

Abstract
Background
Water deficit and soil salinity substantially influence plant growth and productivity. When occurring individually, plants often exhibit reduced growth resulting in yield losses. The simultaneous occurrence of these stresses enhances their negative effects. Unraveling the molecular mechanisms of combined abiotic stress responses is essential to secure crop productivity under unfavorable environmental conditions.
Results
This study examines the effects of water deficit, salinity and a combination of both on growth and transcriptome plasticity of barley seminal roots by RNA-Seq. Exposure to water deficit and combined stress for more than 4 days significantly reduced total seminal root length. Transcriptome sequencing demonstrated that 60 to 80% of stress type-specific gene expression responses observed 6 h after treatment were also present after 24 h of stress application. However, after 24 h of stress application, hundreds of additional genes were stress-regulated compared to the short 6 h treatment. Combined salt and water deficit stress application results in a unique transcriptomic response that cannot be predicted from individual stress responses. Enrichment analyses of gene ontology terms revealed stress type-specific adjustments of gene expression. Further, global reprogramming mediated by transcription factors and consistent over-representation of basic helix-loop-helix (bHLH) transcription factors, heat shock factors (HSF) and ethylene response factors (ERF) was observed.
Conclusion
This study reveals the complex transcriptomic responses regulating the perception and signaling of multiple abiotic stresses in barley.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5634-0) contains supplementary material, which is available to authorized users.
Keywords: Barley, Combined stress, RNA-Seq, Salt stress, Seminal roots, Transcriptome, Water deficit
Background
Natural abiotic stresses such as water deficit and high soil salinity are major factors threatening global crop production [1, 2]. Exposure of plants to these osmotic stresses results in loss of turgor and as a consequence potential disruption of membranes and proteins accompanied by rising levels of reactive oxygen species (ROS) [3]. This, in turn, leads to growth inhibition and loss of yield [4]. While shoot growth is reduced under these stress conditions, roots continue to elongate at a slower rate to ensure survival by extracting water and nutrients from deeper soil layers [5]. Perpetuated root growth is mainly regulated by abscisic acid (ABA), which interacts with auxin, cytokinin, and ethylene in a hormonal network [6]. In addition to physiological alterations, the effects of either water deficit or salinity on gene expression patterns in roots have been studied. For instance, mature chickpea roots displayed several sets of differentially expressed genes in response to either water deficit or salinity at different developmental stages [7]. Microarray experiments in roots and leaves of three-week-old barley plants subjected to both stress conditions individually demonstrated, that the number and function of differentially expressed genes strongly depend on stress type and duration [8]. A study comparing gene expression levels in salt and osmotic-stressed barley leaves and roots came to the same conclusion [9]. While these studies surveyed the transcriptomic response to individual stress types, the simultaneous occurrence of several stress types under field conditions can lead to more severe responses [10]. Combinatorial abiotic stress application typically results in negative and in a few instances in positive physiological interactions between stress types [11]. For instance, a combination of salt and heat stress in Arabidopsis led to a negative effect by significantly reducing biomass and rosette diameter and lower survival rate that exceeded the decreases under single stress conditions [12]. Similarly, tobacco showed reduced respiration under water deficit, while heat shock and combined stress treatments enhanced this response [13]. In barley, plant growth and chlorophyll content reflecting the photosynthetic rate, water, and osmotic potential were reduced when subjected to either water stress, salinity or a combination of both. Yet, plants were more vulnerable to the combinatorial treatment of these stress factors [14]. In contrast, exposing tomato plants to combined heat and salinity had a positive effect leading to a significantly increased protection from the harmful effects of the individual application of salinity by accumulating trehalose and glycine betaine [15].
On the molecular level, it was demonstrated in early cDNA microarray studies in tobacco that the effects of combined water deficit and heat shock cannot be deduced by characterizing responses to single stress treatments [13]. Similarly, in Arabidopsis, the comparison of differentially regulated genes revealed a large overlap between heat and combined stress treatments but also a substantial treatment specificity [12]. In line with observations on the gene expression level, metabolic profiling of maize shoots and leaves treated by a combination of water deficit and salinity stress demonstrated that metabolic adjustments to combined stress were not additive when compared to single stress factors [16].
Barley is better adapted to abiotic stresses than other cereal species such as wheat or maize and can thus be grown in harsher environments [17]. This makes barley an ideal model plant to study abiotic stress adaptions. The usage of high molecular weight organic osmotica such as mannitol or PEG8000 (polyethylene glycol) to establish defined water potentials allows studying plant responses under controlled conditions. It was previously demonstrated that PEG8000 solution can be utilized to mimic water deficit [18–21]. Water deficit treatment of -0.8 MPa is in the mid-range of naturally occurring, plant-usable soil water potentials [22]. Similarly, NaCl concentrations of 150 mM in soil water is considered as moderate salinity and observed in many agricultural regions of the globe [23]. In the present study, we subjected 3-day-old barley seedlings to either PEG8000 solution with a water potential of -0.8 MPa to mimic water deficit, 150 mM NaCl to simulate salt stress or a combination of both. In these seedlings, we monitored root growth for eight consecutive days. Based on the results of these phenotyping experiments, we analyzed samples of seminal roots 6 h and 24 h after stress induction by RNA-Sequencing (RNA-Seq). The aim of this study was to explore the early transcriptomic reprogramming of barley seminal roots exposed to individual and combinatorial stresses at two time points. This study will provide candidate genes for further genetic analyses that might be helpful for marker-assisted barley breeding programs.
Results
Phenotypic response to abiotic stress treatments
Seedlings of the barley spring cultivar Scarlett germinated for 2 days under control conditions were subjected to water deficit (PEG8000: -0.8 MPa), salt stress (NaCl: 150 mM) or a combination of both at T0 for 7 days (Fig. 1). To investigate the effect of the abiotic stress factors on seminal root development, total root length per treatment was determined relative to roots grown under control conditions (Fig. 1). By day four (T4), total seminal root length of seedlings subjected to combined stress treatment was significantly shorter than that of control plants (Fig. 1). By day five (T5), plants subjected to water deficit displayed also significantly reduced total root length relative to control plants. In contrast, although a substantial decrease in total root length was monitored in salt-stressed plants these differences were not statistically significant compared to control plants within 7 days of treatment.
Fig. 1.

Comparison of total root length between control, salt, water deficit and combined stress from stress induction (T0) until 7 days of treatment (T7). Significant differences (α = 0.05) of means at each time point were calculated by ANOVA and indicated with small letters. Means not sharing any letter are significantly different
Mapping of RNA-sequencing reads to the barley reference genome
We monitored global changes in the seminal root transcriptomes of young barley seedlings subjected to water deficit, high salinity and a combination of both for 6 h and 24 h. These treatments correspond to time points T0.25 and T1 in Fig. 1. Hence, at both time points, no morphological differences between control plants and plants subjected to the three types of stress were detectable. Total RNA from four biological replicates per treatment-by-time point combination was extracted from seminal roots, converted into cDNA and subjected to RNA-Seq. The workflow of the RNA-Seq experiment and downstream analyses are summarized in Additional file 1: Figure S1. After quality trimming, between 67 and 76% of the obtained sequences per library mapped to the barley reference genome (Additional file 2: Table S1). After removal of stacked reads, i.e. reads that share identical 5′ coordinates, orientation and length, on average 60% of the remaining reads mapped successfully in pairs to the set of 39,734 high confidence gene models of barley version IBSC v2.0 [63].
