

Abstract
Pepper (Capsicum spp.) is one of the most important vegetable crops; however, pepper genomic studies lag behind those of other important Solanaceae. Here we present the results of a high-throughput genotyping-by-sequencing (GBS) study of a collection of 190 Capsicum spp. accessions, including 183 of five cultivated species (C. annuum, C. chinense, C. frutescens, C. baccatum, and C. pubescens) and seven of the wild form C. annuum var. glabriusculum. Sequencing generated 6,766,231 high-quality read tags, of which 40.7% were successfully aligned to the reference genome. SNP calling yielded 4083 highly informative segregating SNPs. Genetic diversity and relationships of a subset of 148 accessions, of which a complete passport information was available, was studied using principal components analysis (PCA), discriminant analysis of principal components (DAPC), and phylogeny approaches. C. annuum, C. baccatum, and C. chinense were successfully separated by all methods. Our population was divided into seven clusters by DAPC, where C. frutescens accessions were clustered together with C. chinense. C. annuum var. glabriusculum accessions were spread into two distinct genetic pools, while European accessions were admixed and closely related. Separation of accessions was mainly associated to differences in fruit characteristics and origin. Phylogeny studies showed a close relation between Spanish and Mexican accessions, supporting the hypothesis that the first arose from a main genetic flow from the latter. Tajima’s D statistic values were consistent with positive selection in the C. annuum clusters, possibly related to domestication or selection towards traits of interest. This work provides comprehensive and relevant information on the origin and relationships of Spanish landraces and for future association mapping studies in pepper.
Subject terms: Plant domestication, Plant breeding
Introduction
Peppers, chilies, and ajís, among other terms, refer to different forms of Capsicum spp., one of the most important cultivated vegetables in the world1. Thirty-one species are recognised in the genus, of which 26 are wild and five are cultivated2. The latter are: (i) C. annuum L., which is the most diverse, economically relevant, and studied species, and includes ‘bell’, ‘jalapenos’, ‘numex’, and ‘ancho’ types, among others; (ii) C. chinense Jacq., which includes very pungent peppers like the ‘habanero’ type; (iii) C. frutescens L. in which most known form is ‘tabasco’; (iv) C. baccatum L. or ‘ají’, which contains the ‘lemon drop’ and ‘ají escabeche’ as some of its most common forms; and finally; (v) C. pubescens Ruiz et Pav., which contains ‘rocoto’ and ‘manzano’ types3–5. At present, three complexes containing the cultivated peppers are distinguished based on the ability to cross-pollinate: (i) C. annuum complex, which comprises C. annuum, C. chinense, C. frutescens, their wild relatives, and C. galapagoensis Hunziker6, (ii) C. baccatum complex, which contains C. baccatum, C. praetermissum Heiser et Smith, and C. tovarii Eshbaugh, Smith et Nickrent7,8, and (iii) C. pubescens complex with C. pubescens and its wild relatives C. cardenasii Heiser et Smith and C. eximium Hunziker. Despite the fact that there are strong incompatibility barriers for hybridisation among these complexes, the development of viable hybrids, including hybrids between C. annuum and C. baccatum, has been reported in several works9–11.
The cultivated Capsicum species encompass a broad diversity as a result of evolution, domestication, and artificial and natural selection in agricultural environments in different primary and secondary centres of diversity3,12. In this regard, Spain is considered a secondary centre of diversity for peppers, especially for C. annuum which was brought mainly from Mexico just after the discovery of America13. Introduced as an alternative to Asian black pepper (Piper nigrum L.), capsicum peppers rapidly spread across Europe, Africa, and Asia4. In Spain, a process of more than 500 years of selection performed by generations of farmers created a plethora of ecotypes adapted to local conditions, many of which are still cultivated. This is more evident for the fleshy, big-fruited, bell-peppers called ‘Morrón’, named for their similarity to the nose of a sheep or a cow (i.e. morro in Spanish). Amazingly, C. annuum fruits and their derivatives have the largest number of EU Protected Designations of Origin (PDO) and Protected Geographical Indications (PGI) in Spain, such as ‘Arnoia’, ‘Pimiento Asado del Bierzo’, ‘Couto’, ‘Gernika’, ‘Morrón de Fresno’, ‘Riojano’ (PGI), and ‘Bola—Pimentón de Murcia’, ‘Padrón’, ‘Jaranda’, ‘Pimentón de la Vera’, or ‘Piquillo de Lodosa’ as registered PDO4,14. Despite that, most peppers production in Spain is based on F1 hybrids from ‘California Wonder’, ‘Lamuyo’, and ‘Dulce Italiano’ types, which have displaced the traditional and ancient materials mentioned earlier. However, the interest for the “taste of the past” by consumers and the challenge of adapting to climate change are contributing to the enhancement and reintroduction of landraces14–16. In addition, the studies on diversity of these materials are of importance in terms of: (i) genetic fingerprinting of varietal types, (ii) registration of materials, and farmers and communities rights preservation, (iii) genetic relationships in order to provide breeders with information about the available materials for breeding programmes, (iv) conservation of genetic resources, (v) development of non-redundant core collections, and (vi) revert the variability loss due to genetic erosion.
Despite its economical relevance, the development of Capsicum molecular tools lags behind other economically important Solanaceae crops, such as tomato or potato17,18. Its unusually large genome and repetitiveness may be the reasons for this delayed development18–20. In this respect, germplasm diversity analysis is one of the key elements for plant breeding and biodiversity conservation of Capsicum species10,21. However, it is highly dependent on the availability of informative genetic tools such as molecular markers8,22.
Throughout the last decade, high-throughput sequencing technologies development was stimulated by the need for low cost data and by the availability of faster analysis tools23,24. High-throughput genotyping-by-sequencing (GBS) has had an important impact on the scientific community due to its versatile application23,25–27. This approach can provide accurate results independently of the target species or population, and does not require having previous available genomic information23,25,28. GBS has been successfully used in pepper in recent years and an important amount of highly informative genome-wide SNPs were generated in each experiment. Germplasm diversity, population structure, and genomic selective sweeps analysis, as a result of domestication or local adaptation, are common to these works as it is an important first step for latter Genome-Wide Association Studies (GWAS)29,30. GBS-generated SNPs have been proven useful in the detection of trait-associated QTLs for both C. annuum and C. baccatum paving the way for further association studies and for a better understanding of pepper’s evolution31,32. To our knowledge, our work is the first to use GBS to analyse population structure and diversity and to assess selective genomic sweeps in a collection of Spanish landraces.
Herein we present a diversity study of a collection of Capsicum spp., encompassing four cultivated species and the C. annuum wild ancestor C. annuum var. glabriusculum using GBS. Our goals are genotyping a representative collection of Spanish heirlooms and ecotypes encompassing most varietal types of pepper, to shed light into the Spanish landraces phylogenetic relationships, among them and with other materials, to evaluate their molecular diversity and population structure.
Material and methods
Plant material
A diverse collection of 190 Capsicum spp. accessions encompassing five cultivated species, Capsicum annuum var. annuum (from now on C. annuum; 137 accessions), C. chinense (14 accessions), C. frutescens (2 accessions), C. baccatum (28 accessions), C. pubescens (2 accessions), and the wild form C. annuum var. glabriusculum (7 accessions), commonly known as ‘chiltepín’, was considered for GBS sequencing.
