

Abstract
Background
Paracetamol (acetaminophen) is the most widely used non‐prescription analgesic in the world. Paracetamol is commonly taken in overdose either deliberately or unintentionally. In high‐income countries, paracetamol toxicity is a common cause of acute liver injury. There are various interventions to treat paracetamol poisoning, depending on the clinical status of the person. These interventions include inhibiting the absorption of paracetamol from the gastrointestinal tract (decontamination), removal of paracetamol from the vascular system, and antidotes to prevent the formation of, or to detoxify, metabolites.
Objectives
To assess the benefits and harms of interventions for paracetamol overdosage irrespective of the cause of the overdose.
Search methods
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register (January 2017), CENTRAL (2016, Issue 11), MEDLINE (1946 to January 2017), Embase (1974 to January 2017), and Science Citation Index Expanded (1900 to January 2017). We also searched the World Health Organization International Clinical Trials Registry Platform and ClinicalTrials.gov database (US National Institute of Health) for any ongoing or completed trials (January 2017). We examined the reference lists of relevant papers identified by the search and other published reviews.
Selection criteria
Randomised clinical trials assessing benefits and harms of interventions in people who have ingested a paracetamol overdose. The interventions could have been gastric lavage, ipecacuanha, or activated charcoal, or various extracorporeal treatments, or antidotes. The interventions could have been compared with placebo, no intervention, or to each other in differing regimens.
Data collection and analysis
Two review authors independently extracted data from the included trials. We used fixed‐effect and random‐effects Peto odds ratios (OR) with 95% confidence intervals (CI) for analysis of the review outcomes. We used the Cochrane 'Risk of bias' tool to assess the risks of bias (i.e. systematic errors leading to overestimation of benefits and underestimation of harms). We used Trial Sequential Analysis to control risks of random errors (i.e. play of chance) and GRADE to assess the quality of the evidence and constructed 'Summary of findings' tables using GRADE software.
Main results
We identified 11 randomised clinical trials (of which one acetylcysteine trial was abandoned due to low numbers recruited), assessing several different interventions in 700 participants. The variety of interventions studied included decontamination, extracorporeal measures, and antidotes to detoxify paracetamol's toxic metabolite; which included methionine, cysteamine, dimercaprol, or acetylcysteine. There were no randomised clinical trials of agents that inhibit cytochrome P‐450 to decrease the activation of the toxic metabolite N‐acetyl‐p‐benzoquinone imine.
Of the 11 trials, only two had two common outcomes, and hence, we could only meta‐analyse two comparisons. Each of the remaining comparisons included outcome data from one trial only and hence their results are presented as described in the trials. All trial analyses lack power to access efficacy. Furthermore, all the trials were at high risk of bias. Accordingly, the quality of evidence was low or very low for all comparisons. Interventions that prevent absorption, such as gastric lavage, ipecacuanha, or activated charcoal were compared with placebo or no intervention and with each other in one four‐armed randomised clinical trial involving 60 participants with an uncertain randomisation procedure and hence very low quality. The trial presented results on lowering plasma paracetamol levels. Activated charcoal seemed to reduce the absorption of paracetamol, but the clinical benefits were unclear. Activated charcoal seemed to have the best risk:benefit ratio among gastric lavage, ipecacuanha, or supportive treatment if given within four hours of ingestion. There seemed to be no difference between gastric lavage and ipecacuanha, but gastric lavage and ipecacuanha seemed more effective than no treatment (very low quality of evidence). Extracorporeal interventions included charcoal haemoperfusion compared with conventional treatment (supportive care including gastric lavage, intravenous fluids, and fresh frozen plasma) in one trial with 16 participants. The mean cumulative amount of paracetamol removed was 1.4 g. One participant from the haemoperfusion group who had ingested 135 g of paracetamol, died. There were no deaths in the conventional treatment group. Accordingly, we found no benefit of charcoal haemoperfusion (very low quality of evidence). Acetylcysteine appeared superior to placebo and had fewer adverse effects when compared with dimercaprol or cysteamine. Acetylcysteine superiority to methionine was unproven. One small trial (low quality evidence) found that acetylcysteine may reduce mortality in people with fulminant hepatic failure (Peto OR 0.29, 95% CI 0.09 to 0.94). The most recent randomised clinical trials studied different acetylcysteine regimens, with the primary outcome being adverse events. It was unclear which acetylcysteine treatment protocol offered the best efficacy, as most trials were underpowered to look at this outcome. One trial showed that a modified 12‐hour acetylcysteine regimen with a two‐hour acetylcysteine 100 mg/kg bodyweight loading dose was associated with significantly fewer adverse reactions compared with the traditional three‐bag 20.25‐hour regimen (low quality of evidence). All Trial Sequential Analyses showed lack of sufficient power. Children were not included in the majority of trials. Hence, the evidence pertains only to adults.
Authors' conclusions
These results highlight the paucity of randomised clinical trials comparing different interventions for paracetamol overdose and their routes of administration and the low or very low level quality of the evidence that is available. Evidence from a single trial found activated charcoal seemed the best choice to reduce absorption of paracetamol. Acetylcysteine should be given to people at risk of toxicity including people presenting with liver failure. Further randomised clinical trials with low risk of bias and adequate number of participants are required to determine which regimen results in the fewest adverse effects with the best efficacy. Current management of paracetamol poisoning worldwide involves the administration of intravenous or oral acetylcysteine which is based mainly on observational studies. Results from these observational studies indicate that treatment with acetylcysteine seems to result in a decrease in morbidity and mortality, However, further evidence from randomised clinical trials comparing different treatments are needed.
Plain language summary
Interventions for paracetamol (acetaminophen) overdose
Review question: in this review, we looked at the evidence for the interventions (treatments) used to treat people with paracetamol (acetaminophen) poisoning. Mainly, we tried to assess what effects the interventions had on the number of deaths and the need for a liver transplant.
Background: paracetamol is one of the most common drugs taken in overdose. Intentional or accidental poisoning with paracetamol is a common cause of liver injury.
Search date: the evidence is current to January 2017.
Study characteristics: randomised clinical trials (studies where people are randomly put into one of two or more treatment groups) where participants had come to medical attention because they had taken a paracetamol overdose, intentionally or by accident, regardless of the amount of paracetamol taken or the age, sex, or other medical conditions of the person involved.
There are many different interventions that can be used to try to treat people with paracetamol poisoning. These interventions include decreasing the absorption of the paracetamol ingested and hence decreasing the amount absorbed into the bloodstream. The agents include activated charcoal (that binds paracetamol together in the stomach), gastric lavage (stomach washout to remove as much paracetamol as possible), or ipecacuanha (a syrup that is swallowed and causes vomiting (being sick)). Paracetamol once absorbed into the bloodstream goes to the liver where the majority is broken down to harmless products. However, a small amount of the medicine is converted into a toxic product that the liver can normally handle but, when large amounts of paracetamol are taken, the liver is overwhelmed. As a consequence, the toxic product can damage the liver leading to liver failure, kidney failure, and in some cases death. Other interventions to treat paracetamol poisoning include medicines (antidotes) that may decrease the amount of the toxic products (such as a medicine called cimetidine) or breakdown the toxic products (including medicines called methionine, cysteamine, dimercaprol, or acetylcysteine). Finally, attempts can be made to remove paracetamol and its toxic products from the bloodstream using special blood cleansing equipment. All these treatments were examined.
We found 11 randomised clinical trials with 700 participants. Most of these trials looked at different treatments.
Key results: activated charcoal, gastric lavage, and ipecacuanha may reduce absorption of paracetamol if started within one to two hours of paracetamol ingestion, but the clinical benefit was unclear. Activated charcoal seems to be the best choice if the person is able to take it. People may not be able to take charcoal if they are drowsy and some may dislike its taste or texture (or both).
Of the treatments that remove the toxic products of paracetamol, acetylcysteine seems to reduce the rate of liver injury from paracetamol poisoning. Furthermore, it has fewer side effects than some other antidotes such as dimercaprol and cysteamine; its superiority to methionine was unclear. Acetylcysteine should be given to people with paracetamol poisoning at risk of liver damage, risk is determined by the dose ingested, time of ingestion, and investigations.
More recent clinical trials have looked at ways to decrease side effects of intravenous (into a vein) acetylcysteine treatment, by altering the way it is given. These trials have shown that by using a slower infusion and lower initial dose of acetylcysteine, the proportion of side effects such as nausea (feeling sick) and vomiting, and allergy (the body's bad reaction to the medicine such as a rash) may be lowered.
Quality of the evidence: this review of interventions for paracetamol poisoning found surprisingly few published randomised clinical trials for this very common condition. Furthermore, the majority of trials had few participants and all were at high risk of bias. Accordingly, the quality of the evidence should be considered as low or very low.
Summary of findings
Background
Paracetamol (acetaminophen) is a mild analgesic and antipyretic agent which is commonly used worldwide (O'Grady 1997). In therapeutic doses (for adults 500 mg to 1000 mg, three or four times per day), paracetamol has few adverse events (Koch‐Weser 1976). During the late 1960s it was realised that paracetamol poisoning could result in severe hepatotoxicity, liver failure, renal failure, and death (Davidson 1966). Paracetamol is commonly taken in overdose either accidentally or intentionally (Buckley 2007), and in many countries, it is the most common single compound taken in overdose (Prescott 2009). In general, a single dose of more than 10 g or 150 mg/kg to 200 mg/kg of paracetamol carries a risk of liver damage (Buckley 1999a), but smaller doses may also cause liver damage (Kwan 1995), particularly in people with chronic alcohol abuse or anorexia. Paracetamol toxicity is the leading cause of acute liver failure in many high‐income countries (Lee 2004; Morgan 2005; Bernal 2013). One large prospective observational cohort study of 31 liver disease and transplant centres in the US, enrolling 2070 participants with acute liver failure between 1998 and 2013, found that paracetamol poisoning was the cause in half of the participants (Reuben 2016).
It was not until the 1970s that several antidotes that replenish glutathione and detoxify N‐acetyl‐p‐benzoquinone imine (NAPQI) were developed; these included methionine, cysteine, cysteamine, and dimercaprol (Prescott 1976). Oral methionine and intravenous acetylcysteine have been used as antidotes in the UK from this time onwards (McElhatton 1997). In one observational study from Edinburgh, intravenous acetylcysteine first‐line was claimed to be equally as effective as cysteamine and methionine and free of adverse effects (Prescott 1979). Ever since, acetylcysteine has been accepted as an antidote for paracetamol overdose either intravenously or orally. Much of the evidence for its use and efficacy comes from observational studies. Acetylcysteine has now become the mainstay and standard treatment for paracetamol poisoning and can either be administered as a 20‐ to 21‐hour intravenous acetylcysteine regimen or an oral acetylcysteine regimen (Smilkstein 1991; Woo 2000; Williamson 2013).
Before acetylcysteine treatment was available, morbidity following paracetamol overdose was significant. In people with an initial paracetamol concentration above the probable risk nomogram line (200 mg/L at four hours), the reported mortality of untreated people was 5% (Prescott 1979). This rate fell to 0.4% after the introduction of acetylcysteine (Gunnell 1997). Furthermore, the previous Cochrane systematic review of acetylcysteine observational studies found acute liver injury in 58% of people who received no antidote. This decreased to 7% if acetylcysteine was administered within 10 hours of ingestion and 27% if administered beyond 10 hours (Brok 2006). One systematic review of oral and intravenous acetylcysteine treatment following paracetamol poisoning from 1966 to 2009 found similar findings with late (greater than eight to 10 hours) acetylcysteine treatment associated with increased rates of hepatotoxicity (postbaseline aspartate aminotransferase (AST) or alanine aminotransferase (ALT) level above 1000 IU/L) (Green 2013). Green and colleagues included 5164 participants with paracetamol poisoning (definition varied according to the study) in their meta‐analysis; they also compared intravenous and oral acetylcysteine. Rates of hepatotoxicity were similar in both groups at 5% to 6% with early treatment (within eight to 10 hours postingestion) and increasing to 23% to 26% if treatment was given beyond this time.
Observational studies of acetylcysteine since the last Cochrane Review and meta‐analysis by Green and colleagues show very similar results with higher rates of hepatotoxicity in people treated more than eight hours postingestion (Duffull 2013; Marks 2017). More recently, observational studies have found an increased risk of acute liver injury despite early treatment in people with higher plasma paracetamol concentrations at admission. This relationship persists even in people treated within eight hours of acetylcysteine (Cairney 2016; Chiew 2017; Marks 2017). In these observational studies, deaths were uncommon and remained at less than 1%. Observational acetylcysteine studies continue to show low rates of acute liver injury particularly in people treated early. The rate of liver injury and death has improved from historical patient series of no antidote and this has meant randomised clinical trials assessing acetylcysteine versus no treatment have not been considered feasible. Instead, trials have focused on different acetylcysteine regimens dose or duration (or both) to examine the optimal way of administering acetylcysteine. Previous versions of this review have also included and examined observational studies. In this updated review, we excluded observational studies. In the last review and since, nearly all observational studies have examined outcomes with one treatment arm and no comparison groups. Hence, further analysis of observational studies adds little to evidence from previous reviews on treatment effectiveness or comparative effectiveness.
While methionine has fallen out of use in Western countries, it remains on the World Health Organization (WHO) essential drug list, a position that was reviewed and affirmed in 2011 (Shiago 2011). In 1984, the therapeutic guidelines in the British Medical Journal regarded methionine and acetylcysteine as equally effective (Henry 1984). The 2011 WHO panel review of the evidence concluded that acetylcysteine and methionine had equal efficacy and safety (and specifically that there was no evidence against this proposition), and thus, the cheaper methionine was the most cost‐effective antidote. Hence, it is widely used in low‐ to middle‐income countries such as Sri Lanka (Senarathna 2012).
Activated charcoal is a mode of decontamination often used in the management of people who have overdosed. Multiple observational and volunteer studies have investigated the effect of charcoal on paracetamol absorption (Buckley 1999a; Yeates 2000). Buckley and colleagues, in one observational study of 981 participants, found that people receiving activated charcoal within two hours of ingestion were less likely to have a toxic paracetamol concentration. In people receiving activated charcoal within two hours, 15% had a paracetamol concentration above 150 mg/L at the four‐hour nomogram treatment line compared to 41% having a level above the same line who did not receive activated charcoal (Buckley 1999a). Similarly, Duffull and colleagues, in one observational study of 1571 people with acute paracetamol poisoning found that those receiving activated charcoal had a reduced probability of having a paracetamol concentration above 150 mg/L at the four‐hour nomogram line (Duffull 2013). One observational series of 200 participants ingesting greater than 40 g of paracetamol found paracetamol concentrations were markedly reduced in those receiving activated charcoal within four hours and a probable benefit of reducing the risk of hepatotoxicity (ALT greater than 1000 U/L) (Chiew 2017). Healthy volunteer studies similarly showed a reduction in paracetamol absorption when activated charcoal was administered within two hours of ingestion (Yeates 2000; Green 2001; Christophersen 2002).
Paracetamol poisoning treatment protocols vary worldwide (Wolf 2007; MHPRA 2012; Chiew 2015; Heard 2017). People are unlikely to develop hepatotoxicity if they have ingested less than 150 mg/kg to 200 mg/kg of paracetamol (Vale 2004; Dart 2006), or 10 g of paracetamol (whichever is less) (Buckley 1999a), unless people have other risk factors such as chronic ethanol abuse or anorexia. A prediction of a person's risk based on reported dose of paracetamol may be limited as they or their relatives are often unaware of the exact amount ingested and the exact timing. The decision to treat a person with acute paracetamol ingestion with either acetylcysteine or methionine is usually based on their paracetamol concentration taken at a known time since ingestion. This concentration is plotted on a paracetamol nomogram such as the Rumack‐Matthew nomogram, to determine the need for treatment (Smilkstein 1988). Plotting paracetamol concentration versus time since ingestion, there are various 'treatment lines' and 'risk lines' for developing hepatotoxicity that are utilised to guide treatment. These 'nomograms' lines are sometimes referred to as the high risk (300 line; i.e. a line commencing from a paracetamol concentration of 300 mg/L at four hours postingestion), the probable risk (200 line), and the possible risk (150 line) used to guide treatment in such countries as Canada, Australia, New Zealand, and the US (Rumack 1975; Prescott 1979; Smilkstein 1991; Daly 2008). In the UK in 2012, the treatment line was lowered further to the 100 mg/L (660 μmol/L) line (MHPRA 2012). However, other countries do not utilise these nomograms and treat all people with acute paracetamol ingestion with acetylcysteine (Schmidt 2001). If the time of ingestion is unknown, or the treating doctor is not confident of the history of ingestion, or if a paracetamol concentration is not available or not used, treatment with acetylcysteine is commenced (Dart 2006; Daly 2008).
Description of the condition
Paracetamol overdose prior to the 1970s was associated with significant morbidity and mortality. It is still the leading cause of acute liver failure in Western countries (Bernal 2013). Paracetamol is extensively metabolised by the liver; in therapeutic doses in adults, the major non‐toxic metabolites are sulphate and glucuronide conjugates which account for 30% (sulphate) and 55% (glucuronide) of the metabolites. A highly reactive toxic metabolite NAPQI is formed by cytochrome P450 2E1; it is responsible for the hepatocellular injury that occurs when paracetamol is taken in excess (Mitchell 1974). The small amounts of NAPQI produced after therapeutic doses of paracetamol are detoxified by glutathione‐dependent reactions. However, in paracetamol overdose, the formation of NAPQI depletes glutathione; once glutathione is depleted to about one‐third of its normal level, NAPQI starts binding covalently to critical cellular proteins. It is hypothesised that this results in loss of activity and function of critical proteins and eventually hepatic cell death (Mitchell 1974).
Description of the intervention
Many different types of interventions are used to treat paracetamol overdose. These interventions include:
those that decrease paracetamol absorption from the gastrointestinal tract, including gastric lavage, activated charcoal, and ipecacuanha (ipecac syrup, an emetic);
antidotes that prevent the conversion of paracetamol to its hepatotoxic metabolite NAPQI, such as cimetidine;
antidotes to detoxify NAPQI, such as methionine, cysteine, cysteamine, dimercaprol, or acetylcysteine;
those that remove paracetamol from the blood after the drug has entered the bloodstream. This includes intermittent haemodialysis, intermittent haemoperfusion, continuous renal replacement modalities, or charcoal haemoperfusion.
How the intervention might work
There are many different interventions that can be utilised to manage a person with paracetamol poisoning. These interventions work in different ways. First, there are interventions to reduce the absorption of paracetamol once ingested (decontamination), either by binding paracetamol to activated charcoal or removing paracetamol from the stomach by gastric lavage or ipecac syrup (forcing the person to vomit) (Underhill 1990; Buckley 1999a).
Once absorbed into the bloodstream, paracetamol can be removed from the blood, in cases of severe poisoning, with intermittent haemodialysis, intermittent haemoperfusion, continuous renal replacement modalities, or charcoal haemoperfusion (O'Grady 1988; Higgins 1996; Gosselin 2014).
Other treatment options are drugs such as cimetidine that work by inhibiting cytochrome P‐450. The enzyme cytochrome P‐450 breaks down paracetamol into the toxic metabolite NAPQI. By inhibiting cytochrome‐P450 this may reduce the production of NAPQI (Speeg 1995).
Antidotes that detoxify NAPQI, work by replenishing glutathione, and hence preventing the toxic effects due to this metabolite. Several antidotes to NAPQI were developed in the 1970s, including methionine, cysteine, cysteamine, and dimercaprol (Prescott 1976). The amino acid, cysteine, is the main factor limiting the synthesis of glutathione. Acetylcysteine is a cysteine precursor, that is hydrolysed intracellularly to cysteine, thus replenishing glutathione (Olsson 1988). Glutathione can then covalently bind NAPQI in a 1:1 ratio. NAPQI is then detoxified via irreversible glutathione conjugation to two non‐toxic metabolites, mercapturic acid and cysteine conjugates (Prescott 1980). Acetylcysteine also supplies thiol groups, which can directly react with NAPQI in hepatocytes (Jones 1998).
Why it is important to do this review
Paracetamol overdose is common and is still a leading cause of acute liver failure in many countries. This updated systematic review aimed to assess the benefits and harms of interventions for paracetamol overdose.
Objectives
To assess the benefits and harms of interventions for paracetamol overdosage irrespective of the cause of the overdose.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised clinical trials examining the benefits and harms of interventions for people with paracetamol overdose regardless of sources of publication and language.
Types of participants
People who had ingested a paracetamol overdose. The definition of a paracetamol overdose was not clear‐cut and the risk depended on many factors such as age, weight, comorbidities, concomitant medication, and alcohol ingestion. Therefore, all trials on people with paracetamol poisoning were included irrespective of inclusion criteria applied in the trial (e.g. age, time to treatment, comorbidities, etc.).
Types of interventions
Intervention with gastric lavage, ipecacuanha, or activated charcoal at any dose or duration compared with placebo/no intervention or with each other.
Intervention with antidotes (cimetidine, cysteamine, methionine, dimercaprol, and acetylcysteine) compared with each other, with placebo/no interventions, or other interventions for paracetamol overdose.
Intervention with extracorporeal treatments such as charcoal haemoperfusion, intermittent haemodialysis, or continuous renal replacement therapy compared with placebo/no interventions or other interventions for paracetamol overdose.
Different doses, durations, or method of administration (oral or intravenously) of acetylcysteine compared with each other.
Cointerventions were allowed if received equally in all groups of the trial.
We did not want to examine interventions for liver failure, for example different types of liver support systems, or interventions to treat secondary complications of liver failure such as hepatorenal failure, hepatic encephalopathy, coagulopathy, and cerebral oedema. Interventions for acute liver failure would benefit from being addressed in a separate review, with a subgroup analysis of people with paracetamol overdose.
Types of outcome measures
Primary outcomes
Mortality: all‐cause and liver‐related.
Liver transplantation.
Secondary outcomes
Acute hepatitis (elevation of the serum transaminases greater than three times the upper limit of normal (ULN)).
Hepatotoxicity (most commonly defined as number of participants with serum AST or serum ALT greater than 1000 IU/L).
Severe acute hepatitis: transaminitis plus an international normalised ratio (INR) greater than 2.
Acute (fulminant) hepatic failure defined as development of hepatic encephalopathy on a background of severe acute hepatitis (elevation of the serum transaminases plus prolongation of the prothrombin time).
Adverse events.
Plasma paracetamol concentration (e.g. plasma paracetamol above a risk line (nomogram)), fall in plasma paracetamol versus time, absorption of paracetamol measured as area under the curve (AUC) of the plasma (or urine) concentration versus time curve.
Search methods for identification of studies
Electronic searches
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register (Gluud 2017; January 2017), Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library (2016, Issue 11), MEDLINE Ovid (1946 to January 2017), Embase Ovid (1974 to January 2017), and Science Citation Index Expanded (Web of Science; 1900 to January 2017) (Royle 2003). Appendix 1 provides the search strategies and the time spans of the searches.
Searching other resources
We examined the reference lists of relevant papers identified by the search and other published reviews. We searched the WHO International Clinical Trials Registry Platform (www.who.int/ictrp/en/), which includes (among others) the EU Clinical Trials Register and Australian New Zealand Clinical Trials Registry. We searched the ClinicalTrials.gov database, a service of the US National Institute of Health for trials (clinicaltrials.gov).
Data collection and analysis
Two review authors (AC, NB) screened the electronic search results for possibly relevant trials and retrieved the full text. Two review authors (AC and NB) evaluated whether the trials fulfilled the inclusion criteria and extracted data. We resolved disagreements by discussion. We listed included trials (Characteristics of included studies table) and excluded trials (Characteristics of excluded studies table) with the reason for exclusion. We wrote to the principal investigator of included trials to ask for relevant data if such data were not presented in the published reports.
Assessment of risk of bias in included studies
As there were changes in the risk of bias domains since the last review, we reassessed all trials. Two review authors (AC and NB) independently assessed risk of bias of all included studies using Cochrane's tool for assessing domains for risk of bias (Higgins 2011a) according to The Cochrane Hepato‐Biliary Group Module (Gluud 2017) and methodological studies (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b; Lundh 2017). We used the following definitions in the assessment of risk of bias.
Allocation sequence generation
Low risk of bias: sequence generation was achieved using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards, and throwing dice were adequate if performed by an independent person not otherwise involved in the trial.
Unclear risk of bias: the method of sequence generation was not specified.
High risk of bias: the sequence generation method was not randomised or only quasi‐randomised. We only used these studies for the assessments of harms and not for benefits.
Allocation concealment
Low risk of bias: the participant allocations could not have been foreseen in advance of, or during, enrolment. Allocation was controlled by a central and independent randomisation unit, onsite locked computer, identically looking numbered sealed opaque envelopes, or drug bottles or containers prepared by an independent pharmacist or investigator. The allocation sequence was unknown to the investigators.
Unclear risk of bias: the method used to conceal the allocation was not described so that intervention allocations may have been foreseen in advance of, or during, enrolment.
High risk of bias: the allocation sequence was likely to be known to the investigators who assigned the participants. We only used these studies for the assessments of harms and not for benefits.
Blinding of participants and treatment providers (performance bias)
Low risk of bias: it was mentioned that both participants and personnel providing the interventions were blinded and this was described.
Unclear risk of bias: it was not mentioned if the trial was blinded, or the extent of blinding was insufficiently described.
High risk of bias: no blinding or incomplete blinding was performed.
Blinding of outcome assessment (detection bias)
Low risk of bias: it was mentioned that outcome assessors were blinded and this was described.
Unclear risk of bias: it was not mentioned if the trial was blinded, or the extent of blinding was insufficiently described.
High risk of bias: no blinding or incomplete blinding was performed.
Incomplete outcome data
Low risk of bias: missing data were unlikely to make treatment effects depart from plausible values. The study used sufficient methods, such as multiple imputation, to handle missing data.
Unclear risk of bias: there was insufficient information to assess whether missing data in combination with the method used to handle missing data were likely to induce bias on the results.
High risk of bias: the results were likely to be biased due to missing data.
Selective outcome reporting
Low risk of bias: a protocol was published before or at the time the trial was begun and the outcomes called for in the protocol were reported on. If there was no protocol or the protocol was published after the trial had begun, reporting of all‐cause mortality and serious adverse events granted the trial a grade of low risk of bias.
Unclear risk of bias: no protocol was published and the outcomes all‐cause mortality and serious adverse events were not reported on.
High risk of bias: the outcomes in the protocol were not reported on.
For‐profit bias
Low risk of bias: the trial appeared to be free of industry sponsorship or other type of for‐profit support that may have manipulated the trial design, conductance, or results of the trial.
Unclear risk of bias: the trial may or may not have been free of for‐profit bias as no information on clinical trial support or sponsorship was provided.
High risk of bias: the trial was sponsored by industry or received other type of for‐profit support.
Other bias
Low risk of bias: the trial appeared to be free of other bias domains (e.g. academic bias) that could put it at risk of bias.
