

Abstract
Background
The prognosis of people with metastatic cutaneous melanoma, a skin cancer, is generally poor. Recently, new classes of drugs (e.g. immune checkpoint inhibitors and small‐molecule targeted drugs) have significantly improved patient prognosis, which has drastically changed the landscape of melanoma therapeutic management. This is an update of a Cochrane Review published in 2000.
Objectives
To assess the beneficial and harmful effects of systemic treatments for metastatic cutaneous melanoma.
Search methods
We searched the following databases up to October 2017: the Cochrane Skin Group Specialised Register, CENTRAL, MEDLINE, Embase and LILACS. We also searched five trials registers and the ASCO database in February 2017, and checked the reference lists of included studies for further references to relevant randomised controlled trials (RCTs).
Selection criteria
We considered RCTs of systemic therapies for people with unresectable lymph node metastasis and distant metastatic cutaneous melanoma compared to any other treatment. We checked the reference lists of selected articles to identify further references to relevant trials.
Data collection and analysis
Two review authors extracted data, and a third review author independently verified extracted data. We implemented a network meta‐analysis approach to make indirect comparisons and rank treatments according to their effectiveness (as measured by the impact on survival) and harm (as measured by occurrence of high‐grade toxicity). The same two review authors independently assessed the risk of bias of eligible studies according to Cochrane standards and assessed evidence quality based on the GRADE criteria.
Main results
We included 122 RCTs (28,561 participants). Of these, 83 RCTs, encompassing 21 different comparisons, were included in meta‐analyses. Included participants were men and women with a mean age of 57.5 years who were recruited from hospital settings. Twenty‐nine studies included people whose cancer had spread to their brains. Interventions were categorised into five groups: conventional chemotherapy (including single agent and polychemotherapy), biochemotherapy (combining chemotherapy with cytokines such as interleukin‐2 and interferon‐alpha), immune checkpoint inhibitors (such as anti‐CTLA4 and anti‐PD1 monoclonal antibodies), small‐molecule targeted drugs used for melanomas with specific gene changes (such as BRAF inhibitors and MEK inhibitors), and other agents (such as anti‐angiogenic drugs). Most interventions were compared with chemotherapy. In many cases, trials were sponsored by pharmaceutical companies producing the tested drug: this was especially true for new classes of drugs, such as immune checkpoint inhibitors and small‐molecule targeted drugs.
When compared to single agent chemotherapy, the combination of multiple chemotherapeutic agents (polychemotherapy) did not translate into significantly better survival (overall survival: HR 0.99, 95% CI 0.85 to 1.16, 6 studies, 594 participants; high‐quality evidence; progression‐free survival: HR 1.07, 95% CI 0.91 to 1.25, 5 studies, 398 participants; high‐quality evidence. Those who received combined treatment are probably burdened by higher toxicity rates (RR 1.97, 95% CI 1.44 to 2.71, 3 studies, 390 participants; moderate‐quality evidence). (We defined toxicity as the occurrence of grade 3 (G3) or higher adverse events according to the World Health Organization scale.)
Compared to chemotherapy, biochemotherapy (chemotherapy combined with both interferon‐alpha and interleukin‐2) improved progression‐free survival (HR 0.90, 95% CI 0.83 to 0.99, 6 studies, 964 participants; high‐quality evidence), but did not significantly improve overall survival (HR 0.94, 95% CI 0.84 to 1.06, 7 studies, 1317 participants; high‐quality evidence). Biochemotherapy had higher toxicity rates (RR 1.35, 95% CI 1.14 to 1.61, 2 studies, 631 participants; high‐quality evidence).
With regard to immune checkpoint inhibitors, anti‐CTLA4 monoclonal antibodies plus chemotherapy probably increased the chance of progression‐free survival compared to chemotherapy alone (HR 0.76, 95% CI 0.63 to 0.92, 1 study, 502 participants; moderate‐quality evidence), but may not significantly improve overall survival (HR 0.81, 95% CI 0.65 to 1.01, 2 studies, 1157 participants; low‐quality evidence). Compared to chemotherapy alone, anti‐CTLA4 monoclonal antibodies is likely to be associated with higher toxicity rates (RR 1.69, 95% CI 1.19 to 2.42, 2 studies, 1142 participants; moderate‐quality evidence).
Compared to chemotherapy, anti‐PD1 monoclonal antibodies (immune checkpoint inhibitors) improved overall survival (HR 0.42, 95% CI 0.37 to 0.48, 1 study, 418 participants; high‐quality evidence) and probably improved progression‐free survival (HR 0.49, 95% CI 0.39 to 0.61, 2 studies, 957 participants; moderate‐quality evidence). Anti‐PD1 monoclonal antibodies may also result in less toxicity than chemotherapy (RR 0.55, 95% CI 0.31 to 0.97, 3 studies, 1360 participants; low‐quality evidence).
Anti‐PD1 monoclonal antibodies performed better than anti‐CTLA4 monoclonal antibodies in terms of overall survival (HR 0.63, 95% CI 0.60 to 0.66, 1 study, 764 participants; high‐quality evidence) and progression‐free survival (HR 0.54, 95% CI 0.50 to 0.60, 2 studies, 1465 participants; high‐quality evidence). Anti‐PD1 monoclonal antibodies may result in better toxicity outcomes than anti‐CTLA4 monoclonal antibodies (RR 0.70, 95% CI 0.54 to 0.91, 2 studies, 1465 participants; low‐quality evidence).
Compared to anti‐CTLA4 monoclonal antibodies alone, the combination of anti‐CTLA4 plus anti‐PD1 monoclonal antibodies was associated with better progression‐free survival (HR 0.40, 95% CI 0.35 to 0.46, 2 studies, 738 participants; high‐quality evidence). There may be no significant difference in toxicity outcomes (RR 1.57, 95% CI 0.85 to 2.92, 2 studies, 764 participants; low‐quality evidence) (no data for overall survival were available).
The class of small‐molecule targeted drugs, BRAF inhibitors (which are active exclusively against BRAF‐mutated melanoma), performed better than chemotherapy in terms of overall survival (HR 0.40, 95% CI 0.28 to 0.57, 2 studies, 925 participants; high‐quality evidence) and progression‐free survival (HR 0.27, 95% CI 0.21 to 0.34, 2 studies, 925 participants; high‐quality evidence), and there may be no significant difference in toxicity (RR 1.27, 95% CI 0.48 to 3.33, 2 studies, 408 participants; low‐quality evidence).
Compared to chemotherapy, MEK inhibitors (which are active exclusively against BRAF‐mutated melanoma) may not significantly improve overall survival (HR 0.85, 95% CI 0.58 to 1.25, 3 studies, 496 participants; low‐quality evidence), but they probably lead to better progression‐free survival (HR 0.58, 95% CI 0.42 to 0.80, 3 studies, 496 participants; moderate‐quality evidence). However, MEK inhibitors probably have higher toxicity rates (RR 1.61, 95% CI 1.08 to 2.41, 1 study, 91 participants; moderate‐quality evidence).
Compared to BRAF inhibitors, the combination of BRAF plus MEK inhibitors was associated with better overall survival (HR 0.70, 95% CI 0.59 to 0.82, 4 studies, 1784 participants; high‐quality evidence). BRAF plus MEK inhibitors was also probably better in terms of progression‐free survival (HR 0.56, 95% CI 0.44 to 0.71, 4 studies, 1784 participants; moderate‐quality evidence), and there appears likely to be no significant difference in toxicity (RR 1.01, 95% CI 0.85 to 1.20, 4 studies, 1774 participants; moderate‐quality evidence).
Compared to chemotherapy, the combination of chemotherapy plus anti‐angiogenic drugs was probably associated with better overall survival (HR 0.60, 95% CI 0.45 to 0.81; moderate‐quality evidence) and progression‐free survival (HR 0.69, 95% CI 0.52 to 0.92; moderate‐quality evidence). There may be no difference in terms of toxicity (RR 0.68, 95% CI 0.09 to 5.32; low‐quality evidence). All results for this comparison were based on 324 participants from 2 studies.
Network meta‐analysis focused on chemotherapy as the common comparator and currently approved treatments for which high‐ to moderate‐quality evidence of efficacy (as represented by treatment effect on progression‐free survival) was available (based on the above results) for: biochemotherapy (with both interferon‐alpha and interleukin‐2); anti‐CTLA4 monoclonal antibodies; anti‐PD1 monoclonal antibodies; anti‐CTLA4 plus anti‐PD1 monoclonal antibodies; BRAF inhibitors; MEK inhibitors, and BRAF plus MEK inhibitors. Analysis (which included 19 RCTs and 7632 participants) generated 21 indirect comparisons.
The best evidence (moderate‐quality evidence) for progression‐free survival was found for the following indirect comparisons: • both combinations of immune checkpoint inhibitors (HR 0.30, 95% CI 0.17 to 0.51) and small‐molecule targeted drugs (HR 0.17, 95% CI 0.11 to 0.26) probably improved progression‐free survival compared to chemotherapy; • both BRAF inhibitors (HR 0.40, 95% CI 0.23 to 0.68) and combinations of small‐molecule targeted drugs (HR 0.22, 95% CI 0.12 to 0.39) were probably associated with better progression‐free survival compared to anti‐CTLA4 monoclonal antibodies; • biochemotherapy (HR 2.81, 95% CI 1.76 to 4.51) probably lead to worse progression‐free survival compared to BRAF inhibitors; • the combination of small‐molecule targeted drugs probably improved progression‐free survival (HR 0.38, 95% CI 0.21 to 0.68) compared to anti‐PD1 monoclonal antibodies; • both biochemotherapy (HR 5.05, 95% CI 3.01 to 8.45) and MEK inhibitors (HR 3.16, 95% CI 1.77 to 5.65) were probably associated with worse progression‐free survival compared to the combination of small‐molecule targeted drugs; and • biochemotherapy was probably associated with worse progression‐free survival (HR 2.81, 95% CI 1.54 to 5.11) compared to the combination of immune checkpoint inhibitors.
The best evidence (moderate‐quality evidence) for toxicity was found for the following indirect comparisons: • combination of immune checkpoint inhibitors (RR 3.49, 95% CI 2.12 to 5.77) probably increased toxicity compared to chemotherapy; • combination of immune checkpoint inhibitors probably increased toxicity (RR 2.50, 95% CI 1.20 to 5.20) compared to BRAF inhibitors; • the combination of immune checkpoint inhibitors probably increased toxicity (RR 3.83, 95% CI 2.59 to 5.68) compared to anti‐PD1 monoclonal antibodies; and • biochemotherapy was probably associated with lower toxicity (RR 0.41, 95% CI 0.24 to 0.71) compared to the combination of immune checkpoint inhibitors.
Network meta‐analysis‐based ranking suggested that the combination of BRAF plus MEK inhibitors is the most effective strategy in terms of progression‐free survival, whereas anti‐PD1 monoclonal antibodies are associated with the lowest toxicity.
Overall, the risk of bias of the included trials can be considered as limited. When considering the 122 trials included in this review and the seven types of bias we assessed, we performed 854 evaluations only seven of which (< 1%) assigned high risk to six trials.
Authors' conclusions
We found high‐quality evidence that many treatments offer better efficacy than chemotherapy, especially recently implemented treatments, such as small‐molecule targeted drugs, which are used to treat melanoma with specific gene mutations. Compared with chemotherapy, biochemotherapy (in this case, chemotherapy combined with both interferon‐alpha and interleukin‐2) and BRAF inhibitors improved progression‐free survival; BRAF inhibitors (for BRAF‐mutated melanoma) and anti‐PD1 monoclonal antibodies improved overall survival. However, there was no difference between polychemotherapy and monochemotherapy in terms of achieving progression‐free survival and overall survival. Biochemotherapy did not significantly improve overall survival and has higher toxicity rates compared with chemotherapy.
There was some evidence that combined treatments worked better than single treatments: anti‐PD1 monoclonal antibodies, alone or with anti‐CTLA4, improved progression‐free survival compared with anti‐CTLA4 monoclonal antibodies alone. Anti‐PD1 monoclonal antibodies performed better than anti‐CTLA4 monoclonal antibodies in terms of overall survival, and a combination of BRAF plus MEK inhibitors was associated with better overall survival for BRAF‐mutated melanoma, compared to BRAF inhibitors alone.
The combination of BRAF plus MEK inhibitors (which can only be administered to people with BRAF‐mutated melanoma) appeared to be the most effective treatment (based on results for progression‐free survival), whereas anti‐PD1 monoclonal antibodies appeared to be the least toxic, and most acceptable, treatment.
Evidence quality was reduced due to imprecision, between‐study heterogeneity, and substandard reporting of trials. Future research should ensure that those diminishing influences are addressed. Clinical areas of future investigation should include the longer‐term effect of new therapeutic agents (i.e. immune checkpoint inhibitors and targeted therapies) on overall survival, as well as the combination of drugs used in melanoma treatment; research should also investigate the potential influence of biomarkers.
Plain language summary
Systemic treatments (tablets or injections) taken for metastatic melanoma (expanded from its starting point to other parts of the body)
Background
Melanoma is the most dangerous common skin cancer. Early diagnosis offers the best chance of cure. People affected by early stage melanoma represent about 70% to 80% of all those with melanoma and can be treated by surgical removal of the original tumour (known as the primary tumour). However, when a primary melanoma is detected at a later stage, there is a risk of disease spreading to the nearest lymph nodes (glands that are part of the body's immune system) and distant sites, such as the lungs, liver, bone and brain. In this case, systemic chemotherapy (giving drugs that kill cells throughout the body) and biochemotherapy (chemotherapy combined with substances that can improve the immune response, known as immunostimulating cytokines, such as interleukin‐2 and interferon‐alpha) have been the main treatments for over three decades. However, only few people experience spontaneous (i.e. not resulting from therapy) regression of the primary tumour.
Over the past few years, new classes of drugs have been used with promising results. We aimed to look at how new systemic treatments compare with older therapies, as well as with each other, in terms of survival, acceptability, tumour response, and quality of life. We assessed these outcomes in people with metastatic melanoma (AJCC TNM stage IV).
Review question
We aimed to assess the effects of systemic treatments for people with metastatic cutaneous melanoma (melanoma of skin tissue). We searched for relevant trials up to October 2017 and included 122 studies.
We summarised the results of melanoma treatments (delivered systemically), such as conventional chemotherapy, biochemotherapy, as well as newer drug classes, such as immune checkpoint inhibitors (anti‐CTLA4 and anti‐PD1 monoclonal antibodies, which increase the anti‐tumour activity of the immune system), small‐molecule targeted drugs (BRAF inhibitors, which are used only for melanomas containing specific BRAF gene mutations that promote tumour progression, and MEK inhibitors, which work on the same molecular pathway), and anti‐angiogenic drugs (which reduce blood supply to cancer cells). We compared these treatments with conventional chemotherapy.
Study characteristics
All 122 studies were randomised controlled trials that enrolled participants with metastatic cutaneous melanoma and compared different systemic treatments (28,561 participants). Study participants were adults of either sex, with a mean age of 57.5 years. There were 29 studies that included people whose cancer had spread to the brain, which is important because the detection and treatment of brain metastases often present unique challenges. Most treatments were compared with chemotherapy, and all studies were set in hospitals. Frequently, the pharmaceutical company who produced a tested drug also sponsored the study in which it was assessed, especially in the case of new classes of drugs, such as immune checkpoint inhibitors and small‐molecule targeted drugs.
Key results
Compared to conventional chemotherapy, several treatments can improve the progression‐free survival of people with metastatic melanoma. These include biochemotherapy (high‐quality evidence), anti‐CTLA4 monoclonal antibodies plus chemotherapy (moderate‐quality evidence), anti‐PD1 monoclonal antibodies (moderate‐quality evidence), BRAF inhibitors (high‐quality evidence), MEK inhibitors (moderate‐quality evidence), and anti‐angiogenic drugs (moderate‐quality evidence). However, no difference was found for use of a combination of several chemotherapy agents (polychemotherapy) (high‐quality evidence). Moreover, the combination of immune checkpoint inhibitors (anti‐PD1 plus anti‐CTLA4 monoclonal antibodies) performed better than anti‐CTLA4 monoclonal antibodies alone (high‐quality evidence), but anti‐PD1 monoclonal antibodies performed better than anti‐CTLA4 monoclonal antibodies (high‐quality evidence). The combination of small‐molecule inhibitors (BRAF plus MEK inhibitors) lead to better results than BRAF inhibitors alone (moderate‐quality evidence), for people with melanoma that has a BRAF gene change .
Anti‐PD1 monoclonal antibodies improved patients' overall survival compared with either standard chemotherapy (high‐quality evidence) or anti‐CTLA4 monoclonal antibodies (high‐quality evidence). Compared to chemotherapy alone, both BRAF inhibitors (high‐quality evidence), and anti‐angiogenic agents combined with chemotherapy (moderate‐quality evidence) also prolong overall survival, but anti‐CTLA4 monoclonal antibodies plus chemotherapy (low‐quality evidence), MEK inhibitors (low‐quality evidence), combined multiple chemotherapeutic agents (polychemotherapy) (high‐quality evidence), or biochemotherapy (high‐quality evidence) did not lead to significantly improved overall survival. WE also found that the combination of small‐molecule inhibitors performed better than BRAF inhibitors alone (high‐quality evidence). No data on overall survival were available for anti‐CTLA4 monoclonal antibodies alone compared with the combination of anti‐CTLA4 plus anti‐PD1 monoclonal antibodies.
In terms of toxicity (defined as occurrence of high‐grade side effects), biochemotherapy (high‐quality evidence), anti‐CTLA4 monoclonal antibodies (moderate‐quality evidence), polychemotherapy (moderate‐quality evidence), and MEK inhibitors (moderate‐quality evidence) were associated with worse toxicity compared to chemotherapy. In contrast, anti‐PD1 monoclonal antibodies appear to be better tolerated than chemotherapy alone. Anti‐PD1 monoclonal antibodies also appeared to be better tolerated than anti‐CTLA4 monoclonal antibodies. However, evidence quality supporting these findings was assessed as low. Furthermore, the frequency of side effects did not differ significantly between anti‐PD1 plus anti‐CTLA4 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies alone (low‐quality evidence), anti‐angiogenic drugs combined with chemotherapy versus chemotherapy (low‐quality evidence), BRAF inhibitors versus chemotherapy (low‐quality evidence), and BRAF plus MEK inhibitors versus BRAF inhibitors alone (moderate‐quality evidence).
We also conducted an analysis that compared treatments that had not been directly compared in a study. This is known as a network meta‐analysis. For the outcome of progression‐free survival, looking at only the best evidence available, we found the following results (please note that because the highest quality level was moderate, the following results can only be deemed probable): • both combination of immune checkpoint inhibitors and combination of small‐molecule targeted drugs were favoured compared to chemotherapy; • both BRAF inhibitors and combination of small‐molecule targeted drugs were favoured compared to anti‐CTLA4 monoclonal antibodies; • biochemotherapy led to less favourable results than BRAF inhibitors; • the combination of small‐molecule targeted drugs was favoured compared to anti‐PD1 monoclonal antibodies; • both biochemotherapy and MEK inhibitors led to less favourable results than the combination of small‐molecule targeted drugs; and • biochemotherapy led to less favourable results than the combination of immune checkpoint inhibitors
For the outcome of toxicity, looking at only the best evidence available, we found the following results (again, evidence quality was no higher than moderate): • combination of immune checkpoint inhibitors led to less favourable results than chemotherapy; • combination of immune checkpoint inhibitors led to less favourable results than BRAF inhibitors; • the combination of immune checkpoint inhibitors led to less favourable results than anti‐PD1 monoclonal antibodies; and • biochemotherapy was favoured compared to the combination of immune checkpoint inhibitors.
Our results suggest that the combination of small‐molecule targeted drugs (BRAF plus MEK inhibitors) is the most effective treatment strategy, for people with melanoma that has a BRAF gene change, at least in terms of progression‐free survival; however, this combination therapy is burdened by a higher rate of severe toxicity compared to effects observed among people treated with anti‐PD1 monoclonal antibodies, which can be used in all melanoma types, and rank highest in terms of tolerability.
These results need long‐term analysis from randomised trials to be confirmed, with special attention to effects on patients' overall survival.
Quality of the evidence
GRADE findings showed that most evidence was high‐ to moderate‐quality for three (overall survival, progression‐free survival and tumour response) out of four outcomes (toxicity). Evidence quality was reduced due to small numbers of participants in some comparisons, differences between the studies, and poor reporting of trials.
Summary of findings
Background
A glossary of terms used is provided in Table 11.
1. Glossary of terms used.
| Term | Explanation |
| Actinomycin‐D | A polypeptide used as an antibiotic and antineoplastic agent as a result of its ability to inhibit transcription |
| AJCC TNM staging | This is the most widely used tumour staging classification system, which has been developed and constantly updated by the American Joint Committee on Cancer (AJCC) for describing the extent of disease progression in people with cancer. It uses in part the TNM scoring system: tumour size, lymph nodes affected, metastases. Individuals affected by specific tumour type are assigned to categories describing risk of death |
| AJCC TNM stage III | People at this disease stage have melanoma metastasis in their regional lymph node (i.e. the first lymph nodes draining the skin area affected by the melanoma) |
| AJCC TNM stage IIIC | Stage IIIC is a higher risk subgroup among people with lymph node metastasis. The category includes people with all primary tumour stages (T stages) and those with clinically positive lymph nodes, or 4 or more positive lymph nodes |
| AJCC TNM stage IV | People with this disease stage have melanoma metastasis to distant sites (e.g. lung, liver, brain, bone) |
| Anti‐angiogenic agents | Drugs aimed to disrupt tumour vascularisation and reduce blood supply to malignant cells; examples include bevacizumab and endostar |
| Antigen | A substance that invokes the body's immune response |
| Aranoza | An alkylating agent that is used as a chemotherapy drug for various cancers including melanoma as part of combination chemotherapy regimens |
| Bacille Calmette‐Guérin (BCG) | BCG is a vaccine used in the prevention of tuberculosis. However, it is also a form of cancer immunotherapy with established effects in superficial (non‐muscle invading) bladder cancer |
| Bevacizumab | Bevacizumab (Avastin) is an angiogenesis inhibitor approved for use for people with various metastatic cancers. Bevacizumab acts through blockade of vascular endothelial growth factor A (VEGF‐A) that prevents development of new vessels necessary for tumours to grow |
| Bleomycin | An antineoplastic agent used in chemotherapy regimens for various tumours. Belomycin acts through cleavage of DNA within cells |
| Biochemotherapy | A combination of chemotherapy plus immunostimulating cytokines, such as interleukin‐2 and interferon‐alpha |
| Bosentan | An endothelin receptor inhibitor that causes reduced DNA synthesis and promotes apoptosis through competitive antagonism with the anti‐apoptotic factor endothelin‐1, often secreted by cancer cells in an autocrine or paracrine manner |
| BRAF | A gene that makes a protein called B‐Raf. BRAF is involved in sending signals within cells that direct their growth. In some cancers, this gene has mutated (Melanoma Institute Australia 2017) |
| Carmustine | An alkylating agent that prevents DNA replication and cell proliferation used in chemotherapy for various cancers |
| Cobimetinib | An inhibitor of MAPK kinase (MEK) approved for use in metastatic melanoma with BRAF V600E/K mutation usually in combination with a BRAF inhibitor |
| Corynebacterium parvum | C parvum is an aerobic, gram positive bacterium that has been reported to have antineoplastic potential |
| Cyclophosphamide | An alkylating agent used in auto‐immune diseases and various tumours as a chemotherapy drug |
| Cytokine | Small proteins produced by a broad range of cells that are important in cell signalling; they are immunostimulating agents |
| Cytotoxic | Cell killing |
| CTLA4 (cytotoxic T‐cell lymphocyte‐associated antigen‐4) | CTLA4 is a receptor located on the surface of T‐cells that down regulates the immune system (an immune checkpoint). The inhibition of this receptor with monoclonal antibodies, such as ipilimumab and tremelimumab, 'unleashes' the immune response to fight against malignant cells |
| Dabrafenib | An inhibitor of the BRAF kinase that has been approved for people with advanced melanoma carrying the BRAF V600E mutation |
| Dacarbazine | A chemotherapy drug that belongs to the family of alkylating agents that is used in the treatment of various cancers, including melanoma |
| Dendritic cell | These are antigen‐presenting cells that link the innate to the adaptive immune systems via processing antigens and presenting them to T‐lymphocytes. Their role is crucial for proper functioning of vaccines, including cancer vaccines |
| Elesclomol | A drug that causes the accumulation of reactive oxygen species to trigger apoptosis in cancer cells via oxidative stress. It is approved for use for people with metastatic melanoma |
| Endostar | A modified recombinant human endostatin that acts as an anti‐angiogenic agent to prevent the formation of new blood vessels that are necessary for tumour growth and survival |
| Fotemustine | A chemotherapy drug that belongs to the family of alkylating agents and has been approved for the treatment of metastatic melanoma |
| G3 and G4 | G3 (grade 3) and G4 (grade 4) toxicity refers to the highest degree of adverse events due to a systemic treatment. This system grades the toxicity related to a given system or organ (e.g. hepatic, cardiac, haematologic) |
| gp100 | A known melanoma antigen that can be applied to develop a cancer vaccine through processing and presentation by dendritic cells to lymphocytes |
| Granulocyte macrophage ‐ colony‐stimulating factor (GM‐CSF) | A cytokine that stimulates stem cells to give rise to granulocytes and monocytes and boosts the immune system |
| Hydroxyurea | A chemotherapy agent that acts through reducing the generation of deoxyribonucleotides, the building blocks of DNA, to inhibit adequate synthesis of DNA. It is used as a chemotherapy drug for people with myeloproliferative disorders |
| Immune checkpoints | Signalling proteins that protect against auto‐immunity and regulate the immune response; these checkpoints can be hijacked by cancer cells to evade T‐cell‐mediated death, i.e. stopping an immune response to the tumour. CTLA4 and PD1 are both immune checkpoints |
| Immune checkpoint inhibitors | Drugs that override the signalling/activation of immune checkpoints to encourage cytotoxic T‐cell recognition of cancer (i.e. an immune response). These are monoclonal antibodies blocking either CTLA4 or PD1 (two immune checkpoints), known as anti‐CTLA4 and anti‐PD1 monoclonal antibodies |
| Immunomodulating | Stimulates or suppresses the immune system |
| Immunostimulating | Stimulates an immune response |
| Interferon‐alpha | Interferon‐alpha is used for the postoperative treatment of people with AJCC TNM stages II (primary tumour at high risk of disease progression with negative lymph nodes) and III (positive lymph nodes) and to enhance the efficacy of chemotherapy in those who have metastatic melanoma |
| Interleukin‐2 | Interleukin‐2 is a protein that regulates the activities of leucocytes (particularly lymphocytes) that are responsible for immunity. The receptor for interleukin‐2 is expressed by lymphocytes. A recombinant form of human interleukin‐2 has been approved by the FDA for the treatment of melanoma and renal cell cancer |
| Lomustine | An oral alkylating chemotherapeutic agent used mainly to treat brain tumours because it crosses the blood‐brain barrier |
| MEK | Mitogen‐activated protein kinase (MEK) is part of the MAPK signalling pathway (see 'RAS‐RAF‐MEK‐ERK pathway' below), which is activated in melanoma |
| Monoclonal antibodies | Monoclonal antibodies are a type of targeted drug therapy; they work by recognising and finding specific proteins on cancer cells (they work in different ways depending on the protein they are targeting) (Cancer Research UK 2017) |
| Oblimersen | A bcl‐2 antisense oligodeoxynucleotide that reduces cancer cell survival and proliferation by blocking the generation of the anti‐apoptotic protein bcl‐2 thus promoting programmed cell death in cancer cells |
| Oncogene | A gene thats activation or over expression favours cancer growth |
| Paclitaxel | A chemotherapy agent targeting the protein tubulin. The drug interferes with the dynamics of microtubule formation and breakdown leading to problems during cell division and triggering of apoptosis. DHA‐ and nab‐paclitaxel are modified forms of the drug |
| PD1 (programmed cell death protein‐1) | PD1 is a receptor located on the surface of the T‐cells that down regulates the immune system (an immune checkpoint). The inhibition of this receptor with monoclonal antibodies, such as nivolumab and pembrolizumab, 'unleashes' immune response to fight against malignant cells |
| PF‐3512676 | An synthetic oligonucleotide that acts as a Toll‐like receptor‐9 (TLR‐9) agonist. It is used as an immunomodulatory agent alone, or in combination with chemotherapy, to boost anti‐tumour effects by enhancing B‐cell proliferation and antigen‐specific antibody production and cytokine secretion |
| Polychemotherapy | A combination of multiple chemotherapeutic agents |
| Procarbazine | An alkylating agent used as an antineoplastic chemotherapy drug in various tumours such as glioblastoma multiforme and Hodgkin's lymphoma |
| Programmed death‐1 (PD‐1) | PD‐1 is an inhibitory receptor located on the surface of the T‐cells that down regulates the immune system when bound by its ligands (PD‐L1 and PD‐L2, often found on cancer cells). The inhibition of this receptor with monoclonal antibodies, such as pembrolizumab and nivolumab, releases the brake on immune cells thus allowing them to freely fight malignant cells |
| Ramucirumab | A human monoclonal antibody that targets the vascular endothelial growth factor receptor 2 (VEGFR2) to block VEGF binding and thus inhibit angiogenesis. It is approved for use in advanced gastric adenocarcinoma and metastatic non‐small cell lung carcinoma |
| RAS‐RAF‐MEK‐ERK pathway | This is also known as 'MAPK/ERK pathway', which is a chain of proteins in the cell that communicates a signal from a receptor on the surface of the cell to the nucleus of the cell (where DNA is located). When one of the proteins in the pathway is mutated, it can be stuck in the 'on' or 'off' position, which is a necessary step in the development of many cancers, including melanoma. Drugs, such as BRAF and MEK inhibitors, can reverse this switch |
| Small‐molecule inhibitors | Low molecular weight drugs targeting molecules mutated or overexpressed in tumours; examples include BRAF inhibitors (which block the BRAF protein) or MEK inhibitors (which block the MEK protein) |
| Sorafenib | An inhibitor of various tyrosine protein kinases including RAF |
| Selumetinib | An inhibitor of the MAPK kinase (MEK) downstream of BRAF |
| T‐cell | A white blood cell type, which plays a key role in immunity |
| Tasisulam | A small‐molecule agent that induces apoptosis through the intrinsic mitochondrial pathway |
| Tamoxifen | A cytostatic hormonal therapeutic agent used mainly as a treatment for oestrogen receptor positive breast cancer. Tamoxifen acts through competing with oestrogen for its receptor thus reducing oestrogen‐related effects in breast tissue such as DNA synthesis and cell proliferation |
| Temozolomide | An oral alkylating agent that can be used in chemotherapy regimens for various cancers such as glioblastoma multiforme |
| Trametinib | An inhibitor of MAPK kinase (MEK) 1 and 2 approved for use in people with V600E‐mutated metastatic melanoma |
| Vemurafenib | A small‐molecule inhibitor of mutated BRAF, an oncogene involved in cell survival or proliferation |
| Vincristine | An anti‐mitotic agent that binds tubulin thus preventing cell proliferation and triggering apoptosis |
| Vindesine | An anti‐mitotic agent that acts by targeting microtubules and preventing cell division thus useful as a chemotherapy drug in various cancers |
| Vitespen | A tumour‐derived heat shock protein that is used as an adjuvant in cancer immunotherapy |
Description of the condition
Cutaneous melanoma is one of the deadliest forms of skin cancer. According to epidemiological data provided by the International Agency for Research on Cancer (IARC), its worldwide incidence in 2008 was estimated to be 199,627 new cases, with 46,372 deaths (Ferlay 2010). In the USA, cutaneous melanoma ranked fifth in men (44,250 new cases per year, representing 5% of all cancers) and sixth in women (32,000 new cases per year, representing 4% of all cancers) among all tumour histotypes (Siegel 2012). The highest incidence is observed in Australia and New Zealand where melanoma is the fourth most commonly diagnosed cancer (Australian and New Zealand 2008).
Melanoma incidence differs widely across Europe, ranging from 19.2/100,000 persons per year in Switzerland to 2.2/100,000 persons per year in Greece (Forsea 2012). As well as geographical differences, melanoma incidence has been increasing worldwide over the past 30 years at a greater pace than any other malignancy (Little 2012; Siegel 2012), which makes its management a key issue for national healthcare systems. Melanoma is potentially curable in the early stages with the surgical removal of the primary tumour (McKinnon 2005; Mocellin 2011; Pasquali 2013; Sladden 2009).
Once melanoma metastasises (i.e. spreads to lymph nodes, distant organs or both) due to its intrinsic biological aggressiveness and its typical resistance to medical therapy (both chemotherapy and radiotherapy) (Serrone 1999), survival is poor or very poor, with a median overall survival of 24 months for those with American Joint Committee on Cancer (AJCC) TNM stage IIIC disease (unresectable lymph node metastasis), and nine months for people with AJCC TNM stage IV disease (distant metastasis) (Balch 2001; Balch 2009). Overall, fewer than 35% (AJCC TNM stage IIIC) and 12% (AJCC TNM stage IV) of these people are still alive five years after their diagnosis (Balch 2001; Balch 2009).
Metastatic cutaneous melanoma (unresectable AJCC TNM stage IIIC and stage IV) is usually treated with systemic medical therapy (Garbe 2011), and is characterised by a dismal prognosis (median overall survival usually ranges between 10 and 16 months, Balch 2009). Surgery is feasible only in very few select cases showing a very limited tumour burden (Gyorki 2013; Wevers 2013), and radiotherapy is considered only for symptom palliation (Stevens 2006; Testori 2009).
New insights into the prognosis of people with metastatic melanoma come from molecular profiling of primary tumour and distant metastases. Recently, molecular studies have identified aberrant activation of the mitogen‐activated protein kinase (MAPK) pathway and mutations in proteins along the RAS‐RAF‐MEK‐ERK pathway (Figure 1) in cutaneous (50% BRAF‐mutated, 15% NRAS‐mutated, and up to 17% c‐Kit‐mutated in chronically sun damaged people) and mucosal melanoma (11% BRAF‐mutated, 5% NRAS‐mutated, 21% c‐Kit‐mutated) (Scolyer 2011). Determination of the mutational status of a melanoma enables identification of those who may be suitable for new treatments, such as BRAF and c‐Kit inhibitors.
1.
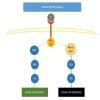
RAS‐RAF‐MEK‐ERK pathway. Copyright © 2018 Claire Gorry: reproduced with permission.
Description of the intervention
Until 2011, conventional treatment for those who have metastatic melanoma included the chemotherapeutic alkylating agent dacarbazine (and its orally available derivative, temozolomide) and the immunostimulatory cytokine, interleukin‐2 (approved for metastatic melanoma treatment only in the USA). However, neither drug has been shown to provide any significant survival benefit in a randomised controlled trial (RCT) (Garbe 2011). When dacarbazine was associated with other chemotherapeutic agents (polychemotherapy) or immunostimulatory cytokines such as interferon‐alpha or interleukin‐2 (biochemotherapy), only some improvement in tumour response without any survival advantage was reported (Ives 2007).
Different immunotherapy regimens (including biotherapy and vaccination regimens) can lead to tumour shrinkage and confer a durable and complete response in some people who have this condition. This prompted investigators to test newer immunomodulating agents including the immune checkpoint inhibitor ipilimumab, a monoclonal antibody blocking the T‐cell lymphocyte‐associated antigen‐4 (i.e. CTLA4, a co‐inhibitory molecule involved in the control of immune responses mediated by T‐lymphocytes) (Kirkwood 2008; Kirkwood 2012; Mocellin 2013b). In 2010, the anti‐CTLA4 strategy was the first treatment demonstrated to be associated with a survival advantage for people with metastatic melanoma (Hodi 2010).
The breakthrough results obtained with anti‐CTLA4 monoclonal antibodies have changed the perspective of melanoma therapy along with another pivotal discovery, which is the impressive tumour response rates (up to 90%) observed with vemurafenib (a small‐molecule inhibitor of mutated BRAF, an oncogene involved in cell survival or proliferation) (Arkenau 2011) in participants with metastatic melanoma harbouring BRAF activating mutations (Flaherty 2012; Long 2012; Sosman 2012).
Agents that have been tested in RCTs for the systemic treatment of metastatic melanoma can be categorised into five main groups based on their predominant mechanism of action (Garbe 2011; Ives 2007; Kirkwood 2012; Arkenau 2011):
conventional chemotherapy (which act mainly through DNA damage);
biochemotherapy (combination of chemotherapy plus immunostimulating cytokines);
immune checkpoint inhibitors (which override the signalling/activation of immune checkpoints, which have been hijacked by cancer cells to evade T‐cell‐mediated death, thus stimulating the immune system against malignant cells);
small‐molecule targeted drugs (which inhibit the protein products of oncogenes specifically activated in malignant cells); and
a miscellany of other treatments (such as anti‐angiogenic drugs, which inhibit cancer vascularisation).
Conventional chemotherapy
Dacarbazine has been the mainstay of metastatic melanoma therapy (and thus the reference drug for this disease) for over three decades. Dacarbazine was approved for the treatment of metastatic melanoma by the USA Food and Drug Administration (FDA) in 1975, although its efficacy in terms of survival has never been proven in a RCT (Crosby 2000; Huncharek 2001). Dacarbazine is an alkylating agent that produces DNA damage by adding a methyl group to the guanine base in the O6 position. Ultimately, the DNA damage caused by dacarbazine is believed to prompt programmed cell death (apoptosis) (National Toxicology Program 2011). Several trials have tested the hypothesis that dacarbazine‐based polychemotherapy regimens might be more effective than dacarbazine alone; however, these trials showed only some improvement in tumour response rates without showing any convincing survival benefit (Bajetta 2006; Ridolfi 2002). These disappointing results led people to consider cutaneous melanoma as one of the most chemoresistant tumours in humans (La Porta 2007; La Porta 2009).
Biochemotherapy
In the oncology field, the term 'biotherapy' generally refers to the use of cytokines to treat cancer. We focused on two cytokines that have been extensively tested for the treatment of people with melanoma: interferon‐alpha and interleukin‐2.
Interferon‐alpha was the first cytokine that demonstrated activity in metastatic melanoma, with 10% to 20% tumour response being observed (Belardelli 2002; Schadendorf 2009). The main mechanism of action of interferon‐alpha is immunostimulation, although other mechanisms have been hypothesised (antiproliferative, differentiation‐inducing, pro‐apoptotic, and anti‐angiogenic) (Pasquali 2010). Interferon‐alpha is the only drug currently approved for the adjuvant (i.e. postoperative) treatment of melanoma after radical removal of regional lymph‐node metastasis by surgery (AJCC TNM stage III) (Eggermont 2009; Garbe 2011; Mocellin 2010; Mocellin 2013).
Interleukin‐2 is an immunostimulant cytokine mainly involved in T‐cell proliferation (Kirkwood 2012). When tested in people with metastatic melanoma, interleukin‐2 showed a 15% to 20% response rate (4% of long‐term responses) (Schwartzentruber 2011; Tarhini 2005). Interleukin‐2 treatment is burdened by a remarkable (although reversible) toxicity usually requiring hospitalisation (and sometimes admission to an intensive care unit) for management.
Biotherapy agents have been coupled with chemotherapy agents (a combination called biochemotherapy) and compared to chemotherapy alone (Ives 2007). Generally, biochemotherapy has shown higher tumour response rates compared to chemotherapy, but significant improvement in survival of people with metastatic melanoma does not appear to be achievable with this approach (Hamm 2008; Keilholz 2002).
Immune checkpoint inhibitors
Melanoma is considered to be a form of immunogenic tumour (able to produce an immune response) on the basis of some spontaneously occurring melanoma regressions and some durable tumour responses observed after treatment with a variety of immunostimulating agents (Kirkwood 2008; Kirkwood 2012). The higher mutation rate observed in primary and metastatic melanoma compared with other tumour types has been suggested as the mechanism behind melanoma immunogenicity (Mocellin 2003). In particular, mutated proteins might represent tumour‐specific antigens (a substance that invokes the body's immune response) that can be selectively recognised by the immune system on melanoma cells. Moreover, melanoma cells often express epitopes derived from proteins involved in melanin synthesis, which makes them suitable for tumour‐selective immune treatment (Mocellin 2009).
Several attempts have been made to activate the immune system against cancer cells. However, it appears evident that tumours can easily elude both naturally occurring and vaccine‐elicited immune surveillance (Mocellin 2008) and metastasise to distant sites. Therefore, investigators have turned their attention to these mechanisms of tumour‐immune escape. It has been found that malignant cells can evade the body's natural immune response through immunosuppressive circuits whose activity is mediated by specific molecules (such as CTLA4 and PD1) collectively named immune checkpoints (Hamid 2013; Mocellin 2013a; Ribas 2013).
Therefore, a new paradigm in cancer treatment emerged when investigators found that anti‐CTLA4 monoclonal antibodies (e.g. ipilimumab) can improve the survival of people with metastatic melanoma by inhibiting the CTLA4 checkpoint and ultimately unleashing the immune response against malignant cells (Hodi 2010). Since then, several RCTs have been conducted or are under way out to test the efficacy of this new strategy in melanoma (Robert 2011) as well as in non‐melanoma cancers (Kirkwood 2012).
Small‐molecule targeted drugs
Although the expression 'targeted therapy' usually refers to a variety of therapeutic strategies selectively targeting cancer‐specific molecular derangements, for the sake of clarity regarding treatment classification, we exclusively referred to the use of small‐molecule inhibitors of oncogenes specifically activated in malignant melanoma cells (Mocellin 2010a; Thompson 2009).
Molecular biological studies have demonstrated that melanoma cells harbour a range of gene or protein alterations that can be targeted to develop tumour‐specific therapies (Thompson 2009). For instance, about 65% of melanomas harbour mutations affecting the RAS‐RAF‐MEK‐ERK pathway (Davies 2002; Long 2011). The drugs (small‐molecule inhibitors) targeting this pathway, such as sorafenib (a RAF inhibitor) and selumetinib (a MEK inhibitor), showed limited antitumour activity in participants with metastatic melanoma (Flaherty 2013; Hauschild 2009; Kirkwood 2012a). In contrast, high tumour response rates (up to 90%) were observed when BRAF inhibitors (with or without MEK inhibitors) were tested in people with metastatic melanoma harbouring activating mutations of the BRAF gene (the most common is known as V600E because the amino acid valine (V) is substituted by glutamic acid (E) at position 600 of the protein BRAF) (Hauschild 2012; McArthur 2014). These mutations constitutionally activate the BRAF kinase, which ultimately stimulates cell proliferation and opposes apoptosis (therefore, mutated BRAF acts as an oncogene). Although complete responses are uncommon (< 5%), these drugs prolong the survival of those who have BRAF‐mutated metastatic melanoma (compared to traditional dacarbazine treatment) (Sosman 2012). After this breakthrough discovery, several RCTs have been completed and others are under way to test the efficacy of this new strategy in melanoma as well as in non‐melanoma cancers harbouring the mutated version of BRAF as well as other molecular derangements (Klein 2013; Menzies 2013). Similarly, c‐Kit inhibitors have been tested in people with metastatic melanoma harbouring activating mutations of the c‐Kit protein kinase (Guo 2011; Scolyer 2011).
Other treatments (including anti‐angiogenic drugs)
Other strategies have been investigated to treat metastatic melanoma, which cannot be classified to the nominated five drug classes. For instance, as in the field of infectious diseases, vaccines (such as those targeting gp100, a melanoma associated antigen) can be used to manipulate the host immune system to elicit a tumour‐specific immune response against malignant tumours (Mocellin 2005). This strategy, known as active‐specific immunotherapy because it chiefly involves the adaptive immune response, has long been tested in oncology, mainly in people with cutaneous melanoma (Mocellin 2004). Despite the promising preclinical evidence and the variety of vaccination regimens tested so far, no vaccine formulation has been proven to significantly change the natural history of metastatic melanoma (Chi 2011). However, in 2011, a RCT showed that the combination of a gp100‐based vaccine with interleukin‐2 provided a survival advantage for people who have metastatic melanoma (Schwartzentruber 2011). Other immunostimulating agents, such as naturally occurring growth factors (e.g. granulocyte and macrophage colony stimulating factor (GM‐CSF)) and bioproducts from bacteria (e.g. Bacillus Calmette‐Guérin (BCG) and Corynebacterium parvum), have been tested in clinical trials, usually in combination with other agents, but results have generally been unsatisfactory (Mocellin 2008).
Promising results have been recently reported with anti‐angiogenic agents, a class of drugs aimed to reduce blood supply to malignant cells (Ashour 2017). This approach has been proven to be effective against a variety of tumour types, such as colorectal cancer (Jayson 2016), but investigation in those with melanoma is still in its infancy (Cui 2013; Kim 2012).
A miscellany of anticancer agents have also been tested in association with chemotherapy to increase the efficacy of conventional cytotoxic drugs. Among these agents there are anti‐oestrogenic drugs (e.g. tamoxifen, a medication widely used against breast cancer) (Jager 2015), multi‐kinase inhibitors (e.g. sorafenib, a small‐molecule inhibitor approved for the treatment of different solid tumours such as kidney carcinoma) (Gentile 2017), and drugs with pro‐apoptotic properties (e.g. elesclomol, a compound supposed to increase the activity of chemotherapy by generating reactive oxygen species) (Caino 2016).
Why it is important to do this review
