

Summary
The basal ganglia regulates motor, cognitive, and emotional behaviors. Dysfunction of dopamine system in this area is implicated in the pathophysiology of schizophrenia characterized by positive symptoms, negative symptoms, and cognitive deficits. Medium spiny neurons (MSNs) are principal output neurons of striatum in the basal ganglia. Similar to current antipsychotics with dopamine D2 receptor antagonism or partial agonism, phosphodiesterase 10A (PDE10A) inhibitors activate indirect pathway MSNs, leading to the expectation of therapeutic potential for the treatment of psychosis. PDE10A inhibitors also activate direct pathway MSNs which may be associated with cognitive functions. These pathways have competing effects on antipsychotic‐like activities and extrapyramidal symptoms in rodents. Therefore, careful consideration of activation pattern of these pathways by a PDE10A inhibitor is critical to produce potent efficacy and superior safety profiles. In this review, we outline the pharmacological profile of TAK‐063, a novel PDE10A selective inhibitor. Our study revealed that off‐rates of PDE10A inhibitors may characterize their pharmacological profiles via regulation of each MSN pathway. TAK‐063, with a faster off‐rate property, could provide a unique opportunity as a novel therapeutic approach to treatment of psychosis and cognitive deficits in schizophrenia. TAK‐063 also has a therapeutic potential in other basal ganglia disorders.
Keywords: medium spiny neuron, phosphodiesterase 10A, psychosis, schizophrenia, TAK‐063
1. INTRODUCTION
Schizophrenia is a devastating neuropsychiatric disorder that typically strikes in late adolescence or early adulthood.1 Positive symptoms in psychosis, such as hallucinations and delusions, are the most apparent manifestation of this disorder. These symptoms emerge episodically and usually trigger the first hospitalization in early adulthood. Chronic aspects of the disorder include negative symptoms, such as social withdrawal, flattened affect, and anhedonia, as well as pervasive cognitive deficits. Accumulating clinical evidence has indicated that negative symptoms and cognitive deficits in schizophrenia are closely related to poor functional outcomes and long‐term prognosis.2, 3, 4
It is generally acknowledged that psychotic symptoms of schizophrenia are associated with hyperactivity of mesolimbic dopaminergic projections resulting in excessive stimulation of dopamine D2 receptors in the striatum complex.5 Indeed, the baseline occupancy of striatal D2 receptors by dopamine is higher in subjects with schizophrenia that in controls,6 and typical and atypical antipsychotics with D2 receptor antagonistic activity or partial agonistic activity are primary medications for the treatment of schizophrenia. However, typical antipsychotics are known to cause extrapyramidal symptoms (EPS) and hyperprolactinemia due to excessive antagonism of D2 receptors in the striatum and pituitary gland, respectively.7 Although atypical antipsychotics produce a lower incidence of EPS than typical antipsychotics, these drugs are still associated with hyperprolactinemia and serious metabolic side effects, including weight gain, diabetes, and an abnormal lipid profile.7 Thus, better‐tolerated therapeutic agents with the potential to address multiple symptoms of schizophrenia would be of considerable therapeutic value.
Phosphodiesterase 10A (PDE10A) is a dual‐substrate enzyme that hydrolyzes both cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP).8, 9, 10 PDE10A is highly expressed in medium spiny neurons (MSNs) of the mammalian striatum.11, 12, 13 Striatal MSNs are divided into the dopamine D1 receptor‐expressing direct pathway and dopamine D2 receptor‐expressing indirect pathway, regulating behavioral responses on integration of cortical glutamatergic and midbrain dopaminergic input.14, 15 These pathways have competing effects on the movement such as EPS.16 Thus, activation of both MSN pathways by PDE10A inhibition is thought to be a promising approach to regulate basal ganglia function and produce an antipsychotic effect with reducing a risk of EPS caused by excessive antagonism of D2 receptors. However, we found that excess activation of direct pathway MSNs also canceled antipsychotic‐like effects by activation of indirect pathway MSNs in rodents. Therefore, balanced activation of the direct and indirect pathways should be carefully considered to achieve both potent efficacy and better tolerability by PDE10A inhibitors. Activation of direct pathway MSNs has been also considered to be associated with cognitive functions via striato‐thalamo‐cortical circuit.17, 18, 19, 20 PDE10A inhibitors may have potential therapeutic utility in the treatment of cognitive impairment associated with schizophrenia.
Extensive investigations and drug discovery research efforts have been conducted to develop PDE10A inhibitors for the treatment of schizophrenia. Despite such enormous efforts, so far no PDE10A inhibitor has reached the market. Therefore, development of a well‐differentiated PDE10A inhibitor was required to further explore the potential of PDE10A inhibition in clinical studies. In this review, we describe a pharmacological profile of a novel PDE10A inhibitor TAK‐063 1‐[2‐fluoro‐4‐(1H‐pyrazol‐1‐yl)phenyl]‐5‐methoxy‐3‐(1‐phenyl‐1H‐pyrazol‐5‐yl)pyridazin‐4(1H)‐one, which has demonstrated potent antipsychotic‐like and pro‐cognitive effects in animal models of schizophrenia. TAK‐063 also provided a therapeutic potential for other basal ganglia disorders, such as Huntington's disease (HD), Parkinson's disease (PD), functional impairment after striatal stroke, and addiction.
2. POTENTIAL THERAPEUTIC APPROACH FOR PSYCHOSIS BY ACTIVATION OF MSNS
The basal ganglia consists of several functionally related nuclei involved in cortical‐subcortical circuits that modulate motor, cognitive, and emotional behaviors.21 Dysregulation of the dopamine system in this area plays a critical role in pathophysiology of schizophrenia. Dopamine D1 receptors and D2 receptors are exclusively expressed in direct and indirect pathway MSNs, respectively.22 Current antipsychotics have D2 receptor antagonism or partial agonism.23, 24, 25, 26 D2 receptors are coupled with an inhibitory G protein (Gi) that impedes the enzyme activity of adenylate cyclase. Adenylate cyclase synthesizes cAMP from adenosine triphosphate (ATP)27; thus, activation of the indirect pathway by the blockade of D2 receptors and concomitant upregulation of cAMP level in the indirect pathway MSNs is thought to be a common mechanism of action of current antipsychotics, although excessive D2 receptor antagonism in the striatum causes EPS such as Parkinsonism. D1 receptors in direct pathway MSNs are coupled with stimulatory G protein (Gs) and stimulate adenylate cyclase. Cyclic nucleotide levels are an integral part of the cell signaling cascade and influence excitability of the MSNs. Thus, upregulation of cyclic nucleotide levels in indirect pathway MSNs could be an alternative approach for novel antipsychotics.