Unclear risk of bias: the trial may or may not have been free of other domains that could put it at risk of bias.
High risk of bias: there were other factors in the trial that could have put it at risk of bias (e.g. authors had conducted trials on the same topic).
Overall risk of bias
We judged trials to be at a low risk of bias if they were assessed as at a low risk of bias in all the above domains. We judged trials to be at a high risk of bias if they were assessed as having an unclear risk of bias or a high risk of bias in one or more of the above domains. We assessed the domains 'blinding of outcome assessment' and 'incomplete outcome data' for each outcome. Thus, we were able to assess the bias risk for each result in addition to each trial. The results of our primary outcomes with a low risk of bias should have been our primary analyses.
Two review authors (AC and NB) independently assessed the risk of bias of each included trial against these criteria. Review authors were not blinded with respect to trial authors, institution, or journal. The authors resolved disagreements by consensus, with a third review author (CG) to be consulted if disagreements persisted.
Where the method of allocation concealment was not reported, or where additional information was required to appropriately assess study quality, we contacted the authors of these trials for clarification. We contacted the authors of three studies and received two replies; however, the responses did not uniformly clarify our questions.
Measures of treatment effect
Measures of treatment effect
We performed the analyses in Review Manager 5 (RevMan 2014). Where possible, we analysed data by intention‐to‐treat including all participants irrespective of compliance or follow‐up.
Dichotomous outcomes
We expressed binary outcomes as odds ratios (OR) with 95% confidence intervals (CI). We estimated rare events (mortality and liver transplantation) by Peto ORs (Bradburn 2007). We used both a random‐effects model (DerSimonian 1986) and a fixed‐effect model meta‐analysis to assess data analysed by OR (DeMets 1987). We explored heterogeneity using the Chi2 test with significance set at P value of 0.10 or less and we measured heterogeneity using the I2 statistic (Higgins 2002). Where conclusions were different, we favoured a random‐effects model if there was a high degree of heterogeneity.
Continuous outcomes
The main outcomes assessed in this systematic review were analysed as dichotomous outcomes as this is how ALT/AST and INR are consistently reported. Paracetamol pharmacokinetic data such as paracetamol concentration where possible was analysed as a continuous outcome. Data on participants were collected until discharge, death, or liver transplantation. We calculated the mean differences (MD; if trials used the same methods of measurement) and the standardised mean difference (SMD; if trials used different methods of measurement) with 95% CI for continuous outcomes.
Mortality data
We analysed mortality data using hospital death (mortality) or liver transplantation as outcomes. We used estimates of log hazard ratios and standard errors. If the trialists did not report these data, we calculated the log hazard ratios and standard errors if possible (Higgins 2011a). We used the generic inverse‐variance method to meta‐analyse survival data (see Section 9.4.3.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b).
Dealing with missing data
Dichotomous outcomes
If the trialists used the correct methodology (e.g. multiple imputation) to deal with missing data, we used these data in our primary analysis.
Continuous outcomes
The only continuous outcomes intended to be analysed were the secondary outcomes of pharmacokinetic data such as paracetamol concentrations or area under the paracetamol curve. If trialists used correct methodology (e.g. multiple imputation) to deal with missing data, we intended to use these data in our primary analysis. If standard deviations (SD) were not reported, we intended to calculate the SDs using data from the trial if possible. Missing pharmacokinetic data were calculated by using non‐linear mixed effects modelling, provided all individual participant data were available.
Assessment of heterogeneity
We assessed the presence of statistical heterogeneity using the Chi2 test with significance set at P < 0.10 and measure the level of heterogeneity using the I2 statistic (Higgins 2002; Higgins 2011b).
Assessment of reporting biases
We planned to use a funnel plot to assess reporting bias had we included 10 or more trials per comparison. Using the asymmetry of the funnel plot, we planned to assess the risk of bias. For dichotomous outcomes, we planned to test asymmetry using the Harbord test (Harbord 2006). For continuous outcomes, we planned to use the regression asymmetry test (Egger 1997) and the adjusted rank correlation (Begg 1994).
Data synthesis
We planned to base our primary conclusions on the results of the primary outcomes with a low risk of bias at the end of intervention. We considered the results of primary outcomes with high risk of bias, and secondary outcomes, outcomes at maximum follow‐up, sensitivity analyses, and subgroup analyses as hypothesis‐generating tests (Jakobsen 2014).
Meta‐analysis
We conducted the meta‐analyses according to the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We used the statistical software Review Manager 5 (RevMan 2014) provided by Cochrane to analyse data (RevMan 2014).
Assessment of significance
We planned to assess our intervention effects using both random‐effects model meta‐analyses (DerSimonian 1986) and fixed‐effect model meta‐analyses (DeMets 1987). We used the more conservative point estimate of the two (Jakobsen 2014). The more conservative point estimate was the estimate closest to zero effect. If the two estimates were equal, we used the estimate with the widest CI. We used three primary outcomes and, therefore, we considered a P value of 0.025 or less as statistically significant (Jakobsen 2014). We used the eight‐step procedure to assess if the thresholds for significance are crossed (Jakobsen 2014).
Trial Sequential Analysis
Traditional meta‐analysis runs the risk of random errors due to sparse data and repetitive testing of accumulating data when updating reviews. Therefore, we performed Trial Sequential Analysis (Thorlund 2011; TSA 2011; Wetterslev 2017) on the outcomes to calculate the required information size and assess the potential breach of the cumulative Z‐curve of the relevant trial sequential monitoring boundaries for benefit, harm, or futility to control for risks of type I errors and type II errors (Brok 2008; Wetterslev 2008; Brok 2009; Thorlund 2009; Wetterslev 2009; Thorlund 2010). A more detailed description of Trial Sequential Analysis can be found at www.ctu.dk/tsa/ (Thorlund 2011).
For dichotomous outcomes, we planned to estimate the required information size based on the proportion of participants with an outcome in the control group, a relative risk reduction of 20%, an alpha of 2.5% (Jakobsen 2014), a beta of 20%, and an assumed diversity of 20% as we had only one or two trials included in each Trial Sequential Analysis without any observable heterogeneity. For continuous outcomes, we planned to estimate the required information size based on the SD observed in the control group of trials with low risk of bias and a minimal relevant difference of 50% of this SD, an alpha of 2.5%, a beta of 20%, and the diversity suggested by the trials in the meta‐analysis.
Subgroup analysis and investigation of heterogeneity
We intended to conduct the following subgroup analyses.
Outcomes at a low risk of bias compared to outcomes at a high risk of bias.
Age of participants categorised into 10‐year groups.
Risk of hepatotoxicity at baseline (according to paracetamol concentration data).
However, due to insufficient data, these analyses could not be conducted.
Sensitivity analysis
To assess the potential impact of missing data for dichotomous outcomes, we intended to perform the following two sensitivity analyses.
'Best‐worst case' scenario: we assumed that all participants lost to follow‐up in the intervention group survived, had no serious adverse event, and had no morbidity; and all those participants with missing outcomes in the control group did not survive, had a serious adverse event, and had morbidity.
'Worst‐best case' scenario: we assumed that all participants lost to follow‐up in the intervention group survived, had a serious adverse event, and had morbidity; and that all those participants lost to follow‐up in the control group had survived, had no serious adverse event, and had no morbidity.
To assess the potential impact of missing SDs for continuous outcomes, we intended to perform the following sensitivity analysis.
Where SDs were missing and it was not possible to calculate them, we planned to impute SDs from trials with similar populations and low risk of bias. If we found no such trials, we intended to impute SDs from trials with a similar population. As the final option, we planned to impute SDs from all trials.
'Summary of findings' tables
We used GRADE to assess the quality of the evidence (Guyatt 2008) associated with each of the major outcomes in our review constructing 'Summary of findings' tables using GRADE software (ims.cochrane.org/revman/other‐resources/gradepro). The GRADE approach appraises the quality of a body of evidence based on the extent to which one can be confident that an estimate of effect or association reflects the item being assessed. The quality measure of a body of evidence considers the within‐study risk of bias, indirectness of the evidence, heterogeneity of data, imprecision of effect estimates, and risk of publication bias.
Results
Description of studies
Results of the search
See flow chart (Figure 1). An updated search was performed in October 2013 that identified 220 references. We excluded 63 as they were animal studies or studies that did not involve paracetamol ingestion. A further 61 were non‐randomised clinical studies, 31 were healthy volunteer studies and 25 were duplicate references. We reviewed 40 full‐text articles. Of these, we included 10 clinical trials and one quasi‐randomised clinical trial. One abandoned randomised clinical trial was identified in the clinical trials database. The search was updated in June 2015 and identified 44 new references of which two were randomised clinical trials, one was previously identified and one was reviewed and excluded as it was a randomised clinical trial of albumin dialysis with the Molecular Adsorbent Recirculating System (MARS) used for the treatment of fulminant liver failure. The search was updated again in January 2017, identifying 49 new references, four of which were randomised clinical trials that had been identified previously. See: Characteristics of included studies; Characteristics of excluded studies tables. Below, we describe the trials according to the assessed interventions.
1.
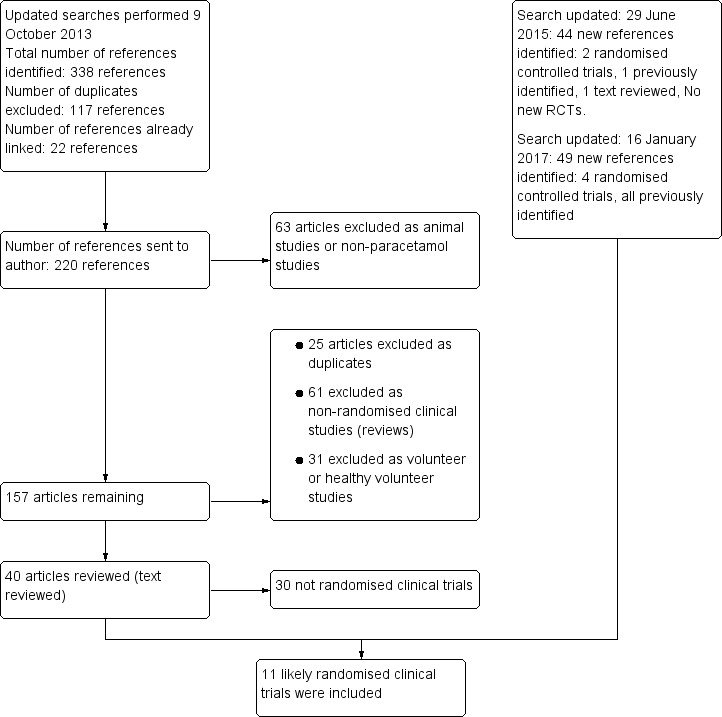
Flow chart: search strategy and results.
Included studies
Prevention of absorption
One trial allocated participants to activated charcoal, ipecacuanha, gastric lavage, or no intervention (Underhill 1990). The primary outcome was the mean percentage fall in paracetamol concentration (from the first to the last sample (150 minutes).
Antidotes
Three trials randomised participants to different antidotes (methionine, cysteine, cysteamine, or dimercaprol) (Douglas 1976a; Hughes 1977; Hamlyn 1981). One trial randomised participants with paracetamol‐induced fulminant hepatic failure to acetylcysteine versus placebo (Keays 1991).
Some randomised clinical trials looked at differing acetylcysteine regimens to decrease the rate of adverse effects from acetylcysteine treatment (Kerr 2005; Bateman 2014). One trial randomised participants to receive either intravenous or oral acetylcysteine (Arefi 2013). Another trial randomised participants to intravenous or intravenous plus oral acetylcysteine. This trial excluded post hoc 40% of participants who vomited twice after oral acetylcysteine was given, and it was unclear what the treatment and outcomes were for these randomised participants (Eizadi‐Mood 2013). Two trials randomised participants to different infusion rates of intravenous acetylcysteine compared with the traditional 20.25‐hour intravenous regimen (Kerr 2005; Bateman 2014).
One multicentre randomised, blind clinical trial, started in 2010, was registered in the US by Cumberland Pharmaceuticals, and compared an intravenous acetylcysteine regimen with a two‐bag regimen (200 mg/kg over four hours followed by 100 mg/kg over 16 hours) versus the traditional acetylcysteine regimen (NCT01118663). This trial was terminated early after enrolling only 17 participants, although the number to be recruited was not reported (NCT01118663).
There was one quasi‐randomised trial that studied cimetidine plus acetylcysteine (Burkhart 1995).
Extracorporeal treatments
One trial randomised participants with acute paracetamol overdose to charcoal haemoperfusion versus no intervention (Gazzard 1974a).
Other interventions
One trial looked at ondansetron to decrease the risk of vomiting as an adjunct to acetylcysteine treatment (Bateman 2014). This was a part of the trial that assessed a modified 12‐hour acetylcysteine regimen.
Excluded studies
We excluded 33 for reasons given in the Characteristics of excluded studies table.
Studies awaiting classification
We found no studies awaiting classification.
Ongoing studies
We found no ongoing studies.
Risk of bias in included studies
Allocation (selection bias)
Generation of the allocation sequence was often not specified, and only five trials described details of randomisation (Douglas 1976a; Hamlyn 1981; Kerr 2005; Eizadi‐Mood 2013; Bateman 2014). Allocation concealment was similarly not described in detail, with most trials at unclear risk of bias, except for two randomised trials (Keays 1991; Bateman 2014).
Blinding (performance bias and detection bias)
All randomised trials were conducted unblinded except one, which used placebo but failed to mask the aroma (Keays 1991). Accordingly, we judged most trials to be at high risk of bias due to lack of blinding. In one trial, the control group was given supportive treatment in a different hospital, which may have seriously affected the value of this comparison group and questions how the randomisation was carried out (Underhill 1990). One trial of intravenous versus a combination of oral plus intravenous acetylcysteine had nausea and vomiting from acetylcysteine treatment as a primary outcome. However, participants were excluded and not analysed if they vomited twice after oral acetylcysteine, which resulted in exclusion of 40% of the participants from the analysis and biased the results (Eizadi‐Mood 2013).
Incomplete outcome data (attrition bias)
The trials varied in their reporting of missing or incomplete data; four trials were judged to be at low risk of bias (Gazzard 1974a; Douglas 1976a; Hughes 1977; Bateman 2014). Four trials did not mention if they had missing outcome data or how missing data were handled, so we judged them to be at unclear risk of attrition bias (Hamlyn 1981; Underhill 1990; Keays 1991; Arefi 2013). We judged two trials to be at high risk of attrition bias because both of them excluded a large number of participants from the analysis (Kerr 2005; Eizadi‐Mood 2013).
Selective reporting (reporting bias)
We judged six trials to be at low risk of reporting bias (Gazzard 1974a; Douglas 1976a; Hughes 1977; Hamlyn 1981; Keays 1991; Bateman 2014). We judged one trial to be at high risk of reporting bias because the trial authors did not report on their planned outcomes (Eizadi‐Mood 2013). In three trials, the risk of reporting bias was unclear (Underhill 1990; Kerr 2005; Arefi 2013). Arefi 2013 was not a registered trial and the primary outcome was unclear. Underhill 1990 did not report on the relevant clinical outcomes such as need for treatment with antidote, and Kerr 2005 had two investigators to make a judgement on the attribution of an event, and it is unclear whether bias might have been introduced in the process of adjudicating on events.
Other potential sources of bias
We judged all but two trials at unclear risk of bias for potential other sources of bias because these trials did not have a sample size calculation, did not perform intention to treat analysis, or did not report on the number of participants screened for randomisation or number of participants excluded from the trial or analysis. Eizadi‐Mood 2013 was at high risk of bias because of the lack of detail regarding the high number of participants excluded from the trial. Thus, only one trial was judged to be at low risk of bias for this domain (Bateman 2014).
In conclusion, the trials varied considerably for each risk of bias domain. Figure 2 shows the percentages across all included trials of each risk of bias item as judged by the review authors. Figure 3 shows the risk of bias in each study as judged by the review authors.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10
Summary of findings for the main comparison. Methionine and supportive treatment compared with supportive treatment for paracetamol (acetaminophen) overdose.
| Methionine and supportive treatment compared with supportive treatment (randomised trials) for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: UK Intervention: methionine and supportive treatment Comparison: supportive treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Supportive treatment | Methionine and supportive treatment | |||||
| Mortality | Study population | Peto OR 0.14 (0.00 to 6.82) | 26 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | The Trial Sequential Analysis‐adjusted CI could not be estimated due to the paucity of data. | |
| 77 per 1000 | 12 per 1000 (0 to 362) | |||||
| Hepatotoxicity | Study population | OR 0.05 (0.01 to 0.53) | 26 (1 RCT) | ⊕⊕⊝⊝ Low1,3 | ‐ | |
| 615 per 1000 | 74 per 1000 (16 to 459) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one level for risk of bias (concerns regarding randomisation sequence generation and allocation concealment probably compromised). 2Downgraded two levels because of serious imprecision (due to small sample studied, low number of deaths, and wide confidence intervals). 3Downgraded one level because of imprecision (due to small sample studied).
Summary of findings 2. Cysteamine compared with no intervention for paracetamol (acetaminophen) overdose.
| Cysteamine compared with no intervention (randomised trials) for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: Royal Victoria Infirmary, Newcastle, UK Intervention: cysteamine Comparison: no intervention | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| No intervention | Cysteamine | |||||
| Mortality | Study population | Peto OR 0.53 (0.05 to 5.22) | 65 (2 RCTs) | ⊕⊝⊝⊝ Very low1,2 | ‐ | |
| 61 per 1000 | 33 per 1000 (3 to 252) | |||||
| Hepatotoxicity (aspartate aminotransferase > 1000 IU/L) | Study population | OR 0.09 (0.02 to 0.35) | 65 (2 RCTs) | ⊕⊕⊝⊝ Low1,3 | Trial Sequential Analysis‐adjusted CI ranged from 0.00 to 24.0. | |
| 545 per 1000 | 97 per 1000 (23 to 290) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one level because of risk of bias (method of randomisation had potential for bias and allocation concealment not specified). 2Downgraded two levels because of serious imprecision (due to small sample studied, low number of deaths, and confidence intervals are wide). 3Downgraded one level because of imprecision (due to small sample studied).
Summary of findings 3. Cysteamine compared with dimercaprol for paracetamol (acetaminophen) overdose.
| Cysteamine compared with dimercaprol (randomised trials) for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: UK Intervention: cysteamine Comparison: dimercaprol | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Dimercaprol | Cysteamine | |||||
| Mortality | Study population | Peto OR 0.14 (0.00 to 6.82) | 52 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | ‐ | |
| 38 per 1000 | 6 per 1000 (0 to 214) | |||||
| Mean maximum alanine aminotransferase (IU/L) | The mean maximum alanine aminotransferase (IU/L) in the dimercaprol was 754 | The mean maximum alanine aminotransferase (IU/L) in the cysteamine group was 722 (IU/L) | ‐ | 52 (1 RCT) | ⊕⊕⊝⊝ Low1,3 | Difference ‐32.00 (95% CI ‐512.9 to 448.9). The difference between the 2 groups was not significant. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded two levels because of serious imprecision (due to small sample studied, low number of deaths, and confidence intervals wide). 2Downgraded one level because of risk of bias (method of randomisation by envelopes and allocation not concealed). 3Downgraded one level because of imprecision (due to small sample studied).
Summary of findings 4. Cysteamine compared with methionine (randomised trials) for paracetamol (acetaminophen) overdose.
| Cysteamine compared with methionine (randomised trials) for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: Newcastle (Royal Victoria Infirmary) and London (Guy's Hospital) Intervention: cysteamine Comparison: methionine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Methionine | Cysteamine | |||||
| Mortality | Study population | Not estimable | 27 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Hepatotoxicity (aspartate aminotransferase > 1000 U/L) | Study population | OR 0.92 (0.05 to 16.46) | 27 (1 RCT) | ⊕⊝⊝⊝ Very low1,3 | ‐ | |
| 77 per 1000 | 71 per 1000 (4 to 578) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one level because of risk of bias (concerns regarding randomisation and allocation concealment not specified). 2Downgraded two levels because of serious imprecision (due to small sample studied and low number of deaths). 3Downgraded two levels because of serious imprecision (due to small sample studied and wide confidence intervals).
Summary of findings 5. Standard intravenous acetylcysteine regimen (20.5 hour) compared with shorter intravenous acetylcysteine regimen (12 hour) for paracetamol (acetaminophen) overdose.
| Standard intravenous acetylcysteine regimen (20.5 hours) compared with shorter (12 hours) protocol for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: 3 acute clinical units in the UK Intervention: standard intravenous acetylcysteine regimen (20.25 hours) Comparison: shorter (12 hours) modified protocol | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Standard intravenous acetylcysteine regimen (20.25 hours) | Shorter (12‐hour protocol) | |||||
| Mortality | Study population | Not estimable | 222 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Hepatotoxicity | Study population |
OR 0.67 (0.11 to 4.08) |
202 (1 RCT) |
⊕⊝⊝⊝ Very low1,3 | ‐ | |
| 30 per 1000 | 20 per 1000 (3 to 111) |
|||||
| Vomiting, retching, or antiemetics from 0‐2 hours | Study population |
OR 0.30 (0.17 to 0.53) |
217 (1 RCT) | ⊕⊕⊝⊝ Low1,4 | ‐ | |
| 651 per 1000 | 359 per 1000 (241 to 498) | |||||
| Vomiting, retching, or antiemetics 0‐12 hours | Study population |
OR 0.40 (0.22 to 0.75) |
203 (1 RCT) | ⊕⊕⊝⊝ Low1,4 | ‐ | |
| 784 per 1000 | 593 per 1000 (444 to 732) | |||||
| Anaphylactoid symptoms | Study population |
OR 0.39 (0.21 to 0.70) |
208 (1 RCT) | ⊕⊕⊝⊝ Low1,4 | ‐ | |
| 750 per 1000 | 539 per 1000 (387 to 677) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one level because of indirectness (a large number of prospective participants excluded prior to randomisation: 1539 judged suitable for treatment, only 222 randomised). 2Downgraded two levels because of very serious imprecision (due to small sample studied and no deaths). 3Downgraded two levels because of very serious imprecision (due to small sample studied, small numbers who developed hepatotoxicity, and wide confidence intervals). 4Downgraded one level because of imprecision (due to small sample studied).
Summary of findings 6. Oral compared with intravenous acetylcysteine for paracetamol (acetaminophen) overdose.
| Oral compared with intravenous acetylcysteine for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: Baharloo Hospital (Tehran) Intervention: oral acetylcysteine Comparison: intravenous acetylcysteine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Intravenous acetylcysteine | Oral acetylcysteine | |||||
| Mortality | Study population | Not estimable | 66 (1 RCT) | ⊕⊝⊝⊝ Very low1,2,3 | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Hepatotoxicity | ‐ | ‐ | ‐ | ‐ | ‐ | Rates of hepatotoxicity not reported, only mean alanine aminotransferase between the 2 study groups. |
| Nausea | Study population | OR 2.71 (1.00 to 7.38) | 66 (1 RCT) | ⊕⊝⊝⊝ Very low1,2,3 | ‐ | |
| 333 per 1000 | 575 per 1000 (333 to 787) | |||||
| Vomiting | Study population | OR 2.10 (0.62 to 7.12) | 66 (1 RCT) | ⊕⊝⊝⊝ Very low1,2,3 | ‐ | |
| 152 per 1000 | 273 per 1000 (100 to 560) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one level because of risk of bias (due to randomisation details or concealment allocation were not specified, participants were excluded from IV group if they developed an anaphylactoid reaction unresponsive to decreasing the administration rate. Unclear whether these participants were analysed and should have been included as intention‐to‐treat). 2Downgraded one level because of risk of imprecision (due to small sample studied). 3Downgraded one level because of indirectness (amount of paracetamol ingested mean dose of 160 mg/kg to 170 mg/kg is below the toxic dose that often requires treatment).
Summary of findings 7. Intravenous acetylcysteine compared with placebo in people with fulminant hepatic failure for paracetamol (acetaminophen) overdose.
| Intravenous acetylcysteine compared with placebo in people with fulminant hepatic failure (randomised trials) for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with fulminant hepatic failure secondary to paracetamol (acetaminophen) overdose Settings: Liver Failure Unit, King's College Hospital Intervention: intravenous acetylcysteine Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Intravenous acetylcysteine | |||||
| Mortality | Study population | Peto OR 0.29 (0.09 to 0.94) | 50 (1 RCT) | ⊕⊕⊝⊝ Low1,2 | Trial Sequential Analysis‐adjusted CI ranged from 0.01 to 15.8. | |
| 800 per 1000 | 537 per 1000 (265 to 790) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one level because of risk of bias (randomisation and allocation concealment unclear). 2Downgraded one level because of imprecision (small sample studied).
Summary of findings 8. Initial infusion rate of intravenous acetylcysteine over 15 minutes compared with 60 minutes for paracetamol (acetaminophen) overdose.
| Initial infusion rate of intravenous acetylcysteine over 15 minutes compared with 60 minutes (randomised trials) for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: multicentre study conducted in tertiary referral hospitals in Australia Intervention: initial infusion of acetylcysteine over 15 minutes Comparison: initial infusion of acetylcysteine over 60 minutes | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Initial infusion over 15 minutes | Initial infusion over 60 minutes | |||||
| Mortality | Study population | Not estimable | 180 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Hepatotoxicity | Study population |
OR 1.34 (0.39 to 4.56) |
175 (1 RCT) | ⊕⊝⊝⊝ Very low1,3 | ‐ | |
| 56 per 1000 | 74 per 1000 (23 to 213) | |||||
| Any adverse event | Study population |
OR 0.51 (0.27 to 0.96) |
180 (1 RCT) | ⊕⊕⊝⊝ Low1,4 | Trial Sequential Analysis‐adjusted CI ranged from 0.36 to 11.0. | |
| 752 per 1000 | 608 per 1000 (451 to 745) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one level because of risk of bias (possible bias due to method of randomisation via "randomisation slips" in a "closed box," many participants lost to follow‐up, and uneven numbers between the 2 treatment groups with many more participants in the 15‐minute infusion group). 2Downgraded two levels because of serious imprecision (due to small sample studied and no deaths). 3Downgraded two levels because of serious imprecision (due to small sample studied, low rate of hepatotoxicity, and wide confidence intervals). 4Downgraded one level because of imprecision (due to small sample).
Summary of findings 9. Oral plus intravenous acetylcysteine compared with intravenous acetylcysteine for paracetamol (acetaminophen) overdose.
| Oral and intravenous acetylcysteine compared with intravenous acetylcysteine for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: poisoning referral centre in Iran Intervention: oral and intravenous acetylcysteine Comparison: intravenous acetylcysteine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Intravenous acetylcysteine | Oral and intravenous acetylcysteine | |||||
| Mortality | Study population | Not estimable | 40 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | Primary outcome for this study was anaphylactoid reaction. Unable to analyse these results due to large number excluded from one arm.1 |
|
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Hepatotoxicity | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded two levels because of serious risk of bias (randomisation sequence generation and allocation concealment not recorded, and a large number of participants excluded (10 excluded from the 25 randomised)). 2Downgraded two levels because of serious imprecision (due to small sample studied and no deaths).
Summary of findings 10. Charcoal haemoperfusion compared with no intervention for paracetamol (acetaminophen) overdose.
| Charcoal haemoperfusion compared with no intervention (randomised trials) for paracetamol (acetaminophen) overdose | ||||||
| Patient or population: people with paracetamol (acetaminophen) overdose Settings: The Liver Unit, King's College Hospital, London UK Intervention: charcoal haemoperfusion Comparison: no intervention | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| No intervention | Charcoal haemoperfusion | |||||
| Mortality | Study population | Peto OR 7.39 (0.15 to 372.38) | 16 (1 RCT) | ⊕⊝⊝⊝ Very low1,2,3 | Note very small numbers in this trial; only 8 in each group. With only 1 death in the charcoal haemoperfusion arm. The Trial Sequential Analysis‐adjusted CI could not be calculated. |
|
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised clinical trial. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded one level because of risk of bias (randomisation sequence generation and allocation concealment not detailed). 2Downgraded two levels because of serious imprecision (due to small sample studied and confidence intervals are very wide). 3Downgraded one level because of risk of indirectness (imbalance between the two groups at baseline).
The 11 included randomised clinical trials differed substantially in inclusion criteria, interventions, and outcome measures. Therefore, it was only possible to perform one meta‐analysis that addressed two of our outcomes. For an overview, we presented single trials in the meta‐analyses and Trial Sequential Analysis. Planned subgroup analyses was not performed due to the small number of trials and inadequate data to perform such analyses.
Activated charcoal, gastric lavage and ipecacuanha
Plasma paracetamol concentration
One small trial (60 participants) found that the mean percentage fall in plasma paracetamol concentration was significantly greater with activated charcoal (52.3%, SD 13.6%) compared with gastric lavage (39.3%, SD 14.7%) or ipecacuanha (40.7%, SD 18.3%) if given within four hours after ingestion (P = 0.03) (Underhill 1990). There was no significant difference between gastric lavage and ipecacuanha (P = 0.081), although both were more effective than no treatment at limiting the absorption of paracetamol. However, the time interval between ingestion and intervention in the different groups was not clearly reported. There were potential areas of bias with the control group. First, the control group was given supportive treatment in a different hospital, which questions how randomisation was carried out. Furthermore, supportive treatment was stopped early due to "ethical reasons," there was an increase in paracetamol levels in four out of five participants in this group between the first and the last sample compared to paracetamol levels falling in the intervention groups. Therefore, the value of this trial was limited.
Antidotes
Methionine versus no intervention
Mortality
Based on one trial, there was no beneficial effect of methionine compared with no intervention on mortality (OR not reported by authors, OR for mortality calculated using Fisher's exact test, OR 0.31, 95% CI 0.01 to 8.31; P = 1.00) (for comparison, see Analysis 1.1: Peto OR 0.14, 95% CI 0.00 to 6.82, that is less valid as we only have one trial) (Hamlyn 1981).
1.1. Analysis.

