

Abstract
An efficient method for chemoselective cysteine arylation of unprotected peptides and proteins using Au(III) or- ganometallic complexes is reported. The bioconjugation reactions proceed rapidly (<5 min) at ambient temperature in various buffers and within a wide pH range (0.5–14). This approach provides access to a diverse array of S-aryl bioconjugates including fluorescent dye, complex drug molecule, affinity label, poly(ethylene glycol) tags and a stapled peptide. A library of Au(111) arylation reagents can be prepared as air-stable, crystalline solids in one step from commercial reagents. The selective and efficient arylation procedures presented in this work broaden the synthetic scope of cysteine bioconjugation and serve as promising routes for the modification of complex biomolecules.
Graphical Abstract
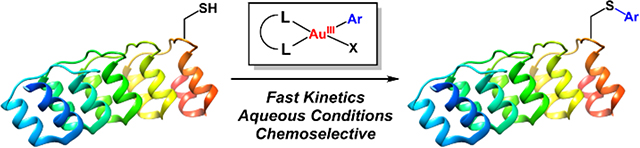
Cysteine bioconjugation is a powerful tool that allows for the introduction of a diverse array of substrates to biomolecules via the formation of covalent linkages.1–5 Transition-metal mediated reactions have emerged recently as attractive methods for the modification of complex biomolecules due to the high functional group tolerance, chemoselectivity, and rapid reaction kinetics associated with these metal-based transformations.6–13 Additionally, the rational choice of transition-metal ion and ligand platform can provide the ability to design organ-ometallic-based bioconjugation reagents with highly tailored reactivity, solubility, and stability properties. This concept is embodied by the versatile and efficient palladium-mediated cysteine arylation methods reported by Buchwald, Pentelute et al. in the past three years that enable access to a broad range of bioconjugates under mild reaction conditions (Scheme 1).9–13
Scheme 1.

Previous work utilizing PdII reagents (references 9–13) and this work detailing AuIII-mediated cysteine S-arylation of biomolecules.
Herein, we expand the scope, generality, and utility of transition-metal mediated cysteine arylation via C-S bond forming, reductive elimination process occurring from a class of robust organometallic Au(III) complexes.
The reluctance of Au(I) to undergo oxidative addition14–19 potentially provides gold-based species with high functional-group tolerance, minimizing the propensity for background reactivity with the variety of functional groups present in complex biomolecules. Furthermore, the thiophilic nature of gold renders Au(III)-based complexes prime candidates for reduction by cysteine thiols, engendering such systems as potential reagents for bioconjugation.20,21 Surprisingly, there are currently no auxiliary-free methods using gold-based complexes for cysteine arylation.22 Here we introduce such a methodology using easily accessed, air-stable Au(III)-aryl complexes. The resulting methodology provides rapid access to a diverse array of protein and peptide bioconjugates with high conversion under mild reaction conditions. The broad scope and utility of the described method is highlighted by the variety of well-defined, air-stable, crystalline Au(III)-based arylation reagents that were prepared in one synthetic step from commercial reagents. The present strategy is positioned at the interface of organometallic and bioconjugation chemistry and aims to provide new and efficient tools for biomolecule modification.
Bourissou et al. have recently described an elegant approach to enhance the reactivity of Au(I) species toward oxidative addition by using preorganized ligand architectures that support the square planar geometry of the ensuing Au(111) products.14,23,24 These systems appeared ideal to us to accommodate the elementary steps of oxidative addition, transmetalation, and reductive elimination17,19,25–27 required for C-S bond formation in cysteine arylation processes (Scheme 1).
Optimization of cysteine-arylation reaction conditions with 1 were carried out using L-glutathione (GSH) as the model peptide substrate (Figure 1). Full conversion to the S-tolyl GSH-conjugate was observed in <5 min upon treatment of GSH with 1 (5 equiv) at 25 °C in the presence of Tris buffer (pH 8) as determined by LC-MS analysis of the crude reaction mixture. Reaction solutions containing up to 80% H2O in H2O/MeCN mixtures were tolerated (Figure 1), whereas larger ratios of H2O led to reduced reaction conversion. This limited efficiency led us to seek another Au(III)-based system that would exhibit improved compatibility in biologically relevant reaction media.