3. PDE10A AS A PROMISING TARGET FOR NOVEL ANTIPSYCHOTICS
PDE10A is a dual‐substrate PDE28 and has been reported to be genetically associated with schizophrenia and bipolar disorders.29, 30, 31 PDE10A is selectively expressed in both direct and indirect pathway MSNs13, 32; thus, PDE10A inhibition and the resulting elevation of striatal cyclic nucleotide levels in indirect pathway MSNs would potentially have the effects of D2 receptor antagonism, the standard treatment for psychosis, along with D1 receptor agonism. Because a recent PET study using a selective PDE10A tracer, [11C]IMA107, suggests that patients with schizophrenia have normal availability of PDE10A in the striatum,33 intracellular signaling changed by PDE10A inhibition is expected to activate both MSN pathways, even under disease conditions. Given the competing effects of both MSN pathways on the striatal output for movement, activation of direct pathway MSNs can lead to a lower EPS liability caused by excessive activation of indirect pathway MSNs. Moreover, the MSN‐specific expression of PDE10A may result in lower risks for the nonstriatal side effects associated with current antipsychotics, such as hyperprolactinemia and metabolic dysfunction. Recent preclinical studies have proposed PDE10A inhibitors as a novel type of antipsychotics with a potent efficacy and reduced side effect profile.34, 35, 36, 37 However, we found that D1 agonist and D2 antagonist also had a competing effect on antipsychotic‐like activity.38 Thus, the activation pattern of direct and indirect pathway MSNs by PDE10A inhibitors should be carefully considered. The direct and indirect pathways are also considered critical for modulating learning behavior via striato‐thalamo‐cortical circuits.17, 18 Integrated striatal outputs following inhibition of PDE10A in both MSN pathways may improve cognitive function by modifying cortical activity. Accordingly, several PDE10A inhibitors have demonstrated improved cognitive function in animal models.39, 40, 41, 42 PDE10A inhibitors may also have potential therapeutic utility in the treatment of cognitive impairment associated with schizophrenia.
4. GENERAL PHARMACOLOGICAL CHARACTERIZATION OF TAK‐063
TAK‐063 is a novel PDE10A inhibitor with a potent inhibitory activity (hPDE10A IC50 = 0.30 nmol/L; Figure 1A) and high selectivity (>15 000‐fold) over other PDE families (PDE1–PDE11) in in vitro assays using recombinant human enzymes.35, 38, 42, 43, 44 Further assessment of in vitro selectivity of TAK‐063 showed that TAK‐063 at 10 μmol/L did not show more than 50% inhibition or stimulation of 91 target molecules, except for PDEs.44 In vitro autoradiography (ARG) studies using [3H]TAK‐063 and rat or mouse brain sections showed that [3H]TAK‐063 accumulated in the caudate putamen, nucleus accumbens, globus pallidus, and substantia nigra, where PDE10A is highly expressed (Figure 1B).44 The selective accumulation of [3H]TAK‐063 was mostly blocked by 1 μmol/L of MP‐10, a Pfizer's PDE10A selective inhibitor with different chemical structure. This specific accumulation of [3H]TAK‐063 was not observed in brain sections from PDE10A knock‐out mice (Figure 1B). These results demonstrate the specific binding of [3H]TAK‐063 to native PDE10A.
Figure 1.
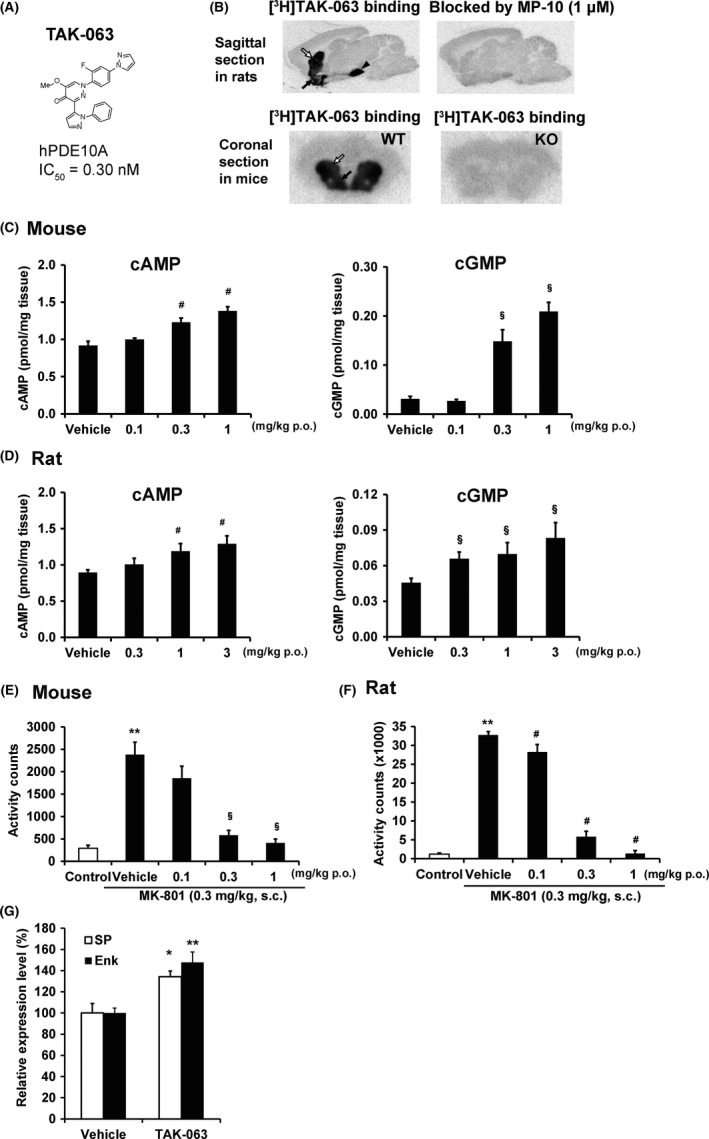
(A) Chemical structure of TAK‐063; (B) Selective binding of [3H]TAK‐063 to native phosphodiesterase 10A (PDE10A) in the brain measured by in vitro autoradiography study. Caudate putamen (white arrow); nucleus accumbens (black arrow); substantia nigra (black arrowhead); wild type (WT); PDE10A knockout (KO); (C, D) Elevation of cAMP and cGMP levels in the mouse (C) and rat (D) striatum 60 min after administration of TAK‐063 (n = 8). Antipsychotic‐like effects of TAK‐063 on MK‐801‐induced hyperactivity in mice (E) rats (F) (n = 6‐8). Data are presented as mean ± SEM. **P ≤ 0.01 vs control by Aspin–Welch test or Student's t‐test. #P ≤ 0.025 vs the vehicle‐treated group by one‐tailed Williams’ test. §P ≤ 0.025 vs the vehicle‐treated group by one‐tailed Williams’ test; (G) Activation of direct and indirect pathway MSNs by TAK‐063 (1 mg/kg p.o.) in the rat striatum (n = 4‐5). Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01 vs vehicle by Student's t‐test. SEM, standard error of mean. Reproduced from Harada et al,44 and Suzuki et al35
To ensure target engagement to PDE10A in rodent brain after oral administration, PDE10A occupancy of TAK‐063 in the rat brain was measured using T‐773, a PDE10A selective PET tracer,45 as a ligand. TAK‐063 was estimated to produce 50% PDE10A occupancy at 0.49 and 0.88 mg/kg p.o. in mice and rats, respectively.38, 44 PDE10A inhibition and the resulting elevation of cAMP and cGMP levels (Figure 1C,D) are known to activate cAMP‐dependent protein kinase (PKA) and cGMP‐dependent PKG. Activated PKA and PKG increase the phosphorylation of key substrates, such as cAMP response element‐binding protein (CREB) and (±)‐α‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐proprionic acid (AMPA) receptor GluR1 subunit. To evaluate in vivo PDE10A inhibition by TAK‐063, the effects of TAK‐063 on cAMP and cGMP levels and phosphorylation levels of CREB and GluR1 were assessed. Oral administration of TAK‐063 increased cAMP and cGMP levels in the mouse and rat striatum in a dose‐dependent manner. The minimum effective dose of TAK‐063 for the upregulation of cAMP and cGMP in the mouse striatum was 0.3 mg/kg p.o.35 TAK‐063 dose‐dependently increased phosphorylation levels of CREB and GluR1 in the mouse striatum, in the same dose range of TAK‐063 that upregulated striatal cAMP and cGMP levels.35 Immunohistochemical study also revealed that TAK‐063 activated both direct and indirect pathway to a similar extent; TAK‐063 at 0.3 and 3 mg/kg p.o. increased in the number of c‐Fos expression immunoreactive cells, as a marker of neural activation, in both direct and indirect pathway MSNs.46 These results suggest that TAK‐063 inhibited PDE10A activity and activated downstream cAMP and cGMP signals in both pathway MSNs in the striatum.
