

Abstract
The biosynthetic route to the napyradiomycin family of bacterial meroterpenoids has been fully described 32 years following their original isolation and 11 years after their gene cluster discovery. The antimicrobial and cytotoxic natural products napyradiomycins A1 and B1 are produced using three organic substrates (1,3,6,8-tetrahydroxynaphthalene, dimethylallyl pyrophosphate, and geranyl pyrophosphate), and catalysis via five enzymes: two aromatic prenyltransferases (NapT8 and T9); and three vanadium dependent haloperoxidase (VHPO) homologues (NapH1, H3, and H4). Building upon the previous characterization of NapH1, H3, and T8, we herein describe the initial (NapT9, H1) and final (NapH4) steps required for napyradiomycin construction. This remarkably streamlined biosynthesis highlights the utility of VHPO enzymology in complex natural product generation, as NapH4 efficiently performs a unique chloronium-induced terpenoid cyclization to establish two stereocenters and a new carbon−carbon bond, and dual-acting NapH1 catalyzes chlorination and etherification reactions at two distinct stages of the pathway. Moreover, we employed recombinant napyradiomycin biosynthetic enzymes to chemoenzymatically synthesize milligram quantities in one pot in 1 day. This method represents a viable enantioselective approach to produce complex halogenated metabolites, like napyradiomycin B1, that have yet to be chemically synthesized.
The napyradiomycins are a diverse set of meroterpenoid natural products originally isolated from the actinomycete Streptomyces ruber (formerly Chainia rubra) in 1986,1,2 and today number in excess of 50 members.3 Initially isolated for their broad-spectrum Gram-positive antibacterial activities, additional studies of these hybrid polyketide-terpenoid compounds have identified promising anticancer and antiangiogenic activities. Select few napyradiomycins have been synthesized in a racemic manner,4,5 and only one enantioselective synthetic route to napyradiomycin A1 has been achieved to date (Figure 1).6 No chemical syntheses of cyclohexane-containing napyradiomycins have however been reported. The main challenge relating to the construction of the napyradiomycins, and similar molecules, is the limited methodology for stereospecific installation of their halogenated stereocenters. Recent pioneering advances have dramatically accelerated the field of asymmetric halogenation.7 As recently reported, over 2000 natural products that possess a chiral chlorinated or brominated stereocenter have been isolated, while only 12 have been synthesized via a catalytic, enantioselective halogenation strategy.8 Many of these chiral halogenated metabolites have biological activities with significant therapeutic and chemical interest,9 necessitating development of viable strategies to perform this challenging chemical transformation.
Figure 1.
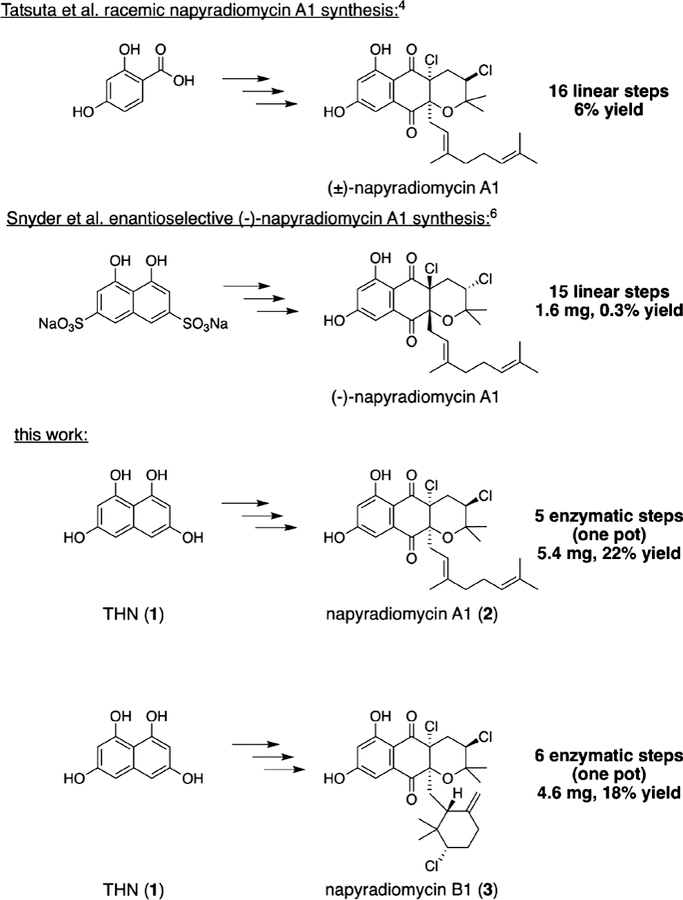
Total chemical syntheses of napyradiomycin A1 and comparison with chemoenzymatic approaches described herein.