Many systemic treatments have been and continue to be tested for the management of metastatic cutaneous melanoma, although only recent results appear to provide affected people with new hope to improve life expectancy. No systematic reviews or meta‐analyses have been performed on all systemic therapies tested so far for the treatment of metastatic skin melanoma. Two previous Cochrane Reviews (Crosby 2000; Sasse 2007) partially covered the chemotherapy (chemotherapy versus best supportive care) and the biochemotherapy (biochemotherapy versus chemotherapy) fields, respectively. This review updates both previous Cochrane Reviews and broadened the scope. Since the reviews were published, many trials have been conducted to test new chemotherapeutic regimens based on conventional cytotoxic chemotherapeutics; traditional immunotherapy (e.g. interleukin‐2, interferon‐alpha); and most of all, new agents, including co‐inhibitory molecular inhibitors (such as the anti‐CTLA4 or anti‐PD1 monoclonal antibodies) and small molecular inhibitors (such as BRAF and MEK inhibitors).
Therefore, it is of utmost importance to provide physicians (especially oncologists and dermatologists) and investigators involved in melanoma treatment and research with a systematic assessment, and where feasible, meta‐analysis of the available evidence regarding the therapeutic regimens tested in RCTs to date. We planned to descriptively and quantitatively summarise the evidence in this field and provide readers with coverage of the therapeutic efficacy as well as toxicity, quality of life, and economic burden issues.
A protocol for this review has been published (Pasquali 2014). Gorry 2018 (currently at protocol stage) will assess neoadjuvant treatment for malignant and metastatic cutaneous melanoma.
Objectives
To assess the beneficial and harmful effects of systemic treatments for metastatic cutaneous melanoma.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) testing systemic therapies for the treatment of metastatic cutaneous melanoma.
Types of participants
People with unresectable lymph node metastasis (AJCC TNM stage IIIC) and distant metastatic (AJCC TNM stage IV) cutaneous melanoma. No restrictions in terms of age, sex, drug dosage, radiologic examination, or treatment duration were applied.
Types of interventions
We considered all comparisons of systemic therapies for the treatment of metastatic cutaneous melanoma, including:
polychemotherapy (experimental arm) versus single‐agent chemotherapy (comparator arm);
biochemotherapy (experimental arm) versus chemotherapy (comparator arm);
immune checkpoint inhibitors (experimental arm) versus any other agent (comparator arm);
small‐molecule targeted drugs (experimental arm) versus any other agent (comparator arm);
chemotherapy plus other agents (e.g. anti‐angiogenic drugs) (experimental arm) versus chemotherapy alone (comparator arm); and
other comparisons (e.g. single agent chemotherapy verus other single agent chemotherapy).
Types of outcome measures
Primary outcomes
Overall survival: defined as time from randomisation until death from any cause (effect measure: hazard ratio (HR)).
Progression‐free survival: defined as time from randomisation until diagnosis of disease recurrence (local or distant/metastatic) (effect measure: HR).
Toxicity: defined as the occurrence of grade 3 (G3) or higher adverse events according to the World Health Organization (WHO) scale (Brundage 1993) (effect measure: relative risk (RR)).
Secondary outcomes
Tumour response: defined as incidence of complete plus partial tumour response according to WHO or Response Evaluation Criteria In Solid Tumors (RECIST) criteria (Therasse 2002) (effect measure: RR).
Quality of life (since there are no standardised disease‐specific scales and questionnaires to assess the quality of life of people with cutaneous melanoma, we described findings from studies).
Economic evaluation (expressed as cost‐utility analysis with the quality‐adjusted life years (QALYs)).
Search methods for identification of studies
We aimed to identify all relevant RCTs regardless of language or publication status (published, unpublished, in press, or in progress).
Electronic searches
We searched the following databases up to 4 October 2017:
the Cochrane Skin Group Specialised Register using the search strategy 'melanoma and (metastatic or metastas* or "stage iv" or "stage 4")';
the Cochrane Central Register of Controlled Trials (CENTRAL) 2017, Issue 9, in the Cochrane Library using the strategy in Appendix 1;
MEDLINE via Ovid (from 1946) using the strategy in Appendix 2;
Embase via Ovid (from 1974) using the strategy in Appendix 3; and
LILACS (Latin American and Caribbean Health Science Information database, from 1982) using the strategy 'melanoma and metasta$'.
We also searched the American Society of Clinical Oncology (ASCO) database up to February 2017 using the terms "melanoma", "randomised" and "metastatic".
Trials registers
We searched the following trials registers up to February 2017 using the key words "melanoma" and "randomised":
ISRCTN registry (www.isrctn.com);
ClinicalTrials.gov (www.clinicaltrials.gov);
Australian New Zealand Clinical Trials Registry (www.anzctr.org.au);
World Health Organization International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/); and
EU Clinical Trials Register (www.clinicaltrialsregister.eu).
Searching other resources
References from included studies
We checked the references of included studies for further references to relevant trials.
Adverse effects
We did not perform a separate search for adverse effects of the target interventions. However, we examined data on adverse effects from the included studies we identified.
Data collection and analysis
Selection of studies
Two review authors (SM and SP) selected trials independently by checking the titles and abstracts identified using the search methods described. The same two review authors retrieved the full text of all possibly relevant studies and assessed the eligibility of each study. We resolved discordant evaluations by discussion to reach consensus. We included trials with mixed disease stages if they reported outcomes separately for metastatic disease.
Data extraction and management
Two review authors (SM and SP) independently compared similarity among studies eligible for inclusion in terms of interventions and outcomes. The same two review authors also extracted relevant data for colation in a database. Review authors extracted the following details were extracted using a data extraction form that had been piloted previously:
Trial methods, sequence generation, method of concealment of allocation, masking of participants, trialists, and outcome assessors, exclusion of participants after randomisation, proportion and reasons for losses at follow up.
Participants' country of origin and study setting, sample size, tumour stage, inclusion and exclusion criteria.
Intervention group, type of treatment, dose and frequency, duration of intervention and follow up.
Control group, type of treatment, dose and frequency, duration of intervention and follow up.
Outcomes: primary and secondary outcomes as specified in Types of outcome measures.
A third review author (AH) independently verified the extracted data. We resolved discordant evaluations on all data necessary for the final analysis by discussion and final consensus. The review authors were not blinded to the names of trial authors, journals where the trial results were published, or institutions where the trials were conducted. In case of multiple publications reporting on the same RCT, we chose the most recent and complete publication.
Assessment of risk of bias in included studies
Two review authors (SM and SP) independently assessed the included studies in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The review authors compared their evaluations and resolve possible inconsistencies.
We assessed the risk of bias in included trials by considering the following aspects:
the method of generation of the randomisation sequence;
the method of allocation concealment;
the blinding of participants, clinicians, and outcome assessors;
the presence of incomplete outcome data; and
selective outcome reporting.
This information is recorded in a 'Risk of bias' table, which is part of the Characteristics of included studies table for each study.
We reported the risk of bias for each study in accordance with the Cochrane Handbook for Systematic Reviews of Interventions:
low risk of bias (plausible bias unlikely to seriously alter the results) if all criteria were met;
unclear risk of bias (plausible bias that raises some doubt about the results) if one or more criteria were assessed as unclear; or
high risk of bias (plausible bias that seriously weakens confidence in the results) if one or more criteria were not met.
Measures of treatment effect
Overall survival and progression‐free survival
We measured the treatment effect on participant survival as hazard ratios (HR), which is defined as the ratio between the risk of event in the experimental arm and the same risk in the comparator arm participants. We reported each HR along with its 95% confidence interval (CI). HR values lower or greater than one indicate a favourable or unfavourable effect of the experimental versus the comparator treatment, respectively.
We extracted all available summary statistics from all reports of the included trials for the outcome measures considered. We extracted HRs directly from original studies when reported; if unreported, we calculated HRs from Kaplan‐Meier survival curves using dedicated methods (Parmar 1998; Tierney 2007). Whenever feasible, unadjusted HRs were used.
As well as HRs (which is a relative measure of treatment effect), we also provided readers with an absolute measure of treatment effect. To achieve this aim, we used the calculated summary HRs (obtained from meta‐analysis of eligible trials) and the one‐year overall (or progression‐free) survival rate in the control population of participants with metastatic cutaneous melanoma; we then calculated the mortality (or progression) rates in the experimental and control groups (reported in 'Summary of findings' tables) using methods described by Altman (Altman 1999; Altman 2002). Briefly, if at some specified time (t) the survival probability in the control group is Sc(t), then the survival probability in the active group is [Sc(t)]h, where h is the meta‐analysis HR comparing the treatment groups: mortality rates are then simply calculated as 1‐S. These absolute risks (events rates) can be used to simply calculate the absolute risk reduction (ARR = event rate for experimental treatment minus event rate for comparison treatment), which can be in turn used to calculate the number needed to treat for an additional beneficial outcome (NNTB = 1/ARR) (Higgins 2011).
In the event that some studies presented their findings as odds ratios (OR) for death at different time points (rather than reporting the preferred measure HR) (Case 2002), we considered the reported OR as surrogate measure of treatment effect on the survival outcome of interest; we then used sensitivity analysis to investigate the potential influence of this suboptimal measure of treatment effect on the results of meta‐analysis of time‐to‐event (survival) data.
Tumour response
We measured the treatment effect on tumour response as risk ratio (RR), that is, the ratio between the overall response rate in the experimental arm and that in the comparator arm. According to this definition, the RR corresponds to the rate of complete or partial responses in the experimental treatment compared to the comparator. We reported each RR along with its 95% CI. RR values higher or lower than one indicate a favourable or unfavourable effect of the experimental versus the comparator treatment, respectively.
Toxicity
We measured the treatment effect on treatment‐related side‐effects (toxicity) as RR, that is, the ratio between the toxicity rate in the experimental arm and that in the comparator arm. We reported each RR along with its 95% CI. RR values lower or higher than one indicate a favourable or unfavourable effect of the experimental versus the comparator treatment, respectively.
Quality of life and economic analysis
We expected that no homogeneous data would be available from the literature for quality of life because of the lack of a melanoma‐specific questionnaire. Lack of homogeneity may prevent pooling of data; in this case, we descriptively reported data.
When dealing with economic analysis, we considered cost‐utility analysis with quality‐adjusted life years.
Unit of analysis issues
Cross‐over and cluster‐design trials
Because cross‐over trials (where each participant is allocated not to a single intervention ‐ as happens in parallel group trials ‐ but to a sequence of treatments) are typically used to assess treatments with a temporary effect in the management of stable (i.e. chronic) disease, we did not expect to find cross‐over trials dedicated to the treatment of metastatic melanoma, usually (and unfortunately) a rapidly evolving condition. However, we did not want to exclude these types of studies a priori, should any have been found. Such trials would require special methods to be included in a meta‐analysis (e.g. considering the findings specific for the first treatment, if available) to avoid the 'carry over' effect (i.e. the impact of the second treatment may be affected by a the effect of the first treatment), as recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Moreover, sensitivity analysis to asses the impact of such design trials on summary effects would be performed.
Similarly, although we were unaware of cluster design trials, we did not want to exclude these types of studies a priori, should any have been found. In this case, sensitivity analysis to asses the impact of such design trials on summary effects would have been performed.
Studies with multiple treatment groups
For multiple‐arm trials that compared two (or more) experimental arms with the same control arm, we took within‐study correlation into consideration as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We computed a composite effect size for the comparison of each experimental arm versus the control arm; we then calculated the correlation factor (r) based on the number of cases in each arm, which enabled us to compute the variance (V) of the composite effect size, as suggested by Borenstein and Higgins (Borenstein 2009). Using this variance, we computed the standard error and then the 95% CI of the composite effect.
Network meta‐analysis
Given that direct comparisons between key therapies had not been published (e.g. immune checkpoint inhibitors versus small‐molecule targeted drugs), we used the network meta‐analysis methodology to compute estimates of indirect comparisons and generate treatment ranking (Cipriani 2013; Mills 2013). To perform this network meta‐analysis, studies need to satisfy the principle of transitivity. For instance, indirect comparisons can be performed when different trials share the same participant population in terms of first‐ or second‐line treatment and presence or absence of severe clinical conditions, such as brain metastasis. We then evaluated consistency (i.e. heterogeneity) within loops (e.g. for a comparison between therapies A and B, the included study must have directly compared A and B and both treatments with a third common comparator, C) using the methods for assessing heterogeneity as described. We used a random‐effects model to estimate HR (progression‐free survival and overall survival) and RR (tumour response and toxicity). We also used multivariate random‐effects meta‐regression to estimate consistency and inconsistency. We performed analyses using the 'mvmeta' package (Chaimani 2013; White 2011) for Stata (Stata 2017).
Dealing with missing data
We contacted trial authors for clarification where data were missing or unclear.
We extracted results for intention‐to‐treat analysis whenever provided. In studies reporting per‐protocol analysis results only, we performed an available‐case analysis.
Assessment of heterogeneity
We assessed the consistency of results (effect sizes) among studies using the two standard heterogeneity tests: the Chi² based Cochran Q‐test and the I² statistic (Higgins 2011). To be more conservative, we considered that heterogeneity was statistically substantial when the Cochran Q‐test P value was less than 0.1 (i.e. the alpha level of significance for this test was set at 10%). In addition, we considered inconsistency across studies as low, moderate, and high for I² statistic values lower than 25%, between 25% and 50%, and greater than 50%, respectively. We considered heterogeneity as significant when the I² statistic was greater than 50%, the Q‐test P value was less than 0.1, or both. We applied the random‐effects model to calculate the overall effect (which assumes that studies do not share the same common effect and assigns a weight to each study taking into account both within‐study and between‐study variance), using the inverse‐variance method.
Assessment of reporting biases
We planned to construct funnel plots to detect publication and small study effect biases if we included at least 10 studies in meta‐analysis (Borenstein 2009; Higgins 2011). We planned to investigate funnel plot asymmetry with the Egger linear regression approach and the Begg rank correlation test (these tests will be considered statistically significant for P values less than 0.1). To avoid duplicate study bias, we only considered the study with the longest follow‐up length when multiple reports for the same trial were available.
Data synthesis
Two review authors (SM and SP) performed all meta‐analyses according to the guidelines reported in Chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
For time‐to‐event (i.e. survival) outcomes, we used RevMan 5.3 (RevMan 2014) to estimate pooled HRs and 95% CIs using the random effects model (Borenstein 2009; Higgins 2011).
For binary outcomes, we used RevMan 5.3 to estimate pooled RRs and 95% CIs using the random effects model.
For the network meta‐analysis we used the 'mvmeta' package (Chaimani 2013; White 2011) for Stata (Stata 2017).
We planned to include at least one 'Summary of findings' table for the primary outcomes for the most important comparison. We also planned inclusion of further 'Summary of findings' tables where there were several major comparisons or need to summarise findings for different populations. We used the GRADE approach to assess the quality of evidence for all primary and key secondary outcomes for all main comparisons. We considered downgrading evidence based on five domains: risk of bias, inconsistency, imprecision, indirectness; and publication bias. Overall quality of evidence could be assessed as high, moderate, low or very low (Guyatt 2008; Higgins 2011).
Subgroup analysis and investigation of heterogeneity
We performed subgroup analysis and meta‐regression to investigate potential sources of between‐study heterogeneity. Planned subgroups or covariates included: year of publication, untreated or previously treated distant metastasis, inclusion or exclusion of brain metastasis, and duration of follow‐up. Further details of investigation of heterogeneity are presented in Assessment of heterogeneity.
Sensitivity analysis
We investigated potential sources of between‐study heterogeneity by excluding trials at high risk of bias and each single trial to ascertain their role in affecting summary statistics.
Results
Description of studies
Results of the search
The database searches (see Electronic searches) retrieved 4303 records. We also identified 19 ongoing studies (see Characteristics of ongoing studies). We excluded 4157 references based on titles and abstracts. We obtained the full text of the remaining 146 studies. We excluded 24 studies (see Characteristics of excluded studies), and included 122 studies (Characteristics of included studies). See the study flow diagram for a full description of our screening process (Figure 2).
2.
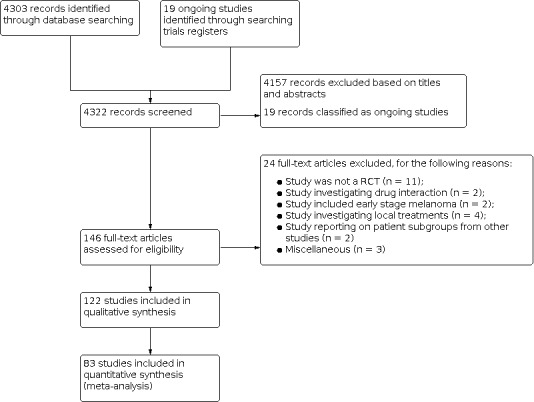
Study flow diagram.
Included studies
Review findings were based on data reported in the full‐text reports of the 122 included randomised controlled trials (RCTs). Descriptions of studies are presented in Characteristics of included studies.
Design
Most included studies were phase III RCTs (n = 76, 62%) or phase II RCTs (n = 41, 34%). We also included one phase I RCT and RCTs with mixed designs (n = 4, 3%). All trials were designed as parallel‐group studies (neither cross‐over trials nor cluster design trials were found for inclusion).
Double‐blinding design was employed in 23 trials (19%) (Cui 2013; Eisen 2010; Flaherty 2013a; Glaspy 2009; Gupta 2014; Hauschild 2009a; Hodi 2010a; Kefford 2010; Kim 2012; Larkin 2015; Lawson 2015; Long 2015; McDermott 2008; Middleton 2015; O'Day 2009; O'Day 2011; O'Day 2013; Postow 2015; Robert 2011; Robert 2013; Robert 2015a; Rusthoven 1996; Wolchok 2010). The remaining 99 studies (81%) were open label design.
In many cases, trials were sponsored by pharmaceutical companies producing the tested drug: this was especially true for new classes of drugs, such as immune checkpoint inhibitors and small‐molecule targeted drugs.
Sample sizes
There was significant variation in sample size among the included RCTs, ranging from 30 (Gorbonova 2000) to 945 (Larkin 2015) participants.
Participants
Overall, the 122 RCTs randomised 28,561 participants. Eighty‐nine trials (73%) were conducted in untreated participants (N = 20,737). Previously treated participants (N = 3450) were enrolled in 30 trials (25%): in 20 of these RCTs both untreated and previously treated participants were enrolled. In three trials systemic treatments were administered after surgery for distant metastasis (2%). Included studies were conducted in adults with no restriction for enrolling both men and women (mean men:women ratio = 1.38). Mean age was 57.5 years (range: 18 to 87 years). Participants with brain metastasis (N = 741) were included in 29 studies (24%), although definitions for allowing inclusion of this condition differed across trials (Characteristics of included studies). All trials enrolled participants from a hospital, with unresectable locoregional disease (AJCC TNM stage IIIC) or metastatic cutaneous melanoma (AJCC TNM stage IV). Many reports stated “metastatic or locoregionally advanced disease”, but then did not report data separately.
Interventions
All studies investigated systemic treatments as per eligibility criteria. Several drugs and schedules were tested. Description of drugs and scheduled for each study are reported in Characteristics of included studies tables. Overall, dacarbazine was the most used drug across the trials (n = 50, 46%). The following treatment comparisons were investigated:
polychemotherapy (experimental arm) versus single‐agent chemotherapy (comparator arm): 21 RCTs;
biochemotherapy (experimental arm) versus chemotherapy (comparator arm): 34 RCTs;
immune checkpoint inhibitors (experimental arm) versus any other agent (comparator arm): 11 RCTs;
small‐molecule targeted drugs (experimental arm) versus any other agent (comparator arm): 9 RCTs;
chemotherapy plus other agents (e.g. anti‐angiogenic drugs, tamoxifen, elesclomol) (experimental arm) versus chemotherapy alone (comparator arm): 34 RCTs; and
other comparisons (e.g. single agent chemotherapy versus other single agent chemotherapy): 13 RCTs.
Outcomes
We evaluated the following outcomes for each study:
progression‐free survival: 89 RCTs (73%);
overall survival: 105 RCTs (94%);
tumour response: 117 RCTs (96%);
toxicity: 118 RCTs (97%);
participants' quality of life: 12 RCTs (11%); and
cost analysis: 1 RCT (< 1%).
Excluded studies
We reported the reasons for exclusion of 24 studies in the Characteristics of excluded studies. The reasons for exclusion were that the study: was not a randomised trial (n = 11); investigated mechanisms of action of a drug (or drug interaction with other drugs) (n = 2); investigated early stage melanoma (not advanced/metastatic melanoma) (n = 2); investigated either local or loco‐regional therapies (n = 4); investigated subgroups of participants of particular interest from RCTs already included in this review (n = 2); investigated both melanoma and other tumour types, but melanoma‐specific data could not be extracted (n = 1); gathered data from three RCTs already included in this review (n = 1); and reported the preliminary results of a RCT already included in this review (n = 1).
Ongoing studies
We searched for phase III RCTs, either open to recruitment or following up participants, investigating participants with metastatic melanoma. We identified open studies in 'recruiting and 'not yet recruiting' phases and active studies not yet recruiting.
We identified 19 phase III RCTs (see Characteristics of ongoing studies). These studies will investigate two new classes of anticancer drugs for melanoma (i.e. immune checkpoint inhibitors ipilimumab, nivolumab, and pembrolizumab; and the targeted drugs dabrafenib, vemurafenib, and trametinib) in tumours harbouring mutations in proteins other than BRAF, such as NRAS, which is also believed to play a role in melanoma progression. Studies also investigate combinations of these drugs and in association with other agents, such as interferon‐alpha and interleukin‐2.
Studies awaiting classification
There are no studies awaiting classification.
Risk of bias in included studies
Figure 3 and Figure 4 summarise the risk of bias for included studies.
3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Overall, the risk of bias of included studies can be considered as limited. Considering the 122 included studies and the seven bias domains assessed, we performed 854 evaluations (Figure 4): only seven evaluations (< 1%) assigned high risk of bias for six trials (Beretta 1976; Carvajal 2014; Hamid 2014; Hofmann 2011; Ranson 2007; Richtig 2004). We assessed that only 21 studies (17%) were at low risk of bias for all domains (Bedikian 2006; Cui 2013; Eisen 2010; Flaherty 2012b; Flaherty 2013a; Glaspy 2009; Hauschild 2009a; Hersh 2015; Hodi 2010a; Larkin 2015; Lawson 2015; Long 2015; McDermott 2008; O'Day 2013; Ribas 2015; Robert 2013; Robert 2015a; Schadendorf 2006; Schwartzentruber 2011a; Weber 2015; Wolchok 2010). We assessed a further 22 trials (18%) at low risk of bias for four domains and one domain at unclear risk of bias (Atkins 2008; Bajetta 2006a; Bedikian 2011; Chiarion‐Sileni 2011; Eigentler 2008; Gupta 2014; Hauschild 2001; Hauschild 2012; Hodi 2014; Kaufmann 2005; Keilholz 2005; Larkin 2014; Maio 2010; McArthur 2014; Middleton 2007; Middleton 2015; O'Day 2009; Patel 2011; Ribas 2013; Robert 2015; Robert 2015b; Testori 2008). Most included studies (n = 73, 60%) were assessed at unclear risk of bias for two or more domains.
Allocation
Random sequence generation
In most included RCTs (n = 62, 51%), the risk of selection bias due to issues linked to random sequence generation was judged to be low. Information regarding random sequence generation was lacking so the risk was assessed as unclear in 59 studies (48%). One study (Hofmann 2011) that compared dacarbazine to best supportive care in pre‐treated participants with metastatic melanoma was assessed at high risk of bias: initially enrolled participants were randomly assigned to either chemotherapy or best supportive care, but enrolment was slow and allocation appeared to be based on physician's choice.
Allocation concealment
In most included RCTs (n = 69, 56%) the risk of selection bias due to issues linked to allocation concealment was judged to be unclear, which was mainly due to the lack of information reported in published study reports. In 52 studies (43%), we judged this domain at low risk of bias. One study (Hofmann 2011) was assessed at high risk of selection bias due to lack of allocation concealment (see 'Random sequence generation' risk of bias assessment).
Blinding
Performance bias
All included RCTs were deemed at low risk of performance bias. In particular, 23 studies (19%) (Cui 2013; Eisen 2010; Flaherty 2013a; Glaspy 2009; Gupta 2014; Hauschild 2009a; Hodi 2010a; Kefford 2010; Kim 2012; Larkin 2015; Lawson 2015; Long 2015; McDermott 2008; Middleton 2015; O'Day 2009; O'Day 2011; O'Day 2013; Postow 2015; Robert 2011; Robert 2013; Robert 2015a; Rusthoven 1996; Wolchok 2010) were designed as double‐blinded trials, and were assessed at low risk of bias for this domain. The remaining 99 trials (81%) were designed as open label studies, with no blinding of participants or personnel. However, we judged that in this setting (metastatic melanoma), with the treatments tested and the outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judge the risk of performance bias as low for these RCTs.
No studies were assessed at high risk of performance bias.
Detection bias
The risk of detection bias was found to be low in 31 RCTs (25%). There was insufficient information reported in the remaining 91 studies (75%) to permit judgement and were assessed at unclear risk of bias for this domain.
No studies were assessed at high risk of detection bias.
Incomplete outcome data
Most included RCTs (n = 99, 81%) were judged to be at low risk of attrition bias. There was insufficient information reported in the remaining 23 (19%) studies to permit judgement and were assessed at unclear risk of bias for this domain.
No studies were assessed at high risk of bias of attrition detected.
Selective reporting
Most included RCTs (n = 62, 51%) were found to be at low risk of reporting bias. There was insufficient information reported in 59 studies (48%) to permit judgement and were assessed at unclear risk of selective reporting bias. One study (Beretta 1976) was assessed at high risk of reporting bias because data from one of the four trial arms were not analysed for unclear reasons.
Other potential sources of bias
We did not find any other sources of bias in most included RCTs (n = 111, 91%). There was insufficient available information to permit judgement for seven studies (6%). We detected a high risk of bias in four trials (3%); Carvajal 2014 and Hamid 2014 showed a potential conflict of interest between some authors and the funding body; drug dosage was amended in Ranson 2007; and Richtig 2004 was stopped when approximately 50% of planned participants were enrolled.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10
Summary of findings 1. Anti‐PD1 monoclonal antibodies versus chemotherapy.
| Anti‐PD1 monoclonal antibodies compared with chemotherapy for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: anti‐PD1 monoclonal antibodies Comparison: chemotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chemotherapy | Anti‐PD1 | |||||
| Overall survival† | 600 per 1000† | 320 per 1000† (290 to 360) |
HR 0.42 (0.37 to 0.48) |
N = 418 (n = 1) | ⊕⊕⊕⊕ higha | ‐ |
| Progression‐free survival† | 850 per 1000† | 610 per 1000† (520 to 690) | HR 0.49 (0.39 to 0.61) | N = 957 (n = 2) | ⊕⊕⊕⊝ moderateb | ‐ |
| Tumour response | 81 per 1000 | 277 per 1000 (193 to 398) |
RR 3.42 (2.38 to 4.92) |
N = 1367 (n = 3) | ⊕⊕⊕⊕ higha | ‐ |
| Toxicity (≥ G3) | 300 per 1000 | 165 per 1000 (93 to 291) | RR0.55 (0.31 to 0.97) | N = 1360 (n = 3) | ⊕⊕⊝⊝ lowc | ‐ |
| * The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI: confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 40%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 15%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Not downgraded: high‐quality evidence.
b Downgraded by one level: inconsistency (between‐study heterogeneity).
c Downgraded by two levels: inconsistency (between‐study heterogeneity) and imprecision (CI includes both a meaningful benefit (relative risk reduction > 25%) and a small/null benefit (relative risk reduction < 10%)).
Summary of findings 2. Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies.
| Anti‐PD1 monoclonal antibodies compared with anti‐CTLA4 monoclonal antibodies for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: anti‐PD1 monoclonal antibodies Comparison: anti‐CTLA4 monoclonal antibodies | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Anti‐CTLA4 | Anti‐PD1 | |||||
| Overall survival† | 600 per 1000† | 438 per 1000† (423 to 454) |
HR 0.63 (0.60 to 0.66) |
N = 764 (n = 1) | ⊕⊕⊕⊕ higha | ‐ |
| Progression‐free survival† | 850 per 1000† | 641 per 1000† (612 to 679) |
HR 0.54 (0.50 to 0.60) |
n = 1465 (n = 2) | ⊕⊕⊕⊕ higha | ‐ |
| Tumour response | 157 per 1000 | 388 per 1000 (315 to 477) |
RR 2.47 (2.01 to 3.04) |
N = 1465 (n = 2) | ⊕⊕⊕⊕ higha | ‐ |
| Toxicity (≥ G3) | 398 per 1000 | 278 per 1000 (215 to 362) |
RR 0.70 (0.54 to 0.91) |
N = 1465 (n = 2) | ⊕⊕⊝⊝ lowb | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI: confidence interval; RR: risk ratio; HR: hazard ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 40%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 15%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Not downgraded: high‐quality evidence.
b Downgraded by two levels: inconsistency (between‐study heterogeneity) and imprecision (CI includes both a meaningful benefit (relative risk reduction > 25%) and a small/null benefit (relative risk reduction < 10%).
Summary of findings 3. Anti‐CTLA4 monoclonal antibodies plus chemotherapy versus chemotherapy.
| Anti‐CTLA4 monoclonal antibodies plus chemotherapy compared with chemotherapy for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: anti‐CTLA4 monoclonal antibodies plus chemotherapy (combo) Comparison: chemotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative Effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chemotherapy | Combo | |||||
| Overall survival† | 600 per 1000† | 524 per 1000† (449 to 604) | HR 0.81 (0.65 to 1.01) | N = 1157 (n = 2) | ⊕⊕⊝⊝ lowa | ‐ |
| Progression‐free survival† | 850 per 1000† | 763 per 1000† (697 to 825) | HR 0.76 (0.63 to 0.92) | N = 502 (n = 1) | ⊕⊕⊕⊝ moderateb | ‐ |
| Tumour response | 100 per 1000 | 128 per 1000 (92 to 177) | RR 1.28 (0.92 to 1.77) | N = 1157 (n = 2) | ⊕⊕⊕⊝ moderatec | ‐ |
| Toxicity (≥ G3) | 352 per 1000 | 595 per 1000 (419 to 852) | RR 1.69 (1.19 to 2.42) | N = 1142 (n = 2) | ⊕⊕⊕⊝ moderated | ‐ |
| * The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI: confidence interval; HR: hazard ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 40%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 15%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Downgraded by two levels: inconsistency (between‐study heterogeneity) and imprecision (CI includes both a meaningful benefit (relative risk reduction > 25%) and a harmful effect).
b Downgraded by one level: imprecision (CI includes both a meaningful benefit (relative risk reduction > 25%) and a small/null benefit (relative risk reduction < 10%)).
c Downgraded by one level: imprecision (CI includes both a meaningful benefit (relative risk increase > 25%) and a harmful effect).
d Downgraded by one level: inconsistency (between‐study heterogeneity).
Summary of findings 4. Anti‐CTLA4 monoclonal antibodies with versus without anti‐PD1 monoclonal antibodies.
| Anti‐CTLA4 plus anti‐PD1 monoclonal antibodies compared with anti‐CTLA4 monoclonal antibodies for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: Anti‐CTLA4 plus Anti‐PD1 monoclonal antibodies (combo) Comparison: Anti‐CTLA4 monoclonal antibodies | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Anti‐CTLA4 | Combo | |||||
| Overall survival | See comment | See comment | See comment | See comment | See comment | Outcome not measured |
| Progression‐free survival† | 750 per 1000† | 425 per 1000† (375 to 478) |
HR 0.40 (0.35 to 0.46) |
N = 738 (n = 2) | ⊕⊕⊕⊕ higha | ‐ |
| Tumour response | 182 per 1000 | 636 per 1000 (376 to 1073) | RR 3.50 (2.07 to 5.92) | N = 738 (n = 2) | ⊕⊕⊕⊕ higha | ‐ |
| Toxicity (≥ G3) | 521 per 1000 | 818 per 1000 (442 to 1521) | RR 1.57 (0.85 to 2.92) | N = 764 (n = 2) | ⊕⊕⊝⊝ lowb | ‐ |
| * The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. progression rates). CI: confidence interval | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year progression‐free survival rate = 25%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Not downgraded: high‐quality evidence.
b Downgraded by two levels: inconsistency (between‐study heterogeneity) and imprecision (CI includes both a meaningful harm (relative risk increase > 25%) and a beneficial effect)
Summary of findings 5. BRAF inhibitors versus chemotherapy.
| BRAF inhibitors compared with chemotherapy for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: BRAF inhibitors Comparison: chemotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chemotherapy | BRAF inhibitors | |||||
| Overall survival† | 600 per 1000† | 307 per 1000† (226 to 407) |
HR 0.40 (0.28 to 0.57) |
N = 925 (n = 2) | ⊕⊕⊕⊕ higha | ‐ |
| Progression‐free survival† | 850 per 1000† | 401 per 1000† (328 to 475) |
HR 0.27 (0.21 to 0.34) |
N = 925 (n = 2) | ⊕⊕⊕⊕ higha | ‐ |
| Tumour response | 82 per 1000 | 556 per 1000 (397 to 778) |
RR 6.78 (4.84 to 9.49) |
N = 925 (n = 2) | ⊕⊕⊕⊕ higha | ‐ |
| Toxicity (≥ G3) | 341 per 1000 | 433 per 1000 (163 to 1135) | RR 1.27 (0.48 to 3.33) | N = 408 (n = 2) | ⊕⊕⊝⊝ lowb | ‐ |
| * The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI: confidence interval; HR: hazard ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 40%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 15%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Not downgraded: high‐quality evidence.
b Downgraded by two levels: inconsistency (between‐study heterogeneity) and imprecision (CI includes both a meaningful harm (relative risk increase > 25%) and a meaningful benefit (relative risk reduction > 25%)).
Summary of findings 6. MEK inhibitors versus chemotherapy.
| MEK inhibitors compared with chemotherapy for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: MEK inhibitors Comparison: chemotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chemotherapy | MEK inhibitors | |||||
| Overall survival† | 600 per 1000† | 541 per 1000† (412 to 682) |
HR 0.85 (0.58 to 1.25) |
N = 496 (n = 3) | ⊕⊕⊝⊝ lowa | ‐ |
| Progression‐free survival† | 850 per 1000† | 667 per 1000† (549 to 781) |
HR 0.58 (0.42 to 0.80) |
N = 496 (n = 3) | ⊕⊕⊕⊝ moderateb | ‐ |
| Tumour response | 138 per 1000 | 277 per 1000 (186 to 413) |
RR 2.01 (1.35 to 2.99) |
N = 496 (n = 3) | ⊕⊕⊕⊕ highc | ‐ |
| Toxicity (≥ G3) | 413 per 1000 | 665 per 1000 (446 to 995) |
RR 1.61 (1.08 to 2.41) |
N = 91 (n = 1) | ⊕⊕⊕⊝ moderated | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI: confidence interval; RR: risk ratio; HR: hazard ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 40%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 15%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Downgraded by two levels: inconsistency (between‐study heterogeneity) and imprecision (CI includes both a meaningful benefit (relative risk reduction > 25%) and a harmful effect).
b Downgraded by one level: inconsistency (between‐study heterogeneity).
c Not downgraded: high‐quality evidence.
d Downgraded by one level: imprecision (sample size lower than optimal information size, calculated to be equal to 400 participants).
Summary of findings 7. BRAF plus MEK inhibitors versus BRAF inhibitors.
| BRAF plus MEK inhibitors compared with BRAF inhibitors for the treatment of metastatic melanoma | ||||||
|
Patient or population: people cutaneous melanoma Settings: hospital (metastatic disease) Intervention: BRAF inhibitor plus MEK inhibitor (combo) Comparison: BRAF inhibitor | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| BRAF inhibitor | Combo | |||||
| Overall survival† | 350 per 1000† | 260 per 1000† (204 to 321) |
HR 0.70 (0.59 to 0.82) |
N = 1784 (n = 4) | ⊕⊕⊕⊕ higha | ‐ |
| Progression‐free survival† | 700 per 1000† | 490 per 1000† (411 to 574) | HR 0.56 (0.44 to 0.71) | N = 1784 (n = 4) | ⊕⊕⊕⊝ moderateb | ‐ |
| Tumour response | 494 per 1000 | 652 per 1000 (593 to 721) |
RR 1.32 (1.20 to 1.46) |
N = 1784 (n = 4) | ⊕⊕⊕⊕ higha | ‐ |
| Toxicity (≥ G3) | 495 per 1000 | 500 per 1000 (421 to 594) | RR 1.01 (0.85 to 1.20) | N = 1774 (n = 4) | ⊕⊕⊕⊝ moderateb | ‐ |
| * The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 65%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 30%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Not downgraded: high‐quality evidence.
b Downgraded by one level: inconsistency (between‐study heterogeneity).
Summary of findings 8. Anti‐angiogenic drugs plus chemotherapy versus chemotherapy.
| Anti‐angiogenic drugs plus chemotherapy compared with chemotherapy for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: Anti‐angiogenic drug plus chemotherapy (combo) Comparison: chemotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chemotherapy | Combo | |||||
| Overall survival† | 600 per 1000† | 423 per 1000† (338 to 524) |
HR 0.60 (0.45 to 0.81) |
N = 324 (n = 2) | ⊕⊕⊕⊝ moderatea | ‐ |
| Progression‐free survival† | 850 per 1000† | 730 per 1000† (627 to 825) |
HR 0.69 (0.52 to 0.92) |
N = 324 (n = 2) | ⊕⊕⊕⊝ moderatea | ‐ |
| Tumour response | 104 per 1000 | 178 per 1000 (100 to 315) | RR 1.71 (0.96 to 3.03) | N = 324 (n = 2) | ⊕⊕⊕⊝ moderatea | ‐ |
| Toxicity (≥ G3) | 272 per 1000 | 185 per 1000 (25 to 1447) | RR 0.68 (0.09 to 5.32) | N = 324 (n = 2) | ⊕⊕⊝⊝ lowb | ‐ |
| * The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI: confidence interval; HR: hazard ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 40%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 15%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Downgraded by one level: imprecision (sample size lower than optimal information size, calculated to be equal to 400 participants).
b Downgraded by two levels: inconsistency (between‐study heterogeneity) and imprecision (sample size lower than optimal information size, calculated to be equal to 400 participants).
Summary of findings 9. Biochemotherapy versus chemotherapy.
| Biochemotherapy compared with chemotherapy for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: biochemotherapy (chemotherapy combined with both interferon‐alpha and interleukin‐2) Comparison: chemotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chemotherapy | Biochemotherapy | |||||
| Overall survival† | 600 per 1000† | 577 per 1000† (537 to 621) |
HR 0.94 (0.84 to 1.06) |
N = 1317 (n = 7) | ⊕⊕⊕⊕ higha | ‐ |
| Progression‐free survival† | 850 per 1000 ° | 818 per 1000† (793 to 847) |
HR 0.90 (0.83 to 0.99) |
N = 964 (n = 6) | ⊕⊕⊕⊕ higha | ‐ |
| Tumour response | 192 per 1000 | 262 per 1000 (214 to 321) |
RR 1.36 (1.12 to 1.66) |
N = 770 (n = 7) | ⊕⊕⊕⊕ higha | ‐ |
| Toxicity (≥ G3) | 631 per 1000 | 852 per 1000 (719 to 1000) |
RR 1.35 (1.14 to 1.61) |
N = 631 (n = 2) | ⊕⊕⊕⊕ higha | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI: confidence interval; RR: risk ratio; HR: hazard ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 40%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 15%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Not downgraded: high‐quality evidence.
Summary of findings 10. Polychemotherapy versus chemotherapy.
| Polychemotherapy compared with chemotherapy for the treatment of metastatic melanoma | ||||||
|
Patient or population: people with cutaneous melanoma Settings: hospital (metastatic disease) Intervention: polychemotherapy Comparison: chemotherapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chemotherapy | Polychemotherapy | |||||
| Overall survival† | 600 per 1000† | 596 per 1000† (541 to 655) |
HR 0.99 (0.85 to 1.16) |
N = 594 (n = 6) | ⊕⊕⊕⊕ higha | ‐ |
| Progression‐freesurvival† | 850 per 1000† |
869 per 1000† (822 to 907) |
HR 1.07 (0.91 to 1.25) |
N = 398 (n = 5) |
⊕⊕⊕⊕ higha | ‐ |
| Tumour response | 143 per 1000 | 182 per 1000 (146 to 226) |
RR 1.27 (1.02 to 1.58) |
N = 1885 (n = 5) | ⊕⊕⊕⊝ moderateb | ‐ |
| Toxicity (≥ G3) | 189 per 1000 | 372 per 1000 (272 to 512) |
RR 1.97 (1.44 to 2.71) |
N = 390 (n = 3) | ⊕⊕⊕⊝ moderatec | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). † Numbers presented refer to event rates (i.e. death rates and progression rates). CI: confidence interval; RR: risk ratio; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Assumed risk in the control population: 1‐year overall survival rate = 40%.
Assumed risk in the control population: 1‐year progression‐free survival rate = 15%.
Assumed risk in the control population: tumour response rate across control arms of included trials.
Assumed risk in the control population: toxicity rate across control arms of included trials.
a Not downgraded: high‐quality evidence.
b Downgraded by one level: imprecision (CI includes both a meaningful benefit (relative risk increase > 25%) and a small/null benefit (relative risk increase < 10%)).
c Downgraded by one level: imprecision (sample size lower than optimal information size, calculated to be equal to 400 participants).
We analysed outcomes according to descriptions in Types of outcome measures. Each outcome was investigated for the pre‐established interventions described in Types of interventions. Findings from included studies were meta‐analysed when a drug (or a drug regimen) was tested in at least two studies. Accordingly, 39 studies were not included in the meta‐analyses. (Table 12 presents reasons for exclusion from meta‐analysis). Quantitative analysis was performed with findings from 83 studies for five different types of interventions: conventional chemotherapy, biochemotherapy, immune checkpoint inhibitors, small‐molecule targeted drugs, and other agents (including anti‐angiogenic drugs) (Table 13).
2. Reasons for excluding 39 studies from meta‐analysis.
| Study ID | Reason for exclusion from meta‐analysis |
| Hamid 2014 | Single study investigating tasisulam |
| Kefford 2010 | Single study investigating bosentan |
| Hofmann 2011 | Single study comparing dacarbazine and best supportive care |
| Schadendorf 2006 | Single study investigating dendritic cells therapy |
| Agarwala 2002 | Single study investigating histamine with interleukin‐2 |
| Bajetta 1985 | Different polychemotherapy regimens not compared in other studies |
| Beretta 1976 | Different polychemotherapy regimens not compared in other studies |
| Cocconi 1992 | Different polychemotherapy regimens not compared in other studies |
| Dummer 2006 | Different PEG‐interferon schedules tested |
| Flaherty 2001 | Inpatient and outpatient interleukin‐2‐based regimens not compared in other studies |
| Glaspy 2009 | Different lenalidomide schedules not compared in other studies |
| Jelic 2002 | Different polychemotherapy regimens not compared in other studies |
| Keilholz 1997 | Study comparing biochemotherapy versus biotherapy |
| Legha 1996 | Study comparing alternating and sequential biochemotherapy and chemotherapy |
| Miller 1989 | Single study investigating Indomethacine with interferon |
| Moon 1975 | Different single‐agent chemotherapy regimens not compared in other studies |
| Presant 1982 | Different polychemotherapy regimens not compared in other studies |
| Richtig 2004 | Different temozolomide and interferon schedules tested |
| Wittes 1978 | Different polychemotherapy regimens not compared in other studies |
| Vuoristo 2005 | Different interferon‐based regimens not compared in other studies |
| Punt 2006 | Different biochemotherapy regimens not compared in other studies |
| Reichle 2007 | Single study investigating chemotherapy and COX‐2 inhibitor |
| Sparano 1993 | Single study comparing interleukin‐2 with versus without interferon‐alpha |
| Wolchok 2010 | Different ipilimumab schedules tested |
| Avril 2004 | Single study comparing fotemustine and dacarbazine |
| O'Day 2011 | Single study testing Intetumumab |
| Ranson 2007 | Single study testing lomeguatrib |
| Hersh 2015 | Single study testing nab‐paclitaxel |
| Bedikian 2006 | Single study testing oblimersen |
| Bedikian 2011 | Single study testing DHA‐paclitaxel |
| Weber 2009 | Single study testing PF‐3512676 |
| Carvajal 2014 | Single study testing ramucirumab |
| Balch 1984 | Single study testing dacarbazine and C parvum after surgery |
| Eigentler 2008 | Single study testing vindesine after surgery |
| Lawson 2015 | Single study testing GM‐CSF and a polypeptide vaccination after surgery |
| Eisen 2010 | Single study testing lenalidomide |
| Middleton 2015 | Single study testing veliparib |
| Testori 2008 | Single study testing vetaspen |
3. Studies included in meta‐analysis.
Hodi 2010a; Hodi 2014; Maio 2010; Schwartzentruber 2011a were included in a meta‐analysis of immunostimulating agents.
We presented 10 comparisons in relation to overall survival, progression‐free survival, tumour response, and toxicity (≥ G3) in 'Summary of findings' tables:
anti‐PD1 monoclonal antibodies compared with chemotherapy (Table 1);
anti‐PD1 monoclonal antibodies compared with anti‐CTLA4 monoclonal antibodies (Table 2);
anti‐CTLA4 monoclonal antibodies plus chemotherapy compared with chemotherapy alone (Table 3);
anti‐PD1 plus Anti‐CTLA4 monoclonal antibodies compared with anti‐CTLA4 monoclonal antibodies (Table 4);
BRAF inhibitors compared with chemotherapy (Table 5);
MEK inhibitors compared with chemotherapy (Table 6);
BRAF plus MEK inhibitors compared with BRAF inhibitors alone (Table 7);
anti‐angiogenic drugs plus chemotherapy compared with chemotherapy alone (Table 8);
biochemotherapy compared with chemotherapy alone (Table 9); and
polychemotherapy compared with chemotherapy alone (Table 10).
Overall survival
Polychemotherapy versus single agent chemotherapy
We included 14 studies that compared cytotoxic polychemotherapy and single agent chemotherapy (Bafaloukos 2005; Bellett 1976; Carter 1975; Chapman 1999; Chauvergne 1982; Chiarion Sileni 2001; Costanza 1972; Costanza 1977; Glover 2003; Kogoniia 1981; Lopez 1984; Luikart 1984; Zimpfer‐Rechner 2003). Hazard ratios (HRs) were directly available or could be extrapolated for six studies (Bafaloukos 2005; Chapman 1999; Chauvergne 1982; Chiarion Sileni 2001; Luikart 1984; Zimpfer‐Rechner 2003). Polychemotherapy and single agent chemotherapy was administered to 312 and 282 participants, respectively. Meta‐analysis suggested a similar risk of death between polychemotherapy and single agent chemotherapy (Analysis 1.1, HR 0.99, 95% CI 0.85 to 1.16; heterogeneity: Tau² = 0.00; Chi² = 3.86, df = 5, P = 0.57; I² = 0%; high‐quality evidence).
1.1. Analysis.

