

Abstract
Dimethylcyclopropanes are valuable synthetic targets that are challenging to access in high yield using Zn carbenoid reagents. Here, we describe a cobalt-catalyzed variant of the Simmons–Smith reaction that enables the efficient dimethylcyclopropanation of 1,3-dienes using a Me2CCl2/Zn reagent mixture. The reactions proceed with high regioselectivity based on the substitution pattern of the 1,3-diene. The products are vinylcyclopropanes, which serve as substrates for transition-metal catalyzed ring-opening reactions, including 1,3-rearrangements and [5 + 2]-cycloadditions. Preliminary studies indicate that moderately activated monoalkenes are also amenable to dimethylcyclopropanation under the cobalt-catalyzed conditions.
Keywords: homogeneous catalysis, cobalt, carbenoids, carbocycles, cycloaddition
Graphical Abstarct
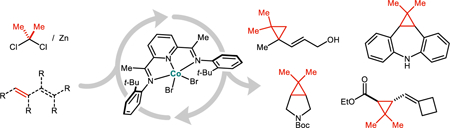
A cobalt catalyst promotes the regioselective dimethylcyclopropanation of 1,3-dienes using Me2CCl2 as an isopropylidene source and Zn as a stoichiometric reductant. The products are gem-dimethylated vinylcyclopropanes, which may be converted into other cyclic systems by strain-induced ring opening reactions. Moderately activated isolated alkenes are also viable substrates, providing access to medicinally relevant building blocks.
Geminal dimethylation is a common substitution pattern in polycyclic terpene natural products.[1] Medicinal chemists have also explored the installation of dimethyl groups in biologically active compounds as a strategy to improve their potency or to eliminate metabolic liabilities.[2] There are now over 50 clinically approved pharmaceutical compounds that feature these motifs. The presence of gem-dimethylation in a target compound introduces significant synthetic complexity in that a hindered tetrasubstituted carbon atom must be generated. The most common routes utilize classical carbonyl chemistry and entail either a double α-alkylation reaction or a double addition of a methyl nucleophile to a carboxylic acid derivative.[3]
It is attractive to consider an alternative approach based on the formation of dimethylcyclopropanes,[4] which are themselves common in biologically active compounds[2, 5] but may also be diversified into other frameworks through strain-induced ring-opening reactions.[6] The Simmons–Smith reaction is potentially well-suited to addressing these structures; however, it is known that the efficiency of Zn carbenoid-based cyclopropanations decreases significantly when using disubstituted gem-dihaloalkanes.[7] For example, the addition of 2,2-diiodopropane under Furukawa conditions has only been demonstrated for two simple substrates, cyclopentene and cyclohexene, which provide yields up to 59% after a reaction time of 5 days.[8] A notable advance in this area was reported by Charette, who showed that directing group effects can be leveraged to achieve a high-yielding dimethylcyclopropanation of allylic alcohols.[9] Additionally, the Corey–Chaykovsky-type dimethylcyclopropanation of α,β-unsaturated carbonyl compounds has been developed.[10]
We recently found that [PDI]Co (PDI = pyridine–diimine) complexes function as catalysts[11],[12] for Simmons–Smith-type cyclopropanation reactions and generate a reactive carbene equivalent of significantly altered selectivity properties from Zn carbenoids.[13] Motivated by this finding, we became interested in using transition metal catalysis to address the dimethylcyclopropanation reaction. Here, we report a cobalt-catalyzed dimethylcyclopropanation of 1,3-dienes using Me2CCl2/Zn as a source of isopropylidene equivalents.