Transcriptomic relationships of RNA-Seq samples
Transcriptomic relationships between the type and duration of stress treatment were determined in a multidimensional scaling (MDS) plot (Fig. 2). Replicated samples of treatment-by-duration combinations clustered closely together. Moreover, samples subjected to short and long-term stress as well as distinct stress treatments are separable in the MDS plot, demonstrating that the observed transcriptomic divergence is driven by stress type and duration. For both stress durations, control and combined stress samples are positioned most distantly apart, while individually salt stressed and water stressed samples cluster between the control and combined treatment.
Fig. 2.
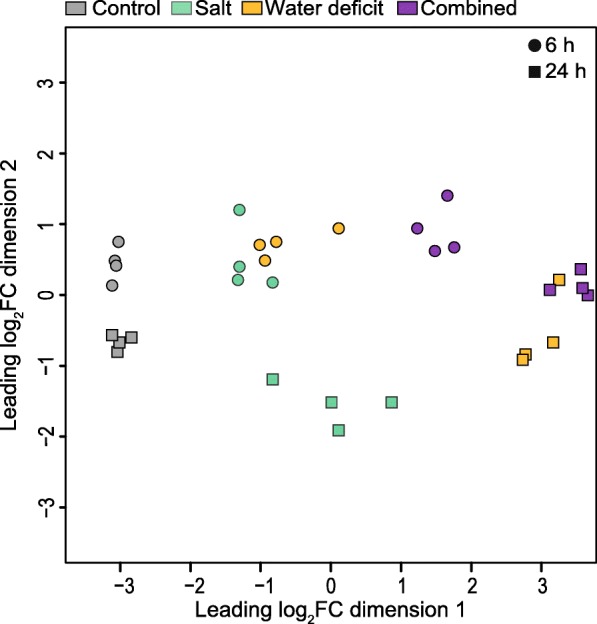
Multidimensional scaling plot of replicated RNA-Seq samples. Features represent libraries from control, water deficit, salt and combined stress treatment after 6 h and 24 h. Samples are arranged based on their calculated distances
Identification of differentially expressed genes
Differentially expressed genes (DEGs) were computed in three pairwise contrasts between control and stress treatment samples for short- and long-term response. The number of DEGs (FDR ≤5% and |log2FC| >1) between control and stress treatment for the three treatment by two time point combinations are depicted as volcano plots (Fig. 3a). A comprehensive list of these DEGs is provided in Additional file 3: Table S2. The number of DEGs varied between treatment-by-time combinations. Under both, short and long-term stress exposure, the salt stress treatment resulted in the smallest number of DEGs (953 at 6 h and 1802 at 24 h). The most severe impact on gene expression was observed in the combined water deficit and salt treatment with 4845 DEGs at 6 h and 8105 DEGs after 24 h. After short-term treatment, the total number of genes differentially regulated in the combined treatment was substantially higher than the sum of genes differentially regulated by water deficit or salt stress alone. Furthermore, the direction of regulation depended on stress type and duration. Between 60% (salt treatment) and 80% (combined treatment) of genes that were differentially expressed after 6 h, were also responsive after 24 h of treatment (Fig. 3b). Among these, 70 to 75% of up-regulated DEGs and 55 to 95% of the down-regulated DEGs were conserved over time. Cross-comparison between the different gene sets after 6 h showed that the highest proportion of genes (65%) was unique to the combination treatment, while water deficit (4%) and salt (2%) treatment resulted in less uniquely expressed genes (Fig. 3c). This indicates that the combined treatment does not only result in the additive regulation of genes differentially expressed in the two single stress treatments. Instead, a substantial number of genes was only regulated by the combined stress but not by the individual stress factors. A set of 623 DEGs (12%) was responsive to all three treatments, pointing towards regulatory changes that were unaffected by stress type. Long-term stress response showed a strong overlap (52%) of genes responsive to water deficit and combined stress treatments, which is in line with the distribution of samples in the MDS plot in Fig. 2. Nevertheless, 20% of DEGs were specific for combined stress (Fig. 3d), while only 9 and 3% of DEGs were unique to water deficit and salt treatment, respectively.
Fig. 3.

Overview of differentially expressed genes (DEGs) between control and stress-treated samples. a Volcano plots depict DEGs for each treatment-by-time combination. Up-regulated DEGs are indicated by blue dots, down-regulated DEGs are indicated by red dots. Total number of DEGs are shown in the upper left and right corner for significant up- and down-regulated DEGs. DEGs that do not exceed the threshold of |log2FC| >1 and FDR ≤1% are depicted in grey. b Overlap of DEGs at 6 h that are also differentially expressed at 24 h for each treatment in percent. Bars show overlaps of all DEGs, up-regulated and down-regulated DEGs separately. c Venn diagram showing the overlap between DEGs responsive to water deficit, salt and combined stress after 6 h of treatment. Arrows indicate number and direction of DEGs. d Venn diagram showing the overlap between DEGs after 24 h of treatment. Arrows indicate number of up- and down-regulated DEGs
Assessment of stress-responsive pathways
Gene Ontology (GO) terms were assigned to DEGs to functionally characterize stress-responsive processes and functions. GO terms were analyzed for singular enrichment and obtained results were cross-compared with the SEACOMPARE tool. A full list of enriched GO terms in all treatment-by-time combinations is provided in Additional file 4: Table S3. In total, 63 GO terms responsive to short-term stress remained after filtering with REVIGO. Half of these terms were treatment-specific, while the other half was shared between two or more treatments (Table 1). The highest number of unique treatment-specific GO terms was observed for the combinatorial treatment. A substantial number of GO terms was commonly enriched between combined stress and each of the single stresses but not between water deficit and salinity. Shared terms of biological processes and molecular functions that were responsive to salt and combined treatment were commonly down-regulated. This included several catalytic activities such as ‘transferase activity’ (GO:0016740) but also metabolic processes like ‘phosphorus metabolic process’ (GO:0006793). Mutual DEGs between water deficit and combined stress were identified to be involved in oxidative and general stimulus responses. While ‘oxidoreductase activity’ (GO:0016491) and ‘oxidation-reduction process’ (GO:0055114) were down-regulated, responses to stimuli were up-regulated. The last set of treatment-independent terms showed up-regulation in all biological processes mainly involved in ‘transcription’ (GO:0006351), ‘regulation of gene expression’ (GO:0010468) and the corresponding functional term ‘transcription factor activity’ (GO:0003700). Genes involved in binding and hydrolase activity were down-regulated.
