in human platelets. Proc Natl Acad Sci U S A 1991;88(17):7844–7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Githaka JM, Vega AR, Baird MA, Davidson MW, Jaqaman K, Touret N. Ligand-induced growth and compaction of CD36 nanoclusters enriched in Fyn induces Fyn signaling. J Cell Sci 2016;129(22):4175–4189. [DOI] [PubMed] [Google Scholar]
- 46.Ishii T, Itoh K, Ruiz E, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res 2004;94(5):609–616. [DOI] [PubMed] [Google Scholar]
- 47.D’Archivio M, Scazzocchio B, Filesi C, et al. Oxidised LDL up-regulate CD36 expression by the Nrf2 pathway in 3T3-L1 preadipocytes. FEBS Lett 2008;582(15):2291–2298. [DOI] [PubMed] [Google Scholar]
- 48.Olagnier D, Lavergne RA, Meunier E, et al. Nrf2, a PPARgamma alternative pathway to promote CD36 expression on inflammatory macrophages: implication for malaria. PLoS Pathog 2011;7(9):e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aubouy A, Olagnier D, Bertin G, et al. Nrf2-driven CD36 and HO-1 gene expression in circulating monocytes correlates with favourable clinical outcome in pregnancy-associated malaria. Malar J 2015;14:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheedy FJ, Grebe A, Rayner KJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 2013;14(8):812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Byon CH, Heath JM, Chen Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol 2016;9:244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chu LY, Ramakrishnan DP, Silverstein RL. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013;122(10):1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramakrishnan DP, Hajj-Ali RA, Chen Y, Silverstein RL. Extracellular Vesicles Activate a CD36-Dependent Signaling Pathway to Inhibit Microvascular Endothelial Cell Migration and Tube Formation. Arterioscler Thromb Vasc Biol 2016;36(3):534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang W, Febbraio M, Silverstein RL. CD9 tetraspanin interacts with CD36 on the surface of macrophages: a possible regulatory influence on uptake of oxidized low density lipoprotein. PLoS One 2011;6(12):e29092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 2010;11(2):155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seimon TA, Nadolski MJ, Liao X, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab 2010;12(5):467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y, Kennedy DJ, Ramakrishnan DP, et al. Oxidized LDL-bound CD36 recruits an Na(+)/K(+)-ATPase-Lyn complex in macrophages that promotes atherosclerosis. Sci Signal 2015;8(393):ra91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaqaman K, Kuwata H, Touret N, et al. Cytoskeletal control of CD36 diffusion promotes its receptor and signaling function. Cell 2011;146(4):593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest 2009;119(1):136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park YM, Drazba JA, Vasanji A, Egelhoff T, Febbraio M, Silverstein RL. Oxidized LDL/CD36 interaction induces loss of cell polarity and inhibits macrophage locomotion. Mol Biol Cell 2012;23(16):3057–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rahaman SO, Li W, Silverstein RL. Vav Guanine nucleotide exchange factors regulate atherosclerotic lesion development in mice. Arteriosclerosis, thrombosis, and vascular biology 2013;33(9):2053–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rahaman SO, Zhou G, Silverstein RL. Vav protein guanine nucleotide exchange factor regulates CD36 protein-mediated macrophage foam cell formation via calcium and dynamin-dependent processes. J Biol Chem 2011;286(41):36011–36019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bustelo XR. Vav family exchange factors: an integrated regulatory and functional view. Small GTPases 2014;5(2):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kotla S, Singh NK, Rao GN. ROS via BTK-p300-STAT1-PPARgamma signaling activation mediates cholesterol crystals-induced CD36 expression and foam cell formation. Redox Biol 2017;11:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feng J, Han J, Pearce SF, et al. Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J Lipid Res 2000;41(5):688–696. [PubMed] [Google Scholar]
- 66.Rao X, Zhong J, Maiseyeu A, et al. CD36-dependent 7-ketocholesterol accumulation in macrophages mediates progression of atherosclerosis in response to chronic air pollution exposure. Circ Res 2014;115(9):770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leonarduzzi G, Gargiulo S, Gamba P, et al. Molecular signaling operated by a diet-compatible mixture of oxysterols in up-regulating CD36 receptor in CD68 positive cells. Mol Nutr Food Res 2010;54 Suppl 1:S31–41. [DOI] [PubMed] [Google Scholar]
- 68.Leonarduzzi G, Gamba P, Gargiulo S, et al. Oxidation as a crucial reaction for cholesterol to induce tissue degeneration: CD36 overexpression in human promonocytic cells treated with a biologically relevant oxysterol mixture. Aging Cell 2008;7(3):375–382. [DOI] [PubMed] [Google Scholar]
