

Abstract
Background
Hepatic stellate cell (HSC) activation induced by transforming growth factor β1 (TGF-β1) plays a pivotal role in fibrogenesis, while the complex downstream mediators of TGF-β1 in such process are largely unknown.
Methods
We performed pharmacoproteomic profiling of the mice liver tissues from control, carbon tetrachloride (CCl4)-induced fibrosis and NPLC0393 administrated groups. The target gene MAT2A was overexpressed or knocked down in vivo by tail vein injection of AAV vectors. We examined NF-κB transcriptional activity on MAT2A promoter via luciferase assay. Intracellular SAM contents were analyzed by LC-MS method.
Findings
We found that methionine adenosyltransferase 2A (MAT2A) is significantly upregulated in the CCl4-induced fibrosis mice, and application of NPLC0393, a known small molecule inhibitor of TGF-β1 signaling pathway, inhibits the upregulation of MAT2A. Mechanistically, TGF-β1 induces phosphorylation of p65, i.e., activation of NF-κB, thereby promoting mRNA transcription and protein expression of MAT2A and reduces S-adenosylmethionine (SAM) concentration in HSCs. Consistently, in vivo and in vitro knockdown of MAT2A alleviates CCl4- and TGF-β1-induced HSC activation, whereas in vivo overexpression of MAT2A facilitates hepatic fibrosis and abolishes therapeutic effect of NPLC0393.
Interpretation
This study identifies TGF-β1/p65/MAT2A pathway that is involved in the regulation of intracellular SAM concentration and liver fibrogenesis, suggesting that this pathway is a potential therapeutic target for hepatic fibrosis.
Fund
This work was supported by National Natural Science Foundation of China (No. 81500469, 81573873, 81774196 and 31800693), Zhejiang Provincial Natural Science Foundation of China (No. Y15H030004), the National Key Research and Development Program from the Ministry of Science and Technology of China (No. 2017YFC1700200) and the Key Program of National Natural Science Foundation of China (No. 8153000502).
Research in context.
Evidence before this study
Hepatic stellate cell (HSC) activation induced by pleiotropic TGF-β1 is a central event during the liver fibrogenesis. The important role of downstream mediator Smad2/3 in this aspect has been well-studied. However, therapies specifically targeting this pathway are not yet available for clinical use. Therefore, other potential mediators of TGF-β1 pathways that exert pro-fibrotic effect are needed to be uncovered.
Added value of this study
In this study, by performing pharmacoproteomic analysis, we identified MAT2A as a new effector for pro-fibrotic stimuli. In vitro experiment showed that MAT2A could mediate TGF-β1-induced HSC activation. We also found that in vivo overexpression of MAT2A promoted HSC activation and abolished anti-fibrotic effect of NPLC0393 via decreasing of SAM contents in liver tissue. Furthermore, we showed that the NF-κB subunit p65, functioning as a transcription factor of MAT2A, was induced by TGF-β1 and contributed to TGF-β1-induced HSC activation.
Implications of all the available evidence
This study found that TGF-β1/p65/MAT2A signaling pathway participates the regulation of intracellular SAM concentration and promotes liver fibrogenesis, suggesting that this pathway is a potential therapeutic target for hepatic fibrosis.
Alt-text: Unlabelled Box
Keywords
Liver fibrosis
TGF-β1
MAT2A
p65
SAM
1. Introduction
Hepatic fibrosis is a reversible wound-healing response that occurs in chronic liver injury caused by viral infection, drugs, metabolic and autoimmune disorders [1]. This disease is characterized by exaggerated and abnormal deposition of extracellular matrix (ECM) components such as type I collagen, which may further lead to cirrhosis accompanied with portal hypertension, liver failure, and/or liver cancer [2]. Transforming growth factor β1 (TGF-β1) mediated activation of hepatic stellate cell (HSC) is regarded as a crucial event, resulting in ECM assembly and remodeling to synergistically promote fibrogenesis [[3], [4], [5], [6]]. However, the intricate mechanisms underlying TGF-β1 mediated HSC activation in hepatic fibrogenesis are not fully clarified. Therefore, there is an urgent need to identify the key mediators in such process for the development of more effective therapy to treat hepatic fibrogenesis.
Despite the well-established role of TGF-β1/Smad signaling in fibrogenesis, therapies specifically targeting this pathway are not yet available for clinical use [7]. Recent trials trying to neutralize TGF-β1 itself or inhibit the secondary messengers of TGF-β1 all failed because of insufficient pharmacokinetics or off-target related toxicity [8]. Furthermore, there exists intricate ‘cross talk’ interactions between canonical TGF-β1 signaling pathway and other pathways to overcome auto-inhibitory feedback loops, leading to constitutive fibrogenesis in HSC. Therefore, other targetable key mediators downstream of TGF-β1 need to be further investigated. Previously, we reported that PP2Cα small molecule activator NPLC0393 exerts anti-fibrotic effect on carbon tetrachloride (CCl4) or bile duct ligation (BDL)-induced animal model of liver fibrosis by inhibiting TGF-β1/Smad3 signal transduction [9].
Methionine adenosyltransferase (MAT) is an essential enzyme that synthesize S-adenosylmethionine (SAM), the principle methyl donor and a precursor of the glutathione (GSH) [10]. The normal level of hepatic SAM is required for maintaining liver health and preventing liver injury. Three MAT-encoding genes, including MAT1A, MAT2A and MAT2B, are expressed in mammalian cells. MAT1A gene encodes for the α1 catalytic subunit (consisting of MAT I or MAT III), while MAT2A and MAT2B genes encode for the α2 catalytic subunit (consisting MATII) and the β regulatory subunit, respectively [11,12]. MAT1A is mainly expressed in adult quiescent hepatocytes, whereas MAT2A and MAT2B are expressed in extrahepatic tissues as well as non-parenchymal cells of the liver, such as HSCs and Kupffer cells [13,14]. Several studies have reported that MAT2A expression was increased in the liver of CCl4- or thioacetamide (TAA)-treated animal models or in the culture-activated HSCs [[15], [16], [17], [18], [19]]. However, whether MAT2A is involved in TGF-β1 activation of HSC remains unclear.
In this study, we analyzed the liver tissues from control, CCl4-induced fibrotic and NPLC0393-treated mice groups using quantitative proteomic approach, and identified MAT2A as a key downstream mediator of TGF-β1 signaling pathway. We further found that phosphorylated NF-κB p65 served as the major mediator that was induced by TGF-β1 and increased MAT2A transcription subsequently. The consequential upregulation of MAT2A reduced SAM concentration, facilitating the liver fibrotic progression. In summary, NPLC0393 could halt this TGF-β1/p65/MAT2A pathway to restore SAM concentration, thereby ameliorating liver fibrosis.
2. Materials and methods
2.1. Animal experiments
In the animal experiment for pharmacoproteomic analysis, 18 male C57BL/6 mice (6 weeks, weighting 18–20 g, Shanghai SLAC Laboratory Animal Co. Ltd. China) were randomly divided into 3 groups: Control group (Con, n = 6), in which the mice were administered intraperitoneally (i.p.) the olive oil daily; Carbon tetrachloride model group (CCl4, n = 6), in which the mice were administered intraperitoneally 0.5 mL/kg CCl4 (diluted 1:10 in olive oil) twice a week for 4 weeks; NPLC0393 treatment group (CCl4 + NPLC0393, n = 6), in which the mice were administered intraperitoneally CCl4 twice weekly and the NPLC0393 (dissolved in DMSO) at a daily dose of 2.5 mg/kg for 4 weeks. For validation experiments, such as Western blot and SAM examination, NPLC0393 alone treatment group (NPLC0393, n = 6) was included, in which mice were only administered NPLC0393 in the same way. NPLC0393 was provided by Prof. Lihong Hu's laboratory, Shanghai Institute of Materia Medica, Chinese Academy of Sciences [20]. All mice were kept under standard conditions, and were sacrificed for the histochemical staining and proteomic experiments. Liver tissues were dissected into small pieces, some were fixed in 10% buffered formalin, and some were snap frozen in liquid nitrogen and stored at −80 °C. The animal experiment was approved by Science and Technology Commission of Shanghai Municipality, and all experimental procedures were performed according to the ethical guidelines of Animal Care and Use Committee, Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
For in vivo gene transfer, mice were received a single tail vein injection of 1011 genome copies of AAV vectors. After 3 weeks, NPLC0393 was peripherally administered in combination with CCl4 for another 4 weeks. Infection efficiency in the liver was verified by immunohistochemistry of either Flag or primary antibody over prefixed liver slides and by Western blot. AAV2/9-shRNA-Negative Control (NC shRNA), AAV2/9-shRNA-MAT2A (5′-UACCUUGAUGAGGAUACAAUU-3′, MAT2A shRNA) and AAV2/9-empty vector (AAV-EV), AAV2/9-promoter of glial fibrillary acidic protein (pGFAP)-MAT2A (AAV-MAT2A) were purchased from Hanbio (Shanghai, China).
