

Summary
Chronic cerebral circulation insufficiency (CCCI) may not be an independent disease; rather, it is a pervasive state of long‐term cerebral blood flow insufficiency caused by a variety of etiologies, and considered to be associated with either occurrence or recurrence of ischemic stroke, vascular cognitive impairment, and development of vascular dementia, resulting in disability and mortality worldwide. This review summarizes the features and recent progress of CCCI, mainly focusing on epidemiology, experimental research, pathophysiology, etiology, clinical manifestations, imaging presentation, diagnosis, and potential therapeutic regimens. Some research directions are briefly discussed as well.
Keywords: chronic cerebral circulation insufficiency, clinical manifestation, diagnosis, experimental research, treatment
1. INTRODUCTION
Cerebral circulation insufficiency consists of 2 types: acute, such as ischemic stroke and transient ischemic attack (TIA), and chronic, such as chronic cerebral circulation insufficiency (CCCI). The former has received great concerns, while the latter has not aroused enough attention so far. As these 2 types of brain circulation dysfunction can mutually be transformed into each other (Figure 1), it is imperative to gain a deep understanding about CCCI.
Figure 1.

Atherosclerotic stenosis–associated chronic cerebral circulation insufficiency. The classification, subtypes, and clinical consequences of intracranial and extracranial atherosclerotic stenosis–associated cerebral circulation insufficiency
CCCI, which was initially proposed by Japanese scholars in the 1990s,1 refers to a state of reduction in cerebral blood flow (CBF) below the physiologically required volume, leading to brain dysfunctions, and this state should, more often than not, last for at least 2 months. CCCI can be secondary to a variety of etiologies, with atherosclerosis predominating.1, 2, 3
Clinical studies have revealed that the symptoms of CCCI, such as dizziness and headache, are actually reversible once cerebral circulation is improved.2, 4 On the contrary, persistent CBF reduction, if not corrected, may evoke stroke, TIA, vascular cognitive impairment, or even dementia.1, 2, 3, 4 Therefore, identifying this condition followed by prompt and effective intervention is enormously valuable.
2. EPIDEMIOLOGY
According to the latest epidemiological statistics in the first Edition of the Annual Epidemiological Report in China 2016, CCCI carries a relatively high incidence in the elderly, occurring in two‐thirds of individuals older than 65 years of age. It is also observed in 50% of individuals aged from 50 to 65 years, and 25% aged from 45 to 50 years. However, the global prevalence of CCCI is undetermined at present.
3. ETIOLOGY
A plethora of factors have been proposed to compromise the blood flow in CCCI, increase susceptibility to this disease, or accelerate disease progression. Major etiologies can be summarized as follows:5, 6 (i) cerebrovascular pathological changes, mainly including vascular spasm, stenosis, or occlusion in either the vertebral or carotid artery system secondary to multiple factors such as atherosclerosis, vasculitic disease, moyamoya disease, and arteriovenous malformation; (ii) cardiovascular factors such as long‐standing hypertension or hypotension, and cerebral hypoperfusion induced by cardiac failure and arrhythmia; (iii) systemic diseases such as obstructive sleep apnea‐hypopnea syndrome (OSAHS), chronic obstructive pulmonary ventilation disorder, anemia, abnormal blood composition, chronic carbon monoxide poisoning, diabetes, smoking, and obesity.
The primary etiology of CCCI in Chinese elderly populations is atherosclerotic stenosis, especially intracranial stenosis, the prevalence of which is far more common than extracranial stenosis (37.5% for intracranial artery occlusive disease alone vs 4.9% for extracranial carotid occlusive artery alone).3 It is estimated that 33%‐50% stroke and more than 50% TIA in Chinese populations were caused by intracranial atherosclerosis.7 In addition, the stroke recurrence rate in these patients is as high as 25%‐30% within 2 years after the first stroke episode.8, 9, 10
4. EXPERIMENTAL RESEARCH
4.1. Animal models
4.1.1. The 2VO model
The two‐vessel occlusion (2VO) model of CCCI was first established by de la Torre JC et al who permanently ligated the bilateral common carotid arteries (CCA) in rats to induce chronic hypoperfusion of the forebrain.11 As the most commonly used model of CCCI, the 2VO model plays a significant role in diverse neuroscience studies such as white matter (WM) damage, neuronal degeneration in the hippocampus, learning and memory dysfunctions, and neural signal transmission disorders after CCCI.
4.1.2. The microcoils model
Shibata et al reported another model of CCCI using selected adult C57Bl/6 male mice subjected to bilateral CCA occlusion with a newly designed microcoil with an inner diameter of 0.16‐0.22 mm.9 Using this model, they successfully observed activation of microglia and astrocytes in the WM following CCCI. Since then, more mechanistic studies have used this approach to explore the pathophysiological mechanisms of CCCI. For instance, it was found that repair of WM damage was interfered by disruptive compensatory mechanisms of oligodendrocyte precursor cells to oligodendrocyte renewal secondary to oxidative stress during prolonged cerebral hypoperfusion stress induced by the microcoil with an inner diameter of 0.18 mm in a mice model, while astrocytes might facilitate oligodendrogenesis through secreting brain‐derived neurotrophic factor (BNDF).12, 13
Compared with the 2VO model, the microcoil model poses a less serious threat to the gray matter (GM) or the visual pathway and is appropriate for the maintenance of CCCI over a relatively long period of time. Nevertheless, the potential drawback of this model includes a rapid reduction in CBF by the microcoil insertion, which may not be consistent with the real clinical setting.
4.1.3. The cervical arteriovenous fistula model
On the basis of different ways of anastomoses between the cervical arteries and veins, end‐to‐end and end‐to‐side anastomosis models were established.14 However, the end‐to‐end model may raise intracranial venous sinus pressure and therefore has been rapidly replaced by the end‐to‐side anastomosis model. Hai et al utilized the end‐to‐side anastomosis model to explore the expression of vascular endothelial growth factor (VEGF) and angiogenesis after CCCI.15 It should be noted that the operation process of this model is complicated and its practical value remains unknown yet.
4.1.4. The rUCCAO model
Right unilateral common carotid artery occlusion (rUCCAO) is a novel experimental animal model developed on the basis of the 2VO model.16 This model also takes advantage of the dysplastic feature of the posterior communicating artery (PCA) in C57B/6 mice and can induce chronic ischemia by ligating the right CCA. Notably, several advantages render the model a promising tool for research investigation. First of all, the mortality rate of this model is very low and a persistent mild reduction in CBF can be guaranteed. In addition, it, to some extent, has an application value in studying the inflammatory reaction, cognitive impairment, and WM damage after CCCI. Meanwhile, it can help clarify the pathological mechanism of vascular dementia (VD), particularly subcortical ischemic dementia.
4.1.5. Combination of the 2VO model with other disease models
Generally, the realistic clinical scenario is quite complex and may not be confined to an isolated disease. Knowing that most patients with CCCI are usually concomitant with some other underlying chronic diseases such as hypertension, diabetes, and hyperlipidemia, researchers have developed animal models by combining the 2VO model with other chronic diseases to imitate clinical settings, thus enabling the obtained data to be more clinically valuable. Some classic examples are listed below:
The spontaneous hypertension rats (SHR) with CCCI: it was firstly reported by Masumura et al17 and is likely to be more meaningful for exploring the pathogenesis of WM lesions (WMLs) in CCCI.
The Alzheimer's disease (AD) transgenic mice (APP23) with bilateral CCA stenosis: it is used to investigate the association between AD and CCCI.18, 19 Compared with APP23 mice without CCCI, cerebral hypoperfusion aggravated cognitive impairment and motor dysfunctions with more accumulation of WMLs and deposition of meningo‐parenchymal amyloid‐β (Aβ).