- 69.Wraith KS, Magwenzi S, Aburima A, Wen Y, Leake D, Naseem KM. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase-signaling pathways. Blood 2013;122(4):580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimman A, Titz B, Komisopoulou E, Biswas S, Graeber TG, Podrez EA. Phosphoproteomic analysis of platelets activated by pro-thrombotic oxidized phospholipids and thrombin. PLoS One 2014;9(1):e84488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen K, Li W, Major J, Rahaman SO, Febbraio M, Silverstein RL. Vav guanine nucleotide exchange factors link hyperlipidemia and a prothrombotic state. Blood 2011;117(21):5744–5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang M, Cooley BC, Li W, et al. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017;129(21):2917–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Magwenzi S, Woodward C, Wraith KS, et al. Oxidised LDL activates blood platelets through CD36-NADPH oxidase-mediated inhibition of the cGMP/Protein kinase G signalling cascade. Blood 2015;125(17):2693–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delaney MK, Kim K, Estevez B, et al. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler Thromb Vasc Biol 2016;36(5):846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walsh TG, Berndt MC, Carrim N, Cowman J, Kenny D, Metharom P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol 2014;2:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qiao J, Arthur JF, Gardiner EE, Andrews RK, Zeng L, Xu K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol 2018;14:126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carnevale R, Loffredo L, Sanguigni V, et al. Different degrees of NADPH oxidase 2 regulation and in vivo platelet activation: lesson from chronic granulomatous disease. J Am Heart Assoc 2014;3(3):e000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Violi F, Pignatelli P. Platelet oxidative stress and thrombosis. Thromb Res 2012;129(3):378–381. [DOI] [PubMed] [Google Scholar]
- 79.Violi F, Pignatelli P, Basili S. Nutrition, supplements, and vitamins in platelet function and bleeding. Circulation 2010;121(8):1033–1044. [DOI] [PubMed] [Google Scholar]
- 80.Pignatelli P, Carnevale R, Di Santo S, et al. Inherited human gp91phox deficiency is associated with impaired isoprostane formation and platelet dysfunction. Arterioscler Thromb Vasc Biol 2011;31(2):423–434. [DOI] [PubMed] [Google Scholar]
- 81.Violi F, Pignatelli P. Platelet NOX, a novel target for anti-thrombotic treatment. Thromb Haemost 2014;111(5):817–823. [DOI] [PubMed] [Google Scholar]
- 82.Pignatelli P, Sanguigni V, Lenti L, et al. gp91phox-dependent expression of platelet CD40 ligand. Circulation 2004;110(10):1326–1329. [DOI] [PubMed] [Google Scholar]
- 83.Choo HJ, Kholmukhamedov A, Zhou C, Jobe S. Inner Mitochondrial Membrane Disruption Links Apoptotic and Agonist-Initiated Phosphatidylserine Externalization in Platelets. Arterioscler Thromb Vasc Biol 2017;37(8):1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berger M, Wraith K, Woodward C, et al. Dyslipidemia-associated atherogenic oxidized lipids induce platelet hyperactivity through phospholipase Cgamma2-dependent reactive oxygen species generation. Platelets 2018:1–6. [DOI] [PMC free article] [PubMed]
- 85.Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc 2008;3(1):8–21. [DOI] [PubMed] [Google Scholar]
- 86.Zhao H, Kalivendi S, Zhang H, et al. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med 2003;34(11):1359–1368. [DOI] [PubMed] [Google Scholar]
- 87.Cameron SJ, Ture SK, Mickelsen D, et al. Platelet Extracellular Regulated Protein Kinase 5 Is a Redox Switch and Triggers Maladaptive Platelet Responses and Myocardial Infarct Expansion. Circulation 2015;132(1):47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001;410(6824):37–40. [DOI] [PubMed] [Google Scholar]
- 89.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005;120(5):649–661. [DOI] [PubMed] [Google Scholar]
- 90.Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood 2009;113(4):893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Adam F, Kauskot A, Nurden P, et al. Platelet JNK1 is involved in secretion and thrombus formation. Blood 2010;115(20):4083–4092. [DOI] [PubMed] [Google Scholar]
- 92.Abe J, Kusuhara M, Ulevitch RJ, Berk BC, Lee JD. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem 1996;271(28):16586–16590. [DOI] [PubMed] [Google Scholar]
- 93.Cooley BC. Murine arterial thrombus induction mechanism influences subsequent thrombodynamics. Thromb Res 2015;135(5):939–943. [DOI] [PubMed] [Google Scholar]
- 94.Li W, McIntyre TM, Silverstein RL. Ferric chloride-induced murine carotid arterial injury: A model of redox pathology. Redox Biol 2013;1:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hui KY, Haber E, Matsueda GR. Monoclonal antibodies to a synthetic fibrin-like peptide bind to human fibrin but not fibrinogen. Science 1983;222(4628):1129–1132. [DOI] [PubMed] [Google Scholar]
- 96.Ciciliano JC, Sakurai Y, Myers DR, et al. Resolving the multifaceted mechanisms of the ferric chloride thrombosis model using an interdisciplinary microfluidic approach. Blood 2015;126(6):817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cooley BC. Collagen-induced thrombosis in murine arteries and veins. Thromb Res 2013;131(1):49–54. [DOI] [PubMed] [Google Scholar]
- 98.Yang M, Kholmukhamedov A, Schulte ML, et al. Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo. Blood Adv 2018;2(21):2848–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Choo HJ, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol 2012;32(12):2946–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jobe SM, Wilson KM, Leo L, et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008;111(3):1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.White MJ, Schoenwaelder SM, Josefsson EC, et al. Caspase-9 mediates the apoptotic death of megakaryocytes and platelets, but is dispensable for their generation and function. Blood 2012;119(18):4283–4290. [DOI] [PubMed] [Google Scholar]
- 102.Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell 2007;128(6):1173–1186. [DOI] [PubMed] [Google Scholar]
- 103.Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol 2003;19:541–564. [DOI] [PubMed] [Google Scholar]
- 104.Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab 2006;4(3):211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]