Table 1.
Enriched functional GO terms among DEGs responding to 6 h short-term treatment

aOnly non-redundant terms (similarity ≤0.5) with FDR ≤5% are shown for identified molecular processes (white background) and molecular functions (grey background)
bIndented terms belong to the same cluster as the above listed higher-ranking term. Numbers in parentheses indicate the level of the GO term
cTreatments are water deficit (D), salt (S) and combined (SD). The direction of regulation is represented by blue (down-regulation), red (up-regulation), and grey (not significantly regulated)
GO terms assigned to DEGs after long-term exposure were mostly shared between two or more treatments (Table 2). A considerable number of commonly enriched terms was identified between water deficit and combined treatment, which is in support with the MDS plot (Fig. 2) and in line with the overlap of differentially expressed genes in Fig. 3d. Among functional terms, several transferase activities and metabolic processes were down-regulated, while ‘regulation of gene expression’ (GO:0010468) and ‘ion binding’ (GO:0043167) were up-regulated. Up-regulation of ‘developmental process’ (GO:0048856) and its child term was shared between water deficit and salt treatment, in contrast to salt and combined treatment, which showed no overlapping terms. Terms covered by ‘catalytic activity’ (GO:0003824) such as oxidoreductases, that were previously only enriched in short-term combined stress were enriched independent of stress type after long-term exposure. Furthermore, if terms were shared between two or more stress types, the direction of regulation was largely preserved. Up-regulation of the term ‘transcription factor activity’ (GO:0003700) was even conserved in all treatment-by-time combinations.
Table 2.
Enriched functional GO terms among DEGs responding to 24 h long-term treatment

aOnly non-redundant terms (similarity ≤0.5) with FDR ≤5% are shown for identified molecular processes (white background) and molecular functions (grey background)
bIndented terms belong to the same cluster as the above listed higher-ranking term. Numbers in parentheses indicate the level of the GO term
cTreatments are water deficit (D), salt (S) and combined (SD). The direction of regulation is represented by blue (down-regulation), red (up-regulation), purple (up and down-regulation), and grey (not significantly regulated)
Distribution of transcription factors in differentially expressed gene sets
Transcription factors (TFs) within the RNA-Seq dataset were identified via the Plant Transcription Factor Database. In total, 924 of 2620 known barley TFs were expressed in the present dataset. The prevalence of these TFs in the 56 TF families was used as a reference distribution to identify deviations in the family distributions of the DEG datasets for each treatment-by-time combination (Fig. 4). Short-term 6 h stress treatment resulted in the enrichment of 12 treatment-by-TF family combinations (Fig. 4a) while 24 h stress led to the enrichment of 18 such combinations (Fig. 4b). At both stress treatment durations, 6 h and 24 h, bHLH, ERF, and HSF TF families were enriched at all three stress treatment combinations (Fig. 4a and b). In addition, the bZIP TF family was over-represented after water deficit and combined treatment at both time points (Fig. 4a and b). At 6 h, only the HD-ZiP TF family was enriched specifically upon water deficit treatment. At 24 h the bZIP, G2-like, HD-ZIP TF families were specifically enriched upon salt stress, while the LBD, MYB, NAC and TALE TF families were enriched upon combined salt and water deficit treatment. Remarkably, while several TF families were particularly enriched after 24 h after salt treatment, no water deficit-specific enrichment of TF families was observed after 24 h.
Fig. 4.

Prevalence of transcription factor (TF) families after salt, water deficit and combined stress for 6 h (a) and 24 h (b). Only families with ≥15 expressed members are shown. Grey background represents the family distribution among all expressed genes. Colored lines represent treatment-specific distributions of differentially expressed TFs as a percentage of all differentially expressed genes in the treatment. Significant deviations from the background distribution were calculated by Fisher‘s exact tests (α ≤0.05) and are indicated by asterisks
Discussion
Under field conditions, crops are often subjected to simultaneous abiotic stresses such as water deficit, heat or high salinity [10, 11]. Understanding the molecular responses to these combined stresses that have detrimental effects on crop productivity is necessary for sustainable agriculture under changing global climatic conditions [11]. In the present study, we surveyed the individual and simultaneous effects of water deficit (PEG8000, -0.8 MPa) and high salinity (150 mM NaCl), on root development and the global transcriptome profiles of barley seminal roots.
Growth arrest as a response to salinity or water deficit in aboveground parts of plants is a common mechanism to conserve carbohydrates and thus maintain the energy supply. However, roots continue to elongate albeit at a lower rate to access water stored in deeper soil layers [24]. In the present study, we also observed a reduction in root growth rate. Roots of plants grown under combined stress and water deficit conditions continued to elongate but were significantly shorter in comparison to the roots of control plants after four or more days of treatment (Fig. 1). These phenotypic adjustments were also monitored in 12-day-old barley seminal roots, which were significantly shorter upon water deficit conditions of -0.8 MPa compared to control conditions [18]. In maize seedlings, phenotypic plasticity of primary roots in response to water deficit was even faster than in barley as demonstrated by a 30% reduced elongation within 24 h under the same -0.8 MPa water deficit regime as applied in the present study [20]. In contrast to water deficit, exposure to 150 mM NaCl solution did not affect root elongation in barley seedlings in the present study (Fig. 1). This supports the notion that salt tolerance of barley is linked to better root growth rates to provide an additional surface for sequestration of toxic ions that accumulate due to raising Na+ level within the plant [25].