Nature’s methodology for installing halogenated stereo-centers has been developed through a few unique approaches.10 Among the families of natural halogenating enzymes, the vanadium-dependent haloperoxidases (VHPOs) are remarkable biological catalysts that perform a two-electron oxidation of aqueous halide ions in the presence of vanadate (VO43−) and H2O2, generating the traceless byproduct water. Moreover, these enzymes have garnered significant interest as potential biocatalysts due to their robust thermostability and oxidative activity without the need for costly cofactors or redox regenerating systems.11 VHPOs can be divided into two major categories: the more extensively studied macroalgal and fungal VHPOs that produce diffusible hypohalous acid capable of reacting with electron-rich substrates;12 and Streptomyces VHPOs that perform regio-and enantioselective halogenations, suggestive of enzymatic capture of halenium ions.13
In 2007, the 43 kb napyradiomycin biosynthetic gene cluster (nap) was reported from the marine isolate Streptomyces sp. CNQ-525.14 Three VHPO homologues (napH1, H3, H4) were annotated within this cluster, suggestive of the first dedicated involvement of this family of enzymes in secondary metabolism.14,15 Moreover, the napH genes were localized in close proximity to those capable of producing napyradiomycin biosynthetic precursors identified from stable isotope incorporation experiments, including mevalonate-derived isoprene pyrophosphates and the polyketide 1,3,6,8-tetrahydroxynaph-thalene (THN, 1).16 Subsequent in vitro validation of NapH1 demonstrated its ability to facilitate a stereospecific chloronium-induced cyclization of the prenyl moiety to form a 7-methylated napyradiomycin A1 derivative.17 Over the past decade, VHPO homologues were discovered and shown to catalyze many key bond-constructing reactions within Streptomyces sp. THN-derived meroterpenoid biosyntheses, including the merochlorins,18–20 naphterpin, and marinone.21 These diverse chemical transformations include chlorination-induced dearomatization, cyclization, and isomerization reactions that proceed in regio-and stereospecific manners. We recently reported two unique transformations within the napyradiomycin biosynthetic pathway: the Mg2+-dependent NapT8 prenylation of monochlorinated 5 with dimethylallyl pyrophosphate (DMAPP); and the subsequent α-hydroxyke-tone rearrangement of NapT8 product 6 to naphthomevalin (7) via VHPO homologue NapH3.5 This result rationalized the vicinal diprenylation motif unique to the napyradiomycins that formerly defied biosynthetic logic. While this study further expanded the repertoire of biological activities from VHPO enzymes, the transformations for the generation of 5 from 1 and the enzymes required for the formation of the cyclized geranyl moiety in napyradiomycin B1 (3) remained unknown. Our studies aimed to understand the total biosynthetic logic of the entire pathway, while identifying these new enzymatic reactions. Herein we report the characterization of VHPO and prenyltransferase enzymology related to the biosynthesis of 3, and subsequent application of this streamlined five-enzyme pathway to enantioselectively synthesize milligram quantities of these complex napyradiomycin meroterpenoids.
Many napyradiomycins contain additional structural complexity via presumed halogenation-induced cyclization of the geranyl moiety. Because of the functional assignments of two napH homologues, we suspected that NapH4 catalyzes this reaction. Previous attempts to obtain soluble protein expression in Escherichia coli were unsuccessful;17 however, reassignment of the napH4 open reading frame, and subsequent recloning and heterologous production in Streptomyces lividans TK23 resulted in soluble NapH4 (Figures S1, S2). Application of the model monochlorodimedone assay to detect hypohalous acid formation indicated that recombinant NapH4 was properly folded and active, albeit only showing activity in the presence of bromide ions; an unusual characteristic of these stereospecific Streptomyces sp. VCPO enzymes (Figure S3).13
We initially surmised that NapH4 would produce the chlorinated cyclohexanol moiety observed in napyradiomycin B4, a 7′-demethylated variant of the major metabolite isolated from heterologous expression of the nap cluster (Figure S4).14 Instead, when incubated with 2 in the presence of Na3VO4, KCl, and H2O2, NapH4 catalyzed the formation of a single product with a dehydrated mass compared to napyradiomycin B4. Following scale up, isolation, and NMR characterization, the product was identified as 3, possessing an exomethylene-containing chlorinated cyclohexane ring (Figure 2), which matched the original isolation spectra.2 Although alteration of the reaction pH can have a significant impact on the biochemistry of Streptomyces VHPOs, as best exemplified with Mcl24 from the merochlorin biosynthetic pathway,5,13,18,19 we only observed a decrease in the relative formation of 3 at a lower pH (Figure S4).