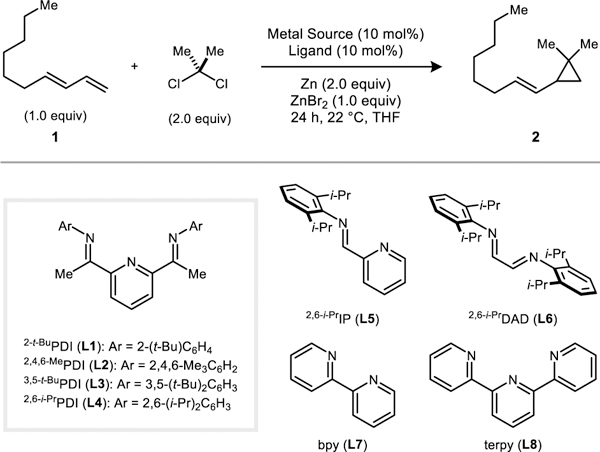
The [2,6-i-PrPDI]CoBr2 complex was previously shown to be an effective catalyst for cyclopropanation reactions using CH2Br2/Zn;[13] however, it afforded negligible yields in the analogous reactions using Me2CCl2/Zn (Table 1, entry 7). We reasoned that the more hindered dimethylcarbene fragment may require the steric profile of the PDI ligand to be correspondingly adjusted. Accordingly, a series of ligands was prepared by varying the size of the flanking aryl substituents (L1–L4). The 2-t-BuPh derived catalyst was identified as being optimal (entry 4), and the dimethylcyclopropanation of a model 1,3-diene (1) provided 2 in 93% yield as a single regioisomer. Zn is not capable of directly activating Me2CCl2, and in the absence of the Co catalyst, there is no conversion (entry 1). Despite the fact that ZnCl2 is generated as a stoichiometric byproduct, the presence of ZnBr2 at the start of the reaction provided a modest beneficial effect on yield (entry 14). ZnBr2 accelerates the initial reduction of the yellow Co(II) complex to a violet Co(I) species, which is then capable of activating Me2CCl2. Finally, other classes of bidentate and tridentate ligands containing combinations of pyridine and imine donors were ineffective relative to PDI (entries 10–13). As a point of comparison, the non-catalytic dimethylcyclopropanation of 1 under Furukawa-type Simmons–Smith conditions (I2CMe2/Et2Zn)[9] provided 2 in a moderate yield of 45% along with several inseparable side products (see Supporting Information for experimental details).
Table 1.
Catalyst Optimization Studies[a]
 | |||
|---|---|---|---|
| Entry | Metal Source | Ligand | Yield 2 [%] |
| 1 | – | – | < 1 |
| 2 | – | 2-t-BuPDI (L1) | < 1 |
| 3 | CoBr2 | – | < 1 |
| 4 | CoBr2 | 2-t-BuPDI (L1) | 93 |
| 5 | CoBr2 | 2,4,6-MePDI (L2) | 77 |
| 6 | CoBr2 | 3,5-t-BuPDI (L3) | 8 |
| 7 | CoBr2 | 2,6-i-PrPDI (L4) | 2 |
| 8 | NiBr2 | 2-t-BuPDI (L1) | 5 |
| 9 | FeBr2 | 2-t-BuPDI (L1) | 4 |
| 10 | CoBr2 | 2,6-i-PrIP (L5) | 2 |
| 11 | CoBr2 | 2,6-i-PrDAD (L6) | <1 |
| 12 | CoBr2 | bpy (L7) | 4 |
| 13 | CoBr2 | terpy (L8) | 1 |
| 14[b] | CoBr2 | 2-t-BuPDI (L1) | 87 |
| 15[c] | CoBr2 | 2-t-BuPDI (L1) | 78 |
| 16[d] | CoBr2 | 2-t-BuPDI (L1) | 79 |
Reactions were run on a 0.14 mmol scale of the 1,3-diene in THF (1 mL). Yields of 2 were determined by GC analysis against an internal standard.
Modifications from standard conditions: without ZnBr2.
Modifications from standard conditions: 1.1 equiv of Me2CCl2.
Modifications from standard conditions: 2.0 equiv of Me2CBr2.
With optimized conditions in hand, we next evaluated the substrate scope of the dimethylcyclopropanation reaction (Figure 3). A variety of 1-substituted, 1,2-disubstituted, and 1,3-disubstituted dienes are viable substrates. In all cases, cyclopropanation occurs at the terminal double bond with high regioselectivity (rr = >19:1). Notably, there is no detectable secondary cyclopropanation of the product alkene despite the presence of excess Me2CCl2 and Zn in the reaction mixture. Common functional handles for further synthetic elaboration are tolerated, including a protected amine (8), BPin group (9), ester (13), and free alcohol (18). The activation of Me2CCl2 by the Co catalyst is relatively facile such that aryl chlorides (4) present in the substrate are tolerated. The TBS-protected variant of Danishefsky’s diene afforded a protected cyclopropanol derivative in high yield (14).
Figure 3.