Hyperactivity induced by MK‐801, an N‐methyl‐D‐aspartate receptor (NMDAR) antagonist, is used as a model for acute psychosis based on the NMDAR hypofunction hypothesis of schizophrenia47 and is often applied to evaluate the antipsychotic‐like effects of PDE10A inhibitors in rodents.48, 49 TAK‐063 (0.3 mg/kg p.o.) produced >70% suppression of MK‐801‐induced hyperactivity in mice and rats at doses that resulted in 26% and 38% PDE10A occupancy, respectively (Figure 1E,F).38 To assess whether the pharmacological effects of TAK‐063 are altered after chronic treatment, we examined striatal molecular marker responses and antipsychotic‐like effects of TAK‐063 after repeated administration in mice. Upregulation of striatal cAMP/cGMP levels and the antipsychotic‐like effect of TAK‐063 (0.5 mg/kg p.o.) were not attenuated after 15 days of pretreatment with TAK‐063 in mice.35 These effects did not differ from those seen after administration of a single dose. These results suggest that the antipsychotic‐like effects by TAK‐063 are maintained after repeated administration.
The potential side effects profile of TAK‐063 was characterized in rats using the current antipsychotics haloperidol, olanzapine, and aripiprazole as controls. TAK‐063 did not affect plasma prolactin or glucose levels at doses up to 3 mg/kg p.o. TAK‐063 at 3 mg/kg p.o. elicited a weak cataleptic response compared with haloperidol and olanzapine.35 Overall, TAK‐063 showed superior profile compared with these antipsychotics in rats.
5. COMBINATION STUDY OF TAK‐063 AND ANTIPSYCHOTICS IN RODENTS
Activation of indirect pathway MSNs via promotion of cAMP production is the principal mechanism of action of current antipsychotics with D2 receptor antagonism. We confirmed that TAK‐063 activated both direct and indirect pathway MSNs by measuring the induction of genes as pathway‐specific markers: enkephalin for the indirect pathway and substance P for the direct pathway (Figure 1G).35 The activation of indirect pathway MSNs through the distinct mechanism of action of these drugs raises the possibility of augmented pharmacological effects by combination therapy. We evaluated the potential of combination therapy with TAK‐063 and current antipsychotics, such as haloperidol or olanzapine. Combined treatment with TAK‐063 (0.1 mg/kg p.o.) and either haloperidol (0.3 mg/kg p.o.) or olanzapine (3 mg/kg p.o.) at subeffective doses produced a potent antipsychotic‐like effect on MK‐801‐induced hyperactivity in rats. TAK‐063 at this dose did not exacerbate plasma prolactin levels and cataleptic response from antipsychotics in rats. Thus, TAK‐063 may produce augmented antipsychotic‐like activities in combination with antipsychotics without exacerbating side effects in rodents.50
6. EFFECT OF TAK‐063 ON CORTICAL ACTIVITY
The striatum and frontal cortex are highly interconnected via neuronal circuitry.51, 52 Augmented striatal outputs following inhibition of PDE10A may modulate cortical activity. To evaluate the effects of TAK‐063 on cortical activity, electroencephalography (EEG) and pharmacological magnetic resonance imaging (phMRI) studies were conducted. NMDAR antagonist ketamine‐induced increase in gamma power was dose dependently and significantly suppressed by TAK‐063 in rats and monkeys.53 In phMRI study, TAK‐063 (0.3 and 3 mg/kg i.p.) inhibited the cortical increases in blood oxygenation level‐dependent (BOLD) signal induced by ketamine. Reduction in this cortical activation is consistent with findings in EEG studies and indicates an effect of TAK‐063 on frontal cortical functions. Immunohistochemical study also supports this idea: TAK‐063 at 0.3 and 3 mg/kg p.o. increased c‐Fos expression in subregions of the medial prefrontal cortex, anterior cingulate cortex, and prelimbic cortex in rats where the specific binding of [3H]TAK‐063 was not observed in the ARG study.46, 53 These data suggest that TAK‐063 modulates activities of cortical regions through activation of neuronal circuits in rats and monkeys.
7. EFFECTS OF TAK‐063 ON COGNITIVE FUNCTIONS IN RODENTS
Activation of direct pathway MSNs has been considered to be associated with cognitive functions via striato‐thalamo‐cortical circuit.17, 18, 19, 20 We evaluated TAK‐063 in various cognitive domains using naïve and drug‐perturbed rodents. In naïve rats, TAK‐063 at 0.1 and 0.3 mg/kg p.o. improved time‐dependent memory decay in object recognition. TAK‐063 at these doses also increased accuracy rate, and TAK‐063 at 0.3 mg/kg p.o. reduced impulsivity in a five‐choice serial reaction time task in naïve rats. NMDAR antagonists such as phencyclidine and MK‐801 were used to induce working memory deficits relevant to schizophrenia in animals.54 TAK‐063 at 0.3 mg/kg p.o. attenuated both phencyclidine‐induced working memory deficits in a Y‐maze test in mice and MK‐801‐induced working memory deficits in an eight‐arm radial maze task in rats. An attentional set‐shifting task using subchronic phencyclidine‐treated rats was used to assess the executive function.55 TAK‐063 at 0.3 mg/kg p.o. reversed cognitive deficits in extradimensional shifts.42 These findings suggest that TAK‐063 has a potential to ameliorate deficits in multiple cognitive domains impaired in schizophrenia. Pro‐cognitive effects of TAK‐063 in these domains were observed at around 0.3 mg/kg p.o. TAK‐063 at this dose also showed a potent antipsychotic‐like effect in rodents.35 Thus, TAK‐063 may improve multiple symptomatic domains, that is, positive symptoms and cognitive deficits, in schizophrenia at similar dose levels.