To characterize the transcriptomic landscape of barley seminal roots and its adaptions in response to different abiotic stresses, seminal roots exposed to stress conditions for 6 h and 24 h were analyzed by RNA-Seq. Although significant developmental differences became only visible after 4 days in barley, previous studies showed, that transcriptomic adaptions are detectable already after a few hours of treatment and preceded the later observed phenotypic effects [8, 26, 27]. The number of identified DEGs, based on pairwise comparisons between control and stress samples, varied substantially between the duration of treatment with 5182 DEGs after 6 h (Fig. 2c) and 9240 DEGs after 24 h (Fig. 2d). This increase over time was for instance also reported in maize [20] and Arabidopsis [28]. Differences in the number of differentially regulated genes were also observed between different stress types. In line with their moderate impact on root elongation, salt treatment resulted in a considerably lower number of DEGs than water deficit at both time points (953 vs 1560 at 6 h and 1802 vs 7094 at 24 h; Fig. 3a). This remarkably low quantity of salt-responsive genes is in line with the previously demonstrated salt tolerance of barley [8]. Previous research suggested a possible link between the number of responsive genes and their association with the complexity and intensity of the imposed stress treatment. For instance, experiments in soybean exposed to different levels of water deficit showed that more severe stress treatment leads to an increased number of DEGs [29]. Furthermore, changes in stress complexity by applying multiple biotic and abiotic stresses to Arabidopsis were also positively correlated with the number of responsive genes [30]. In contrast to this, exposure of Brachypodium distachyon to triple stress (heat, water deficit, and salinity) did not increase the number of DEGs compared to double stress combinations [31]. The results obtained in this study support the notion that the duration of individual or combined stresses increases the number of differentially expressed genes (Fig. 3a). At the same time, genes regulated by these stresses at different time points displayed a substantial degree of conservation of 60 to 80% (Fig. 3b). Similar proportions of conservation of stress-responsive genes were also discovered in maize roots subjected to 6 h and 24 h of water deficit [20]. This finding supports the notion that certain molecular stress responses, which are already established after short-term exposure to stress, are still important after long-term exposure. Comparison of the DEG sets for each time point across treatments revealed that 65% of all short-term stress-responsive genes were unique to the combined stress treatment and as such not predictable by single-stress responses. This is consistent with findings in Arabidopsis ecotypes that also exhibited non-additive effects for plants subjected to combinatorial stress for 61% of the identified DEGs [32]. A similar pattern was observed in Dianthus spiculifolius subjected to cold and water stress, in which approximately half of the stress-responsive genes were unique to the combinatorial treatment after 24 h [33]. In the present study, only 20% of differentially expressed genes were unique to the combinatorial stress after 24 h, while a substantial overlap of regulated genes with water deficit regulated genes was observed. This notion is supported by recent studies in Arabidopsis suggesting that the response to one stress dominates the acclimation responses to a combination of stresses due to an extensive overlap between DEGs [12, 34].
Plant stress responses and adaptive processes to single and multiple stresses are orchestrated by a complex network of cross-talk between signaling pathways and sensors [10]. To gain further insight into biological processes and molecular functions that showed a stress-response, GO terms were assigned to the DEGs and analyzed for enrichment. Enriched GO terms observed in comparable studies in 12-day-old barley roots and young maize roots exposed to the same water deficit treatment are in accordance with the results of the present study. For instance, GO terms related to stress or stimulus responses and oxidoreductase activity were also highly over-represented in both experiments [18, 20, 26]. Moreover, exposure of Arabidopsis and chickpea plants to drought treatment resulted in an identical enrichment [7, 28]. Thus, indicating similar global patterns of stress response across species.
The abundance of enriched GO terms for both time points (Tables 1 and 2) further demonstrates the complexity of stress responses with the involvement of many different pathways. Water deficit and high salinity share a common osmotic component caused by a lowered water potential in the root vicinity [35] leading to identical responses and mutually enriched GO terms. The direction of regulation is highly conserved among these commonly enriched terms except for term GO:0009665 ‘plant-type cell wall organization’ which shows different directions between short and long-term stress and also between salt and other stress types. Within this GO term, expansins are highly represented. Continuous growth and development are based on constant loosening and remodeling of the cell wall that enables expansion [36]. Expansins play an important role in the regulation of these perpetual plasticity changes within cell walls [37]. Extensive studies in maize roots subjected to low water potentials linked enhanced gene expression of several expansins in the root growth zone to the maintenance of root growth under stress. Thus, alterations of root growth triggered by adjustment to water deficit are most likely due to the gene-specific regulation of expansin levels [38]. The regulation of expansin expression is in line with the previously described root growth adaptions. Under salt stress, expansins are up-regulated and thus, roots continue to elongate. This, in turn, can be linked to a better adaptation of barley to salt exposure [25]. In contrast, prolonged exposure to water deficit conditions and combined stress leads to down-regulation of expanins and to decreased root elongation as observed in later time points.
Another group of over-represented GO terms corresponded to antioxidant (GO:0016209) and oxidoreductase activity (GO:0016491) and their respective child terms. Genes associated with these functions are mainly involved in the scavenging of reactive oxygen species (ROS), known to harm plant cells subjected to different stresses [39]. Exposure to salt and water deficit resulted in a significant down-regulation of genes encoding oxidoreductases, glutathione reductases, and peroxidases. Down-regulation of involved enzymes was also observed in abiotically stressed Arabidopsis plants in which the direction of regulation strongly depended on the stress type underscoring the complexity of the ROS-induced network [40]. Several studies have examined expressional changes of genes involved in these processes when subjected to different stresses. In contrast to our study, 6 days of water deficit conditions in young barley roots resulted in the up-regulation of genes involved in oxidoreductase activities [18]. These findings suggest that genes involved in the ROS network are under developmental regulation and thus show different regulation over time.
Transcription factors (TFs) control the activity of downstream target genes. The GO term ‘transcription factor activity’ showed up-regulation in all investigated stress types and time points. In barley, 2620 TFs are classified in 56 families. Among those 924 TFs were active in seminal roots surveyed in the present study. A major proportion of these TFs are located within the bHLH, MYB-related and bZIP families [41].
Heat shock factors (HSFs) were significantly over-represented in all treatment-by-time combinations. HSFs control the expression of Heat-Shock-Proteins (HSPs) [42] that function as chaperones to protect proteins under heat stress [24, 43]. Nevertheless, it has been demonstrated that HSFs also play a role in general stress responses such as water deficit and combinations of non-thermal stresses [42, 44–46]. Experiments in barley showed that exposure to multiple abiotic stresses including water deficit and salinity resulted in the up-regulation of multiple HSFs. Of those, two candidates were subjected to qRT-PCR to validate the expressional changes [47]. We checked for these candidate genes in the present RNA-Seq dataset and found both genes HORVU4Hr1G090090 and HORVU4Hr1G090850 significantly up-regulated in all treatment-by-time combinations and with the highest fold change in the combined treatments. This alteration in modulation severity was also observed in experiments in Arabidopsis, that showed strong induction of HSFA7B by a combination of salt, osmotic and heat stress, while it was induced less severely under single heat stress [34]. The overexpression of AtHSP17.6A in transgenic Arabidopsis plants led to enhanced osmotic stress tolerance [48]. Consequently, the expression of HSFs correlates with osmotic stress tolerance. Therefore, modulating the expression of HSF and HSP encoding genes might be an effective strategy for breeding plants with enhanced tolerance to abiotic stress.
Another over-represented family at all treatment-by-time combinations were ERF TFs. ERFs are involved in many developmental and physiological processes [49] but also act in response to wounding [50] and in abiotic stress response. Like HSF they have the potential to improve crop tolerance to abiotic stresses as demonstrated by transgenic plants overexpressing certain ERFs that are more resistant to salinity, cold and water stress [51, 52]. In the present study, genes identified as ERF were both up and down-regulated. While a swift induction of stress accelerates ethylene production, a moderate change results in inhibition of ethylene biosynthesis which in turn leads to different regulatory directions in gene expression [29]. Exposure of rice to high salinity and water deficit leads to the induction of two ERFs known as DREB1A and DREB2A [53]. In the present study, the barley homolog to DREB2A HORVU1Hr1G060490 was only slightly induced in long-term water deficit and combined stress. The closest barley homolog for DREB1A HORVU5Hr1G080450 was significantly down-regulated in response to both water deficit treatments but not by any other treatment-by-time combination.