The type II diabetes mellitus (T2DM) with bilateral CCA occlusion: Kwon et al selected the Otsuka Long‐Evans Tokushima Fatty (OLETF) rats as a T2DM model, which subsequently underwent permanent bilateral CCA occlusion to induce CCCI.20 The finding from this study revealed an unfavorable interaction between CCCI and T2DM, suggesting that metabolic diseases such as DM could augment cognitive dysfunction in a rat model of VD.
4.1.6. Behavioral cognitive functions and CCCI models
Experimental animal research has provided ample evidence that CCCI is strongly related to cognitive impairments. Several tests have been developed to evaluate behavioral cognitive changes in rodents, such as the water maze test, the T‐maze or Y‐maze test, and the radial arm maze test, the open field test, the novel object recognition test, and the social exploration test.21, 22 In general, the results of relevant animal models indicate alterations in reference learning, spatial working memory, visual or olfactory function, and perceptual learning at different time intervals from the induction of chronic hypoperfusion.23, 24, 25, 26 Notably, these behavioral and cognitive changes are likely to reflect the presence of intracranial pathologies such as disruptions of frontal‐subcortical circuits and axon‐glial integrity, proliferation of microglia, and loss of neuronal cells.
4.2. Pathophysiology
4.2.1. Relationship between CBF and brain function
Normal CBF is around 50‐55 mL/100 g brain tissue/min. A reduction in CBF to 40 mL/100 g brain tissue/min can result in the disruption of glucose utilization in the brain; when CBF drops to a level of <30 mL/100 g brain tissue/min, protein synthesis disorder occurs; once CBF declines to 25‐10 mL/100 g brain tissue/min, neuronal activity starts to become invalid, and neurological dysfunctions turn up, while still in the absence of permanent neuron damage.27, 28, 29 However, cell membrane failure may take place when CBF goes below 8 mL/100 g brain tissue/min, thus leading to cell death and permanent nerve necrosis (Figure 2).
Figure 2.
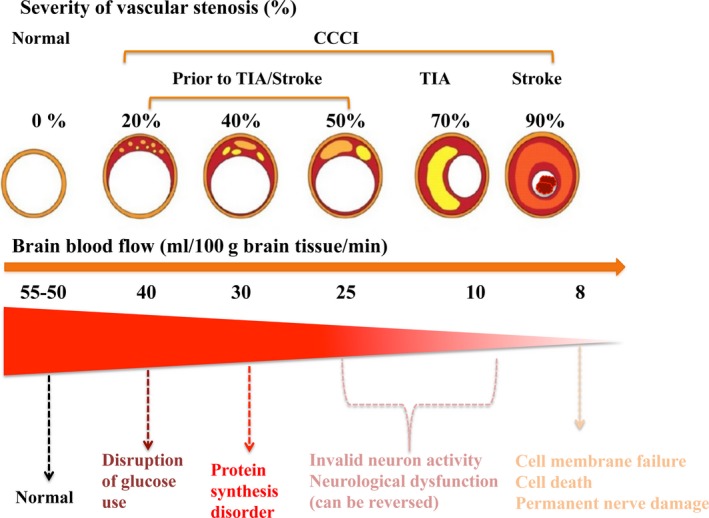
Severity of vascular stenosis, CBF reduction, and degree of neuronal damage. CCCI runs through the whole process of TIA/stroke. CBF around 50‐55ml/100 g brain tissue/min: normal; CBF <40 mL/100 g brain tissue/min: disruption of glucose utilization initiates; CBF <30 mL/100 g brain tissue/min: protein synthesis disorder occurs; reduction in CBF to 10‐25 mL/100 g brain tissue/min: neuronal activity becomes invalid and symptoms of neurological dysfunctions appear; CBF goes below 8 mL/100 g brain tissue/min: cell membrane failure occurs, followed by irreversible nerve damage or cell death. CCCI: chronic cerebral circulation insufficiency; TIA: transient ischemic stroke; CBF: cerebral blood flow
4.2.2. Pathological mechanism
CCCI is increasingly recognized as an alarming condition that can lead to a wide range of neurological deficits, especially neuronal damage and cognitive impairment. Nevertheless, the detailed mechanisms are not entirely clear. A plethora of remarkable pathological changes have been identified during the CCCI process, such as cerebral cortex atrophy, neuronal degeneration, WMLs, and glial cell proliferation.30 The evidence of oxidative stress, apoptosis, enhanced expression of inflammatory factors, and increased Aβ deposition in the brain tissue is also invariably observed.31 Furthermore, disruptive changes in various neurotransmitters such as acetylcholine (Ach), norepinephrine, serotonin, and gamma amino butyric acid (GABA) have been demonstrated as relatively specific features in the animal models of CCCI.32 The aforementioned changes may be responsible for the neurological dysfunctions, resulting in relevant clinical manifestations (Figure 3).
Figure 3.

Proposed pathological mechanisms resulting from chronic cerebral circulation insufficiency
White matter lesions
The WM is mainly composed of myelinated axons, the integrity of which is pivotal for rapid transmission of messages between different areas within the central nervous system (CNS). Although the WM constitutes up to 50% of the total human brain volume, it consumes only 1.7%‐3.6% of blood in nonelderly adults. The inadequate blood flow and scant collateral circulation in the deep WM render this milieu particularly vulnerable to ischemic insults, which jeopardize the function of the WM with unfavorable consequences. For instance, impairment of the WM tracts joining motor regions is considered to be accountable for gait disturbance in a subset of patients with CCCI‐related cerebral small‐vessel disease.33 Compared with the GM, the WM appears to be less vulnerable to ischemic insults, depicted by lower infarction threshold values for CBF, cerebral blood volume (CBF), and apparent diffusion coefficient, as well as slower evolution of ischemic penumbra.34, 35, 36, 37 This different vulnerability is likely attributable to the differing cellular constituents and neurochemical cascades between the WM and the GM. Accordingly, cerebral tissue‐specific thresholds may replace the whole‐brain tissue threshold to predict the likelihood of infarction and tailored strategies for both tissues are recommended.
WMLs can be seen in both human brains with CCCI and animal models with experimental CCCI.38, 39 It has been demonstrated that WMLs secondary to CCCI can be predictive of ischemic events and be correlated with cognitive decline, the degree of which is positively related to the lesion severity. The pathological features in these lesions are typically characterized by diffuse demyelination, pale myelin, axon loss, apoptosis of oligodendrocytes, gliosis, and vacuolization, yet factors contributing to these changes have not been fully elucidated.40, 41 One of the proposed mechanisms is that sustained cerebral ischemia may conduce to oxidative stress and subsequent overexpression of reactive oxygen species (ROS), thus resulting in a detrimental effect on the WM. Furthermore, disruption of blood‐brain barrier (BBB) due to CCCI can prompt the transfer of inflammatory cells as well as other serum substances such as fibrinogens and complement components into the CNS and then release a variety of cytokines, which in turn activate inflammatory responses and cause WM damage.
Notwithstanding, these lesions should not be simply regarded as irreversible or untreatable. There is a growing body of evidence that WMLs could be reduced by modifying vascular risk factors and restoring blood flow prior to the occurrence of permanent axonal damage or demyelination.42, 43 Notably, according to a recent publication, remote ischemic conditioning (RIC) appeared to be a promising approach to alleviate WMLs in patients with cerebral small‐vessel disease.44
Blood‐brain barrier damage
Ultrastructural changes in BBB following cerebral hypoperfusion were investigated by the arteriovenous fistula model, and CCCI was divided into 3 phases: acute (within 3 days), subacute (within 3 weeks), and chronic (within 3 months).45 Electron microscopy did not present any remarkable ultrastructural changes until the subacute phase, which depicted mild vacuolar degeneration in the astroglia around the capillaries of astrocytoma, while the presence of obvious vacuolar degeneration was detected in the chronic phase.