Figure 1.

Gold(III) reagents 1 and 2a (X = CI/I), and glutathione arylation scheme with reaction optimization parameters.
We prepared the Au(III)-tolyl oxidative addition complex, [(Me-DalPhos)Au(tolyl)Cl][SbF6] ([2a][SbF6], Me-DalPhos = (Ad2P(o-C6H4)NMe2)28,29),24 isolated as a crystalline, air-stable solid. We next evaluated the suitability of 2a to serve as a cysteine arylation reagent under a variety of reaction conditions. Quantitative conversion of GSH to the corresponding S-tolyl conjugate was observed in minutes (<5 min) at 25 °C in a 80:20 H2O:MeCN (v,v) mixture, as assayed by LC-MS analysis of the crude reaction mixture. The optimized reaction conditions provided significant improvements upon those employing complex 1, such as lower reagent loading (3 vs. 5 equiv), and a reduced percentage of organic solvent required (20% vs 50%, see Figure 1). Notably, the 2a-mediated cysteine arylation reactions proceeded to completion within a large pH range (0.5–14) and in the presence of several common buffers (Tris, HEPES, Na2CO3). The bioconjugation reactions were also compatible with the disulfide reducing agent, TCEP (tris(2-carboxyethyl)phosphine, 1 equiv), protein denaturing agent, guanidine-HCl (4M) and several other unconventional solvents (SI Figure S79).
We further prepared a library of [(Me-DalPhos)AuArCl][SbF6] oxidative addition complexes (Figure 2A) bearing various biorelevant groups including heterocycles (2m, 2p), an affinity label (2q), fluorescent tag (2r), complex drug molecule (2n), and a poly(ethylene glycol) (PEG) polymer (20). Oxidative addition reactions to generate complexes 2a-2r proceeded rapidly and cleanly upon treatment of CH2Cl2 solutions of (Me-DalPhos)AuCl with the corresponding aryl iodide electrophile in the presence of the halide scavenger, AgSbF6.24 After removal of the liberated Agl byproduct by filtration, the [(Me-DalPhos)AuArCl][SbF6] salts readily crystallized directly from the resulting solution upon standing at 25 °C. This purification procedure afforded single, X-ray diffraction quality crystals of several complexes that enabled their structural determination (see SI section V for all crystallographic data). Notably, iodide to chloride exchange occurs at the gold center during the course of the oxidative addition reaction, as confirmed by X-ray diffraction analysis of several complexes that display CI/I disorder with a 75–100% range of chloride occupancy (SI section V). This observation is in line with X-ray diffraction studies of closely related complexes prepared under similar conditions,24 and is consistent with formation of the more stable gold(III)-chloride derivative.30
Figure 2.

A: Scope of [(Me-DalPhos)AuArCl][SbF6] bioconjugation reagents. B: LC traces of cysteine arylation reaction mixtures with two peptides using reagents 2m (left) and 2n (right). Gold-based species are highlighted in grey. See SI section IV for further experimental details.
Interestingly, despite the exclusion of oxygen and water from the reported preparation of 2a and related complexes previously,24 we found that the synthesis and purification of all [(Me-DalPhos)AuArCl][SbF6] salts presented in this work proceeded cleanly even when performed under open atmosphere conditions using commercial, unpurified solvents. The salts exhibited excellent long-term air and water stability, and no observable degradation was detected after prolonged periods (>3 months) when the reagents were stored on the benchtop at 25 °C as assayed by 1H and 31P NMR spectroscopy. Complex 20 displayed solubility in neat H2O due to the hydrophilicity imparted by the PEG group. This species also demonstrated excellent water stability, showing only limited degradation (ca. 20%) after a sample was allowed to stand for four days at 25 °C in H2O as judged by 31P NMR spectroscopy (see Figure S69).
