

Abstract
The use of pyridinium‐activated primary amines as photoactive functional groups for deaminative generation of alkyl radicals under catalyst‐free conditions is described. By taking advantage of the visible light absorptivity of electron donor–acceptor complexes between Katritzky pyridinium salts and either Hantzsch ester or Et3N, photoinduced single‐electron transfer could be initiated in the absence of a photocatalyst. This general reactivity platform has been applied to deaminative alkylation (Giese), allylation, vinylation, alkynylation, thioetherification, and hydrodeamination reactions. The mild conditions are amenable to a diverse range of primary and secondary alkyl pyridiniums and demonstrate broad functional group tolerance.
Keywords: deamination, electron donor–acceptor complexes, Giese reactions, photochemistry, radical reactions
Visible light photochemistry in organic synthesis has witnessed a surge in research activity over the last decade.1 This is largely due to a growing appreciation of the synthetic utility of photoredox catalysts, which, upon photoexcitation, function as single‐electron or energy transfer catalysts to provide access to free‐radical intermediates.2 An alternative strategy, that circumvents the need for catalysis, is direct photoexcitation of a substrate, which has classically been performed using UV light.3 However, recent developments have taken advantage of the visible light absorptivity of specific functional groups that act as photoactive handles to enable photoinduced electron transfer (PET).4 Although direct photoexcitation is possible with a number of different functional groups,5 such reactions more commonly take advantage of electron donor–acceptor (EDA) complexes, whose absorption spectra display a bathochromic shift relative to their constituent parts, thus enabling photoexcitation with visible light.6
These strategies have enabled the development of a broad range of radical transformations that proceed through visible light‐mediated PET under catalyst‐free conditions. However, such reactions are typically limited to the generation of perfluoroalkyl or stabilized alkyl radicals.5, 7, 8 Access to non‐stabilized alkyl radicals under such conditions is considerably more challenging,9, 10 with only a single report by Melchiorre and co‐workers that generates secondary alkyl radicals by direct photoexcitation of 4‐alkyl‐1,4‐dihydropyridine derivatives.11 We sought an alternative functional group that could act as a versatile photoactive handle for catalyst‐free generation of non‐stabilized carbon‐centered radicals. One possibility was Katritzky N‐alkylpyridinium salts 1, which are easily prepared from primary amines 2 by reaction with 2,4,6‐triphenylpyrylium 3, are air and moisture stable, and allow selective deaminative transformations of abundant amino groups (Scheme 1 A).12 While these redox active amines have recently been applied to a number of radical‐mediated transformations, they usually rely on catalysis to promote single‐electron transfer (SET)‐induced deamination.13, 14
Scheme 1.
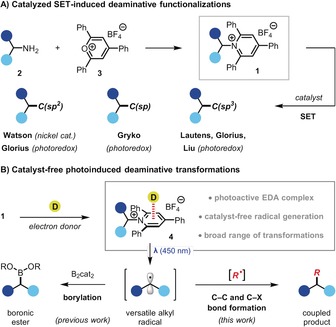
Radical‐mediated transformations of Katritzky pyridinium salts.
We recently reported a catalyst‐free deaminative borylation reaction that proceeds through EDA complex formation between 1 and bis(catecholato)diboron (B2cat2) (Scheme 1 B).15 Subsequent PET and fragmentation provided efficient access to non‐stabilized alkyl radicals that were intercepted by the diboron reagent. We reasoned that the 2,4,6‐triphenylpyridinium moiety in 1 could be complexed with other electron‐donors to generate EDA complex 4,16 thus providing a photoactive handle capable of generating non‐stabilized alkyl radicals for application in a diverse range of C−C or C−X bond forming reactions (Scheme 1 B). Herein, we report that Katritzky pyridinium salts are versatile substrates for photoinduced deaminative functionalizations of primary amines under catalyst‐free conditions.
Our investigations began by studying the use of pyridiniums 1 in Giese reactions with electron‐deficient alkenes (Table 1). Such reactions are well‐developed using photocatalysis, but there are few reports of photoinduced reactions under catalyst‐free conditions.17 Given the overall transformation is reductive, a stoichiometric reductant was required. We selected Hantzsch ester (5) as this would act as a reductant but could also function as an electron‐donor to form the key EDA complex with 1.10c,10d Gratifyingly, irradiation (λ max=450 nm) of a mixture of 4‐aminopiperidine‐derived pyridinium 1 a, Hantzsch ester, and methyl acrylate in DMA yielded the desired Giese adduct 6 in 77 % yield (Table 1). Control experiments confirmed the necessity of light and 5 for successful reaction, and alternative reductants, such as Et3N, gave no desired product.18
Table 1.