Comparison 1: Polychemotherapy versus single agent chemotherapy, Outcome 1: Overall survival
Biochemotherapy versus chemotherapy
Chemotherapy with interferon‐alpha versus without interferon‐alpha
This comparison included 15 studies (Bajetta 1994; Bajetta 2006a; Danson 2003; Daponte 2013; Dorval 1999; Falkson 1991; Falkson 1995; Falkson 1998; Gorbonova 2000; Kaufmann 2005; Kirkwood 1990; Maio 2010; Thomson 1993; Vorobiof 1994; Young 2001). Hazard ratios (HRs) were directly available from or could be extrapolated for 11 studies (Bajetta 1994; Bajetta 2006a; Danson 2003; Daponte 2013; Dorval 1999; Falkson 1991; Falkson 1998; Kaufmann 2005; Thomson 1993; Vorobiof 1994; Young 2001). Overall, 942 participants were allocated to chemotherapy with interferon‐alpha and 843 to chemotherapy alone. Meta‐analysis suggested a lower risk of death for the combination of chemotherapy and interferon‐alpha, although this difference was not statistically significant (Analysis 4.1, HR 0.87, 95% CI 0.73 to 1.04) and between‐study heterogeneity was remarkable (heterogeneity: Tau² = 0.06; Chi² = 37.19, df = 10, P < 0.0001; I² = 73%; low‐quality evidence). We did not identify any particular study driving heterogeneity results in a sensitivity analysis. All participants were previously untreated and without brain metastases. Heterogeneity dropped remarkably (I² = 9%) when only studies published after 2000 were considered (HR 0.95, 95% CI 0.84 to 1.08), but increased (I² = 85%) when only studies published before 2000 were included (HR 0.75, 95% CI 0.52 to 1.07). Heterogeneity also dropped when Vorobiof 1994 was excluded from analysis (heterogeneity: Tau² = 0.02; Chi² = 16.45, df = 9, P = 0.06; I² = 45%), without changing the effect estimate (HR 0.94, 95% CI 0.83 to 1.07).
4.1. Analysis.

Comparison 4: Chemotherapy ± interferon‐alpha, Outcome 1: Overall survival
Chemotherapy with interleukin‐2 versus without interleukin‐2
Two studies provided data for this comparison (Hauschild 2001; Keilholz 2005); it was not possible to extract HR data from Sertoli 1999. Overall, 320 participants were allocated to chemotherapy plus interleukin‐2 and 324 participants to chemotherapy alone. Analysis suggested a small and statistically non‐significant benefit for combination therapy of chemotherapy and interleukin‐2 (Analysis 5.1, HR 0.95, 95% CI 0.82 to 1.11; heterogeneity: Tau² = 0.00; Chi² = 0.45, df = 1, P = 0.50; I² = 0%; high‐quality evidence).
5.1. Analysis.

Comparison 5: Chemotherapy ± interleukin‐2, Outcome 1: Overall survival
Chemotherapy with interferon‐alpha and interleukin‐2 versus without interferon‐alpha and interleukin‐2
Data for this comparison were available from seven studies (Atkins 2008; Atzpodien 2002; Eton 2002; Johnston 1998; Middleton 2007; Ridolfi 2002a; Rosenberg 1999). Overall, 659 participants were allocated to chemotherapy with both interferon‐alpha and interleukin‐2 and 658 participants to chemotherapy alone. Analysis suggested a slightly lower risk of death associated with combination therapy of chemotherapy plus interleukin‐2 and interferon‐alpha, although this difference was not statistically significant (Analysis 6.1, HR 0.94, 95% CI 0.84 to 1.06; heterogeneity: Tau² = 0.01; Chi² = 7.61, df = 6, P = 0.27; I² = 21%; high‐quality evidence). We also analysed those trials enrolling only previously untreated patients with metastatic melanoma (biochemotherapy used as first‐line treatment) (Atkins 2008; Eton 2002; Middleton 2007; Ridolfi 2002a; Rosenberg 1999) and found a similar effect size with higher heterogeneity (Analysis 7.1, HR 0.96, 95% CI 0.83 to 1.10; heterogeneity: Tau² = 0.01; Chi² = 6.64, df = 4, P = 0.16; I² = 40%). The leave‐one‐out procedure suggested Rosenberg 1999 to be the study driving heterogeneity (HR 0.92, 95% CI 0.83 to 1.04; heterogeneity: Tau² = 0.00; Chi² = 1.42, df = 3, P = 0.70; I² = 0%); however, we could not explain why this trial caused heterogeneity.
6.1. Analysis.

Comparison 6: Chemotherapy ± interferon‐alpha and interleukin‐2, Outcome 1: Overall survival
7.1. Analysis.

Comparison 7: Chemotherapy ± interferon‐alpha and interleukin‐2 (first line), Outcome 1: Overall survival
Immune checkpoint inhibitors
Anti‐CTLA4 monoclonal antibodies plus chemotherapy versus chemotherapy alone (first line)
Two studies provided data for this comparison (Ribas 2013; Robert 2011): in Ribas 2013 the anti‐CTLA4 monoclonal antibody tremelimumab did not add any significant advantage to chemotherapy; and in Robert 2011 the anti‐CTLA4 monoclonal antibody ipilimumab significantly increased the efficacy of chemotherapy (HR 0.72, 95% CI 0.59 to 0.88). Overall, 578 participants were allocated to anti‐CTLA4 monoclonal antibodies and chemotherapy and 579 to chemotherapy alone. Meta‐analysis suggested a lower risk of death for combination therapy of anti‐CTLA and chemotherapy, although this difference was not statistically significant (Analysis 10.1, HR 0.81, 95% CI 0.65 to 1.01; heterogeneity: Tau² = 0.02; Chi² = 2.99, df = 1, P = 0.08; I² = 67%; low‐quality evidence). High level heterogeneity detected in this analysis was likely to be linked to the effects caused by participants in Ribas 2013 who failed chemotherapy subsequently being treated with tremelimumab, which potentially nullified the difference between the study arms due to this anti‐CTLA4 monoclonal antibody.
10.1. Analysis.

Comparison 10: Anti‐CTLA4 monoclonal antibodies (first line), Outcome 1: Overall survival
Anti‐CTLA4 monoclonal antibodies with immune stimulating agents versus without immune stimulating agents (second line)
This comparison included two studies (Hodi 2010a; Hodi 2014). Overall, 526 participants were allocated to anti‐CTLA4 monoclonal antibodies with immune stimulating agents: melanoma antigen gp100 (Hodi 2010a) and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) (Hodi 2014), and 259 participants were allocated to anti‐CTLA4 monoclonal antibodies alone. Data from the meta‐analysis suggested a lower risk of death for combination therapy of anti‐CTLA and immune stimulating agents, although this difference was not statistically significant (Analysis 11.1 HR 0.83, 95% CI 0.52 to 1.33; heterogeneity: Tau² = 0.10; Chi² = 5.42, df = 1, P = 0.02; I² = 82%; low‐quality evidence). High level heterogeneity was likely due to a different effect of association between ipilimumab with either gp100 (Hodi 2010a, HR 1.04, 95% CI 0.83 to 1.30) or GM‐CSF (HR 0.64, 95% CI 0.46 to 0.90).
11.1. Analysis.

Comparison 11: Anti‐CTLA4 monoclonal antibodies ± other immunostimulating agents (second line), Outcome 1: Overall survival
Anti‐PD1 monoclonal antibodies versus chemotherapy
This comparison included three studies (Ribas 2015; Robert 2015a; Weber 2015). Overall survival was a study endpoint only for Robert 2015a so meta‐analysis could be performed. In Robert 2015a, 210 participants were allocated to anti‐PD1 monoclonal antibodies and 208 participants to chemotherapy alone. Results from Robert 2015a showed that anti‐PD1 monoclonal antibodies significantly reduced the risk of death from any cause (Analysis 12.1, HR 0.42, 95% CI 0.37 to 0.48; high‐quality evidence).
12.1. Analysis.

Comparison 12: Anti‐PD1 monoclonal antibodies versus chemotherapy, Outcome 1: Overall survival
Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies
This comparison included two studies (Larkin 2015; Robert 2015b). Overall survival was a study endpoint only for Robert 2015b so meta‐analysis could not be performed. In Robert 2015b, 556 participants were allocated to anti‐PD1 monoclonal antibodies and 208 to chemotherapy alone. Results from Robert 2015b suggested a statistically significant lower risk of death for anti‐PD1 monoclonal antibodies (Analysis 13.1; HR 0.63, 95% CI 0.60 to 0.66; high‐quality evidence).
13.1. Analysis.

Comparison 13: Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies, Outcome 1: Overall survival
Anti‐CTLA4 monoclonal antibodies with anti‐PD1 monoclonal antibodies versus without anti‐PD1 monoclonal antibodies
This comparison included two studies (Larkin 2015; Postow 2015) which did not investigate overall survival.
Small‐molecule targeted drugs
BRAF inhibitors versus chemotherapy
This comparison included two studies (Hauschild 2012; McArthur 2014). Overall, 524 participants were allocated to single agent BRAF inhibitor and 401 participants to chemotherapy alone. Data from the meta‐analysis suggested a statistically significant lower risk of death for single agent BRAF inhibitor (Analysis 18.1, HR 0.40, 95% CI 0.28 to 0.57; heterogeneity: Tau² = 0.01; Chi² = 1.04, df = 1, P = 0.31; I² = 4%; high‐quality evidence).
18.1. Analysis.

Comparison 18: Single agent BRAF inhibitor, Outcome 1: Overall survival
MEK inhibitors versus chemotherapy
This comparison included three studies (Flaherty 2012b; Gupta 2014; Robert 2013). Overall, 300 participants were allocated to single agent MEK inhibitor treatment and 196 participants to chemotherapy alone. Data from the meta‐analysis suggested a lower risk of death for single agent MEK inhibitor, although the difference was not statistically significant (Analysis 19.1, HR 0.85, 95% CI 0.58 to 1.25; heterogeneity: Tau² = 0.07; Chi² = 4.63, df = 2, P = 0.10; I² = 57%; low‐quality evidence; downgraded due to inconsistency and imprecision).
19.1. Analysis.

Comparison 19: Single agent MEK inhibitor, Outcome 1: Overall survival
BRAF inhibitors with MEK inhibitors versus without MEK inhibitors
This comparison included four studies (Flaherty 2012a; Larkin 2014; Long 2015; Robert 2015). Overall, 918 participants were allocated to combination therapy of BRAF plus MEK inhibitors and 866 participants to single agent BRAF inhibitor. Data from the meta‐analysis suggested a statistically significant lower risk of death for combination therapy (Analysis 20.1, HR 0.70, 95% CI 0.59 to 0.82, heterogeneity: Tau² = 0.00; Chi² = 0.15, df = 3, P = 0.98; I² = 0%; high‐quality evidence).
20.1. Analysis.

Comparison 20: Combination of BRAF and MEK inhibitors versus single agent BRAF inhibitor, Outcome 1: Overall survival
Chemotherapy with versus without other agents
Chemotherapy with Bacillus Calmette‐Guérin (BCG) versus without BCG
This comparison included six studies (Costanzi 1982; Mastrangelo 1979; Newlands 1976; Ramseur 1978; Veronesi 1984; Verschraegen 1993). HRs were available or extractable for two studies (Newlands 1976; Verschraegen 1993). Overall, 74 participants were allocated to chemotherapy with BCG and 80 to chemotherapy alone. Analysis suggested a lower risk of death for combination of chemotherapy and BCG, although the difference was not statistically significant (Analysis 8.1, HR 0.87, 95% CI 0.61 to 1.25; heterogeneity: Tau² = 0.00; Chi² = 0.50, df = 1, P = 0.48; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
8.1. Analysis.

Comparison 8: Chemotherapy ± Bacille Calmette‐Guérin (BCG), Outcome 1: Overall survival
Chemotherapy with Corynebacterium parvum versus without C parvum
This comparison included seven studies (Clunie 1980; Gough 1978; Kokoschka 1978; Presant 1979; Robidoux 1982; Thatcher 1986; Veronesi 1984). HRs were directly available or could be extrapolated for four RCTs (Clunie 1980; Kokoschka 1978; Presant 1979; Robidoux 1982). Overall, 114 participants were allocated to chemotherapy with C parvum and 128 participants to chemotherapy alone. Analysis suggested a slightly lower risk of death for combination of chemotherapy and C parvum, although this difference was not statistically significant (Analysis 9.1, HR 0.95, 95% CI 0.74 to 1.22; heterogeneity: Tau² = 0.00; Chi² = 0.79, df = 3, P = 0.85; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
9.1. Analysis.

Comparison 9: Chemotherapy ± Corynebacterium parvum, Outcome 1: Overall survival
Chemotherapy with tamoxifen versus without tamoxifen
We included four trials for this comparison (Agarwala 1999; Cocconi 1992; Falkson 1998; Rusthoven 1996). HRs were either directly reported or could be extrapolated. Tamoxifen‐based polychemotherapy was administered to 326 participants and 317 participants received cytotoxic chemotherapy alone. Tamoxifen was associated with a non‐statistically significant slightly higher risk of death (Analysis 2.1, HR 1.03, 95% CI 0.80 to 1.33; heterogeneity: Tau² = 0.04; Chi² = 7.58, df = 3, P = 0.06; I² = 60%; low‐quality evidence; downgraded due to inconsistency and imprecision). Leave‐one‐out analysis suggested that heterogeneity was mainly related to Cocconi 1992 (HR 1.13, 95% CI 0.96 to 1.33, heterogeneity: Tau² = 0.00; Chi² = 1.52, df = 2, P = 0.47; I² = 0%): however, we could not explain why this trial caused heterogeneity.
2.1. Analysis.

Comparison 2: Chemotherapy ± tamoxifen, Outcome 1: Overall survival
Chemotherapy with anti‐angiogenic drugs versus without anti‐angiogenic drugs
This comparison included two studies (Cui 2013; Kim 2012). Overall, 199 participants were allocated to standard chemotherapy plus anti‐angiogenic therapies and 125 participants to chemotherapy alone. Data from the meta‐analysis suggested a statistically significant lower risk of death for combination of chemotherapy and anti‐angiogenic agents (Analysis 17.1, HR 0.60, 95% CI 0.45 to 0.81; heterogeneity: Tau² = 0.00; Chi² = 0.71, df = 1, P = 0.40; I² = 0%; moderate‐quality evidence; downgraded due to imprecision ‐ there were fewer than 400 participants, so the sample size was smaller than optimal information size).
17.1. Analysis.

Comparison 17: Chemotherapy ± anti‐angiogenic drugs, Outcome 1: Overall survival
Chemotherapy with sorafenib versus without sorafenib
This comparison included three studies (Flaherty 2013a; Hauschild 2009a; McDermott 2008). Overall, 596 participants were allocated to standard chemotherapy plus sorafenib and 598 participants to chemotherapy alone. Analysis suggested a similar risk of death for combination of chemotherapy and sorafenib (Analysis 15.1, HR 1.00, 95% CI 0.88 to 1.14; heterogeneity: Tau² = 0.00; Chi² = 0.03, df = 2, P = 0.99; I² = 0%; high‐quality evidence).
15.1. Analysis.

Comparison 15: Chemotherapy ± sorafenib, Outcome 1: Overall survival
Chemotherapy with elesclomol versus without elesclomol
This comparison included two studies (O'Day 2011; O'Day 2013). Overall survival was a study endpoint only for O'Day 2013 so meta‐analysis could not be performed. In O'Day 2013, 325 participants were allocated to chemotherapy plus elesclomol and 326 participants to chemotherapy alone. Results from O'Day 2013 suggested a statistically significant lower risk of death for chemotherapy alone, although the difference was not statistically significant (Analysis 16.1, HR 1.10, 95% CI 0.92 to 1.32; moderate‐quality evidence; downgraded due to imprecision).
16.1. Analysis.

Comparison 16: Chemotherapy ± elesclomol, Outcome 1: Overall survival
Other comparisons
Single agent chemotherapy versus another single agent chemotherapy
Meta‐analysis was feasible for two different single agent drug regimens: dacarbazine and temozolomide. Three trials were included (Chiarion‐Sileni 2011; Middleton 2000; Patel 2011). Overall, 659 and 654 participants were allocated to temozolomide and dacarbazine, respectively. Temozolomide was associated with a small and non statistically significant survival improvement compared to single agent dacarbazine (Analysis 3.1, HR 0.98, 95% CI 0.85 to 1.12; heterogeneity: Tau² = 0.00; Chi² = 2.33, df = 2, P = 0.31; I² = 14%; high‐quality evidence).
3.1. Analysis.

Comparison 3: Temozolomide versus dacarbazine, Outcome 1: Overall survival
Progression‐free survival
Polychemotherapy versus single agent chemotherapy
We included 14 studies that compared cytotoxic polychemotherapy to single agent chemotherapy (Bafaloukos 2005; Bellett 1976; Carter 1975; Chapman 1999; Chauvergne 1982; Chiarion Sileni 2001; Costanza 1972; Costanza 1977; Glover 2003; Kogoniia 1981; Lopez 1984; Luikart 1984; Zimpfer‐Rechner 2003). HRs were either available or extractable for five studies (Bafaloukos 2005; Glover 2003; Chiarion Sileni 2001; Luikart 1984; Zimpfer‐Rechner 2003). Cytotoxic polychemotherapy and single agent chemotherapy were administered for 219 and 179 participants, respectively. Data from the meta‐analysis suggested a slightly higher risk of melanoma progression for polychemotherapy, although this difference did not reach statistical significance (Analysis 1.2, HR 1.07, 95% CI 0.91 to 1.25; heterogeneity: Tau² = 0.00; Chi² = 0.87, df = 4, P = 0.93; I² = 0%; high‐quality evidence).
1.2. Analysis.

Comparison 1: Polychemotherapy versus single agent chemotherapy, Outcome 2: Progression‐free survival
Biochemotherapy versus chemotherapy
Chemotherapy with interferon‐alpha versus without interferon‐alpha
This comparison included 15 studies (Bajetta 1994; Bajetta 2006a; Danson 2003; Daponte 2013; Dorval 1999; Falkson 1991; Falkson 1995; Falkson 1998; Gorbonova 2000; Kaufmann 2005; Kirkwood 1990; Maio 2010; Thomson 1993; Vorobiof 1994; Young 2001). HRs were directly available or could be extrapolated from six studies (Bajetta 1994; Bajetta 2006a; Daponte 2013; Falkson 1991; Falkson 1998; Kaufmann 2005). Overall, 671 participants were allocated to chemotherapy with interferon‐alpha and 610 participants to chemotherapy alone. Data from the meta‐analysis suggested a lower risk of death for combination of chemotherapy and interferon‐alpha, although this difference was not statistically significant (Analysis 4.2, HR 0.87, 95% CI 0.74 to 1.01; heterogeneity: Tau² = 0.02; Chi² = 13.32, df = 5, P = 0.02; I² = 62%; low‐quality evidence; downgraded due to inconsistency and imprecision). High heterogeneity appeared to result from inclusion of Falkson 1991: when this trial was omitted from analysis, heterogeneity dropped to 0% (in this sensitivity analysis the effect size was also reduced: HR 0.92, 95% CI 0.84 to 1.00). However, we could not explain why Falkson 1991 caused heterogeneity.
4.2. Analysis.

Comparison 4: Chemotherapy ± interferon‐alpha, Outcome 2: Progression‐free survival
Chemotherapy with interleukin‐2 versus without interleukin‐2
This comparison included two studies (Hauschild 2001; Keilholz 2005). Progression‐free survival was a study endpoint only for Keilholz 2005 so meta‐analysis could not be performed. Keilholz 2005 randomised 183 participants to receive chemotherapy plus interleukin‐2 and 180 participants to receive chemotherapy alone. Findings reported by Keilholz 2005 suggested a statistically significant lower risk of melanoma progression for chemotherapy alone, although the difference was not statistically significant (Analysis 5.2, HR 0.87, 95% CI 0.70 to 1.08; moderate‐quality evidence; downgraded due to imprecision).
5.2. Analysis.

Comparison 5: Chemotherapy ± interleukin‐2, Outcome 2: Progression‐free survival
Chemotherapy with interferon‐alpha and interleukin‐2 versus without interferon‐alpha and interleukin‐2
This comparison included seven studies (Atkins 2008; Atzpodien 2002; Eton 2002; Johnston 1998; Middleton 2007; Ridolfi 2002a; Rosenberg 1999). HRs either were directly available or could be extrapolated for six studies (Atkins 2008; Atzpodien 2002; Eton 2002; Johnston 1998; Middleton 2007; Ridolfi 2002a). Overall, 488 participants were allocated to chemotherapy with both interferon‐alpha and interleukin‐2 and 476 to chemotherapy alone. Meta‐analysis suggested a statistically significant better progression‐free survival for biochemotherapy (Analysis 6.2, HR 0.90, 95% CI 0.83 to 0.99; heterogeneity: Tau² = 0.00; Chi² = 5.22, df = 5, P = 0.39; I² = 4%; high‐quality evidence). This result was also confirmed when studies investigating first‐line treatment were considered (Analysis 7.2, HR 0.86, 95% CI 0.76 to 0.99).
6.2. Analysis.

Comparison 6: Chemotherapy ± interferon‐alpha and interleukin‐2, Outcome 2: Progression‐free survival
7.2. Analysis.

Comparison 7: Chemotherapy ± interferon‐alpha and interleukin‐2 (first line), Outcome 2: Progression‐free survival
Immune checkpoint inhibitors
Anti‐CTLA4 monoclonal antibodies plus chemotherapy versus chemotherapy alone (first line)
Two studies reported this comparison (Ribas 2013; Robert 2011) but HR data were not extractable from Ribas 2013. Robert 2011 randomised 250 participants to receive anti‐CTLA4 monoclonal antibodies plus chemotherapy and 252 participants to receive chemotherapy alone. Findings suggested a statistically significant better progression‐free survival for combination of anti‐CTLA plus chemotherapy (Analysis 10.2, HR 0.76, 95% CI 0.63 to 0.92; moderate‐quality evidence; downgraded due to imprecision).
10.2. Analysis.

Comparison 10: Anti‐CTLA4 monoclonal antibodies (first line), Outcome 2: Progression‐free survival
Anti‐CTLA4 monoclonal antibodies with immunostimulating agents versus without immunostimulating agents (second line)
This comparison included two studies (Hodi 2010a; Hodi 2014). Overall, 526 participants were allocated to anti‐CTLA4 monoclonal antibodies combined with immunostimulating agents (gp100 in Hodi 2010a and GM‐CSF in Hodi 2014), and 259 to anti‐CTLA4 monoclonal antibodies alone. Meta‐analysis suggested a better progression‐free survival for anti‐CTLA monoclonal antibodies alone, although the difference was not statistically significant (Analysis 11.2, HR 1.06, 95% CI 0.75 to 1.51; heterogeneity: Tau² = 0.05; Chi² = 3.61, df = 1, P = 0.06; I² = 72%; low‐quality evidence; downgraded due to inconsistency and imprecision). The inclusion of trials testing two different immunostimulating agents may explain high between‐study heterogeneity.
11.2. Analysis.

Comparison 11: Anti‐CTLA4 monoclonal antibodies ± other immunostimulating agents (second line), Outcome 2: Progression‐free survival
Anti‐PD1 monoclonal antibodies versus chemotherapy
This comparison included three studies (Ribas 2015; Robert 2015a; Weber 2015). HRs were either available or extractable for Ribas 2015 and Robert 2015a. Overall, 570 participants were allocated to anti‐PD1 monoclonal antibodies and 387 to chemotherapy alone. Meta‐analysis suggested a statistically significant better progression‐free survival for participants allocated to anti‐PD1 monoclonal antibodies (Analysis 12.2, HR 0.49, 95% CI 0.39 to 0.61; heterogeneity: Tau² = 0.01; Chi² = 2.26, df = 1, P = 0.13; I² = 56%; moderate‐quality evidence; downgraded due to inconsistency).
12.2. Analysis.

Comparison 12: Anti‐PD1 monoclonal antibodies versus chemotherapy, Outcome 2: Progression‐free survival
Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies
This comparison included two studies (Larkin 2015; Robert 2015b). Overall, 872 participants were allocated to anti‐PD1 monoclonal antibodies and 593 to anti‐CTLA4 monoclonal antibodies. Meta‐analysis suggested a statistically significant better progression‐free survival for participants treated with anti‐PD1 monoclonal antibodies (Analysis 13.2, HR 0.54, 95% CI 0.50 to 0.60; heterogeneity: Tau² = 0.00; Chi² = 0.13, df = 1, P = 0.72; I² = 0%; high‐quality evidence).
13.2. Analysis.

Comparison 13: Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies, Outcome 2: Progression‐free survival
Anti‐CTLA4 monoclonal antibodies with anti‐PD1 monoclonal antibodies versus without anti‐PD1 monoclonal antibodies
This comparison included two studies (Larkin 2015; Postow 2015). Overall, 386 participants were allocated to combination therapy with anti‐PD1 plus anti‐CTLA4 monoclonal antibodies and 352 to anti‐CTLA4 monoclonal antibodies alone. Meta‐analysis suggested a statistically significant better progression‐free survival for participants treated with combination treatment (Analysis 14.1, HR 0.40, 95% CI 0.35 to 0.46; heterogeneity: Tau² = 0.00; Chi² = 0.08, df = 1, P = 0.78; I² = 0%; high‐quality evidence).
14.1. Analysis.

Comparison 14: Anti‐PD1 monoclonal antibodies and anti‐CTLA4 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies alone, Outcome 1: Progression‐free survival
Small‐molecule targeted drugs
BRAF inhibitors versus chemotherapy
This comparison included two studies (Hauschild 2012; McArthur 2014). Overall, 524 participants were allocated to single agent BRAF inhibitor and 401 to chemotherapy alone. Meta‐analysis showed that single agent BRAF inhibitor was associated with a statistically significant better progression‐free survival (Analysis 18.2, HR 0.27, 95% CI 0.21 to 0.34, heterogeneity: Tau² = 0.00; Chi² = 0.24, df = 1, P = 0.63; I² = 0%; high‐quality evidence).
18.2. Analysis.

Comparison 18: Single agent BRAF inhibitor, Outcome 2: Progression‐free survival
MEK inhibitors versus chemotherapy
This comparison included three studies (Flaherty 2012b; Gupta 2014; Robert 2013). Overall, 300 participants were allocated to single agent MEK inhibitor and 196 to chemotherapy alone. Meta‐analysis suggested a statistically significantly better progression‐free survival for single agent MEK inhibitor (Analysis 19.2, HR 0.58, 95% CI 0.42 to 0.80; heterogeneity: Tau² = 0.05; Chi² = 4.75, df = 2, P = 0.09; I² = 58%; moderate‐quality evidence; downgraded due to inconsistency). The three studies included different participants populations and this may explain high between‐study heterogeneity. Gupta 2014 enrolled participants with wild‐type BRAF melanomas and Flaherty 2012b tested a MEK inhibitor in both pre‐treated and untreated participants. When Flaherty 2012b was excluded from the meta‐analysis, heterogeneity was reduced to 0%, and effect size decreased (HR 0.67, 95% CI 0.53 to 0.85).
19.2. Analysis.

Comparison 19: Single agent MEK inhibitor, Outcome 2: Progression‐free survival
BRAF inhibitors with versus without MEK inhibitors
This comparison was reported in four studies (Flaherty 2012a; Larkin 2014; Long 2015; Robert 2015). Overall, 918 participants were allocated to combination of BRAF and MEK inhibitors and 866 to single agent BRAF inhibitor. Meta‐analysis suggested a statistically significant better progression‐free survival for combination therapy (Analysis 20.2, HR 0.56, 95% CI 0.44 to 0.71); however, despite studies sharing similar designs, between‐study heterogeneity was high (Tau² = 0.04; Chi² = 9.82, df = 3, P = 0.02; I² = 69%; moderate‐quality evidence; downgraded due to inconsistency). Sensitivity analysis showed that Long 2015 determined heterogeneity; the I² value dropped to 9% when this study was excluded from analysis, with only minimal change in effect size (HR 0.52, 95% CI 0.44, 0.61).
20.2. Analysis.

Comparison 20: Combination of BRAF and MEK inhibitors versus single agent BRAF inhibitor, Outcome 2: Progression‐free survival
Chemotherapy with versus without other agents
Chemotherapy with Bacillus Calmette‐Guérin (BCG) versus without BCG
Six studies investigated this comparison (Costanzi 1982; Mastrangelo 1979; Newlands 1976; Ramseur 1978; Veronesi 1984; Verschraegen 1993). However, the studies did not investigate progression‐free survival, nor were HRs available or extractable.
Chemotherapy with Corynebacterium parvum versus without C parvum
Seven studies investigated this comparison (Clunie 1980; Gough 1978; Kokoschka 1978; Presant 1979; Robidoux 1982; Thatcher 1986; Veronesi 1984). However, the studies did not investigate progression‐free survival, nor were HRs available or extractable.
Chemotherapy with versus without tamoxifen
Four studies investigated this comparison (Agarwala 1999; Cocconi 1992; Falkson 1998; Rusthoven 1996). HRs were either available or extractable for Falkson 1998 and Rusthoven 1996. Tamoxifen‐based polychemotherapy was administered to 238 participants and 237 participants received chemotherapy alone. Tamoxifen was associated with a non statistically significant slightly higher risk of melanoma progression (Analysis 2.2, HR 1.06, 95% CI 0.93 to 1.22; heterogeneity: Tau² = 0.00; Chi² = 0.29, df = 1, P = 0.59; I² = 0%; high‐quality evidence).
2.2. Analysis.

Comparison 2: Chemotherapy ± tamoxifen, Outcome 2: Progression‐free survival
Chemotherapy with sorafenib versus without sorafenib
This comparison included three studies (Flaherty 2013a; Hauschild 2009a; McDermott 2008). Overall, 596 participants were allocated to standard chemotherapy plus sorafenib and 598 to chemotherapy alone. Meta‐analysis suggested better progression‐free survival for participants undergoing chemotherapy plus sorafenib, although the difference was not statistically significant (Analysis 15.2, HR 0.89, 95% CI 0.73 to 1.09; heterogeneity: Tau² = 0.01; Chi² = 2.94, df = 2, P = 0.23; I² = 32%; moderate‐quality evidence; downgraded due to imprecision).
15.2. Analysis.

Comparison 15: Chemotherapy ± sorafenib, Outcome 2: Progression‐free survival
Chemotherapy with elesclomol versus without elesclomol
This comparison was reported by two studies (O'Day 2011; O'Day 2013). Overall, 378 participants were allocated to standard chemotherapy plus elesclomol and 354 to chemotherapy alone. Meta‐analysis suggested better progression‐free survival for participants undergoing chemotherapy plus elesclomol, although the difference was not statistically significant (Analysis 16.2, HR 0.75, 95% CI 0.50 to 1.13; heterogeneity: Tau² = 0.06; Chi² = 3.23, df = 1, P = 0.07; I² = 69%; low‐quality evidence; downgraded due to inconsistency and imprecision).
16.2. Analysis.

Comparison 16: Chemotherapy ± elesclomol, Outcome 2: Progression‐free survival
Chemotherapy with anti‐angiogenic drugs versus without anti‐angiogenic drugs
This comparison was reported by two studies (Cui 2013; Kim 2012). Overall, 199 participants were allocated to standard chemotherapy plus anti‐angiogenic therapies and 125 to chemotherapy alone. Meta‐analysis suggested a statistically significant progression‐free survival benefit for combination of chemotherapy and anti‐angiogenic agents (Analysis 17.2, HR 0.69, 95% CI 0.52 to 0.92; heterogeneity: Tau² = 0.01; Chi² = 1.17, df = 1, P = 0.28; I² = 14%; moderate‐quality evidence; downgraded due imprecision ‐ sample size was smaller than optimal information size).
17.2. Analysis.

Comparison 17: Chemotherapy ± anti‐angiogenic drugs, Outcome 2: Progression‐free survival
Other comparisons
Single agent chemotherapy versus other single agent chemotherapy
Meta‐analysis was feasible for two different single agent drug regimens: dacarbazine and temozolomide. Three trials were included (Chiarion‐Sileni 2011; Middleton 2000; Patel 2011). Overall, 659 and 654 participants were allocated to temozolomide and dacarbazine, respectively. Temozolomide was associated with a statistically non‐significant progression‐free survival improvement compared to single agent dacarbazine (Analysis 3.2, HR 0.87, 95% CI 0.74 to 1.03; heterogeneity: Tau² = 0.01; Chi² = 3.08, df = 2, P = 0.21; I² = 35%; moderate‐quality evidence; downgraded due to imprecision).
3.2. Analysis.

Comparison 3: Temozolomide versus dacarbazine, Outcome 2: Progression‐free survival
Toxicity
Polychemotherapy versus single agent chemotherapy
This comparison included 15 studies (Bajetta 1994; Bajetta 2006a; Danson 2003; Daponte 2013; Dorval 1999; Falkson 1991; Falkson 1995; Falkson 1998; Gorbonova 2000; Kaufmann 2005; Kirkwood 1990; Maio 2010; Thomson 1993; Vorobiof 1994; Young 2001). Description of ≥ G3 toxicity, expressed as the number of participants experiencing toxicity, was available from three studies (Costanza 1977; Chauvergne 1982; Glover 2003). Cytotoxic polychemotherapy and single agent chemotherapy were administered in 241 and 149 participants, respectively, with a statistically significant higher rate of high‐grade toxicity among those undergoing polychemotherapy (Analysis 1.4, RR 1.97, 95% CI 1.44 to 2.71; I² = 42%; moderate‐quality evidence).
1.4. Analysis.