We report here the sequencing results for the collection mentioned above, although, since for 42 of the 190 accessions we did not possess a complete passport information those were excluded for downstream analysis. Hence, the 148 accessions subset was provided by the Universitat Politècnica de València Germplasm Bank, the COMAV Capsicum breeding group (112 accessions), several other research institutions (e.g. Institut National de la Recherche Agronomique (INRA-GEVES), Maritsa Vegetable Crops Research Institute (MVCRI), Mexico Chile breeding programme of Universidad Autónoma de Aguascalientes (UAA), Penn State University and United States Department of Agriculture (USDA); 23 accessions) and several seed companies (e.g. Batlle, Franchi Simenti, Intersemillas, Mascarell, Ramiro Arnedo, Reimer Seeds, and Zeraim Ibérica; 13 hybrids and heirloom lines) (Supplementary Data: Table 1).
The considered subset encompassed C. annuum (118) accessions, C. annuum var. glabriusculum (7), C. chinense (12), C. frutescens (2), and C. baccatum (9) (Supplementary Data: Table 1). Most of the considered accessions from C. annuum correspond to sweet, red, bell-shaped Spanish landraces, although a considerable amount of variability for pungency/sweetness, colour, fruit shape, resistances, origin, and varietal types was also included for a better evaluation of the diversity and phylogeny of the Capsicum genus (Supplementary Data: Table 1). Spanish C. annuum heirlooms and traditional materials have been prospected for more than 35 years in all regions of the country and, therefore, they can be considered highly representative of the variation of this centre of diversity in this species.
DNA extraction, library preparation, and sequencing
DNA was extracted from young leaves using a modified CTAB protocol33. Raw and restriction enzyme HindIII (Thermo Fisher Scientific, Wilmington, NC, USA) digested DNA electrophoresis was run on 0.8% agarose gel to assure DNA integrity. Purity was assessed using Nanodrop® (ND-1000, Thermo Fisher Scientific, Wilmington, NC, USA) and quantity was assessed with Qubit™ (2.0 Fluorometer, Invitrogen, Carlsbad, CA, USA). High molecular weight DNA aliquots with 230/260 and 260/280 ratios ranging between 1.8–2.0 and 1.8–2.2, respectively, were then sent to Cornell University sequencing facilities (Ithaka, NY, USA). ApeKI methylation-sensitive restriction enzyme was used for library preparation as described by Elshire et al.28. Illumina sequencing adaptors and sample-specific barcodes were then ligated to the resulting fragments sticky ends and samples were pooled together for multiplexing. Polymerase chain reaction (PCR) was performed for library construction and Illumina HiSeq2500 (Illumina, Inc., San Diego, CA, USA) single-end technology was used for library sequencing. Generated good barcoded reads were captured, collapsed by similarity, and stored into a FASTQ file to generate unique tags. Only tags occurring at minimal count (≥3) were retained to generate a MasterTag file (FASTQ) as described by Elshire et al.28 and Glaubitz et al.24.
Mapping and SNP calling
MasterTag file was aligned against the reference genome CM334 (Criollo de Morellos version 1.55)20 using Burrows–Wheeler Aligner (BWA version 0.7.8-r455)34 set to default settings. Aligned sequence tags were stored into TOPM (TagsOnPhysicallMap; SAM/BAM format35) file. SNP calling was then performed applying TASSEL-GBS Pipeline (version 3.0.173)24,36. Low-quality SNPs were filtered out by minimum minor allele frequency (mnMAF < 0.01) and missing data per site (MDpS > 10%), and finally converted into Variant Call Format file (VCF).
Sequencing and SNP calling statistics
Sequencing, alignment, SNP calling, and population statistics were performed for a better understanding of results quality. SAMtools (version 1.8)35 was used to calculate the number of reads that passed the quality control and aligned successfully against reference. BEDtools (version 2.25.0)37 was used to assess the percentage of sequence tags that overlap with genic regions. Transitions/transversions ratio was obtained by BCFtools (version 1.8)35. And finally, heterozygosity was obtained by VCFtools (version 0.1.14)38.
Population structure analysis
Evolutionary relationships and population structure were analysed implementing Rstudio (version 1.1.383)39. As a first step, VCF file resulting from SNP calling was subjected to another filtering process by SNPRelate package (version 1.12.2)40. Steps included: (1) removing multi-allelic, monomorphic, and low-quality positions; and (2) filter SNPs with a linkage disequilibrium (LD) threshold of 0.2. Initial analysis of population structure was carried out by principal components analysis (PCA) using the SNPRelate package40 and plotted using ggplot2 package (version 2.2.1)41.
To better understand and describe the genetic structure, discriminant analysis of principal components (DAPC) was applied using DAPC function from adegenet package (version 2.1.1)42–44. Most informative SNPs obtained by SNPRelate40 were used as input. DAPC workflow consisted of: (1) dataset transformation based on PCA, (2) determination of the optimal number of clusters by Bayesian information criterion (BIC) for K = 1–20 by k-means clustering with 100,000 iterations and 1000 randomly chosen starting centroids, (3) selection of K with lowest BIC as optimal number of clusters, selection of optimal number of PCs, and discriminant analysis (DA) functions to retain, and lastly (4) DAPC computation and plotting using ggplot241.
For further elucidation of genetic distance between samples and clusters, a phylogenetic tree was constructed. For that, the aboot function from Poppr package (version 2.7.1)45 was run with the parameters bitewise.distance tree with neighbour-joining algorithm46 and 1000 bootstrap replicates. Plot.phylo function loaded from ape package (version 5.0)47 plotted the generated tree.
Genetic diversity and selective sweeps
Population genetic diversity was calculated through Weir and Cockerham’s Fst index48 between all the clusters detected by DAPC. Tajima’s D statistic49 with a bin size of 500 kb was used to identify selective sweeps from our data for all the established clusters and plotted with ggplot2 package41. The mentioned statistics were obtained by VCFTools38.
Results and discussion
Sequencing and SNP calling
A collection of 190 Capsicum spp. accessions was successfully sequenced using the Illumina HiSeq2500 platform and yielding a total of 568,964,449 raw reads of which 524,450,716 (92.18%) represented good barcode reads. FASTQ file containing only collapsed and filtered reads was then aligned against the CM334 reference genome20. From the 6,766,231 unique read tags present in the file, 40.8% uniquely aligned, 7.4% multiply aligned, and 51.8% did not align successfully to the reference genome. Only 40.8% of sequences uniquely aligned to the reference genome, considerably fewer than the ones that do not align. These results could be due to (1) lack of reference for some positions in the reference genome, (2) the repetitiveness inherent to pepper genome contributing to two or more aligning sites for some sequences, (3) several accessions from species other than C. annuum probably could not align correctly, (4) some of the produced sequences could be mitochondrial or chloroplast DNA. The results are consistent with other recent works with this genus. Taranto et al.29 aligned uniquely 43.4% of tags and 9.8% aligned to multiple sites; and Ahn et al.50 obtained 45.5% of uniquely aligned sequence tags for a C. baccatum accession and 39.2% for a C. annuum accession.