Shin et al studied the degree of BBB damage after CCCI via measuring the immunological activity of claudin‐3, a member of the claudin family that serves in the tight junction for maintaining the integrity and permeability of BBB.46 They discovered that the permeability of BBB was found to be increased significantly two weeks after moderate and sustained cerebral ischemia. Besides, distinctive immunostaining of claudin‐3 was observed, and this trend continued even after 6 weeks of ischemia. Therefore, the expression of claudin‐3 is believed to be in association with destruction of BBB during CCCI, although the underlying mechanism still requires further study.
The relationship between BBB and WMLs has been confirmed in both human and animal studies, in which BBB disruption is indicated as a cause rather than the consequence of WMLs.47, 48 Additionally, BBB permeability is closely correlated with the severity of WMLs. It should be noted that other than WMLs, pathological BBB alterations are also recognized in both the normal appearing WM and regions close to the WMLs borders, implying that there is an association between BBB disruption and the progression of WMLs.49
Neuroglial cells
Neuroglial cells are indeed a group of stromal cells that exert critical functions in the nervous system. There are 4 types of neuroglial cells in the CNS: astrocytes, microglial cells, oligodendrocytes, and ependymal cells. It has been recognized that different glial cells do have certain specific effects on the post‐CCCI processes.
Activation of astrocytes is assumed to play a vital role in neurodegeneration and neuronal injury in the hippocampus subjected to chronic cerebral hypoxia.50 The expressions of glial fibrillary acidic protein (GFAP), S100B protein, Nestin protein, and glutamate are often selected as markers for astrocyte activation.51, 52, 53 Schmidt‐Kastner et al observed that expressions of GFAP and Nestin in the new cerebral cortex were found to be increased after 1 week of CCCI.52 Similarly, Vicente et al found that the levels of S100B and GFAP in the hippocampus were elevated markedly after 10 weeks of CCCI.22
Microglia cells are specialized macrophages that pose central functions in removing necrotic tissues and enhancing CNS regeneration under pathological circumstances, such as cerebral ischemia. On the contrary, they can also direct immune or inflammatory responses through the secretion of diverse proinflammatory cytokines, chemokines, and ROS, which hamper the restoration and neurogenesis after brain injury. A strong correlation emerges between the activation of microglia and the magnitude of WMLs during CCCI. In other words, proliferation of microglia is capable of exacerbating WM damage.54, 55 Nonetheless, the exact mechanism regarding the activated microglia under CCCI remains to be settled. In the 2VO model, inhibition of autophagy was able to attenuate microglial activation, decrease WMLs, and improve cognitive deficits of experimental mice.56 In addition, it has been demonstrated that MCP‐1‐mediated microglia activation worsened WMLs and impaired working memory, possibly via p38MAPK/PKC pathway.57
Accumulating evidence has implied that apoptosis of oligodendrocytes, a group of myelin‐forming cells in the brain that are more susceptible to ischemic insults than other glia cells, can contribute to WMLs formation. Previous studies revealed that loss of oligodendrocytes and activation of caspase signaling cascades (such as TNF‐α, caspase‐3, and Bax) might be responsible for the pathogenesis of WMLs in CCCI.58 Notably, in an experimental VD model generated by rUCCAO, the recruitment, rather than the proliferation and differentiation of oligodendrocyte progenitor cells (OPCs), was regarded as a crucial, but compromised step for remyelination following CCCI.59
Neuronal cells
Neuronal loss is another characteristic feature seen after CCCI. Animal models demonstrated that neuronal death was present at 1 week and continued for up to 3 months after CCCI,60 especially in the striatum, hippocampus, and cortex, indicating that neurons in these 3 regions are especially vulnerable to CCCI.61, 62 In addition, CCCI‐induced neuronal loss might be correlated with abnormal phosphorylation of tau protein. The apoptotic morphology and DNA fragmentation in hippocampal pyramidal neurons are presumably responsible for the memory impairment due to CCCI.
Neurotransmitters
Some studies discovered that the cholinergic system underwent changes in CCCI, as reflected by alterations in cholinergic markers such as Ach, acetylcholinesterase (AchE), choline acetyl‐transferase (ChAT) as well as M‐type ACh receptors in both mRNA and protein forms,63 suggesting that central cholinergic system dysfunction poses a significant effect on the pathogenesis of cognitive deficits and neuropathological lesions in CCCI.64 Furthermore, protection or reversal of the significantly decreased expression of vesicular acetylcholine transporter (vAChT) and vAChT‐positive neurons in hippocampal subregions such as CA1 is able to mitigate cognitive impairments related to the possible mechanism in CCCI‐induced VD. Meanwhile, oxidative stress resulting from CCCI is believed to participate in the process of undermining central cholinergic function.65
Substantial downregulation of the hippocampal GABAergic system, including GABA, GABAB receptor 1 (GABABR1), and glutamic acid decarboxylase67 (GAD67), has been found to be present in the 2VO rat models of CCCI. It should be noted that amelioration of cognitive dysfunction and neuronal injury secondary to CCCI is likely to be accomplished via enhancing GABAergic expression and transmission in the hippocampal CA1 area with clonidine or bone marrow mesenchymal stem cells (BMSCs).66, 67 Glutamate is a well‐known excitatory neurotransmitter in the CNS, and impaired activity of glutamate transporters may induce glutamate excitotoxicity, followed by apoptosis of glutamatergic neurons. Vesicular glutamate transporters (vGluT) play a vital role in the modulation of intracellular glutamate expression, among which the levels of vGluT1 and vGluT3 were significantly lowered in the hippocampus at 3 months after the establishment of the ischemic model.65 Consistent with the protein findings, reductions in GluT1‐ and vGluT3‐positive neurons were noticed in the hippocampus, and this descending trend was most evident in CA1 region. Additionally, excitatory amino acid transporter 2, which is accountable for delivering glutamate into neurons, was increased remarkably following CCCI, denoting that the overloaded glutamate in this condition is a culprit for neuronal damage and functional anomalies.68
Existing evidence has also revealed the presence of a series of changes in some other neurotransmitters such as dopamine and histamine.69, 70, 71 For instance, in CCCI animals, the density of tyrosine hydroxylase, along with the expression of dopamine and the number of dopaminergic neurons, was considerably declined compared with controls. Inspiringly, a growing number of studies have been designed to investigate the impact of inhibiting or promoting these neurotransmitters on neurological outcomes in the scenario of CCCI, knowing that the underlying mechanism needs to be explored more comprehensively.
Oxidative stress
Studies have denoted that oxidative stress is involved in brain injury during sustained cerebral hypoperfusion. For instance, altered expressions of prooxidants and antioxidants such as cyclooxygenase‐2 and endothelial nitric oxide synthase were found to take place in the early period of chronic hypoperfusion.72 Another study claimed that increased oxidative damage might correlate with central cholinergic dysfunction, resulting in spatial learning/memory dysfunctions and working memory impairment.65 Accordingly, suppression or elimination of ROS with certain antioxidants displayed a potential protective effect against brain lesions and neurobehavioral deficits induced by CCCI.73, 74 In addition, increased attention has been paid to the mutual adverse relationship between oxidative stress and Aβ protein, assuming that oxidative stress could be triggered by cerebrovascular dysfunction due to an excessive accumulation of Aβ peptide during chronic hypoperfusion; meanwhile, increased oxidative alterations might hinder the clearance or accelerate the formation of Aβ protein.