2.2. Protein extraction and sample preparation
Mouse liver tissues were washed with ice-cold PBS and lysed in SDT lysis buffer (4% SDS (m/v), 100 mM DTT, 100 mM Tris, pH = 7.6). The lysates were homogenized with sonication, denatured and reduced at 95 °C for 5 min and then centrifuged at 13,000 x g for 30 min. The supernatants were collected, and protein concentration was determined by tryptophan fluorescence emission assay as described previously [21]. Peptides were prepared following the Filter Assisted Sample Preparation (FASP) procedure [22].
2.3. Mass spectrometry analysis
All experiments were performed on an Orbitrap Q-Exactive (Thermo Fisher Scientific) platform connected to an online nanoflow EASY-nLC1000 HPLC system (Thermo Fisher Scientific). Peptides were loaded on a self-packed column (75 μm × 150 mm, 3 μm ReproSil-Pur C18 beads, Dr. Maisch GmbH, Ammerbuch, Germany) and separated with a 240 min gradient for each sample at a flow rate of 300 nL/min. The mobile phase A of RP-HPLC was 0.1% formic acid in water, and the mobile phase B was 0.1% formic acid in acetonitrile. Data-dependent acquisition was performed using Xcalibur software in positive ion mode. The MS1 full scan was set at a resolution of 70,000 @ m/z 200 by orbitrap mass analyzer (350–1700 m/z) with an AGC target of 3e6 and maximum injection time of 20 ms. Then ‘top 15’ MS2 scans were generated by HCD fragmentation at a resolution of 17,500 @ m/z 200. Other instrument parameters were set as follows: 27% normalized collision energy (NCE), 1e5 AGC target, 100 ms maximum injection time, 2.0 m/z isolation window.
MS raw data were analyzed with MaxQuant (1.6.0.1) by searching against the UniProt Mouse database (downloaded in November 2017). The false discovery rate (FDR) was set to 0.01 for both peptide and protein identification. Carbamidomethyl cysteine was searched as a fixed modification, oxidized methionine and protein N-term acetylation as variable modifications. Enzyme specificity was set to trypsin/P. The LFQ intensity values were used for the following data analysis. The mass spectrometry proteomics data has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier (Project accession No.: PXD011134).
2.4. Bioinformatic analysis of proteomic data
The STRING database was used to predict protein-protein interactions (PPI) and related biological processes. The medium confidence score (0.4) was chosen for searching more potential interactions. The PPI network was reconstructed in Cytoscape software (3.6.1).
2.5. Histological analysis and immunofluorescence staining
Liver tissues were immediately formalin fixed, paraffin embedded, and 10 μm thick slices were stained with Masson's Trichrome for a histological assessment. Immunohistochemstry and Immunofluorescence staining with α-SMA(at a 1:100 dilution, Abcam, ab21027) and anti-MAT2A (at a 1:50 dilution, NOVUS, NBP1–92100) were also performed to the species. Immunofluorescence staining was examined using the confocal laser scanning microscopy (Leica Microsystems Heidelberg GmbH, Germany). Stained sections were examined using an Olympus FV1000 microscope (Olympus Corporation, Tokyo, Japan). Image-Pro® plus 4.5 image analysis software (Media Cybernetics, USA) was used to quantify the degree of fibrosis. All of these experiments were completed by KCI BioTech (Suzhou, China).
2.6. Western blot analysis
Liver tissues and cell samples were washed 3 times with 5 mL PBS and collected with SDT lysis buffer, followed by centrifugation for 20 min at 4 °C. Protein lysates were separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes, which were then blocked with 5% fat-free milk at room temperature for 1 h and incubated with anti-α-SMA (Abcam, ab124964), anti-MAT2A antibody (Abcam, ab77471), anti-MAT2B antibody (Abcam, ab109484), anti-MAT1A (Abcam, ab174687), anti-COL1A1 antibody (Novus, NBP1-30054), anti-PPM1A/PP2Cα (Abcam, ab14824), anti-RELA/p65 (CST, 8242), anti-phospho-RELA/ p-p65 (Ser536, CST, 3033) anti-TGF-β1 (Abcam, ab92486) at 4 °C overnight. After three washes in TBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Super Signal West Pico Chemiluminescent Substrate (ThermoScientific, 34,094) was used to visualize the antigens. GAPDH (Sigma-Aldrich, G8795) was used as an internal control. The protein bands were scanned and quantified by densitometric analysis using image J software. The experiment was repeated at least three times.
2.7. Quantitative real-time PCR
Total RNA was extracted from the cells or tissues using Trizol reagent (Invitrogen, USA) according to the manufacturer's protocol. One microgram of RNA was reverse transcribed to cDNA using the PrimeScript RT reagent Kit (Takara) and processed in triplicated reactions for qPCR using primers listed below and the Thunderbird SYBR Green qPCR Mix reagent (Toyobo) in an ABI 7500 Real-Time PCR System (Applied Biosystems, USA). Primer sequences used are shown in Table S4.
2.8. Measurement of SAM concentration by LC-MS
SAM was extracted from 30 mg–100 mg liver tissues or in 6 × 105 LX-2 cells as described previously [23]. Briefly, cultured cells and liver tissues were washed thrice with PBS. Then 4 mL and 500 μL of cold extract solvent (acid acetonitrile/methanol/water 40:40:20, containing 0.1 M formic acid) was added to liver tissues and cells, respectively, and was stand on ice for 15 min. The cell-solvent mixture was collected and transferred to Eppendorf tubes, followed by centrifugation at 12,000 rpm for 10 min at 4 °C, and 250 μL supernatant were dried in a vacuum centrifuge and redissolved in 250 μL H2O for high performance liquid chromatography (HPLC) analysis. All of the SAM measurements were performed on a TSQ Quantiva mass spectrometer (Thermo Fischer Scientific, USA). Thermo Xcalibur 3.0.63 was used for system control, data acquisition, and data processing, and the mass spectrometry proteomics data for SAM concentration has been deposited to the ProteomeXchange (Project accession No.: PXD011135, See (“Supplementary materials and methods” for more details).
2.9. Cell culture and treatments
The human HSC cell line LX-2 (provided by institute of Liver Diseases, Shanghai University of Traditional Chinese Medicine) and mouse HSC cell line JS-1 (provided by Prof. Jinsheng Guo, Division of Digestive Diseases, Zhongshan Hospital, Fudan University) were routinely cultured in DMEM medium containing 10% FBS. Primary mouse hepatocytes were cultured in 1640 Medium containing 10% FBS. Cells were treated with different concentration (1, 5 and 10 μM) of NPLC0393 for 24 h and stimulated with 5 ng/mL TGF-β1 (R&D Systems, USA) for another 24 h. PBS was used as vehicle.
For the study of SAM effect, LX-2 cells were incubated with or without 2 mM SAM (Abbott Laboratories, US) for 24 h and then stimulated with TGF-β1 for another 24 h. For the study of SAM role in NPLC0393 anti-fibrotic effect, LX-2 cells were pre-treated with NPLC0393 for 12 h and then stimulated with TGF-β1 in the complete DMEM or L-methionine-free DMEM for another 24 h.
2.10. RNA interference and overexpression experiments
The siRNA targeting human MAT2A (MAT2A siRNA-1: 5′- GUGAGAGAGAGCUAUUAGA-3′, MAT2A siRNA-2: 5′-ACACAUUGGAUAUGAUGAU-3′, MAT2A siRNA-3: 5′- AGCAGUUGUGCCUGCGAAA-3′), human PPM1A/PP2Cα (5′- GAGUUAUGUCAGAGAAGAA-3′) were synthesized by Genepharma Co. (Shanghai, China). LX-2 cells were cultured in six-well plates (105 cells/well) and transfected using Lipofectamine 3000 (Invitrogen, CA) with MAT2A, PP2Cα siRNA or negative control siRNA (NC siRNA) for 48 h following the manufacturer's instructions. On the basis of results knockdown efficiency, siRNA#3 was selected and used in the main experiment.