Figure 2.
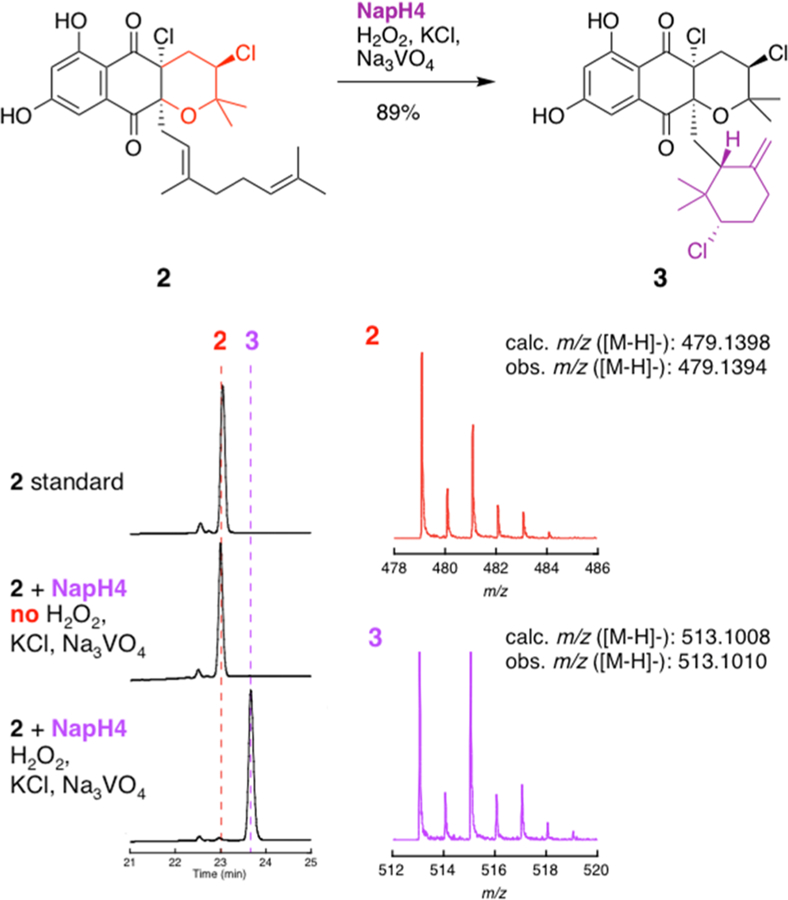
NapH4-catalyzed chloronium-mediated cyclization of the geranyl moiety of 2 to form 3.
An analogous bromonium-based cyclization reaction was previously reported with marine red algal VBPO enzymes when incubated with terpenoid substrates.22 Despite suffering from poor regioselectivities, broad substrate specificities, and the generation of multiple products in low yields, these enzymes showcased the first example of enantioselective halogenation and concomitant carbon−carbon bond formation by a VHPO. This biological reactivity has been paralleled chemically in the halonium-induced polyene cyclizations that have been employed asymmetrically via stereoselective iodonium/bromonium formations.23 Only two racemic polyene cyclizations of chloronium ions have been reported;24 however, these reactions generally occur in much lower yield and diastereoselectivity than bromo-and iodo-cyclizations.25 In contrast, NapH4 catalyzes a remarkably high yielding, diastereoselective, and stereospecific chloronium-induced terpenoid cyclization.
With the completion of the pathway from 5 to 7, we next turned our attention to the start point from 1 to 5. As prenyltransferase (PTase) NapT8 had previously been assigned function in the prenylation of 5,5 we suspected the second predicted ABBA aromatic PTase NapT9 as the enzyme responsible for installing the geranyl moiety. This function would be analogous to the prenylation of 1 by Mcl23 and NphB (originally Orf2) in merochlorin18 and naphterpin26 biosyntheses, respectively. Following heterologous expression and purification of NapT9, we observed the Mg2+-dependent conversion of 1 and geranyl pyrophosphate (GPP) to 4-geranyl 1,3,6,8-tetrahydroxynaphthalene (4-geranyl THN, 4) (Figures 3, S5). The reversed-phase HPLC and extracted ion chromatograms of this reaction exhibited a unique elution profile, likely due to keto−enol tautomers formed under the weakly acidic conditions.27 This unique chromatogram mirrored that of synthetic 4,21 but due to significant oxygen sensitivity, isolation was not attempted.
Figure 3.
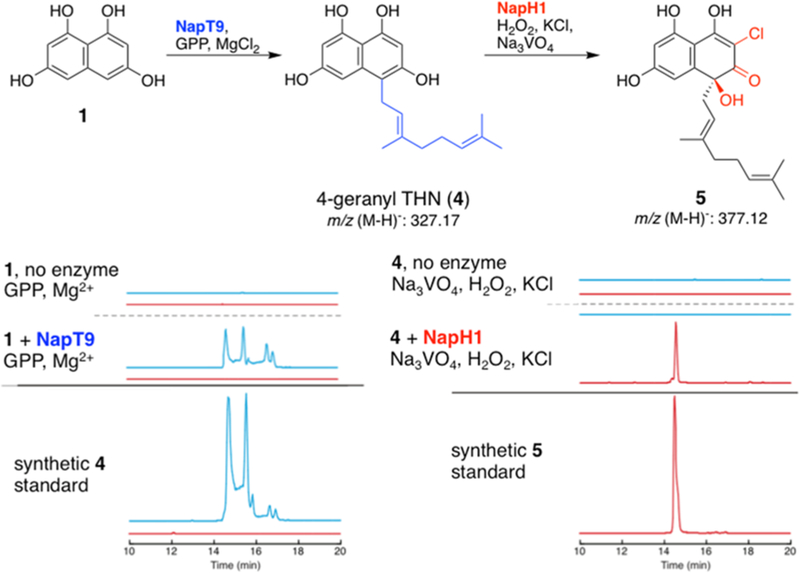
Negative mode extracted ion chromatograms showing conversion of 1 to 4 (blue trace, 327.1 m/z) and 5 (red trace, 377.1 m/z) by NapT9 and NapH1 activities, respectively.
We suspected the conversion of 4 to 5 to be catalyzed by a VCPO based on related transformations in other 1-derived meroterpenoid pathways.5,18,19,21 Incubation of in situ NapT9-generated 4 with nap VCPO enzymes in the presence of Na3VO4, H2O2, and KCl showed that NapH1 catalyzed the production of 5 as the major chlorinated product. This was further confirmed by preparative scale up and NMR characterization (Figures 3, S6). The NapH1 activity was particularly surprising given its previous role in alkene halofunctionalization to cyclize 7 to 2.17 NapH4 was also able to form 5 albeit in a substantially lower quantity compared to NapH1 (Figure S6). Analogous to marinone biosynthetic VCPO enzyme MarH1,21 NapH1 catalyzes the monochlorination of prenylated THN molecules even with excess H2O2. This diverges from the in vitro activities of other VCPO enzymes (MarH3,21 Mcl245) that facilitate dichlorination and subsequent α-hydroxyketone rearrangements with comparable substrates. We are presently exploring the biophysical understanding of these divergent activities.
Identification of the activities of NapT9 and NapH4, and the dual functionality of NapH1, allowed us to complete the napyradiomycin biosynthetic pathway (Scheme 1). Briefly, prenyltransferase NapT9 catalyzes the Mg2+-dependent ger-anylation of 1 at the nucleophilic 4-position. Prenylated product 4 is subsequently oxidatively dearomatized and monochlorinated via NapH1 in the presence of Na3VO4 and H2O2 to afford 5. NapT8 then catalyzes a Mg2+-dependent prenylation with DMAPP to form 6, which undergoes a NapH3-catalyzed α-hydroxyketone rearrangement, producing 7.5 Next, NapH1 performs an enantioselective chlorination-induced cyclization reaction to form 2,5,17 which is further transformed by a novel asymmetric, chlorination-induced terpenoid cyclization by NapH4 to 3.
Scheme 1.

Biosynthetic Pathway from 1,3,6,8-Tetrahydroxynaphthalene (1) to Napyradiomycin B1 (3)
Following complete elucidation of the biosynthetic pathway to 3, analysis of the individual enzymatic activities suggested that only two types of enzymes were present with orthogonal redox and metal cofactor requirements. We hypothesized that a controlled addition of all Nap biosynthetic enzymes to one pot could be employed to generate useful quantities of napyradiomycin family members in an enantioselective and protecting group-free strategy. In vitro interrogation identified maximal activity of NapH4 in HEPES-KOH, pH 8.0 (Figure S4), so this was selected as the one-pot reaction buffer for scale up and optimization. A 5 mM reaction scale (∼1 mg/mL of 1) was chosen to generate maximal quantities of napyradiomycins despite the increasing hydrophobicity of these complex halogenated meroterpenoid products.