The phytohormone ABA mediates gene expression by induction of ABA-dependent TFs from the bZIP and bHLH families. Both have regulatory functions in numerous developmental and physiological processes, including stress response [54, 55]. Enrichment of bHLH among differentially expressed genes was observed in all treatments and durations, while the bZIP family was enriched in all conditions except the salt treatment at 6 h. It was previously demonstrated in Arabidopsis that the large family of bHLH TFs contains members that regulate cell elongation and thus have a direct effect on the root development [56]. The function of these bHLH genes in barley needs to be elucidated in future genetic analyses.
Conclusions
This data provides a starting point to understand the complex molecular mechanisms involved in the perception and signaling of multiple abiotic stresses in barley. Moreover, candidate genes identified here are a resource for further detailed genetic studies. Understanding the molecular networks underlying the signaling of combinatorial stresses will also be helpful for the identification of possible breeding targets for improved barley stress tolerance.
Material and methods
Plant material, growth conditions, and treatment
For phenotyping and transcriptome experiments, seeds of the German spring barley cultivar Scarlett were stratified in Petri dishes on soaked filter paper at 4 °C for 3 days to synchronize germination. Subsequently, seeds were transferred to germination paper (Anchor Paper Co, Saint Paul, USA) and grown in half-strength Hoagland solution [57] in growth cabinets (Conviron, Winnipeg, Manitoba, Canada) at 16 °C at night (8 h) and 20 °C at day (16 h). After 2 days of growth under control conditions, Hoagland solution was replaced and complemented with either PEG8000 solution (-0.8 MPa) to simulate water deficit, NaCl solution (150 mM) to induce salt stress or a combination of both.
Phenotypic evaluation of seedlings
To facilitate image-based phenotyping, stratified seedlings were grown in germination pouches in custom-built boxes that were manufactured in-house for this purpose. Each box consists of 25 slots to fit growth pouches at an angle of 15° to ensure root growth along the germination paper and not the pouch foil. In each pouch, one seedling was grown. This experimental system enables easy handling without disturbing the roots when documenting them. The replicates for each treatment (control, water deficit, high salinity and combined) were allocated to the boxes in a randomized block design. In total, 25 replicates per treatment were measured in four boxes (blocks). To avoid exposure of the roots to light, the boxes were closed with lids until the initiation of shoot growth. Then, the top of the boxes was covered with aluminum foil sparing the emerging shoots. Seedlings were imaged prior to stress induction and after stress induction every 24 h for eight consecutive days. Total root length was measured with WinRHIZO Pro (Version 2009b, Regent Instruments, Canada) based on pixel color classifications. Obtained values were log2-transformed to meet the assumptions for an ANOVA. The block model included boxes as blocks (B) and pouches as plots (P) (B/P = B + B*P) with B*P as the residual error term. The following treatment model was applied: DxS = D + S + D*S with D = water deficit (control vs treated) and S = salt (control vs treated). The R package ggpubr (R Version 3.4.0, [58], ggpubr_0.1.6, [59]) was used for visual representation of the data. Statistical evaluation was performed with the packages car (car_2.1–6, [60]) for ANOVA and agricolae (agricolae_1.2–8, [61]) for Tukey’s tests.
RNA isolation, cDNA library construction and RNA-Sequencing
For RNA extraction from seminal roots, ten seeds were grown in paper rolls as previously described [62]. Samples were harvested after 6 h and 24 h of treatment, immediately frozen in liquid nitrogen and stored at -80 °C until RNA extraction. For each replicate, ten roots were pooled. Total RNA was extracted with the RNeasy Mini Kit (Qiagen, Venlo, The Netherlands), according to the manufacturer’s instructions. RNA quality was checked with a Bioanalyzer (Agilent RNA 6000 Nano Chip, Agilent Technologies, Santa Clara, CA, USA). RIN (RNA integrity number) values ≥7.5 were obtained for all collected samples indicating their high quality and integrity. cDNA libraries for transcriptome sequencing were constructed according to the TruSeq RNA Sample Preparation protocol (Illumina, San Diego CA, USA). Library indexing, cluster preparation, and paired-end sequencing were performed according to the manufacturer’s instructions (Illumina). In total, 32 libraries with four biological replicates per treatment were sequenced on an Illumina HiSeq 4000 sequencer resulting in 100 bp paired-end reads.
Processing of raw sequencing data
RNA-Seq reads were processed with CLC Genomics Workbench (Version 10.0.1; https://www.qiagenbioinformatics.com/products/clc-genomics-workbench/). The raw sequencing data were deposited in NCBI’s sequencing read archive under accession number SRP133479 (https://www.ncbi.nlm.nih.gov/sra/SRP133479). Low-quality reads and adapter sequences were removed from the dataset by trimming. Only reads with a length of ≥40 bp were retained for further analyses. The remaining reads were mapped to the barley reference genome of the genotype morex [63], ftp://ftp.ensemblgenomes.org/pub/plants/release-36/fasta/hordeum_vulgare/dna/; Hv_IBSC_PGSB_v2) allowing large gaps of up to 50 kb to span introns. Reads were mapped successfully when they matched uniquely with ≥80% of their length and ≥ 90% identity to the reference sequences. By mapping RNA-seq reads of the barley genotype Scarlett to the reference genotype Morex we introduce a mapping bias which is reflected by the average mapping rates of 72% in Additional file 3: Table S2. By this approach, we toss out many true positives (Additional file 2: Table S1). However, without a Scarlett reference genome at hand, we cannot decide which of these unmapped reads would map to unique and which would map to multiple positions in the Scarlett genome. Therefore, de novo assembly of unmapped reads could introduce substantial false positive rates that might affect gene expression patterns. We, therefore, decided to exclude reads that do not map to the Morex reference genome from further analyses. Stacked reads, i.e. read pairs that have identical 5′ coordinates, orientation and length, were removed from the dataset. Subsequently, the remaining reads were mapped to the set of high-confidence gene models [63], ftp://ftp.ensemblgenomes.org/pub/plants/release-36/gff3/hordeum_vulgare/; Hv_IBSC_PGS_v2.36). Only reads that matched with ≥90% of their sequence length and ≥ 90% identity to the longest transcripts of the high confidence gene models were considered as mapped. Multi-mapping reads that mapped to more than one position were excluded from subsequent analyses.
Multidimensional scaling analysis
To assess the quality of the data, samples were clustered in a multidimensional scaling plot (MDS plot) using the plotMDS function implemented in the Bioconductor package limma in R (R Version 3.4.0, limma_3.32.2, [64]). Resulting distances between paired samples were displayed as the leading log2-fold change, which is defined as the estimated root-mean-square deviation for the top 500 genes with the largest standard deviation among all samples. This analysis provided a visual representation of sample relationships by spatial arrangement.