8. BALANCED ACTIVATION OF DIRECT AND INDIRECT PATHWAYS BY TAK‐063 IS ASSOCIATED WITH EFFICACY IN PPI AND METH‐INDUCED HYPERACTIVITY
Unlike current antipsychotics, MP‐10 did not show antipsychotic‐like effects in widely used animal models, for example, MP‐10 did not suppress methamphetamine (METH)‐induced hyperactivity and did not improve prepulse inhibition (PPI) in rodents (Figure 2A,B).38 Using the same experimental protocols, we confirmed that TAK‐063 produced significant antipsychotic‐like effects in these experimental paradigms (Figure 2A,B),38 although both TAK‐063 and MP‐10 can increase cellular cAMP/cGMP content in the striatum and suppress hyperactivity induced by MK‐801 in rodents.
Figure 2.

(A) Antipsychotic‐like effects of haloperidol, TAK‐063, MP‐10, and compound 1 on methamphetamine (METH)‐induced hyperactivity in rats (n = 4‐6) and (B) low prepulse inhibition in C57BL/6J mice (n = 6‐15). Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01 vs control by Aspin–Welch test. #P ≤ 0.05 vs the vehicle‐treated group by two‐tailed Williams’ test. §P ≤ 0.05 vs the vehicle‐treated group by two‐tailed Shirley–Williams test; (C) Effect of SKF82958 (SKF, 1, 3, and 10 mg/kg i.p.) on the antipsychotic‐like effect of haloperidol (HAL, 0.3 mg/kg p.o.) in METH‐induced hyperactivity in rats (n = 5). *P ≤ 0.05, **P ≤ 0.01 by Aspin–Welch test. #P ≤ 0.05 by two‐tailed Williams’ test; (D) Activation of the direct and indirect pathways by TAK‐063, MP‐10, and compound 1 (n = 6). Data are presented as mean ± SEM. §P ≤ 0.05 vs the vehicle‐treated group by two‐tailed Williams’ test for substance P (SP) expression. #P ≤ 0.05 vs the vehicle‐treated group by two‐tailed Williams’ test for enkephalin (Enk) expression; (E) Ratio of SP mRNA to Enk mRNA after PDE10A inhibition. PDE10A occupancy at each dose used is shown below the dose. The values in the graph represent expression ratio of SP to Enk (SP/Enk) relative to that of the vehicle‐treated group. Data are presented as mean ± SEM (n = 6). #P ≤ 0.05 vs the vehicle‐treated group by two‐tailed Williams’ test; (F) Effects of TAK‐063 and MP‐10 on dopamine release in the striatum of wild‐type (WT) and PDE10A knockout (KO) mice (n = 3‐5); (G) Off‐rate from phosphodiesterase 10A of TAK‐063, MP‐10, and compound 1, measured by in vitro autoradiography study (n = 3); (H) Comparison of sensitivity to binding inhibition by cyclic nucleotide monophosphates (cNMPs) between TAK‐063 and MP‐10 (n = 4). Data are presented as mean ± SEM. Data were analyzed by two‐way analysis of variance followed by Aspin–Welch test (**P ≤ 0.01). SEM, standard error of mean. Reproduced from Suzuki et al;38 (I) Schematic representation showing the hypothetical mechanisms of action underlying the different properties of fast and slow off‐rate PDE10A inhibitors. Fast off‐rate and competitive PDE10A inhibitor TAK‐063 was more sensitive than slow off‐rate competitive inhibitor MP‐10 to binding inhibition by cyclic nucleotides. In indirect pathway MSNs with lower levels of intracellular cAMP, the fast off‐rate inhibitor (TAK‐063) and the slow off‐rate inhibitors (MP‐10, compound 1) provide similar levels of PDE10A inhibition, that is, similar levels of enkephalin mRNA induction, while direct pathway MSNs with higher levels of intracellular cAMP, the fast off‐rate inhibitor (TAK‐063) shows milder PDE10A inhibition than that by the slow off‐rate inhibitors (MP‐10, compound 1), that is, different levels of substance P mRNA induction. Differential activation pattern of MSNs may also affect cortical activity through the striato‐thalamo‐cortical circuit. cAMP, cyclic adenosine monophosphate; DA, dopamine; Enk, enkephalin; GABA, gamma‐aminobutyric acid; GPe, external segment of globus pallidus; GPi, internal segment of globus pallidus; MSNs, medium spiny neurons; PDE10Ai, PDE10A inhibitor; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; SP, substance P; STN, subthalamic nucleus; VTA, ventral tegmental area
Given our concept that PDE10A inhibitors would produce similar antipsychotic‐like effects to those of current antipsychotics with better safety profiles based on D2 receptor antagonism plus D1 receptor agonism, this pharmacological profile of MP‐10 was contrary to our expectations. One of the major differences between PDE10A inhibition and D2 receptor antagonism is activation of the direct pathway MSNs. PDE10A inhibitors are known to activate both direct and indirect pathways.37, 46 These pathways are considered to have competing effects on movement.16 In fact, activation of the direct pathway by a D1 agonist SKF82958 reduced cataleptic response induced by a D2 antagonist haloperidol.35 Importantly, we found that excess activation of the direct pathway by SKF82958 also canceled the antipsychotic‐like effects of haloperidol in METH‐induced hyperactivity model in rats (Figure 2C).38 Thus, understanding of activation pattern of these pathways caused by PDE10A inhibitors is critical. Gene expression analysis of pathway‐specific markers suggested that TAK‐063 and MP‐10 activated the indirect pathway to similar extent; however, MP‐10 induced more excessive activation of the direct pathway than TAK‐063 did (Figure 2D).38 Calculation of the ratio of substance P mRNA to enkephalin mRNA after administration of these compounds (Figure 2E) also revealed that MP‐10, but not TAK‐063, induced a significant increase in the relative expression of substance P (substance P/enkephalin, P ≤ 0.05) at 85% and 95% occupancies (30 and 100 mg/kg, respectively). This excessive activation of the direct pathway by MP‐10 might counteract its pharmacological effects of D2 antagonism.