An initial division of the NapT9 reaction with subsequent H2O2-dependent VHPO steps was required to maximize the amount of oxygen-sensitive 4 for downstream catalysis. Initial attempts to transform 1 in one pot with excess GPP, H2O2, and multiple enzyme additions were successful in producing 2 albeit as a very minor component of a complex reaction mixture (Figure S7). The high abundance of oxidized byproducts of 1 and 4 in the one pot reaction mixture implied that significant optimization of the NapT9 and NapH1 reactions would be necessary to improve yields to 5. Major contributors to yield improvement (45% isolated yield of 5) included: reducing GPP to 1.1 mol equiv; adding commercial E. coli inorganic pyrophosphatase to minimize PTase inhibition; and sequentially adding H2O2 to the reaction mixture (Figure S7).
Following the two-step optimization to 5, the addition of DMAPP (1.1 equiv), H2O2, and additional biosynthetic enzymes at once enabled production of intermediates along the napyradiomycin biosynthetic pathway that directly correlated to the Nap enzymes added (Figures 4, S8). Multiple 1 mL replicates were set up following identical reaction conditions and times, as larger volume reactions generated an increased number and intensity of byproducts. After the threestep addition of the first four Nap biosynthetic enzymes, inorganic metal cofactors and organic pyrophosphate and hydrogen peroxide cosubstrates to 1 (9.6 mg, 50 μmol, divided over 10 × 1 mL) in one pot, 2 (5.4 mg, 22%) was isolated after a 24 h reaction time; analogous reaction conditions with the inclusion of NapH4 produced 3 (4.6 mg, 18%), which has yet to be chemically synthesized in an enantioselective or racemic strategy. Moreover, the chemoenzymatic application of the napyradiomycin biosynthetic pathway established all five stereocenters of 3, three of which are chlorinated, using 1.1 mol equiv of organic pyrophosphates and 0.2−1 mol % of biological catalysts over a reaction time of 24 h.
Figure 4.
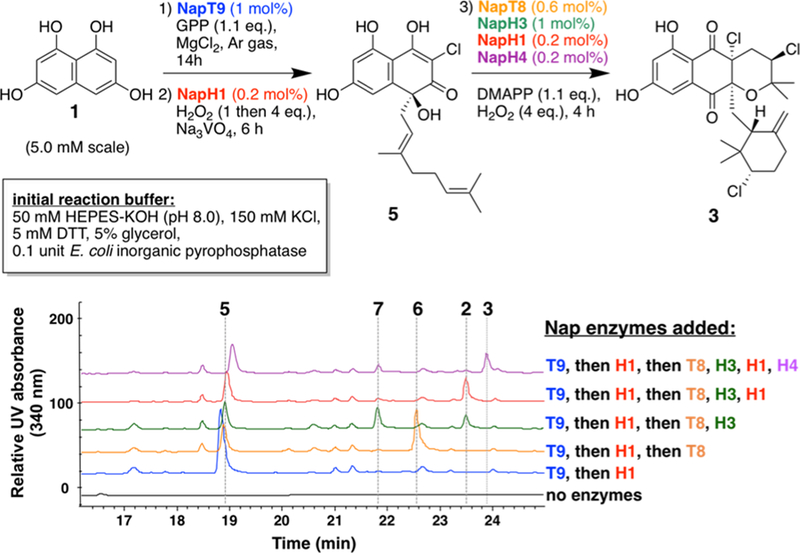
Chemoenzymatic strategy to the one pot, three-step formation of napyradiomycin biosynthetic intermediates from metabolic precursors.