Comparison 1: Polychemotherapy versus single agent chemotherapy, Outcome 4: Toxicity (≥ G3)
Biochemotherapy versus chemotherapy
Chemotherapy with interferon‐alpha versus without interferon‐alpha
This comparison included 13 studies (Bajetta 1994; Bajetta 2006a; Danson 2003; Daponte 2013; Dorval 1999; Falkson 1991; Falkson 1995; Falkson 1998; Gorbonova 2000; Kaufmann 2005; Thomson 1993; Vorobiof 1994; Young 2001). Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was available from three studies (Bajetta 1994; Falkson 1991; Maio 2010). Overall, 579 participants were allocated to chemotherapy plus interferon‐alpha and 212 to chemotherapy alone. Meta‐analysis suggested a non statistically significant higher rate of ≥ G3 toxicity for the combined regimen (Analysis 4.4, RR 1.72, 95% CI 0.37 to 7.95; heterogeneity: Tau² = 1.16; Chi² = 5.51, df = 2, P = 0.06; I² = 64%; low‐quality evidence; downgraded due to inconsistency and imprecision).
4.4. Analysis.

Comparison 4: Chemotherapy ± interferon‐alpha, Outcome 4: Toxicity (≥ G3)
Chemotherapy with interleukin‐2 versus without interleukin‐2
This comparison included two studies (Hauschild 2001; Keilholz 2005). Overall, 320 participants were allocated to chemotherapy plus interleukin‐2 and 324 to chemotherapy alone. Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was unavailable from the studies.
Chemotherapy with interferon‐alpha plus interleukin‐2 versus without interferon‐alpha plus interleukin‐2
This comparison included seven studies (Atkins 2008; Atzpodien 2002; Eton 2002; Johnston 1998; Middleton 2007; Ridolfi 2002a; Rosenberg 1999). Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was available from Johnston 1998 and Middleton 2007. Analysis suggested a statistically significant higher ≥ G3 toxicity for combined chemotherapy, interferon‐alpha and interleukin‐2 (Analysis 6.4, RR 1.35, 95% CI 1.14 to 1.61; heterogeneity: Tau²: 0.00, Chi² = 0.50, df = 1, P = 0.48; I² = 0%; high‐quality evidence). When the analysis was restricted to the first‐line setting, results (based on a single study ‐ Middleton 2007) were similar (Analysis 7.4, RR 1.45, 95% CI 1.12 to 1.87).
6.4. Analysis.

Comparison 6: Chemotherapy ± interferon‐alpha and interleukin‐2, Outcome 4: Toxicity (≥ G3)
7.4. Analysis.

Comparison 7: Chemotherapy ± interferon‐alpha and interleukin‐2 (first line), Outcome 4: Toxicity (≥ G3)
Immune checkpoint inhibitors
Anti‐CTLA4 monoclonal antibodies plus chemotherapy versus chemotherapy alone (first line)
This comparison included two studies (Ribas 2013; Robert 2011). Overall, 578 participants were allocated to anti‐CTLA4 monoclonal antibodies plus chemotherapy and 579 to chemotherapy alone. Meta‐analysis suggested a statistically significant higher rate of ≥ G3 toxicity for combined anti‐CTLA and chemotherapy (Analysis 10.4, RR 1.69, 95% CI 1.19 to 2.42; heterogeneity: Tau² = 0.06; Chi² = 6.51, df = 1, P = 0.01; I² = 85%; moderate‐quality evidence; downgraded due to inconsistency).
10.4. Analysis.

Comparison 10: Anti‐CTLA4 monoclonal antibodies (first line), Outcome 4: Toxicity (≥ G3)
Anti‐CTLA4 monoclonal antibodies with immune stimulating agents versus without immune stimulating agents (second line)
This comparison included two studies (Hodi 2010a; Hodi 2014) Overall, 526 participants were allocated to anti‐CTLA4 monoclonal antibodies plus immune stimulating agents (gp100 in Hodi 2010a and GM‐CSF in Hodi 2014), and 259 to anti‐CTLA4 monoclonal antibodies alone. Meta‐analysis suggested higher rates of ≥ G3 toxicity for the combined regimen, although the difference was not statistically significant (Analysis 11.4, RR 0.87, 95% CI 0.69 to 1.11; heterogeneity: Tau² = 0.02; Chi² = 2.08, df = 1, P = 0.15; I² = 52%; low‐quality evidence; downgraded due to inconsistency and imprecision).
11.4. Analysis.

Comparison 11: Anti‐CTLA4 monoclonal antibodies ± other immunostimulating agents (second line), Outcome 4: Toxicity (≥ G3)
Anti‐PD1 monoclonal antibodies versus chemotherapy
This comparison included three studies (Ribas 2015; Robert 2015a; Weber 2015). Overall, 847 participants were allocated to anti‐PD1 monoclonal antibodies and 520 to chemotherapy alone. Meta‐analysis showed a statistically significant lower ≥ G3 toxicity rate for anti‐PD1 monoclonal antibodies (Analysis 12.4, RR 0.55, 95% CI 0.31 to 0.97; heterogeneity: Tau² = 0.21; Chi² = 14.24, df = 2, P = 0.0008; I² = 86%; low‐quality evidence; downgraded due to inconsistency and imprecision).
12.4. Analysis.

Comparison 12: Anti‐PD1 monoclonal antibodies versus chemotherapy, Outcome 4: Toxicity (≥ G3)
Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies
This comparison included two studies (Larkin 2015; Robert 2015b). Overall, 872 participants were allocated to anti‐PD1 monoclonal antibodies and 593 to anti‐CTLA4 monoclonal antibodies. Meta‐analysis showed a statistically significant lower ≥ G3 toxicity rate for anti‐PD1 monoclonal antibodies (Analysis 13.4, RR 0.70, 95% CI 0.54 to 0.91; heterogeneity: Tau² = 0.02; Chi² = 2.14, df = 1, P = 0.14; I² = 53%; low‐quality evidence; downgraded due to inconsistency and imprecision).
13.4. Analysis.

Comparison 13: Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies, Outcome 4: Toxicity (≥ G3)
Anti‐CTLA4 monoclonal antibodies with anti‐PD1 monoclonal antibodies versus without anti‐PD1 monoclonal antibodies
This comparison included two studies (Larkin 2015; Postow 2015). Overall, 386 participants were allocated to combination therapy with anti‐PD1 and anti‐CTLA4 monoclonal antibodies and 352 to anti‐CTLA4 monoclonal antibodies alone. Meta‐analysis suggested a higher ≥ G3 toxicity rate for anti‐CTLA4 monoclonal antibodies, although the difference was not statistically significant (Analysis 14.3, RR 1.57, 95% CI 0.85 to 2.92; heterogeneity: Tau² = 0.16; Chi² = 5.00, df = 1, P = 0.03; I² = 80%; low‐quality evidence; downgraded due to inconsistency and imprecision).
14.3. Analysis.

Comparison 14: Anti‐PD1 monoclonal antibodies and anti‐CTLA4 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies alone, Outcome 3: Toxicity (≥ G3)
Small‐molecule targeted drugs
BRAF inhibitors versus chemotherapy
This comparison included two studies (Hauschild 2012; McArthur 2014). Overall, 524 participants were allocated to single agent BRAF inhibitor and 401 to chemotherapy alone. Meta‐analysis suggested a higher ≥ G3 toxicity rate for single agent BRAF inhibitor, although the difference was not statistically significant (Analysis 18.4, RR 1.27, 95% CI 0.48 to 3.33; heterogeneity: Tau² = 0.43; Chi² = 8.35, df = 1, P = 0.004; I² = 88%; low‐quality evidence; downgraded due to inconsistency and imprecision).
18.4. Analysis.

Comparison 18: Single agent BRAF inhibitor, Outcome 4: Toxicity (≥ G3)
MEK inhibitors versus chemotherapy
This comparison included three studies (Flaherty 2012b; Gupta 2014; Robert 2013). Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was available only from Robert 2013. There was a statistically significant higher ≥ G3 toxicity rate reported for MEK inhibitor (Analysis 19.4, RR 1.61, 95% CI 1.08 to 2.41; moderate‐quality evidence; downgraded due to imprecision).
19.4. Analysis.

Comparison 19: Single agent MEK inhibitor, Outcome 4: Toxicity (≥ G3)
BRAF inhibitors with versus without MEK inhibitors
This comparison included four studies (Flaherty 2012a; Larkin 2014; Long 2015; Robert 2015). Overall, 918 participants were allocated to combination of BRAF and MEK inhibitors and 866 to single agent BRAF inhibitor. Meta‐analysis suggested a lower ≥ G3 toxicity rate for combination therapy, although the difference was not statistically significant (Analysis 20.4, RR 1.01, 95% CI 0.85 to 1.20; heterogeneity: Tau² = 0.02; Chi² = 8.24, df = 3, P = 0.04; I² = 64%; moderate‐quality evidence; downgraded due to inconsistency).
20.4. Analysis.

Comparison 20: Combination of BRAF and MEK inhibitors versus single agent BRAF inhibitor, Outcome 4: Toxicity (≥ G3)
Chemotherapy with versus without other agents
Chemotherapy with Bacillus Calmette‐Guérin (BCG) versus without BCG
Six studies investigated this comparison (Costanzi 1982; Mastrangelo 1979; Newlands 1976; Ramseur 1978; Veronesi 1984; Verschraegen 1993). Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was unavailable from these studies.
Chemotherapy with Corynebacterium parvum versus without C parvum
Seven studies investigated this comparison (Clunie 1980; Gough 1978; Kokoschka 1978; Presant 1979; Robidoux 1982; Thatcher 1986; Veronesi 1984). Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was unavailable from these studies.
Chemotherapy with tamoxifen versus without tamoxifen
Four studies investigated this comparison; all had either available or extractable HRs (Agarwala 1999; Cocconi 1992; Falkson 1998; Rusthoven 1996). Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was available in only from Falkson 1998. Falkson 1998 administered tamoxifen‐based polychemotherapy and single agent chemotherapy to 134 and 137 participants, respectively. There was a non statistically significant lower rate of ≥ G3 toxicity among participants undergoing tamoxifen‐based polychemotherapy (Analysis 2.4, RR 0.70, 95% CI 0.38 to 1.28; moderate‐quality evidence; downgraded due to imprecision).
2.4. Analysis.

Comparison 2: Chemotherapy ± tamoxifen, Outcome 4: Toxicity (≥ G3)
Chemotherapy with sorafenib versus without sorafenib
This comparison included three studies (Flaherty 2013a; Hauschild 2009a; McDermott 2008). Overall, 596 participants were allocated to standard chemotherapy plus sorafenib and 598 to chemotherapy alone. Meta‐analysis suggested a higher ≥ G3 toxicity rate for chemotherapy plus sorafenib, although the difference was not statistically significant (Analysis 15.4, RR 1.08, 95% CI 0.93 to 1.26; heterogeneity: Tau² = 0.01; Chi² = 3.40, df = 2, P = 0.18; I² = 41%; moderate‐quality evidence; downgraded due to imprecision).
15.4. Analysis.

Comparison 15: Chemotherapy ± sorafenib, Outcome 4: Toxicity (≥ G3)
Chemotherapy with elesclomol versus without elesclomol
This comparison included two studies (O'Day 2011; O'Day 2013). Overall, 378 participants were allocated to standard chemotherapy plus elesclomol and 354 to chemotherapy alone. Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was available in only from O'Day 2013. O'Day 2013 reported a marginally statistically significant higher toxicity for chemotherapy plus elesclomol (Analysis 16.4, RR 1.22, 95% CI 1.00 to 1.50; moderate‐quality evidence; downgraded due to imprecision).
16.4. Analysis.

Comparison 16: Chemotherapy ± elesclomol, Outcome 4: Toxicity (≥ G3)
Chemotherapy with anti‐angiogenic drugs versus without anti‐angiogenic drugs
This comparison included two studies (Cui 2013; Kim 2012). Overall, 199 participants were allocated to standard chemotherapy plus anti‐angiogenic drugs bevacizumab (Kim 2012) and endostar (Cui 2013) and 125 to chemotherapy alone. Meta‐analysis suggested a higher ≥ G3 toxicity rate for chemotherapy alone, although the difference was not statistically significant (Analysis 17.4, RR 0.68, 95% CI 0.09 to 5.32; heterogeneity: Tau² = 1.53; Chi² = 2.34, df = 1, P = 0.13; I² = 57%; low‐quality evidence; downgraded due to inconsistency and imprecision).
17.4. Analysis.

Comparison 17: Chemotherapy ± anti‐angiogenic drugs, Outcome 4: Toxicity (≥ G3)
Other comparisons
Single agent chemotherapy versus other single agent chemotherapy
Meta‐analysis was feasible for the comparison between dacarbazine and temozolomide. Three trials were included (Chiarion‐Sileni 2011; Middleton 2000; Patel 2011). Description of ≥ G3 toxicity, expressed as number of participants experiencing toxicity, was available from two studies (Middleton 2000; Patel 2011). Overall, 585 and 579 participants were allocated to temozolomide and dacarbazine, respectively. Temozolomide was found to be less toxic than dacarbazine, which had higher incidence of ≥ G3 toxicity, although the difference was not statistically significant (Analysis 3.4, RR 1.15, 95% CI 0.98 to 1.35; heterogeneity: Tau²: 0.00, Chi² = 0.62, df = 1, P = 0.43; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
3.4. Analysis.

Comparison 3: Temozolomide versus dacarbazine, Outcome 4: Toxicity (≥ G3)
Objective tumour response
Polychemotherapy versus single agent chemotherapy
This comparison included 15 studies (Bajetta 1994; Bajetta 2006a; Danson 2003; Daponte 2013; Dorval 1999; Falkson 1991; Falkson 1995; Falkson 1998; Gorbonova 2000; Kaufmann 2005; Kirkwood 1990; Maio 2010; Thomson 1993; Vorobiof 1994; Young 2001). Cytotoxic polychemotherapy and single agent chemotherapy was administered in 1124 and 761 participants, respectively. Meta‐analysis showed a statistically significant higher response rate for polychemotherapy (Analysis 1.3, RR 1.27, 95% CI 1.02 to 1.58; heterogeneity: Tau² = 0.00; Chi² = 5.43, df = 7, P = 0.61; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
1.3. Analysis.

Comparison 1: Polychemotherapy versus single agent chemotherapy, Outcome 3: Tumour response
Biochemotherapy versus chemotherapy
Chemotherapy with interferon‐alpha versus without interferon‐alpha
This comparison included 15 studies (Bajetta 1994; Bajetta 2006a; Danson 2003; Daponte 2013; Dorval 1999; Falkson 1991; Falkson 1995; Falkson 1998; Gorbonova 2000; Kirkwood 1990; Kaufmann 2005; Maio 2010; Thomson 1993; Vorobiof 1994; Young 2001). Overall, 1403 participants were allocated to chemotherapy with interferon‐alpha and 1061 to chemotherapy alone. Meta‐analysis suggested a statistically significant higher objective response for combination of chemotherapy and interferon (Analysis 4.3, RR 1.36, 95% CI 1.12 to 1.66; heterogeneity: Tau² = 0.03; Chi² = 16.93, df = 14, P = 0.26; I² = 17%; high‐quality evidence).
4.3. Analysis.

Comparison 4: Chemotherapy ± interferon‐alpha, Outcome 3: Tumour response
Chemotherapy with interleukin‐2 versus without interleukin‐2
This comparison included three studies (Hauschild 2001; Keilholz 2005; Sertoli 1999). Overall, 381 participants were allocated to chemotherapy with interleukin‐2 and 354 to chemotherapy alone. Meta‐analysis suggested a higher response rate for chemotherapy alone, although the difference was not statistically significant (Analysis 5.3, RR 0.85, 95% CI 0.64 to 1.13; heterogeneity: Tau² = 0.00; Chi² = 0.68, df = 2, P = 0.71; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
5.3. Analysis.

Comparison 5: Chemotherapy ± interleukin‐2, Outcome 3: Tumour response
Chemotherapy with interferon‐alpha and interleukin‐2 versus without interferon‐alpha and interleukin‐2
This comparison included seven studies (Atkins 2008; Atzpodien 2002; Eton 2002; Johnston 1998; Middleton 2007; Ridolfi 2002a; Rosenberg 1999). Overall, 474 participants were allocated to chemotherapy with both interferon‐alpha and interleukin‐2 and 296 to chemotherapy alone. Meta‐analysis showed a statistically significant higher response rate for biochemotherapy (Analysis 6.3, RR 1.36, 95% CI 1.11 to 1.67; heterogeneity: Tau² = 0.00; Chi² = 6.16, df = 6, P = 0.41; I² = 3%; high‐quality evidence). When the analysis was restricted to the first‐line setting, results were similar (Analysis 7.3, RR 1.45, 95% CI 1.15 to 1.83; heterogeneity: Tau² = 0.00; Chi² = 4.25, df = 4, P = 0.37; I² = 6%).
6.3. Analysis.

Comparison 6: Chemotherapy ± interferon‐alpha and interleukin‐2, Outcome 3: Tumour response
7.3. Analysis.

Comparison 7: Chemotherapy ± interferon‐alpha and interleukin‐2 (first line), Outcome 3: Tumour response
Immune checkpoint inhibitors
Anti‐CTLA4 monoclonal antibodies plus chemotherapy versus chemotherapy alone (first line)
This comparison included two studies (Ribas 2013; Robert 2011). Overall, 578 participants were allocated to anti‐CTLA4 monoclonal antibodies and chemotherapy and 579 to chemotherapy alone. Meta‐analysis suggested a higher response rate for the combined regimen, although the difference was not statistically significant (Analysis 10.3, RR 1.28, 95% CI 0.92 to 1.77; heterogeneity: Tau² = 0.00; Chi² = 0.68, df = 1, P = 0.41; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
10.3. Analysis.

Comparison 10: Anti‐CTLA4 monoclonal antibodies (first line), Outcome 3: Tumour response
Anti‐CTLA4 monoclonal antibodies with immunostimulating agents versus without immunostimulating agents (second line)
This comparison included two studies (Hodi 2010a; Hodi 2014) Overall, 526 participants were allocated to anti‐CTLA4 monoclonal antibodies and with immunostimulating agents (gp100 in Hodi 2010a and GM‐CSF in Hodi 2014), and 259 to anti‐CTLA4 monoclonal antibodies alone. Meta‐analysis suggested a higher response rate for the combined regimen, although the difference was not statistically significant (Analysis 11.3, RR 0.74, 95% CI 0.38 to 1.47; heterogeneity: Tau² = 0.15; Chi² = 2.53, df = 1, P = 0.11; I² = 60%; low‐quality evidence; downgraded due to inconsistency and imprecision).
11.3. Analysis.

Comparison 11: Anti‐CTLA4 monoclonal antibodies ± other immunostimulating agents (second line), Outcome 3: Tumour response
Anti‐PD1 monoclonal antibodies versus chemotherapy
This comparison included three studies (Ribas 2015; Robert 2015a; Weber 2015). Overall, 847 participants were allocated to anti‐PD1 monoclonal antibodies and 520 to chemotherapy alone. Meta‐analysis showed a statistically significant higher response rate for anti‐PD1 monoclonal antibodies (Analysis 12.3, RR 3.42, 95% CI 2.38 to 4.92; heterogeneity: Tau² = 0.02; Chi² = 2.35, df = 2, P = 0.31; I² = 15%; high‐quality evidence).
12.3. Analysis.

Comparison 12: Anti‐PD1 monoclonal antibodies versus chemotherapy, Outcome 3: Tumour response
Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies
This comparison included two studies (Larkin 2015; Robert 2015b). Overall, 872 participants were allocated to anti‐PD1 monoclonal antibodies and 593 to anti‐CTLA4 monoclonal antibodies. Meta‐analysis showed a statistically significant higher response rate for anti‐PD1 monoclonal antibodies (Analysis 13.3, RR 2.47, 95% CI 2.01 to 3.04; heterogeneity: Tau² = 0.00; Chi² = 0.87, df = 1, P = 0.35; I² = 0%; high‐quality evidence).
13.3. Analysis.

Comparison 13: Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies, Outcome 3: Tumour response
Anti‐CTLA4 monoclonal antibodies with anti‐PD1 monoclonal antibodies versus without anti‐PD1 monoclonal antibodies
This comparison included two studies (Larkin 2015; Postow 2015). Overall, 386 participants were allocated to combination therapy with anti‐PD1 anti‐CTLA4 monoclonal antibodies and 352 to anti‐CTLA4 monoclonal antibodies alone. Meta‐analysis showed a statistically significant higher response rate for the combined regimen (Analysis 14.2, RR 3.50, 95% CI 2.07 to 5.92; heterogeneity: Tau² = 0.08; Chi² = 1.63, df = 1, P = 0.20; I² = 39%; high‐quality evidence).
14.2. Analysis.

Comparison 14: Anti‐PD1 monoclonal antibodies and anti‐CTLA4 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies alone, Outcome 2: Tumour response
Small‐molecule targeted drugs
BRAF inhibitors versus chemotherapy
This comparison included two studies (Hauschild 2012; McArthur 2014). Overall, 524 participants were allocated to single agent BRAF inhibitor and 401 to chemotherapy alone. Meta‐analysis showed a statistically significant higher response rate for single agent BRAF inhibitor (Analysis 18.3, RR 6.78, 95% CI 4.84 to 9.49; heterogeneity: Tau² = 0.00; Chi² = 0.10, df = 1, P = 0.75; I² = 0%; high‐quality evidence).
18.3. Analysis.

Comparison 18: Single agent BRAF inhibitor, Outcome 3: Tumour response
MEK inhibitors versus chemotherapy
This comparison included three studies (Flaherty 2012b; Gupta 2014; Robert 2013). Overall, 300 participants were allocated to single agent MEK inhibitor and 196 to chemotherapy alone. Meta‐analysis showed a statistically significant higher response rate for single agent MEK inhibitor (Analysis 19.3, RR 2.01, 95% CI 1.35 to 2.99; heterogeneity: Tau² = 0.00; Chi² = 1.51, df = 2, P = 0.47; I² = 0%; high‐quality evidence).
19.3. Analysis.

Comparison 19: Single agent MEK inhibitor, Outcome 3: Tumour response
BRAF inhibitors with MEK inhibitors versus without MEK inhibitors
This comparison included four studies (Flaherty 2012a; Larkin 2014; Long 2015; Robert 2015). Overall, 918 participants were allocated to combination of BRAF and MEK inhibitors and 866 to single agent BRAF inhibitor. Meta‐analysis showed a statistically significant higher response rate for combination therapy (Analysis 20.3, RR 1.32, 95% CI 1.20 to 1.46; heterogeneity: Tau² = 0.00; Chi² = 3.90, df = 3, P = 0.27; I² = 23%; high‐quality evidence).
20.3. Analysis.

Comparison 20: Combination of BRAF and MEK inhibitors versus single agent BRAF inhibitor, Outcome 3: Tumour response
Chemotherapy with other agents versus without other agents
Chemotherapy with Bacillus Calmette‐Guérin (BCG) versus without BCG
Six studies investigated this comparison (Costanzi 1982; Mastrangelo 1979; Newlands 1976; Ramseur 1978; Veronesi 1984; Verschraegen 1993). Overall, 658 participants were allocated to chemotherapy with BCG and 649 to chemotherapy alone. Meta‐analysis suggested a higher response rate for chemotherapy alone, although the difference was not statistically significant (Analysis 8.2, RR 0.85, 95% CI 0.65 to 1.12; heterogeneity: Tau² = 0.00; Chi² = 4.76, df = 5, P = 0.45; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
8.2. Analysis.

Comparison 8: Chemotherapy ± Bacille Calmette‐Guérin (BCG), Outcome 2: Tumour response
Chemotherapy with Corynebacterium parvum versus without C parvum
Seven studies investigated this comparison (Clunie 1980; Gough 1978; Kokoschka 1978; Presant 1979; Robidoux 1982; Thatcher 1986; Veronesi 1984). Overall, 247 participants were allocated to chemotherapy with C parvum and 290 to chemotherapy alone. Meta‐analysis suggested a higher response rate for chemotherapy plus C parvum, although the difference was not statistically significant (Analysis 9.2, RR 1.03, 95% CI 0.77 to 1.38; heterogeneity: Tau² = 0.00; Chi² = 5.63, df = 6, P = 0.47; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
9.2. Analysis.

Comparison 9: Chemotherapy ± Corynebacterium parvum, Outcome 2: Tumour response
Chemotherapy with tamoxifen versus without tamoxifen
Four studies investigated this comparison (Agarwala 1999; Cocconi 1992; Falkson 1998; Rusthoven 1996). Tamoxifen‐based polychemotherapy was administered to 326 participants and 317 received cytotoxic chemotherapy alone. Tamoxifen was associated with a non statistically significant higher response rate (Analysis 2.3, RR 1.33, 95% CI 0.94 to 1.89; heterogeneity: Tau² = 0.02; Chi² = 3.44, df = 3, P = 0.33; I² = 13%; moderate‐quality evidence; downgraded due to imprecision).
2.3. Analysis.

Comparison 2: Chemotherapy ± tamoxifen, Outcome 3: Tumour response
Chemotherapy with sorafenib versus without sorafenib
This comparison included three studies (Flaherty 2013a; Hauschild 2009a; McDermott 2008). Overall, 596 participants were allocated to standard chemotherapy plus sorafenib and 598 to chemotherapy alone. Meta‐analysis suggested a higher response rate for chemotherapy plus sorafenib, although the difference was not statistically significant (Analysis 15.3, RR 1.17, 95% CI 0.91 to 1.50; heterogeneity: Tau² = 0.00; Chi² = 1.41, df = 2, P = 0.49; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
15.3. Analysis.

Comparison 15: Chemotherapy ± sorafenib, Outcome 3: Tumour response
Chemotherapy with elesclomol versus without elesclomol
This comparison included two studies (O'Day 2011; O'Day 2013). Overall, 378 participants were allocated to standard chemotherapy plus elesclomol and 354 to chemotherapy alone. Meta‐analysis suggested a higher response rate for chemotherapy plus elesclomol, although the difference was not statistically significant (Analysis 16.3, RR 1.86, 95% CI 0.98 to 3.50; heterogeneity: Tau² = 0.00; Chi² = 0.12, df = 1, P = 0.73; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
16.3. Analysis.

Comparison 16: Chemotherapy ± elesclomol, Outcome 3: Tumour response
Chemotherapy with anti‐angiogenic drugs versus without anti‐angiogenic drugs
This comparison included two studies (Cui 2013; Kim 2012). Overall, 199 participants were allocated to standard chemotherapy plus anti‐angiogenic drugs bevacizumab (Kim 2012) and endostar (Cui 2013) and 125 to chemotherapy alone. Meta‐analysis suggested a statistically significant higher response rate for the combination of chemotherapy plus anti‐angiogenic agents, although the difference was not statistically significant (Analysis 17.3, RR 1.71, 95% CI 0.96 to 3.03; heterogeneity: Tau² = 0.00; Chi² = 0.20, df = 1, P = 0.65; I² = 0%; moderate‐quality evidence; downgraded due to imprecision).
17.3. Analysis.

Comparison 17: Chemotherapy ± anti‐angiogenic drugs, Outcome 3: Tumour response
Other comparisons
Single agent chemotherapy versus other single agent chemotherapy
Meta‐analysis was feasible for the comparison between temozolomide and dacarbazine. Three trials were eligible (Chiarion‐Sileni 2011; Middleton 2000; Patel 2011). Overall, 659 and 654 participants were allocated to temozolomide and dacarbazine, respectively. Temozolomide was associated with a non statistically significant higher response rate compared to single agent dacarbazine (Analysis 3.3, RR 1.21, 95% CI 0.85 to 1.73; heterogeneity: Tau² = 0.03; Chi² = 2.75, df = 2 (P = 0.25); I² = 27%; moderate‐quality evidence; downgraded due to imprecision).
3.3. Analysis.

Comparison 3: Temozolomide versus dacarbazine, Outcome 3: Tumour response
Quality of life
Polychemotherapy versus single agent chemotherapy
No data were available for this comparison.
Biochemotherapy versus chemotherapy
Chemotherapy with interferon‐alpha versus without interferon‐alpha
The effect on quality of life after dacarbazine plus recombinant interferon‐alpha was compared to dacarbazine alone for participants with metastatic malignant melanoma. In Young 2001, no differences in quality of life were observed between treatment groups. The same finding was reported in Thomson 1993 but fatigue and activity, as measured using linear analogue scale of assessment (LASA) scale and functional living index respectively, both improved in the combination treatment group.
Chemotherapy with interferon‐alpha and interleukin‐2 versus without interferon‐alpha and interleukin‐2
Chiarion‐Sileni 2003 used the Rotterdam Symptom Checklist (RSCL) questionnaire to compare quality of life in advanced melanoma participants receiving biochemotherapy or chemotherapy. Deterioration in overall quality of life reported with biochemotherapy was significantly worse than with chemotherapy. Mean scores decreased in all domains in the biochemotherapy group, but in the chemotherapy group, only activity level and physical symptom distress scores showed deterioration.
Interleukin‐2 with histamine versus without histamine
This comparison was assessed in Agarwala 2002 but quality of life was evaluated and reported in an extension study (Beusterien 2003). Three distinct assessments were completed by participants at different time points. Overall State of Health (OSH) and General Health Perception (GHP) scores did not differ significantly between groups. However, Quality of Well Being Scale ‐ Self‐Administered (QWB‐SA) scores deteriorated more quickly over time in the interleukin‐2 only group compared to the interleukin‐2 plus histamine group. This led to a significant difference in median quality‐adjusted survival duration in favour of the interleukin‐2 plus histamine group.
Immune checkpoint inhibitors
Anti‐CTLA4 monoclonal antibodies (first line)
Sherrill 2013 conducted a quality‐adjusted time without symptoms of disease or toxicity of treatment (Q‐TWIST) analysis for participants with untreated stage III/IV melanoma to compare quality of life after ipilimumab plus dacarbazine versus placebo plus dacarbazine. Quality‐adjusted survival was not significantly different between the groups during the first year of study (0.50 months favouring the ipilimumab/dacarbazine group) but after extended follow‐up, this difference gradually increased to 1.5 months, 2.36 months and 3.28 months at 2, 3 and 4 years, respectively.
Anti‐CTLA4 monoclonal antibodies with immunostimulating agents versus without other immunostimulating agents (second line)
This comparison was evaluated in Revicki 2012 where health‐related quality of life (HRQoL) outcomes were assessed during the study's 12 week treatment induction period for participants with stage III or IV melanoma. Ipilimumab with or without gp1000 vaccine was compared to gp100 vaccine alone and was shown to have no significant negative impact on HRQoL compared to gp100 alone. Constipation was reported to be significantly improved in the ipilimumab arms compared to the gp100 alone arm.
Anti‐PD1 monoclonal antibodies versus chemotherapy
In KEYNOTE‐002, a randomised, controlled phase II trial, participants with ipilimumab‐refractory melanoma were treated with either pembrolizumab (anti‐PD1 monoclonal antibody) or chemotherapy (Ribas 2015). In terms of health‐related quality of life, participants treated with pembrolizumab consistently reported less deterioration in individual function and symptoms scales when compared to those treated with chemotherapy. Furthermore, fewer participants in the pembrolizumab group reported decrements of more than 10 points in the global health status quality of life score compared to the chemotherapy group.
Small‐molecule targeted drugs
BRAF inhibitors versus chemotherapy
In Grob 2014, single agent dabrafenib (a BRAF inhibitor) was found to be superior to dacarbazine chemotherapy in improving quality of life for participants with metastatic melanoma in the BREAK‐3 study. More specifically, on the basis of EORTC QLQ‐C30 questionnaires, there was an enhancement of emotional and social functioning as well as an improvement in unwanted symptoms such as nausea and vomiting, appetite loss, diarrhoea, fatigue, dyspnoea and insomnia.
MEK inhibitors versus chemotherapy
In Schadendorf 2014, participants with BRAF mutated metastatic melanoma from the METRIC study were assessed in terms of quality of life after receiving the MEK inhibitor trametinib as a single agent versus chemotherapy. Based on EORTC QLQ‐C30 questionnaires the trametinib group showed improvement from baseline in various parameters including better global health, physical, role, and social functioning as well as reduction in fatigue, pain, insomnia, nausea and vomiting, constipation and dyspnoea.
BRAF inhibitors with versus without MEK inhibitors
Impact on quality of life with the combination of dabrafenib and trametinib versus dabrafenib monotherapy in participants with BRAF mutated metastatic melanoma was evaluated in Schadendorf 2015. Global health dimension scores from baseline were better in the combination therapy group. A trend favouring combination therapy was also observed for pain, insomnia as well as physical, social, role, emotional and cognitive functioning. However, the opposite trend was reported for nausea and vomiting, diarrhoea, dyspnoea and constipation with significant improvements from baseline in the dabrafenib monotherapy group.
Other comparisons
Kiebert 2003 investigated temozolomide versus dacarbazine and assessed quality of life in participants being treated for metastatic melanoma. Kiebert 2003 found that treatment with temozolomide led to functional improvements, improved emotional well‐being and decreased symptoms compared to treatment with dacarbazine. At 12 weeks post‐treatment, participants in the temozolomide group reported better EORTC QLQ‐C30 subscale scores in all but two function and symptom categories with better physical functioning, less fatigue and reduced sleep disturbances. Improvements in all symptoms except diarrhoea were in favour of temozolomide at week 24 and there was near significant enhancement in cognitive functioning.
Fotemustine versus dacarbazine
Avril 2004 assessed fotemustine versus dacarbazine. No significant difference was observed between treatment arms.
Vindesine versus observation
Quality of life after adjuvant treatment with single agent vindesine was compared to observation alone in participants with metastasised melanoma after complete metastasectomy in Eigentler 2008. However, feedback from EORTC‐QLQ questionnaires was insufficient to draw any conclusions.
Polychemotherapy versus best supportive care
Best supportive care plus a polychemotherapy regimen consisting of cisplatin, vindesine and dacarbazine was compared to best supportive care alone for quality of life impact in participants with advanced melanoma in Hofmann 2011. Despite the deterioration in global health status reported in both arms, no statistically significant difference was observed between the treatments in any aspect of quality of life based on EORTC QLQ‐C30 questionnaires.
Economic evaluation
The economic aspects of various treatments were assessed in a single study; therefore no reliable conclusions could be drawn (Middleton 2000). The treatment costs of single agent dacarbazine and single agent temozolomide for advanced malignant melanoma were evaluated by Hillner 2000 and compared as part of a post hoc economic analysis independent from the actual clinical trial (Middleton 2000). Hillner 2000 combined costs and survival duration to analyse the incremental cost‐effectiveness of temozolomide over dacarbazine. Despite dacarbazine displaying a trend toward superior cost‐effectiveness, statistically, temozolomide was deemed to be equally effective, if not better at improving survival, with a higher but acceptable incremental cost per life‐year below the threshold of USD 50,000.
We identified one ongoing phase III RCT (NCT02821013) which plans to evaluate the economic aspects of continuous versus intermittent anti‐PD‐1 therapy in participants with metastatic melanoma.
Network meta‐analysis findings
We focused attention on four drug classes (chemotherapy, biochemotherapy, immune checkpoint inhibitors and small‐molecule targeted drugs) and two primary outcomes (progression‐free survival and toxicity) for the network meta‐analysis. Reasons for this decision are provided in the following sections.
Drug classes
Chemotherapy was chosen as the most common treatment among the included trials, which made chemotherapy the ideal common comparator (a key feature in network meta‐analysis, especially when performed according to the augmented data technique as suggested by White 2015, as we did; see Figure 5). We applied the following principles for other drug classes:
5.
Network plot
We chose drug classes for which high‐quality evidence was available for effects on patient survival based on direct comparison data. This choice was dictated by the need to include high‐quality data in the analysis: network meta‐analysis enables indirect comparisons to be made and generate treatment ranking (information not provided by conventional pair‐wise meta‐analysis). However, reliability of findings unavoidably hinges on the quality of imputed data.
We aimed to reduce the complexity of the network (by decreasing the number of nodes connecting each drug regimen to the common comparator, especially when few trials or only one trial represented a single drug regimen) and increase the robustness of the network (by decreasing the number of drug regimens analysed, especially when few trials or only one trial represented a single drug regimen), and therefore, decrease the likelihood of model instability or lack of model convergence.
We focused our attention on drugs currently approved for melanoma treatment to provide information that is most useful in routine clinical practice.
Outcomes
We chose one survival outcome (progression‐free survival) to represent treatment benefit, and toxicity to represent treatment harm. We chose to investigate progression‐free survival instead of overall survival because:
Progression‐free survival is widely accepted as a surrogate of overall survival, especially in the advanced/metastatic setting (as was the case for this review); progression‐free survival is generally used as the outcome for drug approval in this setting.
Data for overall survival are not yet mature for recent treatments (such as immune checkpoint inhibitors and small‐molecule targeted drugs), which are currently acknowledged as the most effective therapies for people with melanoma.
Progression‐free survival data are available for more studies compared to overall survival data (which is, at least in part, a corollary of the previous consideration).
Progression‐free survival is virtually free from the issue (typical of overall survival) of the cross‐over effect, that is, participants failing one treatment (e.g. less effective reference therapy) are given another treatment (e.g. more effective experimental therapy), which can confound the results of data analysis.
Adopting these criteria, a total of 19 studies were eligible for inclusion in the network meta‐analysis (Atkins 2008; Eton 2002; Flaherty 2012a; Flaherty 2012b; Gupta 2014; Hauschild 2012; Larkin 2014; Larkin 2015; Long 2015; McArthur 2014; Middleton 2007; Postow 2015; Ribas 2013; Ridolfi 2002a; Robert 2011; Robert 2013; Robert 2015; Robert 2015a; Robert 2015b). Studies compared eight treatments: chemotherapy; biochemotherapy (with both interferon‐alpha and interleukin‐2); anti‐CTLA4 monoclonal antibodies; anti‐PD1 monoclonal antibodies; anti‐CTLA4 plus anti‐PD1 monoclonal antibodies; BRAF inhibitors; MEK inhibitors; and BRAF plus MEK inhibitors (see network plot, Figure 5).
A total of 7632 participants were randomised to receive either conventional chemotherapy (N = 1777), biochemotherapy (N = 507), anti‐CTLA4 monoclonal antibodies (N = 886), anti‐PD1 monoclonal antibodies (N = 1407), anti‐CTLA4 plus PD‐1 monoclonal antibodies (N = 408), BRAF inhibitors (N = 1285), MEK inhibitors (N = 259), or BRAF plus MEK inhibitors (N = 918).
Progression‐free survival
Progression‐free survival data were available for all trials (Atkins 2008; Eton 2002; Flaherty 2012a; Flaherty 2012b; Gupta 2014; Hauschild 2012; Larkin 2014; Larkin 2015; Long 2015; McArthur 2014; Middleton 2007; Postow 2015; Ridolfi 2002a; Robert 2011; Robert 2013; Robert 2015; Robert 2015a; Robert 2015b) except Ribas 2013.
Network meta‐analysis, which was conducted to investigate treatment modalities, generated 28 comparisons. Network meta‐analysis results were consistent with standard pair‐wise meta‐analysis for seven comparisons: biochemotherapy versus chemotherapy; anti‐PD1 monoclonal antibodies versus chemotherapy; anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies; anti‐CTLA4 plus anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies; BRAF inhibitors versus chemotherapy; MEK inhibitors versus chemotherapy; and BRAF plus MEK inhibitors versus BRAF inhibitors (Figure 6).
6.
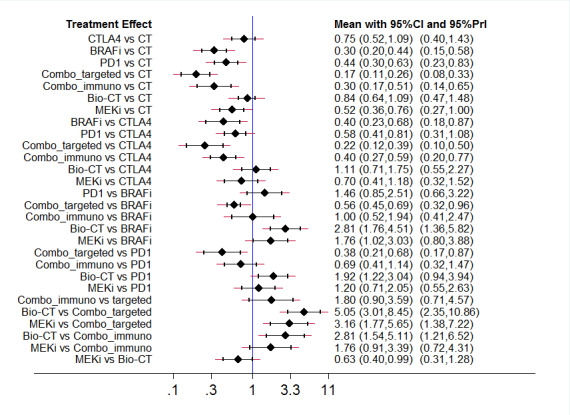
Interval plot: network meta‐analysis results for progression‐free survival. The network included eight treatment modalities. The effect measure is reported as hazard ratio (HR). CI: confidence interval; PrI: predictive interval.
Overall, we did not observe statistically significant network inconsistency: the P value of the design‐by‐treatment interaction model (which addresses both loop and design inconsistency at the global network level) was equal to 0.764. A comparison between findings of conventional pair‐wise meta‐analysis and indirect comparisons generated by network meta‐analysis was feasible only for the anti‐PD1 versus anti‐CTLA4 monoclonal antibodies comparison. The results showed a high correlation between both types of meta‐analysis technique: the HR was 0.54 (95% CI 0.50 to 0.60) for conventional meta‐analysis and 0.58 (95% CI 0.41 to 0.81) for network meta‐analysis (ratio of ratio = 0.93, low risk of loop inconsistency).
Indirect comparisons indicated that (Figure 6):
Compared to chemotherapy, both combination of immune checkpoint inhibitors (HR 0.30, 95% CI 0.17 to 0.51; moderate‐quality evidence, downgraded due to indirectness) and combination of small‐molecule targeted drugs (HR 0.17, 95% CI 0.11 to 0.26; moderate‐quality evidence, downgraded due to indirectness) improved progression‐free survival. Anti‐CTLA4 monoclonal antibodies did not significantly improve progression‐free survival (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness).
Compared to anti‐CTLA4 monoclonal antibodies, both BRAF inhibitors (HR 0.40, 95% CI 0.23 to 0.68; moderate‐quality evidence; downgraded due to indirectness), and combination of small‐molecule targeted drugs (HR 0.22, 95% CI 0.12 to 0.39; moderate‐quality evidence; downgraded due to indirectness) were associated with better progression‐free survival. In contrast, neither biochemotherapy (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness) nor MEK inhibitors (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness) significantly differed from anti‐CTLA4 monoclonal antibodies.
Compared to BRAF inhibitors, both biochemotherapy (HR 2.81, 95% CI 1.76 to 4.51; moderate‐quality evidence, downgraded due to indirectness) and MEK inhibitors (HR 1.76, 95% CI 1.02 to 3.03; very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness) were associated with worse progression‐free survival. Neither anti‐PD1 monoclonal antibodies (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness) nor combination of immune checkpoint inhibitors (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness) significantly differed from BRAF inhibitors.
Compared to anti‐PD1 monoclonal antibodies, the combination of small‐molecule targeted drugs improved progression‐free survival (HR 0.38, 95% CI 0.21 to 0.68; moderate‐quality evidence, downgraded due to indirectness), whereas biochemotherapy was associated with worse progression‐free survival (HR 1.92, 95% CI 1.22 to 3.04; low‐quality evidence, downgraded due to inconsistency and indirectness). Neither combination of immune checkpoint inhibitors (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness) nor MEK inhibitors (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness) significantly differed from anti‐PD1 monoclonal antibodies.
Compared to the combination of small‐molecule targeted drugs, both biochemotherapy (HR 5.05, 95% CI 3.01 to 8.45; moderate‐quality evidence, downgraded due to indirectness) and MEK inhibitors (HR 3.16, 95% CI 1.77 to 5.65; moderate‐quality evidence, downgraded due to indirectness) were associated with worse progression‐free survival. Combination of immune checkpoint inhibitors did not significantly differ from combination of small‐molecule targeted drugs (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness).
Compared to combination of immune checkpoint inhibitors, biochemotherapy was associated with worse progression‐free survival (HR 2.81, 95% CI 1.54 to 5.11; moderate‐quality evidence, downgraded due to indirectness). MEK inhibitors did not significantly differ from combination of immune checkpoint inhibitors (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness).
Compared to biochemotherapy, MEK inhibitors improved progression‐free survival (HR 0.63, 95% CI 0.40 to 0.99; very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness).
Toxicity
Toxicity data were available for all studies included in the network meta‐analysis (Atkins 2008; Eton 2002; Flaherty 2012a; Flaherty 2012b; Gupta 2014; Hauschild 2012; Larkin 2014; Larkin 2015; Long 2015; McArthur 2014; Middleton 2007; Postow 2015; Ribas 2013; Ridolfi 2002a; Robert 2011; Robert 2013; Robert 2015; Robert 2015a; Robert 2015b) (Figure 7).
7.
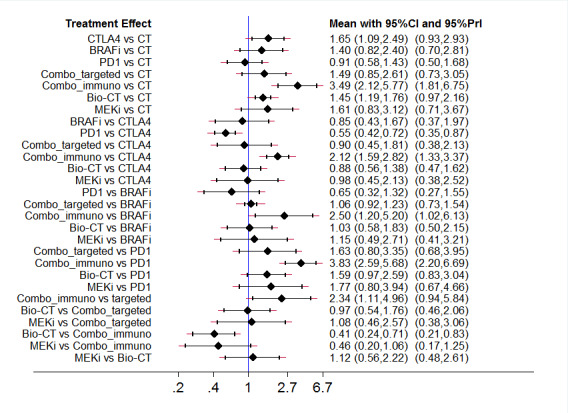
Interval plot: network meta‐analysis results for high grade toxicity. The network included eight treatment modalities. The effect measure is reported as relative risk (RR). CI: confidence interval; PrI: predictive interval.
Network meta‐analysis to investigate treatment modalities generated 28 comparisons. Network meta‐analysis results were consistent with standard pair‐wise meta‐analysis for seven comparisons: biochemotherapy versus chemotherapy; anti‐PD1 monoclonal antibodies versus chemotherapy; anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies; anti‐CTLA4 plus anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies; BRAF inhibitors versus chemotherapy; MEK inhibitors versus chemotherapy; and BRAF plus MEK inhibitors versus BRAF inhibitors) (Figure 6).
A comparison between direct and indirect evidence (findings of conventional pair‐wise meta‐analysis versus findings of indirect comparisons generated by network meta‐analysis) was feasible only for the anti‐PD1 versus anti‐CTLA4 monoclonal antibodies comparison. The results showed a good correlation between types of meta‐analysis technique: the RR was 0.70 (95% CI 0.54 to 0.91) for conventional meta‐analysis and 0.55 (95% CI 0.42 to 0.72) for network meta‐analysis (ratio of ratio = 1.27, low risk of loop inconsistency). However, when we looked at the overall network inconsistency, we found a highly statistically significant inconsistency (treatment by design interaction model P = 0.001), which undermines the reliability of the following findings regarding indirect comparisons (Figure 7):
Compared to chemotherapy, both anti‐CTLA4 monoclonal antibodies (RR 1.65, 95% CI 1.09 to 2.49; very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness) and combination of immune checkpoint inhibitors (RR 3.49, 95% CI 2.12 to 5.77; moderate‐quality evidence, downgraded due to indirectness) increased toxicity. Combination of small‐molecule targeted drugs did not significantly differ from chemotherapy (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness).
None of BRAF inhibitors (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness), combination of small‐molecule targeted drugs (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness), biochemotherapy (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness), or MEK inhibitors (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness) significantly differed from anti‐CTLA4 monoclonal antibodies.
Compared to BRAF inhibitors, combination of immune checkpoint inhibitors increased toxicity (RR 2.50, 95% CI 1.20 to 5.20; moderate‐quality evidence, downgraded due to indirectness). None of anti‐PD1 monoclonal antibodies (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness), biochemotherapy (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness) or MEK inhibitors (very low‐quality evidence; downgraded due to inconsistency, imprecision and indirectness) significantly differed from BRAF inhibitors.
Compared to anti‐PD1 monoclonal antibodies, the combination of immune checkpoint inhibitors increased toxicity (RR 3.83, 95% CI 2.59 to 5.68; moderate‐quality evidence, downgraded due to indirectness). None of combination of small‐molecule targeted drugs (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness), biochemotherapy (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness), or MEK inhibitors (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness) significantly differed from anti‐PD1 monoclonal antibodies.
Compared to the combination of small‐molecule targeted drugs, the combination of immune checkpoint inhibitors increased toxicity (RR 2.34, 95% CI 1.11 to 4.96; low‐quality evidence, downgraded due to inconsistency and indirectness). Neither biochemotherapy (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness) nor MEK inhibitors (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness) significantly differed from the combination of small‐molecule targeted drugs.
Compared to the combination of immune checkpoint inhibitors, biochemotherapy was associated with lower toxicity (RR 0.41, 95% CI 0.24 to 0.71; moderate‐quality evidence, downgraded due to indirectness). MEK inhibitors did not significantly differ from the combination of immune checkpoint inhibitors (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness).
MEK inhibitors did not significantly differ from biochemotherapy (very low‐quality evidence, downgraded due to inconsistency, imprecision and indirectness).
Ranking findings
Results of ranking analysis for progression‐free survival (expressed as surface under the cumulative ranking (SUCRA) values, ranging from 0 (worst case) to 1 (best case)) suggested that the combination of BRAF plus MEK inhibitors is the best treatment option (SUCRA: 0.99), followed by BRAF inhibitors (SUCRA: 0.77) and combination of anti‐CLA4 plus anti‐PD1 monoclonal antibodies (SUCRA: 0.77), anti‐PD1 monoclonal antibodies (SUCRA: 0.56), MEK inhibitors (SUCRA: 0.46), anti‐CTAL4 monoclonal antibodies (SUCRA: 0.25), biochemotherapy (SUCRA: 0.18), and conventional chemotherapy (SUCRA: 0.02).
Ranking analysis results for (high grade) toxicity suggested that anti‐PD1 monoclonal antibodies were associated with the best safety profile (SUCRA: 0.91), followed by chemotherapy (SUCRA: 0.87), BRAF inhibitors (SUCRA: 0.55), biochemotherapy (SUCRA: 48), the combination of BRAF plus MEK inhibitors (SUCRA: 0.42), MEK inhibitors (SUCRA: 0.41), anti‐CTLA4 monoclonal antibodies (SUCRA: 0.36), and the combination of anti‐CTLA4 plus anti‐PD1 monoclonal antibodies (SUCRA: 0.01). However, these results cannot be considered fully reliable due to the finding of network inconsistency as described in the preceding paragraph.
The findings for both efficacy (progression‐free survival) and acceptability (inverse of toxicity) were combined together in a bivariate ranking plot. Noticeably, in this plot toxicity is transformed into acceptability by using the inverse values of the corresponding relative risks: therefore, higher values indicate higher acceptability (due to lower toxicity) (Figure 8): accordingly, the ideal treatment (highest performance = best efficacy + best acceptability) should appear in the upper right corner of the plot. The combination of BRAF plus MEK inhibitors was associated with the highest treatment efficacy, but it was also associated with lower acceptability. In contrast, anti‐PD1 monoclonal antibodies showed the best acceptability performance, but resulted less effective than the combination of small‐molecule targeted drugs. Accordingly, no 'ideal' treatment is available.
8.
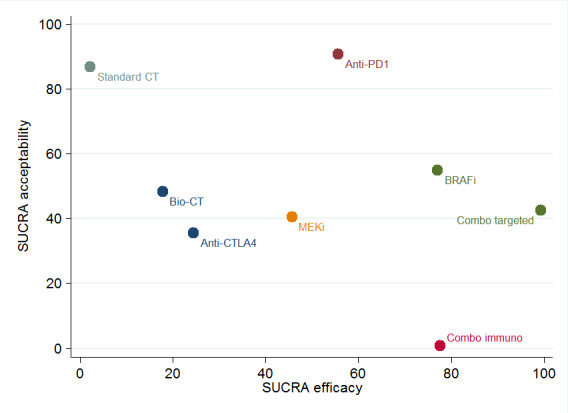
Ranking plot. Ranking plot representing simultaneously the efficacy (progression‐free survival) on the X axis and the acceptability (the inverse of toxicity) on the Y axis. The network included eight treatments for patients with metastatic melanoma. Optimal treatment should be characterised by both high efficacy and acceptability and should be in the right upper corner of this graph.
Quality assessment of trials and evidence grading
None of the studies included in the network meta‐analysis presented a severe risk of bias (as described in Risk of bias in included studies). Furthermore, the analysis of the comparison‐adjusted funnel plot (a funnel plot specifically adapted for network meta‐analysis) did not indicate any evident risk of publication bias (Figure 9). These findings, coupled with the absence of network inconsistency and the lack of violation of the transitivity assumption, enabled us to grade the evidence generated from indirect comparisons for progression‐free survival with confidence.
9.
Comparison adjusted funnel plot for network meta‐analysis of progression‐free survival
In contrast, significant network inconsistency detected during toxicity data analysis add some uncertainty on the findings observed for this outcome.
Other findings
Immunostimulating agents
Immunostimulating agents other than those described above (cytokines (e.g. interferon‐alpha and interleukin‐2), immune checkpoint inhibitors, bioproducts of bacteria such as BCG and Cparvum) have been tested in clinical trials for the treatment of people with metastatic melanoma. In particular, gp100 (a melanoma associated antigen) and granulocyte‐macrophage colony stimulating factor (GM‐CSF) were administered in association with anti‐CTLA4 monoclonal antibody ipilimumab and evaluated in single RCTs (ipilimumab with gp100, Hodi 2010a; ipilimumab plus GM‐CSF, Hodi 2014). The gp100 melanoma antigen was also tested in combination with interleukin‐2 (Schwartzentruber 2011a). Another agent, thymosin‐alpha, was tested in association with interferon and dacarbazine (Maio 2010). In single studies, these combinations, except gp100 plus ipilimumab, resulted in prolonged survival with minimal toxicity. GM‐CSF significantly reduced ipilimumab toxicity.
When these findings were combined in a meta‐analysis, the addition of immunostimulating agents had an impact on participants' overall survival (Analysis 21.1, HR 0.82, 95% CI 0.67 to 0.99). However, this result was characterised by high between‐study heterogeneity (I² = 53%). Sensitivity analysis conducted using the leave‐one‐out procedure suggested that when Hodi 2010a was excluded, heterogeneity dropped to 0% and treatment effect was greater (HR 0.75, 95% CI 0.64 to 0.88): this effect was likely due to adding gp100 to ipilimumab did not add any therapeutic benefit. We also found a non‐significant positive effect of immunostimulating agents on progression‐free survival (HR 0.92, 95% CI 0.74 to 1.14, Analysis 21.2), although this result did not reach statistical significance and heterogeneity was high (I² = 74%). Again, analysis without Hodi 2010a yielded no heterogeneity (I² = 0%) and showed a statistically significant progression‐free survival advantage (HR 0.82, 95% CI 0.73 to 0.92). Analysis for objective tumour response showed better response rates for combined treatment although with high heterogeneity (RR 1.23, 95% CI 0.60 to 2.50; I² = 72%, Analysis 21.3). Unfortunately, we could not identify the source of heterogeneity. Similarly, there was a non‐significant reduction in high‐grade toxicity (RR 0.92, 95% CI 0.77 to 1.08; I² = 45%, Analysis 21.4). We could not identify possible reasons for heterogeneity.
21.1. Analysis.