Giese reaction substrate scope.[a]
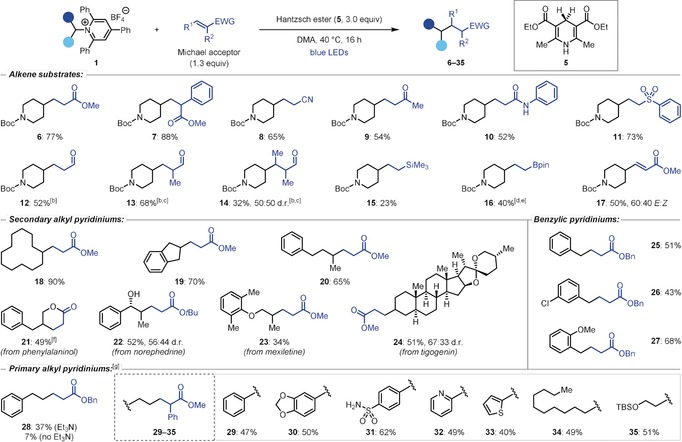
|
[a] General conditions: Pyridinium (0.2 mmol, 1.0 equiv), Michael acceptor (1.3 equiv) and 5 (3.0 equiv) in DMA (0.5 m) at 40 °C for 16 h. Yields are of isolated products after flash column chromatography. [b] Isolated as the alcohol after reduction with NaBH4. [c] Reaction performed at 60 °C for 40 h. [d] Isolated as the alcohol after oxidation with NaBO3. [e] Using 1.8 equiv of vinylboronic acid pinacol ester. [f] Lactonization was promoted by treatment with Amberlyst®. [g] Reactions performed at 60 °C in DMA (0.25 m) with the addition of Et3N (3.0 equiv). DMA=N,N‐dimethylacetamide. TBS=tert‐butyldimethylsilyl.
These optimized conditions were subsequently applied to a broad range of Michael acceptors (Table 1). Giese products from reactions with substituted acrylates (7), acrylonitrile (8), methyl vinyl ketone (9), N‐phenylacrylamide (10), and phenyl vinyl sulfone (11) were formed in good to excellent yields. Aldehydes were tolerated (12–14), although the substituted enals methacrolein (13) and tiglic aldehyde (14) required higher temperatures for successful reaction. Interestingly, vinyl silanes and boronic esters were also suitable substrates, providing products 15 and 16, respectively, albeit in low yield. Finally, methyl propiolate underwent the Giese reaction to give alkene product 17 as a mixture of E and Z isomers.
With respect to the pyridinium salts, a variety of cyclic (18 and 19) and acyclic (20) secondary alkyl substrates reacted efficiently. The Giese product from a γ‐amino alcohol‐derived pyridinium could be cyclized by treatment with acid to generate lactone 21. Alternatively, t‐butyl acrylate could be used in place of methyl acrylate to inhibit lactonization, allowing isolation of norephedrine‐derived alcohol 22. Pharmaceutical and natural product derivatives were also readily accessed, as exemplified by the formation of product 23, from the anti‐arrhythmic drug mexiletine, and 24, from the steroid tigogenin.
While primary benzylic pyridiniums yielded products 25–27 in good yields, primary non‐benzylic substrates failed to undergo the deaminative Giese reaction. However, we found that adding Et3N to the reaction mixture and increasing the reaction temperature to 60 °C had a dramatic effect on the outcome of the reaction and enabled the isolation of adduct 28, albeit in low yield. Switching from benzyl acrylate to the more activated alkene methyl 2‐phenylacrylate provided further improvements and enabled isolation of product 29 in 47 % yield. Despite the yield being moderate, this result is notable as it is a rare example of a photoinduced Giese reaction of a non‐stabilized primary alkyl radical under mild and catalyst‐free conditions. With these new conditions, a range of non‐benzylic primary alkyl pyridiniums reacted to give the Giese products (29–35) in moderate to good yields. Furthermore, the functional group tolerance of the methodology was highlighted by generating products bearing primary sulfonamide (31), pyridine (32), thiophene (33), and silyl ether (35) moieties.