A comprehensive demonstration of the aryl scope was performed with GSH under the optimized bioconjugation conditions (25 °C, 3 equiv AU complex, 80:20 H2O:MeCN, 0.1 M Tris buffer, pH 8.0). Quantitative conversion to the GSH S-aryl conjugates was observed in <5 min for all 17 substrates displayed in Figure 2A (b-r). Notably, the reaction does not inherently necessitate organic co-solvent when the organometallic reagent is soluble in water (20, see SI Figure S93). As a representative example, the S-(C6H4-p-Cl) conjugate was easily separated from small molecule byproducts and Au-based species by reversed-phase HPLC, and ICP-AES analysis of the purified peptide indicated more than 99.9% of gold was removed using this purification procedure (See SI section IV).
The chemoselectivity of oxidative addition was probed through treatment of (Me-DalPhos)AuCl with p-chloro- and p-bromoiodobenzene electrophiles in the presence of AgSbF6 (see SI section II). For both substrates, oxidative addition occurs exclusively across the Ar-I bond, resulting in the formation of complexes 2i and 2j (Figure 2A), respectively. Furthermore, treatment of GSH with 2i and 2j generated the p-Cl-C6H4 and p-Br-C6H4 tagged peptides as the sole products, without any evidence for iodo aryl-based conjugates as judged by LC-MS analyses. The high chemoselectivity of the (Me-DalPhos)Au system is in stark contrast to that observed for reported Pd-based platforms that readily react with all Ar–X (X = CI, Br, I) species with limited selectivity.9 Thus, the functional group tolerance coupled with the high selectivity of the reported Au-based system may provide complementary advantages to the previously developed Pd-based reagents for transferring aryl groups of various complexities to biomolecules.
To further establish the versatility and utility of this methodology, we applied our bioconjugation strategy to more complex peptide substrates. Cysteine arylation of two different peptide sequences was observed in nearly quantitative conversion for a variety of aryl substrates (see SI section IV; representative examples shown in Figure 2B). No reaction was observed using a control peptide where the cysteine residue was mutated to serine, highlighting the chemoselectivity of the Au(III)- mediated bioconjugation method. Additionally, trypsin digest and MS/MS analyses of a (p-Cl-C6H4)-peptide conjugate support modification at the cysteine residue exclusively (see SI section IV experimental details).
We next extended the scope of Au(III)-mediated cysteine bioconjugation to the modification of proteins. Cysteine arylation of DARPin (designed ankyrin repeat protein) was observed using complex 2a within 30 min at 25 °C, as verified by LC-MS analysis of the reaction mixture (Figure 3A). The presence of a small amount of DMF co-solvent (5%) was required for efficient bioconjugation due to the inherent solubility constraints of 2a. However, treatment of fibroblast growth factor 2 (FGF2)31 with water soluble 20 (15 equiv) in neat aqueous buffer resulted in complete conversion to the PEGylated conjugate as confirmed by the deconvoluted mass spectrum from LC-MS analysis of the reaction mixture (see SI Figure S102). The rapid and efficient protein bioconjugation reactions demonstrate the potential generality, and suitability of the described Au(III)-mediated methodology for the modification of complex proteins under mild, biologically-relevant conditions and at low micromolar concentration of protein (36 μM).
Figure 3.

A: DARPin modification using 2a, and deconvoluted mass spectra of the protein before and after conjugation. B: Solid-state structure of peptide stapling reagent, [((Me-DalPhos)AuCl)2(μ2-1,4-C6H4)]2+ (2s), with thermal ellipsoids rendered at the 50% probability level and with hydrogen atoms and two SbF6− anions removed for clarity. C: LC-MS trace of the purified phenylene-stapled peptide. [M+H]+: 670.1965 (calc’d, 670.1968) m/z.