Substrate scope studies. [a] Standard reaction conditions: 0.14 mmol scale of the 1,3-diene (1.0 equiv), Me2CCl2 (2.0 equiv), Zn (2.0 equiv), ZnBr2 (1.0 equiv), [2-t-BuPDI]CoBr2 (3) (10 mol%), and THF (1 mL); 22 °C for 24 h. [b] Simmons–Smith reaction conditions: ZnEt2 (4.0 equiv), I2CMe2 (4.0 equiv), and CH2Cl2 (1.0 mL). [c] Modifications to standard conditions: DCE (1.0 mL) instead of THF, Me2CBr2 (2.0 equiv) instead of Me2CCl2.
Unlike the Simmons–Smith reaction, the cobalt-catalyzed process is relatively insensitive to the electronic properties of the diene.[14] For example, the presence of an electron-withdrawing group, such as a phosphonate ester (11), a ketone (12), or an ester (13), does not significantly decrease the rate of the reaction nor does it result in poor yields of the product. Additionally, the cobalt catalyst does not appear to interact strongly with alcohol directing groups.[15] The diene 16 is selectively monocyclopropanated at the terminal double bond (rr = >19:1) as expected based solely on steric considerations. By contrast, Furukawa-type conditions using I2CMe2/Et2Zn afford selectivity for the internal double bond, which is proximal to the alcohol.[9]
Synthetic ethyl chrysanthemate is prepared on industrial scales by the addition of ethyl diazoacetate to 2,5-dimethyl-2,4-hexadiene.[16] A limitation of this process is that the cyclopropane is formed as a mixture of cis and trans diastereomers, whereas natural pyrethrins are found only with the trans relative stereochemistry. Using the cobalt-catalyzed cyclopropanation, ethyl (E)-5-methyl-2,4-hexadienoate reacts at the less hindered internal alkene with >19:1 trans selectivity (19). This approach was applied to the synthesis of the commercial pesticide phenothrin (20)[17] and unnatural pyrethrin analogues that contain other alkyl (25), cycloalkyl (21–23), or aryl (24) substituents in the place of the terminal methyl groups.
We considered the possibility that the vinyl cyclopropane products generated by this method might serve as substrates for transition metal-catalyzed ring-opening reactions; however, it was unknown at the outset whether the steric hindrance imposed by a gem-dimethyl group would be tolerated. Vinylcyclopropane 26 was prepared from the corresponding 1,3-diene precursor in 98% yield. Heating 26 at 60 °C for 12 h in the presence of a IPr/Ni(COD)2 catalyst provided the rearranged cyclopentene product 27 in 65% yield.[18] The position of the alkene in 27 indicates that only the less hindered C–C bond is activated by the catalyst.
A substrate containing both a 1,3-diene and an isolated alkene was cyclopropanated under the cobalt-catalyzed conditions to yield a single regioisomer of the monocyclopropane product (28). When 28 was subjected to the Rh-catalyzed [5 + 2]-cycloaddition conditions developed by Wender,[19] the bicyclic product 29 was formed in 77% yield. Like the 1,3-rearrangement reaction, the ring-opening proceeds with high regioselectivity to furnish a single isomer of the product.
Finally, it was of interest to test whether the catalytic conditions developed for the dimethylcyclopropanation of 1,3-dienes could be applied to isolated alkenes. Simple, unactivated alkenes such as 1-octene or cyclohexene were unreactive (<2% yield); however, substrates possessing a moderate degree of strain proved to be viable. For example, cyclopentene is cyclopropanated in 87% yield under the standard optimized conditions (30). This result represents a significant improvement in yield and reaction time over the only previously reported attempt to carry out this transformation (45% yield after 5 days using I2CMe2/Et2Zn).[8] Additionally, cyclooctene (31), indene (32), and norbornene (33) react in high yield. In the latter case, the exo-addition product is formed exclusively. N-Boc-2,5-dihydropyrrole (34) is cyclopropanated in 90% yield to generate 35, which is a protected precursor to the HCV protease inhibitor boceprevir.[20] The process-scale route to this bicyclic amine consists of a multi-step sequence in which the gem-dimethylcyclopropane unit is ultimately derived from ethyl chrysanthemate.
Tricyclic antidepressants (TCAs) are a class of compounds that commonly contain a dibenzazepine.[21] Parent 5H-dibenz[b,f]azepine (36), bearing an unprotected N-H group, could be directly dimethylcyclopropanated to provide 37 in 96% yield. Notably, previous attempts to cyclopropanate this substrate under Simmons–Smith conditions (CH2I2 + Zn/Cu) only yielded the product of methylene insertion into the N–H bond.[22] One-step derivatization of 37 using chlorosulfonyl isocyanate[23] yielded a dimethylcyclopropane-containing analogue (38) of carbamazepine,[21b] which is used as a treatment for epilepsy and neuropathic pain.