The adenovirus vector encoding RELA (Ad-p65) and the empty vector (Ad-EV) were purchased from Vigene Biosciences (Jinan, China). LX-2 cells were cultured in 12-well plates and added with 1 μg of target plasmid per well. After 12 h, the transfection medium was changed to normal medium. Effects of overexpression on mRNA and protein levels were examined 36 h later.
2.11. Transient transfections and luciferase activity assays
For testing the NF-κB transcriptional activity, the 1 μg pGL6-NF-κB-Luc plasmid (Beyotime Institute Biotechnology, China) and 1 μg pRL-TK plasmid (Promega) as internal control were transiently co-transfected into LX-2 cells (1 × 105 cells/well) using Lipofectamine 3000 (Invitrogen). After transfection for 12 h, cells were pre-treated with 10 μM NPLC0393, 10 μM SB431542, or 50 ng/mL NF-κB inhibitor SN50 for 12 h and were stimulated with 5 ng/mL TGF-β1 or 25 ng/mL TNF-α (R&D Systems, USA) for another 24 h.
For the measurement of MAT2A promoter activity, the Human MAT2A promoter-luciferase reporter plasmid containing binding sites for NF-κB (Sangon, China) and pRL-TK were transfected into LX-2 cells. After 12 h, cells were pre-treated with 10 μM NPLC0393 or 50 ng/mL SN50, a specific NF-κB translocation inhibitor, for 12 h and were stimulated with 5 ng/mL TGF-β1 or 25 ng/mL TNF-α for another 24 h. Dual luciferase assays were carried out according to the manufacturer's protocol (Promega) and light intensity was measured in a Synergy HT luminometer (BioTek, US). Each experiment was done in triplicate samples and results were normalized against those of cotransfected pRL-TK.
2.12. Data analysis
One way analysis of variance (ANOVA) and Tukey's honestly significant difference (HSD) test were performed with language R. Fold change >1.5 and P value <.01 were set as criterions for a significant change. The raw ratios for CCl4 model and NPLC0393 treatment of all differentially expressed proteins were normalized to obtain a standard deviation of 1 and a mean of 0 for each protein. The transformed profiles were then clustered by fuzzy C method using M fuzz package in R software [24]. For our analysis, the optimal parameters number of clusters “c” and fuzzification parameter “m” were 4 and 2, respectively, and the distance metric was Euclidean distance. Statistical analysis was performed using GraphPad Prism version 5.0 (GraphPad Software, CA). Data were presented as mean ± SEM. For two population comparisons, an unpaired t-test was performed and for multiple comparison test, one-way analysis of variance (ANOVA) followed by Tukey post hoc test was performed. Two-sided P values <.05 was considered statistically significant.
3. Results
3.1. Identify differentially expressed proteins in response to fibrogenesis and NPLC0393 treatment
NPLC0393, known as a specific small molecule activator of PP2Cα, has shown an inhibitory effect on the animal models of liver fibrosis by regulating TGF-β1/Smad3 signaling pathway (Fig. S1a) [9]. In this study, we used this compound as a chemical probe to investigate the downstream mediators in non-canonical TGF-β1 signaling pathway involved in fibrogenesis. By performing histological and biochemical examinations, we first confirmed that NPLC0393 administration was able to exert a significant anti-fibrotic effect on CCl4-induced fibrotic model mice without any toxicity to normal liver (n = 6 per group, Fig. S1b, c).
Next, to profile the proteins and biological pathways in NPLC0393-based anti-fibrotic effect, label-free quantitative proteomic analysis was performed using liver tissue samples from the control, CCl4 model and NPLC0393-treated mice (n = 6 per group, Fig. 1a). A total of 2953 non-redundant proteins were quantified by the LC-MS/MS-based proteomic experiments (Table S1) and the distributions of label-free quantification (LFQ) intensity values in each sample indicated that there were no analytical biases toward any samples (Fig. S2a). Meantime, the Pearson correlation coefficients between any two samples in each group were >0.89, suggesting that good biological reproducibility was achieved in each group. (Fig. S2b). Proteins with both fold change >1.5 and ANOVA P value <.01 between two groups were considered as the differentially expressed proteins. As a result, 507 proteins (i.e., 17.2% of the total 2953 proteins) were significantly changed between CCl4 model and control groups, including 297 proteins upregulated and 210 proteins downregulated (Fig. 1b, Table S2). Hierarchical clustering analysis (HCA) of differentially expressed proteins showed the distinct protein expression patterns across all the control, CCl4 and NPLC0393 treatment groups and samples within each group could be clustered together (Fig. S2c).
Fig. 1.

pharmacoproteomic analysis of the mice livers from control, fibrosis model and NPLC0393 treatment groups. (a) Workflow chart of label-free quantitative proteomic experiment. Total protein was extracted from different mice liver tissues (n = 6 for Control, CCl4 and NPLC0393 groups respectively) and digested with trypsin. Equal amount of resolved peptides were analyzed by LC-MS/MS. Bioinformatic analysis of differentially expressed proteins was carried out using STRING database. (b) Volcano plot illustrating proteins with different abundance in CCl4 and Control groups. The Log2 ratios of protein intensities of CCl4 to Control group were plotted against the negative Log10 P values. Red points represent upregulated proteins in CCl4 group, blue points represent downregulated proteins in CCl4 group (fold change >1.5, P value <.01) and gray points represent unchanged proteins. (c) Clustering of the differentially expressed proteins. The changed proteins were assigned to four clusters by fuzzy c-means (FCM) clustering algorithm. The y axis is log10 transformed and normalized, and the number of proteins in each cluster is indicated in parentheses. Each trace was colored according to its membership value in the corresponding cluster (referred to membership colour bar). (d) Protein-protein interaction analysis of proteins in Cluster 2 and Cluster 3 using STRING database. Interactions between two proteins were indicated with gray edges. Colour of the node indicates clusters (Orange: Cluster 2; Blue: Cluster 3; unconnected proteins were not shown). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Additionally, based on the profiles of protein abundance across the three groups, all of the differentially expressed proteins were assigned to four clusters by the fuzzy c-means (FCM) clustering analysis (Fig. 1c, Table S3). Since we considered that proteins with a reversed expression pattern upon NPLC0393 treatment might be involved in the anti-fibrotic effect of NPLC0393, thus in the present study, we mainly focused on proteins in Cluster 2 and Cluster 3. As shown in Fig. 1c, the expression levels of 210 proteins in Cluster 2 (144 proteins) and Cluster 3 (66 proteins) were significantly changed in CCl4 model and returned to basal levels after NPLC0393 treatment. The PPI analysis showed a complex network with several biological processes that contained highly connected proteins (Fig. 1d). For instance, proteins in Cluster 2 were mainly involved in wound healing (e.g. Anxa2, F2, F13a1), apoptosis (e.g. Bak1, Bax, Stat1) and cell-cell adhesion process (e.g. Itgb1, Cdh2, Cd44), which might be closely linked to liver fibrosis. Proteins in Cluster 3 were involved in different metabolic processes, such as sulfur compound metabolic pathway (Fig. S2d). Notably, the PPI network showed a significant enrichment of proteins belonging to SAM metabolic process. Biosynthesis of SAM requires the enzyme methionine adenosyltransferase (MAT). Previous studies reported that MAT2A was implicated in liver fibrosis [9,16,17]. Our proteomic data also showed that MAT2A was upregulated by CCl4 induction and reversed by NPLC0393 treatment. Therefore, we decided to focus on dissecting the potential link between MAT2A and non-canonical TGF-β1 signaling pathway.
