

Abstract
Breast cancer is the most common cause of cancer death in women worldwide. This malignancy is a complex disease, which is defined by an intrinsic heterogeneity on the histopathological and molecular level as well as response to therapy and outcome. In addition to classical histopathological features, breast cancer can be categorized into at least five major subtypes based on comprehensive gene expression profiling: luminal A, luminal B, basal-like, ERBB2-positive, and normal-like breast cancer. Genetically engineered mouse models can serve as tools to study the molecular underpinnings for this disease. Given the genetic complexity that drives the initiation and progression of individual breast cancer subtypes, it is evident that certain models can reflect only particular aspects of this malignancy. In this book chapter, we will primarily focus on advances in modeling breast cancer at defined stages of carcinogenesis using genetically engineered mice. We will discuss the ability as well as shortcomings of these models to faithfully recapitulate the spectrum of human breast cancer subtypes.
Keywords: Mouse models, Molecular subtypes, Transgene, Tumorigenesis, Cancer progression
1. Introduction
Breast cancer affects a staggering number of 1.7 million women each year on a global scale, and it is the most common cause of cancer-related deaths in this gender. This disease claimed the lives of approximately 521,000 women in 2012, which represents one in seven of all cancer-associated fatalities that year alone (2014 fact sheet of the World Health Organization; http://www.who.int/mediacentre/factsheets/fs297/en/). Unfortunately, these statistical figures might be an underestimation due to the lower screening rates and incomplete reporting in developing countries. It seems to be a general phenomenon that developed countries tend to have higher incidences in breast cancer. For example, more than 230,000 new cases are being diagnosed every year in the Unites States of America alone. Despite earlier detection, this malignancy remains the second leading cause of cancer-related deaths with more than 40,000 fatalities (breast cancer statics of the Susan G. Komen Foundation; http://ww5.komen.org/BreastCancer/Statistics.html)
While in the public eye breast cancer is viewed as one specific disease, there is ample scientific evidence today to suggest that this malignancy represents multiple subtypes that vary significantly in their histopathology, dependence on hormones and local growth factors, and their downstream effectors as well as response to therapy and clinical outcome. Based solely on hierarchical clustering of gene expression profiles, breast cancer can be classified into at least five distinctly different breast cancer types: luminal-like (luminal A and B), basal-like, ERBB2-positive, and normal-like breast cancer [1, 2]. Given the fact that breast cancer is phenotypically and genetically heterogeneous, it should be evident that studying particular tumor subtypes in vivo requires not just one, but multiple genetically defined model systems as scientific tools. Therefore, any claims in the scientific literature or presentations that are intended to generalize or highlight one particular model as “authentic to human breast cancer” should be rejected [1]. Every cancer model replicates particular aspects of disease subtypes such as the requirement of specific genetic lesions that mediate neoplastic transformation and tumorigenesis. These models may also recapitulate certain histopathological characteristics and cancer-associated biological processes (e.g., cancer invasion and metastasis) as well as resistance to cytotoxic and targeted therapy [3].
In addition to the numerous human breast cancer cell lines that exist today, an increasing number of animal models are available to study mammary gland development and tumorigenesis in vivo. In particular, mouse models have become primary tools for cancer research, and those can be further subdivided into three main groups: (1) xenograft models; (2) chemically, virally, or ionizing radiation (IR)-induced models; and (3) genetically engineered mice (GEMs) that include transgenics and knockouts [1]. Classical xenograft models based on the transplantation of well-characterized human breast cancer cell lines into immunocompromised mice are relatively inexpensive and can generate tumors after a short latency. Unfortunately, these models are often poor predictors of response to therapy in humans due to major differences in tumor histopathology [4]. Nonetheless, xenograft models have recently seen a renaissance with a new primary focus on patient-derived cancer samples that are propagated exclusively in immunocompromised mice without culturing ex vivo. Patient-derived xenografts (PDXs) are being collected, characterized, and distributed by major institutions such as the National Cancer Institute and The Jackson Laboratory. Despite much euphoria about the advantages of these new models, it will remain a fact that PDXs have limitations that are, in many aspect, identical to classical xenograft models [1]. First, PDXs do not allow an introduction of specific mutations into endogenous genes of tumor cells, and it is difficult to achieve a homogeneous expression of reporter genes for in vivo imaging. Second, recipient mice are immunocompromised which represents an obstacle in the development and testing of immunotherapies or other cancer therapies that rely directly or indirectly on an intact immune system. Third, any types of tumor cells that are passaged ex vivo or in vivo undergo a selection process. Future studies will show whether PDXs that will be exponentially propagated from mouse to mouse will undergo a genetic drift that is comparable to cultured cells. Fourth, it should be assumed that sequential passaging of breast cancer tissues between animals will eventually result in a partial or even complete substitution of the human tumor stroma with murine cells. With regard to the cellular composition of metastatic lesions, tumor cells from PDXs or classical xenograft should acquire virtually identical properties that facilitate growth and survival in a predominantly murine tumor environment. Finally, PDXs are not fundamentally different from classical xenografts with regard to species-related incompatibilities to hormones and other cytokines. For example, the human prolactin can activate the orthologous mouse receptor, but the human prolactin receptors are insensitive to mouse prolactin [5]. Consequently, cancer cells within PDXs have to undergo a selection process that facilitates engraftment and growth without prolactin and activation of downstream signaling mediators and target genes. Great efforts are currently being made to “humanize” ligand-receptor systems in mice as described by Ueda and colleagues for interleukin 6 (IL-6) signaling [6] or by expressing the genes coding the human variants of M-CSF, interleukin 3 (IL-3), GM-CSF, and human thrombopoietin under the control of the endogenous mouse loci [7].
Over the past quarter century, genetically engineered mouse models (GEMs) have been developed in order to understand the molecular, biochemical, and cellular functions of oncogenes or tumor suppressor genes during the initiation and progression of cancer. The first genetically modified mouse model for breast cancer research was developed in the mid-1980s by Philip Leder and coworkers. This transgenic strain expresses the c-Myc oncogene under the control of the mouse mammary tumor virus long terminal repeat (MMTV-LTR), which resulted in a spontaneous development of adenocarcinomas [8]. Since then, a myriad of transgenic lines have been generated to investigate the role of proto-oncogenes in mammary gland development and tumor onset. Many of these first-generation GEMs have been described in detail in a special issue of the journal Oncogene in 2000 [9]. The second generation of GEMs for cancer research is based on the targeted modification of endogenous tumor susceptibility genes using homologous recombination [10]. They include knockout mice that are deficient in tumor suppressor genes as well as mice that express oncogenic mutants under the control of the endogenous promoter [11–13]. The first and second generations of GEMs have provided novel insights into the biological significance of the gain-of-function of oncogenes or the loss-of-function of tumor suppressor genes in cancer initiation. The third generation of genetically engineered models relies on advanced molecular tools that allow a somatic deletion of endogenous genes as well as temporally and spatially controlled expression of oncogenes in a ligand-controlled manner. These new models are currently being utilized to examine the importance of any gene-of-interest in cancer initiation, progression, and metastasis [14].
In this chapter, we will focus primarily on the use of genetically engineered mice as research tools to study the molecular, biochemical, and cellular mechanisms that govern the genesis of human breast cancer. We are however unable to provide a complete description of all mammary tumor models that have been developed to date and apologize to those investigators whose genetic tools have not been mentioned. Instead, we focus on the more widely used models and their advantages but also shortcomings in modeling particular breast cancer subtypes. We will also highlight the use of particular GEMs to study breast cancer metastasis, i.e., a complex and important cellular process in cancer progression that is responsible for virtually all fatalities following disease recurrence and therapy resistance.
2. Genetically Engineered Mouse Models (GEMs) for Human Breast Cancer
2.1. Transgenic Mice with a Mammary-Specific Expression of Oncogenes
The primary objective to use conventional transgenic mice for breast cancer research is to overexpress the coding region of an oncogene (wild-type or mutant) or tumor-associated microRNA under the regulation of a mammary-specific promoter. By upregulating the levels of these oncogenes in the mammary epithelium, it is possible to assess whether their deregulated expression is sufficient to cause neoplastic transformation and mammary tumor development. Transgenic mice are also being used to target the expression of dominant-negative variants of genes in mammary epithelial cells to validate the role of the tumor suppressor genes in vivo. Tumor latency and penetrance can often vary depending upon the potency of the selected oncogene and the level of expression achieved through the selected promoter. The most commonly used mammary-specific gene regulatory elements include the mouse mammary tumor virus long terminal repeat (MMTV-LTR) and milk protein gene promoters of the whey acidic protein (WAP) or the β-lactoglobulin (BLG) [14].