We also observed that SKF82958, MP‐10, but not TAK‐063, induced dopamine release in the rat striatum and that the MP‐10‐induced striatal dopamine release was absent in PDE10A knockout mice (Figure 2F). Thus, dopamine release by excessive activation of the direct pathway may be associated with pharmacological profiles of these PDE10A inhibitors.
To gain insight into the difference between TAK‐063 and MP‐10, we assessed the binding kinetics of these compounds. ARG studies revealed that TAK‐063 showed faster off‐rate from PDE10A in rat brain sections compared with MP‐10 (Figure 2G). D2 receptors are coupled with Gi and inhibit adenylate cyclase in the indirect pathway, whereas D1 receptors, predominantly expressed in the direct pathway, are coupled with Gs and stimulate adenylate cyclase. Thus, when stimulated by dopamine, cAMP levels in the direct pathway can be higher than those in the indirect pathway. In fact, an immunohistochemistry study suggested that cAMP levels in the direct pathway were higher than those in the indirect pathway.38 Both TAK‐063 and MP‐10 bind to the substrate‐binding site in the catalytic domain of PDE10A; 43, 56 thus, cAMP and cGMP can compete with TAK‐063 and MP‐10 at this domain. Under conditions with higher intracellular cyclic nucleotide concentrations induced by PDE10A inhibition and/or dopamine increase, the binding of these compounds to PDE10A possibly differs depending on their off‐rates. In general, competitive enzyme inhibitors with faster off‐rates to the target enzyme are more readily inhibited by lower concentrations of enzyme substrates (ie, cyclic nucleotides for PDE10A) than those with slower off‐rates. In fact, TAK‐063 was more sensitive than MP‐10 to binding inhibition by higher concentrations of cyclic nucleotides in rat brain sections (Figure 2H), although TAK‐063 and MP‐10 had similar binding affinity to PDE10A; Ki values of TAK‐063 and MP‐10 were 3.2 and 4.3 nmol/L, respectively.38 We hypothesized that when PDE10A occupancy is getting higher, the released dopamine stimulates the Gs‐coupled D1 receptor to produce cyclic nucleotide in the direct pathway. Under the conditions with higher concentrations of cyclic nucleotides, MP‐10, but not TAK‐063, causes sustained PDE10A inhibitory activity and inhibits the degradation of cyclic nucleotides. This synergistic upregulation of the cyclic nucleotide levels by slower off‐rate PDE10A inhibitors would lead to the excessive activation of the direct pathway. Supporting this idea, compound 1, a slower off‐rate PDE10A inhibitor from TAK‐063 chemical series, showed MP‐10‐like pharmacological profile (Figure 2).38 Pharmacological profiles of D2 antagonist haloperidol and PDE10A inhibitors are summarized in Table 1.
Table 1.
Pharmacological profiles of D2 antagonist haloperidol and PDE10A inhibitors
| D2 antagonist | PDE10A inhibitor | |||
|---|---|---|---|---|
| Haloperidol | TAK‐063 | MP‐10 | Compound 1 | |
| Off‐rate from PDE10A | N.D. | Fast | Slow | Slow |
| Activation of direct pathway MSNs | − | + | ++ | ++ |
| Activation of indirect pathway MSNs | + | + | + | + |
| Striatal dopamine release in rodents | N.D. | − | + | + |
| Effect on METH‐induced hyperactivity in rodents | + | + | − | − |
| Effect on low PPI in C57BL/6 mice | + | + | − | − |
−, no significant effect; +, significant effect; ++, significant effect on substance P mRNA induction and significant increase in the expression ratio of substance P mRNA to enkephalin mRNA (substance P/enkephalin); MSN, medium spiny neuron; N.D., not determined; METH, methamphetamine; PPI, prepulse inhibition. Data from Suzuki et al.38
Taken together, off‐rate from PDE10A may characterize the pharmacological profile of PDE10A inhibitors. PDE10A selective inhibitors with slower off‐rate may cause excessive activation of the direct pathway, and this excessive activation in turn may cancel antipsychotic‐like effects produced by their D2 antagonism. Besides antipsychotic‐like effects, differential activation pattern of MSNs may also affect other pharmacological properties of PDE10A inhibitors. Interestingly, different from TAK‐063, MP‐10 did not significantly inhibit ketamine‐induced increase in EEG gamma power and did not produced cognitive improvement in novel object recognition task in rats.37, 42, 53 Differential activation pattern of MSNs may affect the modulation of cortical activity by PDE10A through the striato‐thalamo‐cortical circuit. TAK‐063, with a balanced activation of direct and indirect pathways through faster off‐rate, would provide a unique opportunity for further evaluation of PDE10A inhibition as a therapeutic strategy for schizophrenia.
9. THE PHYSIOLOGICAL ROLE OF PDE10A AND ITS RELEVANCE IN BASAL GANGLIA DISORDERS
Besides schizophrenia, the basal ganglia circuit is involved in a variety of neurological diseases like neurodegenerative diseases, such as HD and PD, functional impairment after striatal stroke, the movement disorders, and also psychiatric conditions like addiction.57 Accumulating preclinical and clinical evidence supports that PDE10A could be a potential therapeutic target for diseases of the basal ganglia.58
In patients with HD and PD, loss of PDE10A levels associated with disease progression and severity was demonstrated with a PDE10A‐specific PET tracer.59, 60, 61, 62 Impairment of cyclic nucleotide signaling has been observed during disease progression of HD and PD, and restoration of this signaling pathway is under investigation as a potential treatment paradigm for these conditions.63 For example, activation of CREB has been suggested to mediate the resistance to cell death in particular neuronal populations in animal models of HD via brain‐derived neurotrophic factor (BDNF) production.64 Supporting the hypothesis of a beneficial effect of elevated cAMP/cGMP levels on neural protection, PDE10A inhibition by TAK‐063 and TP‐10 (developed by Pfizer)34 showed beneficial effects in the R6/2 mouse model of HD,64, 65 whereas Roche's PDE10A selective inhibitor did not lead to disease modifying effects in this mouse model (Prinssen et al, presented at CHDI's Annual HD Therapeutics Conference 2017). Regulation of neural network and its underlying molecular mechanism is complex; thus, pharmacological profile of compounds targeting the same molecule could be different. Careful consideration of the environment around the target and characterization of detailed properties of compounds would be needed for more understanding of their therapeutic potential. In addition to HD, PDE10A has been also discussed as a therapeutic target in PD.66 The efficacy of PDE10A inhibitors for reversing L‐DOPA‐induced dyskinesia in a rat model of PD was recently reported.67 Even though changes in PDE10A levels are observed in HD and PD, it is not clear whether loss of PDE10A is part of the underlying pathology that is causative for the development of the disease or if it is an adaptive change due to reduced cyclic nucleotide levels. Further studies are needed to evaluate a potential beneficial effect of PDE10A inhibition in HD and PD.