ApeKI was selected for genome complexity reduction due to its sensitivity to methylation and consequent frequent cut in gene-rich regions28,51. BAM file containing only mapped reads was compared against the previously available CM334 annotated genes gff3 (General Feature File) file in order to calculate the number of intersected genic regions. From a total of 3,260,848 tags, 39.3% overlapped genic regions. Our results indicate that chromosomes 2 (49%), 3 (48%), and 8 (47%) have the greater percentage of tags on genic regions (Fig. 1). Contrastingly, chromosomes 9 (33%), 10 (32%), and 11 (32%) presented the lowest values of tags overlapping genes, with only a third of total number of read tags being located inside these regions (Fig. 1). Taranto et al.29 using GBS technology and a widely diverse collection reported similar results. These results could be due to the GBS protocol, which targets preferably genic regions and has a lower genomic coverage especially for repetitive regions51,52.
Fig. 1.
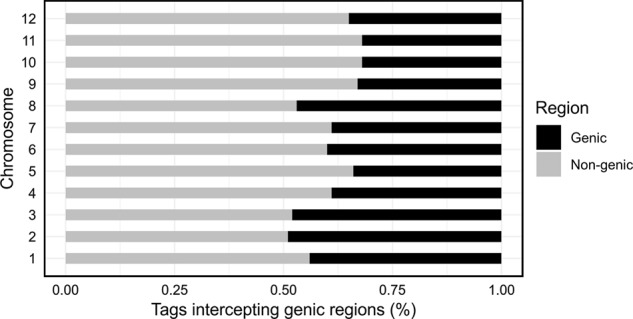
Distribution of filtered Illumina sequencing tags intercepting genic (black) and non-genic (grey) regions per chromosome
Variant calling was performed by TASSEL-GBS Pipeline24,36 on the TOPM (BAM) file containing only uniquely aligned sequences producing 640,377 raw SNPs. Finally, after removing low-quality positions by MAF and MDpS, 531,680 SNPs, distributed along the entire genome, were selected (Supplementary Data: Fig. 1).
Transitions were found in greater proportion (59.7%). Transitions/transversions ratios are in agreement to other previous reports for pepper29. This phenomenon could be explained as an evolutionary advantage in case of mis-pairing because they are more likely to preserve protein structure than transvertions53.
The levels of observed heterozygosity for the 531,680 called SNPs ranged between 2.35% and 6.50% for these pepper accessions and averaged 3.16%. Regarding species, C. annuum var. glabriusculum (2.87%) displayed the lowest mean value, while C. baccatum (5.20%) the highest. And finally, experimental lines (3.10%) presented a lower mean value of observed heterozygote positions, while commercial hybrids (4.15%) presented the highest (Supplementary Data: Table 2). Values as low as the ones found here are not unusual for autogamous species such as pepper54,55. Lower mean values for observed heterozygosity were reported by Taranto et al.29 for a collection of 397 accessions from 8 different species (2.40%). However, Cheng et al.56 and Lee et al.57 reported higher mean values for bigger and more diverse populations than ours, 17.00% (ranging from 1.00% to 23.00%) and 15.00% (ranging from 9.00% to 21.00%), respectively. In another study, Nimmakayala et al.32 reported values in between the ones mentioned above (6.00%, ranging from 3.00% to 18.00%) for a diverse C. annuum population. The literature seems to support the perception that wild accessions have higher heterozygosity values than cultivated57. In this way, Ibiza et al.22 found a higher level of heterozygosity for C. baccatum, possibly indicating a higher level of allogamy than the others (Supplementary Data: Table 2).
SNP filtering
A previous step to remove low quality and monomorphic positions by mnMAF, MDpS, and LD was performed. Original VCF with 531,680 positions was filtered by SNPRelate package40 resulting in a significant decrease to 4083 highly informative and well distributed across genome variants (Supplementary Data—Fig. 1; Supplementary Data—Table 3). Most pepper population diversity and structure analyses have relied on just a dozen to a few dozens of markers due to lack of data resolution of the peppers genome12,57,58. Fortunately, with the NGS technologies, thousands of markers are now easily available for researchers28. Thus, the most recent works used a similar number of markers to our study in order to assess genetic structure30 and for GWAS32.
Population genetic relationships
The set of 4083 SNPs and SNPRelate package47 were used for PCA analysis. The first two principal components (PC) accounted for 45.5% and 6.3% of total variability, respectively (Fig. 2). The population can be divided into three different clusters: (i) one comprising all C. annuum plus four C. annuum var. glabriusculum accessions (mex_v1196, mex_q1078, mex_s1120, and usa_a1003), (ii) a second composed exclusively of C. baccatum accessions, and (iii) a plurispecific cluster formed by C. chinense, C. frutescens, and the remaining three C. annuum var. glabriusculum accessions. The first PC (PC1) separated species by complexes. In this way, the C. annuum complex (C. annuum, C. chinense, and C. frutescens) accessions displayed low and negative values for the PC1 whereas the C. baccatum complex accessions showed positive values. The second PC (PC2) differentiated C. annuum from the other species. Several studies using SSR12,58 and SNP markers29,57 described a similar separation of species. As in our case, Nicolai et al.58 and González-Pérez et al.12 reported a close relationship between C. chinense and C. frutescens. C. annuum var. glabriusculum peculiar distribution was also mentioned in Nicolai et al.58 and Taranto et al.29, where it was positioned near C. annuum, C. chinense, and C. frutescens.
Fig. 2. First and second principal components for both 148 accessions (left) and 118 C. annuum and closely related four C. annuum var. glabriusculum (right) based on 4083 filtered SNPs.

Accession corresponding species (left) and origin (right) information is provided in a colour palette in each graph
In order to visualise in detail the relationships within the C. annuum cluster, a PCA was performed with only those accessions and the four closest C. annuum var. glabriusculum accessions, comprising 122 accessions. The PC1 (14.5%) and PC2 (8.6%) separated the accessions based on origin and fruit traits (Fig. 2). Sweet large-fruited Spanish and other European accessions (Bulgaria, France, Italy, and Serbia) clustered together, whereas pungent small-fruited North American (Mexico and USA) and Indian, Spanish, Turkish, and Sri Lankan accessions represented a much more diverse cluster of accessions. C. annuum var. glabriusculum mex_s1120 and usa_a1003 accessions clustered together and far away from the C. annuum group, mex_q1078 was at mid-distance from groups, and mex_v1196 was the closest to the cluster formed by the pungent C. annuum varieties (Fig. 2). Nicolai et al.58, Lee et al.57, and Taranto et al.29 reported a similar distribution based on fruit traits or geographic origin inside the C. annuum cluster.
Population genetic structure
For further elucidation of the genetic structure DAPC was pursued. K values and lineal components to be retained were pre-determined using the find.clusters function. K = 7 was determined to be the most likely as indicated by BIC value (Supplementary Data—Table 4). The first 100 PCs and the first two DA functions were retained for the analysis, representing more than 90% of total variability. Notwithstanding, both K = 6 and K = 8 showed similar BIC values and cluster formation meaning that could also represent a good fit to our collection (Supplementary Data—Table 4).