Many impressive syntheses of complex natural products have been recently reported using enzymes from their biosynthetic pathways, highlighting the synthetic utility of this methodology.28 While challenges still exist in the application of chemoenzymatic synthesis, it represents a complementary approach to overcome some of the biological challenges in microbial secondary natural product production (long culturing times, low production yields, reliance on naturally occurring biosynthetic precursors), while offering exquisite catalysis of regio-and stereospecific chemical reactions that would be difficult to mirror in a round-bottomed flask. We are particularly encouraged by the application of NapH1 and NapH4 enzymology in the chiral installation of halogenated stereocenters and aim to further extend the utility and scalability of VHPO catalysis within the challenging field of catalytic asymmetric halogenation.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Drs. Brendan Duggan and Anthony Mrse for help with general NMR instrumentation maintenance and guidance and Dr. Yongxuan Su for assistance with mass spectrometry. We thank Dr. Stefan Diethelm for preliminary biochemical contributions and general advice and Prof. Michihiko Kobayashi for kindly providing pHSA81. This research was supported by the US National Institutes of Health (R01-AI047818), the Australian Research Council (DP160103393) and the Natural Sciences and Engineering Research Council of Canada (NSERC-PDF to S.M.K.M.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b10134.
Materials and methods, general procedures, compound characterization and NMR spectra (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Shiomi K; Iinuma H; Hamada M; Naganawa H; Manabe M; Matsuki C; Takeuchi T; Umezawa H Novel antibiotics napyradiomycins. Production, isolation, physico-chemical properties and biological activity. J. Antibiot 1986, 39, 487. [DOI] [PubMed] [Google Scholar]
- (2).Shiomi K; Nakamura H; Iinuma H; Naganawa H; Isshiki K; Takeuchi T; Umezawa H; Iitaka Y Structures of new antibiotics napyradiomycins. J. Antibiot 1986, 39, 494. [DOI] [PubMed] [Google Scholar]
- (3).(a) Shiomi K; Nakamura H; Iinuma H; Naganawa H; Takeuchi T; Umezawa H; Iitaka Y New antibiotic napyradiomycins A2 and B4 and stereochemistry of napyradiomycins. J. Antibiot 1987, 40, 1213. [DOI] [PubMed] [Google Scholar]; (b) Henkel T; Zeeck A Secondary metabolites by chemical screening. 15. Structure and absolute configuration of naphthomevalin, a new dihydro-naphthoquinone antibiotic from Streptomyces sp. J. Antibiot 1991, 44, 665. [DOI] [PubMed] [Google Scholar]; (c) Hori Y; Abe Y; Shigematsu N; Goto T; Okuhara M; Kohsaka M Napyradiomycins A and B1: Non-steroidal estrogen-receptor antagonists produced by a Streptomyces. J. Antibiot 1993, 46, 1890. [DOI] [PubMed] [Google Scholar]; (d) Soria-Mercado IE; Prieto-Davo A; Jensen PR; Fenical W Antibiotic terpenoid chloro-dihydroquinones from a new marine actinomycete. J. Nat. Prod 2005, 68, 904. [DOI] [PubMed] [Google Scholar]; (e) Motohashi K; Sue M; Furihata K; Ito S; Seto H Terpenoids produced by actinomycetes: Napyradiomycins from Streptomyces antimycoticus NT17. J. Nat. Prod 2008, 71, 595. [DOI] [PubMed] [Google Scholar]; (f) Haste NM; Farnaes L; Perera VR; Fenical W; Nizet V; Hensler ME Bactericidal kinetics of marine-derived napyradiomycins against contemporary methicillin-resistant Staphylococcus aureus. Mar. Drugs 2011, 9, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Yamamoto K; Tashiro E; Motohashi K; Seto H; Imoto M Napyradiomycin A1, an inhibitor of mitochrondrial complexes I and II. J. Antibiot 2012, 65, 211. [DOI] [PubMed] [Google Scholar]; (h) Cheng Y-B; Jensen PR; Fenical W Cytotoxic and antimicrobial napyradiomycins from two marine-derived Streptomyces strains. Eur. J. Org. Chem 2013, 2013, 3751. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wu Z; Li S; Li J; Chen Y; Saurav K; Zhang Q; Zhang H; Zhang W; Zhang W; Zhang S; Zhang C Antibacterial and cytotoxic new napyradiomycins from the marine-derived Streptomyces sp. SCSIO 10428. Mar. Drugs 2013, 11, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Farnaes L; Coufal NG; Kauffman CA; Rheingold AL; DiPasquale AG; Jensen PR; Fenical W Napyradiomycin derivatives, produced by a marine-derived actinomycete, illustrate cytotoxicity by induction of apoptosis. J. Nat. Prod 2014, 77, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Lacret R; Peŕez-Victoria I; Oves-Costales D; de la Cruz M; Domingo E; Martin J; Díaz C; Vicente F; Genilloud O; Reyes F MDN-0170, a new napyradiomycin from Streptomyces sp. strain CA-271078. Mar. Drugs 2016, 14, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Hwang JS; Kim GJ; Choi HG; Kim MC; Hahn D; Nam J-W; Nam S-J; Kwon HC; Chin J; Cho SJ; Hwang H; Choi H Identification of antiangiogenic potential and cellular mechanisms of napyradiomycin A1 isolated from the marine-derived Streptomyces sp. YP127. J. Nat. Prod 2017, 80, 2269. [DOI] [PubMed] [Google Scholar]
- (4).(a) Takemura S; Hirayama A; Tokunaga J; Kawamura F; Inagaki K; Hashimoto K; Nakata M A concise total synthesis of (±)-A80915G, a member of the napyradiomycin family of antibiotics. Tetrahedron Lett 1999, 40, 7501. [Google Scholar]; (b) Tatsuta K; Tanaka Y; Kojima M; Ikegami H The first total synthesis of (±)-napyradiomycin A1. Chem. Lett 2002, 31, 14. [Google Scholar]
- (5).Miles ZD; Diethelm S; Pepper HP; Huang DM; George JH; Moore BS A unifying paradigm for naphthoquinone-based meroterpenoid (bio)synthesis. Nat. Chem 2017, 9, 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Snyder SA; Tang Z-Y; Gupta R Enantioselective total synthesis of (−)-napyradiomycin A1 via asymmetric chlorination of an isolated olefin. J. Am. Chem. Soc 2009, 131, 5744. [DOI] [PubMed] [Google Scholar]
- (7).(a) Hu DX; Seidl FJ; Bucher C; Burns NZ Catalytic chemo-, regio-, and enantioselective bromochlorination of allylic alcohols. J. Am. Chem. Soc 2015, 137, 3795. [DOI] [PubMed] [Google Scholar]; (b) Bucher C; Deans RM; Burns NZ Highly selective synthesis of halomon, plocamenone, and isoplocamenone. J. Am. Chem. Soc 2015, 137, 12784. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Denmark SE; Kuester WE; Burk MT Catalytic, asymmetric halofunctionalization of alkenes–a critical perspective. Angew. Chem., Int. Ed 2012, 51, 10938. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chung WJ; Vanderwal CD Stereoselective halogenation in natural product synthesis. Angew. Chem., Int. Ed 2016, 55, 4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Landry ML; Burns NZ Catalytic enantioselective dihalogenation in total synthesis. Acc. Chem. Res 2018, 51, 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gribble GW Biological activity of recently discovered halogenated marine natural products. Mar. Drugs 2015, 13, 4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Agarwal V; Miles ZD; Winter JM; Eustaquio AS; El Gamal AA; Moore BS Enzymatic halogenation and dehalogenation reactions: Pervasive and mechanistically diverse. Chem. Rev 2017, 117, 5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Smith DRM; Grüschow S; Goss RJM Scope and potential of halogenases in biosynthetic applications. Curr. Opin. Chem. Biol 2013, 17, 276. [DOI] [PubMed] [Google Scholar]; (b) Latham J; Brandenburger E; Shepherd SA; Menon BRK; Micklefield J Development of halogenase enzymes for use in synthesis. Chem. Rev 2018, 118, 232. [DOI] [PubMed] [Google Scholar]; (c) Frank A; Seel CJ; Groll M; Gulder T Characterization of a cyanobacterial haloperoxidase and evaluation of its biocatalytic halogenation potential. ChemBioChem 2016, 17, 2028. [DOI] [PubMed] [Google Scholar]; (d) Fernań-dez-Fueyo E; van Wingerden M; Renirie R; Wever R; Ni Y; Holtmann D; Hollmann F Chemoenzymatic halogenation of phenols by using the haloperoxidase from Curvularia inaequalis. ChemCatChem 2015, 7, 4035. [Google Scholar]; (e) Seel CJ; Kraĺík A; Hacker M; Frank A; König B; Gulder T Atom-economic electron donors for photobiocatalytic halogenations. ChemCatChem 2018, 10, 3960. [Google Scholar]
- (12).Wever R; Krenn BE; Renirie R Marine vanadium-dependent haloperoxidases, their isolation, characterization, and application. Methods Enzymol 2018, 605, 141. [DOI] [PubMed] [Google Scholar]