Growth factors
Alterations in many growth factors during CCCI were previously reported, among which the most widely investigated is BDNF. Evidence shows that both mRNA and protein expressions of BDNF were apparently raised in the hippocampus 6 h after 2VO, and descended back to the normal level 24 h after 2VO, suggesting a transient endogenous protective response to CCCI‐induced cerebral ischemic insults.75, 76 Reduction in BDNF level appeared to aggravate CCCI‐induced cognitive impairment.77 Moreover, other growth factors, such as VEGF and nerve growth factor, which are believed to be potential therapeutic strategies to alleviate CCCI‐related injuries, have also been investigated, although all of them are still in the preclinical exploratory phase.78, 79
Aging
It is widely accepted that senescence‐related changes pose gradual, yet tremendous impact on brain homeostasis and function. In particular, aging may weaken the endogenous capacities for regeneration and remodeling, as reflected by diminished neurogenesis, increased glial cell activation, overaccumulation of ROS and proinflammatory cytokines, and impaired mitochondrial function.80 Among the limited studies performed, these have indicated that aging can deteriorate either structural or functional alterations in animal models of CCCI. Miyamoto et al demonstrated that 8‐month‐old mice were more susceptible to prolonged cerebral hypoperfusion than 2‐month‐old mice, with decreased oligodendrogenesis, more severe WMLs, and worse deficits in working memory.81 Furthermore, the differential effects of induced cerebral hypoperfusion between young adult and more aged mice (3 months vs 21 months) were further confirmed in a recent study.82 Compared with young models, more pronounced alterations with regard to the impaired cognitive functions, myelin degradation, and microglia activation occurred in aged mice. More studies are required to gain deeper insights into the interaction between advanced age and cerebral hypoperfusion, and the underlying mechanism as well.
5. CLINICAL MANIFESTATIONS
5.1. Clinical presentations
Patients with CCCI usually complained of motor or sensory dysfunctions, cognitive decline, and mental or emotional abnormalities. Details are listed in Table 1A.1, 83, 84
Table 1.
(A) Clinical presentations of patients with CCCI. (B) Diagnostic criteria for CCCI
| (A) |
| 1. Common clinical presentations |
| (a) Headache, heavy headedness, dizziness, and vertigo |
| (b) Fatigue, feeling drowsy all day |
| (c) Irritability, lack of concentration, sleep disorders, anxiety, and depression |
| (d) Unilateral numbness of face, hand, and foot |
| (e) Limb weakness or inflexibility |
| (f) Uncontrolled spasm in unilateral or one part of a limb |
| (g) Unexplained fainting or tumbling |
| (h) Nausea, vomiting, and fluctuations of blood pressure |
| (i) Sudden but transient loss of vision |
| (j) Sudden change in personality and mentality |
| (k) Torpidity, impaired working ability and memory, difficulties with assimilating new information |
| 2. CCCI of anterior circulation (internal carotid artery system) |
| Forebrain dysfunctions, such as impairment of memory, especially for the details of recent events and newly acquired information; repeating questions, torpidity, poor judgment, decline in working ability, concentration and visuospatial skills; emotional disturbances like labile affect, irritability, apathy, and anxiety; sleeping disorders such as insomnia; personality and even mental changes |
| 3. CCCI of posterior circulation (vertebrobasilar artery system) |
| Posterior circulation ischemia–associated symptoms, such as dizziness, ataxia, unsteadiness, disequilibrium, vertigo with or without nausea and vomiting, headache, tinnitus, dysarthria or dysphagia, diplopia, blurred vision, and other motor or sensory deficits |
| (B) |
| (1) Ages of the patients with CCCI are usually equal to or more than 60 years (the age can also be extended to 45 years on the basis of specific circumstances) |
| (2) Factors that can potentially lead to cerebral arteriosclerosis, such as hypertension, diabetes, hyperhomocysteinemia, hypercholesterolemia, hyperlipidemia, chronic heart failure, coronary heart disease, sustained hypotension, and peripheral arteriosclerosis |
| (3) Symptoms of chronic brain dysfunctions such as dizziness, headache, cognitive decline, memory loss, inattention, emotional instability, working ability reduction, and sleeping disorder; the above subjective feelings persist at least for 2 months |
| (4) With or without neurological positive signs such as hyperactivity of tendon reflex, positive palm‐chin reflex, sucking reflex, and Rossolimo sign |
| (5) Laboratory tests |
| (a) Transcranial Doppler ultrasound examination shows cerebral arterial stenosis or occlusion |
| (b) With or without corresponding lacuna infarction/mild white matter demyelination on CT and/or MRI |
| (c) DSA/CTA/MRA examinations reveal the stenosis or occlusion of cerebral perfusion artery, or arteriosclerosis‐associated changes |
| (d) Perfusion imaging confirms the existence of cerebral blood flow reduction or decreased metabolism |
| (e) Agents that can improve cerebral circulation and brain metabolite are proven to be effective |
| (f) Exclude other diseases that can cause the aforementioned manifestations |
5.2. Classification
Based on the involvement of the cerebral arterial system, CCCI classification denotes 2 subtypes85, 86, 87: CCCI of anterior circulation and CCCI of posterior circulation. There exist several differences in presenting symptoms, clinical evaluation, and diagnostic testing between them.
5.2.1. CCCI of anterior circulation
The target artery of CCCI is located in the internal carotid arterial system, in which forebrain dysfunction is the main clinical manifestation (Table 1A).
No explicit localizing neurological signs, findings such as positive primitive reflexes like sucking reflex or palm‐chin reflex, and active tendon reflexes could be observed.
With or without lacunar infarcts, mild brain atrophy and WMLs on neuroimaging.
5.2.2. CCCI of posterior circulation
The target artery of CCCI is located in the vertebrobasilar arterial system, in which posterior circulation ischemia‐associated symptoms are the main clinical manifestations (Table 1A).
With or without brain stem or cerebellar infarction on neuroimaging.
Evidence of vertebrobasilar artery anomalies such as atherosclerosis, stenosis, distension, and dilation.
6. IMAGING FEATURES
Modern imaging technology provides invaluable information with respect to various aspects, including changes in cerebral perfusion and metabolism, structural integrity of brain parenchyma, severity of vascular lesions, and neurological recovery status over time. In terms of cerebral perfusion assessment in CCCI research, neuroimaging modalities include positron emission tomography (PET), single‐photon emission computed tomography (SPECT), computed tomography (CT), and magnetic resonance imaging (MRI).
PET is regarded as the “gold standard” that affords an advantage of quantitatively assessing multiple parameters related to the cerebral physiological and metabolic conditions. Evaluation of CCCI can be accomplished with PET, which estimates cerebral perfusion via measuring the CBF and oxygen extraction fraction (OEF).88 However, it is not considered as a first‐line option because of a relatively high cost and demand for technique as well as a low penetration rate.
SPECT, being more readily available, is recommended as a promising tool for assessing cerebral hemodynamics semiquantitatively. Currently, there are 2 types of tracers for SPECT, namely 99mTc‐hexamethylprpyleneamine oxime and 99mTc‐ethyl cysteinate dimer, both of which can be used to evaluate brain metabolism and blood flow regionally (Figure 4A). Cerebral vasoreactivity (CVR), reflected as the percentage increase in CBF as a result of vasodilators relative to baseline, has been demonstrated to hold the potential of evaluating the residual vessel dilating capacity and predicting the risk of cerebral hemodynamic disturbances in patients with major cerebral arterial occlusive diseases.64 Among patients with CCCI secondary to various types of vaso‐occlusive diseases, either PET or SPECT combined with acetazolamide challenge has been proven to be advantageous for estimating CVR.89
Figure 4.