Comparison 21: Immunostimulating agents, Outcome 1: Overall survival
21.2. Analysis.

Comparison 21: Immunostimulating agents, Outcome 2: Progression‐free survival
21.3. Analysis.

Comparison 21: Immunostimulating agents, Outcome 3: Tumour response
21.4. Analysis.

Comparison 21: Immunostimulating agents, Outcome 4: Toxicity (≥ G3)
Lenalidomide did not improve tumour response (5.3% versus 5.8%; P = 0.82), time to progression (median 3.0 months versus 2.1 months; P = 0.19), or overall survival (median 5.9 months versus 7.4 months, respectively; P = 0.32) compared to placebo in participants with metastatic melanoma (Eisen 2010).
Taxanes
The taxanes docetaxel and paclitaxel were administered to participants enrolled in the control arm of several studies (Flaherty 2013a; Gupta 2014; Hamid 2014; Hauschild 2009a; Kim 2012; O'Day 2009; O'Day 2013; Weber 2015; Zimpfer‐Rechner 2003). Paclitaxel was the experimental treatment in two studies (Bedikian 2011; Hersh 2015) and tested as docosahexaenoic acid‐paclitaxel by Bedikian 2011 and nab‐paclitaxel by Hersh 2015. Although docosahexaenoic acid‐paclitaxel did not impact participant outcomes, nab‐paclitaxel improved progression‐free survival (the primary study endpoint) compared to dacarbazine (HR 0.79, 95% CI 0.63 to 0.99).
Adjuvant therapies after surgery
Three trials investigated different systemic therapeutic strategies after surgery: chemotherapy with vindesine (Eigentler 2008); chemo‐immunotherapy with dacarbazine and C parvum (Balch 1984); and a polypeptide vaccine or GM‐CSF (Lawson 2015) without showing any difference in either tumour response or prognosis.
Discussion
Summary of main results
This Cochrane Review summarised the available evidence on systemic treatments for people with metastatic melanoma. While effectiveness of conventional chemotherapy alone has never been convincingly proven, our results suggest that more than one treatment is more effective than chemotherapy. For instance, the addition of immunostimulating cytokines (such as interleukin‐2 and interferon‐alpha) to chemotherapy (biochemotherapy) prolongs progression‐free survival (high‐quality evidence) (at the cost of higher rates of toxicity (high‐quality evidence)), although this result does not translate into a significant overall survival benefit (high‐quality evidence) (Table 9).
In recent years, two new classes of therapeutic agents have been implemented in the clinical setting: immune checkpoint inhibitors (anti‐CTLA4 and anti‐PD1 monoclonal antibodies) and small‐molecule targeted drugs (BRAF and MEK inhibitors), which are active exclusively against BRAF‐mutated melanoma. These new treatments have revolutionised the landscape of metastatic melanoma treatment. The results of our meta‐analysis showed that when chemotherapy was combined with anti‐CTLA4 monoclonal antibodies (ipilimumab and tremelimumab), progression‐free survival was likely to be significantly improved compared to chemotherapy alone. However, this benefit is probably associated with higher toxicity rates (moderate‐quality evidence) and comparative effectiveness may not translate into a significant overall survival advantage (Table 3). Compared to conventional chemotherapy, anti‐PD1 monoclonal antibodies (nivolumab and pembrolizumab) improved overall survival (high‐quality evidence), probably leads to longer progression‐free survival (moderate‐quality evidence), and may lead to a lower incidence of high‐grade toxicity (low‐quality evidence) (Table 1). When comparing both immune checkpoint inhibitors (i.e. anti‐PD1 monoclonal antibodies and anti‐CTLA4 monoclonal antibodies) against each other, anti‐PD1 monoclonal antibodies improved overall survival and progression‐free survival more than anti‐CTLA4 monoclonal antibodies (both high‐quality evidence), and the former may result in better toxicity (low‐quality evidence) (Table 2). Moreover, the combination of anti‐PD1 and anti‐CTLA4 monoclonal antibodies yielded better results in terms of progression‐free survival (high‐quality evidence) compared to anti‐CTLA4 monoclonal antibodies alone; there may be no significant difference in toxicity (low‐quality evidence) (Table 4). No data for overall survival were available for this comparison.
Among small‐molecule targeted drugs, BRAF inhibitors for BRAF‐mutated melanoma significantly improved both progression‐free survival and overall survival (both high‐quality evidence) compared to conventional chemotherapy; there may be no significant difference in toxicity (low‐quality evidence) (Table 5). Compared to chemotherapy, MEK inhibitors for BRAF‐mutated melanoma probably increased progression‐free survival (moderate‐quality evidence), but are likely to have higher toxicity rates (moderate‐quality evidence). MEK inhibitors may not significantly improve overall survival (Table 6). Interestingly, when a BRAF inhibitor was combined with a MEK inhibitor the combination therapy for BRAF‐mutated melanoma performed better in terms of overall survival (high‐quality evidence) and probably in terms of progression‐free survival (moderate‐quality evidence) compared to single agent BRAF inhibitor; however, there was likely to be no significant difference in toxicity (moderate‐quality evidence) (Table 7). The results of BRAF inhibitors are exclusively limited to people with a BRAF‐mutated melanoma, because this drug class is only active against this type of melanoma.
Chemotherapy combined with anti‐angiogenic drugs (bevacizumab and endostar, both of which are recently implemented compounds) may also improve both overall survival (moderate‐quality evidence) and progression‐free survival (moderate‐quality evidence) compared to chemotherapy alone (Table 8); the combination may have no difference on toxicity (low‐quality evidence). Polychemotherapy did not result in significantly better survival (either overall or progression‐free survival) than chemotherapy (both high‐quality evidence) and probably burdens people being treated with higher toxicity rates (moderate‐quality evidence) (Table 10).
We also conducted a network meta‐analysis. The results of the network meta‐analysis whose agreed with standard pair‐wise meta‐analysis results in terms of direct comparisons, and enabled us to make indirect comparisons between treatments not formally compared in clinical trials. Network meta‐analysis findings suggested that a combination of BRAF and MEK inhibitors was the most effective treatment strategy for BRAF‐mutated melanoma, at least in terms of progression‐free survival (Figure 8). However, this combination therapy is burdened by a higher rate of severe toxicity compared to as observed among people treated with the anti‐PD1 monoclonal antibodies, which were associated with the best acceptability (Figure 8).
Data on quality of life and costs were quite scarce, so conclusions could be drawn on these concepts (with special regard to the sustainability of newer agents, the cost of which is much higher than conventional chemotherapy agents).
Moreover, future research should focus on direct comparisons of drugs that have not been directly compared in randomised controlled trials (RCTs). The efficacy of combinations of new drug classes such as immune checkpoint inhibitors and small‐molecule targeted drugs (on which no data are yet available) should also be considered.
Overall completeness and applicability of evidence
This Cochrane Review provides an unprecedented overview of systemic treatments for people with metastatic melanoma. Overall, the available evidence was directly relevant and sufficiently comprehensive to appropriately address the review's aims.
Newly introduced classes of drugs (immune check point inhibitors and targeted drugs inhibiting BRAF or MEK) demonstrated significant therapeutic effects. An important aspect to note is that BRAF inhibitors are active only against BRAF‐mutated melanoma, which represents roughly half of all metastatic melanoma. Results from our network meta‐analysis suggest a combination of BRAF and MEK inhibitors to be the most effective treatment strategy for people with BRAF‐mutated melanoma (Figure 8). However, this finding was based on data assessing progression‐free survival only and should be confirmed by mature overall survival data.
Longer follow‐up periods are needed before similar conclusions could be speculated for overall survival. In particular, data for anti‐PD1 monoclonal antibodies combined with anti‐CTLA4 agents are not yet sufficiently mature to inform a definitive overall survival analysis. The relatively short follow‐up periods of trials reporting on immune checkpoint inhibitors and small‐molecule targeted drugs are presented in Characteristics of included studies: long‐term outcomes from these trials should improve the applicability of study results. In the meantime, because progression‐free survival correlates well with overall survival (at least in the metastatic setting), and is therefore considered to be a reliable surrogate for overall survival (which is why many anticancer drugs are approved for clinical use worldwide on the basis of progression‐free survival data only), our results provide useful information to make a reasonably reliable judgement on the usefulness of these therapies for the treatment of people with metastatic melanoma.
Data on quality of life and costs were very limited so conclusions could not be drawn. In particular, cost‐effectiveness of new therapies is yet to be determined for metastatic melanoma (Cashin 2008). As a result, it is unclear how treatment for people living with melanoma can be sustained, particularly from a global point of view (Wise 2016).
Quality of the evidence
The available evidence (based on findings from 122 RCTs that involved 28,561 participants) on systemic treatments for people with metastatic melanoma informed identification of effective classes of drugs for improving objective tumour response, progression‐free survival and overall survival.
Overall, the risk of bias of included studies can be considered as limited. Considering the 122 included studies and the seven bias domains assessed, we performed 854 evaluations (Figure 4): only seven evaluations (< 1%) assigned high risk of bias for six trials (Beretta 1976; Carvajal 2014; Hamid 2014; Hofmann 2011; Ranson 2007; Richtig 2004). Of note, none of the six high risk of bias trials were included in meta‐analyses or contributed to any conclusions on treatment efficacy. We assessed that only 21 studies (17%) were at low risk of bias for all domains (Bedikian 2006; Cui 2013; Eisen 2010; Flaherty 2012b; Flaherty 2013a; Glaspy 2009; Hauschild 2009a; Hersh 2015; Hodi 2010a; Larkin 2015; Lawson 2015; Long 2015; McDermott 2008; O'Day 2013; Ribas 2015; Robert 2013; Robert 2015a; Schadendorf 2006; Schwartzentruber 2011a; Weber 2015; Wolchok 2010). We assessed a further 22 trials (18%) at low risk of bias for four domains and one domain at unclear risk of bias (Atkins 2008; Bajetta 2006a; Bedikian 2011; Chiarion‐Sileni 2011; Eigentler 2008; Gupta 2014; Hauschild 2001; Hauschild 2012; Hodi 2014; Kaufmann 2005; Keilholz 2005; Larkin 2014; Maio 2010; McArthur 2014; Middleton 2007; Middleton 2015; O'Day 2009; Patel 2011; Ribas 2013; Robert 2015; Robert 2015b; Testori 2008). Most included studies (n = 73, 60%) were assessed at unclear risk of bias for two or more domains. Because uncertainty was mainly sustained by lack of information provided in study reports, our findings underscore the importance of mandating key information as a requirement for publishing trial results (and exploiting online repositories for supplemental material). This recommendation has been made many times by international guidelines, such as the CONSORT group (Schulz 2010).
GRADE assessment showed that most evidence was high‐ to moderate‐quality for three of four outcomes (overall survival, progression‐free survival and tumour response). GRADE evaluations of overall survival indicated high‐quality evidence in 50% (9/18) assessments; moderate‐quality evidence in four (22%) and low‐quality evidence in five (28%) assessments. GRADE evaluations for progression‐free survival indicated high‐quality evidence in 35% (6/17) assessments; moderate‐quality evidence in eight (47%) and low‐quality evidence in five (18%) assessments. Assessment for tumour response found high‐quality evidence in 42% (8/19) assessments; moderate‐quality evidence for 53% (10/19) and low‐quality evidence in one (5%) assessment. In contrast, evidence for toxicity was mainly moderate‐ to low‐quality: only one of 16 evaluations was high quality (6%); moderate quality in 59% (8/16) and low‐quality in 44% (7/16) assessments. The main reasons for downgrading evidence were inconsistency of findings (remarkable between‐study heterogeneity) and imprecision of the effect estimate (mostly linked to confidence intervals including both a meaningful effect and a small/null effect or even a meaningful opposite effect). Of note, we could not find reasonable sources of between‐study heterogeneity, and the definition of heterogeneity itself was limited by the often low number of studies available for each comparison and outcome. Formal assessment of publication bias was rarely feasible due to the few studies available for each comparison and outcome (mostly fewer than 10).
Limitations exist when investigating toxicity across trials because this is often reported as incidence of a given event (i.e. rates of study participants who developed an adverse event). consequently, the overall rate of participants who experienced toxicity (and its grade) was missing from several studies. Meta‐analyses of toxicity are characterised by relevant heterogeneity, suggesting challenges in toxicity reporting.
Although eligible trials have similar inclusion criteria, some differences do exist, as shown in the Characteristics of included studies tables. In studies investigating small‐molecule targeted drugs, all participants had BRAF mutated melanoma, but some studies testing immune checkpoint inhibitors enrolled both BRAF mutated and BRAF wild type melanomas, although participants with BRAF mutated disease were in the minority (Larkin 2014; Postow 2015). Theoretically, this may introduce bias when results of targeted therapy and immunotherapy were compared in the network meta‐analysis: people with or without this mutation may have an intrinsically different natural history. However, it should be noted that the association between BRAF mutational status and patient prognosis is quite controversial (Edlundh‐Rose 2006; Long 2011; Meckbach 2014), which may minimises this risk of bias.
Criteria for inclusion of participants with brain metastases differed across trials. People with brain metastases were generally excluded or included only if no active disease was evident at imaging evaluation three months after brain treatment. However, both targeted drugs (Long 2012a) and immune checkpoint inhibitors (Di Giacomo 2012; Margolin 2012) have demonstrated therapeutic activity in this particular subgroup of people with advanced disease, although immune checkpoint inhibitor treatment showed little or no activity in those who were symptomatic.
As expected, the quality of evidence for network meta‐analysis findings was generally lower than observed in direct comparison meta‐analysis due to intrinsic indirectness (which was a reason for downgrading shared for all evaluations). GRADE assessment for progression‐free survival found that 43% (9/21) provided moderate‐quality evidence, 5% (1/21) provided low‐quality evidence and 52% (11/21) provide very low‐quality evidence. In line with evidence quality assessment in direct comparisons, quality of evidence for toxicity was lower than observed for efficacy outcomes. Most GRADE evaluations yielded low‐ (1/21, 5%) and very low‐quality evidence (16/20, 76%); only 19% (4/21) of evaluations found moderate‐quality evidence.
In many cases, trials were sponsored by pharmaceutical companies producing the tested drug: this was especially true for new classes of drugs, such as immune checkpoint inhibitors and small‐molecule targeted drugs.
Potential biases in the review process
Our literature search was likely to detect all relevant randomised controlled trials. Nevertheless, it is always possible that we overlooked some potentially relevant trials; moreover, it is possible that some trials have not been indexed by the databases searched. However, the main conclusions of this review were based on trials that will be widely and well known by melanoma experts worldwide. Therefore, the included studies should represent the current knowledge in this field of cancer medicine reasonable well.
We did not contact the contact relevant individuals and organisations for information about unpublished or ongoing studies. There is a chance that some ongoing studies may have been completed and results may be available.
Agreements and disagreements with other studies or reviews
The present review had wider selection criteria compared to previous Cochrane Reviews on treatments for metastatic melanoma that investigated the effectiveness of chemotherapy (Crosby 2000) and biochemotherapy (Sasse 2007). Crosby 2000 aimed to assess whether conventional chemotherapy was superior to placebo (or best supportive care), but findings were inconclusive because no RCTs addressing this issue were found by the authors. In the present review, there was no formal evidence of superiority for chemotherapy compared to best supportive care or placebo, although this information was based on the findings of one study (Eisen 2010). Chemotherapy (with special regard to dacarbazine) has been the reference treatment in several contemporary trials testing new agents: our analysis showed that biochemotherapy, immune checkpoint inhibitors and small‐molecule targeted drugs are more effective or likely to be more effective than conventional chemotherapy in terms of progression‐free survival (Figure 8), and that the anti‐PD1 antibodies (immune checkpoint inhibitor) and BRAF inhibitors (small‐molecule targeted drugs) performed better than chemotherapy in terms of overall survival. Therefore, although it remains unclear whether or not chemotherapy is beneficial for people with metastatic melanoma, we can state that treatments which are more effective than chemotherapy are available currently.
Two previous reviews could not demonstrate that biochemotherapy was more effective than chemotherapy alone (Ives 2007; Sasse 2007). In this review, we found that biochemotherapy impacted favourably on participant progression‐free survival. Both Ives 2007 and Sasse 2007 included fewer studies than this review; furthermore, they used number of events at fixed time points (using relative risks or odds ratios as effect measures), which we consider a non optimal way of analysing time to event (survival) data (we expressed survival data as hazard ratios).
Some network meta‐analyses have been published recently on the treatment of metastatic melanoma. These have focused on the most recent therapeutic developments in this field, that is, the implementation of immune checkpoint inhibitors (anti‐CTLA4 and anti‐PD1 monoclonal antibodies) and small‐molecule targeted drugs (BRAF and MEK inhibitors). Devji 2017 limited analysis to results obtained for participants with BRAF‐mutated melanoma, and Ciren 2016 analysed only tumour response data (no survival data considered). Pasquali 2017 reported on both efficacy (survival) and toxicity findings. The results of all three network meta‐analyses agree with our findings and results.
Authors' conclusions
Implications for practice.
Based on network meta‐analysis rankings, the review findings support the use of BRAF inhibitors (either alone or combined with MEK inhibitors), and anti‐PD1 monoclonal antibodies (either alone or combined with anti‐CTLA4 monoclonal antibodies) as effective treatments for people with metastatic melanoma in terms of progression‐free survival, with consideration of the following.
BRAF inhibitors are effective only in people with BRAF‐mutated melanoma;
BRAF inhibitors combined with MEK inhibitors are the most effective regimen in people with BRAF‐mutated melanoma (at least in terms of progression‐free survival); and
anti‐PD1 monoclonal antibodies are the least toxic regimen, but the combination of immune checkpoint inhibitors has highest toxicity.
Implications for research.
Randomised controlled trials with longer follow‐up periods (12 to 24 months) for participants treated with new therapeutic agents immune checkpoint inhibitors and targeted therapies are needed to assess impact on overall survival. Other outcomes that need to be assessed include quality of life and issues relating to health economics, such as cost‐effectiveness. More research is also required to test whether combinations of these therapies or their sequential use can increase their effectiveness. This is particularly important for people with BRAF‐mutated melanoma who can benefit from both BRAF inhibitors with or without MEK inhibitors and immune checkpoint inhibitors.
A common reason for downgrading evidence quality was imprecision: recruiting inadequate numbers of participants was an issue in some of the older included studies. This limitation has been recognised, and trials no longer tend to exhibit this problem. Future published trials should guarantee adequate reporting by adhering to guidelines such as CONSORT.
Identification of biomarkers for guide selection of people most responsive to immune checkpoint inhibitors is of paramount importance and should be intensively investigated.
It is also important to understand whether there is a role for combining traditional biochemotherapy (based on interleukin‐2 and interferon‐alpha) with immune checkpoint inhibitors or small‐molecule targeted drugs. This issue is being addressed (at least in part) in ongoing trials.
Results of this Cochrane Review found that some drugs which are not currently used in clinical practice, such as anti‐angiogenic agents (bevacizumab and endostar), oblimersen, and nab‐paclitaxel, deserve further investigation to determine whether or not they can be added to the armamentarium of therapeutic interventions suitable to fight metastatic melanoma. Immune‐stimulating agents, such as gp100 and GM‐CSF, which can enhance the effectiveness of immune checkpoint inhibitors in the second‐line setting, should be tested as first‐line treatments to assess their clinical value as upfront therapy.
What's new
| Date | Event | Description |
|---|---|---|
| 22 February 2019 | Amended | Small amendment to wording of background in PLS after a query via Cochrane Library feedback |
History
Protocol first published: Issue 5, 2014 Review first published: Issue 2, 2018
Notes
Small amendment to wording of background in PLS after a query via Cochrane Library feedback in consulation with lead author.
Acknowledgements
We thank Cochrane Skin, and in particular Professor Hywel Williams, Miss Laura Prescott, and Dr Finola Delamere for their assistance.
The authors also thank Dr Mauro Apostolico for his help in retrieving articles and Dr Alessandra Costa for her assistance with uploading articles into RevMan.
The Cochrane Skin editorial base wishes to thank Bob Boyle, Cochrane Dermatology Editor for this review; Ben Carter, Statistical Editor; Ching‐Chi Chi, Methods Editor; the clinical referees, Laurence Le Cleach and Emilie Sbidian; the consumer referee, Kathie Godfrey; and Ann Jones, who copy‐edited the review.
Disclaimer
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Skin Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health. This project was supported by the Complex Reviews Support Unit, also funded by the National Institute for Health Research (project number 14/178/29).
Appendices
Appendix 1. CENTRAL (Cochrane Library) search strategy
#1 MeSH descriptor: [Melanoma] explode all trees #2 MeSH descriptor: [Skin Neoplasms] explode all trees #3 melanoma:ti,ab #4 #1 or #2 or #3 #5 (metastatic or metastas*):ti,ab #6 ("stage iv" or "stage 4"):ti,ab #7 MeSH descriptor: [Neoplasm Metastasis] explode all trees #8 #5 or #6 or #7 #9 #4 and #8
Appendix 2. MEDLINE (Ovid) search strategy
1. exp Melanoma/ 2. exp Skin Neoplasms/ 3. melanoma.ti,ab. 4. or/1‐3 5. (metastatic or metastas$).ti,ab. 6. exp Neoplasm Metastasis/ 7. ("stage iv" or "stage 4").ti,ab. 8. or/5‐7 9. 4 and 8 10. randomized controlled trial.pt. 11. controlled clinical trial.pt. 12. randomized.ab. 13. placebo.ab. 14. clinical trials as topic.sh. 15. randomly.ab. 16. trial.ti. 17. 10 or 11 or 12 or 13 or 14 or 15 or 16 18. exp animals/ not humans.sh. 19. 17 not 18 20. 9 and 19
[Lines 10‐19: Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision)]
Appendix 3. Embase (Ovid) search strategy
1. exp melanoma/ 2. melanoma.ti,ab. 3. 1 or 2 4. (metastatic or metastas$).ti,ab. 5. metastasis/ or exp skin metastasis/ 6. ("stage iv" or "stage 4").ti,ab. 7. 4 or 5 or 6 8. crossover procedure.sh. 9. double‐blind procedure.sh. 10. single‐blind procedure.sh. 11. (crossover$ or cross over$).tw. 12. placebo$.tw. 13. (doubl$ adj blind$).tw. 14. allocat$.tw. 15. trial.ti. 16. randomized controlled trial.sh. 17. random$.tw. 18. or/8‐17 19. exp animal/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ 20. human/ or normal human/ 21. 19 and 20 22. 19 not 21 23. 18 not 22 24. 3 and 7 and 23
Data and analyses
Comparison 1. Polychemotherapy versus single agent chemotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Overall survival | 6 | 594 | Hazard Ratio (IV, Random, 95% CI) | 0.99 [0.85, 1.16] |
| 1.2 Progression‐free survival | 5 | 398 | Hazard Ratio (IV, Random, 95% CI) | 1.07 [0.91, 1.25] |
| 1.3 Tumour response | 14 | 1885 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.02, 1.58] |
| 1.4 Toxicity (≥ G3) | 3 | 514 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.97 [1.44, 2.71] |
Comparison 2. Chemotherapy ± tamoxifen.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Overall survival | 4 | 643 | Hazard Ratio (IV, Random, 95% CI) | 1.03 [0.80, 1.33] |
| 2.2 Progression‐free survival | 2 | 475 | Hazard Ratio (IV, Random, 95% CI) | 1.06 [0.93, 1.22] |
| 2.3 Tumour response | 4 | 643 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.94, 1.89] |
| 2.4 Toxicity (≥ G3) | 1 | 271 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.38, 1.28] |
Comparison 3. Temozolomide versus dacarbazine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Overall survival | 3 | 1313 | Hazard Ratio (IV, Random, 95% CI) | 0.98 [0.85, 1.12] |
| 3.2 Progression‐free survival | 3 | 1313 | Hazard Ratio (IV, Random, 95% CI) | 0.87 [0.74, 1.03] |
| 3.3 Tumour response | 3 | 1313 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.85, 1.73] |
| 3.4 Toxicity (≥ G3) | 2 | 1164 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.98, 1.35] |
Comparison 4. Chemotherapy ± interferon‐alpha.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Overall survival | 11 | 1785 | Hazard Ratio (IV, Random, 95% CI) | 0.87 [0.73, 1.04] |
| 4.2 Progression‐free survival | 6 | 1272 | Hazard Ratio (IV, Random, 95% CI) | 0.87 [0.74, 1.01] |
| 4.3 Tumour response | 15 | 2419 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.12, 1.66] |
| 4.4 Toxicity (≥ G3) | 3 | 791 | Risk Ratio (M‐H, Random, 95% CI) | 1.72 [0.37, 7.95] |
Comparison 5. Chemotherapy ± interleukin‐2.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Overall survival | 2 | 644 | Hazard Ratio (IV, Random, 95% CI) | 0.95 [0.82, 1.11] |
| 5.2 Progression‐free survival | 1 | 363 | Hazard Ratio (IV, Random, 95% CI) | 0.87 [0.70, 1.08] |
| 5.3 Tumour response | 3 | 735 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.64, 1.13] |
Comparison 6. Chemotherapy ± interferon‐alpha and interleukin‐2.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Overall survival | 7 | 1307 | Hazard Ratio (IV, Random, 95% CI) | 0.94 [0.84, 1.06] |
| 6.2 Progression‐free survival | 6 | 964 | Hazard Ratio (IV, Random, 95% CI) | 0.90 [0.83, 0.99] |
| 6.3 Tumour response | 7 | 1307 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.11, 1.67] |
| 6.4 Toxicity (≥ G3) | 2 | 657 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [1.14, 1.61] |
Comparison 7. Chemotherapy ± interferon‐alpha and interleukin‐2 (first line).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Overall survival | 5 | 1118 | Hazard Ratio (IV, Random, 95% CI) | 0.96 [0.83, 1.10] |
| 7.2 Progression‐free survival | 4 | 775 | Hazard Ratio (IV, Random, 95% CI) | 0.86 [0.76, 0.99] |
| 7.3 Tumour response | 5 | 1118 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [1.15, 1.83] |
| 7.4 Toxicity (≥ G3) | 1 | 241 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [1.12, 1.87] |
Comparison 8. Chemotherapy ± Bacille Calmette‐Guérin (BCG).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Overall survival | 2 | 154 | Hazard Ratio (IV, Random, 95% CI) | 0.87 [0.61, 1.25] |
| 8.2 Tumour response | 6 | 770 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.65, 1.12] |
Comparison 9. Chemotherapy ± Corynebacterium parvum.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Overall survival | 4 | 242 | Hazard Ratio (IV, Random, 95% CI) | 0.95 [0.74, 1.22] |
| 9.2 Tumour response | 7 | 537 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.77, 1.38] |
Comparison 10. Anti‐CTLA4 monoclonal antibodies (first line).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 10.1 Overall survival | 2 | 1157 | Hazard Ratio (IV, Random, 95% CI) | 0.81 [0.65, 1.01] |
| 10.2 Progression‐free survival | 1 | 502 | Hazard Ratio (IV, Random, 95% CI) | 0.76 [0.63, 0.92] |
| 10.3 Tumour response | 2 | 1157 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.92, 1.77] |
| 10.4 Toxicity (≥ G3) | 2 | 1142 | Risk Ratio (M‐H, Random, 95% CI) | 1.69 [1.19, 2.42] |
Comparison 11. Anti‐CTLA4 monoclonal antibodies ± other immunostimulating agents (second line).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 11.1 Overall survival | 2 | 784 | Hazard Ratio (IV, Random, 95% CI) | 0.83 [0.52, 1.33] |
| 11.2 Progression‐free survival | 2 | 785 | Hazard Ratio (IV, Random, 95% CI) | 1.06 [0.75, 1.51] |
| 11.3 Tumour response | 2 | 785 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.38, 1.47] |
| 11.4 Toxicity (≥ G3) | 2 | 785 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.69, 1.11] |
Comparison 12. Anti‐PD1 monoclonal antibodies versus chemotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 12.1 Overall survival | 1 | 418 | Hazard Ratio (IV, Random, 95% CI) | 0.42 [0.37, 0.48] |
| 12.2 Progression‐free survival | 2 | 957 | Hazard Ratio (IV, Random, 95% CI) | 0.49 [0.39, 0.61] |
| 12.3 Tumour response | 3 | 1367 | Risk Ratio (M‐H, Random, 95% CI) | 3.42 [2.38, 4.92] |
| 12.4 Toxicity (≥ G3) | 3 | 1360 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.31, 0.97] |
Comparison 13. Anti‐PD1 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 13.1 Overall survival | 1 | 834 | Hazard Ratio (IV, Random, 95% CI) | 0.63 [0.60, 0.66] |
| 13.2 Progression‐free survival | 2 | 1465 | Hazard Ratio (IV, Random, 95% CI) | 0.54 [0.50, 0.60] |
| 13.3 Tumour response | 2 | 1465 | Risk Ratio (M‐H, Random, 95% CI) | 2.47 [2.01, 3.04] |
| 13.4 Toxicity (≥ G3) | 2 | 1435 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.54, 0.91] |
Comparison 14. Anti‐PD1 monoclonal antibodies and anti‐CTLA4 monoclonal antibodies versus anti‐CTLA4 monoclonal antibodies alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 14.1 Progression‐free survival | 2 | 738 | Hazard Ratio (IV, Random, 95% CI) | 0.40 [0.35, 0.46] |
| 14.2 Tumour response | 2 | 738 | Risk Ratio (M‐H, Random, 95% CI) | 3.50 [2.07, 5.92] |
| 14.3 Toxicity (≥ G3) | 2 | 764 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [0.85, 2.92] |
Comparison 15. Chemotherapy ± sorafenib.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 15.1 Overall survival | 3 | 1194 | Hazard Ratio (IV, Random, 95% CI) | 1.00 [0.88, 1.14] |
| 15.2 Progression‐free survival | 3 | 1194 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.73, 1.09] |
| 15.3 Tumour response | 3 | 1194 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.91, 1.50] |
| 15.4 Toxicity (≥ G3) | 3 | 1194 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.93, 1.26] |
Comparison 16. Chemotherapy ± elesclomol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 16.1 Overall survival | 1 | 651 | Hazard Ratio (IV, Random, 95% CI) | 1.10 [0.92, 1.32] |
| 16.2 Progression‐free survival | 2 | 732 | Hazard Ratio (IV, Random, 95% CI) | 0.75 [0.50, 1.13] |
| 16.3 Tumour response | 2 | 732 | Risk Ratio (M‐H, Random, 95% CI) | 1.86 [0.98, 3.50] |
| 16.4 Toxicity (≥ G3) | 1 | 651 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [1.00, 1.50] |
Comparison 17. Chemotherapy ± anti‐angiogenic drugs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 17.1 Overall survival | 2 | 324 | Hazard Ratio (IV, Random, 95% CI) | 0.60 [0.45, 0.81] |
| 17.2 Progression‐free survival | 2 | 324 | Hazard Ratio (IV, Random, 95% CI) | 0.69 [0.52, 0.92] |
| 17.3 Tumour response | 2 | 324 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.96, 3.03] |
| 17.4 Toxicity (≥ G3) | 2 | 324 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.09, 5.32] |
Comparison 18. Single agent BRAF inhibitor.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 18.1 Overall survival | 2 | 925 | Hazard Ratio (IV, Random, 95% CI) | 0.40 [0.28, 0.57] |
| 18.2 Progression‐free survival | 2 | 925 | Hazard Ratio (IV, Random, 95% CI) | 0.27 [0.21, 0.34] |
| 18.3 Tumour response | 2 | 925 | Risk Ratio (M‐H, Random, 95% CI) | 6.78 [4.84, 9.49] |
| 18.4 Toxicity (≥ G3) | 2 | 925 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.48, 3.33] |
Comparison 19. Single agent MEK inhibitor.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 19.1 Overall survival | 3 | 496 | Hazard Ratio (IV, Random, 95% CI) | 0.85 [0.58, 1.25] |
| 19.2 Progression‐free survival | 3 | 496 | Hazard Ratio (IV, Random, 95% CI) | 0.58 [0.42, 0.80] |
| 19.3 Tumour response | 3 | 496 | Risk Ratio (M‐H, Random, 95% CI) | 2.01 [1.35, 2.99] |
| 19.4 Toxicity (≥ G3) | 1 | 91 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.61 [1.08, 2.41] |
Comparison 20. Combination of BRAF and MEK inhibitors versus single agent BRAF inhibitor.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 20.1 Overall survival | 4 | 1784 | Hazard Ratio (IV, Random, 95% CI) | 0.70 [0.59, 0.82] |
| 20.2 Progression‐free survival | 4 | 1784 | Hazard Ratio (IV, Random, 95% CI) | 0.56 [0.44, 0.71] |
| 20.3 Tumour response | 4 | 1784 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [1.20, 1.46] |
| 20.4 Toxicity (≥ G3) | 4 | 1774 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.85, 1.20] |
Comparison 21. Immunostimulating agents.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 21.1 Overall survival | 4 | 1458 | Hazard Ratio (IV, Random, 95% CI) | 0.82 [0.67, 0.99] |
| 21.2 Progression‐free survival | 4 | 1458 | Hazard Ratio (IV, Random, 95% CI) | 0.92 [0.74, 1.14] |
| 21.3 Tumour response | 4 | 1451 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.60, 2.50] |
| 21.4 Toxicity (≥ G3) | 4 | 1458 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.77, 1.08] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Agarwala 1999.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Single centre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 56. |
|
| Interventions |
Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and the outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Agarwala 2002.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated and previously treated metastatic melanoma. Participants randomised: 305. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: reported in a separate analysis (Beusterien 2003). The addition of subcutaneous histamine dihydrochloride to IL‐2. treatment improved median quality‐adjusted survival duration and did not adversely affect QoL. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | Published reports include all expected outcomes. However, no protocol is available and thus it is unclear if all planned outcomes are reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Atkins 2008.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 395. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Based on permuted blocks within strata, with dynamic balancing within main institutions and their affiliate networks". Comment: This method ensured low risk of selection bias |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Atzpodien 2002.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated and previously treated metastatic melanoma. Participants randomised: 124. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 12. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Two patients were randomized, but did not receive therapy and were evaluated as progressive disease". Comment: There was insufficient information about completeness of outcome data to permit judgement. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Avril 2004.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 229. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: no significant difference was observed between treatment arms. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "An independent centralized radiologic committee (two radiologists not involved in the study) performed a blinded review of all radiologic files of patients who had CR, PR, or stable disease on the investigator’s evaluation. Imaging of patients declared progressive disease (PD) as a best response were not reviewed." Comment: It was unclear if this method was sufficient to ensure low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced in across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Bafaloukos 2005.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 132. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Bajetta 1985.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 37. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: not reported. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomization". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Bajetta 1994.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 266. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 36. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomization". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Unclear risk | There was insufficient information to permit judgment. |
Bajetta 2006a.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 151. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Balch 1984.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Resected advanced regional and distant metastasis from cutaneous melanoma. Number of participants: 136. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: 10 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Bedikian 2006.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 771. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available (24 months minimum follow‐up). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were centrally randomly assigned in a 1:1 ratio in blocks of four". Comment: This method ensured low risk of selection bias. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An independent panel blinded to treatment assignment reviewed all radiologic responses." Comment: outcome assessment was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Bedikian 2011.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 393. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients meeting the enrollment criteria were randomly assigned in blocks within each country." Comment: This method ensured low risk of selection bias |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Bellett 1976.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 50. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Unclear risk | There was insufficient information to permit judgment. |
Beretta 1976.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 450. |
|
| Interventions | Four‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomly allocated". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | High risk | One arm (D) was closed early and participants' data were not analysed. |
| Other bias | Unclear risk | There was insufficient information to permit judgment. |
Carter 1975.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated and previously treated metastatic melanoma. Number of participants:270. |
|
| Interventions | Four‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Unclear risk | There was insufficient information to permit judgment. |
Carvajal 2014.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated and previously treated metastatic melanoma. Randomised participants: 106. | |
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Particpants with brain metastasis: excluded. Median follow‐up: not available. Note: Both progression‐free survival and overall survival appeared longer in the subset of participants who developed an adverse event of hypertension while receiving ramucirumab. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "One hundred and six patients were enrolled and randomised". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | High risk | There was a potential conflict of interest for some authors and the funding body which likely resulted in bias to the study methodology. |
Chapman 1999.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Number of randomised participants: 240. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: cross‐over to polychemotherapy was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Chauvergne 1982.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 51. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Particpants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Chiarion Sileni 2001.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated and previously treated metastatic melanoma. Participants randomised: 60. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 31 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol (provided by the trial principal investigator) and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Chiarion‐Sileni 2011.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated and previously treated metastatic melanoma. Participants randomised: 149. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Incidence of CNS metastasis. Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 46 weeks. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was performed centrally... using a minimisation method". Comment: randomisation method was adequate |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol (provided by the trial principal investigator) and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Clunie 1980.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Metastatic melanoma not previously treated with either Dacarbazine or C. parvum. Randomised participants: 49. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomization data". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Cocconi 1992.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Metastatic melanoma not previously treated with either tamoxifen or dacarbazine. Randomised participants: 117. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The allocation was made on the basis of randomly permuted blocks of two within strata". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Cocconi 2003.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 125. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The allocation was made on the basis of randomly permuted blocks of two within strata". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Costanza 1972.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Previously treated (no treatment in the previous 4 weeks) and untreated metastatic melanoma. Participants randomised: 140. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly allocated". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Costanza 1977.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 415. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Costanzi 1982.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Particpants randomised: 286. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: was not allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Unclear risk | There was insufficient information to permit judgment. |
Cui 2013.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Untreated metastatic melanoma harbouring no mutations in KRAS, NRAS, BRAF, or c‐kit genes. Participants randomised: 114. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Simple stratified randomization with permuted blocks of size 2 was used to create a prospective randomization schedule". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Random assignment of patients was performed by designated personnel at each participating site in a double‐blind fashion such that the investigator and patient did not know the treatment assignment" Comment: Allocation was likely concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Danson 2003.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Number of participants: 181. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: no significant difference was noted between arms. Participants with brain metastasis: included. Median follow‐up: 6 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned, using permuted blocks". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Daponte 2013.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 260. |
|
| Interventions | Four arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized through a computerized procedure of permuted blocks centralized at the coordinating center" Comment: Randomisation method wad adequate. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Dorval 1999.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated and previously treated metastatic melanoma. Number of participants: 117. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Dummer 2006.
| Study characteristics | ||
| Methods | Phase I‐II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 150. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: included. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomly assigned patients". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Eigentler 2008.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Particpants with metastasised melanoma after complete metastasectomy. Randomised participants: 139. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. |
|
| Notes | Cross‐over: not reported. Quality of life: evaluation of the quality of life was insufficient because of the low feedback rate of the questionnaires. Participants with brain metastasis: not reported. Median follow‐up: 46 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...permuted block (size 12) randomization list" Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Eisen 2010.
| Study characteristics | ||
| Methods | Phase II/III parallel‐group RCT. Double‐blind study. Multicentre trial. |
|
| Participants | Previously treated metastatic melanoma. Participants randomised: 306. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not investigated. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized using an interactive voice response system". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method makes low the risk of performance bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method makes low the risk of detection bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Eton 2002.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Number of participants: 183. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 52 months |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomly assigned". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Falkson 1991.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 64. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomized". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Falkson 1995.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 73. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomized". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Falkson 1998.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 258. |