Comparison 1 Methionine versus no intervention, Outcome 1 Mortality.
Hepatotoxicity
Compared with no intervention, methionine reduced the number of people with hepatotoxicity (OR not reported by authors, OR for hepatotoxicity calculated using Fisher's exact test; OR 0.05, 95% CI 0.004 to 0.51; P = 0.01; Analysis 1.2: OR 0.05, 95% CI 0.01 to 0.53). Trial Sequential Analysis (not shown) demonstrated that the diversity‐adjusted required information size (DARIS) was 9731 participants of which they only accrued 26 participants corresponding to 0.26% of DARIS. This DARIS was calculated based on a proportion of deaths of 10% in the control group, a relative risk reduction of 20%, an alpha of 2.5%, a beta of 20% (corresponding to a power of 80%), and an assumed diversity of 20%. The Trial Sequential Analysis‐adjusted CI could not be estimated due to the paucity of data. See Table 1.
1.2. Analysis.

Comparison 1 Methionine versus no intervention, Outcome 2 Hepatotoxicity (aspartate aminotransferase > 1000 IU/L).
Cysteamine versus no intervention or methionine or dimercaprol
Mortality
Compared with no intervention, cysteamine had no effect on mortality (Analysis 2.1: Peto OR 0.53, 95% CI 0.05 to 5.22, 2 trials, 65 participants) (Douglas 1976a; Hamlyn 1981).
2.1. Analysis.

Comparison 2 Cysteamine versus no intervention, Outcome 1 Mortality.
The Hamlyn 1981 trial compared cysteamine to methionine (27 participants). There were no deaths in either group (Analysis 3.1: Peto OR: not estimable).
3.1. Analysis.

Comparison 3 Cysteamine versus methionine, Outcome 1 Mortality.
One trial (52 participants) found no difference between cysteamine and dimercaprol on mortality (OR not reported, OR for mortality calculated using Fisher's exact test, OR 0.32, 95% CI 0.01 to 8.25; P = 1.00) (Analysis 4.1: Peto OR 0.14, 95% CI 0.00 to 6.82) (Hughes 1977). One participant who received dimercaprol died. See Table 3.
4.1. Analysis.

Comparison 4 Cysteamine versus dimercaprol, Outcome 1 Mortality.
Hepatotoxicity
Compared with no intervention, cysteamine seemed to decrease the risk of developing hepatotoxicity (Analysis 2.2: OR 0.09, 95% CI 0.02 to 0.35, 2 trials, 65 participants) (Douglas 1976a; Hamlyn 1981). Douglas and colleagues had a control group with higher paracetamol concentration before treatment (Douglas 1976a), which may have introduced bias into the comparison group. The Hamlyn 1981 trial was essentially a continuation of the Douglas 1976a trial, and results of four of the participants were used in both trials (Douglas 1976a; Hamlyn 1981). As it could not be determined which these participants were, the same participant data were analysed twice for these four people. Trial Sequential Analysis of cysteamine versus control on hepatotoxicity demonstrated that this effect was not statistically significant and the Trial Sequential Analysis‐adjusted CI ranged from 0.00 to 24.0 (Figure 4). See Table 2.
2.2. Analysis.

Comparison 2 Cysteamine versus no intervention, Outcome 2 Hepatotoxicity (aspartate aminotransferase > 1000 IU/L).
4.

Trial Sequential Analysis of cysteamine versus control on hepatotoxicity defined as aspartate aminotransferase (AST) above 1000 IU/L. The diversity‐adjusted required information size (DARIS) was 982 participants based on a proportion of 53% with the outcome in the control group (Pc); a risk reduction of 20%; an alpha (a) of 2.5%; a beta (b) of 20% (equivalent to a power of 80%); and an assumed diversity of 20%. As demonstrated the trial sequential monitoring boundaries for benefit, harm, or futility were crossed by the cumulative Z value.
Hamlyn 1981 found cysteamine and methionine to be equally as effective at reducing hepatotoxicity (OR not reported, OR for hepatotoxicity calculated using Fisher's exact test, OR 0.92, 95% CI 0.05 to 18.86, P = 1.0) (Analysis 3.2: OR 0.92, 95% CI 0.05 to 16.46). See Table 4.
3.2. Analysis.
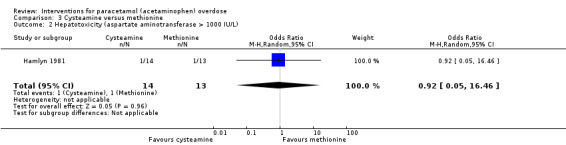
Comparison 3 Cysteamine versus methionine, Outcome 2 Hepatotoxicity (aspartate aminotransferase > 1000 IU/L).
One trial (52 participants) comparing cysteamine and dimercaprol found that cysteamine was superior to dimercaprol in terms of the severity of hepatic necrosis found on liver biopsy (Hughes 1977). However, no evidence of a difference in effect was found when comparison was made of peak AST concentrations with a peak AST of 722 IU/L versus 754 IU/L in those receiving cysteamine and dimercaprol (Wilcoxon's rank sum test P=NS) (Analysis 4.2: mean difference: ‐32.00 95%CI ‐126.33 to 62.33) see Table 3.
4.2. Analysis.

Comparison 4 Cysteamine versus dimercaprol, Outcome 2 Maximum alanine aminotransferase (IU/L).
Adverse events
All trials reported that most participants given cysteamine had nausea and vomiting during its administration. Some participants also had severe headaches, one a transient truncal rash, and one developed severe malaise (Hamlyn 1981). Dimercaprol was given as a deep intramuscular injection, which all participants found painful; 9/26 participants developed severe abdominal pain (Hughes 1977).
Acetylcysteine
Mortality
One trial (50 participants) found that intravenous acetylcysteine compared with placebo in people with paracetamol‐induced fulminant hepatic failure reduced mortality with a difference in survival of 28% (Chi2 test utilised to assess difference in survival, P = 0.037; 95% CI for difference in survival 3% to 53%) (OR not reported, OR for mortality calculated using Fisher's exact test, OR 0.27, 95% CI 0.08 to 0.95; P = 0.07) (Analysis 5.1: Peto OR 0.29, 95% CI 0.09 to 0.94) (Keays 1991). As shown in Figure 5, this effect was also not statistically significant in a Trial Sequential Analysis and the Trial Sequential Analysis‐adjusted CI ranged from 0.01 to 15.8.
5.1. Analysis.

Comparison 5 Intravenous acetylcysteine versus 'placebo' in people with fulminant hepatic failure, Outcome 1 Mortality.
5.

Trial Sequential Analysis of acetylcysteine versus placebo on mortality. The diversity‐adjusted required information size (DARIS) is 375 participants based on a proportion of 80% with the outcome in the control group (Pc); a risk reduction of 20% (Peto OR: POR); an alpha (a) of 2.5%; a beta (b) of 20% (equivalent to a power of 80%); and an assumed diversity of 20%. As demonstrated the trial sequential monitoring boundaries for benefit, harm, or futility were crossed by the cumulative Z value.
One trial (180 participants) found no difference between an initial intravenous dose of acetylcysteine administered over 15 minutes compared with administration for 60 minutes for mortality (OR for mortality not calculated by authors, Analysis 6.1: not estimable) (Kerr 2005).
6.1. Analysis.
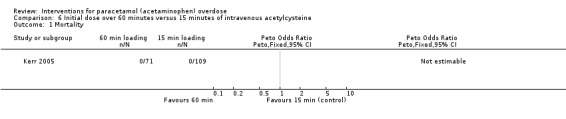
Comparison 6 Initial dose over 60 minutes versus 15 minutes of intravenous acetylcysteine, Outcome 1 Mortality.
Hepatotoxicity
One trial (180 participants) found no difference between an initial intravenous dose of acetylcysteine administered over 15 minutes compared with administration for 60 minutes (Kerr 2005) (OR for hepatotoxicity not calculated by authors, OR for hepatotoxicity using Fisher's exact test, OR 1.31, 95% CI 0.39 to 4.56; P = 0.75) (Analysis 6.2: OR 1.34, 95% CI 0.39 to 4.56).
6.2. Analysis.