Imaging modalities for CCCI evaluation. A, SPECT reveals mild anterior circulation hypoperfusion in the left middle cerebral artery (L‐MCA) territory (red arrows). B, CTP maps indicate remarkable hypoperfusion in the left hemisphere, with decreased CBF, increased CBV, prolonged MTT and TTP. C, MRA shows L‐MCA occlusion; DWI shows an area of restricted diffusion (ischemic brain lesion) in the territory of L‐MCA; PWI shows that the hypoperfusion area is larger than that in DWI, this diffusion/perfusion mismatch suggests the presence of ischemic penumbra. CCCI: chronic cerebral circulation insufficiency; SPECT: single‐photon emission computed tomography; CTP: computed tomography perfusion; CBF: cerebral blood flow; CBV: cerebral blood volume; MTT: mean transit time; TTP: time to peak; MRA: magnetic resonance angiography; DWI: diffusion‐weighted MR; PWI: perfusion‐weighted MR
Perfusion imaging modalities such as computed tomography perfusion (CTP) and perfusion‐weighted MRI (PWI) are noninvasive techniques that enable the documentation of perfusion parameters including CBF, CBV, mean transit time (MTT), and time to peak (TTP) (Figure 4B,C). Both of the 2 modalities need contrast agents for visualization of perfusion parts of the brain and are able to be used in combination with acetazolamide to assess cerebral hemodynamics more accurately.90, 91 Unlike PCT, PWI relies on a gadolinium‐based contrast agent to yield cerebral hemodynamic values and displays a more complete coverage of brain structures without exposure to radiation. The combinatory use of diffusion‐weighted MRI (DWI) and PWI is invariably suggested in an attempt to differentiate ischemic from infarcted area, and this may require more imaging time and a high standard for cooperation patients.
Arterial spin labeling (ASL) is a noninvasive magnetic resonance perfusion technique, which is capable of depicting hyper‐ or hypoperfusion status encountered in a diversity of clinical scenarios. Regardless of the limited reports concerning the feasibility of ASL in patients with CCCI so far, it holds the potential of real‐time detecting CVR quantitatively as well as serially assessing following revascularization in these patients.92
7. DIAGNOSIS
7.1. Diagnostic criteria
The diagnostic criteria for CCCI mainly include (i) ages above 45 years;1 (ii) symptoms of chronic brain dysfunctions for at least 2 months; (iii) presence of cerebral arteriosclerosis‐related factors; (iv) imaging and ultrasound‐confirmed arterial stenosis or occlusion, infarction, and demyelination. Detailed descriptions are summarized in Table 1B.
7.2. Differentiation
The differential diagnosis of CCCI includes all other causes that can lead to manifestations similar to that of cerebral ischemia. A few examples are outlined below.
Focal epilepsy: a history of seizures, epilepsy waves in EEG recording, controlled by antiepileptic drugs.
The inner ear vertigo: more common in young individuals, absence of brain stem signs.
Syncope attack: acute onset of cerebral circulation insufficiency, more common in young women, transient consciousness loss caused by physical factors such as hypoglycemia, hypotension, and alkalosis, with prodromal symptoms such as pale sweating and limb weakness before the acute onset.
Psychiatric anxiety disorder and depression: presence of anxiety and depression performances, while lacking objective evidence of CCCI.
8. TREATMENT AND PREVENTION
Ideal management of CCCI remains a topic of debate as data from randomized control trials with high quality are still lacking.79, 93, 94, 95, 96 Essentially, so far, the primary goal focuses on the enhancement of cerebral circulation and metabolism.
8.1. Treatment of CCCI
Risk factors control, such as arteriosclerosis, hypertension, diabetes, hyperlipidemia, and so forth;
Use of antiplatelet and anticoagulant agents, such as aspirin and/or clopidogrel;
Vessel dilators: the most commonly used are calcium channel blockers, such as flunarizine and nimodipine;
Chinese medicine: Yangxue Qingnao granules and ginkgo leaf preparations, etc., to improve microcirculation;
Surgical intervention: carotid endarterectomy or stenting can be considered when the severity of artery stenosis reaches to 70% or more;
Nonmedication management: RIC.
8.2. Prevention of CCCI
Reasonable diet with fresh vegetables, fruit, fish, black fungus, and red wine, etc.;
Maintain a healthy lifestyle: participate in outdoor activities such as brisk walking and jogging (e.g each time 30‐40 min, 5 days a week); adopt some cultural and sports activities, like singing, dancing, and tennis, to enhance brain thinking activities; avoid emotional and excessive fatigue; early detection of cerebral insufficiency, if necessary, use of drugs to improve brain circulation.
9. CONCERNS AND PROSPECTS
Currently, clinical diagnosis of CCCI faces immense challenges. First of all, considering the unspecific features of core symptoms of CCCI such as dizziness and headache, it is quite difficult to measure or assess them objectively. Awareness of depression or cognitive impairment suggests that these statements are more likely to be “somatization” symptoms, which require more clinical observations to determine whether the depression or cognitive impairment is a central symptom of a patient with CCCI. More importantly, to evaluate whether there is 1 or more brain regions of low perfusion and its relevance with clinical symptoms are crucial for determining the disease as an entity of CCCI. Therefore, more accurate approaches for evaluation are warranted in the future.
Up to now, the majority of CCCI researches are descriptive and based on experimental models, while scant clinical studies have unraveled the underlying mechanisms that contribute to CCCI. Concerning the therapeutic strategies, despite considerable progress achieved in the preclinical stages, few of them have been translated into clinical practice.
Additionally, it should be kept in mind that some clinical presentations such as cognitive decline may be present in the presymptomatic stage for a considerably long period of time, during which certain endogenous protective mechanisms probably become undermined. From our perspective, CCCI is considered as a disorder with multifactorial traits, and no single unifying hypothesis can conclusively explicate all the distinctive features. Hence, therapies targeted against only one element would hardly take effect as expected. Accordingly, attention ought to be focused on the early detection, prevention as well as treatment of cerebral hypoperfusion with strategies directed at 2 or more therapeutic targets.
Recently, some randomized control studies have revealed that RIC is a promising way to ameliorate or even prevent CCCI;95, 96 meanwhile, a corresponding multicenter randomized control trial is currently underway (NCT02534545).
CONFLICT OF INTEREST
All authors declare no conflict of interest.
ACKNOWLEDGEMENTS
This study was sponsored by the National Natural Science Foundation (81371289), the National Key R&D Program (2017YFC1308400), and the Project of Beijing Municipal Top Talent of Healthy Work (2014‐2‐015), of China. The funding agencies had no role in the design and conduct of the study; in the collection, analysis, and interpretation of the data; or in the preparation, review, or approval of the manuscript.
Zhou D, Meng R, Li S‐J, et al. Advances in chronic cerebral circulation insufficiency. CNS Neurosci Ther. 2018;24:5–17. 10.1111/cns.12780
Contributor Information
Ran Meng, Email: Ranmeng2011@pku.org.cn.
Xun‐Ming Ji, Email: jixm@ccmu.edu.cn.