3.2. Upregulation of MAT2A in the model of liver fibrosis
Our present proteomic study revealed that MAT2A (ratio of CCl4: Con =1.72, ANOVA P value: 8.61e-5) was one of the differentially expressed proteins among the control, CCl4 model and NPLC0393 treatment groups. We first verified the in vivo effect of NPLC0393 on the MAT2A expression by immunoblotting analysis. Consistent with proteomic data, the expression level of MAT2A protein detected by immunoblotting was significantly elevated in CCl4 model group compared to control group and was returned to the basal level by NPLC0393 treatment (n = 6 per group, Fig. 2a). Similar expression pattern was also observed in rat BDL liver fibrosis model (Fig. S3a). In addition, the expression level of TGF-β1, α-SMA and COL1A1 proteins were changed in a manner similar to that of MAT2A. Expression of MAT2A protein was also visualized by immunofluorescence. As shown in Fig. 2b, the staining of MAT2A and α-SMA were co-localized in the activated HSCs (aHSCs) within fibrotic septa, and α-SMA as well as MAT2A-positive cells were markedly decreased in NPLC0393 treatment group. Consistent with the protein quantification, we also found that the expression levels of mat2a, col1a1, a-sma mRNA were increased in fibrotic liver and were reduced by NPLC0393 treatment (Fig. 2c). Given that MAT2A is involved in biosynthesis of SAM, we next examined change of SAM concentration in liver tissues. As shown in Fig. 2d, a significant decrease in SAM level was observed in CCl4-induced fibrotic liver, while NPLC0393 treatment led to almost complete recovery of SAM. Furthermore, the level of global DNA methylation was altered in a manner similar to that of SAM (Fig. S3b). We also examined the expression level of MAT1A which is the predominant enzyme responsible for SAM synthesis in parenchymal cells and MAT2B which is recognized as an allosteric regulator of MAT2A activity to confirm whether they were involved in the regulation of SAM production [12]. We found a slight downregulation of MAT1A and an obvious upregulation of MAT2B in CCl4 group and these changes were reversed by NPLC0393 treatment (Fig. S3c). Taken together, these results indicate that upregulation of MAT2A are positively correlated with fibrosis development and negatively correlated with intracellular SAM levels in the liver tissues, and NPLC0393 downregulates the MAT2A expression and increases the cellular SAM concentration in the fibrotic liver.
Fig. 2.

Upregulation of MAT2A reduced SAM concentration in the fibrotic liver tissues. (a) Representative Western blot results of MAT2A, TGF-β1, COL1A1 and α-SMA in mice liver tissues from Control (Con), CCl4 treatment (CCl4) or CCl4 combined with NPLC0393 treatment (CCl4 + NPLC0393) and NPLC0393 alone treatment (NPLC0393) groups. The densitometric changes in fold change over Control group were shown in right panel (n = 6 per group). (b) Immunofluorescence staining of MAT2A (green) and the HSC marker α-SMA (red) in mice liver tissues. Co-localization of MAT2A with α-SMA was shown in yellow (n = 6 per group). Scale bar, 100 μm. (c) qRT-PCR result of mat2a, a-sma and col1a1 mRNA expressions in mice liver tissues from each group. Gene expression was normalized using gapdh as the calibrator gene and fold changes over Control group were shown. Data are presented as means ± S.D. * P < .05, ** P < .01 and *** P < .001 compared with Control group; # P < .05, ## P < .01 ### P < .001 compared with CCl4 group (n = 6 per group). (d) The SAM concentration in mice liver tissues from control, CCl4 model and NPLC0393 treatment groups were measured (n = 5 per group). Data are presented as means ± S.D. * P < .05, ** P < .01 and *** P < .001. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. TGF-β1 induces MAT2A expression in HSCs
Growth factors, such as hepatic growth factor (HGF), epidermal growth factor (EGF), insulin-like growth factor-1 (IGF-1), and leptin have previously been shown to induce the mRNA and protein expression of MAT2A in cultured-hepatocytes and extrahepatic cancer cells [25,26]. Given that the TGF-β1 is recognized as the crucial factor in the progression of liver fibrosis, we attempted to explore the correlation between TGF-β1 and MAT2A expression. We first measured MAT2A expression levels in the absence or presence of TGF-β1 and NPLC0393 in the normal human hepatocyte (L02) and human HSC cell line (LX-2), respectively. As observed in Fig. 3a, the basal expression level of MAT2A in LX-2 was higher than in L02. TGF-β1 treatment markedly induced MAT2A in LX-2 but slightly reduced MAT2A expression in L02. NPLC0393 inhibited TGF-β1-induced MAT2A upregulation in LX-2, whereas it had little effect on MAT2A expression in L02. Moreover, in both of LX-2 and mouse HSC cell line (JS-1), NPLC0393 dose-dependently reversed TGF-β1-induced upregulation of MAT2A (Fig. S3d, e). Next, we also observed that TGF-β1 stimulation resulted in upregulation of mRNA levels for MAT2A, COL1A1 and α-SMA, which was reversed by NPLC0393 pre-treatment (Fig. 3b). Taken together, these results indicate that the change of MAT2A expression in response to TGF-β1 stimulation and NPLC0393 treatment mainly occurs in HSCs.
Fig. 3.

Upregulation of MAT2A led to the decrease of SAM concentration in the TGF-β1-induced activated HSCs. (a) The normal human hepatocyte cell line L02 and human HSC cell line LX-2 were treated with or without 10 μM NPLC0393 for 24 h and then stimulated with 5 ng/mL TGF-β1 for another 24 h. The levels of MAT2A, COL1A1 and α-SMA proteins were measured by Western blot. Data are representative of three independent experiments. (b) LX-2 cells were treated as in (a) and the expression level of MAT2A, COL1A1 and α-SMA mRNA were examined by qRT-PCR. * P < .05, ** P < .01 and *** P < .001 compared with Control group; # P < .05, ## P < .01 ### P < .001 compared with TGF-β1 treatment group (n = 3 per group with duplicates). (c) The SAM concentration in LX-2 cells from Control, TGF-β1 and NPLC0393 treatment groups were measured by LC-MS (n = 6 per group with duplicates). * P < .05, ** P < .01. (d) LX-2 cells were pre-treated with 2 mM SAM or 10 μM NPLC0393 for 24 h and then stimulated with 5 ng/mL TGF-β1 in the complete DMEM or L-methionine-free DMEM for another 24 h. The expression levels of MAT2A, MAT2B, COL1A1 and α-SMA proteins were analyzed by Western blot. Met (+) means normal DMEM culture medium and Met (−) means L-methionine-free DMEM culture medium. Data are representative of three independent experiments.
3.4. SAM level is negatively correlated with MAT2A expression in HSCs
To investigate the correlation between MAT2A expression and intracellular SAM level, we examined SAM concentration in LX-2 cells treated with TGF-β1 and NPLC0393. The SAM concentration was significantly decreased by TGF-β1 but was restored by NPLC0393 treatment (Fig. 3c). Then, we attempt to determine whether SAM itself can inhibit HSC activation and mediate inhibitory effect of NPLC0393 on TGF-β1 signaling. For this purpose, LX-2 cells were treated with TGF-β1 and NPLC0393 in the absence and or presence of L-methionine which mimics SAM function [27,28]. As shown in Fig. 3d, L-methionine depletion could remarkably induce expression of MAT2A, COL1A1and α-SMA proteins, whereas exogenous SAM inhibited the basal and TGF-β1-induced expression of these proteins. Furthermore, the inhibitory effect of NPLC0393 on these proteins was not observed when the cells were cultured in the L-methionine depleted medium. Of note, the change of MAT2B expression is positively correlated with that of MAT2A. Taken together, these results suggest that MAT2A expression level is negatively correlated with intracellular SAM concentration in the context of TGF-β1-induced HSC activation and the stable intracellular SAM concentration is required for the inhibitory effect of NPLC0393 on HSCs activation.
3.5. MAT2A promotes liver fibrogenesis
To investigate the role of MAT2A in liver fibrosis and the anti-fibrotic effect of NPLC0393, we established MAT2A overexpressed mice model by tail vein injection of AAV2/9-GFAP-MAT2A vector (AAV-MAT2A) which specifically expressed ectopic MAT2A in liver. We found that overexpression of MAT2A alone could sufficiently induce liver fibrosis, and almost abolished the anti-fibrotic effect of NPLC0393 (Fig. 4a–c). We also generated AAV-shRNA-MAT2A vector (MAT2A shRNA) to establish liver specific MAT2A knockdown mice model. We found that the depletion of MAT2A in the liver modestly inhibited expression levels of α-SMA, COL1A1 and TGF-β1, and alleviated CCl4-induced liver fibrosis as compare to AAV-shRNA-Negative Control mice (NC shRNA) (Fig. S4a-c). Then, we examined whether modulation of MAT2A expression can induce change in SAM concentration. As shown in Fig. 4d, overexpression of MAT2A resulted in a slight decrease in SAM concentration (P < .05). NPLC0393 treatment did not reverse the reduction of SAM concentration induced by CCl4 in MAT2A-overexpressed mice as compared to AAV-empty vector (AAV-EV) mice. Knockdown of MAT2A did not significantly affect the basal SAM level and slightly reversed CCl4-induced decrease of SAM concentration (P < .05) (Fig. S4d). In addition, in vitro knockdown of MAT2A in LX-2 cells inhibited basal and TGF-β1-induced upregulation of COL1A1 and α-SMA but did not affect the basal level of MAT2B (Fig. 4e, Fig. S4e). Taken together, these results indicate that upregulation of MAT2A and consequent reduction of SAM concentration can mediate HSC activation, and NPLC0393 exerts anti-fibrotic function via reversing such changes.
Fig. 4.

Profibrotic effect of MAT2A on liver fibrogenesis. (a) Mice were injected via tail vein with AAV-MAT2A or AAV-empty vector (AAV-EV) for 3 weeks and subjected to CCl4 administration with or without NPLC0393 for another 4 weeks. Representative image of MAT2A fluorescence, Sirius Red and α-SMA staining of the liver tissues were shown (Scale bars, 100 μm, n = 6 per group). (b) Semi-quantitative scoring of α-SMA staining for each group was shown in a bar graph. IOD: integrated optical density. (c) Representative Western blot result of Flag-MAT2A, α-SMA and COL1A1 proteins in mice livers from different groups. (d) The SAM concentration in mice liver from different groups were measured by LC-MS. * P < .05, ** P < .01 (n = 6 per group). (e) LX-2 cells transfected with the Negative Control siRNA (NC siRNA) or MAT2A siRNA were treated with 5 ng/mL TGF-β1 for 24 h. The expression levels of MAT2A, MAT2B, α-SMA and COL1A1 proteins were analyzed by Western blot. Data are representative of three independent experiments. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.6. TGF-β1 induces MAT2A expression via p65 phosphorylation
Key factors reported in regulating MAT2A transcription were also observed in our proteomic data. A subunit of NF-κB RELA (also known as p65) was significantly upregulated in fibrotic models and was reversed by NPLC0393 treatment (Fig. S5a). As previously reported, p65 could stimulate MAT2A transcription by binding to its promoter in tumor cells [29,30]. Therefore, we hypothesized that p65 may exert a similar role in HSCs. Verification of p65 expression in vivo showed that the expression level of total p65 protein was moderately higher in CCl4 than in control and NPLC0393-treated liver tissues, while the phosphorylated p65 (representing the activated p65 proteins) was changed more significantly across these three groups (Fig. 5a). Similarly, we found that the p65 phosphorylation was significantly upregulated in TGF-β1-stimulated LX-2 cells and inhibited by NPLC0393 as well as TGF-β1 inhibitor SB431542 treatments, but the total p65 protein was barely affected (Fig. 5b). Since PP2Cα was reported to act as IκB kinase β (IKKβ) phosphatases to terminate NF-κB activation [31], we also examined whether NPLC0393 inhibits phosphorylation of IKKβ. Unexpectedly, we found that NPLC0393 did not affect both total and phosphorylation of IKKβ in LX-2 cells (Fig. S5b). Then, we transfected LX-2 cells with NF-κB-Luc reporter plasmid to investigate the effect of TGF-β1 and NPLC0393 on NF-κB transcriptional activity. As indicated, TGF-β1 stimulation induced a significant increase in NF-κB luciferase activity, but this effect was reversed by NPLC0393 treatment (Fig. 5c). We further examined the effect of TGF-β1 and NPLC0393 on the MAT2A promoter activity by transfecting LX-2 cells with MAT2A-Luc reporter plasmid containing a functional NF-κB cis-acting element of the human MAT2A promoter (−571/+60 bp) [30]. As shown in Fig. 5d, similar to TNF-α, TGF-β1 induced a 3.5-fold increase of MAT2A promoter activity, which was reduced by NPLC0393 or NF-κB inhibitor SN50 treatments. Moreover, to determine whether p65 could mediate TGF-β1 induced upregulation of MAT2A and whether the anti-fibrotic effect of NPLC0393 on HSCs was exerted via inhibiting this process, we overexpressed p65 in LX-2 cells. As shown in Fig. 5e, overexpression of p65 enhanced basal and TGF-β1-induced upregulation of MAT2A, COL1A1 and p65 phosphorylation and abolished the inhibitory effect of NPLC0393. In addition, given that NPLC0393 was initially identified as a small molecule activator of PP2Cα, we attempted to examine the role of PP2Cα in MAT2A expression and HSC activation. As shown in Fig. 5f, knockdown of PP2Cα with siRNA almost completely abrogated the inhibitory effect of NPLC0393 on p65 phosphorylation, MAT2A, and α-SMA protein expressions, suggesting that the inhibitory effect of NPLC0393 on TGF-β1-induced HSC activation is dependent on the presence of PP2Cα. Taken together, these results indicate that TGF-β1 induces MAT2A expression via p65 phosphorylation and NPLC0393 inhibits this process by dephosphorylating p65 (Fig. 6).
Fig. 5.

Phosphorylation of p65 mediated TGF-β1-induced MAT2A expression. (a) Representative Western blot result of the total p65 and phospho-p65 (p-p65) in the control, CCl4-treated or NPLC0393-treated mice (n = 6 per group). (b) LX-2 cells were pre-treated with 10 μM NPLC0393 or 10 μM SB431542 for 24 h and then stimulated with 5 ng/mL TGF-β1 for another 2 h. The expression levels of the total p65 and p-p65 were analyzed by Western blot. (c) LX-2 cotransfected with the 1 μg/mL NF-κB reporter gene and 1 μg/mL pRL-TK plasmid were treated with vehicle, NPLC0393 or 10 μM SB431542 for 12 h and then stimulated with 5 ng/mL TGF-β1 for another 24 h. The Luciferase activity of NF-κB was normalized against that of the cotransfected pRL-TK. The result was shown as fold changes over untreated transfectants obtained from three independent tests. (d) LX-2 cells cotransfected with the 1 μg/mL MAT2A promoter construct and 1 μg/mL pRL-TK plasmid were treated with NPLC0393 or 50 ng/mL SN50 for 12 h and then stimulated with 5 ng/mL TGF-β1 for another 24 h. Relative luciferase activities were analyzed as above. (e) LX-2 cells infected with Ad-CMV-p65 (Ad-p65) or Ad-empty vector (Ad-EV) were treated with or without NPLC0393 for 12 h and then stimulated with 5 ng/mL TGF-β1 for another 24 h and the expression levels of p-p65, MAT2A, COL1A1 and α-SMA proteins were analyzed by Western blot. (f) LX-2 cells transfected with NC siRNA or PP2Cα siRNA were treated as (e) and the expression levels of p-p65, MAT2A and α-SMA proteins were analyzed by Western blot. * P < .05, ** P < .01 and *** P < .001. Data are representative of three independent experiments.
Fig. 6.
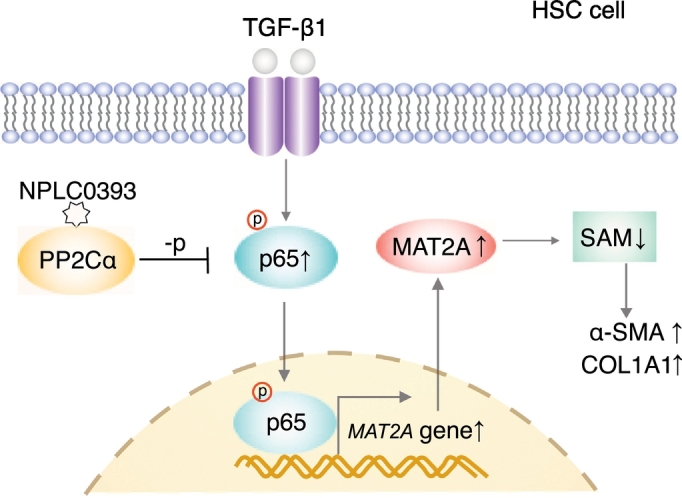
Schematic representation of TGF-β1/p65/MAT2A signaling pathway and inhibitory effect of NPLC0393 on this pathway. In HSCs, TGF-β1 induces MAT2A expression via p65 phosphorylation, which results in decrease of SAM concentration and consequential increase of α-SMA and COL1A1 expressions. PP2Cα small molecular activator NPLC0393 downregulates MAT2A by inhibiting phosphorylation of p65, thereby maintaining intracellular SAM concentration.
4. Discussion
The role of classical TGF-β1/Smad signaling pathway in fibrogenesis has been extensively studied. However, antagonizing only Smad2/3 does not prevent fibrosis completely and results in unexpected effects such as enhanced proliferation and inflammation, indicating additional pathways downstream of TGF-β1 also play important roles in fibrogenesis [32]. In the present study, using a small molecule NPLC0393 as a tool, we found that a novel TGF-β1/p65/MAT2A pathway may regulate intracellular SAM concentration to modulate HSC activation and liver fibrogenesis (Fig. 6).
Our proteomic study and subsequent bioinformatic analysis of differentially expressed proteins indicated that the proteins involved in SAM metabolism exhibited close link with other biological processes, such as wound healing, cell adhesion, apoptosis, lipid regulation. The multiple property of SAM may be due to its regulation of transmethylation, polyamine and some signaling pathways including mTOR signaling [33,34]. In addition, as a precursor of GSH, SAM plays a chemopreventive role in oxidative stress induced by CCl4. Oxidative stress promotes the initial inflammation and its progression to the fibrosis via modulating NF-κB and MAPK signaling pathway and expression of TGF-β1 [35,36]. Consistent with these studies, our LC-MS/MS data demonstrated that three oxidative stress-associated proteins (e.g. Sod1, Gpx1, Mapk3) were altered in the CCl4-intoxicated mouse and reversed by NPLC0393-treatment (Table S2). More importantly, it was also reported that SAM could alleviate liver fibrosis [37,38]. Gnmt (glycine N-methyltransferase), the most abundant SAM-dependent methyl transferases in liver, is implicated in maintaining proper SAM concentration. Children with GNMT mutation or Gnmt knockout mice developed liver fibrosis [[39], [40], [41]]. Bhmt (betaine homocysteine methyltransferase) contributes to conversion of homocysteine into methionine, and reduced mRNA abundance of Bhmt was observed in liver cirrhosis and HCC tissues [42]. Consistent with previous studies, both Gnmt and Bhmt were found to be downregulated in CCl4 group in our proteomic results (Fig. 1d).
Previous studies reported that MAT2A expression was highly induced in the proliferating liver, dedifferentiated cells and tumor cells [43]. Ramani, et al.16 demonstrated that MAT2A is essential for HSC activation. In line with previous findings, the expression of MAT2A was significantly upregulated in both of CCl4 and BDL fibrosis models and was positively correlated with the level of TGF-β1. Although TGF-β1 is recognized as a critical factor for HSC activation, its role in MAT2A modulation has not been investigated. Our study revealed that TGF-β1 stimulation increased mRNA and protein levels of MAT2A in HSCs, which was efficiently blocked by NPLC0393 treatment. However, we observed that the expression of MAT2A in hepatocytes was moderately downregulated by TGF-β1 stimulation, and was not further affected by NPLC0393 pre-treatment. Consistently, it has been reported that TGF-β1 decreased MAT2A mRNA level in hepatoma cells, which can block the cell proliferation to regulate hepato-carcinogenesis [44]. These findings indicate that the TGF-β1 signaling mediated MAT2A regulation varies and is cell type dependent [45].
Importantly, overexpression of MAT2A in mouse liver could sufficiently induce liver fibrosis and abrogate the anti-fibrotic action of NPLC0393 on CCl4-induced fibrosis, while knockdown of MAT2A in vivo and in vitro ameliorated the fibrogenic events induced by CCl4 or TGF-β1, suggesting that MAT2A may be a potential target for the prevention and treatment of liver fibrosis. Of note, the expression pattern of MAT can significantly influence the intracellular steady-state SAM level [46]. In the present study, SAM concentration decreased in CCl4-treated liver tissue as well as in TGF-β1-treated HSCs and these changes were accompanied by abnormal upregulation of MAT2A, which was further validated in MAT2A-overexpression experiments. It is interesting that upregulation of MAT2A did not induce elevation of SAM level, although MAT2A is the unique enzyme catalyzing SAM production in the HSCs [14]. This result can be partially explained that abnormal upregulation of MAT2A can induce the expression of regulatory subunit MAT2B [[47], [48], [49]]. Although the comparison between expression levels of MAT2A and MAT2B has not been reported, it was suggested that one MAT2B chain binds MAT2A homodimer to inhibit catalytic activity of MAT2A [12]. Consistent with this hypothesis, we found that MAT2A upregulation induced by CCl4 or TGF-β1 was accompanied by the elevation of MAT2B while knockdown of MAT2A did not affected expression level of MAT2B. However, we cannot eliminate the possibility that SAM was consumed for polyamine synthesis in the activated HSCs, which supports cell proliferation [50,51]. Taken together, our results suggest that the effect of MAT2A on SAM concentration in HSCs is dependent on the extent of MAT2A expression level and activation status of HSCs.
Previous studies have demonstrated that SAM can attenuate hepatic fibrosis through distinct mechanisms [37,[52], [53], [54]]. We showed that exogenous SAM treatment of aHSCs resulted in reduction of the basal and TGF-β1-induced MAT2A, COL1A1 and α-SMA protein expression, whereas SAM depletion by L-methionine-free media resulted in opposite phenotypes and abolished inhibitory effect of NPLC0393 on HSC activation. This observation is consistent with other reports showing that the induction of MAT2A mRNA expression and MAT activity under limited availability of L-methionine [28,55]. It is well recognized that aHSCs produce TGF-β1 or other cytokines to reinforce HSCs activation [1]. Therefore, we believe that TGF-β1-induced MAT2A upregulation and consequential reduction of SAM concentration further activate TGF-β1 signaling, making it a positive feed-forward loop for maintenance of fibrosis process while NPLC0393 treatment disrupts this loop by reducing MAT2A expression and maintaining stable cellular SAM level.
MAT2A expression is coordinately regulated by complex mechanisms in response to diverse pathological stimuli [56]. Our proteomic data showed that NF-κB subunit p65, a reported transcription factor of MAT2A [29,30,57], was upregulated in CCl4-treated mice liver. Our in vitro studies demonstrated that TGF-β1 increased p65 phosphorylation and enhanced NF-κB transcriptional activity in aHSCs, which was also observed in the TGF-β1-stimulated cancer cells [58]. The pro-fibrogenesis role of p65 is also supported by the previous studies which reported that NF-κB could induce the expression of proinflammatory and profibrotic genes (e.g. IL-1/6, TNF-α and IL-17) [[59], [60], [61]], and also confers resistance to apoptosis [[62], [63], [64]]. These events further exacerbate and maintain activated status. We showed that induction of NF-κB transcriptional activity by TGF-β1 enhanced MAT2A promoter activity and overexpression of p65 enhanced both of the basal and TGF-β1-induced MAT2A and COL1A1 expression. Therefore, although we cannot exclude the possibility that p65 promotes HSC activation via its well-known targets, our results uncovered another mechanism for the pro-fibrogenic properties of p65 during the HSC activation.
PP2Cα has been identified as a direct regulator of NF-κB signaling through dephosphorylation and inactivation of IKKβ or NF-κB in cancer cells [31,65]. However, our observation that PP2Cα activator NPLC0393 inhibits phosphorylation of p65 but not IKKβ, both of which are downstream mediators of TGF-β1, indicates that the effect of PP2Cα on TGF-β1 downstream signaling components is likely cellular context dependent. In addition, we further confirmed that PP2Cα could regulate p65 phosphorylation and downstream fibrotic events and mediate inhibitory effect of NPLC0393 on HSCs activation.
In conclusion, our study demonstrates that TGF-β1 stimulates HSC activation through promoting phosphorylation of p65, transcriptional expression of MAT2A and consequential reduction of SAM concentration, and NPLC0393 suppresses the activation of HSCs partially by blocking such process. These findings link TGF-β1 to SAM metabolism and indicate a novel TGF-β1/p65/MAT2A pathway in HSC activation.
Funding sources
This work was supported by National Natural Science Foundation of China (No. 81500469, 81573873, 81774196 and 31800693), Zhejiang Provincial Natural Science Foundation of China (No. Y15H030004), the National Key Research and Development Program from the Ministry of Science and Technology of China (No. 2017YFC1700200) and the Key Program of National Natural Science Foundation of China (No. 8153000502). These funders supported this study in performing experiment, data collection, data analysis, interpretation or writing of the manuscript.
Declaration of interests
The authors declare no conflict of interests.
Author contributions
H.Z., P.L., S.H.F., K.F.W. and J.S.Z. conceived the project, designed and supervised this study. K.F.W., S.H.F. and Q.L. carried out in vitro and in vivo experiments. H.Z., S.H.F. and Q.L. wrote the manuscript, J.G., S.H.F., Q.L., H.W.Z. and Z.Y.Z. performed the proteomic and SAM LC-MS mass spectrometry experiments and data analysis. S.H.F., K.F.W. and F.H.J. contributed to the animal experiments. L.H.H., X.S. and D.M.G. provided NPLC0393 compound and contributed to advice and scientific discussion. J.S.W. and Y.M.M. performed histological experiments and data analysis. All authors approved the final version of the manuscript.