Statistical assessment of differential gene expression
To meet the assumptions of a linear model, the obtained read counts were normalized by sequencing depth and log2-transformed. The mean-variance relationship was estimated and used to assign precision weights to each observation to adjust for heteroscedasticity [65]. A linear model was fitted to assess differences in gene expression between control and stress treatments at 6 h and 24 h. The model included a fixed effect for treatment and time and the interaction of both terms. To estimate the variability over all genes and to shrink the variances towards a common value, an empirical Bayes approach was applied [66]. The contrasts.fit function of the R package limma was used to compute pairwise comparisons between each stress and control treatment at 6 h and 24 h and between short and long-term stress induction for each treatment. To correct the calculated p-values of the performed pairwise t-tests for multiplicity, the false discovery rate (FDR) was adjusted to ≤5% [67].
Gene ontology (GO) and transcription factor analyses
To gain better insight into stress-responsive pathways, GO categories were assigned to differentially expressed genes with the web-based agriGO v2.0 software [68]. Singular enrichment analysis identified over-represented categories by comparing GO terms of up and down-regulated differentially expressed genes separately to the set of all expressed genes based on Fisher’s exact test. To correct for multiple testing, the resulting p-values were adjusted by controlling the FDR ≤5% [67]. The obtained results were combined and cross-compared with the SEACOMPARE tool implemented in the agriGO v2.0 software [68]. REVIGO [69] was used to filter redundant GO terms based on their similarity. Only terms with a similarity of ≤0.5 were kept.
Transcription factors were identified by sequence similarity searches of proteins from IBSC (The International Barley Sequencing Consortium) v1.0 annotation [70] deposited in the Plant Transcription Factor Database v4.0 [41] versus the barley gene annotation IBSC v2.36 [63] via blastp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins). All expressed transcription factors within the RNA-Seq dataset were separated for short and long-term responsive genes and assigned to 56 families. The same classification was performed for transcription factors identified as differentially expressed in each treatment-by-time combination. Significant shifts between the expected background distribution of all expressed transcription factors and the observed distribution of differentially expressed transcription factors were determined by Fisher’s exact test (α ≤0.05) for 6 h and 24 h separately.
Additional files
Overview of the experimental workflow of the RNA-Seq experiment. (PDF 130 kb)
Overview of RNA-Seq output and mapping results. (XLSX 16 kb)
Comprehensive list of the differential expression analysis. (XLSX 3317 kb)
Overview of all identified over-represented Gene Ontology (GO) terms. (XLSX 28 kb)
Acknowledgments
We thank Professor Dr. Jens Léon from the Department of Plant Breeding (University of Bonn, Germany) for providing the barley seeds used in this study. Moreover, we would like to thank Christa Schulz and Helmut Rehkopf for technical assistance and Lea Bach for taking the root pictures.
Funding
This work was supported by the DFG funded graduate school GRK2064.
Availability of data and materials
The raw sequencing data is deposited in NCBI’s sequencing read archive under accession number SRP133479 (https://www.ncbi.nlm.nih.gov/sra/SRP133479). The datasets supporting the conclusions of this article are included within the article and its Additional files.
Abbreviations
- DEGs
Differentially expressed genes
- GO
Gene ontology
- TFs
Transcription factors
Authors’ contributions
AO performed the phenotyping experiment and wrote the manuscript. AO, PDR, and JAB performed and analyzed the RNA-Seq experiment. HPP supported the statistical analyses. FH conceived and coordinated the study and helped to draft the manuscript. All authors have read and approved the manuscript for publication.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alina Osthoff, Email: aosthoff@uni-bonn.de.
Petra Donà dalle Rose, Email: p.donadallerose@gmail.com.
Jutta A. Baldauf, Email: baldauf@uni-bonn.de
Hans-Peter Piepho, Email: hans-peter.piepho@uni-hohenheim.de.
Frank Hochholdinger, Email: hochholdinger@uni-bonn.de.
References
- 1.Boyer JS. Plant productivity and environment. Science. 1982;218:443–448. doi: 10.1126/science.218.4571.443. [DOI] [PubMed] [Google Scholar]
- 2.Zheng Z, Zhang F, Ma F, Chai X, Zhu Z, Shi J, Zhang S. Spatiotemporal changes in soil salinity in a drip-irrigated field. Geoderma. 2009;149:243–248. doi: 10.1016/j.geoderma.2008.12.002. [DOI] [Google Scholar]
- 3.Larcher W. Physiological plant ecology:: ecophysiology and stress physiology of functional groups. 4th ed. Berlin. New York: Springer; 2003. [Google Scholar]
- 4.Krasensky J, Jonak C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J Exp Bot. 2012;63:1593–1608. doi: 10.1093/jxb/err460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamaguchi M, Sharp RE. Complexity and coordination of root growth at low water potentials: recent advances from transcriptomic and proteomic analyses. Plant Cell Environ. 2010;33:590–603. doi: 10.1111/j.1365-3040.2009.02064.x. [DOI] [PubMed] [Google Scholar]
- 6.Rowe JH, Topping JF, Liu J, Lindsey K. Abscisic acid regulates root growth under osmotic stress conditions via an interacting hormonal network with cytokinin, ethylene and auxin. New Phytol. 2016;211:225–239. doi: 10.1111/nph.13882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garg R, Shankar R, Thakkar B, Kudapa H, Krishnamurthy L, Mantri N, et al. Transcriptome analyses reveal genotype- and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci Rep. 2016;6:19228. doi: 10.1038/srep19228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozturk ZN, Talamé V, Deyholos M, Michalowski CB, Galbraith DW, Gozukirmizi N, et al. Monitoring large-scale changes in transcript abundance in drought- and salt-stressed barley. Plant Mol Biol. 2002;48:551–573. doi: 10.1023/A:1014875215580. [DOI] [PubMed] [Google Scholar]
- 9.Ueda A, Kathiresan A, Inada M, Narita Y, Nakamura T, Shi W, et al. Osmotic stress in barley regulates expression of a different set of genes than salt stress does. J Exp Bot. 2004;55:2213–2218. doi: 10.1093/jxb/erh242. [DOI] [PubMed] [Google Scholar]