|
| Interventions | Four‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...randomized". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Flaherty 2001.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Number of randomised participants: 81. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "All measurements for response were confirmed by one of the coauthors (C.A.), who also was responsible for collection of data from individual centers." Comment: It was unclear if this method was sufficient to ensure low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published report include all expected outcomes. However, no protocol is available and thus it is unclear if all planned outcomes are reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Flaherty 2012a.
| Study characteristics | ||
| Methods | Phase I‐II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma harboring activating mutations of BRAF. Participants randomised: 162. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: Dabrafenib 150 mg twice daily + trametinib 2 mg was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: enrolled if at least a 3‐month history of stable disease. Median follow‐up: 14 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly assigned". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published report include all expected outcomes. However, no protocol is available and thus it is unclear if all planned outcomes are reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Flaherty 2012b.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Previoulsy treated and untreated metastatic melanoma with a V600E or V600K BRAF mutation. Participants randomised: 322. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: Cross‐over to trametinib was allowed at disease progression. Quality of life: QoL analysis was reported in a separated study (Schadendorf 2014). Trametinib was associated with less functional impairment, smaller declines in health status, and less exacerbation of symptoms than dacarbazine. Participants with brain metastasis: included when brain disease was stable. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "A blinded, independent central review of tumor assessments was performed." Comment: This method makes low the risk of detection bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Flaherty 2013a.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blind study. Multicentre trial. |
|
| Participants | Metastatic melanoma not previously treated with either chemotherapy or MAP kinase pathway‐targeted drugs. Participants randomised: 823. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method makes low the risk of performance bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method makes low the risk of detection bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Glaspy 2009.
| Study characteristics | ||
| Methods | Phase II/III parallel‐group RCT. Double‐blind study. Multicentre trial. |
|
| Participants | Untreated or previously treated (dacarbazine, temozolomide, IL‐2, and/or IFN‐α) metastatic melanoma. Randomised participants: 294. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method makes low the risk of performance bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method makes low the risk of detection bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Glover 2003.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 94. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Gorbonova 2000.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 30. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Tumour response | |
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There was insufficient information to permit judgement. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgement. |
| Other bias | Unclear risk | There was insufficient information to permit judgement. |
Gough 1978.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Previuos treatment not reported. Randomised participants: 36. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients randomized". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Gupta 2014.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Untreated metastatic wild‐type BRAF melanoma. Randomised participants: 83 |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...using a variable block size". Comment: This method ensured low risk of selection bias. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "...masking". Comment: There was insufficient information about allocation concealment to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind". Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double blind". Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hamid 2014.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Previosuly treated metastatic melanoma. Randomised participants: 336. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: 5 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned 1:1 to treatment with tasisulam or paclitaxel''. Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | High risk | There is a potential conflict of interest for some authors and the funding body which likely caused bias in the study methodology . |
Hauschild 2001.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 290. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hauschild 2009a.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blind study. |
|
| Participants | Previously treated metastatic melanoma progressing under either temozolomide or dacarbazine. Participants randomised: 270. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Simple, stratified randomization with permuted blocks of size 4 was used to create a prospective randomization schedule that was implemented in a telephone based interactive voice recognition system". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Random assignment of eligible patients was performed by designated personnel at each participating site using the IVRS in a double‐blind fashion such that the investigator, sponsor, and patient did not know the treatment assignment". Comment: Likely that allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind". Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double blind". Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hauschild 2012.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma with BRAF V600E mutation. Participants randomised: 250. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: Cross‐over to dabrafenib 150 mg twice daily was allowed at disease progression. Quality of life: Dabrafenib had functional and symptomatic benefit compared to dacarbazine (Grob 2014). Participants with brain metastasis: excluded unless they were without evidence of active central nervous system metastases for more than 3 months after surgery or stereotactic radiosurgery. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A centrally located, computerised, interactive, voice activated response system controlled assignment of patient treatment". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Although investigators were aware of treatment group when assessing progression‐free survival, a masked independent review committee (IRC) reviewed all scans and, per protocol, had to confirm progression before patients crossed over from dacarbazine to dabrafenib". Comment: Likely that allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hersh 2015.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 529. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized... via a centralized system". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...independent radiologic review". Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hodi 2010a.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blind study |
|
| Participants | HLA‐A*0201–positive metastatic melanoma which had progressed during systemic treatment . Participants randomised: 676. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: Ipilimumab did not have a detrimental effect on QoL during the treatment induction phase (Revicki 2012). Participants with brain metastasis: participants with active, untreated metastases in the central nervous were excluded. Median follow‐up: 21 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hodi 2014.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated and previously treated (1 chemotherapy was allowed) metastatic melanoma. Patients randomised: 245. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 13 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Stratified randomization based on permuted blocks within strata with dynamic institution balancing was used." Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment assignments were obtained from the central randomization desk at the ECOG coordinating center." Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Tumor responses were determined by the investigators using RECIST (Response Evaluation Criteria in Solid Tumors) criteria and were audited as a part of ECOG‐ACRIN (American College of Radiology Imaging Network) standard procedures." Comment: It was unclear if this method was sufficient to ensure low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Hofmann 2011.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Previously treated metastatic melanoma. Participants randomised: 117. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Overall survival Tumour response Toxicity |
|
| Notes | Cross‐over: was not allowed. Quality of life: No significant difference in the quality of life could be found. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "After the first five patients, it was decided... that the centres have the option to enrol patients on a treatment preference basis (patients’ choice) ". Comment: This domain was assessed at high risk of selection bias because initially enrolled participants were randomly assigned to either chemotherapy or best supportive care, but enrolment was slow and allocation appeared to be based on physician's choice. |
| Allocation concealment (selection bias) | High risk | Quote: "...patients' choice". Comment: Unlikely that allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "There was no centralized review of the radiology files provided." Comment: Overall, there was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Jelic 2002.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Single centre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 219. |
|
| Interventions | Four‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Johnston 1998.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Metastatic melanoma untreated or previously treated with no more than one previous systemic chemotherapy. Randomised participants: 65. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients who were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Kaufmann 2005.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 294. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned without stratification". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "There was no centralized review of the radiologic files provided." Comment: It was unclear if this method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Kefford 2010.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 80. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Keilholz 1997.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated and previously treated metastatic melanoma. Randomised participants: 138. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: > 2 years. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients randomized". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Keilholz 2005.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 363. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 3.4 years. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Kim 2012.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blinded study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 214. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 13 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Random assignment was performed using an interactive voice response system". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | No sufficient information to judge |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Kirkwood 1990.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 74. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Kogoniia 1981.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 132. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Tumour response | |
| Notes | Cross‐over: cross‐over was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: not reported. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Low risk | No other sources of bias found. |
Kokoschka 1978.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 34. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Larkin 2014.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma with BRAF V600 mutations. Participants randomised: 495. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: participants with previously treated brain metastases were eligible if they had at least a 3‐week history of stable disease. Median follow‐up: 7 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "We performed a blinded, independent central review of tumor assessments." Comment: It is unclear if this method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Larkin 2015.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blind study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 945. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: participants with inactive brain metastasis were excluded. Median follow‐up: > 9 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Enrolled patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | Quote: "Data on overall survival are insufficiently mature to present". Comment: Low risk of selective reporting. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Lawson 2015.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blind study. Multicentre trial. |
|
| Participants | Participants underwent surgery for locally advanced or metastatic melanoma. Randomised participants: 815. |
|
| Interventions | HLA‐A2–positive (serologically defined)
HLA‐A2–negative group
|
|
| Outcomes | Progression‐free survival. Overall survival. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: participants who underwent surgery for brain metastasis were included. Median follow‐up: 82 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Random assignment was conducted centrally by using permuted blocks within strata". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Placebo‐controlled”. Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “Placebo‐controlled”. Comment: The method ensured low risk of detection bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Legha 1996.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants:102. |
|
| Interventions | Two‐arm study:
Treatment schedules:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: participants with symptomatic brain metastasis were excluded. Median follow‐up: 45 months. Note: Both biochemotherapy schedules were compared with a non‐randomised group of participants who received chemotherapy alone. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly assigned". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Long 2015.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blind study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma with BRAF Val600Glu or Val600Lys mutations. Participants randomised: 423. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: Dabrafenib and trametinib resulted in better preservation of health‐related quality of life and pain improvement compared to dabrafenib monotherapy (Schadendorf 2015). Participants with brain metastasis: participants with previously treated brain metastases were eligible if they had at least a 12‐week history of stable disease. Median follow‐up: 9 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A centrally located, computerised, interactive, voice activated response system controlled the random assignment". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Investigators, site staff, and patients were unaware of assignment throughout the study, and masking was maintained by using tablets and bottles of active drug and placebo that were identical in appearance. At the time of the primary analysis, only the sponsor and those assessing the data were made aware of treatment group assignments." Comment: Allocation likely concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Double blind”. Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “Double blind”. Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Lopez 1984.
| Study characteristics | ||
| Methods | Phase II parallel RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 42. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Tumour response. Toxicity. |
|
| Notes | Cross‐over: not available. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...before randomisation". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Low risk | No other sources of bias found. |
Luikart 1984.
| Study characteristics | ||
| Methods | Phase III parallel RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 57. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: cross‐over to polychemotherapy was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...random table of numbers". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Maio 2010.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 488. |
|
| Interventions | Five‐arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". "The randomization list was produced by the Internal Quality Control Unit of Biostatistics and Data Management". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was blinded and centralized". Comment: This method ensured low risk of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Independent, blinded evaluation of tumor images was performed by Fondazione Biomedica Europea." Comment: It was unclear if this method ensured low the risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Mastrangelo 1979.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated or previously treated (only treatments other than a nitrosurea was allowed) metastatic melanoma. Randomised participants: 62. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomly allocated". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
McArthur 2014.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma with BRAF V600E e V600K mutations. Participants randomised: 675. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: Cross‐over to vemurafenib was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: excluded when metastases to the central nervous system had progressed or required treatment in the previous 3 months. Median follow‐up: 12 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned using an interactive voice recognition system supported by an independent vendor". Comment: Randomisation method adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients and investigators were aware of treatment allocation" Comment: An independent review committee (IRC) had to confirm progression before participants crossed over from dacarbazine to dabrafenib. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | Secondary endpoints not reported will be subject of future publications. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
McDermott 2008.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 101. |
|
| Interventions | Two.arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Simple stratified randomization with permuted blocks of size 4 was used by the sponsor to create a prospective randomization schedule that was provided to the vendor for the telephone‐based interactive voice recognition system". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Random assignment of eligible patients was performed by designated personnel at each participating site using the interactive voice recognition system in a double‐blind fashion" Comment: Likely that allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Middleton 2000.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 305. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: Temozolomide therapy significantly improved health‐related QoL (Kiebert 2003). Participants with brain metastasis: excluded. Median follow‐up: not available. Cost analysis: Temozolomide was associate with incremented cost‐effectiveness (Hillner 2000). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was no sufficient information to judge. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Middleton 2007.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 241. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Site‐specific randomization codes were produced electronically for each stratified group". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "...site personnel called a central randomization desk". Comment: This method ensured low risk of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was no sufficient information to judge. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Middleton 2015.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Untreated and previously treated metastatic melanoma. Randomised participants: 346. |
|
| Interventions | Three‐arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Mediano follow‐up: not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized sequentially 1:1:1 using a computer‐based model". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Miller 1989.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated and previously treated metastatic melanoma. Randomised participants: 53. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Participants with liver metastasis were also excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Treatment assignments were provided to the investigators by a Research Nurse using sealed envelopes." Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Moon 1975.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 120. |
|
| Interventions | Three‐arm study:
|
|
| Outcomes | Tumour response. | |
| Notes | Cross‐over: allowed. Quality of life: not reported. Participants with brain metastasis: included. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomly allocated". Comments: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Newlands 1976.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 56. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomly allocated". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
O'Day 2009.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Untreated and previously treated (1 chemotherapy was allowed) metastatic melanoma. Participants randomised: 81. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: cross‐over to open‐label elesclomol plus paclitaxel was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 3 months (for censored participants). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...using an interactive voice‐response system". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Low risk | Quote: "Investigators and patients were blinded with respect to treatment assignment; unblinded site pharmacists were responsible for reconstituting study drugs at the pharmacy at each site". This method ensured low risk of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind". This method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double blind". This method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
O'Day 2011.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 129. |
|
| Interventions | Four‐arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: quote "Patients in the blinded dacarbazine‐containing arms who could not tolerate dacarbazine were allowed to cross‐over to open‐label 10 mg/kg intetumumab monotherapy, and those on dacarbazine monotherapy who experienced progressive disease (PD) were allowed to cross over to open‐label dacarbazine plus10 mg/kg intetumumab". Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 24 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomisation was stratified". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...blinded". Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...blinded". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
O'Day 2013.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blind study. Multicentre trial. |
|
| Participants | Untreated and previously treated (1 chemotherapy was allowed) metastatic melanoma. Participants randomised: 651. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not reported. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...randomly assigned patients". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind". Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double blind". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Patel 2011.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 859. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 19 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation, performed centrally at the EORTC Headquarters, was stratified by performance status (0 versus 1) and institution, using a minimisation technique". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Postow 2015.
| Study characteristics | ||
| Methods | Phase I dose‐escalation parallel‐group RCT. Double‐blinded study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 142. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: cross‐over was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: > 11 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly assigned". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind trial". Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind trial". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Presant 1979.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 120. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Presant 1982.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 195. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Punt 2006.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 93. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomisation was performed centrally". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ramseur 1978.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated and previously treated metastatic melanoma. Number of randomised participants: 28. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly allocated". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ranson 2007.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 104. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: quote "Patients experiencing disease progression in the TMZ alone arm were permitted to continue study treatment by changing to the LM/TMZ combination". Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were to be randomly assigned". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | High risk | Quote: "In the course of the trial, it became apparent that MGMT persisted in tumor biopsy samples taken 24 to 72 hours after the end of cycle 1 LM/TMZ. Therefore, the trial was extended by 20 patients, with the LM dose in those assigned combination treatment being increased to 60 mg/d, then to 80 mg/d". |
Reichle 2007.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 76. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: cross‐over to combination therapy was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: included (quote: "controlled brain metastasis"). Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ribas 2013.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 655. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: cross‐over to tremelimumab was not allowed for participants who progressed during standard chemotherapy. Cross‐over to ipilimumab was allowed at disease progression. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Tumor data assessed by investigators were reviewed by the sponsor to ensure compliance with RECIST criteria." Comment: It was unclear if this method ensured low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ribas 2015.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Metastatic melanoma progressing after treatment with ipilimumab or BRAF and/or MEK inhibitors. Participants randomised: 540. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: pembrolizumab had smaller decrements in the individual function and symptoms scales. Cross‐over: cross‐over to pembrolizumab after progression under investigation‐choice systemic chemotherapy was allowed. Participants who crossed‐over were randomly assigned to receive either 2 mg/kg or 10 mg/kg pembrolizumab. Participants with brain metastasis: excluded. Median follow‐up: 10 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Block randomisation with a block size of six in each stratum was used". Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Individual treatment assignment between pembrolizumab and chemotherapy was open label; investigators and patients were masked to assignment to pembrolizumab dose". Comment: Allocation likely concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Investigators and patients were masked to assignment to pembrolizumab dose". Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "investigators were masked to assignment to pembrolizumab dose". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Richtig 2004.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated and previously treated metastatic melanoma. Randomised participants: 47. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomised". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no missing data. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | High risk | Quote: "The study was stopped after the inclusion of approximately 50%". Comment: High risk of bias due to the trial stopping after approximately 50% of the planned participants were enrolled. |
Ridolfi 2002a.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 165. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: allowed at disease progression. Quality of life: investigated in a separate analysis (Chiarion‐Sileni 2003). Biochemotherapy worsened significantly quality of life compared to chemotherapy. Participants with brain metastasis: excluded. Median follow‐up: 17 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...system of random permuted blocks within the strata (oncologic center variable) was used with a block size of four." Comment: Adequate randomisation method used. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | No protocol is available and thus it is unclear if all planned outcomes are reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Ringborg 1989.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 119. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients... were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Robert 2011.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blinded study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 502. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: A paper reported on quality‐adjusted time without symptoms of disease or toxicity of treatment (Q‐TWiST) (Sherrill 2013). Particpants treated with ipilimumab had little benefit in quality‐adjusted survival during the first year. The benefits of ipilimumab has increased with extended survival after 2, 3, and 4 years. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Robert 2013.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Untreated metastatic melanoma with BRAF mutations. Randomised participants: 91. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: Participants with either brain or spinal cord metastasis were eligible when asymptomatic, treated, and stable off treatment for > 3 months. Median follow‐up: 12 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned by central interactive voice response system (1:1 ratio, block size four)." Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients, investigators, and the study team were masked to the treatment assigned." Comment: Allocation was likely concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Robert 2015.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma with BRAF V600E e V600K mutations. Participants randomised: 704. |
|
| Interventions | Two arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: This was reported in a separated analysis (Grob 2015). Combination of dabrafenib and trametinib adds a clear benefit over monotherapy with vemurafenib. Participants with brain metastasis: participants with previously treated brain metastases were eligible if they had at least a 12‐week history of stable disease. Median follow‐up: 10 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Robert 2015a.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blinded study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma without BRAF mutations. Participants randomised: 418. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not investigated. Participants with brain metastasis: participants with active brain metastasis were excluded. Median follow‐up: 9 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Double‐blind”. Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “double‐blind”. Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Robert 2015b.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Metastatic melanoma that had no more than one previous systemic therapy for advanced disease (CTLA‐4, PD‐1, or PD‐L1 inhibitors were not allowed). Participants randomised: 834. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported Participants with brain metastasis: participants with brain metastasis were excluded when they had active metastasis. Median follow‐up: > 9 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Robidoux 1982.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 88. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Rosenberg 1999.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 102. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 42 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomization between the two study arms was performed by the central data management office". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Rusthoven 1996.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Double‐blinded study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 204. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: participants were allowed to cross‐over to tamoxifen‐based regimen at disease progression. Participants with brain metastasis: participants with brain metastasis were eligible if they had completed planned surgery/radiotherapy, did not require glucocorticosteroids at study entry, and had stable disease in the brain at a repeat computed tomography (CT) scan 2 weeks before randomisation. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Before randomization, patients were stratified". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐blind". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Schadendorf 2006.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 108. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: 22 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The study was externally monitored". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Schwartzentruber 2011a.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 185. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: 41 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Stratified randomization was performed with the use of random block sizes to ensure balance with respect to a potentially important prognostic feature." Comment: Randomisation method was adequate. |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...blinded central radiologic review". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Sertoli 1999.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 92. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients... were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit judgment. |
| Other bias | Unclear risk | There was insufficient information to permit judgment. |
Sparano 1993.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated and previously treated (only one chemotherapy line was allowed) metastatic melanoma. Participants randomised: 85. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All responses were independently reviewed by the study's principal investigators and by a single radiologist". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Testori 2008.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. Multicentre trial. |
|
| Participants | Untreated metastatic melanoma. Participants randomised: 322. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: 9 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Risk was likely low because this was a multicentre trial with centralised randomisation |
| Allocation concealment (selection bias) | Low risk | Risk was likely low because this was a multicentre trial with centralised randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Thatcher 1986.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 79. |
|
| Interventions | Two‐arm trial:
All participants who had disease progression were treated with dacarbazine 250 mg/m² IV daily on days 1 to 5 and actinomycin D 1.5 mg/m² IV on day 1 every 3 weeks. |
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: > 36 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: However, there was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Thomson 1993.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 170. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: analysis of quality of life was reported in a different article (Coates 1993). There was no statistically significant difference in quality of life between treatment arms. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised centrally using a dynamic randomisation technique." Comment: This method ensured low risk of selection bias. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Veronesi 1984.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 377. |
|
| Interventions | Three‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The composition of the series... was prepared by the coordinating center". Comment: This method ensured low risk of selection bias. |
| Allocation concealment (selection bias) | Low risk | Quote: "The envelopes... were opened at the moment of choice of treatment." Comment: This method ensured low risk of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reasons for exclusions not reported. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Verschraegen 1993.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Randomised participants: 103. Untreated and previously treated metastatic melanoma. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Vorobiof 1994.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 60. |
|
| Interventions | Three‐arm trials:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: 13 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...closed envelope random number technique". Comment: This method ensured low risk of selection bias. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no missing data. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Vuoristo 2005.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated and previously treated (only drugs other than dacarbazine were allowed) metastatic melanoma. Randomised participants: 106. |
|
| Interventions | Four‐arm trial:
|
|
| Outcomes | Overall survival. Progression‐free survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: > 17 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The randomization was performed at the Finnish Cancer Registry and stratified for treatment arm by institution." Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Weber 2009.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 184. |
|
| Interventions | Four‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised." Comment: There was insufficient information about the sequence generation process to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was no information sufficient to judge. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Weber 2015.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Previously treated metastatic melanoma. Both BRAF mutant and non‐mutant tumours were included. Randomised participants: 405. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: excluded when brain metastases were active. Median follow‐up: 8 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "We used permuted blocks (block size of six) within each stratum for randomisation." Comment: This method ensured low risk of selection bias. |
| Allocation concealment (selection bias) | Low risk | Quote: "using an interactive voice response system." Comment: This method ensured low risk of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "those doing tumour assessments were masked to treatment assignment". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Wittes 1978.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 95. |
|
| Interventions | Three‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Wolchok 2010.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Double‐blind study. |
|
| Participants | Previously treated metastatic melanoma. Participants randomised: 217. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Overall survival. Tumour response. Toxicity. |
|
| Notes | Cross‐over: not allowed. Quality of life: not reported. Participants with brain metastasis: included. Median follow‐up: 9 months. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was done with a permuted block procedure" Comment: This method ensured low risk of selection bias. |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients, treating doctors, and doctors’ staff were unaware of the dose to which patients were assigned, whereas pharmacists were unmasked". Comment: This statement makes low the risk of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blinded". Comment: The method ensured low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double blinded". Comment: The method ensured low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | No differences between protocol and published report. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Young 2001.
| Study characteristics | ||
| Methods | Phase III parallel‐group RCT. Open label study. |
|
| Participants | Untreated metastatic melanoma. Randomised participants: 61. |
|
| Interventions | Two‐arm study:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: There were no differences in quality of life between treatment groups. Cross‐over: not allowed. Participants with brain metastasis: excluded. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...random permuted blocks method" Comment: This method ensured low risk of selection bias. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Zimpfer‐Rechner 2003.
| Study characteristics | ||
| Methods | Phase II parallel‐group RCT. Open label study. |
|
| Participants | Previoulsy treated metastatic melanoma. Randomised participants: 40. |
|
| Interventions | Two‐arm trial:
|
|
| Outcomes | Progression‐free survival. Overall survival. Tumour response. Toxicity. |
|
| Notes | Quality of life: not reported. Cross‐over: not allowed. Participants with brain metastasis: included. Median follow‐up: not available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomized". Comment: There was insufficient information to permit judgment. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information to permit judgment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As an open label study, no blinding of participants or personnel was possible. However, we believe that in this setting (metastatic melanoma), with the treatments tested and outcomes assessed, the knowledge of which intervention was received or administered (rather than the intervention itself), could not affect the outcomes under investigation. Therefore, we judged the risk of performance bias as low. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data were balanced across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Unclear risk | Published reports included all expected outcomes. However, no protocol was available so it was unclear if all planned outcomes were reported. |
| Other bias | Low risk | The study appeared to be free of other sources of bias. |
Abbreviations: BCG ‐ Bacillus Calmette‐Guérin; BCNU ‐ 1,3‐bis(2‐chloroethyl)‐1‐nitrosourea; CCNU ‐ lomustine; ECOG ‐ Eastern Cooperative Oncology Group; CR ‐ complete response; G‐CSF ‐ granulocyte‐colony stimulating factor; IFN ‐ interferon‐alpha; IFN‐α ‐ interferon‐alpha; IL‐2 ‐ interleukin‐2; IM ‐ intramuscular; IV ‐ intravenous; MAP ‐ mitogen‐activated protein; MGMT ‐ methylguanine‐DNA methyltransferase; NA ‐ not applicable; PEG‐IFN ‐ pegylated interferon; PR ‐ partial response; QoL ‐ quality of life; RCT ‐ randomised controlled trial; SC ‐ subcutaneous.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Asemissen 2005 | This study investigated mechanisms of interaction between interleukin‐2 and histamine in a subgroup of 19 participants enrolled in a trial. This study was excluded because study endpoints did not match inclusion criteria; the study was about drug interaction and not patient survival or tumour response or toxicity. |
| Atzpodien 1995 | This study is not an RCT. |
| Bleehen 1995 | This study is not an RCT. |
| Buchbinder 2015 | This study is not an RCT. |
| Bukowski 1983 | This study investigated adjuvant therapy following radical resection of lymph node metastasis (participants were not affected with early stage and not advanced/metastatic melanoma). |
| Cashin 2008 | This study is not an RCT. |
| Cormier 1997 | This study investigated the effect of dopamine for reducing renal toxicity caused by interleukin‐2. |
| Curl 2014 | This study is not an RCT. |
| Downey 2007 | This study is not an RCT. |
| Hill 1984 | This study reported a retrospective analysis of participants who had experienced a complete tumour response in RCTs from the Central Oncology Group. |
| Hughes 2016 | This RCT did not investigate systemic treatments for metastatic disease. Participants were randomised to receive a local treatment, liver infusion, for hepatic metastasis. |
| Hwu 2009 | This article is a commentary on preliminary findings of a RCT already included in this review (Schwartzentruber 2011a). |
| Kaufman 2010 | This study did not investigate systemic treatment. It tested direct injection of an oncolytic herpes simplex virus type 1 encoding granulocyte macrophage colony‐stimulating factor into accessible melanoma lesions. |
| Kleeberg 1982 | This study is not an RCT. |
| Lattanzi 1995 | This study is not an RCT. |
| McDermott 2013 | This study analysed selected participants experiencing long‐term survival in Hodi 2010a. |
| Mornex 2003 | This study investigated whole brain radiotherapy associated with fotemustine compared to fotemustine alone, and thus did not test effectiveness of systemic treatment. |