To shed light on the mechanism of this catalyst‐free Giese reaction, we analyzed the reaction components by UV/Vis absorption spectroscopy. DMA solutions of secondary alkyl pyridinium 36 and Hantzsch ester (5) were both found to absorb in the visible region (>400 nm) (Figure 1 A). However, a mixture of 36 and 5 displayed a significant red‐shift in absorbance, confirming formation of the postulated EDA complex. A similar shift was observed with a mixture of primary alkyl pyridinium 37 and 5 (Figure 1 B). Interestingly, a mixture of 37, 5, and Et3N showed a further bathochromic shift, suggesting the formation of a ternary EDA complex, which could contribute to the enhanced reactivity observed with primary alkyl pyridiniums upon addition of Et3N. The formation of alkyl radical intermediates was confirmed by a radical clock experiment with cyclopropylmethyl pyridinium 38, during which ring‐opening occurred to give alkene 39 as the only observable product (Figure 1 C).
Figure 1.
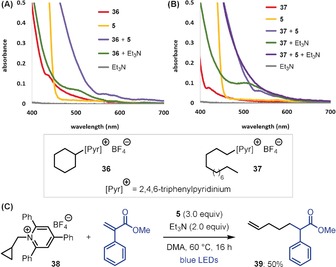
Mechanistic studies. A) Spectrophotometry of pyridinium 36. B) Spectrophotometry of pyridinium 37. C) Radical clock experiment.
These results suggest a mechanism comprised of initial formation of an EDA complex 40 between the electron‐deficient pyridinium 1 and electron‐rich Hantzsch ester (5) (Figure 2). Subsequent PET leads to dihydropyridine radical cation 41 and radical 42, which fragments to triphenylpyridine 43 and alkyl radical 44. Addition of 44 to methyl acrylate generates radical 45, which undergoes hydrogen atom transfer (HAT) with dihydropyridine radical cation 41 (BDFE=31 kcal mol−1)19 or 5 (BDFE=69 kcal mol−1)19 to form Giese product 46 (BDFE≈96 kcal mol−1)20 and pyridinium 47 or dihydropyridine radical 48, respectively.21
Figure 2.
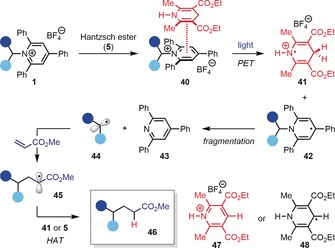
Proposed mechanism.
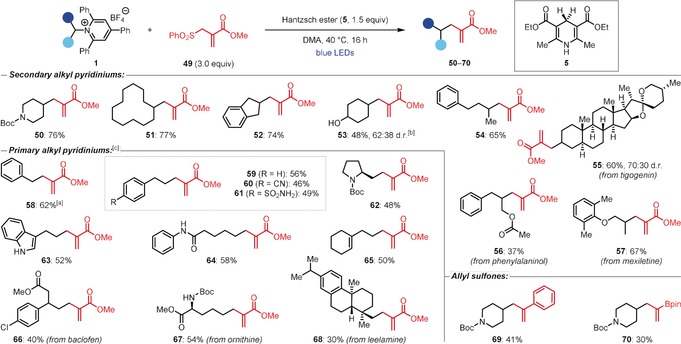
Encouraged by the results of the Giese reaction, we proceeded to investigate other catalyst‐free transformations. Pleasingly, with only slight modification to the reaction conditions,18 allylation reactions with allyl sulfone 49 were also found to be efficient (Table 2).14c A range of secondary alkyl pyridiniums underwent the catalyst‐free deaminative allylation to give products 50–57 in moderate to good yields. As with the Giese reaction, although primary benzylic pyridiniums yielded the allylation product 58 under these conditions, non‐benzylic primary alkyl pyridiniums 59–68 required the addition of Et3N for successful reaction. The allylation reaction was found to tolerate a diverse range of functional groups, including alcohols (53), nitriles (60), sulfonamides (61), unprotected indoles (63), olefins (65), and secondary carbamates (67), and was also applied to various pharmaceuticals (57 and 66) and natural product derivatives (55, 56, 67, and 68). Furthermore, the use of other allyl sulfone reagents enabled the preparation of styrene derivative 69 and alkenylboronic ester 70.
Table 2.
Allylation reaction substrate scope.[a]
|
[a] General conditions: Pyridinium (1.0 equiv), allyl sulfone (3.0 equiv), and 5 (1.5 equiv) in DMA (0.4 m) at 40 °C for 16 h. Yields are of isolated products after flash column chromatography. [b] Isolated after acetyl protection of the alcohol. [c] Reactions performed at 60 °C with 5 (2.5 equiv) and Et3N (3.0 equiv).