The MMTV-LTR is a regulatory element of the mouse mammary tumor virus that is present in the milk of infected mice. It was John J. Bittner who described in 1936 a cancer-inducing component of the milk (“milk factor”) that could be transmitted from a diseased female mouse to its offspring [15]. The virus can also be passed on via the germline in particular mouse strains. To better understand the expression profile of the MMTV-LTR as part of genetically engineered transgenes, it is important to have some basic knowledge about the infection and life cycle of the MMTV. In contrast to its name, the expression the MMTV-LTR is not restricted to mammary epithelial cells. Other secretory tissues in males and females such as the salivary gland and seminal vesicle show a considerable activation of the MMTV-LTR. Moreover, the LTR is expressed in macrophages and lymphocytes that play a role in the life cycle of a virus that is transmitted through the exogenous route (i.e., the milk). Any reference in the literature about a “leaky” expression profile of MMTV-driven transgenes is, in fact, a result of the normal activation of the MMTV-LTR in particular cell types that are essential for the life cycle of MMTV. It is known that the regulatory elements of the MMTV are active throughout the ductal epithelium in nulliparous females, but their expression is elevated by steroid hormones and prolactin in pregnant and lactating mice (see review by Ross [16]). Since these hormones promote the proliferation and functional differentiation of luminal epithelial cells, it is evident that the expression of the LTR is higher in this particular epithelial compartment. Nonetheless, the MMTV promoter is active in basal mammary epithelial cells as demonstrated in cell lineage-tracing experiments using the MMTV-Cre transgene [17]. Smith and colleagues have used the integration of MMTV to genetically label multipotent mammary progenitors [18]. Ductal cells that possess phenotypic properties of early transformation in response to MMTV infection are able to maintain their neoplastic characteristic following serial transplantation, which is clear evidence that the MMTV-LTR is expressed in mammary stem/progenitor cells.
The MMTV-LTR has been widely utilized for the overexpression of oncogenes such as Myc [8], Ha-ras [19], Wnt [20], and the polyomavirus middle T antigen (PyMT) [21], as well as mutant and wild-type ErbB2 (also known as Her2/neu) [22, 23]. Expressions of all these oncogenes result in mammary tumorigenesis at defined latency periods. There are two important facts to consider before designing an MMTV-based transgenic vector or using established MMTV-driven oncogenic models for cancer research: the source of the MMTV-LTR and the genetic background. There are several MMTV-LTRs of different lengths that have been used to make transgenic mice. A shorter promoter variant of 1.2 kb was used in some models such as the MMTV-tTA and MMTV-Cre strains generated in the mid-1990s [24, 25]. This promoter seems to facilitate a more widespread expression of transgenes in tissues other than the mammary and salivary glands, but the activation of the LTR appears to be less affected by the genetic background. This could be viewed as an advantage to assess the function of genes in other organs as well as in different genetic backgrounds. A more frequently used MMTV-LTR construct to make transgenic mice is based on the original MMTV-SV40-BSSK vector from the laboratory of Philip Leder, which is now available under a Uniform Biological Material Transfer Agreement through Addgene (plasmid #1824). This construct is the backbone of many popular transgenic strains that are still being used today in breast cancer research such as the aforementioned lines expressing c-Myc, Ha-ras, PyMT, and ErbB2. The longer LTR (2.4 kb) of the MMTV-SV40-BSSK vector carries a portion of the v-Ha-ras leader sequence and is suggested to mediate an enhanced expression of transgenes in the mammary gland [23]. For example, the expression of Cre recombinase and the tetracycline-controlled transactivator (tTA) under the control of the MMTV-SV40-BSSK vector was more confined to the mammary and salivary glands compared to the MMTV-Cre and MMTV-tTA strains generated earlier using the shorter LTR [26, 27]. However, the expression of MMTV-driven transgenes from the longer LTR seems to vary in different strains of mice. This fact leads into the second important issue to consider when using MMTV-based genetic strains in breast cancer research: the genetic strain background. It has been known for some time that endogenous, replication-competent MMTV is present in few strains of mice, while others that do not carry the virus might be susceptible to infection. Also, mice that carry the virus are not at equal risk for infection. The rate and latency of mammary tumor formation as well as the ability of MMTV-induced cancer cells to metastasize varies greatly among strains [28]. Notably, C57BL/6, which is a preferred genetic background for gene-targeting experiments, confers resistance to MMTV infection and MMTV-induced mammary tumors. This phenomenon was also observed in MMTV-driven transgenic lines that were generated in an FVB background and subsequently crossed with C57BL/6 mice. For example, Rowse and colleagues [29] reported that a single intercross between MMTV-neu transgenics (FVB) with wild-type C57BL/6 mice increased the latency of mammary tumor formation from an average of 7–12 months (FVB) to greater than 18 months (F1: FVBxC57BL/6). Similarly, female mice expressing the polyomavirus middle T antigen (PyMT) exhibit a longer tumor latency and reduced metastatic dissemination of cancer cells when the MMTV-PyMT transgene was carried over from an FVB into a C57BL/6 background [30]. In conclusion, different genetic backgrounds can cause dramatic variations in cancer phenotypes that, when poorly controlled, can lead to an incorrect assessment of the significance of genes in mammary carcinogenesis. For example, the paradigm proposed by Yu et al. [31] that deficiency in Cyclin D1 protects against ErbB2-induced breast cancer has recently been challenged [32]. Following a transfer of the Cyclin D1 null allele from C57BL/6 into FVB and generating intercrosses with MMTV-neu mice, it became apparent that, despite a delay in tumor onset, all Cyclin D1 knockout females overexpressing ErbB2 developed mammary tumors. Consequently, the specific inhibition of Cyclin D1 cannot be therapeutically relevant to treat ErbB2-positive human breast cancers. On a molecular level, this phenomenon is due to a compensatory upregulation of Cyclin D3 in the absence of Cyclin D1 in mammary tumor cells that arise in MMTV-neu mice maintained in the FVB background [32]. This is an important observation with relevance to the human disease. Most human breast cancer cell lines and primary human breast cancers overexpress Cyclin D3 in addition to Cyclin D1, and a knockdown of one of these D-type Cyclins has been demonstrated to lead to a compensatory upregulation of the remaining Cyclin D1 or D3 [32].
In addition to the regulatory elements of the MMTV, promoters from milk protein genes such as WAP (whey acidic protein) and BLG (β-lactoglobulin) have been successfully utilized to target the expression of oncogenes such as TGF-α, c-Myc [33], and Stat5 [34] specifically to the mammary gland (for a more comprehensive list of transgenic lines, please refer to a review by Hennighausen) [9]. In comparison to the MMTV-LTR, these promoters are under more stringent control of lactogenic hormones, and consequently, their expression is influenced to an even greater extent by the reproductive cycle. Given the fact that milk proteins are being primarily synthesized in luminal epithelial cells during pregnancy and lactation, the expression of transgenes under the control of the WAP and BLG promoter is particularly high in the lobuloalveolar compartment of the gland. A suitable alternative to express oncogenes in the ductal epithelium could be the use of the 5′ flanking sequence of the rat prostatic steroid-binding protein C3(1) gene [35]. The most prominent breast cancer model using this promoter is the C3(1)/SV40 T antigen transgenic line that develops sporadic tumors in the prostate and mammary gland epithelium. On the histopathological level, females give rise to neoplastic lesions that closely resemble human ductal carcinoma in situ (DCIS) prior to progressing into invasive carcinomas [36]. Although the C3(1) promoter seems not to be directly regulated by hormones, estrogen is suggested to play a significant role in tumor onset in C3(1)/SV40 T antigen transgenic mice, possibly due to an increase in cell proliferation [37]. To express transgenes in a hormone-independent manner in the mammary gland, some investigators utilized a promoter of the neu-related lipocalin (NRL) gene [38]. Interestingly, a subset of NRL promoter-driven transgenic females expressing prolactin or TGF-α can develop estrogen receptor-positive mammary tumors [39, 40]. Specifically, NRL-TGF-alpha transgenic females have been reported to give rise to ERα+/PR− tumors and hormone receptor-negative lesions when combined with a heterozygous Trp53 knockout.
In summary, there are several promoters that have been utilized to drive the expression of oncogenes to the mammary epithelium. Each of these regulatory elements has advantages and shortcomings with regard to hormonal regulation, expression in tissues other than the mammary gland, mosaic expression profiles, and differences in transgene activation in epithelial subtypes, as well as variations in transgene expression depending on the genetic strain background. The scientific questions and experimental designs should provide the basic rationale for selecting a particular promoter to generate a genetically modified mouse strain.