| Quirt 1983 | This study investigated both participants with early stage and metastatic melanoma. This study was excluded because tumour stage of enrolled participants did not match our inclusion criteria (no separate findings for different stages were reported and thus we could not include even part of the results). |
| Richards 1999 | This study is not an RCT. |
| Spieth 2008 | This study is not an RCT. |
| Van Dyk 1975 | This study was not an RCT. |
| Varker 2007 | This study randomised participants with metastatic melanoma treated with bevacizumab to receive local interleukin‐2 injections. |
| Weber 2013 | This study gathered data from three different RCTs and focused on adverse events. This study was excluded because it is a secondary analysis pooling data from one RCT, Hodi 2010a, already included in this systematic review. |
| Yang 1995 | This study enrolled both participants with metastatic melanoma and metastatic renal cell carcinoma. Information specifically regarding melanoma was not reported for any study endpoint. |
RCT ‐ randomised controlled trial.
Characteristics of ongoing studies [ordered by study ID]
NCT01280565.
| Study name | A phase 3 study to compare efficacy and safety of masitinib to dacarbazine in the treatment of patients with non‐resectable or metastatic stage 3 or stage 4 melanoma carrying a mutation in the juxta membrane domain of C‐Kit. |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 200. |
| Interventions | Two‐arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcome:
|
| Starting date | January 2011. |
| Contact information | Jean Jaques Grob, jean-jacques.grob@mail.ap-hm.fr |
| Notes | ‐ |
NCT01515189.
| Study name | Phase 3 trial in subjects with metastatic melanoma comparing 3 mg/kg ipilimumab versus 10 mg/kg ipilimumab. |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 700. |
| Interventions | Two‐arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | January 2012. |
| Contact information | 88 recruiting sites (available at clinicaltrials.gov). |
| Notes | ‐ |
NCT01763164.
| Study name | Study comparing the efficacy of MEK162 versus dacarbazine in unresectable or metastatic NRAS mutation‐positive melanoma. |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 397. |
| Interventions | Two‐arm study:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | July 2013. |
| Contact information | 167 recruiting sites (available at clinicaltrials.gov). |
| Notes | ‐ |
NCT01909453.
| Study name | Study comparing combination of LGX818 plus MEK162 versus vemurafenib and LGX818 monotherapy in BRAF mutant melanoma (COLUMBUS). |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 900. |
| Interventions | Four‐arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | September 2013. |
| Contact information | 230 recruiting sites (available at clinicaltrials.gov). |
| Notes | ‐ |
NCT01940809.
| Study name | Ipilimumab with or without dabrafenib, trametinib, and/or nivolumab in treating patients with melanoma that is metastatic or cannot be removed by surgery |
| Methods | Phase I RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 40. |
| Interventions | Five‐arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | August 2013. |
| Contact information |
|
| Notes | ‐ |
NCT01943422.
| Study name | Safety and efficacy study of vemurafenib and high‐dose interferon alfa‐2b in melanoma (12‐107) |
| Methods | Phase I/II RCT |
| Participants | Metastatic melanoma. Estimated enrolment: 63. |
| Interventions | Three‐arm study:
|
| Outcomes |
Primary outcomes:
Secondary outcome:
|
| Starting date | September 2013. |
| Contact information | John Kirkwood, MD, kirkwoodjm@upmc.edu |
| Notes | ‐ |
NCT02130466.
| Study name | A phase I/II study to assess the safety and efficacy of MK‐3475 in combination with trametinib and dabrafenib in subjects with advanced melanoma. |
| Methods | Phase I/II RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 177. |
| Interventions | Four‐arm trial:
|
| Outcomes |
Primary outcomes:
Secondary outcome:
|
| Starting date | May 2014. |
| Contact information | Toll Free Number 1‐888‐577‐8839 |
| Notes | ‐ |
NCT02224781.
| Study name | A randomized phase III trial of dabrafenib + trametinib followed by ipilimumab + nivolumab at progression vs. ipilimumab + nivolumab followed by dabrafenib + trametinib at progression in patients with advanced BRAFV600 mutant melanoma. |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 300. |
| Interventions | Four‐arm study:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | July 2015 |
| Contact information | Michael Atkins, ECOG‐ACRIN Cancer Research Group |
| Notes | ‐ |
NCT02278887.
| Study name | Randomized phase III study comparing a non‐myeloablative lymphocyte depleting regimen of chemotherapy followed by infusion of tumor infiltrating lymphocytes and interleukin‐2 to standard ipilimumab treatment in metastatic melanoma. |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 162. |
| Interventions | Two arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcome:
Other outcome measure:
|
| Starting date | September 2014. |
| Contact information | John BAG Haanen, j.haanen@nki.nl |
| Notes | ‐ |
NCT02339571.
| Study name | Randomized phase II/III study of nivolumab plus ipilimumab plus sargramostim versus nivolumab plus ipilimumab in patients with unresectable stage III or stage IV melanoma. |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma with brain metastasis. Estimated enrolment: 400. |
| Interventions | Two‐arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | September 2015. |
| Contact information | Frank Hodi, ECOG‐ACRIN Cancer (Eastern Co‐operative Oncology Group‐American College of Radiology Imaging Network) Research Group |
| Notes | ‐ |
NCT02388906.
| Study name | A phase III, randomized, double‐blind study of adjuvant immunotherapy with nivolumab versus ipilimumab after complete resection of stage IIIb/c or stage IV melanoma in subjects who are at high risk for recurrence (CheckMate 238: CHECKpoint Pathway and nivoluMAb Clinical Trial Evaluation 238). |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma with brain metastasis. Estimated enrolment: 800. |
| Interventions | Two‐arm study:
|
| Outcomes |
Primary outcome:
Secondary outcome:
|
| Starting date | March 2015. |
| Contact information | 136 recruiting sites (available at clinicaltrials.gov). |
| Notes | ‐ |
NCT02416232.
| Study name | An open label non randomized access study of trametinib for patients with advanced unresectable (stage IIIc) or distant metastatic (stage IV) BRAF V600E/K mutation positive cutaneous melanoma. |
| Methods | Phase III non‐RCT. |
| Participants | Metastatic melanoma with brain metastasis. Estimated enrolment: 250. |
| Interventions | Single arm study: participants will receive trametinib 2 mg orally once daily and, where appropriate, in combination with dabrafenib 150 mg orally twice daily. |
| Outcomes |
Primary outcomes:
|
| Starting date | March 2015. |
| Contact information | USA GSK Clinical Trials Call Center, GSKClinicalSupportHD@gsk.com |
| Notes | ‐ |
NCT02460068.
| Study name | A randomized, phase III study of fotemustine versus the combination of fotemustine and ipilimumab or the combination of ipilimumab and nivolumab in patients with metastatic melanoma with brain metastasis (NIBIT‐m²). |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma with brain metastasis. Estimated enrolment: 168. |
| Interventions | Three‐arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | December 2012 |
| Contact information | Anna Maria Di Giacomo, PhD, MD, a.m.digiacomo@ao-siena.toscana.it |
| Notes | ‐ |
NCT02506153.
| Study name | A phase III randomized trial comparing physician/patient choice of either high dose interferon or ipilimumab to MK‐3475 (pembrolizumab) in patients with high risk resected melanoma. |
| Methods | Phase III RCT. |
| Participants | Participants who underwent surgery for distant metastasis. Estimated enrolment: 1378. |
| Interventions | Two‐arm trial:
|
| Outcomes |
Primary outcomes:
Secondary outcomes:
|
| Starting date | October 2015. |
| Contact information | 314 recruiting sites (available at clinicaltrials.gov). |
| Notes | ‐ |
NCT02599402.
| Study name | Clinical trial of nivolumab (BMS‐936558) combined with ipilimumab followed by nivolumab monotherapy as first‐line therapy of subjects with histologically confirmed stage III (unresectable) or stage IV melanoma. CheckMate 401: CHECKpoint Pathway and nivoluMAb Clinical Trial Evaluation 401 |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma with brain metastasis. Estimated enrolment: 615. |
| Interventions | Two‐arm study:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | December 2015. |
| Contact information | 41 recruiting sites (available at clinicaltrials.gov). |
| Notes | ‐ |
NCT02625337.
| Study name | Phase 2 study comparing pembrolizumab with intermittent/short‐term dual MAPK pathway inhibition plus pembrolizumab in patients harboring the BRAFV600 mutation. |
| Methods | Phase II RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 32. |
| Interventions | Four‐arm trial:
|
| Outcomes |
Primary outcomes:
Secondary outcomes:
|
| Starting date | January 2016. |
| Contact information |
|
| Notes | ‐ |
NCT02714218.
| Study name | Phase IIIb/IV, randomized, double blinded, study of nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg vs nivolumab 1 mg/kg in combination with ipilimumab 3 mg/kg in subjects with previously untreated, unresectable or metastatic melanoma. |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 304. |
| Interventions | Two‐arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Starting date | March 2016. |
| Contact information | 52 recruiting sites (available at clinicaltrials.gov). |
| Notes | ‐ |
NCT02752074.
| Study name | A phase III randomized, double‐blind, placebo‐controlled study of pembrolizumab (MK‐3475) in combination with epacadostat or placebo in subjects with unresectable or metastatic melanoma (Keynote‐252 / ECHO‐301). |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 600. |
| Interventions | Two‐arm trial:
|
| Outcomes |
Primary outcomes:
Secondary outcomes:
|
| Starting date | June 2016. |
| Contact information | Merck Sharp & Dohme Corp 1‐888‐577‐8839 |
| Notes | ‐ |
NCT02821013.
| Study name | A randomized phase III trial of the duration of anti‐PD‐1 therapy in metastatic melanoma (STOP‐GAP). |
| Methods | Phase III RCT. |
| Participants | Metastatic melanoma. Estimated enrolment: 550. |
| Interventions | Two‐arm trial:
|
| Outcomes |
Primary outcome:
Secondary outcome:
|
| Starting date | June 2016. |
| Contact information | Janet Dancey, jdancey@ctg.queensu.ca |
| Notes | ‐ |
B‐Raf – a protein; C‐Kit – a protein; CR ‐ complete response; CTCAE ‐ Common Terminology Criteria for Adverse Events; EORTC QLQC30 ‐ European Organization for Research and Treatment quality of life questionnaire (version 3.0); EQ‐5D ‐ EuroQol‐5D; irAEs – immune‐related adverse events; IV ‐ intravenously; MAPK ‐ mitogen‐activated protein kinase; mWHO ‐ modified WHO criteria; NRAS ‐ neuroblastoma RAS viral oncogene; PR ‐ partial response; PD‐1 ‐ an inhibitory receptor located on the surface of the T‐cells; PD‐L1 – programmed death‐ligand 1; RCT – randomised controlled trial; SC ‐ subcutaneously; SD ‐ stable disease; Vs – versus.
Differences between protocol and review
Network meta‐analysis
Given that direct comparisons between key therapies were unavailable (e.g. immune checkpoint inhibitors versus small‐molecule targeted drugs), we conducted a network meta‐analysis to compute estimates of indirect comparisons and to generate treatment rankings (Cipriani 2013; Mills 2013).
Study selection
We used the following criteria to assess randomised controlled trials (RCTs) for inclusion:
studies reporting on the outcomes of interest, that is, progression‐free survival (as an efficacy outcome) and severe toxicity (as a harm outcome); and
studies reporting on treatments for which high quality evidence of efficacy was available from direct comparisons and for which interventions are approved for routine use in clinical practice.
Further details on outcomes and treatments included in the network meta‐analysis are reported in the Effects of interventions section (see Network meta‐analysis findings).
We chose to include phase III and earlier phase studies because early phase trials were more likely to report on tumour response (which was a review secondary outcomes). Furthermore, early phase trials sometimes also describe survival findings (which was a review primary outcome). However, phase II trials are not designed to detect survival differences but rather tumour response differences.
We included trials with mixed disease stages if outcomes for metastatic disease were reported separately.
Evidence grading
We used the GRADE system adapted for network meta‐analysis to assess evidence quality according to four levels: high‐, moderate‐, low‐, and very low‐quality (Salanti 2014).
Quality was downgraded by one level (serious concern) or two levels (very serious concern) for study limitations (risk of bias), evidence for publication bias (assessed by inspecting a funnel plot dedicated to network meta‐analysis (Chaimani 2013)), indirectness (indirect population, intervention, control, outcomes; lack of transitivity assumption), inconsistency (between‐study statistical heterogeneity, as suggested by network meta‐analysis estimate of prediction interval crossing the null value), and imprecision (as suggested by wide confidence intervals estimated by network meta‐analysis).
Statistical analysis
Review primary outcomes were progression‐free survival and high‐grade toxicity. The outcome measure for survival data was hazard ratio (HR) and 95% confidence interval (CI). The outcome measure for toxicity was relative risk (RR) and 95% CI.
Random‐effects network meta‐analysis was carried out within a frequentist setting (Hong 2013). A common heterogeneity parameter (Tau²) was assumed across all comparisons, allowing the inclusion of comparisons based on a single RCT. Summary effects are presented with 95% CIs and predictive interval. Predictive intervals were calculated using between‐study variance (Tau²) and represents the interval where the results of future studies are expected to be, thus providing information on the magnitude of heterogeneity. They are calculated as μ ± (tαdf) x √ (τ² + SE(μ)², where tαdf is the 100 x (1 ‐ α/2)% percentile of the t‐distribution with df degrees of freedom and μ is the meta‐analysis effect estimate (Chaimani 2013).
The key assumption of network meta‐analysis is transitivity (Donegan 2013). If information about comparisons A versus B and A versus C is available, then network meta‐analysis can derive information regarding the BC comparison based on the transitivity equation (A versus B – A versus C = B versus C). Transitivity holds assuming that:
the common treatment, in this case conventional chemotherapy (used to compare different drug schedules indirectly), was similar when it appeared in different trials;
pair‐wise comparisons did not differ substantially with respect to the distribution of effect modifiers; and
in principle, participants could be randomised to any of the treatments compared in the network.
Lack of transitivity can manifest as inconsistency between direct and indirect estimates ('loop inconsistency') or between estimates deriving from different study designs ('design inconsistency', which can occur when the relative effectiveness of treatment A versus B is different when estimated in studies with different designs, such as A versus B and A versus B versus C). We investigated inconsistency using a design‐by‐treatment interaction model, which addresses both loop and design inconsistency (Higgins 2012; White 2012).
Inconsistencies of single loops can be assessed with an inconsistency plot, where a ratio of ratio can be calculated as the ratio between the relative risk estimated by the conventional pair‐wise meta‐analysis and that estimated by the network meta‐analysis. A ratio of ratio value close to the unit indicates that the results of the two techniques are in agreement; in general, values greater than 2 suggest high inconsistency (Chaimani 2013).
Network meta‐analysis also provides a ranking probability curve of each treatment (rankogram) by calculating the probability of each treatment to achieve the best rank amongst all treatments. The surface under the cumulative ranking (SUCRA) line for each treatment, which equals one when a treatment is certain to be the best and zero when a treatment is certain to be the worst, was used for treatment ranking (Chaimani 2013; Salanti 2011). We also generated a bivariate ranking plot including both efficacy (progression‐free survival) and acceptability (the inverse of toxicity: low toxicity rates are associated with high SUCRA values): an ideal treatment should be characterised by both high efficacy and high acceptability so should appear in the right upper corner of the ranking plot.
A dedicated funnel plot (comparison‐adjusted funnel plot) can be used to assess small‐study effects (which includes publication bias) (Chaimani 2013). This plot takes into consideration that included studies estimate effects for different comparisons: therefore, there cannot be a single reference line against which symmetry can be assessed. In the absence of small‐study effect the comparison‐adjusted funnel plot should be symmetrical around the zero line.
All statistical tests were two‐sided. Statistical analysis and graph generation was performed with Stata 11.2 (Stata 2017).
Contributions of authors
Simone Mocellin was the review contact person. Sandro Pasquali and SImone Mocellin co‐ordinated contributions from co‐authors and wrote the final draft of the review. Sandro Pasquali, Andreas V Hadjinicolaou and SImone Mocellin screened studies against eligibility criteria. Sandro Pasquali obtained data on ongoing and unpublished studies. Sandro Pasquali, Andreas V Hadjinicolaou and SImone Mocellin and appraised study quality. Sandro Pasquali, Andreas V Hadjinicolaou and SImone Mocellin extracted data and sought additional information from trial authors. Sandro Pasquali and Simone Mocellin entered data into RevMan. Sandro Pasquali and Simone Mocellin analysed and interpreted data. Sandro Pasquali and Simone Mocellin worked on the methods section. Vanna Chiarion Sileni and Carlo Riccardo Rossi contributed to the writing of the review and critical revision. Sandro Pasquali and Simone Mocellin drafted the clinical sections of the Background and responded to the clinical comments of the referees. Sandro Pasquali and Simone Mocellin responded to methodology and statistics comments from external peer referees. Simone Mocellin is the guarantor of the update.
Sources of support
Internal sources
University of Padova, Italy
External sources
-
The National Institute for Health Research (NIHR), UK
The NIHR, UK, is the largest single funder of the Cochrane Skin Group.
Declarations of interest
Sandro Pasquali: nothing to declare. Andreas V Hadjinicolaou: nothing to declare. Vanna Chiarion Sileni: nothing to declare. Carlo Riccardo Rossi: nothing to declare. Simone Mocellin: nothing to declare.
Edited (no change to conclusions)
References
References to studies included in this review
Agarwala 1999 {published data only}
- Agarwala SS, Ferri W, Gooding W, Kirkwood JM. A phase III randomized trial of dacarbazine and carboplatin with and without tamoxifen in the treatment of patients with metastatic melanoma. Cancer 1999;85(9):1979-84. [PMID: ] [PubMed] [Google Scholar]
Agarwala 2002 {published data only}
- Agarwala SS, Glaspy J, O'Day SJ, Mitchell M, Gutheil J, Whitman E, et al. Results from a randomized phase III study comparing combined treatment with histamine dihydrochloride plus interleukin-2 versus interleukin-2 alone in patients with metastatic melanoma. Journal of Clinical Oncology 2002;20(1):125-33. [PMID: ] [DOI] [PubMed] [Google Scholar]
Atkins 2008 {published data only}
- Atkins MB, Hsu J, Lee S, Cohen GI, Flaherty LE, Sosman JA, et al. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): a trial coordinated by the Eastern Cooperative Oncology Group. Journal of Clinical Oncology 2008;26(35):5748-54. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Atzpodien 2002 {published data only}
- Atzpodien J, Neuber K, Kamanabrou D, Fluck M, Brocker EB, Neumann C, et al. Combination chemotherapy with or without s.c. IL-2 and IFN-alpha: results of a prospectively randomized trial of the Cooperative Advanced Malignant Melanoma Chemoimmunotherapy Group (ACIMM). British Journal of Cancer 2002;86(2):179-84. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Avril 2004 {published data only}
- Avril MF, Aamdal S, Grob JJ, Hauschild A, Mohr P, Bonerandi JJ, et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a phase III study. Journal of Clinical Oncology 2004;22(6):1118-25. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bafaloukos 2005 {published data only}
- Bafaloukos D, Tsoutsos D, Kalofonos H, Chalkidou S, Panagiotou P, Linardou E, et al. Temozolomide and cisplatin versus temozolomide in patients with advanced melanoma: a randomized phase II study of the Hellenic Cooperative Oncology Group. Annals of Oncology 2005;16(6):950-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bajetta 1985 {published data only}
- Bajetta E, Buzzoni R, Viviani S, Vaglini M, Nava M, Bonadonna G. Prospective randomized trial in advanced malignant melanoma with cis-platinum, vindesine, and etoposide vs. cis-platinum, vindesine, and lomustine. American Journal of Clinical Oncology 1985;8(5):401-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bajetta 1994 {published data only}
- Bajetta E, Di Leo A, Zampino MG, Sertoli MR, Comella G, Barduagni M, et al. Multicenter randomized trial of dacarbazine alone or in combination with two different doses and schedules of interferon alfa-2a in the treatment of advanced melanoma. Journal of Clinical Oncology 1994;12(4):806-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bajetta 2006a {published data only}
- Bajetta E, Del Vecchio M, Nova P, Fusi A, Daponte A, Sertoli MR, et al. Multicenter phase III randomized trial of polychemotherapy (CVD regimen) versus the same chemotherapy (CT) plus subcutaneous interleukin-2 and interferon-alpha2b in metastatic melanoma. Annals of Oncology 2006;17(4):571-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Balch 1984 {published data only}
- Balch CM, Murray D, Presant C, Bartolucci AA. Ineffectiveness of adjuvant chemotherapy using DTIC and cyclophosphamide in patients with resectable metastatic melanoma. Surgery 1984;95(4):454-9. [PMID: ] [PubMed] [Google Scholar]
Bedikian 2006 {published data only}
- Bedikian AY, Millward M, Pehamberger H, Conry R, Gore M, Trefzer U, et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group. Journal of Clinical Oncology 2006;24(29):4738-45. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bedikian 2011 {published data only}
- Bedikian AY, DeConti RC, Conry R, Agarwala S, Papadopoulos N, Kim KB, et al. Phase 3 study of docosahexaenoic acid-paclitaxel versus dacarbazine in patients with metastatic malignant melanoma. Annals of Oncology 2011;22(4):787-93. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bellett 1976 {published data only}
- Bellett RE, Mastrangelo MJ, Laucius JF, Bodurtha AJ. Randomized prospective trial of DTIC (NSC-45388) alone versus BCNU (NSC-409962) plus vincristine (NSC-67574) in the treatment of metastatic malignant melanoma. Cancer Treatment Reports 1976;60(5):595-600. [PMID: ] [PubMed] [Google Scholar]
Beretta 1976 {published data only}
- Beretta G, Bonadonna G, Cascinelli N, Morabito A, Veronesi U. Comparative evaluation of three combination regimens for advanced malignant melanoma: results of an international cooperative study. Cancer Treatment Reports 1976;60(1):33-40. [PMID: ] [PubMed] [Google Scholar]
Carter 1975 {published data only}
- Carter RD, Krementz ET, Hill GJ. DTIC and combination therapy for metastatic melanoma: a COG cooperative study. Proceedings of the American Association for Cancer Research 1975;16(66):NO. [Google Scholar]
Carvajal 2014 {published data only}
- Carvajal RD, Wong MK, Thompson JA, Gordon MS, Lewis KD, Pavlick AC et al. A phase 2 randomised study of ramucirumab (IMC-1121B) with or without dacarbazine inpatients with metastatic melanoma. European Journal of Cancer 2014;50(12):2099-107. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chapman 1999 {published data only}
- Chapman PB, Einhorn LH, Meyers ML, Saxman S, Destro AN, Panageas KS, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. Journal of Clinical Oncology 1999;17(9):2745-51. [PMID: ] [DOI] [PubMed] [Google Scholar]
Chauvergne 1982 {published data only}
- Chauvergne J, Bui NB, Cappelaere P, Gary-Bobo J, Guerrin J, Armand JP, et al. [Chemotherapy in advanced malignant melanoma. Results of a controlled trial comparing a combination of dacarbazine (DTIC) and detorubicin with dacarbazine alone] [Chimiotherapie des melanomes malins evolues. Resultats d'un essai controle comparant l'association de detorubicine et de dacarbazine (DTIC) a la dacarbazine seule.]. La Semaine des Hopitaux 1982;58(46):2697-701. [PMID: ] [PubMed] [Google Scholar]
Chiarion Sileni 2001 {published data only}
- Chiarion Sileni V, Nortilli R, Aversa SM, Paccagnella A, Medici M, Corti L, et al. Phase II randomized study of dacarbazine, carmustine, cisplatin and tamoxifen versus dacarbazine alone in advanced melanoma patients. Melanoma Research 2001;11(2):189-96. [PMID: ] [DOI] [PubMed] [Google Scholar]
Chiarion‐Sileni 2011 {published data only}
- Chiarion-Sileni V, Guida M, Ridolfi L, Romanini A, Del Bianco P, Pigozzo J, et al. Central nervous system failure in melanoma patients: results of a randomised, multicentre phase 3 study of temozolomide- and dacarbazine- based regimens. British Journal of Cancer 2011;104(12):1816-21. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Clunie 1980 {published data only}
- Clunie GJ, Gough IR, Dury M, Furnival CM, Bolton PM. A trial of imidazole carboxamide and corynebacterium parvum in disseminated melanoma: clinical and immunologic results. Cancer 1980;46(3):475-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cocconi 1992 {published data only}
- Cocconi G, Bella M, Calabresi F, Tonato M, Canaletti R, Boni C, et al. Treatment of metastatic malignant melanoma with dacarbazine plus tamoxifen. New England Journal of Medicine 1992;327(8):516-23. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cocconi 2003 {published data only}
- Cocconi G, Passalacqua R, Foladore S, Carlini P, Acito L, Maiello E, et al. Treatment of metastatic malignant melanoma with dacarbazine plus tamoxifen, or vindesine plus tamoxifen: a prospective randomized study. Melanoma Research 2003;13(1):73-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Costanza 1972 {published data only}
- Costanza ME, Nathanson L, Lenhard R, Wolter J, Colsky J, Oberfield RA, et al. Therapy of malignant melanoma with an imidazole carboxamide and bis-chloroethyl nitrosourea. Cancer 1972;30(6):1457-61. [PMID: ] [DOI] [PubMed] [Google Scholar]
Costanza 1977 {published data only}
- Costanza ME, Nathanson L, Schoenfeld D, Wolter J, Colsky J, Regelson W, et al. Results with methyl-CCNU and DTIC in metastatic melanoma. Cancer 1977;40(3):1010-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Costanzi 1982 {published data only}
- Costanzi JJ, Al-Sarraf M, Groppe C, Bottomley R, Fabian C, Neidhart J, et al. Combination chemotherapy plus BCG in the treatment of disseminated malignant melanoma: a Southwest Oncology Group Study. Medical and Pediatric Oncology 1982;10(3):251-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cui 2013 {published data only}
- Cui C, Mao L, Chi Z, Si L, Sheng X, Kong Y, et al. A phase II, randomized, double-blind, placebo-controlled multicenter trial of Endostar in patients with metastatic melanoma. Molecular Therapy 2013;21(7):1456-63. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Danson 2003 {published data only}
- Danson S, Lorigan P, Arance A, Clamp A, Ranson M, Hodgetts J, et al. Randomized phase II study of temozolomide given every 8 hours or daily with either interferon alfa-2b or thalidomide in metastatic malignant melanoma. Journal of Clinical Oncology 2003;21(13):2551-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Daponte 2013 {published data only}
- Daponte A, Signoriello S, Maiorino L, Massidda B, Simeone E, Grimaldi AM, et al. Phase III randomized study of fotemustine and dacarbazine versus dacarbazine with or without interferon-alpha in advanced malignant melanoma. Journal of Translational Medicine 2013;11:38. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dorval 1999 {published data only}
- Dorval T, Negrier S, Chevreau C, Avril MF, Baume D, Cupissol D, et al. Randomized trial of treatment with cisplatin and interleukin-2 either alone or in combination with interferon-alpha-2a in patients with metastatic melanoma: a Federation Nationale des Centres de Lutte Contre le Cancer Multicenter, parallel study. Cancer 1999;85(5):1060-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Dummer 2006 {published data only}
- Dummer R, Garbe C, Thompson JA, Eggermont AM, Yoo K, Maier T, et al. Randomized dose-escalation study evaluating peginterferon alfa-2a in patients with metastatic malignant melanoma. Journal of Clinical Oncology 2006;24(7):1188-94. [PMID: ] [DOI] [PubMed] [Google Scholar]
Eigentler 2008 {published data only}
- Eigentler TK, Radny P, Hauschild A, Gutzmer R, Linse R, Pfohler C, et al. Adjuvant treatment with vindesine in comparison to observation alone in patients with metastasized melanoma after complete metastasectomy: a randomized multicenter trial of the German Dermatologic Cooperative Oncology Group. Melanoma Research 2008;18(5):353-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Eisen 2010 {published data only}
- Eisen T, Trefzer U, Hamilton A, Hersey P, Millward M, Knight RD, et al. Results of a multicenter, randomized, double-blind phase 2/3 study of lenalidomide in the treatment of pretreated relapsed or refractory metastatic malignant melanoma. Cancer 2010;116(1):146-54. [PMID: ] [DOI] [PubMed] [Google Scholar]
Eton 2002 {published data only}
- Eton O, Legha SS, Bedikian AY, Lee JJ, Buzaid AC, Hodges C, et al. Sequential biochemotherapy versus chemotherapy for metastatic melanoma: results from a phase III randomized trial. Journal of Clinical Oncology 2002;20(8):2045-52. [PMID: ] [DOI] [PubMed] [Google Scholar]
Falkson 1991 {published data only}
- Falkson CI, Falkson G, Falkson HC. Improved results with the addition of interferon alfa-2b to dacarbazine in the treatment of patients with metastatic malignant melanoma. Journal of Clinical Oncology 1991;9(8):1403-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Falkson 1995 {published data only}
- Falkson CI. Experience with interferon alpha 2b combined with dacarbazine in the treatment of metastatic malignant melanoma. Medical Oncology 1995;12(1):35-40. [PMID: ] [DOI] [PubMed] [Google Scholar]
Falkson 1998 {published data only}
- Falkson CI, Ibrahim J, Kirkwood JM, Coates AS, Atkins MB, Blum RH. Phase III trial of dacarbazine versus dacarbazine with interferon alpha-2b versus dacarbazine with tamoxifen versus dacarbazine with interferon alpha-2b and tamoxifen in patients with metastatic malignant melanoma: an Eastern Cooperative Oncology Group study. Journal of Clinical Oncology 1998;16(5):1743-51. [PMID: ] [DOI] [PubMed] [Google Scholar]
Flaherty 2001 {published data only}
- Flaherty LE, Atkins M, Sosman J, Weiss G, Clark JI, Margolin K, et al. Outpatient biochemotherapy with interleukin-2 and interferon alfa-2b in patients with metastatic malignant melanoma: results of two phase II cytokine working group trials. Journal of Clinical Oncology 2001;19(13):3194-202. [PMID: ] [DOI] [PubMed] [Google Scholar]
Flaherty 2012a {published data only}
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. New England Journal of Medicine 2012;367(18):1694-703. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Flaherty 2012b {published data only}
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. New England Journal of Medicine 2012;367(2):107-14. [PMID: ] [DOI] [PubMed] [Google Scholar]
Flaherty 2013a {published data only}
- Flaherty KT, Lee SJ, Zhao F, Schuchter LM, Flaherty L, Kefford R, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. Journal of Clinical Oncology 2013;31(3):373-9. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Glaspy 2009 {published data only}
- Glaspy J, Atkins MB, Richards JM, Agarwala SS, O'Day S, Knight RD, et al. Results of a multicenter, randomized, double-blind, dose-evaluating phase 2/3 study of lenalidomide in the treatment of metastatic malignant melanoma. Cancer 2009;115(22):5228-36. [PMID: ] [DOI] [PubMed] [Google Scholar]
Glover 2003 {published data only}
- Glover D, Ibrahim J, Kirkwood J, Glick J, Karp D, Stewart J, et al. Phase II randomized trial of cisplatin and WR-2721 versus cisplatin alone for metastatic melanoma: an Eastern Cooperative Oncology Group Study (E1686). Melanoma Research 2003;13(6):619-26. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gorbonova 2000 {published data only}
- Gorbonova VA, Egorov GN, Perevodchikova NI, Orel NF. Combined chemotherapy with or without interferon alpha N1 (IFN) for advanced malignant melanoma--a randomized pilot phase III study. Gan to Kagaku Ryoho. Cancer & Chemotherapy 2000;27(Suppl 2):310-4. [PMID: ] [PubMed] [Google Scholar]
Gough 1978 {published data only}
- Gough IR, Bolton PM, Clunie GJ, Burnett W. Chemoimmunotherapy in disseminated melanoma and colorectal carcinoma. Australian and New Zealand Journal of Surgery 1978;48(3):296-300. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gupta 2014 {published data only}
- Gupta A, Love S, Schuh A, Shanyinde M, Larkin JM, Plummer R, et al. DOC-MEK: a double-blind randomized phase II trial of docetaxel with or without selumetinib in wild-type BRAF advanced melanoma. Annals of Oncology 2014;25(5):968-74. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hamid 2014 {published data only}
- Hamid O, Ilaria R, Garbe C, Wolter P, Maio M, Hutson TE, et al. A randomized, open-label clinical trial of tasisulam sodium versus paclitaxel as second-line treatment in patients with metastatic melanoma. Cancer 2014;120(13):2016-24. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hauschild 2001 {published data only}
- Hauschild A, Garbe C, Stolz W, Ellwanger U, Seiter S, Dummer R, et al. Dacarbazine and interferon alpha with or without interleukin 2 in metastatic melanoma: a randomized phase III multicentre trial of the Dermatologic Cooperative Oncology Group (DeCOG). British Journal of Cancer 2001;84(8):1036-42. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hauschild 2009a {published data only}
- Hauschild A, Agarwala SS, Trefzer U, Hogg D, Robert C, Hersey P, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. Journal of Clinical Oncology 2009;27(17):2823-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hauschild 2012 {published data only}
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380(9839):358-65. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hersh 2015 {published data only}
- Hersh EM, Del Vecchio M, Brown MP, Kefford R, Loquai C, Testori A, et al. A randomized, controlled phase III trial of nab-Paclitaxel versus dacarbazine in chemotherapy-naive patients with metastatic melanoma. Annals of Oncology 2015;26(11):2267-74. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hodi 2010a {published data only}
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New England Journal of Medicine 2010;363(8):711-23. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hodi 2014 {published data only}
- Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA 2014;312(17):1744-53. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hofmann 2011 {published data only}
- Hofmann MA, Hauschild A, Mohr P, Garbe C, Weichenthal M, Trefzer U, et al. Prospective evaluation of supportive care with or without CVD chemotherapy as a second-line treatment in advanced melanoma by patient's choice: a multicentre Dermatologic Cooperative Oncology Group trial. Melanoma Research 2011;21(6):516-23. [PMID: ] [DOI] [PubMed] [Google Scholar]
Jelic 2002 {published data only}
- Jelic S, Babovic N, Kovcin V, Milicevic N, Milanovic N, Popov I, et al. Comparison of the efficacy of two different dosage dacarbazine-based regimens and two regimens without dacarbazine in metastatic melanoma: a single-centre randomized four-arm study. Melanoma Research 2002;12(1):91-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Johnston 1998 {published data only}
- Johnston SR, Constenla DO, Moore J, Atkinson H, A'Hern RP, Dadian G, et al. Randomized phase II trial of BCDT [carmustine (BCNU), cisplatin, dacarbazine (DTIC) and tamoxifen] with or without interferon alpha (IFN-alpha) and interleukin (IL-2) in patients with metastatic melanoma. British Journal of Cancer 1998;77(8):1280-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kaufmann 2005 {published data only}
- Kaufmann R, Spieth K, Leiter U, Mauch C, den Driesch P, Vogt T, et al. Temozolomide in combination with interferon-alfa versus temozolomide alone in patients with advanced metastatic melanoma: a randomized, phase III, multicenter study from the Dermatologic Cooperative Oncology Group. Journal of Clinical Oncology 2005;23(35):9001-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kefford 2010 {published data only}
- Kefford RF, Clingan PR, Brady B, Ballmer A, Morganti A, Hersey P. A randomized, double-blind, placebo-controlled study of high-dose bosentan in patients with stage IV metastatic melanoma receiving first-line dacarbazine chemotherapy. Molecular Cancer 2010;9:69. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Keilholz 1997 {published data only}
- Keilholz U, Goey SH, Punt CJ, Proebstle TM, Salzmann R, Scheibenbogen C, et al. Interferon alfa-2a and interleukin-2 with or without cisplatin in metastatic melanoma: a randomized trial of the European Organization for Research and Treatment of Cancer Melanoma Cooperative Group. Journal of Clinical Oncology 1997;15(7):2579-88. [PMID: ] [DOI] [PubMed] [Google Scholar]
Keilholz 2005 {published data only}
- Keilholz U, Punt CJ, Gore M, Kruit W, Patel P, Lienard D, et al. Dacarbazine, cisplatin, and interferon-alfa-2b with or without interleukin-2 in metastatic melanoma: a randomized phase III trial (18951) of the European Organisation for Research and Treatment of Cancer Melanoma Group. Journal of Clinical Oncology 2005;23(27):6747-55. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kim 2012 {published data only}
- Kim KB, Sosman JA, Fruehauf JP, Linette GP, Markovic SN, McDermott DF, et al. BEAM: a randomized phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma. Journal of Clinical Oncology 2012;30(1):34-41. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kirkwood 1990 {published data only}
- Kirkwood JM, Ernstoff MS, Giuliano A, Gams R, Robinson WA, Costanzi J, et al. Interferon alpha-2a and dacarbazine in melanoma. Journal of the National Cancer Institute 1990;82(12):1062-3. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kogoniia 1981 {published data only}
- Kogoniia LM, Moroz LV, Perevodchikova NI, Platinskii LV, Borisov AI. Comparison of the efficacy of imidazole-carboxamide and of a combination of nitrosomethylurea, vincristine and dactinomycin in disseminated melanoma [Sravnenie effiktivnosti imidazol-karboksamida i kombinatsii nitrozometilmocheviny, vinkristina, daktinomitsina pri disseminirovannoi melanome.]. Voprosy Onkologii 1981;27(4):16-21. [PMID: ] [PubMed] [Google Scholar]
Kokoschka 1978 {published data only}
- Kokoschka EM, Luger T, Micksche M. Immuno-chemotherapy in patients with disseminated metastasizing stage III melanoma. Randomized study with methyl-CCNU versus C. parvum plus methyl-CCNU [Immuno-Chemotherapie bei Patienten mit disseminiert metastasierendem Melanom Stadium III Randomisierte Studie mit Methyl-CCNU versus C. parvum plus Methyl-CCNU]. Onkologie 1978;1(3):98-103. [PMID: ] [DOI] [PubMed] [Google Scholar]
Larkin 2014 {published data only}
- Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. New England Journal of Medicine 2014;371(20):1867-76. [PMID: ] [DOI] [PubMed] [Google Scholar]
Larkin 2015 {published data only}
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New England Journal of Medicine 2015;373(1):23-34. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lawson 2015 {published data only}
- Lawson DH, Lee S, Zhao F, Tarhini AA, Margolin KA, Ernstoff MS, et al. Randomized, placebo-controlled, phase III trial of yeast-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) versus peptide vaccination versus GM-CSF plus peptide vaccination versus placebo in patients with no evidence of disease after complete surgical resection of locally advanced and/or stage IV melanoma: A trial of the Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group (E4697). Journal of Clinical Oncology 2015;33(34):4066-76. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Legha 1996 {published data only}
- Legha SS, Ring S, Bedikian A, Plager C, Eton O, Buzaid AC, et al. Treatment of metastatic melanoma with combined chemotherapy containing cisplatin, vinblastine and dacarbazine (CVD) and biotherapy using interleukin-2 and interferon-alpha. Annals of Oncology 1996;7(8):827-35. [PMID: ] [DOI] [PubMed] [Google Scholar]
Long 2015 {published data only}
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015;386(9992):444-51. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lopez 1984 {published data only}
- Lopez M, Perno CF, Di Lauro L, Papaldo P, Ganzina F, Barduagni A. Controlled study of DTIC versus DTIC plus epirubicin in metastatic malignant melanoma. Investigational New Drugs 1984;2(3):319-22. [PMID: ] [DOI] [PubMed] [Google Scholar]
Luikart 1984 {published data only}
- Luikart SD, Kennealey GT, Kirkwood JM. Randomized phase III trial of vinblastine, bleomycin, and cis-dichlorodiammine-platinum versus dacarbazine in malignant melanoma. Journal of Clinical Oncology 1984;2(3):164-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Maio 2010 {published data only}
- Maio M, Mackiewicz A, Testori A, Trefzer U, Ferraresi V, Jassem J, et al. Large randomized study of thymosin alpha 1, interferon alfa, or both in combination with dacarbazine in patients with metastatic melanoma. Journal of Clinical Oncology 2010;28(10):1780-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mastrangelo 1979 {published data only}
- Mastrangelo MJ, Bellet RE, Berd D. A phase III comparison of methyl-CCNU + vincristine with or without BCG + allogeneic tumor cells in metastatic melanoma. Cancer Immunology, Immunotherapy 1979;6(4):231-6. [EMBASE: 1980000708] [Mastrangelo 1979] [Google Scholar]
McArthur 2014 {published data only}
- McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncology 2014;15(3):323-32. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
McDermott 2008 {published data only}
- McDermott DF, Sosman JA, Gonzalez R, Hodi FS, Linette GP, Richards J, et al. Double-blind randomized phase II study of the combination of sorafenib and dacarbazine in patients with advanced melanoma: a report from the 11715 Study Group. Journal of Clinical Oncology 2008;26(13):2178-85. [PMID: ] [DOI] [PubMed] [Google Scholar]
Middleton 2000 {published data only}
- Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. Journal of Clinical Oncology 2000;18(1):158-66. [PMID: ] [DOI] [PubMed] [Google Scholar]
Middleton 2007 {published data only}
- Middleton M, Hauschild A, Thomson D, Anderson R, Burdette-Radoux S, Gehlsen K, et al. Results of a multicenter randomized study to evaluate the safety and efficacy of combined immunotherapy with interleukin-2, interferon-{alpha}2b and histamine dihydrochloride versus dacarbazine in patients with stage IV melanoma. Annals of Oncology 2007;18(10):1691-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Middleton 2015 {published data only}
- Middleton MR, Friedlander P, Hamid O, Daud A, Plummer R, Falotico N, et al. Randomized phase II study evaluating veliparib (ABT-888) with temozolomide in patients with metastatic melanoma. Annals of Oncology 2015;26(10):2173-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Miller 1989 {published data only}
- Miller RL, Steis RG, Clark JW, Smith JW 2nd, Crum E, McKnight JE, et al. Randomized trial of recombinant alpha 2b-interferon with or without indomethacin in patients with metastatic malignant melanoma. Cancer Research 1989;49(7):1871-6. [PMID: ] [PubMed] [Google Scholar]
Moon 1975 {published data only}
- Moon JH, Gailani S, Cooper MR, Hayes DM, Rege VB, Blom J, et al. Comparison of the combination of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) and vincristine with two dose schedules of 5-(3,3-dimethyl-1-triazino) imidazole 4-carboxamide (DTIC) in the treatment of disseminated malignant melanoma. Cancer 1975;35(2):368-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Newlands 1976 {published data only}
- Newlands ES, Oon CJ, Roberts JT, Elliott P, Mould RF, Topham C, et al. Clinical trial of combination chemotherapy and specific active immunotherapy in disseminated melanoma. British Journal of Cancer 1976;34(2):174-9. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
O'Day 2009 {published data only}
- O'Day S, Gonzalez R, Lawson D, Weber R, Hutchins L, Anderson C, et al. Phase II, randomized, controlled, double-blinded trial of weekly elesclomol plus paclitaxel versus paclitaxel alone for stage IV metastatic melanoma. Journal of Clinical Oncology 2009;27(32):5452-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
O'Day 2011 {published data only}
- O'Day S, Pavlick A, Loquai C, Lawson D, Gutzmer R, Richards J, et al. A randomised, phase II study of intetumumab, an anti-alphav-integrin mAb, alone and with dacarbazine in stage IV melanoma. British Journal of Cancer 2011;105(3):346-52. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
O'Day 2013 {published data only}
- O'Day SJ, Eggermont AM, Chiarion-Sileni V, Kefford R, Grob JJ, Mortier L, et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. Journal of Clinical Oncology 2013;31(9):1211-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Patel 2011 {published data only}
- Patel PM, Suciu S, Mortier L, Kruit WH, Robert C, Schadendorf D, et al. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). European Journal of Cancer 2011;47(10):1476-83. [PMID: ] [DOI] [PubMed] [Google Scholar]
Postow 2015 {published data only}
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. New England Journal of Medicine 2015;372(21):2006-17. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Presant 1979 {published data only}
- Presant CA, Bartolucci AA, Smalley RV, Vogler WR. Cyclophosphamide plus 5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamide (DTIC) with or without Corynebacterium parvum in metastatic malignant melanoma. Cancer 1979;44(3):899-905. [PMID: ] [DOI] [PubMed] [Google Scholar]
Presant 1982 {published data only}
- Presant CA, Bartolucci AA, Balch C, Troner M. A randomized comparison of cyclophosphamide, DTIC with or without piperazinedione in metastatic malignant melanoma. Cancer 1982;49(7):1355-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Punt 2006 {published data only}
- Punt CJ, Suciu S, Gore MA, Koller J, Kruit WH, Thomas J, et al. Chemoimmunotherapy with dacarbazine, cisplatin, interferon-alpha2b and interleukin-2 versus two cycles of dacarbazine followed by chemoimmunotherapy in patients with metastatic melanoma: a randomised phase II study of the European Organization for Research and Treatment of Cancer Melanoma Group. European Journal of Cancer 2006;42(17):2991-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ramseur 1978 {published data only}