With a protocol for Au-mediated cysteine arylation, we envisaged the same route could furnish a stapled peptide through the construction of an intramolecular cysteine-cysteine linkage. There has been significant interest in the development of stapled peptides as therapeutic agents; however, there is still a growing need for easily-accessible peptide macrocyclization methods that allow for modular tuning of crosslinking units.32–36 The straightforward and efficient Au(III)-mediated bioconjugation procedures together with the use of commercially available (Me-DalPhos)AuCl and diiodoaryl reagents provides a versatile and systematic approach to peptide stapling that is complementary to existing state-of-the-art methods.4,10,34–37 The ortho- phenylene-bridged di-gold(III) stapling reagent, 2s, was prepared in a single synthetic step through treatment of (Me-DalPhos)AuCl (2 equiv) with 1,4-diiodobenzene (1 equiv) in the presence of AgSbF6 (2 equiv), and isolated as a crystalline solid in 64% yield. A single crystal X-ray diffraction study confirmed the solid-state structure of [2s][SbF6]2 (Figure 3B). Peptide stapling was observed under optimized conditions (30 min, 50:50 H2O:MeCN, 25 °C, 0.1 M Tris, pH 8) using a two-fold excess of the 2s macrocyclization reagent for peptides containing cysteine residues at the i, i+4 positions (Figure 3C). The o-phenylene i, i+4 stapled peptide was isolated away from metal-based impurities after purification by reversed-phase HPLC.
The apparent exceptionally rapid kinetics of Au(III)-mediated bioconjugation were benchmarked by performing a comparative study against a closely related Pd(II)-based analogue. The (RuPhos)Pd(tolyl)I complex reported previously9 was ideally suited for comparison with the [(Me-DalPhos)Au(p-ethylbenzene)Cl]+ complex 2b on account of its performance as a cysteine arylation reagent and the similar electronic and steric properties of the aryl substituents on both complexes. Treatment of GSH with equimolar amounts of 2b and (RuPhos)Pd(tolyl)I (Scheme 2) under conditions compatible with both systems resulted in 87±3% conversion to the ethylbenzene conjugate, indicating that the Au-mediated conjugation outperformed the Pd-based arylation in nearly 9:1 kinetic ratio. These data suggest that the kinetics for Au-mediated bioconjugation are on the order of, or faster than those estimated for the Pd- based systems (103–104 M−1 s−1).9
Scheme 2.

Competition experiment between (RuPhos)Pd(tolyl)I and [2b][SbF6] with GSH.
In summary, we present a general protocol for cysteine S-arylation of unprotected peptides and proteins using robust, Au(III) oxidative addition complexes bearing a diverse array of aryl substituents. The reported method operates in a large Ph range under mild reaction conditions and displays rapid reaction kinetics, high chemoselectivity, and excellent functional group tolerance. With this work, we expand the scope of biomolecule modification3,5,38–40 by providing tools that interface bond forming processes characteristic of organometallic complexes18,25,41–46 with bioconjugation. The straightforward synthetic procedures and commercially available or otherwise easily accessible reagents presented should expand the bioconjugation space well beyond the substrates and peptides reported in this study. This work also expands on a relatively underrepresented class of organometallic reagents containing metal-carbon bonds capable of withstanding relatively harsh environmental conditions.47
Supplementary Material
ACKNOWLEDGMENT
A.M.S. thanks UCLA Department of Chemistry and Biochemistry for start-up funds, 3M for a Non-Tenured Faculty Award, Alfred P. Sloan Foundation for a Fellowship in Chemistry and the National Institutes of Health (NIH) for a Maximizing Investigators Research Award (MIRA, R35GM124746). H.D.M thanks the National Science Foundation (NSF, CHE-1507735) for funding. M.S.M thanks the NSF for the Bridge-to-Doctorate (HRD-1400789) and the Predoctoral (GRFP) (DGE-0707424) Fellowships and UCLA for the Christopher S. Foote Fellowship. Dr. Jacquelin Kammeyer (UCLA) is acknowledged for helpful discussions and Mr. Nicholas Bernier (UCLA) is thanked for assistance with ICP-AES measurements. The authors thank Dr. Chi Zhang and Prof. Bradley L. Pentelute (MIT) for generously sharing the plasmid used for DARPin expression and the UCLA-DOE Institute Protein Expression Technology Center (PETC) for expression of DARPin and Dr. Yu Chen (UCLA) for help with mass spectrometry.