- (13).McKinnie SMK; Miles ZD; Moore BS Characterization and biochemical assays of Streptomyces vanadium-dependent chloroperoxidases. Methods Enzymol 2018, 604, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Winter JM; Moffitt MC; Zazopoulos E; McAlpine JB; Dorrestein PC; Moore BS Molecular basis for chloroniummediated meroterpene cyclization: cloning, sequencing, and heterologous expression of the napyradiomycin biosynthetic gene cluster. J. Biol. Chem 2007, 282, 16362. [DOI] [PubMed] [Google Scholar]
- (15).Winter JM; Moore BS Exploring the chemistry and biology of vanadium-dependent haloperoxidases. J. Biol. Chem 2009, 284, 18577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shiomi K; Iinuma H; Naganawa H; Isshiki K; Takeuchi T; Umezawa H Biosynthesis of napyradiomycins. J. Antibiot 1987, 40, 1740. [DOI] [PubMed] [Google Scholar]
- (17).Bernhardt P; Okino T; Winter JM; Miyanaga A; Moore BS A stereoselective vanadium-dependent chloroperoxidase in bacterial antibiotic biosynthesis. J. Am. Chem. Soc 2011, 133, 4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Teufel R; Kaysser L; Villaume MT; Diethelm S; Carbullido MK; Baran PS; Moore BS One-pot enzymatic synthesis of merochlorin A and B. Angew. Chem., Int. Ed 2014, 53, 11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Diethelm S; Teufel R; Kaysser L; Moore BS A multitasking vanadium-dependent chloroperoxidase as an inspiration for the chemical synthesis of the merochlorins. Angew. Chem., Int. Ed 2014, 53, 11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kaysser L; Bernhardt P; Nam SJ; Loesgen S; Ruby JG; Skewes-Cox P; Jensen PR; Fenical W; Moore BS Merochlorins A-D, Cyclic meroterpenoid antibiotics biosynthesized in divergent pathways with vanadium-dependent chloroperoxidases. J. Am. Chem. Soc 2012, 134, 11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Murray LAM; McKinnie SMK; Pepper HP; Erni R; Miles ZD; Cruickshank MC; Lopez-Perez B; Moore BS; George JH Total synthesis establishes the biosynthetic pathway to the naphterpin and marinone natural products. Angew. Chem., Int. Ed 2018, 57, 11009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Carter-Franklin JN; Parrish JD; Tschirret-Guth RA; Little RD; Butler A Vanadium haloperoxidase-catalyzed bromination and cyclization of terpenes. J. Am. Chem. Soc 2003, 125, 3688. [DOI] [PubMed] [Google Scholar]; (b) Carter-Franklin JN; Butler A Vanadium bromoperoxidase-catalyzed biosynthesis of halogenated marine natural products. J. Am. Chem. Soc 2004, 126, 15060. [DOI] [PubMed] [Google Scholar]
- (23).Sakakura A; Ukai A; Ishihara K Enantioselective halocyclization of polyprenoids induced by nucleophilic phosphoramidites. Nature 2007, 445, 900. [DOI] [PubMed] [Google Scholar]
- (24).Snyder SA; Treitler DS; Brucks AP Simple reagents for direct halonium-induced polyene cyclizations. J. Am. Chem. Soc 2010, 132, 14303. [DOI] [PubMed] [Google Scholar]
- (25).Arnold AM; Pothig A; Drees M; Gulder T NXS, morpholine, and HFIP: The ideal combination for biomimetic haliranium-induced polyene cyclizations. J. Am. Chem. Soc 2018, 140, 4344. [DOI] [PubMed] [Google Scholar]
- (26).Kuzuyama T; Noel JP; Richard SB Structural basis for the promiscuous biosynthetic prenylation of aromatic natural products. Nature 2005, 435, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Viviani F; Gaudry M; Marquet A Melanin biosynthesis: A study of polyphenol deoxygenation. J. Chem. Soc., Perkin Trans. 1 1990, 1, 1255. [Google Scholar]
- (28).Recent examples of chemoenzymatic syntheses employing native biosynthetic enzymes to establish chemical complexity: (a) Baker Dockrey SA; Lukowski AL; Becker MR; Narayan ARH Biocatalytic site-and enantioselective oxidative dearomatization of phenols. Nat. Chem 2018, 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sib A; Gulder TAM Stereoselective total synthesis of bisorbicillinoid natural products by enzymatic oxidative dearomatization/dimerization. Angew. Chem., Int. Ed 2017, 56, 12888. [DOI] [PubMed] [Google Scholar]; (c) Tanifuji R; Koketsu K; Takakura M; Asano R; Minami A; Oikawa H; Oguri H Chemo-enzymatic total syntheses of jorunnamycin A, saframycin A, and N-Fmoc saframycin Y3. J. Am. Chem. Soc 2018, 140, 10705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.