During our UV/Vis absorbance studies of pyridinium 36 we found that it also forms an EDA complex with Et3N (Figure 1 A). Thus, we were curious as to whether these photoinduced reactions could be performed with Et3N in place of Hantzsch ester. While the Giese reaction proceeded with low yield, the allylation reaction proceeded smoothly to generate 50 in 71 % when using 6.0 equiv of Et3N in place of Hantzsch ester (Scheme 2).18 An identical result was also obtained when Et3N was replaced by iPr2NEt. This result is intriguing given that these conditions are very similar to the photoredox‐catalyzed conditions recently reported by Liu and co‐workers, which differ only by the use of an iridium photocatalyst.14c We also investigated other addition–elimination reactions with unsaturated sulfone reagents and found that alkynylation and vinylation reactions also proceeded under our catalyst‐free conditions, generating alkyne 71 and alkene 72 in good yields. Again, these conditions are similar to previously reported photoredox‐catalyzed protocols by Gryko and co‐workers but proceed efficiently in the absence of a photocatalyst.14b Finally, we found that by replacing the unsaturated sulfones with other sulfur‐based reagents, under otherwise identical conditions, high yielding hydrodeamination and deaminative thioetherification reactions were also possible, providing good yields of N‐Boc‐piperidine 73 and thioether 74, respectively.
Scheme 2.
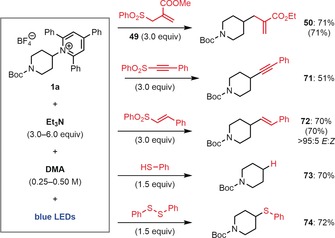
Deaminative transformations promoted by Et3N. Reactions performed at 60 °C. Yields in parentheses are for reactions performed using iPr2NEt in place of Et3N.
In conclusion, we have described the development of a general catalyst‐free deaminative protocol for the generation of non‐stabilized alkyl radicals, proceeding through visible light photoexcitation of EDA complexes of N‐alkylpyridinium salts. The radicals were shown to undergo a range of transformations, including Giese, allylation, vinylation, alkynylation, HAT, and thioetherification reactions. The mild conditions, high functional group tolerance and ease of synthesis of the pyridinium substrates make this a useful catalyst‐free approach to alkyl radical formation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank EPSRC (EP/R004978/1) and H2020 ERC (670668) for financial support. P.S.G thanks the University of Auckland for a Doctoral Scholarship. X.L thanks The Fundamental Research Funds for the Central Universities (grant number GK201603034). We thank Dr. Lin He for helping with the synthesis of pyridinium salt starting materials.
J. Wu, P. S. Grant, X. Li, A. Noble, V. K. Aggarwal, Angew. Chem. Int. Ed. 2019, 58, 5697.
References
- 1.
- 1a. Protti S., Fagnoni M., Photochem. Photobiol. Sci. 2009, 8, 1499; [DOI] [PubMed] [Google Scholar]
- 1b. Yoon T. P., Ischay M. A., Du J., Nat. Chem. 2010, 2, 527; [DOI] [PubMed] [Google Scholar]
- 1c. Buzzetti L., Crisenza G. E. M., Melchiorre P., Angew. Chem. Int. Ed. 2018, 10.1002/anie.201809984; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 10.1002/ange.201809984. [DOI] [Google Scholar]
- 2.
- 2a. Visible Light Photocatalysis in Organic Chemistry (Eds.: C. R. J. Stephenson, T. P. Yoon, D. W. C. MacMillan), Wiley-VCH, Weinheim, 2018; [Google Scholar]
- 2b. Tucker J. W., Stephenson C. R. J., J. Org. Chem. 2012, 77, 1617; [DOI] [PubMed] [Google Scholar]
- 2c. Shaw M. H., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Hoffmann N., Chem. Rev. 2008, 108, 1052; [DOI] [PubMed] [Google Scholar]
- 3b. Bach T., Hehn J. P., Angew. Chem. Int. Ed. 2011, 50, 1000; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1032. [Google Scholar]
- 4. Silvi M., Melchiorre P., Nature 2018, 554, 41. [DOI] [PubMed] [Google Scholar]
- 5.For selected examples, see:
- 5a. Cecere G., König C. M., Alleva J. L., MacMillan D. W. C., J. Am. Chem. Soc. 2013, 135, 11521; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Silvi M., Arceo E., Jurberg I. D., Cassani C., Melchiorre P., J. Am. Chem. Soc. 2015, 137, 6120; [DOI] [PubMed] [Google Scholar]
- 5c. Silvi M., Sandford C., Aggarwal V. K., J. Am. Chem. Soc. 2017, 139, 5736; [DOI] [PubMed] [Google Scholar]
- 5d. Bahamonde A., Melchiorre P., J. Am. Chem. Soc. 2016, 138, 8019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Filippini G., Silvi M., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 4447; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4518; [Google Scholar]
- 5f. Silvi M., Verrier C., Rey Y. P., Buzzetti L., Melchiorre P., Nat. Chem. 2017, 9, 868; [DOI] [PubMed] [Google Scholar]
- 5g. Bonilla P., Rey Y. P., Holden C. M., Melchiorre P., Angew. Chem. Int. Ed. 2018, 57, 12819; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13001; [Google Scholar]
- 5h. Goti G., Bieszczad B., Vega-Peñaloza A., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 1213; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1226. [Google Scholar]
- 6.