DAPC results were similar to those obtained by PCA, although with much more detail. Our population seems to be separated into seven clusters (Fig. 3). Cluster 1 comprised a set of 39 accessions, most of them Spanish landraces and three other European accessions (France, Italy, and Serbia) classified as C. annuum and sharing similar fruits traits: sweet, red, blocky peppers with variable fruit size and flesh thickness. Cluster 4 includes a diverse group of accessions composed of 44 European accessions, mostly C. annuum Spanish landraces and others with European origins (Bulgaria, France, and Italy) representing sweet, big sized, blocky peppers. Both Clusters 1 and 4 showed admixture and a narrow range of diversity between them. This was also observed by Nicolai et al.58, Lee et al.57, and Taranto et al.29 and the explanation probably resides on the fact that most Spanish varieties probably descend from a restricted number of individuals brought since Columbus journeys to the Americas and then spread across several countries which through selection and adaptation to local conditions originated a new range of forms3,4,59. Another reason why it is so difficult to differentiate European accessions may be the introduction of the same commercial lines in many different areas and this might be changing the genetic structure by cross pollinate with local varieties22. Cluster 2 was formed by 22 accessions, all of them C. annuum and encompassing a great diversity of places of origin. Most accessions in this cluster were collected in North American territories (Mexico and USA) with several fruit shapes. Europe was also represented with seven accessions from Bulgaria, France, and Spain, and finally one accession from India and another from Turkey. Bulgarian and French accessions were the only ones non-pungent; however, its fruit shape suggested that they could be improved lines developed from Mexican materials. Besides geographic origin, pungency could be a defining trait for population structure12,29. A group of 14 pungent accessions formed Cluster 3, including 13 C. annuum and one C. annuum var. glabriusculum. Most of them are Mexican varieties with cayenne and jalapeno shaped fruits and is completed by three Spanish accessions, one from Sri Lanka and one from USA, all pungent and with the same fruit shape. C. annuum var. glabriusculum is thought to be an ancestor to the Mexican C. annuum2,60 so its presence in this cluster is plausible and could be affected by the gene flow between domesticated forms and this botanical variety. Another hypothesis is that it could be a misclassification since C. annuum has an important range of phenotypes that could lead to an error during classification.
Fig. 3. Population structure for both 148 considered accessions (top) and subset of 122 C. annuum accessions (bottom) given by DAPC with 4083 informative sites.

Colours represent different assigned clusters. The x-axis provides accession names and respective assigned cluster whereas the y-axis provides the probability of each accession belonging to the assigned cluster
As in the PCA, all nine accessions identified as C. baccatum clustered together into a clearly differentiated group (Cluster 5; Fig. 3). This set of accessions seems to have a particular genetic print that makes them different from the rest of considered species. C. baccatum was domesticated independently from the other species in South America and it is still cultivated in isolated mountain areas that difficult the gene flow with other populations22,61,62. In addition, its crossability with species outside of its cytogenetic complex is difficult63. Ibiza et al. 22 reported a separation between Bolivian and Peru/Ecuador C. baccatum accessions corroborating that geographic isolation is an important factor for genetic structure.
Cluster 6 was composed of only three C. annuum var. glabriusculum accessions (mex_q1078, mex_s1120 and usa_a1003) that seemed to have a similar genetic print and were separated from the rest of accessions classified as the same species (Fig. 3). Nicolai et al.58 and Taranto et al.29 were not able to allocate this species into any group. The former reports three distinct genetic pools for this botanical group.
Finally, C. chinense and C. frutescens, as well as three C. annuum var. glabriusculum accessions (mex_c1333, mex_n1411, and mex_o1430), appeared to be indistinguishable accessions and were assigned to Cluster 7 as also seen in the PCA plot. The group includes 17 accessions from several regions of South and North America. Fruits are typically pungent, small sized, and with several fruit shapes. Many authors agree that C. chinense and C. frutescens should be considered as a single species63,64 and our results may support this hypothesis. However, C. frutescens was represented by only two accessions so solid conclusions could not be drawn. It is believed that a common ancestor through domestication in different locations gave place to C. annuum, C. chinense, and C. frutescens and the close relation seen in this work and others indicates that C. annuum var. glabriusculum could be that link2,58.
DAPC was also performed for C. annuum and the four C. annuum var. glabriusculum closest accessions. K = 5 was determined as the optimal number of clusters and the first 100 PCs and the first two DA functions were retained for the analysis (Fig. 3). Other Ks presented a possible good fit as both K = 4 and K = 6 showed slightly higher BIC values (Supplementary Data—Table 4). Results seem to be in agreement with the ones observed in the DAPC with all the collection and the formed clusters were homologous between analyses (Fig. 3). DAPC clusters 1–5 for the 122 accessions corresponded to the DAPC clusters 6, 2, 3, 4, 1 for 148 accessions, respectively, the only difference being that admixture samples from the first plot are now two clearly distinct groups. The clusters 1–5 of C. annuum are now composed of 3, 22, 14, 44, and 39 accessions, respectively (Supplementary Data—Table 4).
Population phylogenetic relationship
A phylogenetic tree for all the 148 accessions and another one only for the 122 C. annuum accessions were constructed for a better visualisation of sample distribution and relationships (Figs. 4 and 5).
Fig. 4. Clustering tree created from 1000 bootstrap replicates for the 148 accessions considering 4083 filtered SNPs.

Dark green indicates C. annuum accessions, whereas C. annuum var. glabriusculum, C. baccatum, C. chinense, and C. frutescens accessions are represented in orange, purple, pink, and light green, respectively. Node values correspond to bootstrap values
Fig. 5. Clustering tree created from 1000 bootstrap replicates for the 118 C. annuum and closest four C. annuum var. glabriusculum accessions.

Green indicates Asian accessions, orange the European, and purple the North American accessions. Node values correspond to bootstrap values
C. annuum clustered separately from the rest of species, forming a large cluster. This is in agreement with previous results22,57,58. Genetic distance increased from European to North American accessions. As said before, Spanish varieties arose from the ones brought from Mexico since Columbus journeys and were bred into a range of different forms3,4,59. This explains the genetic closeness of many Spanish accessions to Mexican materials. From Spain it spread across Europe and therefore the difficulty to cut apart European lines29,58.
C. annuum var. glabriusculum accessions appeared scattered in the dendrogram. Four out of seven accessions appeared along the annuum–chinense–frutescens complex, of which mex_v1196 and mex_q1078 accessions were especially close, whereas mex_s1120 and usa_a1003 located further to the C. annuum clade. The remaining three accessions (mex_c1333, mex_n1411, and mex_o1430) located more closely to C. chinense and C. frutescens accessions (Fig. 4), hence indicating a possible link between those species and a possible common ancestry2,58.
C. chinense and C. frutescens grouped together into a clearly separated cluster (Fig. 4). In our case, the sample size was not large enough to make strong assumptions, given that C. frutescens was represented by only two accessions, but our findings seem to reinforce that C. chinense and C. frutescens should be considered the same species63,64.