Acknowledgements
We thank Institute of Liver Diseases, Shanghai University of Traditional Chinese Medicine for providing the LX-2 cells and thank Prof. Jinsheng Guo (Zhongshan Hospital, Fudan University, China) for providing the JS-1 cells. We also thank the Institutional Technology Service Center of Shanghai Institute of Materia Medica for technical support.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2019.03.058.
Contributor Information
Jiansheng Zhu, Email: zhujs@enzemed.com.
Ping Liu, Email: liuliver@vip.sina.com.
Hu Zhou, Email: zhouhu@simm.ac.cn.
Appendix A. Supplementary data
Supplementary table 1
Supplementary table 2
Supplementary table 3
Supplementary Figures and methods
References
- 1.Hernandez-Gea V., Friedman S.L. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 2.Wynn T.A., Ramalingam T.R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman S.L. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verrecchia F., Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007;13(22):3056–3062. doi: 10.3748/wjg.v13.i22.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrientos S., Stojadinovic O., Golinko M.S., Brem H., Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 7.Rosenbloom J., Castro S.V., Jimenez S.A. Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Ann Intern Med. 2010;152(3):159–166. doi: 10.7326/0003-4819-152-3-201002020-00007. [DOI] [PubMed] [Google Scholar]
- 8.Beyer C., Distler J.H.W. Tyrosine kinase signaling in fibrotic disorders Translation of basic research to human disease. BBA-Mol Basis Dis. 2013;1832(7):897–904. doi: 10.1016/j.bbadis.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Wang L.R., Wang X., Chen J., Yang Z.Y., Yu L.A., Hu L.H. Activation of protein serine/threonine phosphatase PP2C alpha efficiently prevents liver fibrosis. Plos One. 2010;5(12) doi: 10.1371/journal.pone.0014230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mato J.M., Alvarez L., Ortiz P., Pajares M.A. S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol Ther. 1997;73(3):265–280. doi: 10.1016/s0163-7258(96)00197-0. [DOI] [PubMed] [Google Scholar]
- 11.Kotb M., Mudd S.H., Mato J.M., Geller A.M., Kredich N.M., Chou J.Y. Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet. 1997;13(2):51–52. doi: 10.1016/s0168-9525(97)01013-5. [DOI] [PubMed] [Google Scholar]
- 12.LeGros H.L., Jr., Halim A.B., Geller A.M., Kotb M. Cloning, expression, and functional characterization of the beta regulatory subunit of human methionine adenosyltransferase (MAT II) J Biol Chem. 2000;275(4):2359–2366. doi: 10.1074/jbc.275.4.2359. [DOI] [PubMed] [Google Scholar]
- 13.Mato J.M., Lu S.C. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45(5):1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]
- 14.Shimizu-Saito K., Horikawa S., Kojima N., Shiga J., Senoo H., Tsukada K. Differential expression of S-adenosylmethionine synthetase isozymes in different cell types of rat liver. Hepatology. 1997;26(2):424–431. doi: 10.1002/hep.510260224. [DOI] [PubMed] [Google Scholar]
- 15.Ramani K., Donoyan S., Tomasi M.L., Park S. Role of methionine adenosyltransferase alpha2 and beta phosphorylation and stabilization in human hepatic stellate cell trans-differentiation. J Cell Physiol. 2015;230(5):1075–1085. doi: 10.1002/jcp.24839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramani K., Tomasi M.L. Transcriptional regulation of methionine adenosyltransferase 2A by peroxisome proliferator-activated receptors in rat hepatic stellate cells. Hepatology. 2012;55(6):1942–1953. doi: 10.1002/hep.25594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramani K., Yang H., Kuhlenkamp J., Tomasi L., Tsukamoto H., Mato J.M. Changes in the expression of methionine adenosyltransferase genes and S-adenosylmethionine homeostasis during hepatic stellate cell activation. Hepatology. 2010;51(3):986–995. doi: 10.1002/hep.23411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang H.L., Lai J.J., Lin W.L., Lin W.C. A fermented substance from Aspergillus phoenicis reduces liver fibrosis induced by carbon tetrachloride in rats. Biosci Biotech Bioch. 2007;71(5):1154–1161. doi: 10.1271/bbb.60604. [DOI] [PubMed] [Google Scholar]
- 19.Matsui H., Ikeda K., Nakajima Y., Horikawa S., Imanishi Y., Kawada N. Sulfur-containing amino acids attenuate the development of liver fibrosis in rats through down-regulation of stellate cell activation. J Hepatol. 2004;40(6):917–925. doi: 10.1016/j.jhep.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Yin F., Hu L.H. Six new triterpene saponins with a 21,23-lactone skeleton from Gynostemma pentaphyllum. Helv Chim Acta. 2005;88(5):1126–1134. [Google Scholar]
- 21.Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11(3):319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 22.Wisniewski J.R., Nagaraj N., Zougman A., Gnad F., Mann M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res. 2010;9(6):3280–3289. doi: 10.1021/pr1002214. [DOI] [PubMed] [Google Scholar]
- 23.Rabinowitz J.D., Kimball E. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal Chem. 2007;79(16):6167–6173. doi: 10.1021/ac070470c. [DOI] [PubMed] [Google Scholar]
- 24.Futschik M.E., Carlisle B. Noise-robust soft clustering of gene expression time-course data. J Bioinform Comput Biol. 2005;3(4):965–988. doi: 10.1142/s0219720005001375. [DOI] [PubMed] [Google Scholar]
- 25.Latasa M.U., Boukaba A., Garcia-Trevijano E.R., Torres L., Rodriguez J.L., Caballeria J. Hepatocyte growth factor induces MAT2A expression and histone acetylation in rat hepatocytes: role in liver regeneration. FASEB J. 2001;15(7):1248–1250. doi: 10.1096/fj.00-0556fjev1. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Xia M, Lin M, Yang H, Kuhlenkamp J, Li T, et al. Role of methionine adenosyltransferase 2A and S-adenosylmethionine in mitogen-induced growth of human colon cancer cells. Gastroenterology 2007; 133(5): 1747. [DOI] [PubMed]
- 27.Lin D.W., Chung B.P., Kaiser P. S-adenosylmethionine limitation induces p38 mitogen-activated protein kinase and triggers cell cycle arrest in G1. J Cell Sci. 2014;127:50–59. doi: 10.1242/jcs.127811. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pendleton K.E., Chen B., Liu K., Hunter O.V., Xie Y., Tu B.P. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM Synthetase intron retention. Cell. 2017;169(5):824–35 e14. doi: 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q.Y., Chen J.W., Liu L., Zhang J., Wang D.F., Ma L. The X protein of hepatitis B virus inhibits apoptosis in hepatoma cells through enhancing the methionine adenosyltransferase 2A gene expression and reducing S-Adenosylmethionine production. J Biol Chem. 2011;286(19):17168–17180. doi: 10.1074/jbc.M110.167783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang H., Sadda M.R., Yu V., Zeng Y., Lee T.D., Ou X. Induction of human methionine adenosyltransferase 2A expression by tumor necrosis factor alpha. Role of NF-kappa B and AP-1. J Biol Chem. 2003;278(51):50887–50896. doi: 10.1074/jbc.M307600200. [DOI] [PubMed] [Google Scholar]
- 31.Sun W.J., Yu Y., Dotti G., Shen T., Tan X.J., Savoldo B. PPM1A and PPM1B act as IKK beta phosphatases to terminate TNF alpha-induced IKK beta-NF-kappa B activation. Cell Signal. 2009;21(1):95–102. doi: 10.1016/j.cellsig.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X., Hu H., Yin J.Q. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006;26(1):8–22. doi: 10.1111/j.1478-3231.2005.01192.x. [DOI] [PubMed] [Google Scholar]
- 33.Lu S.C., Mato J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92(4):1515–1542. doi: 10.1152/physrev.00047.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu X., Orozco J.M., Saxton R.A., Condon K.J., Liu G.Y., Krawczyk P.A. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science. 2017;358(6364):813–818. doi: 10.1126/science.aao3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleniewska P., Piechota-Polanczyk A., Michalski L., Michalska M., Balcerczak E., Zebrowska M. Influence of block of NF-kappa B signaling pathway on oxidative stress in the liver homogenates. Oxid Med Cell Longev. 2013;2013 doi: 10.1155/2013/308358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abhilash P.A., Harikrishnan R., Indira M. Ascorbic acid supplementation down-regulates the alcohol induced oxidative stress, hepatic stellate cell activation, cytotoxicity and mRNA levels of selected fibrotic genes in Guinea pigs. Free Radic Res. 2012;46(2):204–213. doi: 10.3109/10715762.2011.647691. [DOI] [PubMed] [Google Scholar]