Recovery of function after stroke involves molecular signaling events that are mediated by cAMP and cGMP pathways, including axonal sprouting, dendritic spine plasticity, and neurogenesis.68 Recent study demonstrated that inhibition of PDE10A by TAK‐063 improved recovery of function after striatal but not cortical stroke, which was probably associated with induction of BDNF in the affected striatum (Birjandi et al, presented at Society for Neuroscience Annual Meeting 2016, Poster 607.20/AA8).
Interestingly, PDE10A is also genetically implicated in movement disorders. Recent human genetic studies reported that mutations in PDE10A caused childhood‐onset chorea.69, 70 These studies independently found evidence that nonsynonymous mutations in the human PDE10A gene underlie a hyperkinetic movement disorder. cAMP signaling in MSNs is crucial for normal activity of basal ganglia circuitry and that disruptions thereof play an important role in the pathophysiology of movement disorders.
The nucleus accumbens, as part of the ventral striatum, is well characterized for its influence mediating reward circuitry and addiction‐related behavior.71 Thus, PDE10A inhibition has been suspected to influence reward behavior and proposed to be beneficial in the management of substance abuse and addictions. In line with this idea, PDE10A inhibitors such as MP‐10 and TP‐10 were reported to prevent morphine‐ and alcohol‐induced addictive behaviors, respectively, in rats.72, 73
10. PHARMACOLOGICAL PROFILES OF TAK‐063 IN CLINICAL STUDIES
The phase 1 clinical studies for TAK‐063 instituted a comprehensive translational development strategy based on preclinical findings. Clinical results were generally consistent with those of the preclinical studies. The main findings in humans included confirmation of target engagement in the PET study,74 the pharmacological effects of TAK‐063 on fMRI BOLD (ketamine‐challenge), and pharmacodynamic effects on EEG and cognition in subjects with schizophrenia (Macek et al, presented at the 5th Biennial Schizophrenia International Research Society Conference 2016). A recent phase 2 study evaluated the efficacy and safety of 20‐mg daily TAK‐063 vs placebo in subjects with acutely exacerbated symptoms of schizophrenia (ClinicalTrials.gov Identifier: NCT02477020). TAK‐063 was safe and well tolerated. Although the primary endpoint was not met in the study, change from baseline in the Positive and Negative Syndrome Scale total score at week 6, the effects of TAK‐063 on secondary endpoints, such as changes in Clinical Global Impression‐Severity scores, and the standardized effect size of 0.308 were suggestive of antipsychotic activity (Macek et al, presented at American Society of Clinical Psychopharmacology Annual Meeting 2017, Poster W63). Although the interpretation of these results is confounded by a relatively high placebo effect, a lack of dose ranging, and an active reference, TAK‐063 exhibited some evidence of efficacy in patients with an acute exacerbation of schizophrenia. Further clinical studies are needed to evaluate the full potential of TAK‐063 for the treatment of schizophrenia.
11. CONCLUSIONS
Herein we have outlined the pharmacological profile of TAK‐063, a highly potent, selective, and orally active PDE10A inhibitor. TAK‐063, but not other PDE10A selective inhibitors such as MP‐10 and compound 1, produced potent antipsychotic‐like effects in multiple rodent models. These findings suggest that the antipsychotic‐like efficacy of TAK‐063 may be attributable to its unique pharmacological properties, resulting in balanced activation of the direct and indirect striatal pathways (Figure 2I). In addition to antipsychotic‐like efficacy, TAK‐063 enhanced cognitive function associated with schizophrenia in rodent models.42 TAK‐063 may provide an opportunity for further evaluation of PDE10A inhibition as a novel therapeutic strategy for psychosis and cognitive deficits in schizophrenia. Besides schizophrenia, recent preclinical and clinical studies support additional therapeutic potentials of PDE10A inhibitors in several basal ganglia disorders.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank Akina Harada for helpful comments regarding the content and organization of this manuscript. This study was funded by Takeda Pharmaceutical Company Limited. The authors are employees of Takeda Pharmaceutical Company Limited. The contents of Figure 1 were reproduced with permission from Refs 35 and 44, and the contents of Figure 2 were reproduced with permission from Ref. 38.
Suzuki K, Kimura H. TAK‐063, a novel PDE10A inhibitor with balanced activation of direct and indirect pathways, provides a unique opportunity for the treatment of schizophrenia. CNS Neurosci Ther. 2018;24:604–614. 10.1111/cns.12798
REFERENCES
- 1. van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635‐645. [DOI] [PubMed] [Google Scholar]
- 2. Sharma T, Antonova L. Cognitive function in schizophrenia. Deficits, functional consequences, and future treatment. Psychiatr Clin North Am. 2003;26:25‐40. [DOI] [PubMed] [Google Scholar]
- 3. Harvey PD, Green MF, Keefe RS, Velligan DI. Cognitive functioning in schizophrenia: a consensus statement on its role in the definition and evaluation of effective treatments for the illness. J Clin Psychiatry. 2004;65:361‐372. [PubMed] [Google Scholar]
- 4. Green MF, Kern RS, Heaton RK. Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res. 2004;72:41‐51. [DOI] [PubMed] [Google Scholar]
- 5. Toda M, Abi‐Dargham A. Dopamine hypothesis of schizophrenia: making sense of it all. Curr Psychiatry Rep. 2007;9:329‐336. [DOI] [PubMed] [Google Scholar]
- 6. Abi‐Dargham A, Rodenhiser J, Printz D, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A. 2000;97:8104‐8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krebs M, Leopold K, Hinzpeter A, Schaefer M. Current schizophrenia drugs: efficacy and side effects. Expert Opin Pharmacother. 2006;7:1005‐1016. [DOI] [PubMed] [Google Scholar]
- 8. Fujishige K, Kotera J, Omori K. Striatum‐ and testis‐specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur J Biochem. 1999;266:1118‐1127. [DOI] [PubMed] [Google Scholar]
- 9. Fujishige K, Kotera J, Michibata H, et al. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J Biol Chem. 1999;274:18438‐18445. [DOI] [PubMed] [Google Scholar]