In summary, cobalt catalysis enables the synthesis of dimethylcyclopropanes that were not previously accessible in high yield using the Simmons–Smith reaction. In particular, the regioselective dimethylcyclopropanation of 1,3-dienes yields polysubstituted vinyl cyclopropanes, which participate in strain-induced ring-opening reactions. Moderately activated monoalkenes are also cyclopropanated efficiently to generate building blocks for medicinal chemistry. These studies collectively highlight the unique properties of transition metal carbenoids over their Zn counterparts as reactive species in cyclopropanation chemistry.
Experimental Section
General procedure for the catalytic dimethylcyclopropanation.
In an N2-filled glovebox, a 2-dram vial was charged with [2-tBuPDI]CoBr2 3 (9.0 mg, 0.014 mmol, 10 mol%), the 1,3-diene or alkene (0.14 mmol, 1.0 equiv), Zn powder (18 mg, 0.28 mmol, 2.0 equiv), ZnBr2 (31 mg, 0.14 mmol, 1.0 equiv), THF (1.0 mL), and a magnetic stir bar. The reaction was stirred at room temperature for approximately 15 min, during which time a deep violet color was observed corresponding to the reduced cobalt catalyst. Me2CCl2 (31.6 mg, 0.28 mmol, 2.0 equiv) was added, and the reaction mixture was stirred at room temperature. After 24 h, the reaction mixture was exposed to ambient atmosphere and concentrated under reduced pressure. The crude residue was directly loaded onto a SiO2 column for purification.
Supplementary Material
Figure 1.

Rings containing gem-dimethyl groups, including dimethylcyclopropanes, are found in biologically active compounds of natural and synthetic origins.
Figure 2.
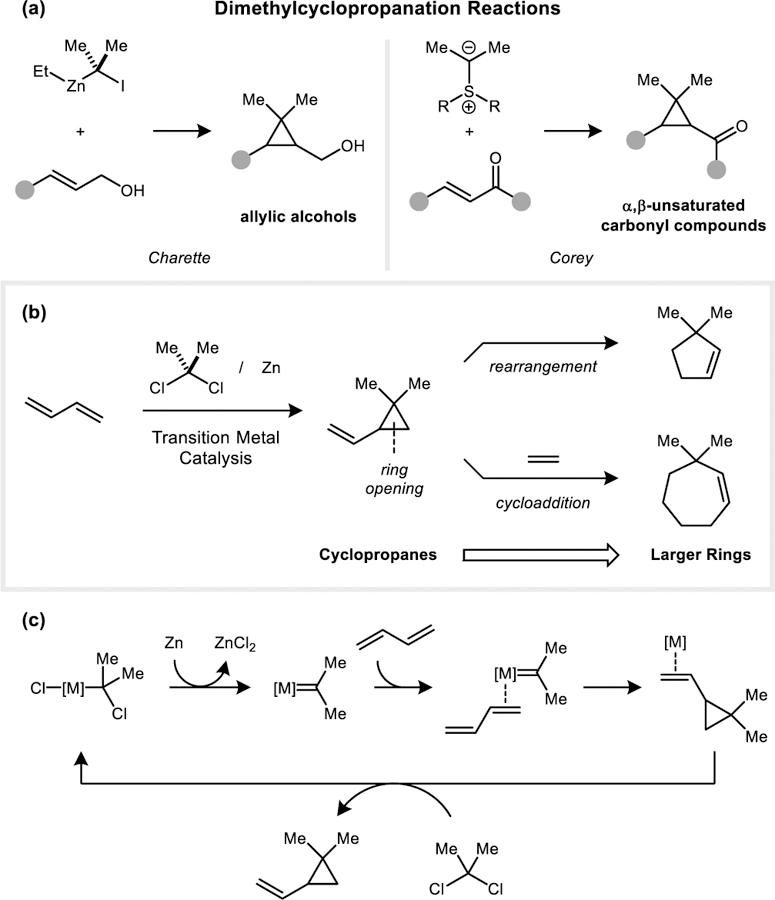
(a) Simmons–Smith-type dimethylcyclopropanation reactions of allylic alcohols and Corey–Chaykovsky dimethylcyclopropanation reactions of α,β-unsaturated carbonyl compounds. (b) Transition metal catalyzed reductive dimethylcyclopropanation reactions of 1,3-dienes and ring-opening reactions to generate 5- and 7-membered rings. (c) Proposed mechanism for a transition metal catalyzed reductive dimethylcyclopropanation reaction.