Comparison 6 Initial dose over 60 minutes versus 15 minutes of intravenous acetylcysteine, Outcome 2 Hepatotoxicity.
One trial (222 participants) compared two different acetylcysteine regimens: a modified 12‐hour intravenous acetylcysteine regimen that had an initial lower loading dose given over two hours versus the standard 20.25‐hour schedule (Bateman 2014). Five participants developed hepatotoxicity, two allocated to the 12‐hour regimen versus three allocated to the 20.5‐hour regimen (OR not reported, calculated using Fisher's exact test, OR 0.67, 95% CI 0.12 to 3.33; P > 0.99) (for comparison see Analysis 7.2: OR 0.67, 95% CI 0.11 to 4.08; which is less valid as we only had one trial). See Table 5.
7.2. Analysis.
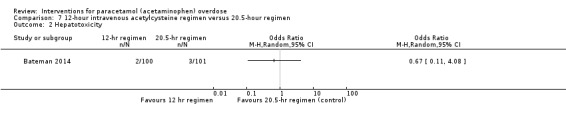
Comparison 7 12‐hour intravenous acetylcysteine regimen versus 20.5‐hour regimen, Outcome 2 Hepatotoxicity.
One trial (66 participants) assessed oral acetylcysteine versus a standard 20.25‐hour intravenous acetylcysteine regimen and found no statistically significant difference in serum AST, ALT, bilirubin, and prothrombin time at 24, 48, and 72 hours but the numbers were small in both groups and the trial was not powered to show a difference in efficacy (Arefi 2013). See Table 6.
Adverse events
There were no adverse events to acetylcysteine reported. See Table 7.
Kerr 2005 found the incidence of drug‐related adverse events within two hours was 45% in the 15‐minute loading group versus 35% in the 60‐minute loading group (95% CI difference between the two groups ‐8% to 22% using the Chi2 test; P = 0.36). However, there seemed to be a decrease in the overall number of participants with occurrence of any adverse events (75% in the 15‐minute loading group versus 61% in the 60‐minute loading group; OR not reported, calculated using Fisher's exact test OR 0.51, 95% CI 0.27 to 0.96; P = 0.05) (Analysis 6.3: OR 0.51, 95% CI 0.27 to 0.96). However, in a Trial Sequential Analysis this effect was not significant and the Trial Sequential Analysis‐adjusted CI ranged from 0.36 to 11.0 (Figure 6). See Table 8.
6.3. Analysis.

Comparison 6 Initial dose over 60 minutes versus 15 minutes of intravenous acetylcysteine, Outcome 3 Any adverse event.
6.

Trial Sequential Analysis of 15‐min infusion of acetylcysteine versus 60‐min infusion of acetylcysteine on any adverse event. The diversity‐adjusted required information size (DARIS) is 820 participants based on a proportion of 60% with the outcome in the control group (Pc); a risk reduction of 20%; an alpha (a) of 2.5%; a beta (b) of 20% (equivalent to a power of 80%); and an assumed diversity of 20%. As demonstrated the trial sequential monitoring boundaries for harm, benefit, or futility were crossed by the cumulative Z value.
One trial (222 participants) compared two different acetylcysteine regimens: a modified 12‐hour intravenous acetylcysteine regimen that had an initial lower loading dose given over two hours versus the standard 20.25‐hour schedule (Bateman 2014). The authors found that there was a reduction in the incidence of vomiting, retching, or need for antiemetics at two hours with the modified 12‐hour regimen (39/108 in the 12‐hour regimen group versus 71/109 in the 20.25‐hour regimen group; adjusted OR 0.26, 97.5% CI 0.13 to 0.52; P < 0.0001) (Analysis 7.3: OR 0.30, 95% CI 0.17 to 0.53) (note difference as Analysis 7.2 was unadjusted). Anaphylactoid reactions were categorised as mild, moderate, and severe, with those receiving the shorter regimen having a reduced rate of reactions (58/108 in the 12‐hour regimen group versus 75/100 in the 20.25‐hour regimen group) (OR for anaphylactoid reactions were not calculated by authors, OR using Fisher's exact test, OR 0.39, 95% CI 0.22 to 0.71; P = 0.002) (Analysis 7.5: unadjusted OR 0.39, 95% CI 0.21 to 0.70). The number of severe anaphylactoid reactions was also reduced with the shorter modified regimen versus the standard regimen (5/108 in the 12‐hour regimen group versus 31/100 in the 20.25‐hour regimen group; adjusted common OR 0.23, 97.5% CI 0.12 to 0.43; P < 0.0001). There was no difference in efficacy between the two regimens, but the trial was underpowered for this outcome. No deaths were recorded and accordingly, mortality could not be estimated (Analysis 7.1).
7.3. Analysis.

Comparison 7 12‐hour intravenous acetylcysteine regimen versus 20.5‐hour regimen, Outcome 3 Vomiting, retching, or antiemetics from 0 to 2 hour.
7.5. Analysis.

Comparison 7 12‐hour intravenous acetylcysteine regimen versus 20.5‐hour regimen, Outcome 5 Anaphylactoid symptoms (all).
7.1. Analysis.
Comparison 7 12‐hour intravenous acetylcysteine regimen versus 20.5‐hour regimen, Outcome 1 Mortality.
One trial (66 participants) assessed oral acetylcysteine versus a standard 20.25‐hour intravenous acetylcysteine regimen (Arefi 2013). Nausea and hypotension were more prevalent in the oral compared with the intravenous acetylcysteine treatment group (nausea: 19/33 (57.6%) participants in the oral group versus 11/33 (33.3%) participants in the intravenous group; P = 0.04) (Arefi 2013). See Table 6.
One trial (40 participants) looked at a combination of intravenous plus oral acetylcysteine versus a standard 20.25‐hour intravenous acetylcysteine regimen (Eizadi‐Mood 2013). The main outcome was the rate of anaphylactoid reactions defined as nausea and vomiting, dyspnoea, and flushing. This trial post hoc excluded participants who vomited twice after oral acetylcysteine and did not include them in the intention‐to treat analysis. The authors found that 13.3% of participants who had intravenous plus oral acetylcysteine vomited versus 28.5% participants in the intravenous acetylcysteine group. However, adding the excluded 10 participants with vomiting increased this rate to 48% and reversed the conclusions of the trial (Eizadi‐Mood 2013). See Table 9.
Ethylenediaminetetraacetic acid free acetylcysteine formulation
One trial registered by Cumberland Pharmaceuticals started in 2010 and was terminated due to lack of recruitment in 2013 (NCT01118663). This trial compared the efficacy and safety of a new acetylcysteine protocol and a changed ethylenediaminetetraacetic acid free acetylcysteine formulation. The new protocol studied was 200 mg/kg of intravenous acetylcysteine in 1 L of fluid over four hours followed by 100 mg/kg in 1 L of fluid over 16 hours versus the standard three‐bag regimen, with an initial 60‐minute loading. The intended primary outcome was the incidence of hepatotoxicity. Secondary outcomes included adverse events and percentage of participants requiring continuation of therapy beyond 21 hours. Preliminary results were reported (clinicaltrials.gov); the trial recruited only 17 participants, with reported adverse event rate of 2/7 participants with the new protocol (four‐hour loading) group versus 1/10 participants from the three‐bag, 60‐minute loading group. The type and severity of these adverse events were not reported. Two participants in each group did not complete the course of intravenous acetylcysteine. The numbers from this trial were too small to analyse (NCT01118663).
Extracorporeal treatment
Mortality
One trial studied charcoal haemoperfusion versus conventional treatment (gastric lavage and fresh frozen plasma and intravenous fluids as clinically indicated) (16 participants) (Gazzard 1974a). One participant allocated to the haemoperfusion group who had ingested 135 g of paracetamol died. There were no deaths in the control group. Accordingly, we found no benefit of charcoal haemoperfusion (Gazzard 1974a) (number of deaths reported only, using Fisher's exact test, OR 3.40, 95% CI 0.12 to 96.8; P = 1.00) (Analysis 8.1: Peto OR 7.39, 95% CI 0.15 to 372.4). The Trial Sequential Analysis‐adjusted CI could not be calculated. See Table 10.
8.1. Analysis.

Comparison 8 Charcoal haemoperfusion versus no intervention, Outcome 1 Mortality.
Plasma paracetamol concentration
One trial studied charcoal haemoperfusion versus conventional treatment (gastric lavage and fresh frozen plasma and intravenous fluids as clinically indicated) (16 participants) (Gazzard 1974a). The mean cumulative amount of paracetamol removed was 1.4 g.
Additional safety data from quasi‐randomised trials
Cimetidine
One quasi‐randomised trial studied cimetidine, with treatment allocation based on month enrolled (Burkhart 1995). As this trial was quasi‐randomised, it could only be used to assess for the risk of harm. This trial found no adverse events.
Discussion
As identified in this review, there are very few randomised clinical trials investigating interventions for paracetamol poisoning, with all the evidence considered to be of low or very low quality. It is important to note that strict bias criteria were utilised and a trial was considered at low risk of bias only if all criteria were met. In many of these studies this was not feasible because, for example, blinding for interventions such as activated charcoal or length of acetylcysteine treatment were not possible. Current practices are often based on observational and non‐randomised studies, with current treatments seeming to have resulted in a decrease in mortality. The management of paracetamol poisoning varies widely between countries; for example, whether treatment is based on dose ingested or nomograms, which nomogram line is used, whether acetylcysteine should be administered orally or intravenously, and length of treatment. Most recommendations advise to measure a plasma paracetamol concentration, and if this level is above a chosen risk‐line, then acetylcysteine is advocated (Dart 2006; Daly 2008). Many factors such as excess alcohol consumption, eating disorder, or use of enzyme‐inducing agents increase the risk of paracetamol hepatotoxicity, and these patient groups may need a lower threshold for treatment. However, most countries use a single nomogram line that has been lowered to essentially treat all people as high risk. Some guidelines suggest that liver biochemistry should be checked after treatment, and this is essential for symptomatic people, when treatment is delayed, with use of modified‐release formulation or large ingestions (Vale 1995; Chiew 2015). If the person has developed, or is at risk of developing, fulminant hepatic failure, acetylcysteine treatment has been suggested to be continued until recovery (Wolf 2007; Chiew 2015; Yoon 2016; Heard 2017). It should be noted that acetylcysteine or paracetamol overdose itself (or both) without evidence of liver injury may increase the INR, and management decisions should be based on the entire liver biochemistry (Whyte 2000; Schmidt 2002).
The methods used for decontamination include gastric lavage, activated charcoal, and ipecacuanha. The results from the one small randomised clinical trial were very low quality evidence that any of these measures can reduce the absorption of paracetamol, if given shortly after ingestion (Underhill 1990). One well‐known complication from all the three interventions is aspiration pneumonia (Liisanantti 2003). However, randomised trials in overdose have reported no increase of adverse events in people receiving activated charcoal (Cooper 2005; Eddleston 2008). Position statements on drug poisonings indicate that serious adverse events seem to be fewer in people receiving activated charcoal compared to ipecacuanha and gastric lavage (Vale 1995; Krenzelok 2004; Chyka 2005). Multiple observational and volunteer studies have investigated the effect of activated charcoal on paracetamol absorption (Buckley 1999a; Yeates 2000). These studies have shown that activated charcoal decreases the initial paracetamol concentration (Buckley 1999a; Duffull 2013; Chiew 2017). Accordingly, weak evidence indicated that activated charcoal is currently the best choice to prevent absorption of paracetamol. It appears most effective when given within two hours of ingestion, but the benefit may extend to four hours especially in larger overdoses (Buckley 1999a; Chiew 2017).
Various antidotes for paracetamol poisoning have been studied, all in small randomised trials (Douglas 1976a; Hughes 1977; Hamlyn 1981; Keays 1991). Acetylcysteine seems preferable to placebo/supportive treatment, dimercaprol, and cysteamine, in terms of adverse effects. However, a randomised clinical trial has not directly compared these treatments in terms of rates of acute liver injury or mortality. Acetylcysteine and methionine have similarly not been compared in a randomised clinical trial; however, the previous Cochrane Review concluded that both drugs demonstrated comparable effectiveness at decreasing the risk of hepatotoxicity (Brok 2006). The initial acetylcysteine study was an observational study that compared intravenous acetylcysteine (100 participants) to three groups, intravenous cysteamine (40 participants), intravenous methionine (20 participants), or supportive treatment alone (57 participants) (Prescott 1979). In this study, the comparison antidote groups were historical control groups treated three years earlier and supportive care groups 10 years earlier. They found intravenous acetylcysteine was highly effective in protecting against severe liver damage, renal failure, and death after paracetamol overdosage when given within eight to 10 hours of ingestion. Since this study was published, acetylcysteine has been recommended for paracetamol poisoning in most countries (Wolf 2007; Chiew 2015; Yoon 2016; Heard 2017). Historical data showed that the overall mortality rate has dropped from 3% (all poisonings (Clark 1973) or 5% (all above the probable risk‐line (Prescott 1979) to 0.4% in the 1980s after the introduction of acetylcysteine (Gunnell 1997)). Similarly, the rates of acute liver injury due to paracetamol toxicity have declined since the routine use of acetylcysteine. The major risk factor for developing acute liver injury following paracetamol ingestion was delayed time to treatment with acetylcysteine (Prescott 1979; Smilkstein 1988). Observational studies and meta‐analyses continue to show similar results with rates of hepatotoxicity reported in people receiving acetylcysteine treatment within eight hours ranging from 0% to 6% compared to people treated more than eight to 10 hours postingestion of 8% to 50% (Buckley 1999b; Kerr 2005; Brok 2006; Doyon 2009; Green 2013; Heard 2014). From the one small randomised clinical trial, survival among people with paracetamol‐induced fulminant hepatic failure was higher if treated with acetylcysteine (Keays 1991).
Acetylcysteine can be administered orally or intravenously. Two small studies in this review compared oral to intravenous acetylcysteine; however, both were at high risk of bias (Arefi 2013; Eizadi‐Mood 2013). One meta‐analysis of over 5000 participants admitted with a paracetamol overdose compared oral to intravenous acetylcysteine and showed similar rates of hepatotoxicity in (12.6% with oral versus 13.2% with intravenous). Treatment delays (beyond 10 hours) were associated with the highest risk of liver injury (Green 2013). Both oral and intravenous acetylcysteine were associated with adverse effects. These reactions range from mild to severe symptoms and include rash, nausea, vomiting, angioedema, tachycardia, bronchospasm, hypotension, and death (Mant 1984; Bailey 1998; Schmidt 2001; Kao 2003). The most common reactions from intravenous acetylcysteine are nausea, vomiting, and cutaneous systemic hypersensitivity reactions (Sandilands 2009). Oral acetylcysteine administration often results in rash, nausea, vomiting, and abdominal pain. The rates of adverse reactions vary greatly between observational studies. Reported rates depend on whether it was a prospective or retrospective study and which adverse effects were measured (e.g. total versus gastrointestinal versus systemic hypersensitivity reactions). There are many observational studies of acetylcysteine, a review that examined some of these larger studies reported that rates of adverse effects from acetylcysteine varied from 8.5% to 77% (Chiew 2016).
Trials of acetylcysteine have looked at different regimens to decrease the rate of adverse effects. There are four trials looking at different acetylcysteine regimens compared with the standard three‐bag intravenous acetylcysteine regimen (Kerr 2005; Arefi 2013; Eizadi‐Mood 2013; Bateman 2014). The primary outcomes of these trials were adverse effects of treatments and none were powered to look at efficacy. Various observational studies have looked at differing acetylcysteine regimens to decrease the rate of adverse reactions or decrease drug administration errors but again these were not powered to look at efficacy (Chiew 2016). Furthermore, there has been increasing concerns that the standard intravenous acetylcysteine doses are inadequate in people with very high initial paracetamol concentrations, who require higher doses of acetylcysteine (Rumack 2012; Chiew 2016). People with increased paracetamol concentration on presentation are at higher risk of liver injury even when intravenous acetylcysteine is administered early (Cairney 2016). Further studies are warranted to compare efficacy between different treatment regimens looking at both the dose and rate of acetylcysteine infusions.
Only one very small study looked at extracorporeal treatment, which showed no benefit from charcoal haemoperfusion (Gazzard 1974a). The Extracorporeal Treatments in Poisoning (EXTRIP) Workgroup, published recommendations in 2014, regarding paracetamol and extracorporeal treatment. EXTRIP searched for randomised clinical trials, observational studies, case reports, case series, and pharmacokinetic studies. From these, the EXTRIP panel concluded that paracetamol was dialysable and suggested extracorporeal treatments could be considered in cases of severe paracetamol poisoning, defined by paracetamol level, acidosis, or coma and also depending on antidote availability. They concluded that intermittent haemodialysis is the preferred extracorporeal treatment in people with paracetamol poisoning. Intermittent haemoperfusion or continuous renal replacement modalities are valid alternatives if intermittent haemodialysis is not available. Furthermore, acetylcysteine treatment should be continued during extracorporeal treatment at an increased rate (Gosselin 2014).
Since the late‐1980s, survival from acute liver failure from any cause has markedly improved, with a significant improvement in paracetamol‐induced acute liver failure. This is because of improved intensive care, earlier illness recognition, and use of emergency liver transplantation when required (Bernal 2013). Over this same period, transplant‐free survival in people with acute liver failure secondary to paracetamol has improved to nearly 70% (Reuben 2016). However, in people who develop irreversible liver damage, the ultimate treatment is liver transplantation.
There are various limitations in this review. First, for most interventions, there were only one or two small trials and most trials were at high risk of bias and at high risk of play of chance. Hence no greater certainty about treatment effects could be determined through meta‐analysis. Second, the interventions studied varied greatly and even those studying similar interventions varied in rates and mode of administration of the drugs meaning comparisons between various intervention options could not be made. Heterogeneity was high due to the varying interventions and participants. Much of the protocol outlined in the methods could not be applied to the studies included.
The results of this systematic review highlighted a lack of randomised clinical trials on interventions for paracetamol overdose, despite it being a very common drug for poisoning worldwide. Most of the included randomised clinical trials were small and some trials assessed interventions not used in the 21st century. Furthermore, no randomised clinical trials investigated the efficacy of acetylcysteine versus commonly available oral antidotes such as methionine. It is unlikely that a trial will be conducted comparing acetylcysteine with placebo, except perhaps in very low risk people. Research has focused on testing different acetylcysteine regimens with the aim to lower adverse effects or to shorten the duration of treatment, while maintaining or improving outcomes.
Summary of main results
The interventions to treat paracetamol poisoning can be divided into two main groups, those treatments that decrease serum paracetamol concentration, either by decontamination or by extracorporeal treatments and antidotes that decrease the amount or detoxify the toxic metabolite NAPQI.
Activated charcoal, gastric lavage and ipecacuanha
One randomised clinical trial found activated charcoal to be more effective than gastric lavage, ipecacuanha, or no intervention in preventing the absorption of paracetamol (Underhill 1990). There were various limitations to this trial.
Antidotes
Cysteamine, methionine, and dimercaprol
Two randomised trials found methionine or cysteamine treatment resulted in lower AST concentrations and less hepatic necrosis on liver biopsy when compared with no intervention (Douglas 1976a; Hamlyn 1981). There was no difference between the two antidotes in one trial (Hamlyn 1981), in terms of peak serum AST and rates of hepatic necrosis. In a further trial, cysteamine was superior to dimercaprol in terms of the severity of hepatic necrosis found on liver biopsy (Hughes 1977). Overall, cysteamine therapy was associated with a high rate of nausea and vomiting. There were few adverse events in participants given methionine and dimercaprol.
Acetylcysteine
One randomised clinical trial found that intravenous acetylcysteine increased survival in participants with paracetamol‐induced fulminant hepatic failure compared with placebo (Keays 1991). However, no randomised trials have assessed the effect of acetylcysteine in the acute treatment of paracetamol overdose. Four completed randomised clinical trials have studied differing regimens of acetylcysteine administration to decrease rates of adverse events but none of these were powered for efficacy (Kerr 2005; Arefi 2013; Eizadi‐Mood 2013; Bateman 2014). One trial was abandoned because of lack of enrolment (NCT01118663).
Extracorporeal treatments
The randomised clinical trial found involved haemoperfusion, we found no evidence to support or refute haemoperfusion for paracetamol overdose.
Acute hepatic failure
Most interventions used to treat acute fulminant liver failure were not investigated in this review, but, based on one small trial, it appeared that acetylcysteine administration improved survival in participants with fulminant hepatic failure from paracetamol (Keays 1991).
Children
There was less evidence on how to manage children who unintentionally ingest paracetamol. Children are rarely able to tell how much and when they have ingested the drug. Furthermore, the applicability of the recommended treatment line in young children has never been proven due to the paucity of data (Vale 1995).
Overall completeness and applicability of evidence
This review looked at many different treatment options for paracetamol overdose, each based on only one or very few randomised clinical trials and the quality of the evidence was found to be low or very low and incomplete. Most of the current recommendations for the treatment of paracetamol overdose are based on observational studies.
Quality of the evidence
In the 11 trials studied, the majority had multiple domains at high risks of bias (systematic errors, i.e. overestimation of benefits and underestimation of harms) as well as very small number of participants giving high risks of play of chance (random errors) and multiple items of high risks of bias. Thus, the quality of the evidence was assessed to be low or very low.
Potential biases in the review process
NB is an author on one of the trials included in this review (Kerr 2005). This risk of bias was minimised as other review authors determined if it could be included and assess risk of bias and extracted data.
Agreements and disagreements with other studies or reviews
We are not aware of any other systematic review of paracetamol overdose that covered all treatment options, besides the earlier versions of this review and the BMJ Clinical Evidence abbreviated reviews (Park 2015). The conclusions from the later review were broadly in line with those of this review. The findings of this review were not in conflict with existing guidelines including acetylcysteine in people at risk of hepatotoxicity or established acute liver injury (Lee 2011; Chiew 2015; Heard 2017).
Authors' conclusions
Implications for practice.
There are few randomised clinical trials for how to treat people with paracetamol overdose, and the quality of evidence is low or very low. Current practices are often based on observational studies. Current treatments seem to have resulted in a decrease in mortality; therefore, there is no reason to abandon current practices without further evidence. Current practices include the use of activated charcoal within one to two hours of ingestion to reduce the absorption of paracetamol and the administration of an antidote such as acetylcysteine (oral or intravenous) or methionine in people at risk of hepatotoxicity. Risk assessment in trials has generally been based on a potentially toxic ingested dose (paracetamol greater than 7.5 g to 10 g or 150 mg/kg bodyweight to 200 mg/kg bodyweight or more) or plasma paracetamol concentration above a chosen risk‐line or based on all people with abnormal liver biochemistry or fulminant hepatic failure after paracetamol overdose. Acetylcysteine can be administered intravenously or orally. Recent studies have focused on decreasing the reaction rates from intravenous acetylcysteine.Slowing the infusion rate may decrease the rate of adverse reactions, and further research is needed into whether these regimens are as efficient.
Implications for research.
People with a paracetamol overdose need to be studied in large multicentre randomised clinical trials with adequate methodology and with relevant clinical outcomes. The substantial fall in mortality from paracetamol overdose since the introduction of acetylcysteine means that it is unlikely that it would be considered ethical to randomise people to this drug versus placebo or no intervention. However, more research is needed comparing different interventions and routes of administration. This review has identified a number of topics that need assessment in randomised clinical trials (e.g. acetylcysteine administered intravenously versus orally; acetylcysteine optimum dosing regimen; acetylcysteine versus methionine: activated charcoal versus no decontamination or other methods for reducing paracetamol absorption). Furthermore, adverse events in relation to the different interventions should be reported systematically. New randomised clinical trials should be designed according to SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) (www.spirit‐statement.org/), registered before inception (www.icmje.org/clin_trialup.htm), and reported according to the CONSORT guidelines (www.consort‐statement.org), as well as with public sharing of depersonalised data to allow individual participant data meta‐analyses (Skoog 2015). Such new trials ought to stratify participants according to perceived risks of liver failure.
What's new
| Date | Event | Description |
|---|---|---|
| 17 January 2017 | New search has been performed | Three new randomised clinical trials were included in this update. Unlike the previously published review version, the current review contains only data from randomised clinical trials. Outcome measures have been changed from a composite outcome measure of mortality and liver transplantation to the individual measures. Furthermore, secondary outcome measures have been changed to standardised definitions of acute liver injury. Changes in the risk of bias domains have been conducted to make them contemporary. |
| 29 June 2015 | New citation required but conclusions have not changed | No change in conclusions. |
Acknowledgements
We are indebted to Dimitrinka Nikolova and Sarah Klingenberg for their helpful assistance. We would like to acknowledge Dr Omid Khakhouie for translating one of the trials into English.
Cochrane Review Group funding acknowledgement: the Danish State is the largest single funder of The Cochrane Hepato‐Biliary Group through its investment in The Copenhagen Trial Unit, Centre for Clinical Intervention Research, Rigshospitalet, Copenhagen University Hospital, Denmark. Disclaimer: the views and opinions expressed in this review are those of the review authors and do not necessarily reflect those of the Danish State or The Copenhagen Trial Unit.
Peer reviewers: Goran Poropat, Croatia; Goran Hauser, Croatia. Contact editors: Vanja Giljaca, UK; Marsha Morgan, UK. Sign‐off editor: Luit Penninga, Denmark.
Appendices
Appendix 1. Search strategies
| Database | Time span | Search strategy |
| Cochrane Hepato‐Biliary Group Controlled Trials Register | January 2017. | (acetaminophen OR paracetamol) AND (overdos* OR poison*) AND (methionin* OR cystein* OR cysteamin* OR dimercaprol OR cimetidin* OR acetylcystein* OR NAC OR 'gastric lavage*' OR 'gastric decontamination*' OR charcoal OR ipecacuanha OR ipecac OR hemoperfusion* OR haemoperfusion*) |
| Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library | 2016, Issue 11. | #1 MeSH descriptor: [Acetaminophen] explode all trees #2 acetaminophen* or paracetamol* #3 #1 or #2 #4 MeSH descriptor: [Drug Overdose] explode all trees #5 overdos* or poison* #6 #4 or #5 #7 methionin* or cystein* or cysteamin* or dimercaprol or cimetidin* or acetylcystein* or NAC or (gastric and (lavage* or decontamination*)) or charcoal or ipecacuanha or ipecac or hemoperfusion* or haemoperfusion* #8 #3 and #6 and #7 |
| MEDLINE Ovid | 1946 to January 2017. | 1. exp Acetaminophen/ 2. (acetaminophen* or paracetamol*).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 3. 1 or 2 4. exp Drug Overdose/ 5. (overdos* or poison*).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 6. 4 or 5 7. (methionin* or cystein* or cysteamin* or dimercaprol or cimetidin* or acetylcystein* or NAC or (gastric and (lavage* or decontamination*)) or charcoal or ipecacuanha or ipecac or hemoperfusion* or haemoperfusion*).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 8. 3 and 6 and 7 9. (random* or blind* or placebo* or meta‐analys*).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] 10. 8 and 9 |
| Embase Ovid | 1974 to January 2017. | 1. exp paracetamol/ 2. (acetaminophen* or paracetamol*).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] 3. 1 or 2 4. exp drug overdose/ 5. exp drug intoxication/ 6. (overdos* or poison*).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] 7. 4 or 5 or 6 8. (methionin* or cystein* or cysteamin* or dimercaprol or cimetidin* or acetylcystein* or NAC or (gastric and (lavage* or decontamination*)) or charcoal or ipecacuanha or ipecac or hemoperfusion* or haemoperfusion*).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] 9. 3 and 7 and 8 10. (random* or blind* or placebo* or meta‐analys*).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] 11. 9 and 10 |
| Science Citation Index Expanded (Web of Science) | 1900 to January 2017 | #6 #5 AND #4 #5 TS=(random* OR blind* OR placebo* OR meta‐analys*) #4 #3 AND #2 AND #1 #3 TS=(methionin* OR cystein* OR cysteamin* OR dimercaprol OR cimetidin* OR acetylcystein* OR NAC OR (gastric AND (lavage* OR decontamination*)) OR charcoal OR ipecacuanha OR ipecac OR hemoperfusion* OR haemoperfusion*) #2 TS=(overdos* OR poison*) #1 TS=(acetaminophen* OR paracetamol*) |
Data and analyses
Comparison 1. Methionine versus no intervention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2 Hepatotoxicity (aspartate aminotransferase > 1000 IU/L) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 2. Cysteamine versus no intervention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 2 | 65 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.53 [0.05, 5.22] |
| 2 Hepatotoxicity (aspartate aminotransferase > 1000 IU/L) | 2 | 65 | Odds Ratio (M‐H, Random, 95% CI) | 0.09 [0.02, 0.35] |
Comparison 3. Cysteamine versus methionine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2 Hepatotoxicity (aspartate aminotransferase > 1000 IU/L) | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.05, 16.46] |
Comparison 4. Cysteamine versus dimercaprol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2 Maximum alanine aminotransferase (IU/L) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 5. Intravenous acetylcysteine versus 'placebo' in people with fulminant hepatic failure.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected |
Comparison 6. Initial dose over 60 minutes versus 15 minutes of intravenous acetylcysteine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2 Hepatotoxicity | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Any adverse event | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 7. 12‐hour intravenous acetylcysteine regimen versus 20.5‐hour regimen.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2 Hepatotoxicity | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Vomiting, retching, or antiemetics from 0 to 2 hour | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Vomiting, retching, or antiemetics 0 to 12 hour | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Anaphylactoid symptoms (all) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
7.4. Analysis.