Finally, the C. baccatum cluster located between the C. annuum main cluster and the C. chinense–C. frutescens group, although considerably closer to the second (Fig. 4). At first sight, these findings disagree with the works from González-Pérez et al.12 and Nicolai et al.58, who reported C. frutescens as the closest species of C. annuum followed by C. chinense and finally C. baccatum, or alternatively, Lee et al. (2016)57, who reported a slightly closer relation between C. annuum and C. chinense. Both groups of species, i.e. C. baccatum and C. chinense–C. frutescens, were domesticated independently but in relatively close regions, i.e. C. baccatum ancestors migrated to the south of Bolivia while the ancestral genetic flow of C. chinense and C. frutescens migrated towards the Amazonian basin where these species were domesticated2,62. By contrast, C. annuum arose farther away, in Mexico65. Thus, this geographical compartmentalisation might explain why our C. chinense and C. frutescens are closer to C. baccatum than C. annuum2,62.
Considering the tree containing only C. annuum, two groups can be identified: one mainly grouping Mexican and USA accessions, including the four chiltepíns (mex_s1120, usa_a1003, mex_q1078, and mex_v1196) and another including mostly C. annuum materials from Spain (Fig. 5).
In the first cluster, we found that ‘Chile Japones’ (usa_jap), several ‘serrano’ forms (mex_scm, mex_serra and mex_serrano), ‘jalapenos’ (usa_cand, mu_espin, mu_jal and sp_jal), and ‘Chile de Arbol’ (mex_arb) grouped the most closely to (wild) chiltepíns, as well as some pungent, thin-flesh cayennes like the Spanish ‘Guindilla Pascual’ (sp_pascual), ‘Picante Largo’ (sp_pic), the breeding line ‘RSW’, and the Indian ‘Torpedo of Bangalore’ (ind_torp) (Fig. 5). Our results seem to agree that ‘serrano’ peppers are close to the ancestral forms within the cultivated C. annuum. In fact they share some wild traits such as pubescence and soft flesh deciduous fruits66 and ‘Jalapeno’ peppers were mainly bred from serrano gene pools, thus the closeness.
In the second cluster, the closest materials to the North American accessions were the Turkish pungent ‘Aci Sivri’ (tur_aci) and sweet numex-like Bulgarian accessions (bul_siv and bul_kap), followed by another small cluster which grouped Basque ‘Guindillas’ (cayennes; sp_11531 and ‘sp_ibarra’), ‘Guernika’ (sp_guer), ‘Padron’ (sp_11205 and sp_pad), and ‘Peperone di Senise’ (ita_senise). These results show a close relationship among Turkish and Balkan materials, suggesting common genetic pool or historic exchange of materials between both countries. The phylogenetic proximity of some cayenne peppers like ‘Aci Sivri’ or ‘Guindilla de Ibarra’ to North American peppers like ‘Chile de Arbol’ was supported by their similarity in the pattern of volatiles67. In addition, these findings are in agreement with the history of Padron peppers, which were brought to Galicia (Spain) by the Franciscans from Mexico in the XVIIth century. This flow of materials from Mexico might have included also the ancestors of many ecotypes from other Northern Spanish regions like ‘Guernika’, with a similar fruit appearance to Padron peppers14.
Triangular-shaped Spanish materials from northern Spain (sp_10183, sp_10185, sp_10186, and sp_11092) and the Mexican ‘Mulato’ (mex_mulato) clustered closely to North American accessions, suggesting that Ancho/Poblano peppers from Mexico might be the ancestors of Piquillo peppers as suggested by Rodriguez-Burruezo et al.67 (Fig. 5).
The rest of C. annuum materials, mainly bell peppers, grouped in several subclusters. ‘Cuneo’ (ita_cuneo and ita_giallo) and ‘Carmagnola’ peppers (ita_carr and ita_carg), from the Italian Piedmont and characterised by large and slightly flattened ‘Morron’ (blocky) peppers, grouped with the French ‘Petit Marsellais’ (fra_petit) (Fig. 5). At first sight this is very surprising because ‘Petit Marsellais’ is a yellow-orange thin-fleshed small-fruited heirloom from the French Provence. However, its fruits look like small blocky peppers and, therefore, a few mutations relative to the fruit size and flesh thickness68 and/or the geographical proximity between the Provence and Piedmont might have enabled some genetic exchange.
Close to the subcluster of Italian-Piedmont peppers we also found other two groups: a small subcluster which includes ‘Pimiento de Mojo’ (sp_mojo), ‘Piquillo’ PGI (sp_piq), and ‘Bola’ PDO (sp_bola), and another larger group of Spanish ‘Morron’ peppers which encompasses materials from accession sp_05057 to accession sp_10582 (Fig. 5). Accessions in the first subcluster shared thin flesh and high dry matter content, useful for their culinary uses: the mojo picon (hot sauce) in Canary Islands, roasted and canned to be stuffed and ground to obtain pepper powder, respectively4,14. The second subcluster can be divided further into two groups of ‘Morron’ peppers. One group (from accessions sp_5057 to sp_11038) of large ‘Morron’ peppers (most fruits ≥150 g) from several Spanish regions; and a second group which comprises most ‘Valenciano’ accessions from Valencia and Murcia regions (sp_4331, sp_5030, sp_05103, sp_5121, sp_5126, sp_vlc, and sp_10582) as well as ‘Largo de Reus’ peppers from Catalonia (sp_reus, sp_01844, and sp_10600) (Fig. 5). Thus, despite a few accessions using these names can be found in other clusters (e.g. accessions sp_01862, sp_05113), these findings suggest a common genetic pool linked to such varieties along the Mediterranean coast of Spain (from Murcia to Catalonia), which can offer the opportunity of finding genetic fingerprints associated to the ‘Valenciano’ or ‘Largo de Reus’ denominations in the next future.
Finally, other four interesting subclusters are worth mentioning: (i) one ranging from accessions sp_cala to sp_10451, (ii) from accessions sp_11630 to sp_04329, (iii) from sp_11751 to sp_01319, (iv) from sp_bier (PGI Pimiento Asado del Bierzo) to sp_10599 (Fig. 5). The first encompasses most accessions from a particular type of ‘Morron’ peppers, commonly called ‘Morron de Conserva’ or ‘Morron de Bola’, with characteristic round/heart-shaped fruits (‘bola’ in Spanish) (Supplementary Data: Table 1). These varieties have been selected for being roasted and canned (‘conserva’ in Spanish) and, despite the lack of nose-shape of the true Morron peppers, they keep the term morron because of their thick flesh14. Calahorra peppers from La Rioja are considered the ancestors of this kind of Spanish peppers, which can be found throughout the country (Fig. 5). The second includes a miscellany of ‘Morron’ peppers from very different origins like breeding lines from seed companies and our research institute from Valencia, i.e. ‘California Wonder’ and ‘Lamuyo’ (modern ‘Morron’), and some ‘Valenciano’, ‘Largo de Reus’ and ‘Trompa de Vaca’ peppers (Fig. 5). With the only exception of sp_11603, all of them are from the Mediterranean coast of Spain, which suggests that at least two different lineages of ‘Morron’ peppers, the one with most ‘Valenciano’ and ‘Largo de Reus’ peppers and this one, are present in this region. The third and the fourth subclusters include ‘Morron’ peppers from the North of the country, but the former from Pyrenees to Cantabric Sea regions, while the latter from Leon and Zamora including two current PGI and ancient related ecotypes: PGI ‘Fresno de la Vega y Benavente’ (sp_fresno) and PGI ‘Pimiento Asado del Bierzo’ (sp_bier) (Supplementary Data: Table 1). Both PGI belong to different Pochard´s types, which suggests that genetic exchanges might have occurred among both varietal types in this region.