Figure 4.
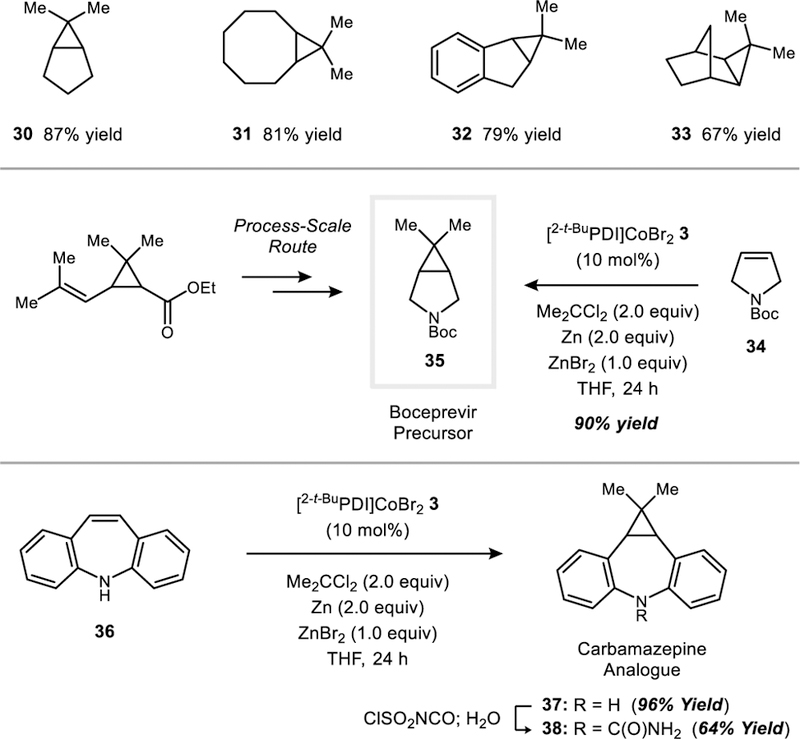
Dimethylcyclopropanations of activated alkenes, including applications to the synthesis of a boceprevir precursor and an analogue of carbamazepine. Standard dimethylcyclopropanation conditions: 0.14 mmol scale of the 1,3-diene (1.0 equiv), Me2CCl2 (2.0 equiv), Zn (2.0 equiv), ZnBr2 (1.0 equiv), [2-t-BuPDI]CoBr2 (3) (10 mol%), and THF (1 mL); 22 °C for 24 h.
Acknowledgements
This work was supported by the National Institutes of Health (R35 GM124791). C.U. is an Alfred P. Sloan Foundation Research Fellow.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Maimone TJ, Baran PS, Nat. Chem. Biol 2007, 3, 396, [DOI] [PubMed] [Google Scholar]; b) Gershenzon J, Dudareva N, Nat. Chem. Biol 2007, 3, 408. [DOI] [PubMed] [Google Scholar]
- [2].Talele TT, J. Med. Chem 2018, 61, 2166–2210. [DOI] [PubMed] [Google Scholar]
- [3].Examples of synthetic routes to gem-dimethyl groups in biologically active compounds: a) Glossop PA, Lane CAL, Price DA, Bunnage ME, Lewthwaite RA, James K, Brown AD, Yeadon M, Perros-Huguet C, Trevethick MA, Clarke NP, Webster R, Jones RM, Burrows JL, Feeder N, Taylor SCJ, Spence FJ, J. Med. Chem 2010, 53, 6640–6652 [DOI] [PubMed] [Google Scholar]; b) Gottumukkala AL, Matcha K, Lutz M, de Vries JG, Minnaard AJ, Chem. Eur. J 2012, 18, 6907–6914; [DOI] [PubMed] [Google Scholar]; c) Wang J, Tong R, J. Org. Chem 2016, 81, 4325–4339; [DOI] [PubMed] [Google Scholar]; d) Kick E, Martin R, Xie Y, Flatt B, Schweiger E, Wang T-L, Busch B, Nyman M, Gu X-H, Yan G, Wagner B, Nanao M, Nguyen L, Stout T, Plonowski A, Schulman I, Ostrowski J, Kirchgessner T, Wexler R, Mohan R, Bioorg. Med. Chem. Lett 2015, 25, 372–377. [DOI] [PubMed] [Google Scholar]
- [4].Recent studies in dimethylcyclopropane synthesis: a) Dian L, Müller DS, Marek I, Angew. Chem., Int. Ed 2017, 56, 6783–6787; Angew. Chem. 2017, 129, 6887–6891; [DOI] [PubMed] [Google Scholar]; b) Cang H, Moss RA, Krogh-Jespersen K, J. Am. Chem. Soc 2015, 137, 2730–2737. [DOI] [PubMed] [Google Scholar]
- [5].Talele TT, J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- [6].a) Meazza M, Guo H, Rios R, Org. Biomol. Chem 2017, 15, 2479–2490; [DOI] [PubMed] [Google Scholar]; b) Souillart L, Cramer N, Chem. Rev 2015, 115, 9410–9464; [DOI] [PubMed] [Google Scholar]; c) Chen P.-h., Billett BA, Tsukamoto T, Dong G, ACS Catal 2017, 7, 1340–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sengmany S, Léonel E, Paugam JP, Nédélec J-Y, Tetrahedron 2002, 58, 271–277. [Google Scholar]