2.2. Ligand-Controlled Oncogene Expression in the Mammary Gland
Transgenic mice representing the first generation of GEMs have provided valuable insights into the role of oncogenes for mammary cancer initiation. However, these mouse models are often unsuitable to assess whether an oncogene is equally important for tumor maintenance and the survival of fully transformed cancer cells at primary and metastatic sites. This information is critical for the selection of genes and their encoded proteins as relevant therapeutic targets for the treatment of patients that have been diagnosed with breast cancer. One way to examine the continued importance of transforming oncogenes in progressing cancers is to unitize mouse models that permit a temporally and spatially controlled regulation of transgenes. Among various ligand-inducible transgenic approaches, tetracycline (Tet)-based systems have been utilized successfully for the regulation of transgene expression in various cell types and tissues in vivo, including the mammary gland (Fig. 1). There are two main types of Tet-controlled transactivator systems: (1) the Tet-OFF using a Tet-responsive transactivator protein (tTA) that becomes transcriptionally inactive in the presence of tetracycline or its more potent derivative, doxycycline (Dox), and (2) the Tet-ON system using the reverse transactivator (rtTA) consisting of a mutated tetracycline repressor domain that becomes active in the presence of Tet or Dox [41, 42]. Regardless of the type of Tet-controlled transactivator, the ligand-controlled expression system requires a second transgene with a Tet operon linked to a minimal promoter (TetO) that initiates the transcription of the gene-of-interest. While the tissue specificity as well as the level of expression of the transactivator (tTA or rtTA) are primarily determined by the promoter of choice, the activation of the downstream TetO-responder transgene is controlled by the transactivator in the presentence or absence of the ligand (i.e., Tet or Dox).
Fig. 1.
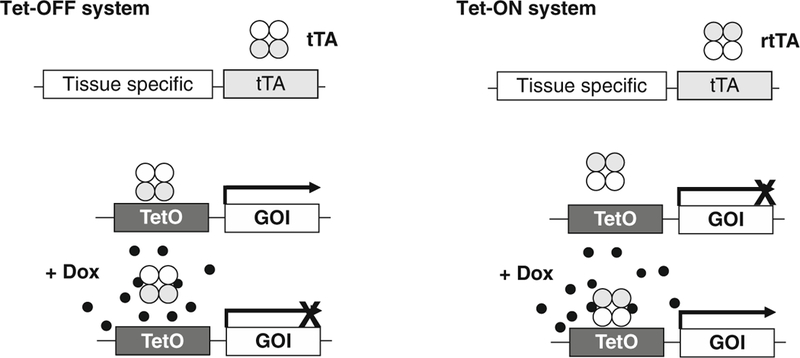
Ligand-controlled expression of oncogenes in the mammary gland using bi-transgenic tetracycline-inducible systems. The Tet-OFF system (left panel) allows for a temporally and spatially controlled expression of a gene-of-interest (GOI) in the absence of the ligand. The tetracycline-controlled transactivator (tTA) is expressed under the regulation of a tissue-specific promoter. The tTA transactivates the expression of the GOI by binding to the Tet-controlled operon in the absence of a ligand. Administration of tetracycline or its more potent derivative doxycycline (Dox) can suppress the expression of the GOI, and withdrawal of the ligand leads to a reactivation of the TetO-driven responder transgene. The Tet-ON system (right panel) is based in the tissue-specific expression of the reverse tetracycline-controlled transactivator (rtTA), which only mediates a transactivation of the TetO promoter-driven GOI in the presence of Dox
Following the pioneering work by Bujard and Gossen [42], Furth and Hennighausen subsequently established the Tet system in genetically engineered mice [43]. They also developed the first MMTV-tTA transgenics with the intent to target the expression of genes to the mammary gland in a ligand-controlled manner [24]. By crossing the MMTV-tTA mice with transgenic animals that carry the simian virus 40 (SV40) T antigen coding sequence under the control of the TetO promoter, these teams of investigators provided the first experimental evidence that early stages of neoplastic transformation can be reversed when the oncogene is silenced [44]. Unfortunately, the original MMTV-tTA strain expresses the transactivator protein in a mosaic fashion in the mammary gland, and these mice exhibit a strong activation of TetO-driven transgenes in other tissues such as the salivary gland and skin. A novel MMTV-tTA transgenic strain has been recently developed using the aforementioned MMTV-SV40-BSSK vector with a longer LTR [27]. These new MMTV-tTA lines show a more defined and enhanced activation of TetO-driven responder genes in the mammary epithelium and salivary gland without noticeable expression in the skin and other tissues. In the absence of Dox, these strains transactivate responder genes in the mammary gland anlagen starting at day 13 of embryonic development and throughout mammogenesis. Studies show that the MMTV-tTA remains active in neoplastic cells that were transformed through overexpression of ErbB2 (MMTV-neu). The observation that the MMTV-tTA is active at the earliest stages of mammary gland differentiation might be evidence for the expression of the transgene in mammary epithelial stem cells. The MMTV-tTA transactivates TetO-driven responder genes in the absence of any ligands (i.e., Tet or Dox), and therefore this strain might be particularly useful for an overexpression of weaker oncogenes that require a constitutive activation over a long period to cause neoplastic transformation in the mammary epithelium.
In comparison to the Tet-OFF system described above, the Tet-ON system utilizes the reverse transactivator (rtTA) and requires the ligand (i.e., Tet or Dox) in order to render the rtTA functional for transactivating a TetO-driven gene-of-interest. The first mammary-specific rtTA lines (MMTV-rtTA) were generated by Gunther and colleagues [45] who have utilized these mice to express oncogenes such as active ErbB2/neu [46], c-Myc [47], and Wnt [48] in a Tet/Dox-inducible manner in the mammary gland. Expression of these potent oncogenes resulted in the development of mammary tumors after a short to medium latency, and a reduction in tumor growth or tumor regression has been observed following oncogene ablation upon Tet/Dox withdrawal. These studies provided clear experimental evidence for the continuous importance of these classical oncogenes for the maintenance and progression of mammary cancer. An alternative mouse model to target an exogenous expression of genes specifically to the mammary gland in a ligand-inducible manner is the WAP-rtTA strain, which expresses the reverse transactivator under the control of the endogenous WAP locus [49]. This genetic strain was used to study the biological significance of a sustained activation of the signal transducer and activator of transcription 5 (STAT5), which leads to extended cell survival and impaired remodeling of the postlactational mammary gland [50, 51].
2.3. Conventional Knockout Models and Mammary Gland Transplantation
The third generation of GEMs is based on the use of homologous recombination to establish conventional knockout mouse models that are deficient in tumor suppressor genes, either by generating a complete null allele (knockout) or through introduction of point mutations that have been identified in human cancers (knockin). In comparison to many mammary-specific transgenics, the gene-targeted knockout/knockin models typically develop mammary cancers after a prolonged latency and frequently in tissues other than the mammary gland. For example, cancer-associated point mutations in the gene encoding the phosphatase and tensin homolog (PTEN) cause inherited predispositions to cancers such as Cowden syndrome (CS). As a negative regulator of the growth-promoting PI3K/Akt signaling pathway, PTEN functions as a classical tumor suppressor. CS patients are frequently diagnosed with breast cancer among other malignancies, and introducing synonymous PTEN mutations into the endogenous locus in mice causes mammary cancers and lymphomas after a long latency [50–52]. A more commonly known cancer model is the conventional knockout of Trp53 [11], which gives rise to a spectrum of neoplasms including mammary tumors. To restrict tumor development to the mammary gland, Jerry and coworkers utilized the conventional Trp53 knockout to transplant the mutant mammary epithelium into the cleared mammary fat pads of wild-type recipient mice [53]. The mammary gland transplantation method is generally applicable for gene-targeted mice that develop mammary gland anlagen either post embryonic day 12 [54]. Hence, in addition to confining the genesis of neoplasms to the mammary gland, this methodology can also be applied to rescue premature lethality of certain knockout models as described in the next section.
Similar to transgenic mice, the genetic background can have a significant effect on the tumor spectrum and latency in knockout mice. For example, C57BL/6 or 129/Sv females that are deficient in Trp53 develop sarcomas and lymphomas but rarely mammary tumors. Backcrossing the Trp53 null allele onto the BALB/c genetic background predisposes Trp53 heterozygous knockout females to develop mammary cancer [55]. Another, more recent example for the effect of the genetic background on mammary tumorigenesis is the Stat1 knockout mouse model. Deficiency in Stat1 in BALB/c females causes mammary tumors in more than 50 % of the mice within 1 year [56, 57]. A unique characteristic of mammary tumors arising in the Stat1-deficient mice is the frequent occurrence of ERα+/PR+ neoplasms that are generally rare in genetically engineered mouse models.
2.4. Mammary-Specific Conditional Knockout Mouse Models
As described in the previous section, a relatively simple methodology to generate a mammary gland-specific knockout is to transplant epithelial cells from a gene-targeted mouse into the cleared mammary fat pad of a wild-type recipient. Approximately 30 % of conventional knockout mice, however, exhibit embryonic or early postnatal lethality and many of those do not survive long enough to retrieve an embryonic mammary gland anlage for transplantation. For example, deficiency in either the breast cancer susceptibility genes 1 or 2, early onset (BRCA1, BRCA2), causes embryonic lethality between day 7.5 and day 9.5 of gestation [12, 58–61]. Similarly, embryos lacking the aforementioned tumor suppressor PTEN die around E9.5 [13, 62]. To bypass early embryonic lethality associated with a conventional knockout and to study gene function in somatic tissues, conditional gene-targeting techniques using the Cre/lox and Flp/frt recombination systems have been developed. In particular, the introduction of the Cre/lox system was a significant technical innovation that subsequently revolutionized the development of entirely new breast cancer models. This technology is based on the combination of two types of genetically engineered mouse strains: (1) a transgenic line expressing the site-specific Cre recombinase under a tissue-specific promoter and (2) a gene-targeted “floxed” mouse, in which two loxP sequences (i.e., Cre recognition sites) are inserted flanking a gene-of-interest using homologous recombination [63, 64]. There are a number of transgenic lines available today that express Cre in mammary epithelial cells. The first two lines, WAP-Cre and MMTV-Cre, were developed in the mid-1990s in the laboratory of Lothar Hennighausen at the National Institutes of Health (NIH) [17, 25]. These transgenic lines have been available since 1999 through various distributors such as The Jackson Laboratory (stock #003551–003553) and the repository of the National Cancer Institute (NCI, Frederick, MD), and consequently they have been widely used to generate mammary-specific knockout models. With regard to mammary tumorigenesis, MMTV-Cre and WAP-Cre mice have been employed to generate the first hereditary breast cancer model by deleting the Brca1 gene in a mammary gland-specific manner [65]. Subsequently, they were utilized to assess the tumor-suppressive role of BRCA2 and PTEN in adult animals [66–68].
Additional MMTV-Cre lines have been generated by other teams using different MMTV-LTRs such as the aforementioned MMTV-SV40-BSSK vector in an effort to better confine the expression of the recombinase to the mammary gland as well as co-express oncogenes and Cre from a bicistronic construct [26, 69]. The expression profile of Cre under the control of the ovine β-lactoglobulin gene (BLG-Cre) in another strain is quite similar to the transgenic WAP-Cre line, mediating the highest levels of gene deletion during pregnancy and lactation [70]. A second WAP-Cre strain was generated by inserting the coding sequence of the recombinase into the endogenous WAP locus [71]. Located close to the centromeric region of chromosome 11, it is apparent that WAP is genetically linked to a number of important genes for mammary gland development and tumorigenesis such as BRCA1, Trp53, STAT5a/b, and STAT3. The simple fact of gene linkage exemplifies the need for the availability of several Cre transgenics that are not only diverse in their expression profile, but also located on different chromosomes to better facilitate the generation of somatic knockout models. The list of Cre transgenics would not be complete without mentioning additional strains that have been used to delete tumor suppressor genes in the mammary gland. Specifically, K14-Cre mice have been applied to generate a mammary tumor model by deleting BRCA2 in combination with Trp53. In addition to several other tissues including the skin, tongue, esophagus, forestomach, and thymus, the human keratin-14 promoter driving Cre was reported to be active in 5–35 % of both luminal epithelial and myoepithelial cells [72]. Similar to K14, keratins 5 and 6 are expressed predominantly in basal mammary epithelial cells, and attempts to generate BRCA1-deficient mammary tumor models in the presence of wild-type Trp53 using the K14-Cre, K5-Cre, and K6-Cre transgenic lines failed [73, 74]. Given the fact that the majority of MMTV-Cre BRCA1fl/fl females develop mammary tumors after a prolonged latency [65, 75], it is evident that an efficient deletion of the tumor suppressor in both basal and luminal epithelial cells facilitates carcinogenesis without the need for genetically manipulating the Trp53 gene. A similar phenomenon was observed in BRCA2 conditional knockouts. While deletion of this tumor suppressor in the presence of wild-type Trp53 using K14-Cre did not result in mammary tumors [72], approximately half of all WAP-Cre-based BRCA2 knockout mice developed mammary cancer after an average latency of 14 months [71]. Collectively, all these observations may support the notion that luminal progenitors are the candidate target population for BRCA1-associated, basal-type breast cancers [76]. This is likely also the case for BRCA2-associated cancers that are largely luminal type in humans based on a higher proportion of steroid receptor-positive and fewer K5/6-positive neoplasms [77].
The main objective to utilize various Cre transgenic lines is to assess the role of tumor susceptibility genes in specific epithelial sub-types. Another major strength of the Cre/lox technology is its broad applicability for cell lineage-tracing experiments using phenotypically neutral reporter genes aimed at identifying the cells-of-origin for neoplastic transformation in various tumor models, including conventional transgenics and knockouts. For example, WAP-Cre transgenic females have been used in combination with various reporter strains expressing LacZ or GFP to determine whether all functionally differentiated alveolar cells following a full-term gestation cycle undergo cell death during the postlactational involution period. Surprisingly, many cells that had expressed the WAP-Cre transgene were still present at the terminal ends of the mammary ductal tree in nonpregnant, multiparous females [78, 79]. Upon transplantation into the epithelial-free mammary fat pads, these parity-induced mammary epithelial cells (PI-MECs) exhibited characteristics of progenitors and contributed to the development of primary and secondary ducts as well as secretory alveoli. Using cell lineage tracing, it was determined that this particular epithelial subtype is a prime target for MMTV-neu-induced neoplastic transformation in single and multiparous female mice [80].
The basic methodology to genetically label cells using the Cre/lox system can also be applied to drive a constitutive expression of oncogenes in particular epithelial subtypes. As described earlier in this book chapter, many mammary-specific promoters are under the control of lactogenic hormones, and their activation varies greatly during different stages of the reproductive cycle. To render the expression of an oncogene independent of lactogenic hormones, a mammary-specific Cre transgenic line can be used to constitutively activate a silent oncogene by removing a transcriptional STOP cassette that is flanked by loxP sites and located between the promoter of a housekeeping gene and an oncogene. This strategy has been used recently to express mutant K-ras in the mammary gland under the control of the endogenous translation elongation factor (Eef1a1) [81]. Interestingly, the WAP-Cre-mediated overexpression of oncogenic K-ras primarily in luminal cells led to the development of mammary tumors with basal-type characteristics. This may suggest that certain histopathological features of a mammary cancer subtype can be greatly influenced by the expression of a particular oncogene. In contrast to this paradigm, activating a constitutive expression of the same oncogene in different epithelial subtypes of ducts and alveoli can lead to heterogeneous mammary neoplasms with distinctly different histopathological characteristic. For example, MMTV-neu transgenic mice are known to develop luminal-type mammary tumors that appear to be homogeneous in their morphology. It has been recently reported, however, that the MMTV-Cre-mediated activation of mutant ErbB2/neu under the control of the endogenous ErbB2 locus resulted in a significant subset of basal-type mammary cancers [82]. Hence, targeting the same oncogene to particular epithelial subtypes could give rise to lesions that are far more heterogeneous and possess distinctly different histopathological features.
In addition to genetically labeling particular epithelial subtypes and constitutively activating oncogenes, the Cre/lox technology and conditional knockout models can be used to discriminate the role of any gene-of-interest during mammary tumor initiation versus cancer progression. For instance, the conditional deletion of the Janus kinase 2 (Jak2) or Stat5a/b in MMTV-neu and NRL-PRL transgenics as well as mutant PTENG129E mice prior to tumor formation demonstrated that the Jak2/Stat5 signaling cascade plays an important role in the initiation of mammary tumorigenesis [51, 83, 84]. In sharp contrast, deleting Jak2 in fully neoplastic cells had no significant impact on the proliferation and survival of these cells in vitro and in vivo. It is therefore evident that molecular targets that are superior for breast cancer prevention may not necessarily be equally important as therapeutic targets to treat breast cancers. With regard to modeling breast cancer in mice, these observations suggest that a mouse model with a gene deletion prior to neoplastic transformation might be a poor predictor for assessing a continuous importance of this gene in transformed cancer cells.
3. Morphological and Molecular Characteristics of GEM-Derived Mammary Tumors That Define Human Breast Cancer Subtypes
Since the development of the first animal models for cancer research, a decade-old central question is still under debate, whether these models faithfully reflect particular human malignancies. Recent advances in genetic engineering allowed the development of mouse models that more closely resemble human breast cancers on the genetic level. Some models carry the precise mutations that have been identified in human breast cancer patients (e.g., PTENG129E). A primary objective of the first NIH Breast Cancer Think Tank and Annapolis Pathology Panel (commonly referred to as “the Annapolis Meeting”) in 1999 was to assess whether genetically engineered mouse mammary tumor models recapitulate histopathological features of human breast cancers. Experts in human breast cancer and comparative pathology assessed the morphological characteristics of 39 murine breast cancer models [85]. In their final report, the panel stated that tumors from GEMs did not completely mimic the common types of breast cancer upon histological analysis, but many similarities between murine and human tumors were identified. It should be noted that the vast majority of the tumor models that were studied by the panel were the first generation of transgenic mice and chemically induced cancer models. More importantly, it should be recognized that mammary glands in mice differ significantly in their composition of the stromal compartment from the human breast [1]. While the normal mouse mammary gland stroma largely consists of adipocytes, the human breast epithelium is surrounded by a thicker sheet of fibroblasts. It has been shown recently that genetic modifications in mouse mammary fibroblasts resulted in a subset of ErbB2-induced mammary cancers with histopathological characteristics that resemble more closely the human breast cancer subtype [86]. In conclusion, the histopathology of mammary neoplasms in GEMs is determined equally by the genetic alterations in cancer cells as well as the tumor microenvironment.
As part of the phenotypic characterization of GEMs that develop mammary neoplasms, individual teams of investigators have used common cellular markers to empirically assess the morphological features. Many of the descriptive attributes of a neoplasm are based primarily on the expression and intracellular location of steroid receptors (e.g., ERα and PR) and receptor tyrosine kinases, in particular ErbB2, as well as certain cytokeratins that are expressed in luminal (CK 8, 18, 19) and basal (CK 5, 6, 14) epithelial cells. Other markers such as Ki-67, cell adhesion proteins such as cadherins, and the intermediate filament vimentin are being used to portray the proliferative state of a neoplasm or cellular processes such as epithelial-mesenchymal transition (EMT) that have been associated with migratory and invasive properties of a tumor. Table 1 lists a number of selected breast cancer models that can be generally classified into luminal-like, basal-like, and ErbB2-associated mammary tumors based on ERa positivity, expression of cytokeratins, as well as over-expression of ErbB2. For this compilation, we have included several newly generated tumor models that develop ERα-positive tumors as well as models that express potent oncogenes in a constitutive manner under the endogenous promoter (mutant ErbB2 knockin) or regulatory elements of housekeeping genes (EF1-K-ras). Some of these models have yet to be further examined on the molecular level using gene expression profiling.
Table 1.
Classification of models that resemble particular human breast cancer subtypes
| Subtypes | GEMs | Strain | Transgene | Features | Ref. |
|---|---|---|---|---|---|
| Luminal-like | Stat1−/− | BALB/c | Stat1 homozygous null | 10–60 % ERα+ depending on MIN grades |
[57] |
| Stat1−/− | C57BL/6 | Stat1 homozygous null | >90 % ERα+/PR+ | [56] | |
|
NRL-PRL lox-stop-lox-Pik3ca (H1047R); MMTV-Cre MMTV-myrAkt1, DMBA treated |
FVB | Rat PRL overexpression Pik3ca knock in R26, CAG, or endogenous Pik3 locus Overexpression of constitutive active Akt1 |
70 % ERα+, ErbB2+ ERα+, CK+/CK14+ mixed population ERα+/ER- mixed, CK+/CK14+ mixed population |
[39, 84] [95–98] [99] |
|
| MMTV-cyclin D1 MMTV-cyclin D1 (T286A) | FVB | Overexpression of cyclin D1 or constitutive active cyclin D1 |
40–50 % ERα+ | [100] | |
| ErbB2 associated | MMTV-nen | FVB | Overexpression of wild-type ErbB2 |
ERα−, ErbB2+ | [22] |
| MMTV-ErbB2 | FVB | Overexpression of constitutively active ErbB2/neu | ERα−, ErbB2+ | [23] | |
| ErbB2KI | FVB | Knock in of active ErbB2 into the endogenous ErbB2 locus | ERα−, ErbB2+ | [26] | |
| Basal-like |
BRCA1fl/fl, MMTV-Cre, p53+/− or p53−/− EF1-lox-stop-lox-K-ras G12D, WAP-Cre |
C56BL/6 | Conditional deletion of BRCA1 in the mammary gland Constitutive overexpression of mutant K-ras |
Triple negative, CK5/6 positive ERα−, CK14, and CK8 mixed |
[75, 65, 101] [81] |
MIN mammary intraepithelial neoplasia
In addition to a morphological description of tumors, more recent studies have focused on the gene expression profiles of mammary neoplasms that originate in various GEMs. In the first comprehensive study, Herschkowitz and colleagues [87] analyzed 13 different mouse models using DNA microarrays and compared the data they obtained to gene expression profiles in human breast cancer subtypes. The results showed that many of the key attributes of human breast cancer subtypes were conserved among the mouse models. Nonetheless, not a single mouse model recapitulated the entire spectrum of gene expression features that are characteristic for a particular human breast cancer subtype. Instead, some distinct murine tumor classes share phenotypes with multiple human subtypes [88]. For example, the gene expression signature of the c-Myc oncogene was a defining feature in both the luminal B and the basal-like category. Interestingly, the pivotal analysis of the gene expression profiles of mouse tumors and their comparison to human malignancies revealed the existence of a potentially novel human breast cancer subtype designated “claudin low.” In a subsequent report by Pfefferle et al. [88], the team of investigators expanded the genomic analysis, and they compared the transcriptomic profiles of 27 murine models of mammary carcinoma and normal mammary tissues to human breast cancer subtypes [88]. The new study also included a few ERα-positive tumors from Stat1 knockout mice and the Pik3ca-H1047R cancer model. Besides the previously discovered similarity between claudin-low subtype in humans and mice, human basal-like breast cancers seem to be recapitulated by three distinct murine tumor classes with gene expression profiles that are associated with the loss of Trp53 or overexpression of c-Myc as well as the SV40 large T antigen. The study also revealed a number of surprising observations such as the finding that the WAP-Cre-Etv6 model mimicked more closely the ErbB2-enriched subtype, which is a group of human breast cancers without a definitive murine counterpart in previous comparative studies. If the author’s assessment is correct, it is likely that the ETS family transcription factor Etv6 controls a transcriptional network that is very similar to human cancers that overexpress ErbB2.
4. Modeling Breast Cancer Metastasis
Significant improvements in the early detection of breast cancer and the availability of novel therapies have resulted in a steady decline in mortality and a prolonged survival rate. Nonetheless, it remains a fact that invasive breast cancer is still a lethal disease (see statistics in Subheading 1 of this chapter). Many patients succumb to complication associated with metastatic disease, even after what appeared to be a complete clinical remission after therapy. Metastasis is a complex process that describes the spread of cancer cells to distant sites. Thus far, the vast majority of studies using GEMs as breast cancer models have primarily focused on the molecular drivers for the initiation of primary mammary neoplasms. Significantly fewer investigations have been carried out in GEMs to dissect the underlying molecular mechanisms that govern cancer cell invasion and metastatic dissemination. One reason is certainly the fact that many mammary tumors in genetically engineered breast cancer models are not highly metastatic or never disseminate. Another possible reason is the insufficient analysis of circulating cancer cells and a thorough examination of the presence of cancer cells in organ systems other than the lung (i.e., a primary site of metastasis in murine cancer models). Finally, regulatory guidelines and laws, intended for a humane treatment of experimental animals, often restrict an examination of larger tumors, the performance of serial surgeries to remove primary tumors, and the study of late-stage, metastatic disease on mice with clear signs of cachexia.
A list of available models to study breast cancer metastasis and their use to examine the mechanisms contributing to the dissemination of cancer cells was given in a comprehensive review by Fantozzi and Christofori [89]. Upon examination of the literature, it is evident that the majority of in vivo studies related to breast cancer metastasis are still being performed in selected cell transplantation models. For example, 4T1 cells (ATCC CRL-2539) are a preferred syngeneic transplantation model in BALB/c mice. These cells, originally derived as a variant of 410.4 cells from a spontaneous mammary tumor, have the ability to spread to the lung, bone, liver, and brain [90]. Other frequently utilized experimental models to study metastasis are xenografts using MDA-MB-231 cells (ATCC HTB-26). Depending on the site of transplantation (orthotopic, ectopic, intravenous, or intracardiac), these cells are capable of forming metastases in the bone, lung, liver, brain, and selected other sites. Among the various genetically engineered strains, the aforementioned MMTV-PyMT transgenic line [21] is, by far, the most frequently used experimental model to study mammary tumor progression and colonization of metastatic cells in the lung. Virtually all females develop mammary tumors after a short latency independent of the reproductive state, and the majority of mice exhibit secondary, pulmonary tumor masses within a few months. When propagated in the FVB genetic background, the kinetics of tumorigenesis and metastasis mediated by the MMTV-PyMT transgene is highly reproducible, but it should be emphasized here again that the metastatic phenotype and tumor latency are greatly influenced by the strain background [30]. Transgenic lines expressing constitutively active or wild-type ErbB2 (MMTV-neu) develop metastatic mammary tumors after a latency period of 4–8 months, and the primary sites for the formation of metastases in these strains are the lymph nodes and the lung. As reviewed by Fantozzi and Christofori [89], there are a number of other transgenic strains in which pulmonary metastases have been reported. These include the classic MMTV-wnt1 model, C3(1)-SV40Tag females, as well as transgenic lines expressing Notch4, Ha-ras, and the hepatocyte growth factor (HGF) under the control of the WAP promoter.
Transgenic models have been used to identify cell-intrinsic mechanisms for metastasis as well as to study the role of the micro-environment on the dissemination of mammary tumor cells. For example, it has been demonstrated in the MMTV-PyMT model that the colony-stimulating factor 1 (CSF-1) plays a critical role as a chemoattractant for macrophages that support tumor cell invasion and metastasis [91, 92]. Using conditional knockout mice that are deficient in signal transducer and activator of transcription 3 (STAT3), it has been demonstrated that active STAT3 controls transcriptional networks that mediate important cell-intrinsic mechanisms for ErbB2-induced mammary cancer cell dissemination [93]. A more recent study showed that the STAT3-responsive gene BCL3 (B-cell lymphoma-3-encoded protein) seems to have a specific effect on tumor cell motility and migration but not the growth of a primary tumor [94].
5. Conclusions
Breast cancer is heterogeneous on the histopathological and genetic level as well as response to therapy. Based solely on gene expression profiling, this malignancy can be classified into at least five subtypes. There are numerous breast cancer cell lines and in vivo models available today to study molecular events that govern the initiation and progression of breast cancer. Models accurately reflect only certain aspects of a complex disease, and therefore there is no one-size-fits-all approach when it comes to carefully selecting a particular cell line or genetic strain for experimental work. This chapter was intended to provide a comprehensive introduction into the main types of genetically engineered mice (GEMs) that are being used today to study molecular and cellular events that are biologically relevant for mammary carcinogenesis. Although the majority of well-characterized GEMs reliably develop mammary lesions at defined latencies, it cannot be emphasized enough that the biology of mammary tumorigenesis in particular transgenic lines or knockout mice is greatly dependent on the genetic strain background. This fact is often overlooked in experiments where a tumor-initiating transgene is crossed with conventional or conditional knockout mice. Various examples for the effects of the genetic background on tumor onset and occurrence of metastatic lesions in the widely used MMTV-neu and MMTV-PyMT transgenic lines have been discussed in this chapter. Moreover, it should always be considered that developmental abnormalities in response to modifying a gene-of-interest may have an indirect effect on cancer initiation. Certain oncogenes under mammary-targeted promoters are known to primarily transform particular epithelial cell subtypes (e.g., alveolar progenitors in MMTV-neu females). However, deficiency in a certain gene might cause impaired cellular differentiation that may lead to the absence of the target cell for transformation. It is also a fact that the absence of mammary tumorigenesis in a well-characterized tumor model that was crossed with a knockout strain to assess the importance of a gene-of-interest (GOI) for breast cancer is often a poor predictor for the potential role of this gene in primary or metastatic cancer cells. In order to more accurately model a therapeutic approach using genetic tools, it is essential to conditionally modify the expression of a GOI after tumor onset or even in metastatic cells. Thus far, the majority of genetic experiments have focused almost exclusively on the role of individual genes in cancer initiation, and fewer studies using GEMs have assessed the molecular events that drive metastatic dissemination of cancer cells. An emerging field of cancer research in genetically modified in vivo model systems is cancer cell dormancy. Not all cancer cells respond equally to radiation or cytotoxic therapy, and recent studies in GEMs have shown that a few cells can survive the ablation of a transforming oncogene leading to disease recurrence. Finally, new genetic tools for the conditional expression or deletion of genes in the mammary epithelium can be employed to better define the cellular origin of certain breast cancer subtypes. These types of experiments will also reveal whether expression of a particular oncogene can cause epithelial subtype transdifferentiation, suggesting that the properties of a cell-of-origin might be very different from the resulting cancer cells (luminal-basal or basal-luminal differentiation).
References
- 1.Wagner KU (2004) Models of breast cancer: quo vadis, animal modeling? Breast Cancer Res 6(1):31–38. doi: 10.1186/bcr723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamen-schikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D (2000) Molecular portraits of human breast tumours. Nature 406:747–752. doi: 10.1038/35021093 [DOI] [PubMed] [Google Scholar]
- 3.Lin WC, Rajbhandari N, Wagner KU (2014) Cancer cell dormancy in novel mouse models for reversible pancreatic cancer: a lingering challenge in the development of targeted therapies. Cancer Res 74(8):2138–2143. doi: 10.1158/0008-5472.CAN-13-3437, 0008–5472.CAN-13–3437 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Talmadge JE, Singh RK, Fidler IJ, Raz A (2007) Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol 170(3):793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Utama FE, LeBaron MJ, Neilson LM, Sultan AS, Parlow AF, Wagner KU, Rui H (2006) Human prolactin receptors are insensitive to mouse prolactin: implications for xenotransplant modeling of human breast cancer in mice. J Endocrinol 188(3):589–601. doi: 10.1677/joe.1.06560, 188/3/589 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Ueda O, Tateishi H, Higuchi Y, Fujii E, Kato A, Kawase Y, Wada NA, Tachibe T, Kakefuda M, Goto C, Kawaharada M, Shimaoka S, Hattori K, Jishage K (2013) Novel genetically-humanized mouse model established to evaluate efficacy of therapeutic agents to human interleukin-6 receptor. Sci Rep 3:1196. doi: 10.1038/srep01196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, Saito Y, Marches F, Halene S, Palucka AK, Manz MG, Flavell RA (2014) Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol 32(4):364–372. doi: 10.1038/nbt.2858, nbt.2858 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart TA, Pattengale PK, Leder P (1984) Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell 38(3):627–637 [DOI] [PubMed] [Google Scholar]
- 9.Hennighausen L (2000) Mouse models for breast cancer. Breast Cancer Res 2(1):2–7. doi: 10.1186/bcr20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Dyke T, Jacks T (2002) Cancer modeling in the modern era: progress and challenges. Cell 108(2):135–144 [DOI] [PubMed] [Google Scholar]
- 11.Donehower LA, Harvey M, Vogel H, McArthur MJ, Montgomery CA Jr, Park SH, Thompson T, Ford RJ, Bradley A (1995) Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinog 14(1):16–22 [DOI] [PubMed] [Google Scholar]
- 12.Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A (1997) Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev 11(10):1226–1241 [DOI] [PubMed] [Google Scholar]
- 13.Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, Fukumoto M, Mak TW (1998) High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol 8(21):1169–1178 [DOI] [PubMed] [Google Scholar]
- 14.Matulka LA, Wagner KU (2005) Models of breast cancer. Drug Discov Today Dis Models 2:1–6 [Google Scholar]
- 15.Bittner JJ (1936) Some possible effects of nursing on the mammary gland tumor incidence in mice. Science 84(2172):162. doi: 10.1126/science.84.2172.162, 84/2172/162 [pii] [DOI] [PubMed] [Google Scholar]
- 16.Ross SR (2010) Mouse mammary tumor virus molecular biology and oncogenesis. Viruses 2(9):2000–2012. doi: 10.3390/v2092000, viruses-02–02000 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner KU, McAllister K, Ward T, Davis B, Wiseman R, Hennighausen L (2001) Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res 10(6):545–553 [DOI] [PubMed] [Google Scholar]
- 18.Kordon EC, Smith GH (1998) An entire functional mammary gland may comprise the progeny from a single cell. Development 125(10):1921–1930 [DOI] [PubMed] [Google Scholar]
- 19.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P (1987) Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49(4):465–475 [DOI] [PubMed] [Google Scholar]
- 20.Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE (1988) Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 55(4):619–625 [DOI] [PubMed] [Google Scholar]
- 21.Guy CT, Cardiff RD, Muller WJ (1992) Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12(3):954–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ (1992) Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci US A 89(22):10578–10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P (1988) Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 54(1):105–115. doi: 10.1016/0092-8674(88)90184-5 [DOI] [PubMed] [Google Scholar]
- 24.Hennighausen L, Wall RJ, Tillmann U, Li M, Furth PA (1995) Conditional gene expression in secretory tissues and skin of transgenic mice using the MMTV-LTR and the tetracycline responsive system. J Cell Biochem 59(4):463–472 [DOI] [PubMed] [Google Scholar]