- 10.Mittler R. Abiotic stress, the field environment and stress combination. Trends Plant Sci. 2006;11:15–19. doi: 10.1016/j.tplants.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki N, Rivero RM, Shulaev V, Blumwald E, Mittler R. Abiotic and biotic stress combinations. New Phytol. 2014;203:32–43. doi: 10.1111/nph.12797. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki N, Bassil E, Hamilton JS, Inupakutika MA, Zandalinas SI, Tripathy D, et al. ABA is required for plant acclimation to a combination of salt and heat stress. PLoS One. 2016;11:e0147625. doi: 10.1371/journal.pone.0147625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rizhsky L, Liang H, Mittler R. The combined effect of drought stress and heat shock on gene expression in tobacco. Plant Physiol. 2002;130:1143–1151. doi: 10.1104/pp.006858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed IM, Dai H, Zheng W, Cao F, Zhang G, Sun D, Wu F. Genotypic differences in physiological characteristics in the tolerance to drought and salinity combined stress between Tibetan wild and cultivated barley. Plant Physiol Biochem. 2013;63:49–60. doi: 10.1016/j.plaphy.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Rivero RM, Mestre TC, Mittler R, Rubio F, Garcia-Sanchez F, Martinez V. The combined effect of salinity and heat reveals a specific physiological, biochemical and molecular response in tomato plants. Plant Cell Environ. 2014;37:1059–1073. doi: 10.1111/pce.12199. [DOI] [PubMed] [Google Scholar]
- 16.Sun C, Gao X, Fu J, Zhou J, Wu X. Metabolic response of maize (Zea mays L.) plants to combined drought and salt stress. Plant Soil. 2015;388:99–117. doi: 10.1007/s11104-014-2309-0. [DOI] [Google Scholar]
- 17.Jogaiah S, Govind SR, Tran L-SP. Systems biology-based approaches toward understanding drought tolerance in food crops. Crit Rev Biotechnol. 2013;33:23–39. doi: 10.3109/07388551.2012.659174. [DOI] [PubMed] [Google Scholar]
- 18.Kreszies Tino, Shellakkutti Nandhini, Osthoff Alina, Yu Peng, Baldauf Jutta A., Zeisler-Diehl Viktoria V., Ranathunge Kosala, Hochholdinger Frank, Schreiber Lukas. Osmotic stress enhances suberization of apoplastic barriers in barley seminal roots: analysis of chemical, transcriptomic and physiological responses. New Phytologist. 2018;221(1):180–194. doi: 10.1111/nph.15351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mickky BM, Aldesuquy HS. Impact of osmotic stress on seedling growth observations, membrane characteristics and antioxidant defense system of different wheat genotypes. Egypt J Basic Appl Sci. 2017;4:47–54. doi: 10.1016/j.ejbas.2016.10.001. [DOI] [Google Scholar]
- 20.Opitz N, Paschold A, Marcon C, Malik WA, Lanz C, Piepho H-P, Hochholdinger F. Transcriptomic complexity in young maize primary roots in response to low water potentials. BMC Genomics. 2014;15:741. doi: 10.1186/1471-2164-15-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng J, Zhao J, Tao Y, Wang J, Liu Y, Fu J, et al. Isolation and analysis of water stress induced genes in maize seedlings by subtractive PCR and cDNA macroarray. Plant Mol Biol. 2004;55:807–823. doi: 10.1007/s11103-004-1969-1. [DOI] [PubMed] [Google Scholar]
- 22.O'Green AT. Soil Water Dynamics. Nature Education Knowledge. 2012;3(6):12. [Google Scholar]
- 23.Wicke B, Smeets E, Dornburg V, Vashev B, Gaiser T, Turkenburg W, Faaij A. The global technical and economic potential of bioenergy from salt-affected soils. Energy Environ Sci. 2011;4:2669. doi: 10.1039/c1ee01029h. [DOI] [Google Scholar]
- 24.Bartels D, Sunkar R. Drought and salt tolerance in plants. Crit Rev Plant Sci. 2005;24:23–58. doi: 10.1080/07352680590910410. [DOI] [Google Scholar]
- 25.Munns R. Comparative physiology of salt and water stress. Plant Cell Environ. 2002;25:239–250. doi: 10.1046/j.0016-8025.2001.00808.x. [DOI] [PubMed] [Google Scholar]
- 26.Opitz N, Marcon C, Paschold A, Malik WA, Lithio A, Brandt R, et al. Extensive tissue-specific transcriptomic plasticity in maize primary roots upon water deficit. J Exp Bot. 2016;67:1095–1107. doi: 10.1093/jxb/erv453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walia H, Wilson C, Wahid A, Condamine P, Cui X, Close TJ. Expression analysis of barley (Hordeum vulgare L.) during salinity stress. Funct Integr Genomics. 2006;6:143–156. doi: 10.1007/s10142-005-0013-0. [DOI] [PubMed] [Google Scholar]
- 28.Coolen S, Proietti S, Hickman R, Davila Olivas NH, Huang P-P, van Verk MC, et al. Transcriptome dynamics of Arabidopsis during sequential biotic and abiotic stresses. Plant J. 2016;86:249–267. doi: 10.1111/tpj.13167. [DOI] [PubMed] [Google Scholar]
- 29.Song L, Prince S, Valliyodan B, Joshi T, Maldonado dos Santos JV, Wang J, et al. Genome-wide transcriptome analysis of soybean primary root under varying water-deficit conditions. BMC Genomics. 2016;17:57. doi: 10.1186/s12864-016-2378-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prasch CM, Sonnewald U. Simultaneous application of heat, drought, and virus to Arabidopsis plants reveals significant shifts in signaling networks. Plant Physiol. 2013;162:1849–1866. doi: 10.1104/pp.113.221044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaar-Moshe L, Blumwald E, Peleg Z. Unique physiological and transcriptional shifts under combinations of salinity, drought, and heat. Plant Physiol. 2017;174:421–434. doi: 10.1104/pp.17.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rasmussen S, Barah P, Suarez-Rodriguez MC, Bressendorff S, Friis P, Costantino P, et al. Transcriptome responses to combinations of stresses in Arabidopsis. Plant Physiol. 2013;161:1783–1794. doi: 10.1104/pp.112.210773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Aimin, Ma Hongping, Liu Enhui, Jiang Tongtong, Feng Shuang, Gong Shufang, Wang Jingang. Transcriptome Sequencing of Dianthus spiculifolius and Analysis of the Genes Involved in Responses to Combined Cold and Drought Stress. International Journal of Molecular Sciences. 2017;18(4):849. doi: 10.3390/ijms18040849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sewelam N, Oshima Y, Mitsuda N, Ohme-Takagi M. A step towards understanding plant responses to multiple environmental stresses: a genome-wide study. Plant Cell Environ. 2014;37:2024–2035. doi: 10.1111/pce.12274. [DOI] [PubMed] [Google Scholar]
- 35.Ahmed IM, Nadira UA, Bibi N, Zhang G, Wu F. Tolerance to combined stress of drought and salinity in barley. Physiological, molecular and biochemical aspects. 2015. pp. 93–121. [Google Scholar]
- 36.Fukuda H. Plant cell wall patterning and cell shape. Hoboken: Wiley; 2015. [Google Scholar]