- 6a. Rosokha S. V., Kochi J. K., Acc. Chem. Res. 2008, 41, 641; [DOI] [PubMed] [Google Scholar]
- 6b. Lima C. G. S., Lima T. de M., Duarte M., Jurberg I. D., Paixão M. W., ACS Catal. 2016, 6, 1389; [Google Scholar]
- 6c. Postigo A., Eur. J. Org. Chem. 2018, 6391. [Google Scholar]
- 7.For selected examples of perfluoroalkyl radical generation, see:
- 7a. Pham P. V., Nagib D. A., MacMillan D. W. C., Angew. Chem. Int. Ed. 2011, 50, 6119; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6243; [Google Scholar]
- 7b. Nappi M., Bergonzini G., Melchiorre P., Angew. Chem. Int. Ed. 2014, 53, 4921; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5021; [Google Scholar]
- 7c. Woźniak Ł., Murphy J. J., Melchiorre P., J. Am. Chem. Soc. 2015, 137, 5678; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Beatty J. W., Douglas J. J., Miller R., McAtee R. C., Cole K. P., Stephenson C. R. J., Chem 2016, 1, 456; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. Spell M. L., Deveaux K., Bresnahan C. G., Bernard B. L., Sheffield W., Kumar R., Ragains J. R., Angew. Chem. Int. Ed. 2016, 55, 6515; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6625; [Google Scholar]
- 7f. Cheng Y., Yu S., Org. Lett. 2016, 18, 2962; [DOI] [PubMed] [Google Scholar]
- 7g. Liu Y.-Y., Yu X.-Y., Chen J.-R., Qiao M.-M., Qi X., Shi D.-Q., Xiao W.-J., Angew. Chem. Int. Ed. 2017, 56, 9527; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9655; [Google Scholar]
- 7h. Tu H.-Y., Zhu S., Qing F.-L., Chu L., Chem. Commun. 2018, 54, 12710; [DOI] [PubMed] [Google Scholar]
- 7i. Guo Q., Wang M., Liu H., Wang R., Xu Z., Angew. Chem. Int. Ed. 2018, 57, 4747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4837. [Google Scholar]
- 8.For stabilized alkyl radical generation, see:
- 8a. Arceo E., Jurberg I. D., Álvarez-Fernández A., Melchiorro P., Nat. Chem. 2013, 5, 750; [DOI] [PubMed] [Google Scholar]
- 8b. Arceo E., Bahamonde A., Bergonzini G., Melchiorro P., Chem. Sci. 2014, 5, 2438; [Google Scholar]
- 8c. Kandukuri S. R., Bahamonde A., Chatterjee I., Jurberg I. D., Escudero-Adán E. C., Melchiorre P., Angew. Chem. Int. Ed. 2015, 54, 1485; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1505; [Google Scholar]
- 8d. Cao Z.-Y., Ghosh T., Melchiorre P., Nat. Commun. 2018, 9, 3274; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Hsu C.-W., Sundén H., Org. Lett. 2018, 20, 2051. [DOI] [PubMed] [Google Scholar]
- 9.For aryl radical generation, see:
- 9a. Fox M. A., Younathan J., Fryxell G. E., J. Org. Chem. 1983, 48, 3109; [Google Scholar]
- 9b. Tobisu M., Furukawa T., Chatani N., Chem. Lett. 2013, 42, 1203; [Google Scholar]
- 9c. Marzo L., Wang S., König B., Org. Lett. 2017, 19, 5976; [DOI] [PubMed] [Google Scholar]
- 9d. Liu B., Lim C.-H., Miyake G. M., J. Am. Chem. Soc. 2017, 139, 13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For heteroatom-centered radical generation, see:
- 10a. Davies J., Booth S. G., Essafi S., Dryfe R. A. W., Leonori D., Angew. Chem. Int. Ed. 2015, 54, 14017; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14223; [Google Scholar]
- 10b. Reina D. F., Dauncey E. M., Morcillo S. P., Svejstrup T. D., Popescu M. V., Douglas J. J., Sheikh N. S., Leonori D., Eur. J. Org. Chem. 2016, 2108; [Google Scholar]