- 25.Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L (1997) Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res 25(21):4323–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ (2000) Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci U S A 97(7):3444–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakamoto K, Schmidt JW, Wagner KU (2012) Generation of a novel MMTV-tTA transgenic mouse strain for the targeted expression of genes in the embryonic and postnatal mammary gland. PLoS One 7(8):e43778. doi: 10.1371/journal.pone.0043778, PONE-D-12–16928 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Medina D (2000) Mouse models for mammary cancer In: Ip M, Asch B (eds) Methods in mammary gland biology and breast cancer research. Springer, New York, pp 3–17 [Google Scholar]
- 29.Rowse GJ, Ritland SR, Gendler SJ (1998) Genetic modulation of neu proto-oncogene-induced mammary tumorigenesis. Cancer Res 58(12):2675–2679 [PubMed] [Google Scholar]
- 30.Davie S, Maglione J, Manner C, Young D, Cardiff R, MacLeod C, Ellies L (2007) Effects of FVB/NJ and C57Bl/6J strain backgrounds on mammary tumor phenotype in inducible nitric oxide synthase deficient mice. Transgenic Res 16(2):193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu Q, Geng Y, Sicinski P (2001) Specific protection against breast cancers by cyclin D1 ablation. Nature 411(6841):1017–1021. doi: 10.1038/35082500 [DOI] [PubMed] [Google Scholar]
- 32.Zhang Q, Sakamoto K, Liu C, Triplett AA, Lin WC, Rui H, Wagner KU (2011) Cyclin D3 compensates for the loss of cyclin D1 during ErbB2-induced mammary tumor initiation and progression. Cancer Res 71(24):7513–7524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandgren EP, Schroeder JA, Qui TH, Palmiter RD, Brinster RL, Lee DC (1995) Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res 55(17):3915–3927 [PubMed] [Google Scholar]
- 34.Eilon T, Groner B, Barash I (2007) Tumors caused by overexpression and forced activation of Stat5 in mammary epithelial cells of transgenic mice are parity-dependent and developed in aged, postestropausal females. Int J Cancer 121(9):1892–1902 [DOI] [PubMed] [Google Scholar]
- 35.Maroulakou IG, Anver M, Garrett L, Green JE (1994) Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci USA 91(23):11236–11240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green JE, Shibata MA, Yoshidome K, Liu ML, Jorcyk C, Anver MR, Wigginton J, Wiltrout R, Shibata E, Kaczmarczyk S, Wang W, Liu ZY, Calvo A, Couldrey C (2000) The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene 19(8):1020–1027. doi: 10.1038/sj.onc.1203280 [DOI] [PubMed] [Google Scholar]
- 37.Yoshidome K, Shibata MA, Couldrey C, Korach KS, Green JE (2000) Estrogen promotes mammary tumor development in C3(1)/SV40 large T-antigen transgenic mice: paradoxical loss of estrogen receptoralpha expression during tumor progression. Cancer Res 60(24):6901–6910 [PubMed] [Google Scholar]
- 38.Stoesz SP, Gould MN (1995) Overexpression of neu-related lipocalin (NRL) in neu-initiated but not ras or chemically initiated rat mammary carcinomas. Oncogene 11(11):2233–2241 [PubMed] [Google Scholar]
- 39.Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA (2003) Prolactin induces ERalpha-positive and ERalpha-negative mammary cancer in transgenic mice. Oncogene 22(30):4664–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rose-Hellekant TA, Schroeder MD, Brockman JL, Zhdankin O, Bolstad R, Chen KS, Gould MN, Schuler LA, Sandgren EP (2007) Estrogen receptor-positive mammary tumorigenesis in TGFalpha transgenic mice progresses with progesterone receptor loss. Oncogene 26(36):5238–5246. doi: 10.1038/sj.onc.1210340, 1210340 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H (1995) Transcriptional activation by tetracyclines in mammalian cells. Science 268(5218):1766–1769 [DOI] [PubMed] [Google Scholar]
- 42.Gossen M, Bujard H (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 89(12):5547–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furth PA, St OL, Boger H, Gruss P, Gossen M, Kistner A, Bujard H, Hennighausen L (1994) Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc Natl Acad Sci U S A 91(20):9302–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ewald D, Li M, Efrat S, Auer G, Wall RJ, Furth PA, Hennighausen L (1996) Time-sensitive reversal of hyperplasia in transgenic mice expressing SV40 T antigen. Science 273(5280):1384–1386 [DOI] [PubMed] [Google Scholar]
- 45.Gunther EJ, Belka GK, Wertheim GB, Wang J, Hartman JL, Boxer RB, Chodosh LA (2002) A novel doxycycline-inducible system for the transgenic analysis of mammary gland biology. FASEB J 16(3):283–292 [DOI] [PubMed] [Google Scholar]
- 46.Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD, Innocent N, Cardiff RD, Schnall MD, Chodosh LA (2002) Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2(6):451–461 [DOI] [PubMed] [Google Scholar]
- 47.Boxer RB, Jang JW, Sintasath L, Chodosh LA (2004) Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell 6(6):577–586 [DOI] [PubMed] [Google Scholar]
- 48.Gunther EJ, Moody SE, Belka GK, Hahn KT, Innocent N, Dugan KD, Cardiff RD, Chodosh LA (2003) Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumorigenesis. Genes Dev 17(4):488–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Creamer BA, Triplett AA, Wagner KU (2009) Longitudinal analysis of mammogenesis using a novel tetracycline-inducible mouse model and in vivo imaging. Genesis 47(4):234–245. doi: 10.1002/dvg.20480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Creamer BA, Sakamoto K, Schmidt JW, Triplett AA, Moriggl R, Wagner KU (2010) Stat5 promotes survival of mammary epithelial cells through transcriptional activation of a distinct promoter in Akt1. Mol Cell Biol 30(12):2957–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt JW, Wehde BL, Sakamoto K, Triplett AA, Anderson SM, Tsichlis PN, Leone G, Wagner KU (2014) Stat5 regulates the phosphatidylinositol 3-kinase/Akt1 pathway during mammary gland development and tumorigenesis. Mol Cell Biol 34(7):1363–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, Karikomi M, Naidu S, Rajmohan R, Caserta E, Chen HZ, Rawahneh M, Moffitt J, Stephens JA, Fernandez SA, Weinstein M, Wang D, Sadee W, La Perle K, Stromberg P, Rosol TJ , Eng C, Ostrowski MC, Leone G (2010) Allele-specific tumor spectrum in Pten knockin mice. Proc Natl Acad Sci U S A 107(11):5142–5147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jerry DJ, Kittrell FS, Kuperwasser C, Laucirica R, Dickinson ES, Bonilla PJ, Butel JS, Medina D (2000) A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene 19(8):1052–1058. doi: 10.1038/sj.onc.1203270 [DOI] [PubMed] [Google Scholar]
- 54.Hennighausen L, Robinson GW (2005) Information networks in the mammary gland. Nat Rev Mol Cell Biol 6(9):715–725. doi: 10.1038/nrm1714 [DOI] [PubMed] [Google Scholar]
- 55.Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, Naber SP, Jerry DJ (2000) Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice: a model for Li-Fraumeni syndrome. Am J Pathol 157(6):2151–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan S, Vermi W, Luo J, Lucini L, Rickert C, Fowler A, Lonardi S, Arthur C, Young L, Levy D, Welch M, Cardiff R, Schreiber R (2012) STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res 14(1):R16. doi: 10.1186/bcr3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneckenleithner C, Bago-Horvath Z, Dolznig H, Neugebauer N, Kollmann K, Kolbe T, Decker T, Kerjaschki D, Wagner KU, Muller M, Stoiber D, Sexl V (2011) Putting the brakes on mammary tumorigenesis: loss of STAT1 predisposes to intraepithelial neoplasias. Oncotarget 2(12):1043–1054, 371 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, Firpo E, Hui CC, Roberts J, Rossant J, Mak TW (1996) The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 85(7):1009–1023 [DOI] [PubMed] [Google Scholar]
- 59.Liu CY, Flesken-Nikitin A, Li S, Zeng Y, Lee WH (1996) Inactivation of the mouse Brca1 gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. Genes Dev 10(14):1835–1843 [DOI] [PubMed] [Google Scholar]
- 60.Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A (1997) Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 386(6627):804–810. doi: 10.1038/386804a0 [DOI] [PubMed] [Google Scholar]
- 61.Suzuki A, de la Pompa JL, Hakem R, Elia A, Yoshida R, Mo R, Nishina H, Chuang T, Wakeham A, Itie A, Koo W, Billia P, Ho A, Fukumoto M, Hui CC, Mak TW (1997) Brca2 is required for embryonic cellular proliferation in the mouse. Genes Dev 11(10):1242–1252 [DOI] [PubMed] [Google Scholar]
- 62.Di CA, Pesce B, Cordon-Cardo C, Pandolfi PP (1998) Pten is essential for embryonic development and tumour suppression. Nat Genet 19(4):348–355. doi: 10.1038/1235 [DOI] [PubMed] [Google Scholar]
- 63.Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K (1994) Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science 265(5168):103–106 [DOI] [PubMed] [Google Scholar]
- 64.Kuhn R, Torres RM (2002) Cre/loxP recombination system and gene targeting. Methods Mol Biol 180:175–204. doi: 10.1385/1-59259-178-7:175, 1–59259-178–7-175 [pii] [DOI] [PubMed] [Google Scholar]