- 10. Soderling SH, Bayuga SJ, Beavo JA. Isolation and characterization of a dual‐substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci U S A. 1999;96:7071‐7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soderling SH, Beavo JA. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr Opin Cell Biol. 2000;12:174‐179. [DOI] [PubMed] [Google Scholar]
- 12. Seeger TF, Bartlett B, Coskran TM, et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985:113‐126. [DOI] [PubMed] [Google Scholar]
- 13. Xie Z, Adamowicz WO, Eldred WD, et al. Cellular and subcellular localization of PDE10A, a striatum‐enriched phosphodiesterase. Neuroscience. 2006;139:597‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Siuciak JA, McCarthy SA, Chapin DS, et al. Genetic deletion of the striatum‐enriched phosphodiesterase PDE10A: evidence for altered striatal function. Neuropharmacology. 2006;51:374‐385. [DOI] [PubMed] [Google Scholar]
- 15. Siuciak JA, Chapin DS, Harms JF, et al. Inhibition of the striatum‐enriched phosphodiesterase PDE10A: a novel approach to the treatment of psychosis. Neuropharmacology. 2006;51:386‐396. [DOI] [PubMed] [Google Scholar]
- 16. Obeso JA, Rodríguez‐Oroz MC, Rodríguez M, Arbizu J, Giménez‐Amaya JM. The basal ganglia and disorders of movement: pathophysiological mechanisms. News Physiol Sci. 2002;17:51‐55. [DOI] [PubMed] [Google Scholar]
- 17. Graybiel AM. The basal ganglia. Curr Biol. 2000;10:R509‐R511. [DOI] [PubMed] [Google Scholar]
- 18. Hikida T, Kimura K, Wada N, Funabiki K, Nakanishi S. Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron. 2010;66:896‐907. [DOI] [PubMed] [Google Scholar]
- 19. Darvas M, Palmiter RD. Specific contributions of N‐methyl‐d‐aspartate receptors in the dorsal striatum to cognitive flexibility. Neuroscience. 2015;284:934‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366‐375. [DOI] [PubMed] [Google Scholar]
- 21. Perez‐Costas E, Melendez‐Ferro M, Roberts RC. Basal ganglia pathology in schizophrenia: dopamine connections and anomalies. J Neurochem. 2010;113:287‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bertran‐Gonzalez J, Bosch C, Maroteaux M, et al. Opposing patterns of signaling activation in dopamine D1 and D2 receptor‐expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671‐5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1081‐1090. [DOI] [PubMed] [Google Scholar]
- 24. Kapur S, Remington G, Jones C, et al. High levels of dopamine D2 receptor occupancy with low‐dose haloperidol treatment: a PET study. Am J Psychiatry. 1996;153:948‐950. [DOI] [PubMed] [Google Scholar]
- 25. Nordstrom AL, Farde L, Wiesel FA, et al. Central D2‐dopamine receptor occupancy in relation to antipsychotic drug effects: a double‐blind PET study of schizophrenic patients. Biol Psychiatry. 1993;33:227‐235. [DOI] [PubMed] [Google Scholar]
- 26. Pani L, Pira L, Marchese G. Antipsychotic efficacy: relationship to optimal D2‐receptor occupancy. Eur Psychiatry. 2007;22:267‐275. [DOI] [PubMed] [Google Scholar]
- 27. Stoof JC, Kebabian JW. Opposing roles for D‐1 and D‐2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature. 1981;294:366‐368. [DOI] [PubMed] [Google Scholar]
- 28. Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov. 2006;5:660‐670. [DOI] [PubMed] [Google Scholar]
- 29. Tam GW, van de Lagemaat LN, Redon R, et al. Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochem Soc Trans. 2010;38:445‐451. [DOI] [PubMed] [Google Scholar]
- 30. McDonald ML, MacMullen C, Liu DJ, Leal SM, Davis RL. Genetic association of cyclic AMP signaling genes with bipolar disorder. Transl Psychiatry. 2012;2:e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kerner B, Lambert CG, Muthen BO. Genome‐wide association study in bipolar patients stratified by co‐morbidity. PLoS ONE. 2011;6:e28477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sano H, Nagai Y, Miyakawa T, Shigemoto R, Yokoi M. Increased social interaction in mice deficient of the striatal medium spiny neuron‐specific phosphodiesterase 10A2. J Neurochem. 2008;105:546‐556. [DOI] [PubMed] [Google Scholar]
- 33. Marques TR, Natesan S, Niccolini F, et al. Phosphodiesterase 10A in Schizophrenia: A PET Study Using [11C]IMA107. Am J Psychiatry. 2016;173:714‐721. [DOI] [PubMed] [Google Scholar]
- 34. Schmidt CJ, Chapin DS, Cianfrogna J, et al. Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther. 2008;325:681‐690. [DOI] [PubMed] [Google Scholar]
- 35. Suzuki K, Harada A, Shiraishi E, Kimura H. In vivo pharmacological characterization of TAK‐063, a potent and selective phosphodiesterase 10A inhibitor with antipsychotic‐like activity in rodents. J Pharmacol Exp Ther. 2015;352:471‐479. [DOI] [PubMed] [Google Scholar]
- 36. Arakawa K, Nakao K, Maehara S. Dopamine D1 signaling involvement in the effects of the phosphodiesterase 10A inhibitor, PDM‐042 on cognitive function and extrapyramidal side effect in rats. Behav Brain Res. 2017;317:204‐209. [DOI] [PubMed] [Google Scholar]
- 37. Grauer SM, Pulito VL, Navarra RL, et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther. 2009;331:574‐590. [DOI] [PubMed] [Google Scholar]
- 38. Suzuki K, Harada A, Suzuki H, Miyamoto M, Kimura H. TAK‐063, a PDE10A inhibitor with balanced activation of direct and indirect pathways, provides potent antipsychotic‐like effects in multiple paradigms. Neuropsychopharmacology. 2016;41:2252‐2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones PG, Hewitt MC, Campbell JE, et al. Pharmacological evaluation of a novel phosphodiesterase 10A inhibitor in models of antipsychotic activity and cognition. Pharmacol Biochem Behav. 2015;135:46‐52. [DOI] [PubMed] [Google Scholar]
- 40. Rodefer JS, Murphy ER, Baxter MG. PDE10A inhibition reverses subchronic PCP‐induced deficits in attentional set‐shifting in rats. Eur J Neurosci. 2005;21:1070‐1076. [DOI] [PubMed] [Google Scholar]
- 41. Smith SM, Uslaner JM, Cox CD, et al. The novel phosphodiesterase 10A inhibitor THPP‐1 has antipsychotic‐like effects in rat and improves cognition in rat and rhesus monkey. Neuropharmacology. 2013;64:215‐223. [DOI] [PubMed] [Google Scholar]
- 42. Shiraishi E, Suzuki K, Harada A, Suzuki N, Kimura H. The phosphodiesterase 10A selective inhibitor TAK‐063 improves cognitive functions associated with schizophrenia in rodent models. J Pharmacol Exp Ther. 2016;356:587‐595. [DOI] [PubMed] [Google Scholar]