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Supporting Information
Detailed experimental procedures, characterization data, and crystallographic data are available in the Supporting Information. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Kalia J; Raines RT CurrOrg Chem. 2010, 14, 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Boutureira O; Bernardes GJL Chem. Rev 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]
- (3).Chalker JM; Bernardes GJL; Lin YA; Davis BG Chem. - An Asian J 2009, 4, 630–640. [DOI] [PubMed] [Google Scholar]
- (4).Spokoyny AM; Zou Y; Ling JJ; Yu H; Lin YS; Pentelute BL J. Am. Chem. Soc 2013, 135, 5946–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).DeGruyter JN; Malins LR; Baran PS Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vinogradova EV Pure Appl. Chem 2017, 89, 1619–1640. [Google Scholar]
- (7).Jbara M; Maity SK; Brik A Angew. Chem. - Int. Ed 2017, 56, 10644–10655. [DOI] [PubMed] [Google Scholar]
- (8).Vara BA; Li X; Berritt S; Walters CR; Petersson EJ; Molander GA Chem. Sci 2018, 9, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vinogradova EV; Zhang C; Spokoyny AM; Pentelute BL; Buchwald SL Nature 2015, 526, 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rojas AJ; Zhang C; Vinogradova EV; Buchwald NH; Reilly J; Pentelute BL; Buchwald SL Chem. Sci 2017, 8, 4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lee HG; Lautrette G; Pentelute BL; Buchwald SL Angew. Chem. - Int. Ed 2017, 56, 3177–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rojas AJ; Pentelute BL; Buchwald SL Org. Lett 2017, 19, 4263–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kubota K; Dai P; Pentelute BL; Buchwald SL J. Am. Chem. Soc 2018, 140, 3128–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Joost M; Amgoune A; Bourissou D Angew. Chem. - Int. Ed 2015, 54, 15022–15045. [DOI] [PubMed] [Google Scholar]
- (15).Harper MJ; Arthur CJ; Crosby J; Emmett EJ; Falconer RL; Fensham-Smith AJ; Gates PJ; Leman T; Mcgrady JE; Bower JF; Russell CA J. Am. Chem. Soc 2018, 140, 4440–4445. [DOI] [PubMed] [Google Scholar]
- (16).Livendahl M; Goehry C; Maseras F; Echavarren AM Chem. Commun 2014, 50, 1533–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Scott VJ; Labinger JA; Bercaw JE Organometallics 2010, 29, 4090–4096. [Google Scholar]
- (18).Hopkinson MN; Gee AD; Gouverneur V Chem. - A Eur. J 2011, 17, 8248–8262. [DOI] [PubMed] [Google Scholar]
- (19).Wegner HA; Auzias M Angew. Chem. - Int. Ed 2011, 50, 8236–8247. [DOI] [PubMed] [Google Scholar]
- (20).On-Yee Chan A; Lui-Lui Tsai J; Kar-Yan Lo V; Li GL; Wong M-K; Che C-M Chem. Commun 2013, 49, 1428–1430. [DOI] [PubMed] [Google Scholar]
- (21).Zou T; Lum CT; Lok C-N; Zhang J-J; Che C-M Chem. Soc. Rev 2015, 44, 8786–8801. [DOI] [PubMed] [Google Scholar]
- (22).Kung KK-Y; Ko H-M; Cui J-F; Chong H-C; Leung Y-C; Wong M-K Chem. Commun 2014, 50, 11899–11902. [DOI] [PubMed] [Google Scholar]
- (23).Joost M; Zeineddine A; Estévez L; Mallet-Ladeira S; Miqueu K; Amgoune A; Bourissou DJ Am. Chem. Soc 2014, 136, 14654–14657. [DOI] [PubMed] [Google Scholar]
- (24).Zeineddine A; Estévez L; Mallet-Ladeira S; Miqueu K; Amgoune A; Bourissou D Nat. Commun 2017, 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wolf WJ; Winston MS; Toste FD Nat. Chem 2014, 6, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kang K; Liu S; Xu T; Wang D; Leng X; Bai R; Lan Y; Shen Q Organometallics 2017, 36, 4727–4740. [Google Scholar]
- (27).Bachman RE; Bodolosky-Bettis SA; Pyle CJ; Gray MA J. Am. Chem. Soc 2008, 130, 14303–14310. [DOI] [PubMed] [Google Scholar]
- (28).Hesp KD; Stradiotto MJ Am. Chem. Soc 2010, 132, 18026–18029. [DOI] [PubMed] [Google Scholar]
- (29).Lundgren RJ; Sappong-Kumankumah A; Stradiotto M Chem. - A Eur. J 2010, 16, 1983–1991. [DOI] [PubMed] [Google Scholar]
- (30).Winston MS; Wolf WJ; Toste FD J. Am. Chem. Soc 2015, 137, 7921–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Itoh N; Ornitz DM J. Biochem 2011, 149, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yudin AK Chem. Sci 2015, 6, 30–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kaspar AA; Reichert JM Drug Discov. Today 2013, 18, 807–817. [DOI] [PubMed] [Google Scholar]
- (34).White CJ; Yudin AK Nat. Chem 2011, 3, 509–524. [DOI] [PubMed] [Google Scholar]
- (35).Blackwell HE; Grubbs RH Angew. Chem. - Int. Ed 1998, 37, 3281–3284. [DOI] [PubMed] [Google Scholar]
- (36).Verdine GL; Hilinski GJ Stapled peptides for intracellular drug targets, 1st ed.; Elsevier Inc, 2012; Vol. 503. [DOI] [PubMed] [Google Scholar]
- (37).Lau YH; de Andrade P; Wu Y; Spring DR Chem. Soc. Rev 2015, 44, 91–102. [DOI] [PubMed] [Google Scholar]
- (38).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; lavarone AT; Wells JA; Toste FD; Chang CJ Science 2017, 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Stephanopoulos N; Francis MB Nat. Chem. Biol 2011, 7, 876–884. [DOI] [PubMed] [Google Scholar]
- (40).Cal PMSD; Bernardes GJL; Gois PMP Angew. Chem. Int. Ed 2014, 53, 10585–10587. [DOI] [PubMed] [Google Scholar]
- (41).Fors BP; Watson DA; Biscoe MR; Buchwald SL J. Am. Chem. Soc 2008, 3, 3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wu CY; Horibe T; Jacobsen CB; Toste FD Nature 2015, 517, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mankad NP; Toste FD J. Am. Chem. Soc 2010, 2, 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tellis JC; Primer DN; Molander GA Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Biffis A; Centomo P; Del Zotto A; Zecca M Chem. Rev 2018, 118, 2249–2295. [DOI] [PubMed] [Google Scholar]
- (46).Stahl SS; Labinger JA; Bercaw JE Angew. Chem. - Int. Ed 1998, 37, 2180–2192. [DOI] [PubMed] [Google Scholar]
- (47).(a) For selected examples see: Crabtree RH Organomet. Chem. Transit. Met 2005, 491–520. For selected example see: [Google Scholar]; (b) Hull JF; Balcells D; Blakemore JD; Incarvito CD; Brudvig GW; Crabtree RH; Haven NJ Am. Chem. Soc 2009, 131, 8730–8731. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Breno KL; Ahmed TJ; Pluth MD; Balzarek C; Tyler DR Coord. Chem. Rev 2006, 250, 1141–1151. [Google Scholar]; (d) Therrien B In Chemistry of Nanocontainers; 2012; Vol. 319, pp 35–56. [Google Scholar]; (e) Allardyce CS; Dorcier A; Scolaro C; Dyson PJ Appl. Organomet. Chem 2005, 19, 1–10. [Google Scholar]; (f) Mohr F; Cerrada E; Laguna M Organometallics 2006, 25, 644–648. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.