The three methods considered were able to detect, although with different levels of detail, the complex relations among the collection, and seemed to be coherent with each other. C. annuum, C. baccatum, and C. chinense separation was observed in all, as well as the incorporation of C. frutescens accessions into C. chinense cluster, and C. annuum var. glabriusculum distribution into two distinct genetic pools. Both PCA (Fig. 2) and DAPC (Fig. 3) offered an idea of the genetic structure behind the collection, however, phylogenetic tree (Figs. 4 and 5) gave a greater level of details on the relations among species and even among closely related accessions.
Genetic diversity among clusters
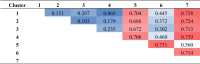
The average weighed Fst value between the seven clusters was 0.486. The highest Fst value was observed between clusters 4 and 7 (0.739) and the lowest between 1 and 4 (0.069) (Table 1). Low values indicate a larger genetic difference among accessions intra-population than between populations, suggesting genetic flow between populations. High values suggest low genetic flow between populations and many genetic differences48,69.
Table 1.
Weighed pairwise Fst values for the seven previously determined clusters. The increasing number of genetic differences between clusters are indicated with bold (low), bold italic (medium), and italic (high)
|
Spanish and other European accessions (Clusters 1 and 4) presented a lower level of genetic differentiation, suggesting a genetic flow between populations (Table 1). Similar results were reported by Nicolai et al.58, Lee et al. 57, and Taranto et al.29. This is probably due to the founder effect by a narrow diversity brought from the New World3,4,59.
Clusters 4 and 7 had the highest Fst value among all cluster combinations (Table 1). The first is mainly composed of European C. annuum accessions with low genetic variability whereas the second is an interspecific cluster composed of C. chinense, C. frutescens and C. annuum var. glabriusculum. Both clusters are geographically separated and the accessions have no genetic flow so the high Fst value is consistent with that scenario.
Cluster 5 also presented high Fst values regarding all combinations, which reveals that the population is isolated and has none or low genetic flow with other clusters, as expected for its geographic origin (Supplementary Data: Table 1). C. baccatum cultivation usually takes place in isolated areas that difficult crosspollinations. Furthermore, crosses outside its botanical complex are extremely difficult22,63,64.
Our data is consistent with previous published data. Taitano et al.30 reported a mean Fst of 0.821, ranging from 0.199 to 0.952, between 5 C. annuum landraces and C. frutescens. Nimmakayala et al.31 presented a fixation index of 0.780 between cultivated C. annuum and C. baccatum and 0.660 between wild accessions of those same species. Nimmakayala et al.32 reported for an only exclusively C. annuum with distinct levels of pungency and fruit weight an Fst between 0.020 and 0.150.
Scans for selective sweeps
Tajima’s D statistic was used to assess possible genomic sweeps associated with selection for each cluster formed. Genomic regions with low or negative Tajima’s D values indicate an unusually high number of high-frequency variants due to a balanced selection. On the other hand, high positive values are due to an excess of rare variants which can be result of a positive selection49. Cluster 1 displayed the highest weighed mean Tajima’s D value (0.854), while cluster 5 presented the lowest value (0.356) (Supplementary Data: Fig. 2).
Clusters 1–4, composed mostly by cultivated C. annuum accessions presented several regions with positive Tajima’s D values indicating a positive selection, possibly related to domestication and/or the pressure to achieve a specific phenotype resulting in an accumulation of trait related mutations31,32,58. High Tajima’s D values for a region spanning 7.5 Mb in the final part of the chromosome 1 for clusters 1 to 4 was found. QTLs implied in fruit weight, length and diameter, and pedicel length were described for this region so this could be an indication of selection for such traits70–72. Chromosome 5 showed a possibly purified region in the last positions for clusters 1 and 4, spanning 6 Mb and possibly linked to fruit diameter72,73. Finally, Clusters 2–4 showed high Tajima’s D values for a 1 Mb region at the end of chromosome 6. This region is linked to the control of several fruit traits such as weight, diameter, pericarp thickness, length, and shape72–75. Bear in mind that the resolution is insufficient to make strong assumptions however it sheds light into future association mapping studies.
Contrarily, clusters comprising only C. annuum wild ancestor C. annuum var. glabriusculum and closely related C. chinense and C. frutescens species presented a Tajima’s D distribution closer to the neutral or balanced selection (Supplementary Data: Fig. 2). This was expected due to the biological status of these accessions (Supplementary Data: Table 1). They are not as exploited as the first four clusters and are often cultivated in open-pollination conditions60,76. Therefore rare alleles are maintained at low frequencies.
C. baccatum cluster (Cluster 5) presented the lowest values, indicating a patron of none or little positive selection as Fst had predicted (Supplementary Data: Fig. 2). Our data is in agreement with previous results by other authors22,77. As mentioned before, C. baccatum is usually cultivated in isolated areas of South America which makes genetic exchanges rare and it has not been subjected to intensive breeding programmes as C. annuum. Other works used this statistical tool to successfully detect genomic selective sweeps in Capsicum spp. Taitano et al.30 reported a purified region on chromosome 6 possibly due to positive selection that confers the phenotype of the Chile de Agua ecotype. Nimmakayala et al.31 detected a positive selection for chromosome 4 of C. baccatum and low values for the other 11 chromosomes suggesting a neutral selection. Same authors, for C. annuum, observed positive values across the entire genome, except for chromosome 831.
Conclusions
Our study confirms the utility of genomic tools, such as GBS in the identification of highly informative SNPs and its application in the study of the genetic relations between Capsicum germplasm accessions. Availability of these tools is of great relevance to Capsicum breeders. Here we explore the genetic diversity, genetic structure, and genetic relationships of a collection of Spanish landraces and several foreign controls using a set of 4083 genome-wide SNPs. Population structure seems to be defined mainly by geographic origin and fruit traits. Sweet bell-shaped, blocky or thick-fleshed Spanish landraces and other European varieties located separately from pungent small American accessions. C. annuum var. glabriusculum seems to have two genetic pools, one closer to C. annuum and another closer to C. chinense and C. frutescens. C. annuum accessions have a higher level of positive selection and purified genomic regions according to Tajima’s D statistics and lower diversity among them. This study sheds light on the origin of Spanish landraces origins and their genetic structure and genetic relations, and provides important information for future association studies and for breeding programmes that contribute to the enhancement and protection of these materials.
Supplementary information
Acknowledgements
This work has been financed by INIA projects RTA2013-00022-C02, RTA2014-00041-C02-02, and RF2010-00025-00-00, FEDER funds. Authors are also grateful to the different Research Institutions, scientists, and breeders, and PDOs and GPIs Regulatory Boards, included on Supplementary Data: Table 1 for providing part of the materials studied here.
Authors' contributions
Conception and Design of study: A.R.-B., S.V., and A.F. Sequencing and SNP Calling analysis: L.P.-D. and S.V. Population diversity and structure analysis: L.P.-D. Analysis and validation of results: A.R.-B., S.V., A.F., and J.P. Wrote the manuscript: L.P.-D. All authors read and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41438-019-0132-8).
References
- 1.FAO. FAOSTAT Statistics Database. http://www.fao.org/faostat/ (2018). Accessed 20 Aug 2018.