- [8].Stahl K-J, Hertzsch W, Musso H, Liebigs Ann. Chem 1985, 1985, 1474–1484. [Google Scholar]
- [9].Charette AB, Wilb N, Synlett 2002, 2002, 0176–0178. [Google Scholar]
- [10].Corey EJ, Jautelat M, J. Am. Chem. Soc 1967, 89, 3912–3914. [Google Scholar]
- [11].Chirik PJ, Angew. Chem., Int. Ed 2017, 56, 5170–5181; Angew. Chem. 2017, 129, 5252–5265. [DOI] [PubMed] [Google Scholar]
- [12].Kanai H, Matsuda H, J. Mol. Catal 1985, 29, 157–164. [Google Scholar]
- [13].Werth J, Uyeda C, Chem. Sci 2018, 9, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Charette AB, Beauchemin A, Org. React 2001, 58, 1–415.; [Google Scholar]; b) Motherwell WB, Nutley CJ, Contemp. Org. Synth 1994, 1, 219–241. [Google Scholar]
- [15].a) Hoveyda AH, Evans DA, Fu GC, Chem. Rev 1993, 93, 1307–1370; [Google Scholar]; b) Poulter CD, Friedrich EC, Winstein S, J. Am. Chem. Soc 1969, 91, 6892–6894. [Google Scholar]
- [16].Doyle MP, Chem. Rev 1986, 86, 919–939. [Google Scholar]
- [17].Fujmoto K, Itaya N, Okuno Y, Kadota T, Yamaguchi T, Agric. Biol. Chem 1973, 37, 2681–2682. [Google Scholar]
- [18].Zuo G, Louie J, Angew. Chem., Int. Ed 2004, 43, 2277–2279; Angew. Chem. 2004, 116, 2327–2329. [DOI] [PubMed] [Google Scholar]
- [19].Wender PA, Husfeld CO, Langkopf E, Love JA, J. Am. Chem. Soc 1998, 120, 1940–1941. [Google Scholar]
- [20].Li T, Liang J, Ambrogelly A, Brennan T, Gloor G, Huisman G, Lalonde J, Lekhal A, Mijts B, Muley S, Newman L, Tobin M, Wong G, Zaks A, Zhang X, J. Am. Chem. Soc 2012, 134, 6467–6472. [DOI] [PubMed] [Google Scholar]
- [21].a) Singh H, Gupta N, Kumar P, Dubey SK, Sharma PK, Org. Process Res. Dev 2009, 13, 870–874; [Google Scholar]; b) Tian M, Abdelrahman A, Weinhausen S, Hinz S, Weyer S, Dosa S, El-Tayeb A, Müller CE, Bioorg. Med. Chem 2014, 22, 1077–1088. [DOI] [PubMed] [Google Scholar]
- [22].Kawashima K, Kawano Y, Chem. Pharm. Bull 1976, 24, 2751–2760. [Google Scholar]
- [23].Ravinder B, Rajeshwar Reddy S, Sridhar M, Murali Mohan M, Srinivas K, Panasa Reddy A, Bandichhor R, Tetrahedron Lett 2013, 54, 2841–2844. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.