- 37.Nieto N., Cederbaum A.I. S-adenosylmethionine blocks collagen I production by preventing transforming growth factor-beta induction of the COL1A2 promoter. J Biol Chem. 2005;280(35):30963–30974. doi: 10.1074/jbc.M503569200. [DOI] [PubMed] [Google Scholar]
- 38.Simile M.M., Banni S., Angioni E., Carta G., De Miglio M.R., Muroni M.R. 5′-Methylthioadenosine administration prevents lipid peroxidation and fibrogenesis induced in rat liver by carbon-tetrachloride intoxication. J Hepatol. 2001;34(3):386–394. doi: 10.1016/s0168-8278(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 39.Augoustides-Savvopoulou P., Luka Z., Karyda S., Stabler S.P., Allen R.H., Patsiaoura K. Glycine N-methyltransferase deficiency: a new patient with a novel mutation. J Inherit Metab Dis. 2003;26(8):745–759. doi: 10.1023/B:BOLI.0000009978.17777.33. [DOI] [PubMed] [Google Scholar]
- 40.Mudd S.H., Cerone R., Schiaffino M.C., Fantasia A.R., Minniti G., Caruso U. Glycine N-methyltransferase deficiency: a novel inborn error causing persistent isolated hypermethioninaemia. J Inherit Metab Dis. 2001;24(4):448–464. doi: 10.1023/a:1010577512912. [DOI] [PubMed] [Google Scholar]
- 41.Varela-Rey M., Martinez-Lopez N., Fernandez-Ramos D., Embade N., Calvisi D.F., Woodhoo A. Fatty liver and fibrosis in glycine N-methyltransferase knockout mice is prevented by nicotinamide. Hepatology. 2010;52(1):105–114. doi: 10.1002/hep.23639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avila M.A., Berasain C., Torres L., Martin-Duce A., Corrales F.J., Yang H. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33(6):907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 43.Mato J.M., Corrales F.J., Lu S.C., Avila M.A. S-adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16(1):15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 44.Paneda C., Gorospe I., Herrera B., Nakamura T., Fabregat I., Varela-Nieto I. Liver cell proliferation requires methionine adenosyltransferase 2A mRNA up-regulation. Hepatology. 2002;35(6):1381–1391. doi: 10.1053/jhep.2002.32538. [DOI] [PubMed] [Google Scholar]
- 45.Meindl-Beinker N.M., Matsuzaki K., Dooley S. TGF-beta signaling in onset and progression of hepatocellular carcinoma. Dig Dis. 2012;30(5):514–523. doi: 10.1159/000341704. [DOI] [PubMed] [Google Scholar]
- 46.Cai J., Mao Z., Hwang J.J., Lu S.C. Differential expression of methionine adenosyltransferase genes influences the rate of growth of human hepatocellular carcinoma cells. Cancer Res. 1998;58(7):1444–1450. [PubMed] [Google Scholar]
- 47.Halim A.B., LeGros L., Geller A., Kotb M. Expression and functional interaction of the catalytic and regulatory subunits of human methionine adenosyltransferase in mammalian cells. J Biol Chem. 1999;274(42):29720–29725. doi: 10.1074/jbc.274.42.29720. [DOI] [PubMed] [Google Scholar]
- 48.Bissell D.M., Roulot D., George J. Transforming growth factor beta and the liver. Hepatology. 2001;34(5):859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 49.Quinlan C.L., Kaiser S.E., Bolanos B., Nowlin D., Grantner R., Karlicek-Bryant S. Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat Chem Biol. 2017;13(7):785–792. doi: 10.1038/nchembio.2384. [DOI] [PubMed] [Google Scholar]
- 50.Tomasi M.L., Ryoo M., Skay A., Tomasi I., Giordano P., Mato J.M. Polyamine and methionine adenosyltransferase 2A crosstalk in human colon and liver cancer. Exp Cell Res. 2013;319(12):1902–1911. doi: 10.1016/j.yexcr.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramani K., Yang H., Xia M., Ara A.I., Mato J.M., Lu S.C. Leptin's mitogenic effect in human liver cancer cells requires induction of both methionine adenosyltransferase 2A and 2 beta. Hepatology. 2008;47(2):521–531. doi: 10.1002/hep.22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gasso M., Rubio M., Varela G., Cabre M., Caballeria J., Alonso E. Effects of S-adenosylmethionine on lipid peroxidation and liver fibrogenesis in carbon tetrachloride-induced cirrhosis. J Hepatol. 1996;25(2):200–205. doi: 10.1016/s0168-8278(96)80074-2. [DOI] [PubMed] [Google Scholar]
- 53.Karaa A., Thompson K.J., McKillop I.H., Clemens M.G., Schrum L.W. S-adenosyl-L-methionine attenuates oxidative stress and hepatic stellate cell activation in an ethanol-LPS-induced fibrotic rat model. Shock. 2008;30(2):197–205. doi: 10.1097/shk.0b013e318160f417. [DOI] [PubMed] [Google Scholar]
- 54.Thompson K.J., Lakner A.M., Cross B.W., Tsukada S., Rippe R.A., McKillop I.H. S-adenosyl-L-methionine inhibits collagen secretion in hepatic stellate cells via increased ubiquitination. Liver Int. 2011;31(6):891–901. doi: 10.1111/j.1478-3231.2011.02512.x. [DOI] [PubMed] [Google Scholar]
- 55.Martinez-Chantar M.L., Latasa M.U., Varela-Rey M., Lu S.C., Garcia-Trevijano E.R., Mato J.M. L-methionine availability regulates expression of the methionine adenosyltransferase 2A gene in human hepatocarcinoma cells: role of S-adenosylmethionine. J Biol Chem. 2003;278(22):19885–19890. doi: 10.1074/jbc.M211554200. [DOI] [PubMed] [Google Scholar]
- 56.Maldonado L.Y., Arsene D., Mato J.M., Lu S.C. Methionine adenosyltransferases in cancers: mechanisms of dysregulation and implications for therapy. Exp Biol Med. 2018;243(2):107–117. doi: 10.1177/1535370217740860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phuong N.T.T., Kim S.K., Im J.H., Yang J.W., Choi M.C., Lim S.C. Induction of methionine adenosyltransferase 2A in tamoxifen-resistant breast cancer cells. Oncotarget. 2016;7(12):13902–13916. doi: 10.18632/oncotarget.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freudlsperger C., Bian Y., Wise S.C., Burnett J., Coupar J., Yang X. TGF-beta and NF-kappa B signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene. 2013;32(12):1549–1559. doi: 10.1038/onc.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bahcecioglu I.H., Koca S.S., Poyrazoglu O.K., Yalniz M., Ozercan I.H., Ustundag B. Hepatoprotective effect of infliximab, an anti-TNF-alpha agent, on carbon tetrachloride-induced hepatic fibrosis. Inflammation. 2008;31(4):215–221. doi: 10.1007/s10753-008-9067-1. [DOI] [PubMed] [Google Scholar]
- 60.Diaz J.A., Booth A.J., Lu G., Wood S.C., Pinsky D.J., Bishop D.K. Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am J Transplant. 2009;9(8):1773–1783. doi: 10.1111/j.1600-6143.2009.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tan Z.M., Qian X.F., Jiang R.Q., Liu Q.H., Wang Y.J., Chen C. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol. 2013;191(4):1835–1844. doi: 10.4049/jimmunol.1203013. [DOI] [PubMed] [Google Scholar]
- 62.Tergaonkar V. NFkappaB pathway: a good signaling paradigm and therapeutic target. Int J Biochem Cell Biol. 2006;38(10):1647–1653. doi: 10.1016/j.biocel.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 63.Watson M.R., Wallace K., Gieling R.G., Manas D.M., Jaffray E., Hay R.T. NF-kappa B is a critical regulator of the survival of rodent and human hepatic myofibroblasts. J Hepatol. 2008;48(4):589–597. doi: 10.1016/j.jhep.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 64.Lin X., Sime P.J., Xu H.D., Williams M.A., LaRussa L., Georas S.N. Yin Yang 1 is a novel regulator of pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(12):1689–1697. doi: 10.1164/rccm.201002-0232OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu X., An H., Jin R., Zou M., Guo Y., Su P.F. PPM1A is a RelA phosphatase with tumor suppressor-like activity. Oncogene. 2014;33(22):2918–2927. doi: 10.1038/onc.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1
Supplementary table 2
Supplementary table 3
Supplementary Figures and methods