- 2.Moscone EA, et al. The evolution of chili peppers (Capsicum—Solanaceae): a cytogenetic perspective. Acta Hortic. 2007;745:137–169. doi: 10.17660/ActaHortic.2007.745.5. [DOI] [Google Scholar]
- 3.DeWitt, D. & Bosland, P. W. Peppers of the World: An Identification Guide (Ten Speed Press, Berkeley, California, US, 1996).
- 4.Nuez, F., Ortega, R. G. & García, J. C. El Cultivo de Pimientos, Chiles y Ajies (Mundi-Prensa, Madrid, Spain, 2003).
- 5.Kraft, K. H. The Domestication of the Chile Pepper, Capsicum annuum: Genetic, Ecological, and Anthropogenic Patterns of Genetic Diversity. ProQuest Dissertations and Theses (2009).
- 6.Onus AN, Pickersgill B. Unilateral incompatibility in Capsicum (Solanaceae): occurrence and taxonomic distribution. Ann. Bot. 2004;94:289–295. doi: 10.1093/aob/mch139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong N, Bosland PW. Capsicum tovarii, a new member of the Capsicum baccatum complex. Euphytica. 1999;109:71–77. doi: 10.1023/A:1003421217077. [DOI] [Google Scholar]
- 8.Ince AG, Karaca M, Onus AN. Genetic relationships within and between Capsicum species. Biochem. Genet. 2010;48:83–95. doi: 10.1007/s10528-009-9297-4. [DOI] [PubMed] [Google Scholar]
- 9.Zijlstra S, Purimahua C, Lindhout P. Pollen tube growth in interspecific crosses between Capsicum species. Euphytica. 1991;26:585–586. [Google Scholar]
- 10.Yoon J, Cheol Yang D, Wahng Do J, Guen Park H. Overcoming two post-fertilization genetic barriers in interspecific hybridization between Capsicum annuum and C. baccatum for introgression of anthracnose resistance. Breed. Sci. 2006;56:31–38. doi: 10.1270/jsbbs.56.31. [DOI] [Google Scholar]
- 11.Manzur JP, Fita A, Prohens J, Rodríguez-Burruezo A. Successful wide hybridization and introgression breeding in a diverse set of common peppers (Capsicum annuum) using different cultivated ají (Capsicum baccatum) accessions as donor parents. PLoS ONE. 2015;10:e0144142. doi: 10.1371/journal.pone.0144142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.González-Pérez S, et al. New insights into Capsicum spp. relatedness and the diversification process of Capsicum annuum in Spain. PLoS ONE. 2014;9:e116276. doi: 10.1371/journal.pone.0116276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crosby, K. M. in VegetablesII (eds. Prohens, J., Nuez, F. & Carena, M. J.) 221–248 (Springer US, 2008).
- 14.Rodríguez-Burruezo, A., Pereira-Dias, L. & Fita, A. in Variedades Locales de Pimiento en España y Su Mejora Genética (eds. Galarreta de, J. I. R., Prohens, J. & Tierno, R.) 405–426 (Gráficas Irudi, Vitoria-Gasteiz, Spain, 2016).
- 15.Hammer K, Arrowsmith N, Gladis T. Agrobiodiversity with emphasis on plant genetic resources. Naturwissenschaften. 2003;90:241–250. doi: 10.1007/s00114-003-0433-4. [DOI] [PubMed] [Google Scholar]
- 16.Brugarolas M, Martinez-Carrasco L, Martinez-Poveda A, Ruiz JJ. A competitive strategy for vegetable products: traditional varieties of tomato in the local market. Span. J. Agric. Res. 2009;7:294–304. doi: 10.5424/sjar/2009072-420. [DOI] [Google Scholar]
- 17.Ashrafi H, et al. De novo assembly of the pepper transcriptome (Capsicum annuum): a benchmark for in silico discovery of SNPs, SSRs and candidate genes. BMC Genom. 2012;13:571. doi: 10.1186/1471-2164-13-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin C, et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl Acad. Sci. USA. 2014;111:5135–5140. doi: 10.1073/pnas.1400975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park M, et al. Evolution of the large genome in Capsicum annuum occurred through accumulation of single-type long terminal repeat retrotransposons and their derivatives. Plant J. 2012;69:1018–1029. doi: 10.1111/j.1365-313X.2011.04851.x. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014;46:270–278. doi: 10.1038/ng.2877. [DOI] [PubMed] [Google Scholar]
- 21.Prohens J, et al. Introgressiomics: a new approach for using crop wild relatives in breeding for adaptation to climate change. Euphytica. 2017;213:158. doi: 10.1007/s10681-017-1938-9. [DOI] [Google Scholar]
- 22.Ibiza VP, Blanca J, Cañizares J, Nuez F. Taxonomy and genetic diversity of domesticated Capsicum species in the Andean region. Genet. Resour. Crop Evol. 2012;59:1077–1088. doi: 10.1007/s10722-011-9744-z. [DOI] [Google Scholar]
- 23.He J, et al. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014;5:484. doi: 10.3389/fpls.2014.00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glaubitz JC, et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One. 2014;9:e90346. doi: 10.1371/journal.pone.0090346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poland J, Rife T. Genotyping-by-Sequencing for plant breeding and genetics. Plant Genome. 2012;5:92–102. doi: 10.3835/plantgenome2012.05.0005. [DOI] [Google Scholar]
- 26.Gardner KM, et al. Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3. 2014;4:1681–1687. doi: 10.1534/g3.114.011023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung YS, Choi SC, Jun TH, Kim C. Genotyping-by-sequencing: a promising tool for plant genetics research and breeding. Hortic. Environ. Biotechnol. 2017;58:425–431. doi: 10.1007/s13580-017-0297-8. [DOI] [Google Scholar]
- 28.Elshire RJ, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taranto F, D’Agostino N, Greco B, Cardi T, Tripodi P. Genome-wide SNP discovery and population structure analysis in pepper (Capsicum annuum) using genotyping by sequencing. BMC Genom. 2016;17:943. doi: 10.1186/s12864-016-3297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taitano, N. et al. Genomewide genotyping of a novel Mexican Chile Pepper collection illuminates the history of landrace differentiation after Capsicum annuum L. domestication. Evol. Appl. 10.1111/eva.12651 (2018). [DOI] [PMC free article] [PubMed]
- 31.Nimmakayala P, et al. Genome-wide divergence and linkage disequilibrium analyses for Capsicum baccatum revealed by genome-anchored single nucleotide polymorphisms. Front. Plant Sci. 2016;7:1646. doi: 10.3389/fpls.2016.01646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nimmakayala P, et al. Genome-wide diversity and association mapping for capsaicinoids and fruit weight in Capsicum annuum L. Sci. Rep. 2016;6:38081. doi: 10.1038/srep38081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doyle, J. J. & Doyle, J. L. Isolation of plant DNA from fresh tissue. Focus12, 13–15 (1990).
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradbury PJ, et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 37.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.R Development Core Team. R: A Language and Environment for Statistical Computing (2009).
- 40.Zheng X, et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickham, H ggplot2: Elegant Graphics for Data Analysis (Springer, US, 2016).