- 43. Kunitomo J, Yoshikawa M, Fushimi M, et al. Discovery of 1‐[2‐Fluoro‐4‐(1H‐pyrazol‐1‐yl)phenyl]‐5‐methoxy‐3‐(1‐phenyl‐1H‐pyrazol‐5‐yl)pyri dazin‐4(1H)‐one (TAK‐063), a highly potent, selective, and orally active phosphodiesterase 10A (PDE10A) Inhibitor. J Med Chem. 2014;57:9627‐9643. [DOI] [PubMed] [Google Scholar]
- 44. Harada A, Suzuki K, Kamiguchi N, et al. Characterization of binding and inhibitory properties of TAK‐063, a novel phosphodiesterase 10A inhibitor. PLoS ONE. 2015;10:e0122197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harada A, Suzuki K, Miura S, et al. Characterization of the binding properties of T‐773 as a PET radioligand for phosphodiesterase 10A. Nucl Med Biol. 2015;42:146‐154. [DOI] [PubMed] [Google Scholar]
- 46. Nakatani A, Nakamura S, Kimura H. The phosphodiesterase 10A selective inhibitor, TAK‐063, induces c‐Fos expression in both direct and indirect pathway medium spiny neurons and sub‐regions of the medial prefrontal cortex in rats. Neurosci Res. 2017;125:29‐36. [DOI] [PubMed] [Google Scholar]
- 47. Forrest AD, Coto CA, Siegel SJ. Animal models of psychosis: current state and future directions. Curr Behav Neurosci Rep. 2014;1:100‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Malamas MS, Stange H, Schindler R, et al. Novel triazines as potent and selective phosphodiesterase 10A inhibitors. Bioorg Med Chem Lett. 2012;22:5876‐5884. [DOI] [PubMed] [Google Scholar]
- 49. Hofgen N, Stange H, Schindler R, et al. Discovery of imidazo[1,5‐a]pyrido[3,2‐e]pyrazines as a new class of phosphodiesterase 10A inhibitors. J Med Chem. 2010;53:4399‐4411. [DOI] [PubMed] [Google Scholar]
- 50. Suzuki K, Harada A, Suzuki H, et al. Combined treatment with a selective PDE10A inhibitor TAK‐063 and either haloperidol or olanzapine at subeffective doses produces potent antipsychotic‐like effects without affecting plasma prolactin levels and cataleptic responses in rodents. Pharmacol Res Perspect. 2018;6:e00372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci. 2013;14:278‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haber SN. The primate basal ganglia: parallel and integrative networks. J Chem Neuroanat. 2003;26:317‐330. [DOI] [PubMed] [Google Scholar]
- 53. Tomimatsu Y, Cash D, Suzuki M, et al. TAK‐063, a phosphodiesterase 10A inhibitor, modulates neuronal activity in various brain regions in phMRI and EEG studies with and without ketamine challenge. Neuroscience. 2016;339:180‐190. [DOI] [PubMed] [Google Scholar]
- 54. Coyle JT. NMDA receptor and schizophrenia: a brief history. Schizophr Bull. 2012;38:920‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McLean SL, Beck JP, Woolley ML, Neill JC. A preliminary investigation into the effects of antipsychotics on sub‐chronic phencyclidine‐induced deficits in attentional set‐shifting in female rats. Behav Brain Res. 2008;189:152‐158. [DOI] [PubMed] [Google Scholar]
- 56. Verhoest PR, Chapin DS, Corman M, et al. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2‐[4‐(1‐methyl‐4‐pyridin‐4‐yl‐1H‐pyrazol‐3‐yl)‐phenoxymethyl]‐quinoline (PF‐2545920) for the treatment of schizophrenia. J Med Chem. 2009;52:5188‐5196. [DOI] [PubMed] [Google Scholar]
- 57. DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol. 2007;64:20‐24. [DOI] [PubMed] [Google Scholar]
- 58. Wilson LS, Brandon NJ. Emerging biology of PDE10A. Curr Pharm Des. 2015;21:378‐388. [DOI] [PubMed] [Google Scholar]
- 59. Pagano G, Niccolini F, Politis M. Current status of PET imaging in Huntington's disease. Eur J Nucl Med Mol Imaging. 2016;43:1171‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ahmad R, Bourgeois S, Postnov A, et al. PET imaging shows loss of striatal PDE10A in patients with Huntington disease. Neurology. 2014;82:279‐281. [DOI] [PubMed] [Google Scholar]
- 61. Niccolini F, Haider S, Reis Marques T, et al. Altered PDE10A expression detectable early before symptomatic onset in Huntington's disease. Brain. 2015;138:3016‐3029. [DOI] [PubMed] [Google Scholar]
- 62. Niccolini F, Foltynie T, Reis Marques T, et al. Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson's disease. Brain. 2015;138:3003‐3015. [DOI] [PubMed] [Google Scholar]
- 63. Threlfell S, West AR. Review: modulation of striatal neuron activity by cyclic nucleotide signaling and phosphodiesterase inhibition. Basal Ganglia. 2013;3:137‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Giampà C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS ONE. 2010;5:e13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Harada A, Suzuki K, Kimura H. TAK‐063, a novel phosphodiesterase 10A inhibitor, protects from striatal neurodegeneration and ameliorates behavioral deficits in the R6/2 mouse model of Huntington's disease. J Pharmacol Exp Ther. 2017;360:75‐83. [DOI] [PubMed] [Google Scholar]
- 66. Garcia AM, Redondo M, Martinez A, et al. Phosphodiesterase 10 inhibitors: new disease modifying drugs for Parkinson's disease? Curr Med Chem. 2014;21:1171‐1187. [DOI] [PubMed] [Google Scholar]
- 67. Padovan‐Neto FE, West AR. Regulation of striatal neuron activity by cyclic nucleotide signaling and phosphodiesterase inhibition: implications for the treatment of Parkinson's disease. Adv Neurobiol. 2017;17:257‐283. [DOI] [PubMed] [Google Scholar]
- 68. Kehler J, Nielsen J. PDE10A inhibitors: novel therapeutic drugs for schizophrenia. Curr Pharm Des. 2011;17:137‐150. [DOI] [PubMed] [Google Scholar]
- 69. Diggle CP, Sukoff Rizzo SJ, Popiolek M, et al. Biallelic mutations in PDE10A Lead to loss of striatal PDE10A and a hyperkinetic movement disorder with onset in infancy. Am J Hum Genet. 2016;98:735‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mencacci NE, Kamsteeg EJ, Nakashima K, et al. De novo mutations in PDE10A cause childhood‐onset chorea with bilateral striatal lesions. Am J Hum Genet. 2016;98:763‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mu Y, Ren Z, Jia J, et al. Inhibition of phosphodiesterase10A attenuates morphine‐induced conditioned place preference. Mol Brain. 2014;7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Logrip ML, Vendruscolo LF, Schlosburg JE, Koob GF, Zorrilla EP. Phosphodiesterase 10A regulates alcohol and saccharin self‐administration in rats. Neuropsychopharmacology. 2014;39:1722‐1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Takano A, Stenkrona P, Stepanov V, et al. A human [11C]T‐773 PET study of PDE10A binding after oral administration of TAK‐063, a PDE10A inhibitor. NeuroImage. 2016;141:10‐17. [DOI] [PubMed] [Google Scholar]