- Ramseur WL, Richards F 2nd, Muss HB, Rhyne L, Cooper MR, White DR, et al. Chemoimmunotherapy for disseminated malignant melanoma: a prospective randomized study. Cancer Treatment Reports 1978;62(7):1085-7. [PMID: ] [PubMed] [Google Scholar]
Ranson 2007 {published data only}
- Ranson M, Hersey P, Thompson D, Beith J, McArthur GA, Haydon A, et al. Randomized trial of the combination of lomeguatrib and temozolomide compared with temozolomide alone in chemotherapy naive patients with metastatic cutaneous melanoma. Journal of Clinical Oncology 2007;25(18):2540-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Reichle 2007 {published data only}
- Reichle A, Vogt T, Coras B, Terheyden P, Neuber K, Trefzer U, et al. Targeted combined anti-inflammatory and angiostatic therapy in advanced melanoma: a randomized phase II trial. Melanoma Research 2007;17(6):360-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ribas 2013 {published data only}
- Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. Journal of Clinical Oncology 2013;31(5):616-22. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ribas 2015 {published data only}
- Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncology 2015;16(8):908-18. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Richtig 2004 {published data only}
- Richtig E, Hofmann-Wellenhof R, Pehamberger H, Forstinger Ch, Wolff K, Mischer P, et al. Temozolomide and interferon alpha 2b in metastatic melanoma stage IV. British Journal of Dermatology 2004;151(1):91-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ridolfi 2002a {published data only}
- Ridolfi R, Chiarion-Sileni V, Guida M, Romanini A, Labianca R, Freschi A, et al. Cisplatin, dacarbazine with or without subcutaneous interleukin-2, and interferon alpha-2b in advanced melanoma outpatients: results from an Italian multicenter phase III randomized clinical trial. Journal of Clinical Oncology 2002;20(6):1600-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ringborg 1989 {published data only}
- Ringborg U, Rudenstam CM, Hansson J, Hafstrom L, Stenstam B, Strander H. Dacarbazine versus dacarbazine-vindesine in disseminated malignant melanoma: a randomized phase II study. Medical Oncology and Tumor Pharmacotherapy 1989;6(4):285-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Robert 2011 {published data only}
- Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. New England Journal of Medicine 2011;364(26):2517-26. [PMID: ] [DOI] [PubMed] [Google Scholar]
Robert 2013 {published data only}
- Robert C, Dummer R, Gutzmer R, Lorigan P, Kim KB, Nyakas M, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncology 2013;14(8):733-40. [PMID: ] [DOI] [PubMed] [Google Scholar]
Robert 2015 {published data only}
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. New England Journal of Medicine 2015;372(1):30-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Robert 2015a {published data only}
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. New England Journal of Medicine 2015;372(4):320-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Robert 2015b {published data only}
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. New England Journal of Medicine 2015;372(26):2521-32. [PMID: 25891173] [DOI] [PubMed] [Google Scholar]
Robidoux 1982 {published data only}
- Robidoux A, Gutterman JU, Bodey GP Sr, Hersh EM. Actinomycin-D plus 5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamine (DTIC) with or without intravenous Corynebacterium parvum in metastatic malignant melanoma. Cancer 1982;49(11):2246-51. [PMID: ] [DOI] [PubMed] [Google Scholar]
Rosenberg 1999 {published data only}
- Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, et al. Prospective randomized trial of the treatment of patients with metastatic melanoma using chemotherapy with cisplatin, dacarbazine, and tamoxifen alone or in combination with interleukin-2 and interferon alfa-2b. Journal of Clinical Oncology 1999;17(3):968-75. [PMID: ] [DOI] [PubMed] [Google Scholar]
Rusthoven 1996 {published data only}
- Rusthoven JJ, Quirt IC, Iscoe NA, McCulloch PB, James KW, Lohmann RC, et al. Randomized, double-blind, placebo-controlled trial comparing the response rates of carmustine, dacarbazine, and cisplatin with and without tamoxifen in patients with metastatic melanoma. National Cancer Institute of Canada Clinical Trials Group. Journal of Clinical Oncology 1996;14(7):2083-90. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schadendorf 2006 {published data only}
- Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Brocker EB, et al. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Annals of Oncology 2006;17(4):563-70. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schwartzentruber 2011a {published data only}
- Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. New England Journal of Medicine 2011;364(22):2119-27. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sertoli 1999 {published data only}
- Sertoli MR, Queirolo P, Bajetta E, Del Vecchio M, Comella G, Barduagni L, et al. Multi-institutional phase II randomized trial of integrated therapy with cisplatin, dacarbazine, vindesine, subcutaneous interleukin-2, interferon alpha2a and tamoxifen in metastatic melanoma. BREMIM (Biological Response Modifiers in Melanoma). Melanoma Research 1999;9(5):503-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Sparano 1993 {published data only}
- Sparano JA, Fisher RI, Sunderland M, Margolin K, Ernest ML, Sznol M, et al. Randomized phase III trial of treatment with high-dose interleukin-2 either alone or in combination with interferon alfa-2a in patients with advanced melanoma. Journal of Clinical Oncology 1993;11(10):1969-77. [PMID: ] [DOI] [PubMed] [Google Scholar]
Testori 2008 {published data only}
- Testori A, Richards J, Whitman E, Mann GB, Lutzky J, Camacho L, et al. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician's choice of treatment for stage IV melanoma: the C-100-21 Study Group. Journal of Clinical Oncology 2008;26(6):955-62. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thatcher 1986 {published data only}
- Thatcher N, Wagstaff J, Mene A, Smith D, Orton C, Craig P. Corynebacterium parvum followed by chemotherapy (actinomycin D and DTIC) compared with chemotherapy alone for metastatic malignant melanoma. European Journal of Cancer & Clinical Oncology 1986;22(8):1009-14. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thomson 1993 {published data only}
- Thomson DB, Adena M, McLeod GR, Hersey P, Gill PG, Coates AS, et al. Interferon-alpha 2a does not improve response or survival when combined with dacarbazine in metastatic malignant melanoma: results of a multi-institutional Australian randomized trial. Melanoma Research 1993;3(2):133-8. [PMID: ] [PubMed] [Google Scholar]
Veronesi 1984 {published data only}
- WHO. Controlled study with imidazole carboxamide (DTIC), DTIC + bacillus Calmette-Guerin (BCG), and DTIC + corynebacterium parvum in advanced malignant melanoma. W.H.O. Collaborating Centres for Evaluation of Methods of Diagnosis and Treatment of Melanoma. Tumori 1984;70(1):41-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Verschraegen 1993 {published data only}
- Verschraegen CF, Legha SS, Hersh EM, Plager C, Papadopoulos N, Burgess MA. Phase II study of vindesine and dacarbazine with or without non-specific stimulation of the immune system in patients with metastatic melanoma. European Journal of Cancer 1993;29A(5):708-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Vorobiof 1994 {published data only}
- Vorobiof DA, Bezwoda WR. A randomised trial of vindesine plus interferon-alpha 2b compared with interferon-alpha 2b or vindesine alone in the treatment of advanced malignant melanoma. European Journal of Cancer 1994;30A(6):797-800. [PMID: ] [DOI] [PubMed] [Google Scholar]
Vuoristo 2005 {published data only}
- Vuoristo MS, Hahka-Kemppinen M, Parvinen LM, Pyrhonen S, Seppa H, Korpela M, et al. Randomized trial of dacarbazine versus bleomycin, vincristine, lomustine and dacarbazine (BOLD) chemotherapy combined with natural or recombinant interferon-alpha in patients with advanced melanoma. Melanoma Research 2005;15(4):291-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Weber 2009 {published data only}
- Weber JS, Zarour H, Redman B, Trefzer U, O'Day S, den Eertwegh AJ, et al. Randomized phase 2/3 trial of CpG oligodeoxynucleotide PF-3512676 alone or with dacarbazine for patients with unresectable stage III and IV melanoma. Cancer 2009;115(17):3944-54. [PMID: ] [DOI] [PubMed] [Google Scholar]
Weber 2015 {published data only}
- Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncology 2015;16(4):375-84. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wittes 1978 {published data only}
- Wittes RE, Wittes JT, Golbey RB. Combination chemotherapy in metastatic malignant melanoma: a randomized study of three DTIC-containing combination. Cancer 1978;41(2):415-21. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wolchok 2010 {published data only}
- Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncology 2010;11(2):155-64. [PMID: ] [DOI] [PubMed] [Google Scholar]
Young 2001 {published data only}
- Young AM, Marsden J, Goodman A, Burton A, Dunn JA. Prospective randomized comparison of dacarbazine (DTIC) versus DTIC plus interferon-alpha (IFN-alpha) in metastatic melanoma. Clinical Oncology 2001;13(6):458-65. [PMID: ] [DOI] [PubMed] [Google Scholar]
Zimpfer‐Rechner 2003 {published data only}
- Zimpfer-Rechner C, Hofmann U, Figl R, Becker JC, Trefzer U, Keller I, et al. Randomized phase II study of weekly paclitaxel versus paclitaxel and carboplatin as second-line therapy in disseminated melanoma: a multicentre trial of the Dermatologic Co-operative Oncology Group (DeCOG). Melanoma Research 2003;13(5):531-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Asemissen 2005 {published data only}
- Asemissen AM, Scheibenbogen C, Letsch A, Hellstrand K, Thoren F, Gehlsen K, et al. Addition of histamine to interleukin 2 treatment augments type 1 T-cell responses in patients with melanoma in vivo: immunologic results from a randomized clinical trial of interleukin 2 with or without histamine (MP 104). Clinical Cancer Research 2005;11(1):290-7. [PMID: ] [PubMed] [Google Scholar]
Atzpodien 1995 {published data only}
- Atzpodien J, Lopez Hanninen E, Kirchner H, Franzke A, Korfer A, Volkenandt M, et al. Chemoimmunotherapy of advanced malignant melanoma: sequential administration of subcutaneous interleukin-2 and interferon-alpha after intravenous dacarbazine and carboplatin or intravenous dacarbazine, cisplatin, carmustine and tamoxifen. European Journal of Cancer 1995;31A(6):876-81. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bleehen 1995 {published data only}
- Bleehen NM, Newlands ES, Lee SM, Thatcher N, Selby P, Calvert AH, et al. Cancer Research Campaign phase II trial of temozolomide in metastatic melanoma. Journal of Clinical Oncology 1995;13(4):910-3. [PMID: ] [DOI] [PubMed] [Google Scholar]
Buchbinder 2015 {published data only}
- Buchbinder EI, Sosman JA, Lawrence DP, McDermott DF, Ramaiya NH, Van den Abbeele AD, et al. Phase 2 study of sunitinib in patients with metastatic mucosal or acral melanoma. Cancer 2015;121(22):4007-15. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bukowski 1983 {published data only}
- Bukowski RM, Deodhar S, Hewlett JS, Greenstreet R. Randomized controlled trial of transfer factor in Stage II malignant melanoma. Cancer 1983;51(2):269-72. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cashin 2008 {published data only}
- Cashin RP, Lui P, Machado M, Hemels ME, Corey-Lisle PK, Einarson TR. Advanced cutaneous malignant melanoma: a systematic review of economic and quality-of-life studies. Value in Health 2008;11(2):259-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cormier 1997 {published data only}
- Cormier JN, Hurst R, Vasselli J, Lee D, Kim CJ, McKee M, et al. A prospective randomized evaluation of the prophylactic use of low-dose dopamine in cancer patients receiving interleukin-2. Journal of Immunotherapy 1997;20(4):292-300. [PMID: ] [DOI] [PubMed] [Google Scholar]
Curl 2014 {published data only}
- Curl P, Vujic I, 't Veer LJ, Ortiz-Urda S, Kahn JG. Cost-effectiveness of treatment strategies for BRAF mutated metastatic melanoma. PLoS One 2014;9(9):e107255. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Downey 2007 {published data only}
- Downey SG, Klapper JA, Smith FO, Yang JC, Sherry RM, Royal RE, et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade. Clinical Cancer Research 2007;13(22 Pt 1):6681-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hill 1984 {published data only}
- Hill GJ 2nd, Krementz ET, Hill HZ. Dimethyl triazeno imidazole carboxamide and combination therapy for melanoma. IV. Late results after complete response to chemotherapy (Central Oncology Group protocols 7130, 7131, and 7131A). Cancer 1984;53(6):1299-305. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hughes 2016 {published data only}
- Hughes MS, Zager J, Faries M, Alexander HR, Royal RE, Wood B, et al. Results of a randomized controlled multicenter phase III trial of percutaneous hepatic perfusion compared with best available care for patients with melanoma liver metastases. Annals of Surgical Oncology 2016;23(4):1309-19. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hwu 2009 {published data only}
- Hwu P. Promising results from phase III clinical trial of a peptide vaccine for advanced melanoma. Immunotherapy 2009;1(4):521. [Google Scholar]
Kaufman 2010 {published data only}
- Kaufman HL, Bines SD. OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncology 2010;6(6):941-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kleeberg 1982 {published data only}
- Kleeberg UR, Mulder JH, Rumke P, Thomas D, Rozencweig M. N-(phosphonacetyl)-L-aspartate (PALA) in advanced malignant melanoma: a phase II trial of the EORTC Malignant Melanoma Cooperative Group. European Journal of Cancer & Clinical Oncology 1982;18(8):723-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lattanzi 1995 {published data only}
- Lattanzi SC, Tosteson T, Chertoff J, Maurer LH, O'Donnell J, LeMarbre PJ, et al. Dacarbazine, cisplatin and carmustine, with or without tamoxifen, for metastatic melanoma: 5-year follow-up. Melanoma Research 1995;5(5):365-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
McDermott 2013 {published data only}
- McDermott D, Haanen J, Chen TT, Lorigan P, O'Day S. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20). Annals of Oncology 2013;24(10):2694-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mornex 2003 {published data only}
- Mornex F, Thomas L, Mohr P, Hauschild A, Delaunay MM, Lesimple T, et al. A prospective randomized multicentre phase III trial of fotemustine plus whole brain irradiation versus fotemustine alone in cerebral metastases of malignant melanoma. Melanoma Research 2003;13(1):97-103. [PMID: ] [DOI] [PubMed] [Google Scholar]
Quirt 1983 {published data only}
- Quirt IC, De Boer G, Kersey PA, Baker MA, Bodurtha AJ, Norvell ST, et al. Randomized controlled trial of adjuvant chemoimmunotherapy with DTIC and BCG after complete excision of primary melanoma with a poor prognosis or melanoma metastases. Canadian Medical Association Journal 1983;128(8):929-33. [PMID: 6339024 ] [PMC free article] [PubMed] [Google Scholar]
Richards 1999 {published data only}
- Richards JM, Gale D, Mehta N, Lestingi T. Combination of chemotherapy with interleukin-2 and interferon alfa for the treatment of metastatic melanoma. Journal of Clinical Oncology 1999;17(2):651-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Spieth 2008 {published data only}
- Spieth K, Kaufmann R, Dummer R, Garbe C, Becker JC, Hauschild A, et al. Temozolomide plus pegylated interferon alfa-2b as first-line treatment for stage IV melanoma: a multicenter phase II trial of the Dermatologic Cooperative Oncology Group (DeCOG). Annals of Oncology 2008;19(4):801-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Van Dyk 1975 {published data only}
- Van Dyk JJ, Falkson G. A clinical trial of procarbazine plus vincristine plus bis-chloroethyl-nitrosourea plus imidazole carboxamide dimethyl triazeno in metastatic malignant melanoma. Medical and Pediatric Oncology 1975;1(2):107-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Varker 2007 {published data only}
- Varker KA, Biber JE, Kefauver C, Jensen R, Lehman A, Young D, et al. A randomized phase 2 trial of bevacizumab with or without daily low-dose interferon alfa-2b in metastatic malignant melanoma. Annals of Surgical Oncology 2007;14(8):2367-76. [PMID: ] [DOI] [PubMed] [Google Scholar]
Weber 2013 {published data only}
- Weber JS, Dummer R, Pril V, Lebbe C, Hodi FS. Patterns of onset and resolution of immune-related adverse events of special interest with ipilimumab: detailed safety analysis from a phase 3 trial in patients with advanced melanoma. Cancer 2013;119(9):1675-82. [PMID: ] [DOI] [PubMed] [Google Scholar]
Yang 1995 {published data only}
- Yang JC, Topalian SL, Schwartzentruber DJ, Parkinson DR, Marincola FM, Weber JS, et al. The use of polyethylene glycol-modified interleukin-2 (PEG-IL-2) in the treatment of patients with metastatic renal cell carcinoma and melanoma. A phase I study and a randomized prospective study comparing IL-2 alone versus IL-2 combined with PEG-IL-2. Cancer 1995;76(4):687-94. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCT01280565 {published data only}
- NCT01280565. A phase 3 study to compare efficacy and safety of masitinib to dacarbazine in the treatment of patients with non-resectable or metastatic stage 3 or stage 4 melanoma carrying a mutation in the juxta membrane domain of C-Kit. clinicaltrials.gov/ct2/show/NCT01280565 (first received 6 August 2010).
NCT01515189 {published data only}
- NCT01515189. Phase 3 trial in subjects with metastatic melanoma comparing 3 mg/kg ipilimumab versus 10 mg/kg ipilimumab. clinicaltrials.gov/ct2/show/NCT01515189 (first received 18 January 2012).
NCT01763164 {published data only}
- NCT01763164. Study comparing the efficacy of MEK162 versus dacarbazine in unresectable or metastatic NRAS mutation-positive melanoma. clinicaltrials.gov/ct2/show/NCT01763164 (first received 4 January 2013).
NCT01909453 {published data only}
- NCT01909453. Study comparing combination of LGX818 plus MEK162 versus vemurafenib and LGX818 monotherapy in BRAF mutant melanoma (COLUMBUS). clinicaltrials.gov/ct2/show/NCT01909453 (first received 24 July 2013).
NCT01940809 {published data only}
- NCT01940809. Ipilimumab with or without dabrafenib, trametinib, and/or nivolumab in treating patients with melanoma that is metastatic or cannot be removed by surgery. clinicaltrials.gov/ct2/show/NCT01940809 (first received 9 September 2013).
NCT01943422 {published data only}
- NCT01943422. Safety and efficacy study of vemurafenib and high-dose interferon alfa-2b in melanoma (12-107). clinicaltrials.gov/ct2/show/NCT01943422 (first received 27 August 2013).
NCT02130466 {published data only}
- NCT02130466. A study of the safety and efficacy of pembrolizumab (MK-3475) in combination with trametinib and dabrafenib in participants with advanced melanoma (MK-3475-022/KEYNOTE-022). clinicaltrials.gov/ct2/show/NCT02130466 (first received 1 May 2014).
NCT02224781 {published data only}
- NCT02224781. Dabrafenib and trametinib followed by ipilimumab and nivolumab or ipilimumab and nivolumab followed by dabrafenib and trametinib in treating patients with stage III-IV BRAFV600 melanoma. clinicaltrials.gov/ct2/show/NCT02224781 (first received 22 August 2014).
NCT02278887 {published data only}
- NCT02278887. Study comparing TIL to standard ipilimumab in patients with metastatic melanoma (TIL). clinicaltrials.gov/ct2/show/NCT02278887 (first received 3 June 2014).
NCT02339571 {published data only}
- NCT02339571. Nivolumab and ipilimumab with or without sargramostim in treating patients with stage III-IV melanoma that cannot be removed by surgery. clinicaltrials.gov/ct2/show/NCT02339571 (first received 12 January 2015).
NCT02388906 {published data only}
- NCT02388906. Efficacy study of nivolumab compared to ipilimumab in prevention of recurrence of melanoma after complete resection of stage IIIb/c or stage IV melanoma (CheckMate 238). clinicaltrials.gov/ct2/show/NCT02388906 (first received 10 March 2015).
NCT02416232 {published data only}
- NCT02416232. Access study of trametinib for subjects with advanced unresectable (Stage IIIc) or distant metastatic (Stage IV) BRAF V600E/K mutation positive cutaneous melanoma. clinicaltrials.gov/ct2/show/NCT02416232 (first received 9 April 2015).
NCT02460068 {published data only}
- NCT02460068. A study of fotemustine(FTM) vs FTM and ipilimumab (IPI) or IPI and nivolumab in melanoma brain metastasis (NIBIT-M2). clinicaltrials.gov/ct2/show/NCT02460068 (first received 22 May 2015).
NCT02506153 {published data only}
- NCT02506153. High-dose recombinant interferon alfa-2B, ipilimumab, or pembrolizumab in treating patients with stage III-IV high risk melanoma that has been removed by surgery. clinicaltrials.gov/ct2/show/NCT02506153 (first received 22 July 2015).
NCT02599402 {published data only}
- NCT02599402. Nivolumab combined with ipilimumab followed by nivolumab monotherapy as first-line treatment for patients with advanced melanoma (CheckMate 401). clinicaltrials.gov/ct2/show/NCT02599402 (first received 5 November 2015).
NCT02625337 {published data only}
- NCT02625337. Study comparing pembrolizumab with dual MAPK pathway inhibition plus pembrolizumab in melanoma patients (IMPemBra). clinicaltrials.gov/ct2/show/NCT02625337 (first received 1 December 2015).
NCT02714218 {published data only}
- NCT02714218. A study of two different dose combinations of nivolumab in combination with ipilimumab in subjects with previously untreated, unresectable or metastatic melanoma. clinicaltrials.gov/ct2/show/NCT02714218 (first received 16 March 2016).
NCT02752074 {published data only}
- NCT02752074. A phase 3 study of pembrolizumab + epacadostat or placebo in subjects with unresectable or metastatic melanoma (Keynote-252 / ECHO-301). clinicaltrials.gov/ct2/show/NCT02752074 (first received 22 April 2016).
NCT02821013 {published data only}
- NCT02821013. Duration of anti-PD-1 therapy in metastatic melanoma (STOP-GAP). clinicaltrials.gov/ct2/show/NCT02821013 (first received 29 June 2016).
Additional references
Altman 1999
- Altman DG, Andersen PK. Calculating the number needed to treat for trials where the outcome is time to an event. BMJ 1999;319(7223):1492-5. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 2002
- Altman DG, Deeks JJ. Meta-analysis, Simpson's paradox, and the number needed to treat. BMC Medical Research Methodology 2002;2:3. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Arkenau 2011
- Arkenau HT, Kefford R, Long GV. Targeting BRAF for patients with melanoma. British Journal of Cancer 2011;104(3):392-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ashour 2017
- Al-Abd AM, Alamoudi AJ, Abdel-Naim AB, Neamatallah TA, Ashour OM. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies - A review. Journal of Advanced Research 2017;8(6):591-605. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Australian and New Zealand 2008
- Australian Cancer Network Melanoma Guidelines Revision Working Party. Clinical Practice Guidelines for the Management of Melanoma in Australia and New Zealand (CP111). Wellington 2008. [www.nhmrc.gov.au/_files_nhmrc/publications/attachments/cp111.pdf]
Bajetta 2006
- Bajetta E, Del Vecchio M, Nova P, Fusi A, Daponte A, Sertoli MR, et al. Multicenter phase III randomized trial of polychemotherapy (CVD regimen) versus the same chemotherapy (CT) plus subcutaneous interleukin-2 and interferon-alpha2b in metastatic melanoma. Annals of Oncology 2006;17(4):571-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Balch 2001
- Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS, Cascinelli N, et al. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. Journal of Clinical Oncology 2001;19(16):3622-34. [PMID: ] [DOI] [PubMed] [Google Scholar]
Balch 2009
- Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. Journal of Clinical Oncology 2009;27(36):6199-206. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Belardelli 2002
- Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon-alpha in tumor immunity and immunotherapy. Cytokine & Growth Factor Reviews 2002;13(2):119-34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Beusterien 2003
- Beusterien KM, Ackerman SJ, Plante K, Glaspy J, Naredi P, Wood D, et al. The health-related quality-of-life impact of histamine dihydrochloride plus interleukin-2 compared with interleukin-2 alone in patients with metastatic melanoma. Supportive Care in Cancer 2003;11(5):304-12. [PMID: ] [DOI] [PubMed] [Google Scholar]
Borenstein 2009
- Borenstein M, Hedges LV, Higgins JPT, Rothstein HR. Multiple comparisons within a study. In: Introduction to Meta-analysis. 1st edition. Wiley-Blackwell, 2009:239-242. [Google Scholar]
Brundage 1993
- Brundage MD, Pater JL, Zee B. Assessing the reliability of two toxicity scales: implications for interpreting toxicity data. Journal of the National Cancer Institute 1993;85(14):1138-48. [PMID: ] [DOI] [PubMed] [Google Scholar]
Caino 2016
- Caino MC, Altieri DC. Molecular pathways: mitochondrial reprogramming in tumor progression and therapy. Clinical Cancer Research 2016;22(3):540-5. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cancer Research UK 2017
- Cancer Research UK 2017. General cancer information. www.about-cancer.cancerresearchuk.org/about-cancer/cancer-in-general/treatment/targeted-cancer-drugs/types/monoclonal-antibodies (accessed 13 December 2017).
Case 2002
- Case LD, Kimmick G, Paskett ED, Lohman K, Tucker R. Interpreting measures of treatment effect in cancer clinical trials. Oncologist 2002;7(3):181-7. [PMID: 12065789 ] [DOI] [PubMed] [Google Scholar]
Chaimani 2013
- Chaimani A, Higgins JP, Mavridis D, Spyridonos P, Salanti G. Graphical tools for network meta-analysis in STATA. PloS One 2013;8(10):e76654. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chi 2011
- Chi M, Dudek AZ. Vaccine therapy for metastatic melanoma: systematic review and meta-analysis of clinical trials. Melanoma Research 2011;21(3):165-74. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Chiarion‐Sileni 2003
- Chiarion-Sileni V, Del Bianco P, De Salvo GL, Lo Re G, Romanini A, Labianca R, et al. Quality of life evaluation in a randomised trial of chemotherapy versus bio-chemotherapy in advanced melanoma patients. European Journal of Cancer 2003;39(11):1577-85. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cipriani 2013
- Cipriani A, Higgins JP, Geddes JR, Salanti G. Conceptual and technical challenges in network meta-analysis. Annals of Internal Medicine 2013;159(2):130-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ciren 2016
- CiRen B, Wang X, Long Z. The evaluation of immunotherapy and chemotherapy treatment on melanoma: a network meta-analysis. Oncotarget 2016;7(49):81493-81511. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Coates 1993
- Coates A, Thomson D, McLeod GR, Hersey P, Gill PG, Olver IN, et al. Prognostic value of quality of life scores in a trial of chemotherapy with or without interferon in patients with metastatic malignant melanoma. European Journal of Cancer 1993;29A(12):1731-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Davies 2002
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417(6892):949-54. [PMID: ] [DOI] [PubMed] [Google Scholar]
Devji 2017
- Devji T, Levine O, Neupane B, Beyene J, Xie F. Systemic therapy for previously untreated advanced BRAF-mutated melanoma: a systematic review and network meta-analysis of randomized clinical trials. JAMA Oncology 2017;3(3):366-73. [PMID: ] [DOI] [PubMed] [Google Scholar]
Di Giacomo 2012
- Di Giacomo AM, Ascierto PA, Pilla L, Santinami M, Ferrucci PF, Giannarelli D, et al. Ipilimumab and fotemustine in patients with advanced melanoma (NIBIT-M1): an open-label, single-arm phase 2 trial. Lancet Oncology 2012;13(9):879-86. [PMID: ] [DOI] [PubMed] [Google Scholar]
Donegan 2013
- Donegan S, Williamson P, D'Alessandro U, Tudur Smith C. Assessing key assumptions of network meta-analysis: a review of methods. Research Synthesis Methods 2013;4(4):291-323. [PMID: ] [DOI] [PubMed] [Google Scholar]
Edlundh‐Rose 2006
- Edlundh-Rose E, Egyhazi S, Omholt K, Mansson-Brahme E, Platz A, Hansson J, et al. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Research 2006;16(6):471-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Eggermont 2009
- Eggermont AM, Testori A, Marsden J, Hersey P, Quirt I, Petrella T, et al. Utility of adjuvant systemic therapy in melanoma. Annals of Oncology 2009;20(Suppl 6):vi30-4. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ferlay 2010
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International journal of cancer 2010;127(12):2893-917. [PMID: ] [DOI] [PubMed] [Google Scholar]
Flaherty 2012
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. The New England journal of medicine 2012;367(18):1694-703. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Flaherty 2013
- Flaherty KT, Lee SJ, Zhao F, Schuchter LM, Flaherty L, Kefford R, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. Journal of Clinical Oncology 2013;31(3):373-9. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Forsea 2012
- Forsea AM, Del Marmol V, Vries E, Bailey EE, Geller AC. Melanoma incidence and mortality in Europe: new estimates, persistent disparities. British Journal of Dermatology 2012;167(5):1124-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Garbe 2011
- Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist 2011;16(1):5-24. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gentile 2017
- Gentile C, Martorana A, Lauria A, Bonsignore R. Kinase inhibitors in multitargeted cancer therapy. Current Medicinal Chemistry 2017;24(16):1671-86. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gorry 2018
- Gorry C, McCullagh L, O'Donnell H, Barrett S, Schmitz S, Barry M, et al. Neoadjuvant treatment for malignant and metastatic cutaneous melanoma. Cochrane Database of Systematic Reviews in press. [DOI] [PMC free article] [PubMed]
Grob 2014
- Grob JJ, Amonkar MM, Martin-Algarra S, Demidov LV, Goodman V, Grotzinger K, et al. Patient perception of the benefit of a BRAF inhibitor in metastatic melanoma: quality-of-life analyses of the BREAK-3 study comparing dabrafenib with dacarbazine. Annals of Oncology 2014;25(7):1428-36. [PMID: ] [DOI] [PubMed] [Google Scholar]
Grob 2015
- Grob JJ, Amonkar MM, Karaszewska B, Schachter J, Dummer R, Mackiewicz A, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncology 2015;16(13):1389-98. [PMID: ] [DOI] [PubMed] [Google Scholar]
Guo 2011
- Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. Journal of Clinical Oncology 2011;29(21):2904-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE; an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gyorki 2013
- Gyorki DE, Yuan J, Mu Z, Zaidi B, Pulitzer M, Busam K, et al. Immunological insights from patients undergoing surgery on ipilimumab for metastatic melanoma. Annals of Surgical Oncology 2013;20(9):3106-11. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamid 2013
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. New England Journal of Medicine 2013;369(2):134-44. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamm 2008
- Hamm C, Verma S, Petrella T, Bak K, Charette M, Melanoma Disease Site Group of Cancer Care Ontario's Program in Evidence-based Care. Biochemotherapy for the treatment of metastatic malignant melanoma: a systematic review. Cancer Treatment Reviews 2008;34(2):145-56. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hauschild 2009
- Hauschild A, Agarwala SS, Trefzer U, Hogg D, Robert C, Hersey P, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. Journal of Clinical Oncology 2009;27(17):2823-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [Updated March 2011]. The Cochrane Collaboration 2011. Available from www.cochrane-handbook.org.
Higgins 2012
- Higgins JP, Jackson D, Barrett JK, Lu G, Ades AE, White IR. Consistency and inconsistency in network meta-analysis: concepts and models for multi-arm studies. Research Synthesis Methods 2012;3(2):98-110. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hillner 2000
- Hillner BE, Agarwala S, Middleton MR. Post hoc economic analysis of temozolomide versus dacarbazine in the treatment of advanced metastatic melanoma. Journal of Clinical Oncology 2000;18(7):1474-80. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hodi 2010
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New England Journal of Medicine 2010;363(8):711-23. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hong 2013
- Hong H, Carlin BP, Shamliyan TA, Wyman JF, Ramakrishnan R, Sainfort F, et al. Comparing Bayesian and frequentist approaches for multiple outcome mixed treatment comparisons. Medical Decision Making 2013;33(5):702-14. [PMID: ] [DOI] [PubMed] [Google Scholar]
Huncharek 2001
- Huncharek M, Caubet JF, McGarry R. Single-agent DTIC versus combination chemotherapy with or without immunotherapy in metastatic melanoma: a meta-analysis of 3273 patients from 20 randomized trials. Melanoma Research 2001;11(1):75-81. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ives 2007
- Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2,621 patients. Journal of Clinical Oncology 2007;25(34):5426-34. [PMID: ] [DOI] [PubMed] [Google Scholar]
Jager 2015
- Jager NG, Linn SC, Schellens JH, Beijnen JH. Tailored tamoxifen treatment for breast cancer patients: a perspective. Clin Breast Cancer 2015;15(4):241-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Jayson 2016
- Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet 2016;388(10043):518-29. [PMID: ] [DOI] [PubMed] [Google Scholar]
Keilholz 2002
- Keilholz U, Gore ME. Biochemotherapy for advanced melanoma. Seminars in Oncology 2002;29(5):456-61. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kiebert 2003
- Kiebert GM, Jonas DL, Middleton MR. Health-related quality of life in patients with advanced metastatic melanoma: results of a randomized phase III study comparing temozolomide with dacarbazine. Cancer Investigation 2003;21(6):821-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kirkwood 2008
- Kirkwood JM, Tarhini AA, Panelli MC, Moschos SJ, Zarour HM, Butterfield LH, et al. Next generation of immunotherapy for melanoma. Journal of Clinical Oncology 2008;26(20):3445-55. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kirkwood 2012
- Kirkwood JM, Butterfield LH, Tarhini AA, Zarour H, Kalinski P, Ferrone S. Immunotherapy of cancer in 2012. CA: a Cancer Journal for Clinicians 2012;62(5):309-35. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kirkwood 2012a
- Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clinical Cancer Research 2012;18(2):555-67. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Klein 2013
- Klein O, Clements A, Menzies AM, O'Toole S, Kefford RF, Long GV. BRAF inhibitor activity in V600R metastatic melanoma. European Journal of Cancer 2013;49(5):1073-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
La Porta 2007
- La Porta CA. Drug resistance in melanoma: new perspectives. Current Medicinal Chemistry 2007;14(4):387-91. [PMID: ] [DOI] [PubMed] [Google Scholar]
La Porta 2009
- La Porta CA. Mechanism of drug sensitivity and resistance in melanoma. Current Cancer Drug Targets 2009;9(3):391-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Little 2012
- Little EG, Eide MJ. Update on the current state of melanoma incidence. Dermatologic Clinics 2012;30(3):355-61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Long 2011
- Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. Journal of Clinical Oncology 2011;29(10):1239-46. [PMID: ] [DOI] [PubMed] [Google Scholar]
Long 2012
- Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncology 2012;13(11):1087-95. [PMID: ] [DOI] [PubMed] [Google Scholar]
Long 2012a
- Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncology 2012;13(11):1087-95. [PMID: ] [DOI] [PubMed] [Google Scholar]
Margolin 2012
- Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncology 2012;13(5):459-65. [PMID: ] [DOI] [PubMed] [Google Scholar]
McKinnon 2005
- McKinnon JG, Starritt EC, Scolyer RA, McCarthy WH, Thompson JF. Histopathologic excision margin affects local recurrence rate: analysis of 2681 patients with melanomas < or =2 mm thick. Annals of Surgery 2005;241(2):326-33. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Meckbach 2014
- Meckbach D, Bauer J, Pflugfelder A, Meier F, Busch C, Eigentler TK, et al. Survival according to BRAF-V600 tumor mutations--an analysis of 437 patients with primary melanoma. PloS One 2014;9(1):e86194. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Melanoma Institute Australia 2017
- Melanoma Institute Australia. Glossary. www.melanoma.org.au/understanding-melanoma/glossary/braf/ (accessed 13 December 2017).
Menzies 2013
- Menzies AM, Kefford RF, Long GV. Paradoxical oncogenesis: are all BRAF inhibitors equal? Pigment Cell & Melanoma Research 2013;26(5):611-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mills 2013
- Mills EJ, Thorlund K, Ioannidis JP. Demystifying trial networks and network meta-analysis. BMJ (Clinical research ed.) 2013;346:f2914. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mocellin 2003
- Mocellin S, Rossi CR, Nitti D, Lise M, Marincola FM. Dissecting tumor responsiveness to immunotherapy: the experience of peptide-based melanoma vaccines. Biochim Biophys Acta. 2003 Dec 5;1653(2):61-71 2003;1653:61-71. [DOI] [PubMed] [Google Scholar]
Mocellin 2004
- Mocellin S, Mandruzzato S, Bronte V, Lise M, Nitti D. Part I: Vaccines for solid tumours. Lancet Oncology 2004;5(11):681-9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mocellin 2005
- Mocellin S. Cancer vaccines: the challenge of developing an ideal tumor killing system. Frontiers in Bioscience 2005;10:2285-305. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mocellin 2008
- Mocellin S, Nitti D. Therapeutics targeting tumor immune escape: towards the development of new generation anticancer vaccines. Medicinal Research Reviews 2008;28(3):413-44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mocellin 2009
- Mocellin S, Pilati P, Nitti D. Peptide-based anticancer vaccines: recent advances and future perspectives. Curr Med Chem 2009;16:4779-96. [DOI] [PubMed] [Google Scholar]
Mocellin 2010
- Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. Journal of the National Cancer Institute 2010;102(7):493-501. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mocellin 2010a
- Mocellin S, Shrager J, Scolyer R, Pasquali S, Verdi D, Marincola FM, et al. Targeted Therapy Database (TTD): a model to match patient's molecular profile with current knowledge on cancer biology. PloS One 2010;5(8):e11965. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mocellin 2011
- Mocellin S, Pasquali S, Nitti D. The impact of surgery on survival of patients with cutaneous melanoma: revisiting the role of primary tumor excision margins. Annals of Surgery 2011;253(2):238-43. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mocellin 2013
- Mocellin S, Lens MB, Pasquali S, Pilati P, Chiarion Sileni V. Interferon alpha for the adjuvant treatment of cutaneous melanoma. Cochrane Database of Systematic Reviews 2013, Issue 6. Art. No: CD008955. [DOI: 10.1002/14651858.CD008955] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mocellin 2013a
- Mocellin S, Benna C, Pilati P. Coinhibitory molecules in cancer biology and therapy. Cytokine & Growth Factor Reviews 2013;24(2):147-61. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mocellin 2013b
- Mocellin S, Nitti D. CTLA-4 blockade and the renaissance of cancer immunotherapy. Biochimica et Biophysica Acta 2013;1836(2):187-96. [DOI: 10.1016/j.bbcan.2013.05.003] [PMID: ] [DOI] [PubMed] [Google Scholar]
National Toxicology Program 2011
- National Toxicology Program. Dacarbazine. Report on carcinogens : carcinogen profiles / U.S. Dept. of Health and Human Services, Public Health Service, National Toxicology Program 2011;12:127-8. [PMID: ] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815-34. [PMID: ] [DOI] [PubMed] [Google Scholar]
Pasquali 2010
- Pasquali S, Mocellin S. The anticancer face of interferon alpha (IFN-alpha): from biology to clinical results, with a focus on melanoma. Current Medicinal Chemistry 2010;17(29):3327-36. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pasquali 2013
- Pasquali S, Haydu LE, Scolyer RA, Winstanley JB, Spillane AJ, Quinn MJ, et al. The importance of adequate primary tumor excision margins and sentinel node biopsy in achieving optimal locoregional control for patients with thick primary melanomas. Annals of Surgery 2013;258(1):152-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Pasquali 2014
- Pasquali S, Kefford R, Chiarion Sileni V, Nitti D, Rossi CR, Pilati P, et al. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database of Systematic Reviews 2014, Issue 5. Art. No: CD011123. [DOI: 10.1002/14651858.CD011123] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pasquali 2017
- Pasquali S, Chiarion-Sileni V, Rossi CR, Mocellin S. Immune checkpoint inhibitors and targeted therapies for metastatic melanoma: A network meta-analysis. Cancer Treatment Reviews 2017;54:34-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
Revicki 2012
- Revicki DA, den Eertwegh AJ, Lorigan P, Lebbe C, Linette G, Ottensmeier CH, et al. Health related quality of life outcomes for unresectable stage III or IV melanoma patients receiving ipilimumab treatment. Health and Quality of Life Outcomes 2012;10:66. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Ridolfi 2002
- Ridolfi R, Chiarion-Sileni V, Guida M, Romanini A, Labianca R, Freschi A, et al. Cisplatin, dacarbazine with or without subcutaneous interleukin-2, and interferon alpha-2b in advanced melanoma outpatients: results from an Italian multicenter phase III randomized clinical trial. Journal of Clinical Oncology 2002;20(6):1600-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Salanti 2011
- Salanti G, Ades AE, Ioannidis JP. Graphical methods and numerical summaries for presenting results from multiple-treatment meta-analysis: an overview and tutorial. Journal of Clinical Epidemiology 2011;64(2):163-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Salanti 2014
- Salanti G, Del Giovane C, Chaimani A, Caldwell DM, Higgins JP. Evaluating the quality of evidence from a network meta-analysis. PloS One 2014;9(7):e99682. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sasse 2007
- Sasse AD, Sasse EC, Clark LG, Ulloa L, Clark OA. Chemoimmunotherapy versus chemotherapy for metastatic malignant melanoma. Cochrane Database of Systematic Reviews 2007, Issue 1. Art. No: CD005413. [DOI: 10.1002/14651858.CD005413.pub2] [DOI] [PubMed] [Google Scholar]
Schadendorf 2009
- Schadendorf D, Algarra SM, Bastholt L, Cinat G, Dreno B, Eggermont AM, et al. Immunotherapy of distant metastatic disease. Annals of Oncology 2009;20(Suppl 6):vi41-50. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schadendorf 2014
- Schadendorf D, Amonkar MM, Milhem M, Grotzinger K, Demidov LV, Rutkowski P, et al. Functional and symptom impact of trametinib versus chemotherapy in BRAF V600E advanced or metastatic melanoma: quality-of-life analyses of the METRIC study. Annals of Oncology 2014;25(3):700-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schadendorf 2015
- Schadendorf D, Amonkar MM, Stroyakovskiy D, Levchenko E, Gogas H, Braud F, et al. Health-related quality of life impact in a randomised phase III study of the combination of dabrafenib and trametinib versus dabrafenib monotherapy in patients with BRAF V600 metastatic melanoma. European Journal of Cancer 2015;51(7):833-40. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c332. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schwartzentruber 2011
- Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. New England Journal of Medicine 2011;364(22):2119-27. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Scolyer 2011
- Scolyer RA, Long GV, Thompson JF. Evolving concepts in melanoma classification and their relevance to multidisciplinary melanoma patient care. Molecular Oncology 2011;5(2):124-36. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Serrone 1999
- Serrone L, Hersey P. The chemoresistance of human malignant melanoma: an update. Melanoma Research 1999;9(1):51-8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sherrill 2013
- Sherrill B, Wang J, Kotapati S, Chin K. Q-TWiST analysis comparing ipilimumab/dacarbazine vs placebo/dacarbazine for patients with stage III/IV melanoma. British Journal of Cancer 2013;109(1):8-13. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Siegel 2012
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a Cancer Journal for Clinicians 2012;62(1):10-29. [PMID: ] [DOI] [PubMed] [Google Scholar]
Sladden 2009
- Sladden MJ, Balch C, Barzilai DA, Berg D, Freiman A, Handiside T, et al. Surgical excision margins for primary cutaneous melanoma. Cochrane Database of Systematic Reviews 2009, Issue 4. Art. No: CD004835. [DOI: 10.1002/14651858.CD004835.pub2] [PMID: ] [DOI] [PubMed] [Google Scholar]
Sosman 2012
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. New England Journal of Medicine 2012;366(8):707-14. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stata 2017 [Computer program]
- Stata. Version 11.2. College Station, TX, USA: StataCorp, 2017. Available at www.stata.com.
Stevens 2006
- Stevens G, McKay MJ. Dispelling the myths surrounding radiotherapy for treatment of cutaneous melanoma. Lancet Oncology 2006;7(7):575-83. [PMID: ] [DOI] [PubMed] [Google Scholar]
Tarhini 2005
- Tarhini AA, Agarwala SS. Interleukin-2 for the treatment of melanoma. Current opinion in Investigational Drugs 2005;6(12):1234-9. [PMID: ] [PubMed] [Google Scholar]
Testori 2009
- Testori A, Rutkowski P, Marsden J, Bastholt L, Chiarion-Sileni V, Hauschild A, et al. Surgery and radiotherapy in the treatment of cutaneous melanoma. Annals of Oncology 2009;20(Suppl 6):vi22-9. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Therasse 2002
- Therasse P. Measuring the clinical response. What does it mean? European Journal of Cancer 2002;38(14):1817-23. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thompson 2009
- Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma in the era of molecular profiling. Lancet 2009;374(9687):362-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8:16. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wevers 2013
- Wevers KP, Hoekstra HJ. Stage IV melanoma: completely resectable patients are scarce. Annals of Surgical Oncology 2013;20(7):2352-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
White 2011
- White IR. Multivariate random-effects meta-regression: Updates to mvmeta. Stata Journal 2011;11(2):255-70. [www.stata-journal.com/sj11-2.html] [Google Scholar]
White 2012
- White IR, Barrett JK, Jackson D, Higgins JP. Consistency and inconsistency in network meta-analysis: model estimation using multivariate meta-regression. Research Synthesis Methods 2012;3(2):111-25. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
White 2015
- White I. Network meta-analysis. Stata Journal 2015;15(4):951-85. [Google Scholar]
Wise 2016
- Wise J. NICE approves immunotherapy combination for advanced melanoma. BMJ 2016;353:i3421. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Crosby 2000
- Crosby T, Fish R, Coles B, Mason M. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database of Systematic Reviews 2000, Issue 2. Art. No: CD001215. [DOI: 10.1002/14651858.CD001215] [DOI] [PubMed] [Google Scholar]