- 65.Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX (1999) Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet 22(1):37–43. doi: 10.1038/8743 [DOI] [PubMed] [Google Scholar]
- 66.Backman SA, Ghazarian D, So K, Sanchez O, Wagner KU, Hennighausen L, Suzuki A, Tsao MS, Chapman WB, Stambolic V, Mak TW (2004) Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proc Natl Acad Sci U S A 101(6):1725–1730. doi: 10.1073/pnas.0308217100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheung AMY, Elia A, Tsao MS, Done S, Wagner KU, Hennighausen L, Hakem R, Mak TW (2004) Brca2 deficiency does not impair mammary epithelium development but promotes mammary adenocarcinoma formation in p53+/GêAE mutant mice. Cancer Res 64(6):1959–1965 [DOI] [PubMed] [Google Scholar]
- 68.Li G, Robinson GW, Lesche R, Martinez-Diaz H, Jiang Z, Rozengurt N, Wagner KU, Wu DC, Lane TF, Liu X, Hennighausen L, Wu H (2002) Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development 129(17):4159–4170 [DOI] [PubMed] [Google Scholar]
- 69.UrsiniGÇÉSiegel J, Hardy WR, Zuo D, Lam SH, Sanguin Gendreau V, Cardiff RD, Pawson T, Muller WJ (2008) ShcA signalling is essential for tumour progression in mouse models of human breast cancer. EMBO J 27(6):910–920. doi: 10.1038/emboj.2008.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Selbert S, Bentley DJ, Melton DW, Rannie D, Lourenco P, Watson CJ, Clarke AR (1998) Efficient BLG-Cre mediated gene deletion in the mammary gland. Transgenic Res 7(5):387–396 [DOI] [PubMed] [Google Scholar]
- 71.Ludwig T, Fisher P, Murty V, Efstratiadis A (2001) Development of mammary adenocarcinomas by tissue-specific knockout of Brca2 in mice. Oncogene 20(30):3937–3948. doi: 10.1038/sj.onc.1204512 [DOI] [PubMed] [Google Scholar]
- 72.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A (2001) Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 29(4):418–425. doi: 10.1038/ng747, ng747 [pii] [DOI] [PubMed] [Google Scholar]
- 73.Smart C, Clarke C, Brooks K, Raghavendra A, Brewster B, French J, Hetherington R, Fleming J, Rothnagel J, Wainwright B, Lakhani S, Brown M (2008) Targeted disruption of Brca1 in restricted compartments of the mouse mammary epithelia. Breast Cancer Res Treat 112(2):237–241 [DOI] [PubMed] [Google Scholar]
- 74.Berton TR, Matsumoto T, Page A, Conti CJ, Deng CX, Jorcano JL, Johnson DG (2003) Tumor formation in mice with conditional inactivation of Brca1 in epithelial tissues. Oncogene 22(35):5415–5426. doi: 10.1038/sj.onc.1206825, 1206825 [pii] [DOI] [PubMed] [Google Scholar]
- 75.Triplett AA, Montagna C, Wagner KU (2008) A mammary-specific, long-range deletion on mouse chromosome 11 accelerates Brca1-associated mammary tumorigenesis. Neoplasia 10(12):1325–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, Feleppa F, Huschtscha LI, Thorne HJ, Fox SB, Yan M, French JD, Brown MA, Smyth GK, Visvader JE, Lindeman GJ (2009) Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 15(8):907–913 [DOI] [PubMed] [Google Scholar]
- 77.Honrado E, Benitez J, Palacios J (2006) Histopathology of BRCA1- and BRCA2-associated breast cancer. Crit Rev Oncol Hematol 59(1):27–39. doi: 10.1016/j.critrevonc.2006.01.006 [DOI] [PubMed] [Google Scholar]
- 78.Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH (2002) An adjunct mammary epithelial cell population in parous females: its role in functional adaptation and tissue renewal. Development 129(6):1377–1386 [DOI] [PubMed] [Google Scholar]
- 79.Matulka LA, Triplett AA, Wagner KU (2007) Parity-induced mammary epithelial cells are multipotent and express cell surface markers associated with stem cells. Dev Biol 303(1):29–44 [DOI] [PubMed] [Google Scholar]
- 80.Henry MD, Triplett AA, Oh KB, Smith GH, Wagner KU (2004) Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene 23(41):6980–6985 [DOI] [PubMed] [Google Scholar]
- 81.Klinakis A, Szabolcs M, Chen G, Xuan S, Hibshoosh H, Efstratiadis A (2009) Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc Natl Acad Sci U S A 106(7):2359–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schade B, Lesurf R, Sanguin-Gendreau V, Bui T, Deblois G, O’Toole SA, Millar EK, Zardawi SJ, Lopez-Knowles E, Sutherland RL, Giguere V, Kahn M, Hallett M, Muller WJ (2013) beta-Catenin signaling is a critical event in ErbB2-mediated mammary tumor progression. Cancer Res 73(14):4474–4487. doi: 10.1158/0008-5472.CAN-12-3925 [DOI] [PubMed] [Google Scholar]
- 83.Sakamoto K, Lin WC, Triplett AA, Wagner KU (2009) Targeting janus kinase 2 in Her2/neu-expressing mammary cancer: implications for cancer prevention and therapy. Cancer Res 69(16):6642–6650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sakamoto K, Triplett AA, Schuler LA, Wagner KU (2010) Janus kinase 2 is required for the initiation but not maintenance of prolactin-induced mammary cancer. Oncogene 29(39):5359–5369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, Rehm S, Russo J, Tavassoli FA, Wakefield LM, Ward JM, Green JE (2000) The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene 19(8):968–988 [DOI] [PubMed] [Google Scholar]
- 86.Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, Naidu S, Wei G, Sharma SM, Stephens JA, Fernandez SA, Gurcan MN, Weinstein MB, Barsky SH, Yee L, Rosol TJ, Stromberg PC, Robinson ML, Pepin F, Hallett M, Park M, Ostrowski MC, Leone G (2009) Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 461(7267):1084–1091. doi: 10.1038/nature08486, nature08486 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herschkowitz J, Simin K, Weigman V, Mikaelian I, Usary J, Hu Z, Rasmussen K, Jones L, Assefnia S, Chandrasekharan S, Backlund M, Yin Y, Khramtsov A, Bastein R, Quackenbush J, Glazer R, Brown P, Green J, Kopelovich L, Furth P, Palazzo J, Olopade O, Bernard P, Churchill G, Van Dyke T, Perou C (2007) Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 8(5):R76. doi: 10.1186/gb-2007-8-5-r76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pfefferle A, Herschkowitz J, Usary J, Harrell J, Spike B, Adams J, Torres-Arzayus M, Brown M, Egan S, Wahl G, Rosen J, Perou C (2013) Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol 14(11):R125. doi: 10.1186/gb-2013-14-11-r125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fantozzi A, Christofori G (2006) Mouse models of breast cancer metastasis. Breast Cancer Res 8(4):212. doi: 10.1186/bcr1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aslakson CJ, Miller FR (1992) Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 52(6):1399–1405 [PubMed] [Google Scholar]
- 91.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J (2004) A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 64(19):7022–7029 [DOI] [PubMed] [Google Scholar]
- 92.Lin EY, Nguyen AV, Russell RG, Pollard JW (2001) Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 193(6):727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ranger JJ, Levy DE, Shahalizadeh S, Hallett M, Muller WJ (2009) Identification of a Stat3-dependent transcription regulatory network involved in metastatic progression. Cancer Res 69(17):6823–6830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wakefield A, Soukupova J, Montagne A, Ranger J, French R, Muller WJ, Clarkson RWE (2013) Bcl3 selectively promotes metastasis of ERBB2-driven mammary tumors. Cancer Res 73(2):745–755 [DOI] [PubMed] [Google Scholar]
- 95.Adams JR, Xu K, Liu JC, Agamez NMR, Loch AJ, Wong RG, Wang W, Wright KL, Lane TF, Zacksenhaus E, Egan SE (2011) Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res 71(7):2706–2717 [DOI] [PubMed] [Google Scholar]
- 96.Meyer DS, Brinkhaus H, Müller U, Müller M, Cardiff RD, Bentires-Alj M (2011) Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res 71(13): 4344–4351 [DOI] [PubMed] [Google Scholar]
- 97.Tikoo A, Roh V, Montgomery KG, Ivetac I, Waring P, Pelzer R, Hare L, Shackleton M, Humbert P, Phillips WA (2012) Physiological levels of Pik3ca(H1047R) mutation in the mouse mammary gland results in ductal hyperplasia and formation of ERalpha-positive tumors. PLoS One 7(5):e36924. doi: 10.1371/journal.pone.0036924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yuan W, Stawiski E, Janakiraman V, Chan E, Durinck S, Edgar KA, Kljavin NM, Rivers CS, Gnad F, Roose-Girma M, Haverty PM, Fedorowicz G, Heldens S, Soriano RH, Zhang Z, Wallin JJ, Johnson L, Merchant M, Modrusan Z, Stern HM, Seshagiri S (2013) Conditional activation of Pik3ca(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene 32(3):318–326. doi: 10.1038/onc.2012.53, onc201253 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Blanco-Aparicio C, Perez-Gallego L, Pequeno B, Leal JF, Renner O, Carnero A (2007) Mice expressing myrAKT1 in the mammary gland develop carcinogen-induced ER-positive mammary tumors that mimic human breast cancer. Carcinogenesis 28(3):584–594. doi: 10.1093/carcin/bgl190 [DOI] [PubMed] [Google Scholar]
- 100.Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA (2008) Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene 27(9):1231–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McCarthy A, Savage K, Gabriel A, Naceur C, Reis-Filho JS, Ashworth A (2007) A mouse model of basal-like breast carcinoma with metaplastic elements. J Pathol 211(4): 389–398 [DOI] [PubMed] [Google Scholar]