REFERENCES
- 1. Wu C, Liao L, Yan X, et al. Effects of Yangxue Qingnao Granules on chronic cerebral circulation insufficiency: a randomized, double‐blind, double‐dummy, controlled multicentre trial. Psychogeriatrics. 2013;13:29‐34. [DOI] [PubMed] [Google Scholar]
- 2. Safouris A, Hambye AS, Sculier C, et al. Chronic brain hypoperfusion due to multi‐vessel extracranial atherosclerotic disease: a potentially reversible cause of cognitive impairment. J Alzheimers Dis. 2015;43:23‐27. [DOI] [PubMed] [Google Scholar]
- 3. Wang Y, Zhao X, Liu L, et al. Prevalence and outcomes of symptomatic intracranial large artery stenoses and occlusions in China: the Chinese Intracranial Atherosclerosis (CICAS) Study. Stroke. 2014;45:663‐669. [DOI] [PubMed] [Google Scholar]
- 4. Calabrese V, Giordano J, Signorile A, et al. Major pathogenic mechanisms in vascular dementia: roles of cellular stress response and hormesis in neuroprotection. J Neurosci Res. 2016;94:1588‐1603. [DOI] [PubMed] [Google Scholar]
- 5. Sarti C, Pantoni L, Bartolini L, Inzitari D. Cognitive impairment and chronic cerebral hypoperfusion: what can be learned from experimental models. J Neurol Sci. 2002;203–204:263‐266. [DOI] [PubMed] [Google Scholar]
- 6. Vagal AS, Leach JL, Fernandez‐Ulloa M, Zuccarello M. The acetazolamide challenge: techniques and applications in the evaluation of chronic cerebral ischemia. AJNR Am J Neuroradiol. 2009;30:876‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wong LK. Global burden of intracranial atherosclerosis. Int J Stroke. 2006;1:158‐159. [DOI] [PubMed] [Google Scholar]
- 8. Kasner SE, Chimowitz MI, Lynn MJ, et al. Predictors of ischemic stroke in the territory of a symptomatic intracranial arterial stenosis. Circulation. 2006;113:555‐563. [DOI] [PubMed] [Google Scholar]
- 9. Liu L, Wong KS, Leng X, et al. Dual antiplatelet therapy in stroke and ICAS: subgroup analysis of CHANCE. Neurology. 2015;85:1154‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gorelick P, Wong KS, Liu L. Epidemiology. Front Neurol Neurosci. 2016;40:34‐46. [DOI] [PubMed] [Google Scholar]
- 11. de la Torre JC, Nelson N, Sutherland RJ, Pappas BA. Reversal of ischemic‐induced chronic memory dysfunction in aging rats with a free radical scavenger‐glycolytic intermediate combination. Brain Res. 1998;779:285‐288. [DOI] [PubMed] [Google Scholar]
- 12. Miyamoto N, Maki T, Pham LD, et al. Oxidative stress interferes with white matter renewal after prolonged cerebral hypoperfusion in mice. Stroke. 2013;44:3516‐3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miyamoto N, Maki T, Shindo A. Astrocytes promote oligodendrogenesis after white matter damage via brain‐derived neurotrophic factor. J Neurosci. 2015;35:14002‐14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hai J, Ding M, Guo Z, Wang B. A new rat model of chronic cerebral hypoperfusion associated with arteriovenous malformations. J Neurosurg. 2002;97:1198‐1202. [DOI] [PubMed] [Google Scholar]
- 15. Hai J, Li ST, Lin Q, Pan QG, Gao F, Ding MX. Vascular endothelial growth factor expression and angiogenesis induced by chronic cerebral hypoperfusion in rat brain. Neurosurgery. 2003;53:963‐970; discussion 970‐962. [DOI] [PubMed] [Google Scholar]
- 16. Yoshizaki K, Adachi K, Kataoka S, et al. Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp Neurol. 2008;210:585‐591. [DOI] [PubMed] [Google Scholar]
- 17. Masumura M, Hata R, Nagai Y, Sawada T. Oligodendroglial cell death with DNA fragmentation in the white matter under chronic cerebral hypoperfusion: comparison between normotensive and spontaneously hypertensive rats. Neurosci Res. 2001;39:401‐412. [DOI] [PubMed] [Google Scholar]
- 18. Zhai Y, Yamashita T, Nakano Y, et al. Chronic cerebral hypoperfusion accelerates Alzheimer's disease pathology with cerebrovascular remodeling in a novel mouse model. J Alzheimers Dis. 2016;53:893‐905. [DOI] [PubMed] [Google Scholar]
- 19. Choi BR, Lee SR, Han JS, et al. Synergistic memory impairment through the interaction of chronic cerebral hypoperfusion and amyloid toxicity in a rat model. Stroke. 2011;42:2595‐2604. [DOI] [PubMed] [Google Scholar]
- 20. Kwon KJ, Lee EJ, Kim MK, et al. Diabetes augments cognitive dysfunction in chronic cerebral hypoperfusion by increasing neuronal cell death: implication of cilostazol for diabetes mellitus‐induced dementia. Neurobiol Dis. 2015;73:12‐23. [DOI] [PubMed] [Google Scholar]
- 21. Jiwa NS, Garrard P, Hainsworth AH. Experimental models of vascular dementia and vascular cognitive impairment: a systematic review. J Neurochem. 2010;115:814‐828. [DOI] [PubMed] [Google Scholar]
- 22. Vicente E, Degerone D, Bohn L, et al. Astroglial and cognitive effects of chronic cerebral hypoperfusion in the rat. Brain Res. 2009;1251:204‐212. [DOI] [PubMed] [Google Scholar]
- 23. Shibata M, Yamasaki N, Miyakawa T, et al. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke. 2007;38:2826‐2832. [DOI] [PubMed] [Google Scholar]
- 24. Choi BR, Kim DH, Back DB, et al. Characterization of white matter injury in a rat model of chronic cerebral hypoperfusion. Stroke. 2016;47:542‐547. [DOI] [PubMed] [Google Scholar]
- 25. Kitamura A, Manso Y, Duncombe J, et al. Long‐term cilostazol treatment reduces gliovascular damage and memory impairment in a mouse model of chronic cerebral hypoperfusion. Sci Rep. 2017;7:4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nishio K, Ihara M, Yamasaki N, et al. A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke. 2010;41:1278‐1284. [DOI] [PubMed] [Google Scholar]
- 27. Victor M, Ropper AH. Adams and Victor's Principles of Neurology. New York: McGraw‐Hill Companies; 2002. [Google Scholar]
- 28. Heiss WD. Experimental evidence of ischemic thresholds and functional recovery. Stroke. 1992;23:1668‐1672. [DOI] [PubMed] [Google Scholar]
- 29. Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557‐565. [DOI] [PubMed] [Google Scholar]
- 30. Yatomi Y, Tanaka R, Shimada Y, et al. Type 2 diabetes reduces the proliferation and survival of oligodendrocyte progenitor cells in ischemic white matter lesions. Neuroscience. 2015;289:214‐223. [DOI] [PubMed] [Google Scholar]
- 31. Li WX, Deng YY, Li F, et al. Icariin, a major constituent of flavonoids from Epimedium brevicornum, protects against cognitive deficits induced by chronic brain hypoperfusion via its anti‐amyloidogenic effect in rats. Pharmacol Biochem Behav. 2015;138:40‐48. [DOI] [PubMed] [Google Scholar]
- 32. Duan W, Chun‐Qing Z, Zheng J, Gui L, Huang HQ, Chen KN. Relief of carotid stenosis improves impaired cognition in a rat model of chronic cerebral hypoperfusion. Acta Neurobiol Exp (Wars). 2011;71:233‐243. [DOI] [PubMed] [Google Scholar]
- 33. Chui HC. Subcortical ischemic vascular dementia. Neurol Clin. 2007;25:717‐740, vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arakawa S, Wright PM, Koga M, et al. Ischemic thresholds for gray and white matter: a diffusion and perfusion magnetic resonance study. Stroke. 2006;37:1211‐1216. [DOI] [PubMed] [Google Scholar]
- 35. Falcao AL, Reutens DC, Markus R, et al. The resistance to ischemia of white and gray matter after stroke. Ann Neurol. 2004;56:695‐701. [DOI] [PubMed] [Google Scholar]
- 36. Koga M, Reutens DC, Wright P, et al. The existence and evolution of diffusion‐perfusion mismatched tissue in white and gray matter after acute stroke. Stroke. 2005;36:2132‐2137. [DOI] [PubMed] [Google Scholar]
- 37. Bristow MS, Simon JE, Brown RA, et al. MR perfusion and diffusion in acute ischemic stroke: human gray and white matter have different thresholds for infarction. J Cereb Blood Flow Metab. 2005;25:1280‐1287. [DOI] [PubMed] [Google Scholar]
- 38. Fernando MS, Simpson JE, Matthews F, et al. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke. 2006;37:1391‐1398. [DOI] [PubMed] [Google Scholar]
- 39. Thiebaut de Schotten M, Tomaiuolo F, Aiello M, et al. Damage to white matter pathways in subacute and chronic spatial neglect: a group study and 2 single‐case studies with complete virtual “in vivo” tractography dissection. Cereb Cortex. 2014;24:691‐706. [DOI] [PubMed] [Google Scholar]
- 40. Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke. 2004;35:2598‐2603. [DOI] [PubMed] [Google Scholar]
- 41. Fazekas F, Kleinert R, Offenbacher H, et al. Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology. 1993;43:1683‐1689. [DOI] [PubMed] [Google Scholar]
- 42. Yamada K, Sakai K, Owada K, Mineura K, Nishimura T. Cerebral white matter lesions may be partially reversible in patients with carotid artery stenosis. AJNR Am J Neuroradiol. 2010;31:1350‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wardlaw JM, Valdes Hernandez MC, Munoz‐Maniega S. What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc. 2015;4:001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y, Meng R, Song H, et al. Remote ischemic conditioning may improve outcomes of patients with cerebral small‐vessel disease. Stroke. 2017;48:3064‐3072. [DOI] [PubMed] [Google Scholar]
- 45. Wu JS, Chen XC, Chen H, Shi YQ. A study on blood‐brain barrier ultrastructural changes induced by cerebral hypoperfusion of different stages. Neurol Res. 2006;28:50‐58. [DOI] [PubMed] [Google Scholar]
- 46. Shin JS, Hyun SY, Kim DH, et al. Chronic hypoperfusion increases claudin‐3 immunoreactivity in rat brain. Neurosci Lett. 2008;445:144‐148. [DOI] [PubMed] [Google Scholar]
- 47. Topakian R, Barrick TR, Howe FA, Markus HS. Blood‐brain barrier permeability is increased in normal‐appearing white matter in patients with lacunar stroke and leucoaraiosis. J Neurol Neurosurg Psychiatry. 2010;81:192‐197. [DOI] [PubMed] [Google Scholar]
- 48. Young VG, Halliday GM, Kril JJ. Neuropathologic correlates of white matter hyperintensities. Neurology. 2008;71:804‐811. [DOI] [PubMed] [Google Scholar]
- 49. Huisa BN, Caprihan A, Thompson J, Prestopnik J, Qualls CR, Rosenberg GA. Long‐term blood‐brain barrier permeability changes in binswanger disease. Stroke. 2015;46:2413‐2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Castillo‐Melendez M, Yawno T, Allison BJ, Jenkin G, Wallace EM, Miller SL. Cerebrovascular adaptations to chronic hypoxia in the growth restricted lamb. Int J Dev Neurosci. 2015;45:55‐65. [DOI] [PubMed] [Google Scholar]
- 51. Kuang X, Du JR, Liu YX, Zhang GY, Peng HY. Postischemic administration of Z‐Ligustilide ameliorates cognitive dysfunction and brain damage induced by permanent forebrain ischemia in rats. Pharmacol Biochem Behav. 2008;88:213‐221. [DOI] [PubMed] [Google Scholar]
- 52. Schmidt‐Kastner R, Aguirre‐Chen C, Saul I, et al. Astrocytes react to oligemia in the forebrain induced by chronic bilateral common carotid artery occlusion in rats. Brain Res. 2005;1052:28‐39. [DOI] [PubMed] [Google Scholar]
- 53. Chen L, Chen L, Lv Y, et al. Tetrandrine ameliorates cognitive impairment via inhibiting astrocyte‐derived S100B activation in a rat model of chronic cerebral hypoperfusion. Neurol Res. 2013;35:614‐621. [DOI] [PubMed] [Google Scholar]
- 54. Kaur C, Rathnasamy G, Ling EA. Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J Neuroimmune Pharmacol. 2013;8:66‐78. [DOI] [PubMed] [Google Scholar]
- 55. Vontell R, Supramaniam V, Thornton C, et al. Toll‐like receptor 3 expression in glia and neurons alters in response to white matter injury in preterm infants. Dev Neurosci. 2013;35:130‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang Z, Zhang N, Shen H, Lin C, Lin L, Yuan B. Microglial activation with reduction in autophagy limits white matter lesions and improves cognitive defects during cerebral hypoperfusion. Curr Neurovasc Res. 2014;11:223‐229. [DOI] [PubMed] [Google Scholar]
- 57. Yuan B, Shi H, Zheng K, et al. MCP‐1‐mediated activation of microglia promotes white matter lesions and cognitive deficits by chronic cerebral hypoperfusion in mice. Mol Cell Neurosci. 2017;78:52‐58. [DOI] [PubMed] [Google Scholar]
- 58. Tomimoto H, Ihara M, Wakita H, et al. Chronic cerebral hypoperfusion induces white matter lesions and loss of oligodendroglia with DNA fragmentation in the rat. Acta Neuropathol. 2003;106:527‐534. [DOI] [PubMed] [Google Scholar]
- 59. Zhou Y, Zhang J, Wang L, et al. Interleukin‐1beta impedes oligodendrocyte progenitor cell recruitment and white matter repair following chronic cerebral hypoperfusion. Brain Behav Immun. 2017;60:93‐105. [DOI] [PubMed] [Google Scholar]
- 60. Cechetti F, Pagnussat AS, Worm PV, et al. Chronic brain hypoperfusion causes early glial activation and neuronal death, and subsequent long‐term memory impairment. Brain Res Bull. 2012;87:109‐116. [DOI] [PubMed] [Google Scholar]
- 61. Jing Z, Shi C, Zhu L, et al. Chronic cerebral hypoperfusion induces vascular plasticity and hemodynamics but also neuronal degeneration and cognitive impairment. J Cereb Blood Flow Metab. 2015;35:1249‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao Y, Gu JH, Dai CL, et al. Chronic cerebral hypoperfusion causes decrease of O‐GlcNAcylation, hyperphosphorylation of tau and behavioral deficits in mice. Front Aging Neurosci. 2014;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhao Q, Murakami Y, Tohda M, Obi R, Shimada Y, Matsumoto K. Chotosan, a kampo formula, ameliorates chronic cerebral hypoperfusion‐induced deficits in object recognition behaviors and central cholinergic systems in mice. J Pharmacol Sci. 2007;103:360‐373. [DOI] [PubMed] [Google Scholar]
- 64. Cao Y, Gou Z, Du Y, et al. Glutamatergic and central cholinergic dysfunction in the CA1, CA2 and CA3 fields on spatial learning and memory in chronic cerebral ischemia‐Induced vascular dementia of rats. Neurosci Lett. 2016;620:169‐176. [DOI] [PubMed] [Google Scholar]
- 65. Xi Y, Wang M, Zhang W, et al. Neuronal damage, central cholinergic dysfunction and oxidative damage correlate with cognitive deficits in rats with chronic cerebral hypoperfusion. Neurobiol Learn Mem. 2014;109:7‐19. [DOI] [PubMed] [Google Scholar]
- 66. Lu Y, Li C, Zhou M, et al. Clonidine ameliorates cognitive impairment induced by chronic cerebral hypoperfusion via up‐regulation of the GABABR1 and GAD67 in hippocampal CA1 in rats. Pharmacol Biochem Behav. 2015;132:96‐102. [DOI] [PubMed] [Google Scholar]
- 67. Long Q, Hei Y, Luo Q, et al. BMSCs transplantation improves cognitive impairment via up‐regulation of hippocampal GABAergic system in a rat model of chronic cerebral hypoperfusion. Neuroscience. 2015;311:464‐473. [DOI] [PubMed] [Google Scholar]
- 68. Yatomi Y, Tanaka R, Shimura H, et al. Chronic brain ischemia induces the expression of glial glutamate transporter EAAT2 in subcortical white matter. Neuroscience. 2013;244:113‐121. [DOI] [PubMed] [Google Scholar]
- 69. Stasiak A, Mussur M, Unzeta M, Samadi A, Marco‐Contelles JL, Fogel WA. Effects of novel monoamine oxidases and cholinesterases targeting compounds on brain neurotransmitters and behavior in rat model of vascular dementia. Curr Pharm Des. 2014;20:161‐171. [DOI] [PubMed] [Google Scholar]
- 70. Zhang N, Miyamoto N, Tanaka R, Mochizuki H, Hattori N, Urabe T. Activation of tyrosine hydroxylase prevents pneumonia in a rat chronic cerebral hypoperfusion model. Neuroscience. 2009;158:665‐672. [DOI] [PubMed] [Google Scholar]
- 71. Stasiak A, Mussur M, Unzeta M, Lazewska D, Kiec‐Kononowicz K, Fogel WA. The central histamine level in rat model of vascular dementia. J Physiol Pharmacol. 2011;62:549‐558. [PubMed] [Google Scholar]
- 72. Mracsko E, Hugyecz M, Institoris A, Farkas E, Bari F. Changes in pro‐oxidant and antioxidant enzyme levels during cerebral hypoperfusion in rats. Brain Res. 2010;1321:13‐19. [DOI] [PubMed] [Google Scholar]
- 73. He XL, Wang YH, Gao M, Li XX, Zhang TT, Du GH. Baicalein protects rat brain mitochondria against chronic cerebral hypoperfusion‐induced oxidative damage. Brain Res. 2009;1249:212‐221. [DOI] [PubMed] [Google Scholar]
- 74. Ueno Y, Zhang N, Miyamoto N, Tanaka R, Hattori N, Urabe T. Edaravone attenuates white matter lesions through endothelial protection in a rat chronic hypoperfusion model. Neuroscience. 2009;162:317‐327. [DOI] [PubMed] [Google Scholar]
- 75. Gray JD, Milner TA, McEwen BS. Dynamic plasticity: the role of glucocorticoids, brain‐derived neurotrophic factor and other trophic factors. Neuroscience. 2013;239:214‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tonchev AB. Brain ischemia, neurogenesis, and neurotrophic receptor expression in primates. Arch Ital Biol. 2011;149:225‐231. [DOI] [PubMed] [Google Scholar]
- 77. Kermani P, Hempstead B. Brain‐derived neurotrophic factor: a newly described mediator of angiogenesis. Trends Cardiovasc Med. 2007;17:140‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang F, Chang G, Geng X. NGF and TERT co‐transfected BMSCs improve the restoration of cognitive impairment in vascular dementia rats. PLoS ONE. 2014;9:e98774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang J, Fu X, Yu L, et al. Preconditioning with VEGF enhances angiogenic and neuroprotective effects of bone marrow mononuclear cell transplantation in a rat model of chronic cerebral hypoperfusion. Mol Neurobiol. 2016;53:6057‐6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. von Bernhardi R, Eugenin‐von Bernhardi L, Eugenin J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci. 2015;7:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Miyamoto N, Pham LD, Hayakawa K, et al. Age‐related decline in oligodendrogenesis retards white matter repair in mice. Stroke. 2013;44:2573‐2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wolf G, Lotan A, Lifschytz T, et al. Differentially severe cognitive effects of compromised cerebral blood flow in aged mice: association with myelin degradation and microglia activation. Front Aging Neurosci. 2017;9:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hirai S. MRI in patients with cerebral infarct and chronic cerebral circulation insufficiency. No To Shinkei. 1991;43:811‐816. [PubMed] [Google Scholar]
- 84. Kalita J, Das BK, Misra UK. SPECT studies of regional cerebral blood flow in 8 patients with Japanese encephalitis in subacute and chronic stage. Acta Neurol Scand. 1999;99:213‐218. [DOI] [PubMed] [Google Scholar]
- 85. Tao WD, Liu M, Fisher M, et al. Posterior versus anterior circulation infarction: how different are the neurological deficits? Stroke. 2012;43:2060‐2065. [DOI] [PubMed] [Google Scholar]
- 86. Roth W, Morgello S, Goldman J, et al. Histopathological differences between the anterior and posterior brain arteries as a function of aging. Stroke. 2017;48:638‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nouh A, Remke J, Ruland S. Ischemic posterior circulation stroke: a review of anatomy, clinical presentations, diagnosis, and current management. Front Neurol. 2014;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Heiss WD. PET imaging in ischemic cerebrovascular disease: current status and future directions. Neurosci Bull. 2014;30:713‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ogasawara K, Ito H, Sasoh M, et al. Quantitative measurement of regional cerebrovascular reactivity to acetazolamide using 123I‐N‐isopropyl‐p‐iodoamphetamine autoradiography with SPECT: validation study using H2 15O with PET. J Nucl Med. 2003;44:520‐525. [PubMed] [Google Scholar]
- 90. Kamath A, Smith WS, Powers WJ, et al. Perfusion CT compared to H(2) (15)O/O (15)O PET in patients with chronic cervical carotid artery occlusion. Neuroradiology. 2008;50:745‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ma J, Mehrkens JH, Holtmannspoetter M, et al. Perfusion MRI before and after acetazolamide administration for assessment of cerebrovascular reserve capacity in patients with symptomatic internal carotid artery (ICA) occlusion: comparison with 99mTc‐ECD SPECT. Neuroradiology. 2007;49:317‐326. [DOI] [PubMed] [Google Scholar]
- 92. Deibler AR, Pollock JM, Kraft RA, Tan H, Burdette JH, Maldjian JA. Arterial spin‐labeling in routine clinical practice, part 2: hypoperfusion patterns. AJNR Am J Neuroradiol. 2008;29:1235‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim MS, Bang JH, Lee J, Han JS, Baik TG, Jeon WK. Ginkgo biloba L. extract protects against chronic cerebral hypoperfusion by modulating neuroinflammation and the cholinergic system. Phytomedicine. 2016;23:1356‐1364. [DOI] [PubMed] [Google Scholar]
- 94. Chen C, Zheng Y, Wu T, Wu C, Cheng X. Oral administration of grape seed polyphenol extract restores memory deficits in chronic cerebral hypoperfusion rats. Behav Pharmacol. 2017;28:207‐213. [DOI] [PubMed] [Google Scholar]
- 95. Meng R, Asmaro K, Meng L, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology. 2012;79:1853‐1861. [DOI] [PubMed] [Google Scholar]
- 96. Meng R, Ding Y, Asmaro K, et al. Ischemic conditioning is safe and effective for octo‐ and nonagenarians in stroke prevention and treatment. Neurotherapeutics. 2015;12:667‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]