- 37.Marowa P, Ding A, Kong Y. Expansins: roles in plant growth and potential applications in crop improvement. Plant Cell Rep. 2016;35:949–965. doi: 10.1007/s00299-016-1948-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Y, Thorne ET, Sharp RE, Cosgrove DJ. Modification of expansin transcript levels in the maize primary root at low water potentials. Plant Physiol. 2001;126:1471–1479. doi: 10.1104/pp.126.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waszczak C, Carmody M, Kangasjärvi J. Reactive oxygen species in plant signaling. Annu Rev Plant Biol. 2018;69:209–236. doi: 10.1146/annurev-arplant-042817-040322. [DOI] [PubMed] [Google Scholar]
- 40.Mittler R, Vanderauwera S, Gollery M, van Breusegem F. Reactive oxygen gene network of plants. Trends Plant Sci. 2004;9:490–498. doi: 10.1016/j.tplants.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Jin J, Tian F, Yang D-C, Meng Y-Q, Kong L, Luo J, Gao G. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45:D1040–D1045. doi: 10.1093/nar/gkw982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rizhsky L, Liang H, Shuman J, Shulaev V, Davletova S, Mittler R. When defense pathways collide. The response of Arabidopsis to a combination of drought and heat stress. Plant Physiol. 2004;134:1683–1696. doi: 10.1104/pp.103.033431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashoub A, Müller N, Jiménez-Gómez JM, Brüggemann W. Prominent alterations of wild barley leaf transcriptome in response to individual and combined drought acclimation and heat shock conditions. Physiol Plant. 2018;163:18–29. doi: 10.1111/ppl.12667. [DOI] [PubMed] [Google Scholar]
- 44.Guo M, Liu J-H, Ma X, Luo D-X, Gong Z-H, Lu M-H. The plant heat stress transcription factors (HSFs): structure, regulation, and function in response to abiotic stresses. Front Plant Sci. 2016;7:114. doi: 10.3389/fpls.2016.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Z, Xin M, Qin J, Peng H, Ni Z, Yao Y, Sun Q. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.) BMC Plant Biol. 2015;15:152. doi: 10.1186/s12870-015-0511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swindell WR, Huebner M, Weber AP. Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics. 2007;8:125. doi: 10.1186/1471-2164-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsumoto T, Morishige H, Tanaka T, Kanamori H, Komatsuda T, Sato K, et al. Transcriptome analysis of barley identifies heat shock and HD-zip I transcription factors up-regulated in response to multiple abiotic stresses. Mol Breeding. 2014;34:761–768. doi: 10.1007/s11032-014-0048-9. [DOI] [Google Scholar]
- 48.Sun W, Bernard C, van de Cotte B, van Montagu M, Verbruggen N. At-HSP17.6A, encoding a small heat-shock protein in Arabidopsis, can enhance osmotolerance upon overexpression. Plant J. 2001;27:407–415. doi: 10.1046/j.1365-313X.2001.01107.x. [DOI] [PubMed] [Google Scholar]
- 49.Nakano T, Suzuki K, Fujimura T, Shinshi H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 2006;140:411–432. doi: 10.1104/pp.105.073783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heyman Jefri, Canher Balkan, Bisht Anchal, Christiaens Fien, De Veylder Lieven. Emerging role of the plant ERF transcription factors in coordinating wound defense responses and repair. Journal of Cell Science. 2017;131(2):jcs208215. doi: 10.1242/jcs.208215. [DOI] [PubMed] [Google Scholar]
- 51.Morran S, Eini O, Pyvovarenko T, Parent B, Singh R, Ismagul A, et al. Improvement of stress tolerance of wheat and barley by modulation of expression of DREB/CBF factors. Plant Biotechnol J. 2011;9:230–249. doi: 10.1111/j.1467-7652.2010.00547.x. [DOI] [PubMed] [Google Scholar]
- 52.Xu Z-S, Chen M, Li L-C, Ma Y-Z. Functions of the ERF transcription factor family in plants. Botany. 2008;86:969–977. doi: 10.1139/B08-041. [DOI] [Google Scholar]
- 53.Dubouzet JG, Sakuma Y, Ito Y, Kasuga M, Dubouzet EG, Miura S, et al. OsDREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. Plant J. 2003;33:751–763. doi: 10.1046/j.1365-313X.2003.01661.x. [DOI] [PubMed] [Google Scholar]
- 54.Corrêa Luiz Gustavo Guedes, Riaño-Pachón Diego Mauricio, Schrago Carlos Guerra, Vicentini dos Santos Renato, Mueller-Roeber Bernd, Vincentz Michel. The Role of bZIP Transcription Factors in Green Plant Evolution: Adaptive Features Emerging from Four Founder Genes. PLoS ONE. 2008;3(8):e2944. doi: 10.1371/journal.pone.0002944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004;5:226. doi: 10.1186/gb-2004-5-6-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makkena Srilakshmi, Lamb Rebecca S. The bHLH transcription factor SPATULA regulates root growth by controlling the size of the root meristem. BMC Plant Biology. 2013;13(1):1. doi: 10.1186/1471-2229-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoagland DR, Arnon DI. The water-culture method for growing plants without soil. Berkeley: College of Agriculture, University of California; 1938. [Google Scholar]
- 58.R Core Team (2017). R: A language and environment for statistical computing. Vienna, Austria.
- 59.Kassambara A. ggpubr: 'ggplot2' Based publication ready plots. 2017. [Google Scholar]
- 60.Fox J, Weisberg S. An R companion to applied regression: second edition. 2nd ed. Los Angeles, London, New Delhi, Singapore, Washington DC: Sage; 2011.
- 61.de Mendiburu F. agricolae: Statistical Procedures for Agricultural Research. 2017. [Google Scholar]
- 62.Ludwig Y, Zhang Y, Hochholdinger F. The maize (Zea mays L.) AUXIN/INDOLE-3-ACETIC ACID gene family: phylogeny, synteny, and unique root-type and tissue-specific expression patterns during development. PLoS One. 2013;8:e78859. doi: 10.1371/journal.pone.0078859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, et al. A chromosome conformation capture ordered sequence of the barley genome. Nature. 2017;544:427–433. doi: 10.1038/nature22043. [DOI] [PubMed] [Google Scholar]
- 64.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 67.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- 68.Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, et al. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;45:W122–W129. doi: 10.1093/nar/gkx382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mayer KFX, Waugh R, Brown JWS, Schulman A, Langridge P, Platzer M, et al. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491:711–716. doi: 10.1038/nature11543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overview of the experimental workflow of the RNA-Seq experiment. (PDF 130 kb)
Overview of RNA-Seq output and mapping results. (XLSX 16 kb)
Comprehensive list of the differential expression analysis. (XLSX 3317 kb)
Overview of all identified over-represented Gene Ontology (GO) terms. (XLSX 28 kb)
Data Availability Statement
The raw sequencing data is deposited in NCBI’s sequencing read archive under accession number SRP133479 (https://www.ncbi.nlm.nih.gov/sra/SRP133479). The datasets supporting the conclusions of this article are included within the article and its Additional files.