Comparison 7 12‐hour intravenous acetylcysteine regimen versus 20.5‐hour regimen, Outcome 4 Vomiting, retching, or antiemetics 0 to 12 hour.
Comparison 8. Charcoal haemoperfusion versus no intervention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Arefi 2013.
| Methods | "Parallel randomised clinical trial." | |
| Participants |
Inclusion criteria: > 18 years, paracetamol ingestion > 7.5 g over the preceding 24 hr. Exclusion criteria: presentation later than 24 hr after ingestion, coingestion of cholinergic drugs, decreased level of consciousness at presentation, primary hepatic encephalopathy, status epilepticus after acetylcysteine administration, history of asthma and anaphylactoid reactions. Oral group vs IV group: Number of participants randomised: 33 vs 33. Age (mean (SD)) (years): 27.76 (9.52) vs 24.61 (5.95). Interval between ingestion and treatment (mean (SD) (hr): 11.88 (7.04) vs 12.21 (7.02). Paracetamol plasma level on admission (mean (SD)) (μg/mL): 78.09 (64.12) vs 72.06 (61.26). Amount of paracetamol ingested (mean (SD)) (mg/kg): 160.78 (28.61) vs 170.81 (17.73). Additional characteristics: no difference between serum AST, ALT, bilirubin or creatinine between the 2 groups. Not reported in either group: number of participants taking additional drugs or consuming additional alcohol, number of participants excluded before randomisation. |
|
| Interventions |
IV group: 20‐hr protocol: first dose 150 mg/kg over 15 min, second dose 50 mg/kg over 4 hr and third dose 100 mg/kg over 16 hr. Oral group: 72‐hr protocol: first dose 140 mg/kg followed by 17 maintenance doses of 70 mg/kg every 4 hr. |
|
| Outcomes | Outcomes: liver enzymes (AST, ALT, bilirubin, PT; measured daily). Adverse effects: nausea, vomiting, flushing, rash, pruritus, dyspnoea, tachycardia, cough, wheeze, hypotension (systolic BP < 100 mmHg within 2 hr of administration), and bronchospasm. |
|
| Notes | No statistically significant difference in AST, ALT, bilirubin, and PT in oral and IV group at 24, 48, and 72 hr. Nausea and hypotension were significantly more prevalent in oral compared to IV treatment group. Nausea: 19 (57.6%) in oral group vs 11 (33.3%) in IV group. Translated from Persian (Farsi). Author contacted but no reply to verify issues with translation. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: translation: "sampling was conducted using randomised blocks of four." Comment: no mention of sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | Not recorded. Comment: no mention of whether there was knowledge of the fixed block randomisation, which might have revealed what the next allocation had to be for the last 1 or 2 people in each block. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not feasible given nature of intervention by 2 different routes. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | High risk of bias for reporting adverse effects such as nausea or vomiting. Low risk of bias for reporting of LFTs. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Mean values shown for several tests for 72 hr, unclear if all participants were still in trial or if some had been discharged. |
| Selective reporting (reporting bias) | Unclear risk | Trial not registered. Unclear what the primary and secondary outcomes of the study were, although there were no significant differences except in adverse effects which related to route of administration. |
| Other bias | Unclear risk | Power: translation quote: "using the sample size formula for comparing difference in means the sample size was set at 30 in each group and 10% added to each group." Unclear what test(s) and time point(s) this referred to and what data were used for determining variance and what difference was considered significant. Not recorded were number assessed for randomisation or number excluded. Participants were 'excluded' from IV arm if anaphylactoid reactions unresponsive to decreasing the administration rate and given oral acetylcysteine (unclear if these were still included based on intention to treat analysis principles when examining outcomes). |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | High risk | Judged as high risk. |
Bateman 2014.
| Methods | Double‐blind randomised clinical trial. | |
| Participants | 3 hospitals: Royal Infirmary (Edinburgh), Royal Victoria Infirmary (Newcastle), Aberdeen Royal Infirmary. Inclusion criteria: acute paracetamol overdose and needed treatment with acetylcysteine on the basis of standard UK guidance for management. Exclusion criteria: people aged < 16 years; detained under Mental Health Act; known permanent cognitive impairment; life‐threatening illness; pregnant women; previous participation in study; considered to have unreliable history of paracetamol overdose; presenting > 36 hr after overdose (24 hr up to May 2011) of a single acute paracetamol overdose; presenting after taking staggered paracetamol overdose (defined as when overdose of paracetamol was taken over a period > 2 hr (1 hr up May 2011); anticoagulants (e.g. warfarin) in therapeutic doses or in overdose; people who, in the opinion of the responsible clinician/ nurse, were unlikely to complete the full course of acetylcysteine e.g. expressing wish to self‐discharge: people who, in the opinion of the responsible clinician/nurse, were unable to complete the initial questionnaire themselves or with nurse assistance; history of hypersensitivity to 5HT3 antagonists; non‐English speaking people. Number assessed for randomisation: 1539 suitable for acetylcysteine treatment. Number excluded before randomisation: 1170. Total number randomised: 369. Ondansetron‐modified: Number randomised: 55 (54 analysed). Age (median) (years): 29. Weight (median) (kg): 70. Number (%) of participants with interval between ingestion and treatment, < 8 hr: 32 (58%). Number of participants with paracetamol plasma level on admission (mean): not reported instead % of participants in a set range. Number (%) of participants with ingested paracetamol ≥ 16 g: 28 (51%). Number (%) of participants taking additional drugs: 25 (45%). Number (%) of participants consuming additional alcohol: 28 (51%). Number of participants excluded after randomisation: 1 withdrawn pretreatment. Ondansetron‐standard: Number randomised: 56 (55 analysed). Age (median) (years): 32. Weight (median) (kg): 68. Number (%) of participants with interval between ingestion and treatment, < 8 hr: 33 (59%). Number (%) of participants with ingested paracetamol ≥ 16 g: 29 (52%). Number (%) of participants taking additional drugs: 32 (57%). Number (%) of participants consuming additional alcohol: 30 (54%). Number of participants excluded after randomisation: 1. Placebo‐modified: Number of participants randomised: 55 (54 analysed). Age (median) (years): 36. Weight (median) (kg): 70. Number (%) of participants with interval between ingestion and treatment, < 8 hr: 32 (58%). Number (%) of participants with ingested paracetamol ≥ 16 g: 30 (55%). Number (%) of participants taking additional drugs: 31 (56%). Number (%) of participants consuming additional alcohol: 24 (44%). Number of participants excluded after randomisation: 1. Placebo‐standard: Number of participants randomised: 56 (54 analysed). Age (median) (years): 33. Weight (kg): 70. Number (%) of participants with interval between ingestion and treatment, < 8 hr: 31 (55%). Number (%) of participants with ingested paracetamol ≥ 16 g: 29 (52%). Number (%) of participants taking additional drugs: 39 (70%). Number (%) of participants consuming additional alcohol: 29 (52%). Number of participants excluded after randomisation: 2. |
|
| Interventions |
Ondansetron‐modified group: ondansetron 4 mg IV pretreatment and the modified (shorter) acetylcysteine regimen. Ondansetron‐standard group: ondansetron 4 mg IV pretreatment and the standard acetylcysteine regimen. Placebo‐modified group: placebo IV pretreatment and modified (shorter) acetylcysteine regimen. Placebo‐standard group: placebo IV pretreatment and standard acetylcysteine regimen. Acetylcysteine regimens used: UK standard schedule (20.25 hr): 150 mg/kg in 200 mL over 15 min, then 50 mg/kg in 500 mL over 4 hr, then 100 mg/kg in 1000 mL over 16 hr. Modified (shorter) protocol (12 hr): 100 mg/kg in 200 mL over 2 hr, then 200 mg/kg in 1 L over 10 hr, then 0.5 L of 5% dextrose to 20.25 hr. |
|
| Outcomes |
Primary outcome: absence of vomiting, retching, or need for rescue antiemetic at 2 hr. Secondary outcomes: up to 12 hr: proportion of participants without nausea (Likert scale), vomiting or retching up to 12 hr and anaphylactoid reactions > 50% increase in ALT over admission. |
|
| Notes | Vomiting, or retching, or rescue antiemetics were significantly lower in participants receiving modified regimen compared to standard regimen and in participants treated with ondansetron versus placebo. Secondary outcome of nausea, vomiting, or retching up to 12 hr was less common in the shorter modified regimen and participants pretreated with ondansetron. Fewer people in the modified regimen had severe reactions requiring interruption to treatment. Participants pretreated with ondansetron had increased frequency of 50% increase in ALT. 2 protocol adjustments: extended time for paracetamol ingested from 1 hr to 2 hr to assist recruitment and second change in new UK guidance in September 2010 changed to 100 mg/L paracetamol nomogram line for recruitment. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "2x2 factorial trial design." "Randomisation by minimisation to achieve balance (1:1:1:1 allocation), according to the following prognostic factors: reported paracetamol dose (<16g or ≥16g); risk factors for paracetamol – induced hepatic toxic effects, and time to presentation." |
| Allocation concealment (selection bias) | Low risk | Quote: "Online program for randomisation." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Ondansetron and saline placebo ampoules identical in appearance." "Acetylcysteine not masked due to ethical and practical concerns." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No blinding of acetylcysteine regimens hence, high risk of bias for the standard vs modified regimens for detection of adverse reactions such as anaphylactoid reaction and nausea and vomiting. But low risk for mortality or liver injury. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "if any patient had missing data for an outcome variable, we removed them from formal statistical analysis at that time point." Only 5/222 participants unable to provide primary outcome data. |
| Selective reporting (reporting bias) | Low risk | Outcome measures published in trial protocol, subsequently reported in final paper. |
| Other bias | Low risk | Intention to treat: "analysis was done according to randomised treatment group, irrespective of adherence to treatment." Power: "To achieve at least 80% power to detect a relative risk of 0.6 for the proportion of patients with retching or vomiting within 2 hours (from 60% in the treated group to 36% in the placebo group), 91 patients needed to be enrolled in each group"… "to allow for a potential higher drop‐outs/noncompliance rate … planned to include 250 patients, 125 randomised to ondansetron and 125 to placebo. This was to ensure 50 patients in each of the four groups." Note: NOT powered for efficacy or non‐inferiority: modified vs conventional regimen IV acetylcysteine. Note: 2 protocol amendments: "Extended the time allowed for ingestion of paracetamol from 1h to 2h to assist recruitment…most patients found to ingest large single overdoses over a period of 2h." "Second, after new UK guidance was issued in September 2012, we used the 100mg/l paracetamol nomogram line for recruitment in all patients." |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | Low risk | Judged as low risk. |
Douglas 1976a.
| Methods | Randomised clinical trial. | |
| Participants |
Inclusion criteria: all participants admitted within 17 hr of paracetamol ingestion, who were hepatitis B surface antigen negative, had no history of pre‐existing liver disease and paracetamol level > 200 mg/L (4 hr) line. Cysteamine group vs non‐cysteamine (control) group: Number of participants randomised: 18 vs 20. Paracetamol "index": concentration by which the participant exceeded, the theoretical "safe" upper limit, indicated by the line at the time when plasma‐paracetamol was measured. Paracetamol "index" (mean) (mg/L): 72 vs 98 (difference between the 2 groups significant). Further analysis of participants under 30 years of age: Early cysteamine treatment (< 9 hr postingestion) (mean) (mg/L): 43 vs 138 (P < 0.01). Late cysteamine treatment (> 9 hr postingestion) (mean) (mg/L): 75 vs 67. Amount of ingested paracetamol (mean) (g): 28 vs 32. Following data not reported in either group: age, male:female ratio, number of participants taking additional drugs or consuming additional alcohol, number of participants excluded after randomisation. |
|
| Interventions |
Cysteamine group: cysteamine given as described by Prescott 1973 except that it was dissolved in 5% dextrose and injected, or added to 5% dextrose infusion, using a Millipore filter attached to a syringe. Control group: supportive treatment: 5% dextrose, 2 L to 3 L daily, with added vitamins and potassium if necessary. |
|
| Outcomes | Mortality, maximum AST, maximum serum bilirubin, maximum PT, liver biopsy findings, maximum serum‐ferritin, renal function, serum amylase, and adverse events. | |
| Notes | 1 death in each treatment group. No difference between the 2 groups in maximum bilirubin and minimum PT. Difference for PT in subgroup of participants aged < 30 years, treated in < 9 hr, was statistically significance. Cysteamine group: late and early presenters had significantly lower serum AST compared to the control group. Liver biopsy: more grade III changes in the control group; however, participants with high plasma paracetamol concentrations regardless of treatment were more likely to have grade III changes. Douglas and colleagues did not provide the time interval between ingestion of paracetamol and treatment in either groups. 4 of the participants were also included in Hamlyn 1981 study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | First stratified into 1 of 4 groups and then "Patients were randomly allocated to receive cysteamine or not using a table of random numbers. Using a table of random numbers, it was possible that one or other group could become weighted with cases receiving only one of the treatment regimens. Adjustment was made therefore, so that every six patients in each group included three who had received cysteamine." Comment: process outlined suggested there may have been rejection of certain patterns of random numbers. |
| Allocation concealment (selection bias) | Unclear risk | As above. Comment: unclear how the adjustment was done and whether this would have maintained allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding described. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Low risk as outcomes measured mortality and LFT values. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data seemed complete with individual data points presented for most participants. |
| Selective reporting (reporting bias) | Low risk | 2 groups different at baseline but noted by authors. Quote: "However, the difference for prothrombin in the young early group just reaches statistical significance (P=0.05)." Comment: there was some focus on subgroup analysis that did not seem justified given how small the study was and that it was examining effects not significant across all participants. |
| Other bias | Low risk | Intention to treat/power/premature stopping: Power: not recorded but authors noted. Quote: "our trial is on too small a scale to permit any conclusions about the effect of cysteamine on mortality rates." Comment: multiple statistical tests were done on various subgroups, without any statistical adjustment for multiple comparisons. |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | High risk | Judged as high risk. |
Eizadi‐Mood 2013.
| Methods | Randomised clinical trial. April 2009 to September 2010. |
|
| Participants |
Inclusion criteria: people with paracetamol poisoning aged ≥ 18 years, with time from ingestion to admission < 8 hr. Exclusion criteria: people who vomited twice after oral acetylcysteine was given (these people were excluded and were managed with IV acetylcysteine only), pregnant women, and risk factors for hepatic toxicity (e.g. hepatic cirrhosis, chronic ethanol ingestion, usage of substances that induce cytochrome P450). IV group vs oral + IV group: Number of participants randomised: 25 vs 25 (10 excluded presumably as vomited more than twice as per exclusion criteria). Age (mean) (years): 23.78 vs 24.46. Amount of ingested paracetamol (mean) (mg): 12,337.5 vs 11,290. Percentage of participants taking additional drugs: 42.3% vs 60%. Number of participants excluded after randomisation: 0 vs 10. Additional information: Percentage of participants vomited pre hospital: 60.7% vs 26.7%. Percentage of participants with no signs or symptoms before acetylcysteine: 10.7% vs 40%. Not recorded in either group: paracetamol plasma level on admission, interval between ingestion and treatment, number of participants consuming additional alcohol. |
|
| Interventions | Ingestion < 4 hr received gastric evacuation and charcoal 1 g/kg in 200 mL water. IV group: IV acetylcysteine with 150 mg/kg infused in 200 mL of 5% dextrose over 30 min, followed by a 4‐hr infusion of 50 mg/kg of acetylcysteine in 500 mL of 5% dextrose and 16 hr of 100 mg/kg in 1 L of 5% dextrose. Oral + IV group: acetylcysteine 140 mg/kg in 200 mL of 5% dextrose orally then IV acetylcysteine 50 mg/kg in 500 mL of 5% dextrose every 4 hr then 100 mg/kg in 1 L of 5% dextrose in 16 hr. If vomiting occurred in any participant within 1 hr after the ingestion of the oral acetylcysteine, then metoclopramide 10 mg IM and oral acetylcysteine given at the same dose again. Oral acetylcysteine in the form of a 600 mg tablet. |
|
| Outcomes | Anaphylactoid reaction defined as nausea and vomiting, dyspnoea, flushing, > 1 symptoms: | |
| Notes |
IV group vs oral + IV group: Anaphylactoid reaction defined as nausea and vomiting, dyspnoea, flushing, > 1 symptoms: No signs or symptoms: 86.7% vs 39.3%. At least 1 sign of anaphylactoid reaction: 60.7% vs 13.3%. Most common symptom was nausea and vomiting: 28.5% vs 13.3%. Flushing or dyspnoea: 3.6% vs 0%. Nausea and vomiting was noted as a symptom of acetylcysteine administration but participants were excluded from oral + IV group if they vomited twice after oral acetylcysteine (10 participants excluded) and should have been analysed. Author contacted to get further details, particularly given the outcome data could not be extracted from the report. However, responses did not clarify any of the above issues including what the absolute numbers were with adverse events for randomised participants. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "allocated in two groups randomly." Comment: randomisation sequence generation not recorded. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "allocated in two groups randomly." Comment: process of randomisation not recorded. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not blinded, primary outcome adverse reactions so high potential for bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 40% of enrolled participants in one group excluded; unclear whether data from these participants were presented. Percentages of most outcomes reported (not numbers); however, these were not the multiples of 4 expected if data on all 25 participants randomised were included in the denominator. Therefore, there appears to be a missing data for most outcomes but it was not apparent how much were missing and how these were reported. |
| Selective reporting (reporting bias) | High risk | Quote: "patients who vomit two times after oral acetylcysteine was given (these patients were excluded and were managed with IV NAC only)." 10/25 participants oral + IV group postrandomisation. Outcomes for these participants not reported. |
| Other bias | High risk | Intention to treat: "group B [oral + IV group] (25 patients). 10 patients of group B were excluded from our study." Results given for 25 (IV group) and 15 (oral + IV group). Absolute numbers not given in results and unclear if percentages in oral + IV group results were out of 25 or 15. |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | High risk | Judged as high risk. |
Gazzard 1974a.
| Methods | Randomised clinical trial. | |
| Participants |
Inclusion criteria: all participants seen in the Liver Unit with a plasma paracetamol level > 200 µg/L at any time in the first 12 hr after overdosage. Exclusion criteria: not reported. Haemoperfusion group vs supportive group: Number of participants: 8 vs 8. Age (mean) (years): 31 vs 35. Time elapsed between ingestion and presentation at the department (mean) (min): 300 vs 180. Paracetamol plasma level on admission (mean (SE)) (mg/L): 305 (46). Amount of ingested paracetamol (g): 56 vs 34. Additional characteristics: Initial plasma half‐life (hr): 7 vs 5. Initial AST level (U/L): 827 vs 142. Initial PT (seconds): 12 vs 2.2. Initial plasma bilirubin (mg/100 mL): 1.85 vs 1.27. Not reported in either group: male:female ratio, number of participants taking additional drugs or consuming additional alcohol. |
|
| Interventions | All participants were treated by gastric lavage when first seen and fresh frozen plasma and fluid were administrated as clinically indicated. Haemoperfusion group: 2 catheters (14 French gauge 50 cm length) positioned in saphenous vein (under local anaesthesia and x‐ray guided) and attached to a perfusion column. Charcoal used was covered with a thin coating of polyhydroxyethyl‐methacrylate. Participants were heparinised before with an IV loading dose of 2000 units 10 min before the procedure, and thereafter a constant infusion pump delivered 1500 heparin units/ hr to 2000 heparin units/hr. Haemoperfusion was continued until participant's paracetamol level was < 30 µg/mL. Supportive group: gastric lavage when first seen, and fresh frozen plasma and fluid administrated as clinically indicated. |
|
| Outcomes | Mortality. Fall in paracetamol level vs time after ingestion. Number of participants experiencing any adverse events. |
|
| Notes | 1 death in the haemoperfusion group. Plasma clearance of paracetamol by charcoal column was variable and small. No clinical problems. Liver damage in most participants was mild but the haemoperfusion group had more evidence of hepatic dysfunction with a higher mean bilirubin. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly allocated" Comment: method for generating sequence not detailed. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "allocated by a system of sealed envelopes." "Although the two groups were randomly allocated those receiving supportive therapy alone had ingested fewer tablets, were first seen earlier following the overdose, had a lower mean level of plasma paracetamol and a shorter initial drug half‐life." Comment: not specified if sequentially numbered, opaque or any other process used to prevent subversion. There was a marked imbalance in severity. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Not feasible given the nature of the interventions. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Not blinded, but low risk for primary outcomes of mortality and paracetamol concentration. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Does not appear to have any missing data (very small trial). |
| Selective reporting (reporting bias) | Low risk | No selective reporting apparent. |
| Other bias | Unclear risk | No power calculation provided. Very small trial which as written focused largely on kinetic outcomes. Intention to treat not detailed. |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | Unclear risk | Judged as high risk. |
Hamlyn 1981.
| Methods | Prospective, randomised, controlled trial. | |
| Participants | 2 hospitals: Newcastle (Royal Victoria Infirmary) and London (Guy's Hospital). Continuation of previously reported trial (Douglas 1976a). 40 participants: 9 (London), 31 (Newcastle) (4 from Newcastle in previous trial Douglas 1976a). Inclusion criteria: paracetamol level above semi‐logarithmic '200' line and treatment within 10 hr of ingestion. Exclusion criteria: children, alcohol dependency, known liver disease, and pregnant woman. No differences between the 3 treatment groups in terms of age, paracetamol ingested, delay to treatment, or paracetamol index (natural logarithm of the perpendicular distance from the blood paracetamol value to the 200‐line). Cysteamine and supportive group: 14 randomised. Age (mean (SD)) (years): 29.3 (14.9). Interval between ingestion and treatment (mean (SD)) (hr): 7.9 (1.9). Paracetamol index (mean (SD)): 0.444 (0.223). Amount of ingested paracetamol (mean (SD)) (g): 33.6 (16.0). Methionine and supportive group: 13 randomised. Age (mean (SD)) (years): 28.4 (14.2). Interval between ingestion and treatment (mean (SD)) (hr): 7.3 (1.6). Paracetamol index (mean (SD)): 0.525 (0.378). Amount of ingested paracetamol (mean (SD)) (g): 42.4 (25.2). Supportive treatment group: 13 participants. Age (mean (SD)) (years): 25.5 (10.8). Interval between ingestion and treatment (mean (SD)) (hr): 6.7 (2.2), this is the interval between ingestion and gastric lavage/treatment. Paracetamol index (mean (SD)): 0.671 (0.297). Amount of ingested paracetamol (mean (SD)) (g): 27.9 (13.0). Not reported in all groups: number of participants taking additional drugs, number of participants consuming additional alcohol, number of participants excluded after randomisation. |
|
| Interventions |
3 treatment groups: all participants received gastric lavage and supportive treatment (10% dextrose, added vitamins with potassium). Cysteamine + supportive group (N): supportive therapy + cysteamine in Newcastle. Cysteamine + supportive group (L): supportive therapy + cysteamine in London. Methionine + supportive group (N): supportive therapy + methionine in Newcastle. Methionine + supportive group (L): supportive therapy + methionine in London. Supportive treatment group (N): supportive therapy only in Newcastle. They did not treat any participant with only supportive care in London. Cysteamine IV as an immediate loading dose through a Millipore filter, followed by slow IV infusion for 20 hr up to a total base‐equivalent dose of 3.6 g. Methionine orally, 2.5 g every 4 hr to a total dose of 10 g. Supportive treatment: IV 10% dextrose with vitamins. Metoclopramide 10 mg IM administrated for severe or persistent vomiting. |
|
| Outcomes | Peak serum AST (LFTs for at least 4 days), maximum serum bilirubin, maximum PT. Renal function, amylase, electrocardiogram, lactate dehydrogenase and creatinine kinase measured daily. Liver biopsy in 20 participants. Number of participants experiencing any adverse events. |
|
| Notes | 1 death in the supportive group. Significant difference in favour of active treatment for AST. Significantly lower numbers of participants with grade III (severe) necrosis with both cysteamine + supportive and methionine + supportive groups. Continuation of Douglas 1976a, now restricted to 10 hr postoverdose. Hamlyn and colleagues did not provide the SD of the mean in any of their results. 4 participants included were from the earlier trial reported by Douglas 1976a. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "We employed, nevertheless, a balanced block randomisation to facilitate frequent trial monitoring. Patients were admitted to the trial by sealed envelope allocation, based upon random number tables, to one of three treatment groups in Newcastle, or one of two treatment groups in London. These comprised: supportive therapy only (S‐Newcastle only), supportive therapy + cysteamine (C) and supportive therapy + methionine (M). In order to avoid age bias, the randomisation also included adjustment for the numbers of under‐ and over‐30s in each group." Comment: 3 different methods for generating random allocation appear to be mentioned (simple randomisation, blocked randomisation, and minimisation). |
| Allocation concealment (selection bias) | High risk | Quote: "admitted to the trial by sealed envelope allocation." 1 centre randomised to only 2 groups of the study. Yet the numbers in each group were similar. Unclear how the method described above would be consistent with the randomisation process outlined above. Not mentioned if envelopes were sequentially numbered. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not mentioned and 2 different routes of administration (oral and IV) with a substance with a strong odour suggests this would be impossible. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not blinded but outcomes measured included mortality and biochemical markers. Biochemical markers detecting difference may be biased by frequency of testing. Regarding outcome of liver biopsy unclear risk of bias, how it was determined which participants to biopsy. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not mentioned of participants had missing outcome data, but only 1 site performed liver biopsies. |
| Selective reporting (reporting bias) | Low risk | No selective reporting apparent. |
| Other bias | Unclear risk | Intention to treat/power/premature stopping: Power: quote: "in patients with severe hepatic necrosis the arithmetic mean peak AST is approximately 3000U/l +/‐ SD 1000U/l and it may be shown that, to show a reduction with treatment to the mild liver damage level of 35 U/l (Hamlyn et al 1978) significant at the two‐tailed 5% level and type II error 5%, 12 paired comparison are needed." "end point of this trial coincided with the commercial introduction of intravenous N‐acetylcysteine." Unclear if the trial terminated prematurely in response to the introduction of IV acetylcysteine. A sample size calculation based on paired comparisons seemed inappropriate for a parallel group study with 3 groups. Information on intention to treat not provided. Numbers excluded before randomisation not reported. |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | Unclear risk | Judged as high risk. |
Hughes 1977.
| Methods | Randomised clinical trial. | |
| Participants |
Inclusion criteria: plasma paracetamol level that fell above a line on a semilog graph joining values of 1.3 mmol/L (200 mg/mL) 2 hr after ingestion and 0.5 mol/L (80 mg/mL) 12 hr after ingestion. Seen within 10 hr of paracetamol overdose. Exclusion criteria: not reported. Males: 18 (all participants). Females: 34 (all participants). Cysteamine group vs dimercaprol group: Number of participants randomised: 26 vs 26. Interval between ingestion and treatment (mean) (hr): 7.7 vs 7.9. Paracetamol plasma level on admission (mean) (mg/L): 295 vs 269. In both groups following data not reported: age, amount of ingested paracetamol, number of participants taking additional drugs, number of participants consuming additional alcohol, number of participants excluded after randomisation. Noted in earlier published data Gazzard 1975b, they noted no difference between the 2 groups in terms of initial paracetamol level. |
|
| Interventions |
Interventions: all received gastric lavage and supportive treatment. Cysteamine group: infusion of cysteamine hydrochloride freshly prepared for each participant, IV through a Millipore filter 0.22 mm in a dose of 2 g in 20 mL of water. A further 1.2 g dissolved in 1500 mL 5% dextrose was given over the next 20 hr. Dimercaprol group: deep IM injection in a dose of 4 mg/kg bodyweight every 4 hr for 24 hr, then 3 mg/kg every 4 hr for 24 hr. Liver biopsy was performed on 16 participants when PT had returned to normal. |
|
| Outcomes | Mortality, maximum AST, maximum serum bilirubin, maximum PT, liver biopsy finding (16 participants) and adverse effects of treatment. | |
| Notes | Peak abnormalities in serum bilirubin and PT were greater in participants treated with dimercaprol as was the severity of hepatic necrosis found on liver biopsy. 1 participant died in the dimercaprol group. Noted in earlier published data (Gazzard 1975b), no difference between the 2 groups in term of initial paracetamol level. Note earlier published results: Gazzard 1975b and Hughes 1976. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were allocated at random to treatment with cysteamine or dimercaprol." Sequence generation not mentioned. |
| Allocation concealment (selection bias) | High risk | Author correspondence: randomisation by envelopes, not concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No mention of blinding and 2 different routes of administration (IM and IV). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not blinded but outcomes measured were mortality and biochemical markers. Potential for detection bias depending on frequency of measuring biochemical markers. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Author correspondence: outcomes were prospectively collected by the researchers. |
| Selective reporting (reporting bias) | Low risk | No selective reporting apparent. |
| Other bias | Unclear risk | Powering or intention to treat not specified. The first 2 reports of the study were interim analyses, the trial did not seem to have had any prespecified power analysis and stopped when the authors deemed other treatments were more promising. |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | Unclear risk | Judged as high risk. |
Keays 1991.
| Methods | Randomised clinical trial. | |
| Participants |
Inclusion criteria: participants with paracetamol‐induced fulminant hepatic failure who had not already received acetylcysteine. Exclusion criteria: not reported. Acetylcysteine) group vs 'placebo' group: Number of participants randomised: 25 vs 25. Age (mean) (years): 33 vs 34. Male:female ratio: 12:13 vs 9:16. Interval between ingestion and admission to Liver Unit (mean) (hr): 53 vs 56. Additional characteristics: Serum creatinine (mean) (mmol/L): 246 vs 247. Arterial pH (mean): 7.39 vs 7.39. PT (mean) (seconds, control time 15 seconds):115 vs 140. Not reported in either group: paracetamol plasma level on admission, amount of ingested paracetamol, number of participants taking additional drugs, number of participants consuming additional alcohol, number of participants excluded after randomisation. |
|
| Interventions |
Acetylcysteine group: acetylcysteine IV infusion 150 mg/kg in 200 mL 5% dextrose over 15 min, followed by 50 mg/kg in 500 mL 5% dextrose over 4 hr, then 100 mg/L over 16 hr. Final infusion rate continued until recovery from encephalopathy or death. 'Placebo' group: equivalent amount of 5% dextrose without acetylcysteine. If needed, all participants received additional intensive liver care: maintenance of intravascular pressures, renal support (haemodialysis), treatment (mannitol, hyperventilation, and thiopentone) for raised intracranial pressure, elective ventilation, and muscle relaxant for grade 4 encephalopathy. |
|
| Outcomes | Mortality, cerebral oedema, hypotension requiring inotropic support, and renal failure. Liver function as assessed by PT and degree of encephalopathy. |
|
| Notes | Rate of survival significantly higher in acetylcysteine group (12/25 (48%)) vs placebo group (5/25 (20%)). Treatment group had a lower incidence of cerebral oedema, fewer participants developed hypotension needing inotropic support. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomised on admission to the liver failure unit by opening one of 50 identical sealed envelopes containing an allocation." No mention of sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | Not specified if sealed envelopes were sequentially numbered, opaque, or any other process to prevent subversion. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Multiple other interventions, not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "treatment with acetylcysteine could not be blind because the solution has an easily pungent aroma." Outcomes measured were mortality, inotrope requirement and biochemical markers, so high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not detailed. |
| Selective reporting (reporting bias) | Low risk | Quote: "two woman who had been randomised to acetylcysteine group underwent orthotropic liver transplantation, for the statistical analysis we assumed that they would have died." |
| Other bias | Unclear risk | Quote: Power "in order to detect a 40% difference in survivals … we had calculated that we would need to recruit 25 patients in each group to give a 90% power of achieving 5% significance. The retrospective study of late treatment with acetylcysteine in patients after paracetamol overdose who subsequently developed fulminant hepatic failure had suggested that such a difference in survival might be achieved." (Harrison 1990). Harrison 1990: "Mortality was 37% in patients who received acetylcysteine 10‐36 h after the overdose, compared with 58% in patients not given the antidote." Comment: a power calculation actually based on the referenced study would thus have had approximately 4 times as many participants. This suggests the power calculation may be post hoc or the study was stopped early. |
| Overall bias assessment (mortality) | Unclear risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | Unclear risk | Judged as high risk. |
Kerr 2005.
| Methods | Multicentre randomised clinical trial. | |
| Participants |
Inclusion criteria: people with paracetamol poisoning who required the administration of acetylcysteine, as assessed by acceptable practice guidelines, and included criteria such as serum paracetamol level, amount ingested, tests of liver injury, or a combination of the 3. Exclusion criteria: hypersensitivity to acetylcysteine. Number assessed: Number randomised: 223. Number excluded after randomisation: 43 excluded; 42 as incomplete study data or notes, or both. 15‐min acetylcysteine infusion group vs 60‐min acetylcysteine infusion group: Number of participants randomised: 109 vs 71. Age (mean) (years): female: 30.2; male: 33.6 vs female: 26.4; male: 31.9. Weight (mean) (kg): female: 63.7; male: 80.0 vs female: 63.6; male: 80.7. Number of participants where acetylcysteine started < 8 hr: 33 vs 25. Number of participants where acetylcysteine started > 8 hr: 74 vs 38. Not reported in either group: interval between ingestion and treatment, paracetamol plasma level on admission, amount of ingested paracetamol, number of participants taking additional drugs or consuming additional alcohol. |
|
| Interventions |
15‐min regimen: 150 mg/kg IV acetylcysteine in 200 mL of 5% dextrose over 15 min (loading). 60‐min regimen: 150 mg/kg IV acetylcysteine in 200mL of 5% dextrose over 60 min (loading). Both groups received the same 4 hr (50 mg/kg IV acetylcysteine in 500 mL of 5% dextrose) and 16 hr infusion (100 mg/kg IV acetylcysteine in 1000 mL of 5% dextrose). |
|
| Outcomes | Adverse events during and after IV acetylcysteine, in particular anaphylactoid reaction, 30 min intervals for the first 4 hr and subsequently 2, 4, and 8 hr intervals, ceasing at 24 hr after acetylcysteine administration. Blood samples: LFT, paracetamol, and coagulation test baseline and 12 hr intervals until the participant was discharged. |
|
| Notes | An adverse event occurred during or after acetylcysteine administration for 82 (75%) participants in the 15‐min group and 43 (61%) participants in the 60‐min treatment group. There were drug‐related adverse events in 49 (45%) participants in the 15‐min group and 27 (38%) participants in the 60‐min group; P = 0.36. Comparison of the 15‐min and 60‐min groups for ALT, AST, and INR revealed no statistically significant difference. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "The 500 randomization slips (250 with "15‐minute" and 250 with "60‐minute") were placed in a closed box. When an eligible patient was enrolled, the duty pharmacist allocated the listed treatment to that site and patient, which resulted in unblocked random allocation." |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Of the 180 evaluable patients, 109 patients were randomised to the 15‐minute treatment arm, and 71 patients were randomised to the 60‐minute treatment arm." The third‐party randomising (the poisons centre duty pharmacist) were not adequately concealed; however, those collecting the treatment presumably were unable to determine what the allocation assigned to their participant would be. The study allocation was quite unbalanced raising concerns about bias in allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Not feasible given nature of intervention. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Study was unblinded, outcomes included adverse events and biochemical markers. Potential for bias as assessors not blinded to treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: potential for bias as 42 participants excluded after randomisation. "223 patients were randomised; of these, 181 patients had evaluable hospital notes and study data.", "42 patients were excluded because of incomplete medical records" and "Further 3 excluded for efficacy as incomplete data." A large number excluded because of incomplete hospital notes. Comment: analysis not able to be done by intention to treat on all randomised participants and a large number of participants excluded for missing data. |
| Selective reporting (reporting bias) | Unclear risk | "Judgments of the attribution of an event to the study drug were made independently by 2 of the investigators, with consideration of the clinical events and assessments surrounding the event, whether medication was administered to treat the event, and whether any other action was implemented." Comment: unclear whether bias might have been introduced in this process of adjudicating on events. |
| Other bias | Unclear risk | Power: quote: "Initial sample size estimation indicated that 249 patients were required for each arm of the study to detect an approximate halving of the rate, with 80% power. An incidence of 9% was estimated using the literature evidence available at study design." Early cessation of study: quote: "The initial research plan included 500 patients. The study was terminated in 2003 with 180 evaluable patients because of the difficulty in obtaining data in a reasonable time frame." "At a formal consensus meeting, the investigators concluded that a reduction in the observed rate of anaphylactoid reaction (from 25% to 10%) was required to justify a change in the guidelines for the initial reaction in 180 patients was only 4.3% (standard error = 5.5%). A sample size of more than 1,000 patients in each arm would be required to show the observed difference to be significant at an equal to 0.05 with 80% power." Comment: early cessation for futility is not best practice but unlikely to have led to a biased estimate of treatment differences. Not recorded mean paracetamol level or mean paracetamol dose ingested, in each group. Known that increased adverse events from IV acetylcysteine at lower paracetamol concentrations, unknown if difference between the 2 groups. |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | Unclear risk | Judged as high risk. |
NCT01118663.
| Methods | Multicentre, double‐blind, randomised clinical study. | |
| Participants |
Inclusion: aged ≥ 12 years; any person requiring treatment with acetylcysteine for acute acetaminophen toxicity. Exclusion criteria: history of allergy or hypersensitivity to acetylcysteine or any component of Acetadote, exposed to investigational drugs within 30 days before Clinical Trial Material (CTM) administration, pregnant or nursing, baseline ALT or AST > 1000 U/L or INR > 2, on dialysis, had congestive heart failure or renal failure such that the volume of the study drug administration would render the participant unsuitable for the study. Acetylcysteine without ethylenediaminetetraacetic acid vs control: Number randomised: 7 vs 10. Male:female ratio: 5:2 vs 7:3. |
|
| Interventions |
Acetylcysteine without EDTA group: Acetadote EF (ethylenediaminetetraacetic acid ‐ free) (new formulation) 200 mg/kg in 1000 mL diluent over 4 hr; then 100 mg/kg in 1000 mL diluent over 16 hr. Control group: Acetadote (old formulation) 150 mg/kg in 200 mL diluent over 60 min; then Acetadote 50 mg/kg in 500 mL diluent over 4 hr; then Acetadote 100 mg/kg in 1000 mL diluent over 16 hr. |
|
| Outcomes |
Primary outcome: hepatotoxicity ALT or AST > 1000 U/L. Secondary outcomes: need for continued treatment beyond the standard 21‐hr dosing regimen, adverse events, anaphylactoid reaction in the first 1 hr. Acetylcysteine without EDTA vs control: 7 started 5 completed vs 10 started 8 completed. Adverse events: 2 vs 1. Number of participants withdrawn: 0 vs 1. |
|
| Notes | Risk of bias table could not be completed as "study was terminated prematurely due to lack of enrolment." Limited study results published on the ClinicalTrials.gov database. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unable to assess as "study was terminated prematurely due to lack of enrolment", limited protocol published on ClinicalTrials.gov database. |
| Allocation concealment (selection bias) | Unclear risk | Unable to assess as study was terminated prematurely. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unable to assess as study was terminated prematurely. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unable to assess as study was terminated prematurely. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unable to assess as "study was terminated prematurely due to lack of enrolment," limited results published on ClinicalTrials.gov database. |
| Selective reporting (reporting bias) | Unclear risk | Unable to assess as "study was terminated prematurely due to lack of enrolment," limited results published on ClinicalTrials.gov database. |
| Other bias | Unclear risk | Unable to assess as study was terminated prematurely. |
| Overall bias assessment (mortality) | Unclear risk | Risk of bias table could not be completed as "study was terminated prematurely due to lack of enrolment." Limited study results published on the ClinicalTrials.gov database. Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | Unclear risk | Risk of bias table could not be completed as "study was terminated prematurely due to lack of enrolment." Limited study results published on the ClinicalTrials.gov database. Judged as high risk. |
Underhill 1990.
| Methods | Randomised clinical trial. | |
| Participants |
Inclusion criteria: people aged ≥ 16 years who had ingested paracetamol ≥ 5 g within 4 hr of admission. Exclusion criteria: depressed conscious level, or with a condition such as previous gastric surgery that might preclude the use of any 1 of the treatment methods. Number of participants randomised: 60. Age (mean) (years): 25.7 (range 16‐62). Male:female ratio: 16:44 (numbers not given for each group but said to be similar age and sex). Time elapsed between ingestion and presentation at the department (mean) (min): 123 (range 30‐240). Paracetamol plasma level on admission (mean) (mg/L): supportive group: 90 vs activated charcoal group: 135 vs gastric lavage group: 160 vs ipecacuanha group: 110. Amount of ingested paracetamol (g): not reported. Number of participants taking additional drugs: 12. Number of participants consuming additional alcohol: 21. Number of participant excluded that were assessed: not reported. |
|
| Interventions |
Gastric lavage group: gastric lavage using a 36 FG tube. Activated charcoal group: activated charcoal carried out with Carbomix to drug ratio of 10:1. Ipecacuanha group: ipecacuanha syrup 30 mL, was repeated after 30 min if there was no response. No intervention group: no intervention to limit absorption (this group was treated in a different hospital in Derby, UK). Group was stopped for ethical reasons after only 5 participants were treated. |
|
| Outcomes | Plasma paracetamol levels measured on samples taken from an indwelling cannula prior to any treatment, and following treatment at 60, 90, 150 min after the first sample. Percentage change between first and last plasma level used as a measure of effectiveness. Fall in plasma paracetamol concentration vs time. |
|
| Notes | Activated charcoal more effective in lowering plasma paracetamol levels than either gastric lavage or ipecacuanha. In the intervention group, the plasma paracetamol concentration increased during treatment in 4/5 participants and led to cessation of the supportive treatment group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly allocated into one of four treatment groups." Not documented how randomisation sequence was generated. |
| Allocation concealment (selection bias) | High risk | "Ethical committee approval was obtained at both hospitals and the inclusion of a group who did not receive absorption limiting treatment was also approved at Derby." The above makes it clear that for allocation to the no intervention group there was no concealment that this was only possible at 1 hospital. Otherwise randomisation process not detailed enough to determine if there was allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not mentioned. Unclear whether there were missing data on randomised participants. |
| Selective reporting (reporting bias) | Unclear risk | Further, multiple methods could be used for the outcome of change in paracetamol level over time. They did not report if this resulted in any clinically relevant difference; for example, in the number requiring treatment with IV acetylcysteine or clinical outcome. |
| Other bias | Unclear risk | Power: not mentioned. Early cessation: quote: "Group 4 (no treatment group, Derby) was stopped for ethical reasons when the serum paracetamol levels increased between the first and last samples in four out of five patients." Intention‐to‐treat analysis: not mentioned. |
| Overall bias assessment (mortality) | High risk | Judged as high risk. |
| Overall bias assessment (non‐mortality outcomes) | High risk | Judged as high risk. |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; BP: blood pressure; hr: hour; IM: intramuscular; INR: international normalised ratio; IU: international units; IV: intravenous; min: minute; LFT: liver function test; NAC: N‐acetylcysteine; PT: prothrombin time; SD: standard deviation; SE: standard error; vs: versus.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bartels 2008 | Human volunteer study. |
| Bastaki 2006 | Review article. |
| Buckley 1999b | Observational study and meta‐analysis. |
| Burkhart 1995 | Quasi‐randomised study, randomised according to month of the year. Rates of adverse effects used. |
| Clark 1996 | Single armed study, all participants given ondansetron. |
| Cooper 2005 | Trial in people taking overdose of any medications, no subgroup analysis of paracetamol. |
| Critchley 1983 | Human volunteer study. |
| Dordoni 1973 | Not randomised. Study including human volunteers. |
| Douglas 1976b | Commentary. |
| Eguia 1997 | Case report and meta‐analysis. |
| Eyer 1991 | Human volunteer study. |
| Ferner 2001 | Editorial. |
| Gawarammana 2006 | Letter regarding Kerr 2005. |
| Gazzard 1974b | Randomised clinical trial of heparin in people with raised INR secondary to paracetamol‐induced liver necrosis. Excluded as per method of search, not to examine interventions that were treating secondary complication of liver failure, such as coagulopathy. |
| Gazzard 1975a | Abstract from meeting confirmed with author same data as Hughes 1977. |
| Gazzard 1975b | Randomised clinical trial of fresh frozen plasma in people with a raised INR . Excluded as per method of search, not to examine interventions that are treating secondary complication of liver failure, such as coagulopathy. |
| Guay 2003 | Update. |
| Hamlyn 1980 | Abstract for the Hamlyn 1981 trial that was included. |
| Hayes 2008 | Retrospective chart review. |
| Hershkovitz 1996 | Case report. |
| Hughes 1976 | Same data as Hughes 1977, correspondence with author confirms this. |
| Jalan 2006 | Review article. |
| Keays 1989 | Abstract, early data from Keays 1991. |
| Koch 2010 | Participants are liver failure from a non‐paracetamol cause. |
| Kulig 1985 | A study of gastric emptying in people with overdose, no subgroup analysis of individual drugs, so unable to obtain paracetamol data. |
| MacDonald 2006 | Commentary. |
| Mann 1992 | Review. |
| Mitchell 1984 | Animal rat study and human volunteer study. |
| Montoya‐Cabrera 1999 | Observational case series. |
| O'Grady 1988 | Randomised clinical trial of charcoal haemoperfusion in people with acute liver failure. Excluded as per method of search, not to examine interventions that are treating secondary complication of liver failure. |
| Renzi 1985 | Human volunteer study. |
| Saliba 2013 | Randomised clinical trial of an albumin dialysis system, Molecular Adsorbent Recirculating System (MARS). Excluded as per method not to investigate liver support devices. |
| Spiller 2006 | Observational prospective case series. |
INR: international normalised ratio.
Contributions of authors
AC: designed, drafted, and revised the protocol; performed the searches; selected trials and studies; extracted data; rated studies; and drafted and revised the review. CG: revised the protocol, conducted the Trial Sequential Analyses, and revised the review. JB: selected the trial Nicholas Buckley was involved in, assessed risks of bias, extracted data, and revised the review. NB: extracted data, rated studies, and revised the protocol and the review.
Sources of support
Internal sources
Copenhagen Trial Unit, Denmark.
External sources
The 1991 Pharmacy Foundation, Denmark.
Copenhagen Hospital Corporation's Medical Research Council's Grant on Getting Research into Practice (GRIP), Denmark.
Danish Medical Research Council's Grant on Getting Research into Practice (GRIP), Denmark.
Copenhagen Hospital Corporation's Medical Research Council, Denmark.
Declarations of interest
AC: none known. CG: none known. JB: none known. NB: none known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Arefi 2013 {published data only}
- Arefi M, Behnoush B, Pouraziz H, Yousefinejad V. Comparison of oral and intravenous N‐acetylcysteine administration in the treatment of acetaminophen poisoning. Scientific Journal of Kurdistan University of Medical Sciences 2013;18(2):36‐43. [] [Google Scholar]
Bateman 2014 {published data only}
- Bateman DN, Dear JW, Thanacoody HK, Thomas SH, Eddleston M, Sandilands EA, et al. Reduction of adverse effects from intravenous acetylcysteine treatment for paracetamol poisoning: a randomised controlled trial. Lancet 2014;383(9918):607‐704. [DOI: 10.1016/S0140-6736(13)62062-0] [DOI] [PubMed] [Google Scholar]
Douglas 1976a {published data only}
- Douglas AP, Hamlyn AN, James O. Controlled trial of cysteamine in treatment of acute paracetamol poisoning. Lancet 1976;2:111‐5. [DOI] [PubMed] [Google Scholar]
Eizadi‐Mood 2013 {published data only}
- Eizadi‐Mood N, Sabzghabaee AM, Siadat S, Yaraghi A, Shariati M, Gheshlaghi F, et al. Intravenous acetylcysteine versus oral and Intravenous acetylcysteine: does a combination therapy decrease side effects of acetylcysteine?. Pakistan Journal of Medical Sciences 2013;29(1 Suppl):308‐11. [] [Google Scholar]
Gazzard 1974a {published data only}
- Gazzard BG, Willson RA, Weston MJ, Thompson RPH, Williams R. Charcoal haemoperfusion for paracetamol overdose. British Journal of Clinical Pharmacology 1974;1:271‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamlyn 1981 {published data only}
- Hamlyn AN, Lesna M, Record CO, Smith PA, Path FRC, Watson AJ. Methionine and cysteamine in paracetamol overdose, prospective controlled trial of early therapy. Journal of International Medical Research 1981;9:226‐31. [DOI] [PubMed] [Google Scholar]
Hughes 1977 {published data only}
- Hughes RD, Gazzard BG, Hanid MA, Trewby PN, Murray‐Lyon IM, Davis M, et al. Controlled trial of cysteamine and dimercaprol after paracetamol overdose. BMJ (Clinical Research Ed.) 1977;2:1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Keays 1991 {published data only}
- Keays R, Harrison PM, Wendon JA, Forbes A, Gove C, Alexander GJM, et al. Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure. BMJ (Clinical Research Ed.) 1991;303:1026‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kerr 2005 {published data only}
- Kerr F, Dawson A, Whyte IM, Buckley N, Murray L, Graudins A, et al. The Australasian Clinical Toxicology Investigators Collaboration randomized trial of different loading infusion rates of N‐acetylcysteine. Annals of Emergency Medicine 2005;45(4):402‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
NCT01118663 {published data only}
- Cumberland Pharmaceuticals. A multi‐center, double‐blind, randomized, controlled study to determine the efficacy and safety of a new formulation of acetylcysteine injection. clinicaltrials.gov/ct2/show/study/NCT01118663 Date first received: 4 August 2014.
Underhill 1990 {published data only}
- Underhill TJ, Greene MK, Dove AF. A comparison of the efficacy of gastric lavage, ipecacuanha and activated charcoal in the emergency management of paracetamol overdose. Archives of Emergency Medicine 1990;7:148‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Bartels 2008 {published data only}
- Bartels S, Sivilotti M, Crosby D, Richard J. Are recommended doses of acetaminophen hepatotoxic for recently abstinent alcoholics? A randomized trial. Clinical Toxicology (Philadelphia, Pa.) 2008;46(3):243‐9. [DOI] [PubMed] [Google Scholar]
Bastaki 2006 {published data only}
- Bastaki SMA. Drugs update. Emirates Medical Journal 2006;24(3):245‐50. [] [Google Scholar]
Buckley 1999b {published data only}
- Buckley NA, Whyte IM, O'Connell DL, Dawson AH. Oral or intravenous N‐acetylcysteine: which is the treatment of choice for acetaminophen (paracetamol) poisoning?. Journal of Toxicology. Clinical Toxicology 1999;37(6):759‐67. [; JC‐‐NLM: Journal ID:kan, 8213460] [DOI] [PubMed] [Google Scholar]
Burkhart 1995 {published data only}
- Burkhart KK, Janco N, Kulig KW, Rumack BH. Cimetidine as adjunctive treatment for acetaminophen overdose. Human & Experimental Toxicology 1995;14(3):299‐304. [DOI] [PubMed] [Google Scholar]
Clark 1996 {published data only}
- Clark RF, Chen R, Williams SR, Johnson CL, Harchelroad F. The use of ondansetron in the treatment of nausea and vomiting associated with acetaminophen poisoning. Journal of Toxicology. Clinical Toxicology 1996;34(2):163‐7. [DOI] [PubMed] [Google Scholar]
Cooper 2005 {published data only}
- Cooper GM, Couteur DG, Richardson D, Buckley NA. A randomized clinical trial of activated charcoal for the routine management of oral drug overdose. QJM : Monthly Journal of the Association of Physicians 2005;98(9):655‐60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Critchley 1983 {published data only}
- Critchley JA, Dyson EH, Scott AW, Jarvie DR, Prescott LF. Is there a place for cimetidine or ethanol in the treatment of paracetamol poisoning?. Lancet 1983;1(8338):1375‐6. [DOI] [PubMed] [Google Scholar]
Dordoni 1973 {published data only}
- Dordoni B, Willson RA, Thompson RPH, Williams R. Reduction of absorption of paracetamol by activated charcoal and cholestyramine. BMJ (Clinical Research Ed.) 1973;3:86‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Douglas 1976b {published data only}
- Douglas AP, Hamlyn AN, James O. Controlled trial of cysteamine in treatment of acute paracetamol (acetaminophen) poisoning. Lancet 1976;1(7951):111‐5. [DOI] [PubMed] [Google Scholar]
Eguia 1997 {published data only}
- Eguia L, Materson BJ. Acetaminophen‐related acute renal failure without fulminant liver failure. Pharmacotherapy 1997;17(2):363‐70. [PubMed] [Google Scholar]
Eyer 1991 {published data only}
- Eyer P, Sprenger M. Oral administration of activated charcoal‐sorbitol suspension as first aid in prevention of poison resorption?. Klinische Wochenschrift 1991;69(19):887‐94. [DOI] [PubMed] [Google Scholar]
Ferner 2001 {published data only}
- Ferner RE. Our poisoned patients. QJM : Monthly Journal of the Association of Physicians 2001;94(3):117‐20. [DOI] [PubMed] [Google Scholar]
Gawarammana 2006 {published data only}
- Gawarammana IB, Greene SL, Dargan PI, Jones AL. Australian clinical toxicology investigators collaboration randomized trial of different loading infusion rates of N‐acetylcysteine. Annals of Emergency Medicine 2006;47(1):124. [DOI] [PubMed] [Google Scholar]
Gazzard 1974b {published data only}
- Gazzard BG, Clark R, Borirakchanyavat V, Williams R. A controlled trial of heparin therapy in the coagulation defect of paracetamol‐induced hepatic necrosis. Gut 1974;15(2):89‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gazzard 1975a {published data only}
- Gazzard BG, Hughes RD, Chhibber AD, Bennett JR, Murray‐Lyon IM, Dordoni B, et al. Proceedings: controlled trial of cysteamine and dimercaprol in the prevention of liver damage after paracetamol overdose. Gut 1975;16(10):839. [PubMed] [Google Scholar]
Gazzard 1975b {published data only}
- Gazzard BG, Henderson JM, William R. Early changes in coagulation following a paracetamol overdose and a controlled trial of fresh frozen plasma therapy. Gut 1975;16(8):617‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guay 2003 {published data only}
- Guay DRP, Hajjar ER. Geriatric pharmacotherapy updates. American Journal Geriatric Pharmacotherapy 2003;1(2):96‐100. [Google Scholar]
Hamlyn 1980 {published data only}
- Hamlyn AN, Lesna M, Record CO, Smith PA, Watson AJ, Meredith T, et al. Prevention of hepatic necrosis in severe paracetamol (acetaminophen) poisoning: prospective controlled trial of early treatment with cysteamine or methionine [abstract]. Gut 1980;21(2):A448. [Google Scholar]
Hayes 2008 {published data only}
- Hayes BK. Frequency of medication errors with intravenous acetylcysteine for acetaminophen overdose. Annals of Pharmacotherapy 2008;42(6):776‐70. [DOI] [PubMed] [Google Scholar]
Hershkovitz 1996 {published data only}
- Hershkovitz E, Shorer Z, Levitas A, Tal A. Status epilepticus following intravenous N‐acetylcysteine therapy. Israel Journal of Medical Sciences 1996;32(11):1102‐4. [] [PubMed] [Google Scholar]
Hughes 1976 {published data only}
- Hughes RD, Gazzard BG, Murray‐Lyon IM, Williams RS. The use of cysteamine and dimercaprol. Journal of International Medical Research 1976;4(4 Suppl):123‐9. [; CN‐00702516] [DOI] [PubMed] [Google Scholar]
Jalan 2006 {published data only}
- Jalan R, Williams R, Bernuau J. Paracetamol: are therapeutic doses entirely safe?. Lancet 2006;368(9554):2195‐6. [DOI] [PubMed] [Google Scholar]
Keays 1989 {published data only}
- Keays RT, Cove C, Forbes A, Alexander GJM, Williams R. Use of late N‐acetyl cysteine in severe paracetamol overdose [abstract]. Gut 1989;30(Suppl 10):A1512. [; CN‐00255461] [Google Scholar]
Koch 2010 {published data only}
- Koch A, Trautwein C. N‐acetylcysteine on its way to a broader application in patients with acute liver failure. Hepatology (Baltimore, Md.) 2010;51(1):338‐40. [DOI] [PubMed] [Google Scholar]
Kulig 1985 {published data only}
- Kulig K, Bar‐Or D, Cantrill SV, Rosen P, Rumack BH. Management of acutely poisoned patients without gastric emptying. Annals of Emergency Medicine 1985;14(6):562‐7. [DOI] [PubMed] [Google Scholar]
MacDonald 2006 {published data only}
- MacDonald TM. Acetaminophen: risk‐management urgently require. Pharmacoepidemiology and Drug Safety 2006;15(6):406‐9. [DOI] [PubMed] [Google Scholar]
Mann 1992 {published data only}
- Mann NS. Acetylcysteine for paracetamol‐induced hepatic failure. Annals of Internal Medicine 1992;116(Suppl 2):1992. [Google Scholar]
Mitchell 1984 {published data only}
- Mitchell MC, Schenker S, Speeg KV Jr. Selective inhibition of acetaminophen oxidation and toxicity by cimetidine and other histamine H2‐receptor antagonists in vivo and in vitro in the rat and in man. Journal of Clinical Investigation 1984;73(2):383‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Montoya‐Cabrera 1999 {published data only}
- Montoya‐Cabrera MA, Escalante‐Galindo P, Nava‐Juaez A, Terroba‐Larios VM, Teran‐Hernandez JA. Evaluation of the efficacy of N‐acetylcysteine administrated alone or in combination with activated charcoal in the treatment of acetaminophen overdose [Evaluacion de la eficacia de la N‐acetilcisteina administrada sola o conbinada con carbon activado en el tratamiento de la sobredosis por acetaminofen]. Gaceta Medica de Mexico 1999;135(3):239‐43. [PubMed] [Google Scholar]
O'Grady 1988 {published data only}
- O'Grady JG, Gimson AE, O'Brien CJ, Pucknell A, Hughes RD, Williams R. Controlled trials of charcoal hemoperfusion and prognostic factors in fulminant hepatic failure. Gastroenterology 1988;94(5):1186‐92. [DOI] [PubMed] [Google Scholar]
Renzi 1985 {published data only}
- Renzi FP, Donovan JW, Martin TG. Concomitant use of activated charcoal and N‐acetylcysteine. Annals of Emergency Medicine 1985;14(6):568‐72. [DOI] [PubMed] [Google Scholar]
Saliba 2013 {published data only}
- Saliba F, Camus C, Durand F, Mathurin P, Letierce A, Delafosse B, et al. Albumin dialysis with a non cell artificial liver support device in patients with acute liver failure: a randomized, controlled trial. Annals of Internal Medicine 2013;159(8):522‐31. [DOI] [PubMed] [Google Scholar]
Spiller 2006 {published data only}
- Spiller HA, Winter ML, Klein‐Schwartz W, Bangh SA. Efficacy of activated charcoal administered more than four hours after acetaminophen overdose. Journal of Emergency Medicine 2006;30(1):1‐5. [DOI] [PubMed] [Google Scholar]
Additional references
Bailey 1998
- Bailey B, McGuigan MA. Management of anaphylactoid reactions to intravenous N‐acetylcysteine. Annals of Emergency Medicine 1998;31:710‐5. [DOI] [PubMed] [Google Scholar]
Begg 1994
- Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics 1994;50(4):1088‐101. [PUBMED: 7786990] [PubMed] [Google Scholar]
Bernal 2013
- Bernal W, Hyyrylainen A, Gera A, Audimoolam VK, McPhail MJW, Auzinger G, et al. Lessons from look‐back in acute liver failure? A single centre experience of 3300 patients. Journal of Hepatology 2013;59:74‐80. [DOI] [PubMed] [Google Scholar]
Bradburn 2007
Brok 2008
- Brok J, Thorlund K, Gluud C, Wetterslev J. Trial sequential analysis reveals insufficient information size and potentially false positive results in many meta‐analyses. Journal of Clinical Epidemiology 2008;61(8):763‐9. [PUBMED: 18411040] [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive. Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [PUBMED: 18824466] [DOI] [PubMed] [Google Scholar]
Buckley 1999a
- Buckley NA, Whyte IM, O'Connell DL, Dawson AH. Activated charcoal reduces the need for N‐acetylcysteine treatment after acetaminophen (paracetamol) overdose. Journal of Toxicology. Clinical Toxicology 1999;37(6):753‐7. [DOI] [PubMed] [Google Scholar]
Buckley 2007
- Buckley N, Eddleston M. Paracetamol (acetaminophen) poisoning. BMJ Clinical Evidence 2007;2007:2101. [PMC free article] [PubMed] [Google Scholar]
Cairney 2016
- Cairney DG, Beckwith HK, Al‐Hourani K, Eddleston M, Bateman DN, Dear JW. Plasma paracetamol concentration at hospital presentation has a dose‐dependent relationship with liver injury despite prompt treatment with intravenous acetylcysteine. Clinical Toxicology (Philadelphia, Pa.) 2016;54(5):405‐10. [DOI] [PubMed] [Google Scholar]
Chiew 2015
- Chiew AL, Fountain JS, Graudins A, Isbister GK, Reith D, Buckley NA. Summary statement: new guidelines for the management of paracetamol poisoning in Australia and New Zealand. Medical Journal of Australia 2015;203(5):215‐8. [DOI] [PubMed] [Google Scholar]
Chiew 2016
- Chiew AL, Isbister GK, Duffull SB, Buckley NA. Evidence for the changing regimens of acetylcysteine. British Journal of Pharmacology 2016;81(3):471‐81. [DOI: 10.1111/bcp.12789] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chiew 2017
- Chiew AL, Isbister GK, Kirby KA, Page CB, Chan BSH, Buckley NA. Massive paracetamol overdose: an observational study of the effect of activated charcoal and increased acetylcysteine dose (ATOM‐2). Clinical Toxicology (Philadelphia, Pa.) 2017;55(10):1055‐65. [DOI] [PubMed] [Google Scholar]
Christophersen 2002
- Christophersen AB, Levin D, Hoegberg LCG, Angelo HR, Kampmann JP. Activated charcoal alone or after gastric lavage: a simulated large paracetamol intoxication. British Journal of Clinical Pharmacology 2002;53:312‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chyka 2005
- Chyka PA, Seger D, Krenzelok EP, Vale JA, American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists. Position paper: single‐dose activated charcoal. Clinical Toxicology 2005;43(2):61‐87. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Clark 1973
- Clark R, Borirakchanyavat V, Davidson AR, Thompson RP, Widdop B, Goulding R, et al. Hepatic damage and death from overdose of paracetamol. Lancet 1973;1(7794):66‐70. [DOI] [PubMed] [Google Scholar]
Daly 2008
- Daly FF, Fountain JS, Murray L, Graudins A, Buckley NA, Panel of Australian and New Zealand clinical toxicologists. Guidelines for the management of paracetamol poisoning in Australia and New Zealand ‐ explanation and elaboration. A consensus statement from clinical toxicologists consulting to the Australasian poisons information centres. Medical Journal of Australia 2008;188(5):296‐301. [DOI] [PubMed] [Google Scholar]
Dart 2006
- Dart R, Erdman A, Olson K, Christianson G, Manoguerra A, Chyka P, et al. Acetaminophen poisoning: an evidence‐based consensus guideline for out‐of‐hospital management. Clinical Toxicology 2006;44(1):1‐18. [] [DOI] [PubMed] [Google Scholar]
Davidson 1966
- Davidson DG, Eastham WN. Acute liver necrosis following overdose of paracetamol. British Medical Journal 1966;2(5512):497‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
DeMets 1987
- DeMets DL. Practical aspects in monitoring: a brief review. Statistics in Medicine 1987;6(7):753‐60. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Doyon 2009
- Doyon S, Klein‐Schwartz W. Hepatotoxicity despite early administration of intravenous N‐acetylcysteine for acute acetaminophen overdose. Academic Emergency Medicine 2009;16(1):34‐9. [DOI] [PubMed] [Google Scholar]
Duffull 2013
- Duffull SB, Isbister GK. Predicting the requirement for N‐acetylcysteine in paracetamol poisoning from reported dose. Clinical Toxicology (Philadelphia, Pa.) 2013;51(8):772‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Eddleston 2008
- Eddleston M, Juszczak E, Buckley NA, Senarathna L, Mohamed F, Dissanayake W, et al. Multiple‐dose activated charcoal in acute self‐poisoning: a randomised controlled trial. Lancet 2008;371(9612):579‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey GD, Schneider M, Minder C. Bias in meta‐analysis detected by a simple graphical test. BMJ (Clinical Research Ed.) 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gluud 2017
- Gluud C, Nikolova D, Klingenberg SL. Cochrane Hepato‐Biliary Group. About Cochrane (Cochrane Review Groups (CRGs)) 2017, Issue 5. Art. No.: LIVER.
Gosselin 2014
- Gosselin S, Juurlink DN, Kielstein JT, Ghannoum M, Lavergne V, Nolin TD, EXTRIP Workgroup. Extracorporeal treatment for acetaminophen poisoning: recommendations from the EXTRIP workgroup. Clinical Toxicology (Philadelphia, Pa.) 2014;52(852):856‐67. [DOI] [PubMed] [Google Scholar]
Green 2001
- Green R, Grierson R, Sitar DS, Tenenbbein M. How long after drug ingestion is activated charcoal still effective?. Journal of Toxicology. Clinical Toxicology 2001;39(6):601‐5. [DOI] [PubMed] [Google Scholar]
Green 2013
- Green JL, Heard KJ, Reynolds KM, Albert D. Oral and intravenous acetylcysteine for treatment of acetaminophen toxicity: a systematic review and meta‐analysis. Western Journal of Emergency Medicine 2013;14(3):218‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gunnell 1997
- Gunnell D, Hawton K, Murray V, Garnier R, Bismuth C, Fagg J, et al. Use of paracetamol for suicide and non‐fatal poisoning in the UK and France: are restrictions on availability justified?. Journal of Epidemiology and Community Health 1997;51:175‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ (Clinical Research Ed.) 2008;336:924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harbord 2006
- Harbord RM, Egger M, Sterne JAC. A modified test for small‐study effects in meta‐analyses of controlled trials with binary endpoints. Statistics in Medicine 2006;25(20):3443‐57. [DOI] [PubMed] [Google Scholar]
Harrison 1990
- Harrison PM, Keays R, Bray GP, Alexander GJ, Williams R. Improved outcome of paracetamol‐induced fulminant hepatic failure by late administration of acetylcysteine. Lancet 1990;335(8705):1572‐3. [DOI] [PubMed] [Google Scholar]
Heard 2014
- Heard K, Rumack BH, Green JL, Bucher‐Bartelson B, Heard S, Bronstein AC, et al. A single‐arm clinical trial of a 48‐hour intravenous N‐acetylcysteine protocol for treatment of acetaminophen poisoning. Clinical Toxicology 2014;52:512‐8. [DOI] [PubMed] [Google Scholar]
Heard 2017
- Heard K, Newton A. Paracetamol overdose. BMJ Best Practice: bestpractice.bmj.com last updated: 28 March 2017:1‐46.
Henry 1984
- Henry J, Volans G. ABC of poisoning. Analgesics: II ‐ paracetamol. British Medical Journal (Clinical Research Ed.) 1984;289(6449):907‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 1996
- Higgins RM, Goldsmith DJ, MacDiarmid‐Gordon A, Taberner D, Venning MC, Ackrill P. Treating paracetamol overdose by charcoal haemoperfusion and long‐hours high‐flux dialysis. QJM : Monthly Journal of the Association of Physicians 1996;89(4):297‐306. [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21:1539‐58. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011 Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org. [Available from www.cochrane‐handbook.org.]
Jakobsen 2014
- Jakobsen J, Wetterslev J, Winkel P, Lange T, Gluud C. Thresholds for statistical and clinical significance in systematic reviews with meta‐analytic methods. BMC Medical Research Methodology 2014;14:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jones 1998
- Jones AL. Mechanism of action and value of N acetylcysteine in the treatment of early and late acetaminophen poisoning: a critical review. Journal of Toxicology. Clinical Toxicology 1998;36:277‐85. [DOI] [PubMed] [Google Scholar]
Kao 2003
- Kao LW, Kirk MA, Furbee RB, Mehta NH, Skinner JR, Brizendine EJ. What is the rate of adverse events after oral N‐acetylcysteine administered by the intravenous route to patients with suspected acetaminophen poisoning?. Annals of Emergency Medicine 2003;42(6):741‐50. [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [PUBMED: 11730399] [DOI] [PubMed] [Google Scholar]
Koch‐Weser 1976
- Koch‐Weser J. Acetaminophen. New England Journal of Medicine 1976;295(23):1297‐300. [DOI] [PubMed] [Google Scholar]
Krenzelok 2004
- Krenzelok EP, McGuigan M, Lheur P, Manoguerra AS. Position paper: Ipecac syrup. Journal of Toxicology. Clinical Toxicology 2004;42(2):133‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kwan 1995
- Kwan D, Bartle WR, Walker SE. Abnormal serum transaminases following therapeutic doses of acetaminophen in the absence of known risk factors. Digestive Diseases and Sciences 1995;40(9):1951‐5. [DOI] [PubMed] [Google Scholar]
Lee 2004
- Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology (Baltimore, Md.) 2004;40(1):6‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lee 2011
- Lee WM, Larson AM, Todd Stravitz R. AASLD position paper: the management of acute liver failure: update 2011. www.aasld.org/practiceguidelines/Documents/AcuteLiverFailureUpdate2011.pdf (accessed June 2017).
Liisanantti 2003
- Liisanantti J, Kaukoranta P, Martikainen M, Ala‐Kokko T. Aspiration pneumonia following severe self‐poisoning. Resuscitation 2003;56(1):49‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lundh 2017
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub3; PUBMED: 23235689] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mant 1984
- Mant TG, Tempowski JH, Volans GN, Talbot JC. Adverse reactions to acetylcysteine and effects of overdose. British Medical Journal (Clinical Research Ed.) 1984;289(6439):217‐9. [PMID: 6234965] [DOI] [PMC free article] [PubMed] [Google Scholar]
Marks 2017
- Marks DJB, Dargan PI, Archer JRH, Davies CL, Dines AM, Wood DM, et al. Outcomes from massive paracetamol overdose: a retrospective observational study. British Journal of Clinical Pharmacology 2017;83:1163‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
McElhatton 1997
- McElhatton PR, Sullivan FM, Volans GN. Paracetamol overdose in pregnancy analysis of the outcomes of 300 cases referred to the Teratology Information Service. Reproductive Toxicology (Elmsford, N.Y.) 1997;11(1):85‐94. [DOI] [PubMed] [Google Scholar]
MHPRA 2012
- Medicines and Healthcare products Regulatory Agency. Treating paracetamol overdose with intravenous acetylcysteine: new guidance, 2012. www.gov.uk/drug‐safety‐update/treating‐paracetamol‐overdose‐with‐intravenous‐acetylcysteine‐new‐guidance (accessed 20 May 2016).
Mitchell 1974
- Mitchell JR, Thorgeirrson SS, Potter WZ. Acetaminophen induced hepatic injury: protective role of glutathione in man and rationale for therapy. Clinical Pharmacology and Therapeutics 1974;16(4):676‐84. [] [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [DOI] [PubMed] [Google Scholar]
Morgan 2005
- Morgan O, Griffiths C, Majeed A. Impact of paracetamol pack size restrictions on poisoning from paracetamol in England and Wales: an observational study. Journal of Public Health 2005;27(1):19‐24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
O'Grady 1997
- O'Grady JG. Paracetamol‐induced acute liver failure. Journal of Hepatology 1997;26 (Suppl):41‐6. [DOI] [PubMed] [Google Scholar]
Olsson 1988
- Olsson B, Johansson M, Gabrielsson J, Bolme P. Pharmacokinetics and bioavailability of reduced and oxidized N‐acetylcysteine. Journal of Clinical Pharmacology 1988;34(1):77‐82. [DOI] [PubMed] [Google Scholar]
Park 2015
- Park BK, Dear JW, Antoine DJ. Paracetamol (acetaminophen) poisoning. BMJ Clinical Evidence 2015;2015:pii: 2101. [PMC free article] [PubMed] [Google Scholar]
Prescott 1973
- Prescott LF, Wright N. The effects of hepatic and renal damage on paracetamol metabolism and excretion following overdosage.: A pharmacokinetic study. British Journal of Pharmacology 1973;49(4):602‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prescott 1976
- Prescott LF, Sutherland GR, Park J, Smith IJ, Proudfoot AT. Cysteamine, methionine, and penicillamine in the treatment of paracetamol poisoning. Lancet 1976;2(7977):109‐13. [DOI] [PubMed] [Google Scholar]
Prescott 1979
- Prescott LF, Illinngworth RN, Chrichley JAJ, Stewart MJ, Adam RD, Proudfoot AT. Intravenous N‐acetylcysteine: the treatment of choice for paracetamol poisoning. British Medical Journal 1979;2:1097‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prescott 1980
- Prescott LF. Kinetics and metabolism of paracetamol and phenacetin. British Journal of Clinical Pharmacology 1980;10:291S‐8S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prescott 2009
- Prescott K, Stratton R, Freyer A, Hall I, Jeune I. Detailed analyses of self‐poisoning episodes presenting to a large regional teaching hospital in the UK. British Journal of Clinical Pharmacology 2009;68(2):260‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Reuben 2016
- Reuben A, Tillman H, Fontana RJ, Davern T, McGuire B, Stravitz RT, et al. Outcomes in adults with acute liver failure between 1998 and 2013: an observational cohort study. Annals of Internal Medicine 2016;164(11):724‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [DOI] [PubMed] [Google Scholar]
Rumack 1975
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975;55:871‐6. [PubMed] [Google Scholar]
Rumack 2012
- Rumack BH, Bateman DN. Acetaminophen and acetylcysteine dose and duration: past, present and future. Clinical Toxicology 2012;50:91‐8. [DOI] [PubMed] [Google Scholar]
Sandilands 2009
- Sandilands EA, Bateman DN. Adverse reactions associated with acetylcysteine. Clinical Toxicology (Philadelphia, Pa.) 2009;47(2):81‐8. [DOI] [PubMed] [Google Scholar]
Savović 2012a
- Savović J, Jones HE, Altman DG, Harris RJ, Juni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Annals of Internal Medicine 2012;157(6):429‐38. [PUBMED: 22945832] [DOI] [PubMed] [Google Scholar]
Savović 2012b
- Savović J, Jones H, Altman D, Harris R, Juni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomised controlled trials: combined analysis of meta‐epidemiological studies. Health Technology Assessment 2012;16(35):1‐82. [PUBMED: 22989478] [DOI] [PubMed] [Google Scholar]
Schmidt 2001
- Schmidt LE, Dalhoff K. Risk factors in the development of adverse reactions to N‐acetylcysteine in patients with paracetamol poisoning. British Journal of Pharmacology 2001;51(1):87‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schmidt 2002
- Schmidt LE, Knudsen TT, Dalhoff K, Bendtsen F. Effect of acetylcysteine on prothrombin index in paracetamol poisoning without hepatocellular injury. Lancet 2002;360(9340):1151‐2. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Senarathna 2012
- Senarathna SG, Sri RS, Buckley N, Fernandopulle R. A cost effectiveness analysis of the preferred antidotes for acute paracetamol poisoning patients in Sri Lanka. BMC Clinical Pharmacology 2012;12(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shiago 2011
- Shiago K, Watson I, Reidenberg MM. Application to change the status of methionine or N‐acetylcysteine on the model list, 2011. www.who.int/selection_medicines/committees/expert/18/applications/Delete_Methionine.pdf (accessed 20 May 2016).
Skoog 2015
- Skoog M, Saarimäki JM, Gluud C, Sheinin M, Erlendsson K, Aamdal S, et al. Transparency and Registration in Clinical Research in the Nordic Countries. Oslo (Norway): Nordic Trial Alliance, NordForsk, 2015. [Google Scholar]
Smilkstein 1988
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N‐acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). New England Journal of Medicine 1988;319(24):1557‐62. [DOI] [PubMed] [Google Scholar]
Smilkstein 1991
- Smilkstein MJ, Bronstein AC, Linden C, Augenstein WL, Kulig KW, Rumack BH. Acetaminophen overdose: a 48‐hour intravenous N‐acetylcysteine treatment protocol. Annals of Emergency Medicine 1991;20(10):1058‐63. [DOI] [PubMed] [Google Scholar]
Speeg 1995
- Speeg KV, Bay MK. Prevention and treatment of drug‐induced liver disease. Gastroenterology Clinics of North America 1995;24(4):1047‐64. [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta‐analyses?. International Journal of Epidemiology 2009;38(1):276‐86. [PUBMED: 18824467] [DOI] [PubMed] [Google Scholar]
Thorlund 2010
- Thorlund K, Anema A, Mills E. Interpreting meta‐analysis according to the adequacy of sample size. An example using isoniazid chemoprophylaxis for tuberculosis in purified protein derivative negative HIV‐infected individuals. Clinical Epidemiology 2010;2:57‐66. [PUBMED: 20865104] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User manual for Trial Sequential Analysis (TSA), 2011. ctu.dk/tsa/files/tsa_manual.pdf (accessed 20 May 2016).
TSA 2011 [Computer program]
- Copenhagen Trial Unit. TSA ‐ Trial Sequential Analysis. Version 0.9 Beta. Copenhagen: Copenhagen Trial Unit, 2011.
Vale 1995
- Vale JA, Proudfoot AT. Paracetamol (acetaminophen) poisoning. Lancet 1995;346(8974):547‐52. [DOI] [PubMed] [Google Scholar]
Vale 2004
- Vale JA, Kulig K, American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists. Position statement: gastric lavage. Journal of Toxicology. Clinical Toxicology 2004;42(7):933‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [PUBMED: 18083463] [DOI] [PubMed] [Google Scholar]
Wetterslev 2009
- Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in random‐effects model meta‐analyses. BMC Medical Research Methodology 2009;9:86. [PUBMED: 20042080] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wetterslev 2017
- Wetterslev J, Jakobsen JC, Gluud C. Trial Sequential Analysis in systematic reviews with meta‐analysis. BMC Medical Research Methodology 2017;17(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Whyte 2000
- Whyte IM, Buckley NA, Reith DM, Goodhew I, Seldon M, Dawson AH. Acetaminophen causes an increased international normalized ratio by reducing functional factor VII. Therapeutic Drug Monitoring 2000;22(6):742‐8. [DOI] [PubMed] [Google Scholar]
Williamson 2013
- Williamson K, Wahl MS, Mycyk MB. Direct comparison of 20‐hour IV, 36‐hour oral, and 72‐hour oral acetylcysteine for treatment of acute acetaminophen poisoning. American Journal of Therapeutics 2013;20(1):37‐40. [DOI] [PubMed] [Google Scholar]
Wolf 2007
- Wolf SJ, Heard K, Sloan EP, Jagoda AS. Clinical policy: critical issues in the management of patients presenting to the emergency department with acetaminophen overdose. Annals of Emergency Medicine 2007;50:292‐313. [DOI] [PubMed] [Google Scholar]
Woo 2000
- Woo OF, Mueller PD, Olson KR, Anderson IB, Kim SY. Shorter duration of oral N‐acetylcysteine therapy for acute acetaminophen overdose. Annals of Emergency Medicine 2000;35(4):363‐8. [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Juni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ (Clinical Research Ed.) 2008;336(7644):601‐5. [PUBMED: 18316340] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yeates 2000
- Yeates PJA, Thomas SHL. Effectiveness of delayed activated charcoal administration in simulated paracetamol (acetaminophen) overdose. British Journal of Clinical Pharmacology 2000;49(1):11‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yoon 2016
- Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N. Acetaminophen‐induced hepatotoxicity: a comprehensive uUpdate. Journal of Clinical and Translational Hepatology 2016;4(2):131‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Brok 2002
- Brok J, Buckley N, Gluud C. Interventions for paracetamol (acetaminophen) overdoses. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD003328] [DOI] [PubMed] [Google Scholar]
Brok 2006
- Brok J, Buckley N, Gluud C. Interventions for paracetamol (acetaminophen) overdose. Cochrane Database of Systematic Reviews 2006, Issue 2. [; DOI: 10.1002/14651858.CD003328.pub2; JC‐‐NLM: Journal ID:100909747] [DOI] [PubMed] [Google Scholar]