- 42.Jombart T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 43.Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jombart T, Ahmed I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27:3070–3071. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamvar ZN, Tabima JF, Grünwald NJ. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ. 2014;2:e281. doi: 10.7717/peerj.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saitou N, Nei M. The Neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 47.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 48.Weir B, Clark Cockerham C. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 49.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahn YK, et al. Whole genome resequencing of Capsicum baccatum and Capsicum annuum to discover single nucleotide polymorphism related to powdery mildew resistance. Sci. Rep. 2018;8:5188. doi: 10.1038/s41598-018-23279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonah H, et al. An improved Genotyping by Sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE. 2013;8:e54603. doi: 10.1371/journal.pone.0054603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hulse-Kemp AM, et al. Reference quality assembly of the 3.5-Gb genome of Capsicum annuum from a single linked-read library. Hortic. Res. 2018;5:4. doi: 10.1038/s41438-017-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wakeley J. The excess of transitions among nucleotide substitutions: new methods of estimating transition bias underscore its significance. Tree. 1996;11:158–163. doi: 10.1016/0169-5347(96)10009-4. [DOI] [PubMed] [Google Scholar]
- 54.Eshbaugh WH. Genetic and biochemical systematic studies of chili peppers (Capsicum - Solanaceae) Bull. Torre. Bot. Club. 1975;102:396. doi: 10.2307/2484766. [DOI] [Google Scholar]
- 55.Raw A. Foraging behavior of wild bees at hot pepper flowers (Capsicum annuum) and its possible influence on cross pollination. Ann. Bot. 2000;85:487–492. doi: 10.1006/anbo.1999.1090. [DOI] [Google Scholar]
- 56.Cheng J, et al. Development of a SNP array and its application to genetic mapping and diversity assessment in pepper (Capsicum spp.) Sci. Rep. 2016;6:33293. doi: 10.1038/srep33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee HY, et al. Genetic diversity and population structure analysis to construct a core collection from a large Capsicum germplasm. BMC Genet. 2016;17:142. doi: 10.1186/s12863-016-0452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicolaï M, Cantet M, Lefebvre V, Sage-Palloix AM, Palloix A. Genotyping a large collection of pepper (Capsicum spp.) with SSR loci brings new evidence for the wild origin of cultivated C. annuum and the structuring of genetic diversity by human selection of cultivar types. Genet. Resour. Crop Evol. 2013;60:2375–2390. doi: 10.1007/s10722-013-0006-0. [DOI] [Google Scholar]
- 59.Andrews, J. Peppers: The Domesticated Capsicums (University of Texas Press, Austin, Texas, US, 1995).
- 60.Hayano-Kanashiro C, Gámez-Meza N, Medina-Juárez LAacute. Wild pepper Capsicum annuum L. var. glabriusculum: taxonomy, plant morphology, distribution, genetic diversity, genome sequencing, and phytochemical compounds. Crop Sci. 2016;56:1–11. doi: 10.2135/cropsci2014.11.0789. [DOI] [Google Scholar]
- 61.McLeod MJ, Guttman SI, Eshbaugh WH. Early evolution of chili peppers (Capsicum) Econ. Bot. 1982;36:361–368. doi: 10.1007/BF02862689. [DOI] [Google Scholar]
- 62.Pickersgill B. Domestication of plants in the Americas: insights from Mendelian and molecular genetics. Ann. Bot. 2007;100:925–940. doi: 10.1093/aob/mcm193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walsh BM, Hoot SB. Phylogenetic relationships of Capsicum (Solanaceae) using DNA sequences from two noncoding regions: the chloroplast atpB‐rbcL spacer region and nuclear waxy introns. Int. J. Plant Sci. 2001;162:1409–1418. doi: 10.1086/323273. [DOI] [Google Scholar]
- 64.McLeod MJ, Eshbaugh WH, Guttman SI. An electrophoretic study of Capsicum (Solanaceae): the purple flowered taxa. Bull. Torre. Bot. Club. 1979;106:326. doi: 10.2307/2560359. [DOI] [Google Scholar]
- 65.Kraft KH, et al. Multiple lines of evidence for the origin of domesticated chili pepper, Capsicum annuum, in Mexico. Proc. Natl Acad. Sci. USA. 2014;111:6165–6170. doi: 10.1073/pnas.1308933111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bosland, P. W. & Votava, E. J. Peppers: Vegetable and Spice Capsicums (CABI, New York, US, 2012).
- 67.Rodríguez-Burruezo A, Kollmannsberger H, González-Mas MC, Nitz S, Nuez F. HS-SPME comparative analysis of genotypic diversity in the volatile fraction and aroma-contributing compounds of Capsicum fruits from the annuum–chinense–frutescens complex. J. Agric. Food Chem. 2010;58:4388–4400. doi: 10.1021/jf903931t. [DOI] [PubMed] [Google Scholar]
- 68.Wang L, Li J, Zhao J, He C. Evolutionary developmental genetics of fruit morphological variation within the Solanaceae. Front. Plant Sci. 2015;6:248. doi: 10.3389/fpls.2015.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting Fst. Nat. Rev. Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barchi L, Lefebvre V, Sage-Palloix AM, Lanteri S, Palloix A. QTL analysis of plant development and fruit traits in pepper and performance of selective phenotyping. Theor. Appl. Genet. 2009;118:1157–1171. doi: 10.1007/s00122-009-0970-0. [DOI] [PubMed] [Google Scholar]
- 71.Han K, et al. An ultra-high-density bin map facilitates high-throughput QTL mapping of horticultural traits in pepper (Capsicum annuum) DNA Res. 2016;23:81–91. doi: 10.1093/dnares/dsv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hill, T. A. et al. Regions underlying population structure and the genomics of organ size determination in Capsicum annuum. Plant Genome10, 10.3835/plantgenome2017.03.0026 (2017). [DOI] [PubMed]
- 73.Yarnes SC, et al. Identification of QTLs for capsaicinoids, fruit quality, and plant architecture-related traits in an interspecific Capsicum RIL population. Genome. 2013;56:61–74. doi: 10.1139/gen-2012-0083. [DOI] [PubMed] [Google Scholar]
- 74.Rao GU, Ben Chaim A, Borovsky Y, Paran I. Mapping of yield-related QTLs in pepper in an interspecific cross of Capsicum annuum and C. frutescens. Theor. Appl. Genet. 2003;106:1457–1466. doi: 10.1007/s00122-003-1204-5. [DOI] [PubMed] [Google Scholar]
- 75.Chaim A, Borovsky Y, De Jong W, Paran I. Linkage of the A locus for the presence of anthocyanin and fs10.1, a major fruit-shape QTL in pepper. Theor. Appl. Genet. 2003;106:889–894. doi: 10.1007/s00122-002-1132-9. [DOI] [PubMed] [Google Scholar]
- 76.Moses M, Umaharan P. Genetic structure and phylogenetic relationships of Capsicum chinense. J. Am. Soc. Hortic. Sci. 2012;137:250–262. doi: 10.21273/JASHS.137.4.250. [DOI] [Google Scholar]
- 77.Albrecht E, Zhang D, Saftner RA, Stommel JR. Genetic diversity and population structure of Capsicum baccatum genetic resources. Genet. Resour. Crop Evol. 2012;59:517–538. doi: 10.1007/s10722-011-9700-y. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.