- 10c. Zhang J., Li Y., Xu R., Chen Y., Angew. Chem. Int. Ed. 2017, 56, 12619; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12793; [Google Scholar]
- 10d. Li Y., Zhang J., Li D., Chen Y., Org. Lett. 2018, 20, 3296. [DOI] [PubMed] [Google Scholar]
- 11. Buzzetti L., Prieto A., Roy S. R., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 15039; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2017, 129, 15235. [Google Scholar]
- 12. Bapat J. B., Blade R. J., Boulton A. J., Epsztajn J., Katritzky A. R., Lewis J., Molina-Buendia P., Nie P.-L., Ramsden C. A., Tetrahedron Lett. 1976, 17, 2691. [Google Scholar]
- 13.For nickel-catalyzed reactions, see:
- 13a. Basch C. H., Liao J., Xu J., Piane J. J., Watson M. P., J. Am. Chem. Soc. 2017, 139, 5313; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Liao J., Guan W., Boscoe B. P., Tucker J. W., Tomlin J. W., Garnsey M. R., Watson M. P., Org. Lett. 2018, 20, 3030; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Guan W., Liao J., Watson M. P., Synthesis 2018, 50, 3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For photoredox-catalyzed reactions, see:
- 14a. Klauck F. J. R., James M. J., Glorius F., Angew. Chem. Int. Ed. 2017, 56, 12336; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12505; [Google Scholar]
- 14b. Ociepa M., Turkowska J., Gryko D., ACS Catal. 2018, 8, 11362; [Google Scholar]
- 14c. Zhang M.-M., Liu F., Org. Chem. Front. 2018, 5, 3443; [Google Scholar]
- 14d. Klauck F. J. R., Yoon H., James M. J., Lautens M., Glorius F., ACS Catal. 2019, 9, 236. [Google Scholar]
- 15.
- 15a. Wu J., He L., Noble A., Aggarwal V. K., J. Am. Chem. Soc. 2018, 140, 10700; [DOI] [PubMed] [Google Scholar]
- 15b. Hu J., Wang G., Li S., Shi Z., Angew. Chem. Int. Ed. 2018, 57, 15227; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15447; [Google Scholar]
- 15c. Sandfort F., Strieth-Kalthoff F., Klauck F. J. R., James M. J., Glorius F., Chem. Eur. J. 2018, 24, 17210; For a mechanistically related decarboxylative borylation, see: [DOI] [PubMed] [Google Scholar]
- 15d. Fawcett A., Pradeilles J., Wang Y., Mutsuga T., Myers E. L., Aggarwal V. K., Science 2017, 357, 283. [DOI] [PubMed] [Google Scholar]
- 16.For EDA complexes of Katritzky pyridiniums, see:
- 16a. Katritzky A. R., de Ville G. Z., Patel R. C., Tetrahedron Lett. 1980, 21, 1723; [Google Scholar]
- 16b. Katritzky A. R., De Ville G., Patel R. C., Tetrahedron 1981, 37, 25. [Google Scholar]
- 17.
- 17a. Okada K., Okamoto K., Morita N., Okubo K., Oda M., J. Am. Chem. Soc. 1991, 113, 9401; [Google Scholar]
- 17b. Lackner G. L., Quasdorf K. W., Pratsch G., Overman L. E., J. Org. Chem. 2015, 80, 6012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Pratsch G., Lackner G. L., Overman L. E., J. Org. Chem. 2015, 80, 6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.See Supporting Information for details.
- 19. Zhu X.-Q., Li H.-R., Li Q., Ai T., Lu J.-Y., Yang Y., Cheng J.-P., Chem. Eur. J. 2003, 9, 871.12584702 [Google Scholar]
- 20. Brocks J. J., Beckhaus H.-D., Beckwith A. L. J., Rüchardt C., J. Org. Chem. 1998, 63, 1935. [Google Scholar]
- 21.See Supporting Information for further mechanistic discussions and evidence for the formation of radicals 42 and 48.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary