

Summary
Caffeine is the most widely used psychostimulant in Western countries, with antioxidant, anti‐inflammatory and anti‐apoptotic properties. In Alzheimer's disease (AD), caffeine is beneficial in both men and women, in humans and animals. Similar effects of caffeine were observed in men with Parkinson's disease (PD); however, the effect of caffeine in female PD patients is controversial due to caffeine's competition with estrogen for the estrogen‐metabolizing enzyme, CYP1A2. Studies conducted in animal models of amyotrophic lateral sclerosis (ALS) showed protective effects of A2 AR antagonism. A study found caffeine to be associated with earlier age of onset of Huntington's disease (HD) at intakes >190 mg/d, but studies in animal models have found equivocal results. Caffeine is protective in AD and PD at dosages equivalent to 3‐5 mg/kg. However, further research is needed to investigate the effects of caffeine on PD in women. As well, the effects of caffeine in ALS, HD and Machado‐Joseph disease need to be further investigated. Caffeine's most salient mechanisms of action relevant to neurodegenerative diseases need to be further explored.
Keywords: adenosine receptor, Alzheimer disease, amyotrophic lateral sclerosis, caffeine, dosage, Huntington disease, neurodegenerative disease, neuroprotection, Parkinson disease
1. Caffeine
Caffeine (1,3,7‐trimethylxanthine) is the most widely used psychostimulant in Western countries.1, 2 It is contained in coffee, tea, energy drinks, several soft drinks and cocoa.2, 3 In a cup containing 437 mL of brewed coffee, there is on average 188 mg of caffeine (range 147‐259 mg).4 The range of caffeine concentration is about 0.01 mg/g of coffee beans (decaffeinated coffee) to 19.9 mg/g (Italian coffee).5 However, most coffee beans contain about 10.0‐12.0 mg of caffeine/g of coffee bean. According to the Centre for Addiction and Mental Health, the average Canadian consumes 210‐238 mg/d of caffeine.6 After consumption, caffeine is quickly absorbed through the gastrointestinal tract and the highest blood caffeine concentration is reached 30‐60 minutes after intake.2, 7 Similar caffeine concentrations are found in the brain, suggesting that caffeine can readily cross the blood‐brain barrier, due to its hydrophobic nature. Average levels of caffeine consumption cause alertness and reduce fatigue, leading to better performance in psychomotor tasks requiring fast reactions.8 A two‐ to four‐year follow‐up study of 4197 women (74 years) without dementia found that caffeine consumption at 200‐300 mg (odds ratio (OR)=0.82, confidence interval (CI)=0.67‐1.01) and >300 mg (OR=0.66, CI=0.52‐0.83) was associated with significant reduction in cognitive decline.9 As well, the effects of coffee are stronger in women above 80 years (OR=0.30, CI=0.14‐0.63), compared to younger women (OR=0.73, CI=0.53‐1.02). The same effect was not observed in 2820 men (74 years).9 However, another study in men (75‐77 years) from Finland (volume not reported), Netherlands (125 mL/cup) and Italy (volume not reported) found that those who consumed coffee had a cognitive decline of 1.2 points, whereas those that did not consume coffee had an additional decline of 1.4 points on the mini‐mental state examination, which assesses global cognitive function.10 Specifically, nonusers experienced 2.6 points of cognitive decline, but users of 1, 2, 3 and 4 cups experienced 1.4, 1.3, 0.6, and 1.6 points of cognitive decline, respectively. The same was not true for men who consumed >4 cups of coffee, concluding that three cups/d of coffee were the most effective in reducing cognitive decline.10 In addition, a 21‐year follow‐up study found that moderate consumption (3‐5 cups/d) was associated with lower risk of dementia in men (OR=0.27, CI=0.08‐0.89) and women (OR=0.51, CI=0.17‐1.52), compared to low consumption (0‐2 cups/d). Among men, the risk of developing dementia was lower when consuming high levels of coffee (>5 cups) compared to low coffee consumption (OR=0.36, CI=0.13‐0.97).11
Caffeine can inhibit lipid peroxidation and reduce reactive oxygen species (ROS) production.12 In fact, chronic caffeine intake ameliorates oxidative stress and improves mitochondrial function in several neurotoxic situations.13 A study in rats showed that caffeine reversed oxidative stress and attenuated inflammation induced by d‐galactose, a compound that can induce aging in rat brains.14 As well, caffeine increases glutathione S‐transferase activity and inhibits red blood cell membrane derangement and apoptosis.15, 16 It is also a strong scavenger of hydroxyl radicals. Therefore, the effect of caffeine in neurodegenerative disorders has been grossly investigated over the last decade. It has been shown that caffeine affects the pathophysiology of neurodegenerative disorders, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD) and Machado‐Joseph disease (MJD). This review investigates the effects of caffeine on these neurodegenerative diseases, as well as the mechanisms involved.
1.1. Caffeine, adenosine and adenosine receptors
As shown in Figure 1, caffeine and adenosine have very similar basic structures. Both caffeine and adenosine have purine backbones, allowing for caffeine to also bind to adenosine receptors as a competitive antagonist of adenosine.17
Figure 1.
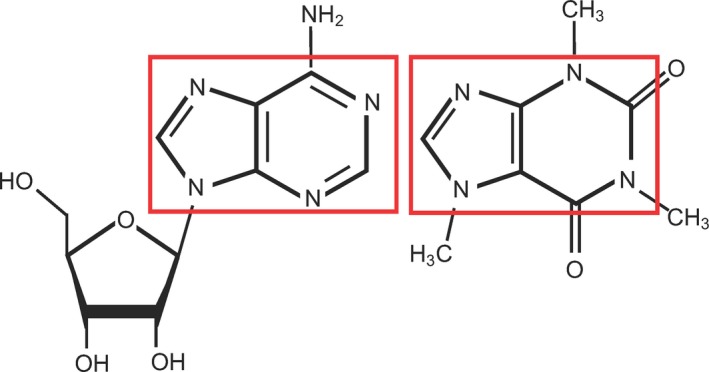
The chemical structure of adenosine (left), an endogenous adenosine receptor agonist, and caffeine (right), an exogenous adenosine receptor antagonist17
Adenosine receptors (A1R, A2AR, A2BR and A3R) are G‐protein‐coupled receptors expressed in a variety of different cells, such as endothelial cells, immune cells, blood vessels, astrocytes, microglia, and the striatum and the spinal cord of the central nervous system.18, 19 The interaction of adenosine with its receptors and the downstream effects are shown in Figure 2. When adenosine binds to A1R and A3R, the inhibitory G‐protein is activated, which inhibits adenylyl cyclase (AC) activation, reducing the conversion of AMP to cyclic AMP (cAMP), causing a decrease in protein kinase A (PKA) activation, leading to lower downstream phosphorylation.20, 21 When PKA is not activated, calcium channels on the plasma membrane will not be phosphorylated, causing a reduction in calcium flow into the cell. However, when adenosine binds to A2AR and A2BR, the stimulatory G‐protein is activated, increasing AC activity, cAMP and PKA levels and calcium entry into the cell.20, 21 The binding affinities of adenosine to adenosine receptors and the KD (equilibrium dissociation constant) values for caffeine are shown in Table 1. At 300 mg of consumption, caffeine affects all adenosine receptors, but it has the highest interaction with A1R and A2AR.
Figure 2.
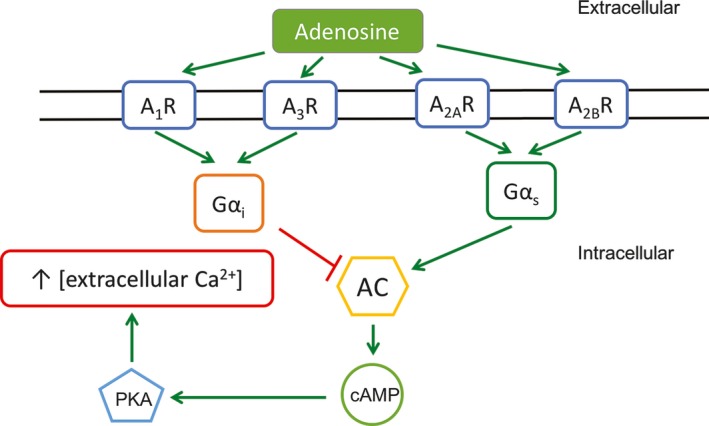
The interaction of adenosine with its receptors and the downstream mechanisms are outlined in this figure. When adenosine binds to A1R and A3R, Gαi is activated, lowering AC activity, cAMP production, PKA activation, and calcium entry into the cell. When adenosine binds to A2 AR and A2 BR, Gαs is activated, increasing AC activity, cAMP production, PKA activation, and calcium entry into the cell. Gαi: inhibitory G‐protein, Gαs: stimulatory G‐protein, AC: adenylyl cyclase, cAMP: cyclic adenosine monophosphate, PKA: protein kinase A20, 21
Table 1.
The binding affinity of adenosine to its receptors and the KD (equilibrium dissociation constant) values for caffeine binding to human adenosine receptors22, 38
| A1R | A2AR | A2BR | A3R | |
|---|---|---|---|---|
| Adenosine | 0.3‐3 nmol/L | 1‐20 nmol/L | 0.5‐5 μmol/L | <10 nmol/L |
| Caffeine (KD) | 12 μmol/L | 2.4 μmol/L | 13 μmol/L | 80 μmol/L |
1.2. Adenosine A1 receptor (A1R)
A1R is abundant throughout different parts of the brain, including the cerebellum and the cerebral cortex.23 Adenosine, through the activation of the inhibitory A1R, acts as a neuromodulator in the nervous system that reduces neuronal excitability.24 Targeted deletion of A1R gene does not produce any drastic changes in basal or caffeine‐induced motor function, and therefore caffeine's effects on motor activity are not mediated by A1R inhibition.23 However, chronic treatment, and blockade of these receptors, with caffeine causes an upregulation of A1R in rodents via an increase in the concentration of A1R.7, 25 Interestingly, an increase in the expression of A1R causes a decrease in pro‐inflammatory cytokines, including tumor necrosis factor alpha (TNF‐α).26 Direct evidence of the neuroprotective effects of inhibiting A1R has also been found in a model of methylmercury poisoning.27
1.3. Adenosine A2A receptor (A2AR)
A2AR is located mainly in the striatum, basal ganglia, olfactory cortex and the hippocampus.23, 24, 28 These receptors are present pre‐ and postsynaptically and are also expressed on the glia.29 Extracellular adenosine levels increase dramatically in response to ischemia, excitotoxicity, inflammation, and other brain insults.30 A2AR activation, via increased adenosine, protects against brain injury by modulating neurotransmission processes that are implicated in neuron‐glia communication. As well, A2AR is upregulated in microglia, where it potentiates the inflammatory cascade,30 and hence its blockade offers robust protection against noxious brain conditions.27, 28, 31 Blocking A2AR downregulates glutamate release, direct calcium entry into the neurons, and inflammatory reactivity of microglia.24 A2AR mediates the inhibition of glutamate reuptake by glutamate transporter 1.32 Also, A2AR mediates the upregulation of phospho‐extracellular signal‐reductase kinases (pERK) 1 and 2, which cause a rapid and dramatic increase in glutamate release, leading to microglial activation.30 Hence, a vicious cycle of excitotoxicity is instigated leading to increased ROS production and inflammatory mediator production. Glutamate release can cause calcium release in the cytosol, leading to further inflammatory response, and eventually to neuronal death.28 Therefore, inhibiting A2AR can have anti‐inflammatory and anti‐apoptotic effects. Indeed, in a model of ischemia reperfusion (IR), A2AR antagonist reduced pERK activation and glutamate protein levels, lowering the downstream inflammatory response in the hippocampus.30 A2AR antagonist also reduced the levels of pro‐inflammatory biomarkers nuclear factor (NF)‐κB, TNF‐α, interleukin (IL)‐6 and prostaglandin E2, and increased the anti‐inflammatory biomarker IL‐10, matching those of the control group.28, 30 The pro‐apoptotic markers caspase 3 and cytochrome C were also downregulated upon A2AR antagonist administration in this model of IR.
1.4. Adenosine A1 receptor and adenosine A2A receptor
A1R activation can offer protective mechanism via lowering Ca2+ influx, thus lowering presynaptic release of excitatory neurotransmitters, namely less glutamate release.32 A2AR releases glutamate, which has excitatory effects that counteract the inhibitory effects of A1R. A main biochemical characteristic of A1R/A2AR heteromer is the ability of A2AR to decrease the affinity of A1R for its agonists with an ultimate switch mechanism, meaning that high levels of adenosine will cause excitatory effects and low levels of adenosine will have inhibitory effects, as shown in Figure 3.32 Since caffeine does not allow adenosine to bind to the A2AR, it abrogates the switch mechanism, resulting in less calcium influx into the cell and hence less glutamate release.
Figure 3.
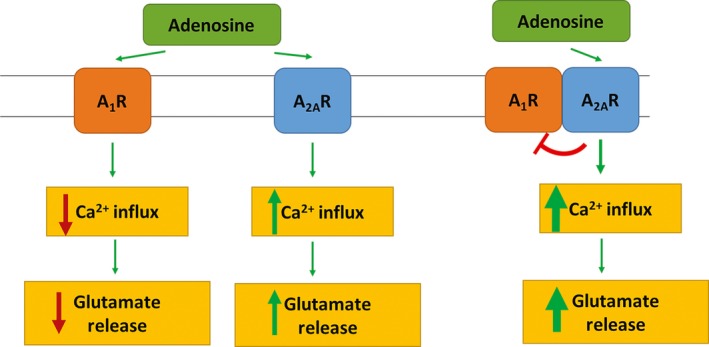
When adenosine binds to, and activates, the A1R, it causes a reduction in the calcium influx into the cell and glutamate release. However, when adenosine binds to, and activates, A2 AR, it increases calcium influx and glutamate release. Adenosine binding to A2 AR inhibits A1R binding to its agonists, therefore causing an increase in calcium influx and glutamate release. When adenosine is readily available, calcium influx and glutamate release into the cell are increased32
1.5. Caffeine, phosphodiesterase and calcium release
Caffeine is a ryanodine receptor agonist.33, 34 Ryanodine receptors are Ca2+ releasing channels on the endoplasmic reticulum (ER).35 Therefore, caffeine can increase the amount of calcium released from the ER by binding to and activating ryanodine receptors.36 However, caffeine consumption at average dosages of 210‐238 mg/d does not affect the ryanodine receptors. The levels of caffeine needed to initiate its effect on the sensitivity of ryanodine receptors are at plasma concentration of 100 μmol/L, which is equivalent to about 1520 mg/d of caffeine intake.37, 38, 39
Caffeine is an inhibitor of phosphodiesterase, which inactivates cAMP.40 However, to inhibit phosphodiesterase, caffeine consumption has to be 20 times higher than the dose obtained from a single cup (100 mg of caffeine) of coffee, whereas to mobilize intracellular calcium depots it has to be 100 times higher.38 While modulation of phosphodiesterase and activation of ryanodine receptors may play a role in the effect of caffeine on Ca2+ levels in neurodegenerative diseases, concentrations of caffeine needed to act on these are difficult to reach at nontoxic doses of consumption.41
2. Caffeine and ALZHEIMER'S Disease
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disorder that leads to cognitive, behavioral, and memory impairments.42 The histopathological features of AD include: the extracellular depositions of diffuse and neuritic plaques that are composed of amyloid‐β (Aβ) peptide, the intracellular accrual of neurofibrillary tangles that consist of hyperphosphorylated aggregates of the microtubule‐associated protein Tau, and selective neuronal loss restricted to the hippocampus and the neocortex.42, 43 Recently, caffeine has been of scientific interest because of its potential as an antioxidant compound, able to protect against oxidative stress in AD.
2.1. Human studies
A 21‐year follow‐up study (875 women and 534 men, age 50 years at the beginning of the study) found that moderate consumption (3‐5 cups, volume not identified) of coffee substantially reduced the risk of AD (62%‐64%) and dementia (65%‐70%) later in life, compared to low coffee consumers (0‐2 cups).11 The Canadian Study of Health and Aging, examining 10 263 men and women over the age of 65 years, observed coffee consumption to be associated with a 31% lower risk of AD in the Canadian population (OR=0.69, CI=0.5‐0.96).44 Another study found that 54 patients with AD had an average daily caffeine intake of 74±98 mg during the 20 years that preceded their diagnosis, whereas the age‐matched controls had an average daily intake of 199±136 mg during the corresponding 20 years of their life.45 Caffeine exposure was found to be significantly associated with a 60% reduced risk of AD (OR=0.40, CI=0.25‐0.67).45
A recent meta‐analysis found that caffeine intake from tea or coffee does not have a statistically reliable protective association, despite a trend of an 18% reduced risk of cognitive disorders such as dementia and AD (relative risk (RR)=0.82, CI=0.67‐1.01).46 However, this meta‐analysis had several limitations. The studies included in this meta‐analysis had different study designs and outcomes, as well as different caffeine intakes. Therefore, the variations between the studies were high. Hence, it is possible that the overall 18% reduction in the risk of cognitive disorders, including AD, is not significant because of a type II error. As well, this study did not compare caffeinated coffee or tea with decaffeinated coffee or tea. We cannot conclude that caffeine is not protective in cognitive disorders based on coffee or tea studies, unless there is a direct comparison of regular tea or coffee, with their respective decaffeinated drinks.
2.2. In vitro and in vivo animal studies
2.2.1. Caffeine improves functional outcomes in Alzheimer's disease animal models
Caffeine consumption, as well as A2AR antagonism or deletion, significantly improved the performance of APPsw (a mouse model of AD) in Morris water maze, ascertaining its protective properties against cognitive impairment and in favor of improved memory retention.7, 32, 47, 48, 49 After 4‐5 weeks of caffeine administration at 1.5 mg/d (human equivalence of 500 mg/d) in 18‐ to 19‐month APPsw mice, there was a significant improvement in memory compared to control (4 weeks: 217%, 5 weeks: 198%).50 In fact, caffeine‐treated mice were significantly better in overall cognitive performance vs the APPsw control group. When administered in young adulthood (4‐9 months) at similar doses, caffeine provided complete protection in all cognitive tasks that were previously impaired in APPsw mice.7 Caffeine's protective effects were global, protecting the working memory, spatial learning, and recognition.7
2.2.2. Caffeine reduces amyloid β production and increases amyloid β clearance
Caffeine's beneficial effects in AD are through its interaction with β‐ and γ‐secretase.51 In a study conducted in 2009, Arendash et al.50 found that caffeine treatment at 1.5 mg/d in APPsw mice reduced Aβ deposition in the hippocampus (40%) and the entorhinal cortex (46%). With caffeine, Aβ1‐40 and Aβ1‐42 levels were reduced in the cortex (25% and 51%, respectively) and hippocampus (37% and 59%, respectively).50 Arendash et al.7 also found similar results upon caffeine treatment at similar doses administered to APPsw mice in young adulthood (4‐9 months). Caffeine treatment at 1.5 mg/d for 5.5 months in APPsw mice significantly reduced both soluble Aβ1‐40 (37%) and insoluble Aβ1‐42 (32%) in the hippocampus.7 Caffeine treatment at 40 mg/kg (~0.8 mg/mouse, 41% lower than that used by Arendash et al.) significantly enhanced Aβ clearance (20%) in the brain endothelial cells of C57BL/6 mice.52 In fact, at plasma concentrations of 20±5.4 μg/mL (103±28 μmol/L), caffeine caused a 20% increase in Aβ clearance. However, it is important to note that this dosage is very close to toxic dosages of caffeine in humans.38 Another study found that crude caffeine treatment at human equivalence of 292 mg/d suppressed Aβ1‐42 levels (52%) and decreased plaque number (67%) in APPsw mice.49 As well, Aβ‐treated neurons exposed to crude or pure caffeine had reductions in the number of caspase‐3 positive neurons of approximately 48% and 43%, respectively.
2.2.3. Effect of caffeine on signaling pathways
Treatment with caffeine at 1.5 mg/d for 5.5 months in APPsw mice normalized PKA levels, otherwise downregulated in APPsw mice.50 As shown in Figure 4, PKA is responsible for inactivating c‐Raf‐1, which is responsible for activating NF‐κB pathway, leading to the production of β‐secretase‐1 (BACE‐1) and other AD‐related proteins. Hence, caffeine consumption at doses equivalent to 500 mg in humans causes a reduction in c‐Raf‐1, NF‐κB pathway activation, and BACE‐1 production in APPsw mice. Furthermore, GSK‐3 is known to regulate Aβ production. It is also involved in presenilin (PS)‐1 and γ‐secretase activity as well as tau hyperphosphorylation; caffeine concentration at 10‐20 μmol/L decreases the GSK‐3α and GSK‐3β, proposing a mechanism for decreasing Aβ levels.50 Also, caffeine normalized PS‐1 levels and caused a 50% reduction in BACE levels in young mice (4‐9 months).7 In human neuroblastoma cells treated with aluminum chloride (AlCl3), pretreatment with caffeine (10 μmol/L) reduced phosphorylated IκBα and NF‐κB levels and nuclear translocation of NF‐κB down to control levels.27
Figure 4.
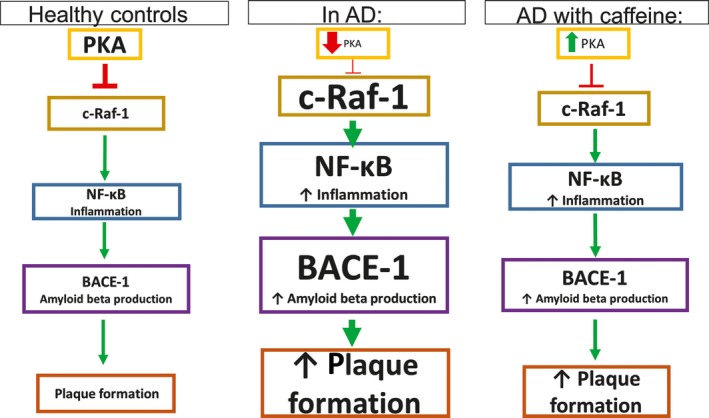
Caffeine normalizes the otherwise reduced PKA levels in APPsw mice, therefore inhibiting c‐Raf‐1 activation, NF‐kB pathway activation, and BACE‐1 production, leading to lower levels of plaque formation50
In mice, caffeine treatment for 8 weeks (0.75 mg/d and 1.5 mg/d) normalized the reduced levels of brain‐derived neurotrophic factor (BDNF) and its receptor (TrkB) responsible for growth, survival and neuronal cell differentiation.53 The effects of caffeine on BDNF were dose dependent, increasing with higher doses of caffeine, but the same was not observed for the levels of TrkB.53, 54 cAMP response element binding protein (CREB), a transcription factor associated with memory and neuronal survival, is downregulated in APPsw mice, but caffeine at 1.5 mg/d significantly increased CREB levels by 126%.55 Caffeine also reduced the upregulated levels of pERK in the striatum (70%) and the cortex (59%) of APPsw mice down to normal levels.
2.2.4. Caffeine reduces oxidative stress and apoptosis, through increasing antioxidant capacity
In human neuroblastoma cells, caffeine (10 μmol/L) lowered ROS production (51%) in the cells treated with Aβ and AlCl3, increased superoxide dismutase (SOD) levels (50%), and lowered malondialdehyde (MDA) levels (50%).27 Also, pro‐apoptotic Bax was upregulated and anti‐apoptotic Bcl‐2 was downregulated in the cells treated with Aβ and AlCl3, whereas pretreatment with caffeine normalized both protein levels. When these cells were treated with A2AR‐specific antagonist (10 μmol/L of SCH58261), cell death was prevented only partially; however, caffeine treatment provided full protection, recognizing the important neuroprotective effects of A1R and A2AR antagonism.27 As well, a study showed that caffeine (0.6 mg/d) increased hippocampal mitochondrial respiration (25%) and ATP levels (46%) in the APPsw mice; and by a much greater degree, caffeine increased hippocampal mitochondrial membrane potential (78%) and decreased ROS production (100%).56 As well, caffeine increased adenosine levels; adenosine is a vasodilator57 responsible for increased blood flow to the brain, contributing to caffeine's protective effects.7
2.2.5. Caffeine may reduce the risk of Alzheimer's disease in individuals carrying ApoE ε4 allele
Those who carry one or two copies of apolipoprotein E (ApoE) ε4 allele have an increased risk of developing AD.58 ApoE is linked to cholesterol transport, because it carries cholesterol from the blood into the brain and shuttles cholesterol from astrocytes to neurons.59 Therefore, presence of ApoE is associated with increased cholesterol levels.60 Indeed, a recent epidemiological study reported that increasing levels of cholesterol were associated with an increased risk of AD, but only for individuals with the ApoE ε4 allele.61 Hypercholestrolemia has been linked with oxidative stress, through the increased production of ROS.59 Prasanthi et al. demonstrated that a 2% cholesterol‐enriched diet increased Aβ levels, tau phosphorylation, and oxidative stress in rabbit hippocampus. However, treatment with caffeine (0.5 and 30 mg) reduced Aβ production, tau phosphorylation, ROS generation, and glutathione depletion and increased the levels of A1R receptor.59 Therefore, treatment with dietary antioxidants, such as caffeine, may be effective in reducing the risk of AD in individuals carrying the ApoE ε4 allele. Further research on the effect of caffeine in these individuals is warranted.
3. Caffeine and Parkinson's Disease
Parkinson's disease (PD) is characterized by bradykinesia (slowness of movement), rigidity, and postural instability.62 The etiology of PD is not fully understood, but it is thought to be the consequence of the loss of dopaminergic neurons of the substantia nigra pars compacta and striatum, resulting in deficit of striatal dopamine that leads to the impairment of the corticostriatal‐thalamo‐cortical or the nigrostriatal pathway of movement.63, 64 Neuronal insult in PD is caused primarily by excessive oxidative stress, leading to damaged proteins, DNA, and lipids. The majority of ROS are initiated from the inflammatory microglial cells, which are activated by certain inflammatory or genetic factors.65 Convergent epidemiological and preclinical data suggest that caffeine can confer neuroprotection against the underlying dopaminergic neuron degeneration and can influence the onset and progression of PD.
3.1. Human studies
3.1.1. Caffeine consumption reduces the risk of developing Parkinson's disease in men
A randomized controlled trial in 61 patients found that treatment with caffeine (200 mg/d for the first 3 weeks and 400 mg/d for the second 3 weeks) improved the total unified Parkinson's disease rating scale by 4.7 points and the motor manifestation by 3.2 points.66 In a 27‐year follow‐up study of 8004 American Japanese men (45‐68 years), those who drank ≥28 oz (794 g of coffee, equivalent to 421 mg of caffeine) of coffee had five times less risk of developing PD vs non‐drinkers and had progressively lower risk of PD with increasing coffee consumption.67 Another study examining 318 260 elderly men and women (61 years) found that consumption of ≥5 cups of coffee in 1995‐1996 was associated with lower risk of PD in 2004‐2006 in men (OR=0.70, CI=0.47‐1.04) and women (OR=0.74, CI=0.42‐1.29), vs non‐users.68 A meta‐analysis of 1 394 488 participants found that the risk of PD decreased by 17% for every 200 mg/d increment of caffeine consumption, and coffee consumption at approximately three cups/d (volume not identified) provided the maximum protection against the risk of developing PD (RR=0.72, CI=0.65‐0.81).69 The association of coffee consumption (3 cups/d) with PD risk was stronger for men (RR=0.68, CI=0.59‐0.78), compared to women (RR=0.76, CI=0.63‐0.93). In men (71 years), consuming 120 mg/d of caffeine resulted in a significant 38% (RR=0.62, CI=0.40‐0.95) lower risk of PD compared to those that consumed very little caffeine (9.2 mg/d), whereas consuming 478 mg/d of caffeine resulted in an even lower risk of PD compared to those who consumed very little caffeine (RR=0.43, CI=0.26‐0.71).70 Men who reported consuming two cups/d (274 mg/d of caffeine) or more of coffee had about 50% lower risk (RR=0.54, CI=0.37‐0.80) of PD than those who did not drink coffee. Women (69 years) who reported consuming 3.2 cups/d (435 mg/d of caffeine) of coffee had 40% lower risk of PD (RR=0.61, CI=0.34‐1.09) than those who consumed very little caffeine (5.6 mg/d).70 During a 12.9‐year follow‐up study of 14 293 men (62.2 years), the hazard ratio of PD in people who drank 0, 1‐4 cups and ≥5 cups of coffee (cup=100 mL) was 1.00, 0.55 (CI=0.26‐1.15) and 0.41 (CI=0.19‐0.88), respectively.71
3.1.2. The effect of caffeine on Parkinson's disease is equivocal in women
In postmenopausal women who consumed caffeine, the relative risk of PD was lower in hormone users vs non‐users.69 A relationship between hormone users and coffee consumption was observed in postmenopausal women.68 Women (61‐62 years) who used hormone therapy had lower risk of PD development upon caffeine consumption (129‐511 mg/d, OR=0.66, CI=0.42‐1.05; 511‐590 mg/d, OR=0.64, CI=0.39‐1.04; and >590 mg/d, OR=0.53, CI=0.28‐0.98) compared to intakes <17.4 mg/d.68 A large prospective cohort study of 77 713 female nurses (30‐55 years), after an 18‐year follow‐up, found that the use of postmenopausal hormones was associated with a 34% reduction in risk of PD among women consuming ~1/2 a cup of coffee/d (68 mg/d of caffeine), but with a 55% increase in risk among women consuming five cups of coffee/d (688 mg/d of caffeine).72 It is important to note that the risk of PD is lower in women versus men, and is inversely associated with circulating levels of estrogen.73 In 15 042 women (64.0 years), after 12.9‐year follow‐up, the hazard ratio of PD in people who drank 0, 1‐4 cups and ≥5 cups of coffee (100 mL/cup) was 1.00, 0.50 (CI=0.22‐1.12) and 0.39 (CI=0.17‐0.89), respectively.71 As a matter of fact, PD patients are often non‐caffeine users.66 However, one study showed that coffee consumption had no association with PD risk, but drinking more than two cups/d of cola (34 mg of caffeine/cup, OR=0.6, CI=0.3‐1.4) or tea (38 mg of caffeine/cup, OR=0.4, CI=0.2‐0.9) was associated with lower risk of developing PD.74 The beneficial effects of these drinks may be attributed to components other than caffeine. Although tolerance can increase for caffeine's stimulant effects, its neuroprotective effects are maintained regardless of the amount of caffeine consumed.64
3.2. In vitro and in vivo animal studies
1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) is a neurotoxin used in mice and nonhuman primates. MPTP, converted to MPP+, can cross the blood‐brain barrier where it is taken up specifically by dopaminergic receptors.65 There, it inhibits complex I of the electron transport chain, causing a severe energy crisis, leading to cell death and hence dopamine insufficiency in the striatum.63 Accompanying the insult from MPTP, there is a peak in microglial proliferation, leading to severe neuroinflammation. Therefore, MPTP‐induced damage is an excellent model of neurodegeneration in PD.
3.2.1. Caffeine reduces MPTP‐induced neuron damage in models of Parkinson's disease
Caffeine treatment at 30 mg/kg for 8 days (0.9 mg/d) 30 minutes prior to MPTP administration attenuated neuron damage and improved motor function (60.6% improvement in grip strength) in male PD mice.63 Another study found that caffeine pretreatment attenuated MPTP‐induced dopamine loss (38% of the control dopamine levels without caffeine) in a dose‐dependent manner in young (10 weeks) male mice, with maximal effects achieved at 10 mg/kg (78% of the control dopamine levels with caffeine).75 In old male mice (6‐9 months), MPTP‐induced dopamine loss (3% of control dopamine levels without caffeine) was attenuated by caffeine pretreatment in a dose‐dependent manner, with 10 mg/kg of caffeine offering the most neuroprotective effect (~30% of control dopamine levels with caffeine).75 In male Sprague Dawley rats, oral caffeine (1 g/L in drinking water) from the onset of MPP+ infusions prevented the loss of nigral dopamine cell bodies.76 As well, supplying caffeine at 1 week (early stages of loss of nigrostriatal dopamine) or 3 weeks (late stages of loss of nigrostriatal dopamine) after initiating MPP+ infusion reduced the loss of nigral cells (1 week: 94% reduction and 3 weeks: 69% reduction). Furthermore, caffeine treatment attenuated microglia activation in the substantia nigra. Therefore, caffeine administration blocked the nigral neurodegenerative process in the model of PD rats.76
3.2.2. Caffeine's protective effects in Parkinson's disease may be due to adenosine antagonism
The neuroprotective effects of caffeine in the substantia nigra could be due to caffeine's competitive inhibition with adenosine. Caffeine can prevent the adenosine‐mediated neuroinflammatory actions via competitively inhibiting adenosine binding to A2AR on microglia, as shown in Figure 5.76 As an adenosine receptor antagonist, caffeine reduces glutamate release and attenuates excitotoxicity.63, 66 Upon MPTP neurointoxication, excessive glutamate is released into the extracellular space, leading to the activation of microglia and neuroinflammation. Midbrain glial cells express A2AR, which are activated by their agonists as well as neuroinflammation caused by MPTP. Caffeine reduces microglial activation and the release of cytokines and free radicals by blocking the activation of A2AR and reducing glutamate release, thereby preventing further damage in the substantia nigra pars compacta and striatal neurons.63, 66
Figure 5.
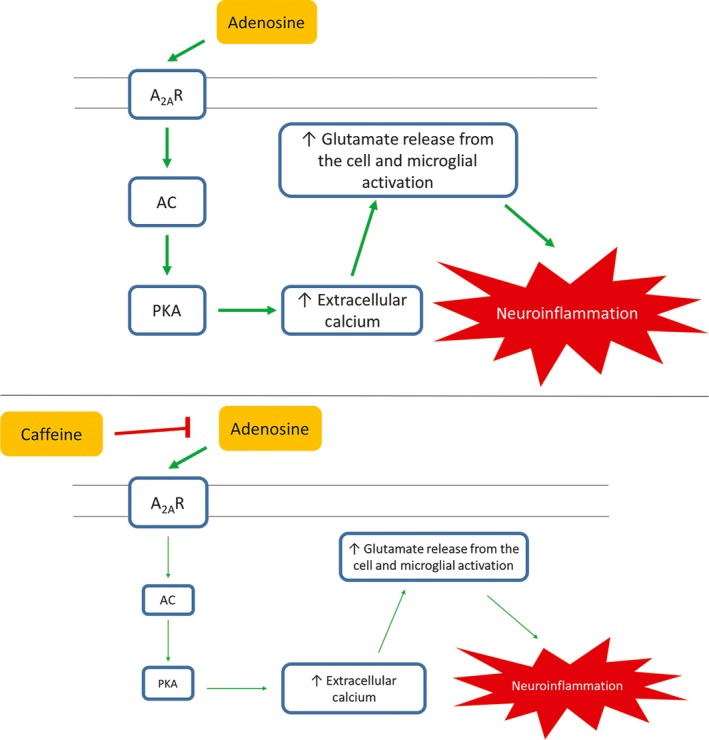
Through activation of the A2 AR, adenosine increases the activation of adenylyl cyclase, leading to protein kinase A (PKA) activation, which causes increase extracellular calcium inside the cell, resulting in an increase in glutamate release from the cell, which leads to neuroinflammation. However, by inhibiting the binding of adenosine to its receptor, caffeine downregulates the activation of PKA, causing a lesser increase in extracellular calcium inside the cell, decreasing the glutamate release from the cell, hence reducing neuroinflammation76
When exposed to caffeine (10 mg/kg) 10 minutes before MPTP administration, the residual dopamine level in mice was 40% of the control, whereas without caffeine it was 15% of the control.77 At 20 mg/kg, caffeine nearly reversed the dopamine depletion caused by MPTP in mice, but at higher levels caffeine caused excessive systemic toxicity. The effects of A2AR antagonists were also investigated and compared to those of caffeine. A2AR antagonism significantly reduced striatal dopamine depletion. As well, when treated with MPTP, A2AR knockout mice experienced attenuated dopamine depletion compared to mice that had A2AR. Therefore, A2AR antagonists mimicked the neuroprotection offered by caffeine. However, it is important to note that A1R antagonism did not mimic the neuroprotection offered by caffeine.77 In human dopaminergic neuroblastoma cell lines, caffeine (100 μmol/L) significantly prevented cell death and decreased the number of apoptotic nuclei from 13.1% to 9.7% under MPP+‐exposed conditions.78 However, this dosage of caffeine exposure is very close to toxic doses of caffeine consumption.38 Caffeine (10 and 100 μmol/L) also decreased the levels of caspase 3 (21% and 23%, respectively).78 Akt phosphorylation significantly increased with caffeine treatment (10 and 100 μmol/L) dose dependently (1.96‐fold and 2.96‐fold, respectively), whereas the inhibition of the PI3K/Akt pathway abolished the effects of caffeine. Therefore, it can be concluded that the neuroprotective effects of caffeine involve the PI3K/AKT pathway.78
3.2.3. Caffeine may not be as effective in female Parkinson's disease models due to estrogen
In female mice, MPTP depleted striatal dopamine levels to 62% of the control, which is a much smaller reduction compared to their male counterparts, whose striatal dopamine levels were 38% of control.75 However, in contrast to their male counterparts, female mice showed no attenuation of MPTP toxicity following caffeine consumption at lower doses (5, 10, 20 mg/kg). Caffeine attenuated MPTP‐induced dopamine loss (~22% of dopamine levels in control without caffeine) in female mice only at the higher dose of 40 mg/kg (~85% of dopamine levels in control with caffeine). In ovariectomized mice treated with placebo, striatal dopamine content was 27% of the control, but lower doses (5, 10, 20 mg/kg) of caffeine offered neuroprotection. However, in ovariectomized mice treated with estrogen, striatal dopamine content was 39% of the control, and only higher doses (40 mg/kg) of caffeine offered neuroprotection. Male mice treated with estrogen also experienced the neuroprotective effects of estrogen; however, no dose of caffeine attenuated their striatal dopamine loss.75 Caffeine is largely metabolized by CYP1A2, which also metabolizes estrogen; by competing for the same enzyme, estrogen inhibits the metabolism of caffeine in premenopausal women or postmenopausal women on hormone therapy.72, 79, 80, 81 Therapeutic estrogen administration has an inhibitory effect on the metabolism of caffeine by CYP1A2, as shown in Figure 6.82 Thus, it can be concluded that caffeine offers protection against PD in male mice, through antagonism of A2AR, among other pathways. The interaction between caffeine and estrogen needs further investigation; however, human studies have shown that caffeine provides protection against PD in both men and women. Indeed, factors such as genetics and smoking habits affect CYP1A2 enzyme activity,83, 84, 85, 86 potentially explaining the interindividual variability observed in different studies. Furthermore, studies have shown the protective effects of paraxanthine and theophylline, two metabolites of caffeine, against MPTP‐induced striatal dopamine depletion, warranting further research investigating the potential neuroprotective effects of caffeine metabolites, in male and female animal models and humans.87
Figure 6.
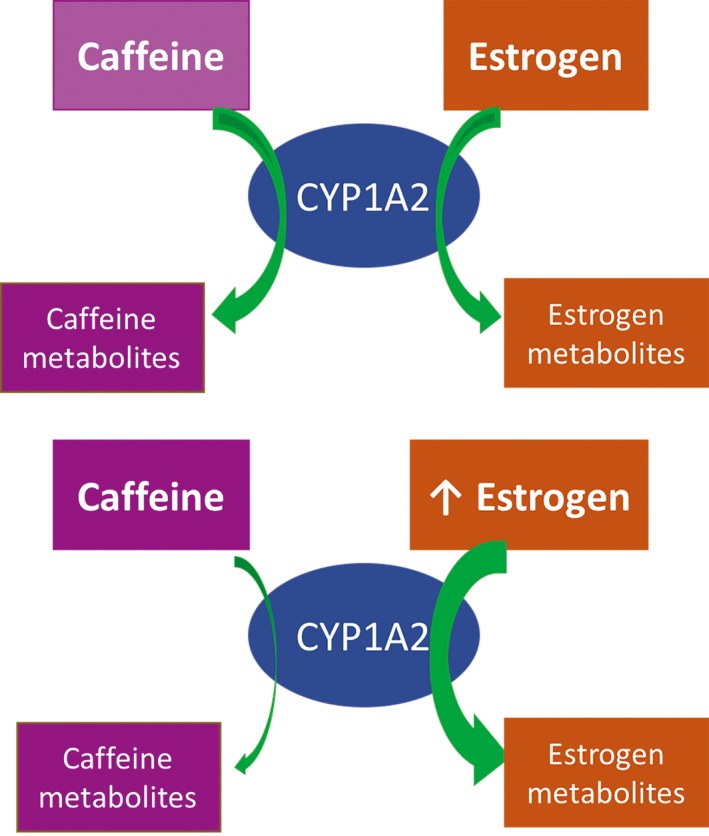
Caffeine is metabolized by CYP1A2, which also metabolizes estrogen. Therefore, metabolism of estrogen has an inhibitory effect on metabolism of caffeine72, 82
4. Caffeine and Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a rapidly progressing neurodegenerative disease, characterized by degeneration of the upper and lower motor neurons, resulting in skeletal muscle atrophy and death by respiratory failure within 3‐5 years of initial symptoms.88, 89, 90 Pathological hallmarks of this disease include the following: progressive muscle weakness, atrophy, and spasticity.91 On a cellular level, excessive stimulation of glutamate receptors leads to a large influx of calcium ions into the postsynaptic neuron, resulting in oxidative stress, oxidative damage, inflammation, and apoptosis.90
4.1. Human studies
In a meta‐analysis incorporating five large cohort studies that examined 1 010 000 men and women (60 years) after an 18‐year follow‐up, caffeine was found not to be associated with the risk of ALS; the results were similar among men and women.92 None of the participants were diagnosed with ALS at the beginning of this study; some developed ALS during the study period. Men consumed 347 mg/d of caffeine and women consumed 250 mg/d of caffeine. As well, consumption of two cups of coffee (274 mg of caffeine) was associated with the same risk of developing ALS as nondrinkers (RR=1.01, CI=0.85‐1.19).
4.2. In vitro and in vivo animal studies
4.2.1. Adenosine A2A receptor ablation and inhibition are beneficial in amyotrophic lateral sclerosis mouse models
A study found that inhibiting A2AR is beneficial in the SOD1G93A mouse model of ALS.88 Adenosine levels are upregulated in the cerebrospinal fluid of progressing human ALS patients, perhaps partially due to upregulated AMPK levels in ALS.88, 93 A2AR is highly expressed in the spinal cord, specifically the nonglial cells, including motor neurons.88 A2AR in the spinal cord of G93A mice is increased threefold compared to wild type. Daily treatment with A2AR antagonist significantly and consistently increased motor neuron survival (71%), slowed down the progressive loss of forelimb grip strength, delayed disease onset (6%), and extended the overall survival (5%). As well, 50% A2AR ablation delayed disease progression and attenuated the progressive loss of forelimb grip strength.88 In addition, co‐activation of A2AR and A1R attenuates the inhibitory effects of A1R.93 In presymptomatic ALS mice, the cross talk between the two receptors is impaired. Thus, in presymptomatic mice, A2AR can be activated by a lower dose of its agonist. A2AR activation results in an increase in intracellular Ca2+ levels, whereas A1R decreases it. An exacerbated A2AR‐mediated action, along with the reported loss of A2AR/A1R interaction, could induce the neuromuscular transmission toward a hyperexcitable adenosinergic tonus that could contribute to the Ca2+‐mediated excitotoxicity in presymptomatic ALS.93 In the presymptomatic mouse model of ALS, the A2AR‐mediated excitatory effects are exacerbated, but this effect disappears in postsymptomatic mice.89 A2AR signaling is also terminated in aged, wild‐type mice, suggesting that disease‐induced early aging of the A2AR influences neuromuscular transmission. Therefore, A2AR inhibition and ablation are in fact beneficial in ALS mouse models.
4.2.2. Caffeine may inhibit Riluzole clearance
Riluzole is the only licensed drug for ALS, and its mechanism of action is through the inhibition of the deleterious effects of an overload of glutamate and other neurotransmitters in the CNS.94 Riluzole is rapidly absorbed, with maximum concentration 1‐2 hours after administration. Its elimination is assumed to be mainly through oxidative metabolism by the CYP1A2 enzyme. There is a significant positive ratio between the expression of CYP1A2 and the clearance of Riluzole.94 Caffeine, another substrate of the CYP1A2 enzyme, prevents N‐hydroxylation of Riluzole via CYP1A2.95 Therefore, although some studies suggest a lack of correlation between ALS risk and caffeine consumption, the effect of caffeine in ALS should be further investigated, especially with the concomitant intake of Riluzole.
5. Caffeine and HUNTINGTON'S Disease
Huntington's disease (HD), an inherited neurodegenerative disorder caused by expanded CAG repeats, is characterized by motor, cognitive, and psychiatric disturbances.41 HD is a hyperkinetic disorder characterized by chorea (jerky, involuntary movements), tremor, dystonia (abnormal muscle tone, resulting in muscle spasm, and abnormal posture), and prominent neuropsychiatric and cognitive changes. Although several cerebral regions of the brain show signs of neurodegeneration, the primary neuropathological hallmark is atrophy of the striatum, specifically the striato‐pallidal neurons that express dopamine receptors.96
5.1. Human studies
One study showed that in 80 HD male and female patients (50 years), caffeine consumption at >190 mg/d in the last 10 years was associated with a 1.6 years earlier age of onset of HD.41
5.2. In vitro and in vivo animal studies
5.2.1. Caffeine treatment improves functional outcome in rat models of Huntington's disease
Chronic quinolinic acid (QA) lesions in rats closely resemble the neurodegeneration in HD.13, 47 Intrastriatal QA administration causes NMDA receptor overstimulation, causing an influx of Ca2+, eventually leading to oxidative stress.13 The mitochondria of the cell take up the excess Ca2+, and excessive Ca2+ can lead to mitochondrial release of pro‐apoptotic factors that cause neuron death. At 40 mg/kg treatment for 7, 14, and 21 days, caffeine completely restored motor function, and when measured at 21 days completely restored body weight in male Sprague Dawley rats administered with QA.13 Furthermore, striato‐pallidal neurons are a population of medium‐sized neurons that degenerate early in HD.47 A2AR is specifically expressed in this region. A2AR antagonist (0.01 mg/kg, but not 1 mg/kg) reduced the loss of motor function in QA‐treated mice, provided neuroprotective effects, and inhibited the QA‐induced increase in glutamate levels.47
5.2.2. Caffeine treatment reduces oxidative stress through increasing antioxidant capacity
In QA administered rats, treatment with caffeine 40 mg/kg for 21 days restored MDA, nitrite, and SOD levels back to levels of the healthy control rats.13 At 10 mg/kg and 20 mg/kg, caffeine reduced the levels of MDA (15% and 42%, respectively) and nitrite (5% and 11%, respectively) and increased the levels of SOD (19% and 62%, respectively) and catalase (19% and 65%, respectively). Therefore, caffeine administration significantly reduced oxidative stress in QA‐treated rats by increasing endogenous antioxidant capacity and decreasing oxidative damage in a dose‐dependent manner. Administration with QA reduced the levels of complex I, II, and III in the mitochondria, but treatment with caffeine (40 mg/kg) restored these levels.13
5.2.3. High dosages of caffeine, as well as adenosine A2A receptor ablation, are detrimental in Huntington's disease animal models
A study that investigated the effects of A2AR gene deletion in a transgenic mouse model of HD found that A2AR knockout moderately, but significantly, worsens motor performance (36%) and reduces survival (15.6%) of these mice.96 As a matter of fact, high dosages of A2AR antagonists or A2AR gene deletion will wipe out the positive effects of A2AR on blood pressure and platelet aggregation, among other effects.47 Presynaptic A2AR controls glutamate release, and blocking these receptors decreases glutamate release.97 However, blocking postsynaptic A2AR is deleterious.96 Interestingly, another study found that blocking A1R exacerbated, whereas blocking A2AR attenuated, the damage to nigrostriatal dopaminergic and GABAergic neurons in mice and rats exposed to mitochondrial inhibition.98 When caffeine (2.5 mmol/L) and glutamate (2.5 mmol/L) were administered together in a model of HD mice, Ca2+ sensitivity to glutamate increased, allowing for glutamate excitotoxicity. However, the blockade of ryanodine receptors attenuated the glutamate‐induced toxicity.99 Since caffeine can act as a ryanodine receptor agonist at high dosages of consumption, this could partially explain the adverse effects of caffeine associated with HD.38, 99 However, these dosages are considered toxic.38 Therefore, the effect of caffeine at dosages close to average intake needs to be further investigated, as caffeine's effect on ryanodine receptors begins to occur at much higher dosages (1520 mg/d) than average consumption (210‐238 mg of caffeine).6, 39 The effect of caffeine and adenosine receptor antagonism in HD is highly dose dependent and needs further investigation.
6. Caffeine and Machado‐Joseph Disease
Machado‐Joseph disease (MJD) results from an increase in the number of CAG codon repeats. It is characterized by an adult age of onset and causes premature death associated with unstable expansion of CAG stretch in the MJD1 gene that encodes polyQ repeat in the corresponding ataxin‐3 protein.100 The clinical hallmarks of the disease are progressive ataxia (loss of control of bodily movements), dysfunction of motor coordination, postural instability, and parkinsonism. Glutamate overstimulation promotes the proteolysis and aggregation of ataxin‐3.101 One study showed that chronic caffeine consumption and genetic A2AR deletion reduced progressive degeneration, neuronal death, neuronal dysfunction, and reactive gliosis, sequestered noxious ataxin‐3, and prevented ataxin‐3‐induced synaptotoxicity.102 Further studies need to explore the effects of caffeine in MJD, at different doses and in men and women.
7. Caffeinated and Decaffeinated Coffee
Several studies have shown that decaffeinated coffee is not associated with the risk of developing neurodegenerative diseases.70, 92, 103 For example, Arendash et al. found that plasma Aβ levels did not decrease with administration of decaffeinated coffee to AD mice, suggesting that caffeine is critical to decreasing plasma Aβ levels in AD.103 As well, several studies have shown that consumption of decaffeinated coffee is not associated with the risk of PD.51, 64, 70, 74 One study investigating the effects of caffeine and ALS also found no effect of decaffeinated coffee on the risk of developing ALS.92 However, other studies show that the protective effects of caffeinated coffee are also present in decaffeinated coffee in non‐neurodegenerative diseases, suggesting a major role of other compounds (polyphenols) in coffee. For example, decaffeinated coffee consumption, as well as caffeinated coffee, had protective effects against heart disease, respiratory disease, stroke, injuries and accidents, diabetes, and infections in 229 119 men and 173 141 women.104
8. Limitations
The dosage of caffeine expressed as cups of coffee in epidemiological studies present challenges in interpretation. In their questionnaires, some epidemiological studies did not specify the actual volume that constitutes a cup, and therefore there is inconsistency in this field when calculating caffeine intake based on an ill‐defined measure of a ‘cup'. Furthermore, meta‐analysis studies also equate a cup of coffee between all studies, even though different studies may have differing volumes for a cup. Therefore, the results drawn from these studies should question the exact daily dosage of caffeine when it is not specified by the investigators. As well, the caffeine content of coffee may vary based on origin, processing methodologies, and brewing techniques, which adds variability to the daily dosage of caffeine.
Epidemiological studies cannot control for some factors, such as emotional, regional, and dietary factors, that may influence the development of neurodegenerative diseases. In addition, in diseases such as ALS and HD, the findings from animal studies do not echo the findings from epidemiological studies. Animal models, such as rats and mice, may be pathophysiologically different compared to humans. Therefore, the effects of caffeine observed at the dosages administered in mice may not accurately depict the effects expected in humans.
Caffeine has differing effects in women vs men, because of the presence of estrogen. There are not enough studies investigating the effect of caffeine at different dosages in women. Due to the interaction of caffeine, estrogen, and CYP1A2, the effects of caffeine observed in men may not accurately reflect the effects of caffeine in women.
9. Future Directions
Epidemiological studies have shown that caffeine consumption is associated with lower risk of developing AD in both men and women. As well, studies in animal models have shown that caffeine has protective effects against oxidative stress, inflammation, and apoptosis. Clinical studies need to investigate the effective, nontoxic, therapeutic dosage of caffeine for patients with AD. Furthermore, studies in humans and mice showed that caffeine administration prior to the onset of AD is beneficial. Hence, clinical studies should aim to investigate caffeine's efficaciousness in those at higher risk of developing AD, prior to diagnosis. It is important to note that the dosages administered in mice resulted in plasma concentrations near toxic levels. Attention should be given to studying caffeine's influence on AD at average consumption levels.
Epidemiological and animal studies have ascertained the beneficial effects of caffeine on the development and progression of PD. Clinical studies need to confirm these findings. Some epidemiological studies showed that PD is inversely associated with caffeine consumption in women, whereas other studies showed no association. It is possible that in PD, caffeine may either be ineffectual in women or that there is a sex‐based differential therapeutic dose given the interaction between estrogen, caffeine, and CYP1A2. Particularly, studies in postmenopausal women who undergo hormone replacement therapy and in premenopausal women with high estrogen levels may inform us about the dynamics of caffeine within an altered hormonal milieu. Furthermore, CYP1A2 enzyme activity varies with factors such as genetics and smoking habits, warranting further research regarding interindividual variability observed upon caffeine consumption. As well, caffeine metabolites are protective in animal models of PD. Therefore, further research on caffeine metabolites can give us a more in‐depth insight into caffeine's protective mechanisms in PD.
Research investigating caffeine intake in patients diagnosed with ALS is needed to ascertain the results of the meta‐analysis of the association of ALS incidence with caffeine intake prediagnosis. Long‐term epidemiological and clinical studies will inform us about caffeine's role on disease onset and progression, survival, and quality of life in ALS patients. A2AR antagonism and partial ablation have been shown to be protective in ALS, suggesting that caffeine may mitigate the progressive decline in functional outcomes and quality of life. Hence, further research is needed to explore the effects of caffeine on the brain and spinal cord of animal models of ALS. These studies can inform us of the effects of caffeine on oxidative stress, antioxidant capacity, inflammation, neurotransmitters, blood‐brain and spinal cord‐brain barrier integrity, calcium homeostasis, and apoptosis, as well as functional outcomes in this disease model.
One epidemiological study showed that caffeine consumption at levels >190 mg/d is associated with a higher risk of developing HD.41 However, studies in animal models show caffeine can restore motor function.13 The discrepancy between the epidemiological and animal studies needs to be further investigated. Perhaps, caffeine has neuroprotective effects at different dosages. Further research needs to educate us about the exact therapeutic dosage of caffeine in the CNS. Additional animal studies examining the effect of caffeine on HD may delineate caffeine's exact mechanism of action in this disease model. This may then inform us further about caffeine's effect on the brain and motor performance in HD.
In terms of MJD, epidemiological studies are needed to advise us on the chronic effects of caffeine consumption on disease development and/or progression. Also, more studies are needed in animal models to understand caffeine's underlying mechanisms associated with MJD. Currently, no conclusion can be drawn about caffeine's effect on MJD, but the results are encouraging.
Caffeine is associated with reduced pain through its inhibition of the adenosine receptors, namely A2AR.29 Chronic caffeine treatment is associated with the upregulation of A1R, which is associated with reducing pain.25, 29 PD and ALS are both associated with higher levels of pain, and hence caffeine's pain‐reducing effects may contribute to improving these patients' quality of life and warrant further investigation.105, 106
Caffeine can also enhance exercise performance in healthy individuals,107 due to its stimulatory effects and its pain‐reducing mechanisms. Caffeine may also enhance the ability to partake in daily activities and improve quality of life for PD, ALS, HD, and MJD patients. Focus needs to be directed toward examining caffeine's potential to improve the quality of life in patients with neurodegenerative diseases with respect to mobility and functional outcomes.
10. Conclusion
Caffeine exerts its effects through different pathways, including the antagonism of adenosine receptors (A1R, A2AR, A2BR, and A3R), inhibition of phosphodiesterase, and activation of ryanodine receptors. However, at average levels of caffeine consumption (~210‐238 mg/d), caffeine's main mechanism of action is antagonism of adenosine receptors. Caffeine consumption, at dosages 3‐5 mg/kg, is associated with a lower risk of AD and PD in both epidemiological and preclinical studies. No such conclusion can be drawn about the effects of caffeine in ALS, HD, and MJD, since there are studies that show beneficial, neutral, or harmful effects of caffeine in all three diseases. Tables 2, 3, 4 summarize the effect of caffeine for each neurodegenerative disease. The epidemiological studies in humans suggest caffeine's preventative role in developing neurodegenerative diseases, namely PD and AD. However, the in vitro and in vivo animal studies suggest that caffeine may also have therapeutic role for patients already diagnosed with the neurodegenerative disease. Further clinical studies are needed to investigate the therapeutic dosage of caffeine in AD and PD patients, as well as those at risk of developing these diseases. Also, further animal studies are needed to understand the underlying mechanisms of caffeine in the CNS of all models of neurodegenerative diseases.
Table 2.
Summary of the human epidemiological studies and the in vitro and in vivo animal studies investigating the effect of caffeine on Alzheimer's disease (AD)
| Alzheimer's disease (AD)—human epidemiological studies | ||||
|---|---|---|---|---|
| Reference | Participants | Duration | Main results | Conclusion |
| van Gelder et al., 2007 |
676 healthy men from Finland, Italy and the Netherlands 75‐77 y |
10 y Mini‐mental state examination to assess global cognitive function |
Coffee: ↓ cognitive decline (54%) Without coffee: 2.6 points of cognitive decline, 1 cup: 1.4 point of cognitive decline 2 cups: 1.3 points of cognitive decline 3 cups: 0.6 points of cognitive decline 4 cups: 1.6 points of cognitive decline Cognitive decline was not reduced for men who consumed >4 cups. |
Consuming coffee was associated with slower cognitive decline in men. Consumption of 3 cups/d was most beneficial. |
| Eskelinen et al., 2009 |
1409 healthy participants 875 women 534 men Midlife: 50.4 y Later in life: 70.1 y |
21 y |
3‐5 cups of coffee: ↓ 65%‐70% risk of dementia and ↓ 62‐64% risk of AD vs 0‐2 cups 3‐5 cups/d of coffee: ↓ risk of dementia in men (OR=0.27, CI=0.08‐0.89) and women (OR=0.51, CI=0.17‐1.52) vs 0‐2 cups/d. In men, >5 cups: ↓ risk of dementia vs low coffee consumption (OR=0.36, CI=0.13‐0.97). |
Moderate coffee consumption at midlife may decrease the risk of developing AD and dementia later in life. |
| Maia & Mendoca, 2002 |
54 patients with probable AD 26 women 28 men 71.2 y 54 healthy controls 26 women 28 men 70.4 y |
20 y preceding diagnosis |
AD patients: average caffeine intake of 74±98 mg Healthy controls: average caffeine intake 199±136 mg Caffeine exposure: ↓ 60% risk of AD (OR=0.40, CI=0.25‐0.67) |
There is an inverse association between caffeine intake and AD |
| Lindsay et al., 2002 |
10 263 Canadian women and men >65 y |
5 y | Daily coffee consumption: ↓ 31% risk of AD (OR=0.69, CI=0.5‐0.96) | Coffee consumption is associated with lower risk of AD in Canadian population. |
| Kim et al., 2015 | 31 479 participants | RR of Caffeine intake from coffee or tea for cognitive disorders (dementia, AD, cognitive impairment, and cognitive decline) was 0.82 (CI=0.67‐1.01) | Meta‐analysis found that caffeine intake from coffee or tea was not associated with the risk of cognitive disorders. May be due to type II error. | |
| Alzheimer's disease—in vitro and in vivo animal studies | ||||
|---|---|---|---|---|
| Reference | Subjects | Treatment | Main results | Conclusion |
| Arendash et al., 2006 |
APPsw mice n=41 transgenic mice n=16 NT mice Females and males 4‐9 mo |
1.5 mg/d (human equivalence of 500 mg/d) of caffeine 5.5 mo of treatment |
Caffeine: complete protection in all cognitive tasks Caffeine: ↓ soluble Aβ1‐40 (37%) and insoluble Aβ1‐42 (32%) in the hippocampus Caffeine: normalized PS‐1 levels and ↓ 50% in BACE levels |
Daily caffeine intake may reduce or delay the risk of developing AD. |
| Arendash et al., 2009 |
APPsw mice n=6 transgenic‐caffeine‐treated n=7 transgenic control n=8 NT control Females and males 18‐19 mo |
1.5 mg/d (human equivalence of 500 mg/d) of caffeine 2 mo of treatment |
Caffeine: ↑ memory vs control (4 wk: ~217%, 5 wk: ~198%) Caffeine: ↓ Aβ deposition in the hippocampus (40%) and the entorhinal cortex (46%) Caffeine: ↓ Aβ1‐42 in cortex (51%) and hippocampus (59%) Caffeine: ↓ Aβ1‐40 in the cortex (25%) and the hippocampus (37%) Caffeine: ↑ PKA levels, and ↓ c‐Raf‐1, NF‐kB pathway activation and BACE‐1 production |
Even with pre‐existing Aβ in APPsw mice, caffeine administration restored memory function and reversed AD pathology, suggesting a therapeutic role of caffeine in well‐established cases of AD. |
| Chu et al. 2012 |
J20 mice n=17 wild type n=14 APP‐control n=9 APP‐CC n=11 APP‐caffeine Males 3 mo |
1.84 mg/d of crude caffeine (CC) or pure caffeine 2 mo of treatment |
Pure caffeine and CC: enhance Morris maze performance CC: ↓ Aβ1‐42 (52%) and plaque number (67%) Aβ with 0.5, 5 and 50 ng/mL CC: 57%, 68%, and 77% ↑ ATP levels vs controls Aβ with pure caffeine and CC: ↓ number of caspase‐3 positive neurons 43% and 48%, respectively |
Pure and crude caffeine can protect against cell death and memory impairment in male mice. |
| Qosa et al., 2012 |
C57BL/6 mice Males n=4 per group 7‐8 wk |
0.8 mg/d of caffeine 3 wk of treatment Plasma concentration: 103±28 μmol/L |
Caffeine: ↑ 20% Aβ clearance | Caffeine enhanced Aβ clearance from the brain of mice, partially explaining the protective effects of caffeine in AD. |
| Dragicevic et al., 2012 |
APPsw mice n=3 transgenic and caffeine n=4 transgenic control n=2 NT and caffeine n=2 NT control Sex not identified 11‐12 mo |
0.6 mg/d of caffeine 1 mo of treatment |
Caffeine: ↑ hippocampal mitochondrial respiration (25%) and ATP levels (46%) Caffeine: ↑ hippocampal mitochondrial membrane potential (78%) and ↓ ROS production (100%). |
Caffeine increases mitochondrial function in APPsw mice. |
| Han et al., 2013 |
APPsw mice n=8 per group Sex not identified 24 mo |
0.75 mg/d or 1.5 mg/d of caffeine 8 wk of treatment |
Caffeine: ↓ escape time (46%‐62%) and ↑ time spent in the target quadrant (32‐46%). Caffeine (0.75 mg/d and 1.5 mg/d): ↑ brain‐derived neurotrophic factors (BDNF) and its receptors (TrkB) |
Chronic caffeine treatment may reverse memory impairment in APPsw mice, and BDNF and its receptor TrkB may be partially responsible. |
| Prasanthi et al., 2011 |
New Zealand white rabbits Male 1.5‐2 y |
Treatment with 0.5 or 30 mg caffeine/d and 2% cholesterol‐enriched diet |
Caffeine (0.5 and 30 mg): ↓ cholesterol‐induced increase in Aβ polypeptide and ↑ cholesterol‐induced decrease in A1R Caffeine (30 mg): ↓ cholesterol‐induced increase in BACE1, tau phosphorylation, ROS generation, and glutathione depletion |
The protective effect of caffeine was dose dependent, and associated with increases in A1R, not decrease in cholesterol levels. |
| Zeitlin et al., 2011 |
APPsw mice n=5‐8 per experimental group Sex not identified 9.5 mo |
1.5 mg/d of caffeine 2 wk of treatment |
Caffeine: ↑ PKA activity in the striatum and ↑ CREB levels by 126% Caffeine: ↓ pERK (striatum: 70%, cortex: 59%) and PJNK (striatum:60%, cortex: 54%) |
Caffeine stimulates pro‐survival pathways and inhibits pro‐apoptotic pathways in the striatum and the cortex. |
| Giunta et al., 2014 | Human neuroblastoma cells |
Pretreatment with 10 μmol/L of caffeine Treatment with AlCl3 |
Caffeine: ↓ phosphorylated IκBα and NF‐κB levels and nuclear translocation of NF‐κB back Caffeine: ↓ ROS production (51%), ↑ SOD (50%) and ↓ MDA (50%) Caffeine: ↓ pro‐apoptotic Bax and ↑ anti‐apoptotic Bcl‐2 levels |
Caffeine has a potential beneficial role in preventing AD in those exposed to aluminum. |
Table 3.
Summary of the human epidemiological studies and the in vitro and in vivo animal studies investigating the effect of caffeine on Parkinson's disease (PD)
| Parkinson's disease (PD)—human epidemiological studies | ||||
|---|---|---|---|---|
| Reference | Participants | Duration | Main results | Conclusion |
| Postuma et al., 2012 |
61 PD patients Placebo: n=31 (19 M, 12 F) Caffeine: n=30 (25 M, 5 F) 65‐68 y |
6 wk 1st three wk: 100 mg caffeine, 2×/d 2nd three wk: 200 mg caffeine, 2×/d |
Improved the total UPDRS (unified Parkinson's disease rating scale) by 4.7 points Improved the motor manifestation by 3.2 points |
Caffeine treatment in PD patients has potential motor benefits |
| Ross et al., 2000 |
8004 American Japanese men 53 y |
27 y | Caffeine >421 mg of caffeine: 5× ↓ risk of developing PD vs nondrinkers, 2.6× ↓ risk vs 124‐208 mg/d, 3.8× ↓ risk vs 209‐287 mg/d, and 2× ↓ risk vs 288‐420 mg/d. | Caffeine has an inverse association with the risk of developing PD. |
| Liu et al., 2012 |
318 260 participants 187 499 women 130 761 men 61 y |
9‐11 y |
Coffee at >5 cups/d: ↓ risk of PD in men (OR=0.70, CI=0.47‐1.04) and women (OR=0.74, CI=0.42‐1.29) vs nonusers Women on hormone therapy: ↓ risk of PD development upon caffeine consumption 129‐511 mg/d OR=0.66 (CI=0.42‐1.05) vs intakes <17.4 mg/d 511‐590 mg/d OR=0.64 (CI=0.39‐1.04) vs intakes <17.4 mg/d >590 mg/d OR=0.53 (CI=0.28‐0.98) vs intakes <17.4 mg/d |
Caffeine has an inverse association with the risk of developing PD. |
| Qi et al., 2014 |
492 722 participants for caffeine Women and men 901 764 participants for coffee Women and men |
For every 200 mg/d increment of caffeine, risk of PD ↓ by 17% Coffee at ~ three cups/d (volume not identified): ↓ risk of PD (RR=0.72, CI=0.65‐0.81) Coffee at two cups/d: 26% ↓ risk of PD vs nonusers Coffee consumption (3 cups/d) ↓ PD risk in men (RR=0.68, CI=0.59‐0.78) and women (RR=0.76, CI=0.63‐0.93) vs nonusers |
Coffee and caffeine consumption have inverse associations with the risk of developing PD. | |
| Palacios et al., 2012 |
63 590 women 69 y 48 532 men 71 y |
8 y |
Men—caffeine at 120 mg/d: ↓ risk of PD by 38% (RR=0.62, CI=0.40‐0.95) vs 9.2 mg/d Men—caffeine at ≥274 mg/d (≥2 cups coffee/d) ↓ risk of PD by ~50% (RR=0.54, CI=0.37‐0.80) vs 9.2 mg/d Men—caffeine at 478 mg/d ↓ risk of PD (RR=0.43, CI=0.26‐0.71) vs 9.2 mg/d Women—caffeine at 435 mg/d (3.2 cups coffee/d) ↓ risk of PD by 40% (RR=0.61, CI=0.34‐1.09) vs 5.6 mg/d. |
Caffeine has a protective effect against the risk of developing PD. |
| Hu et al., 2007 |
15 042 women 64.0 y 14 293 men 62.2 y |
12.9 y |
In men, 0, 1‐4 cups, and >5 cups of coffee (100 mL/cup) had a hazard ratio of 1.00, 0.55 (CI=0.26‐1.15) and 0.41 (CI=0.19‐0.88), respectively, of PD In women, 0, 1‐4 cups and >5 cups of coffee (100 mL/cup) had a hazard ratio of 1.00, 0.50 (CI=0.22‐1.12) and 0.39 (CI=0.17‐0.89), respectively, for PD. |
Coffee drinking is associated with lower risk of developing PD |
| Ascherio et al., 2003 |
77 713 women 30‐55 y |
18 y |
Postmenopausal hormone users + ~1/2 a cup of coffee/d (68 mg/d of caffeine): ↓ 34% risk of PD Postmenopausal hormone users + five cups of coffee/d (688 mg/d of caffeine): ↑ 55% risk among |
Use of postmenopausal hormone therapy was associated with a lower risk of PD in women with low caffeine intake, but it was associated with higher risk of PD in women with high caffeine intake. |
| Parkinson's disease—in vitro and in vivo animal studies | ||||
|---|---|---|---|---|
| Reference | Subjects | Treatment | Main results | Conclusion |
| Bagga et al., 2015 |
PD mice n=10 control n=7 caffeine n=10 MPTP n=8 MPTP and caffeine Males 4 mo |
0.9 mg of caffeine 8 d of caffeine administration 30 min prior to MPTP treatment |
Caffeine: ↓ neuron damage in the striatum Caffeine: ↑ motor function (60.6% improvement in grip strength) |
Pretreatment with caffeine provides partial neuroprotection against severe striatal degeneration in PD. |
| Sonsalla et al., 2012 |
Sprague Dawley rats n=5 per group Males Age not identified |
Oral caffeine (1 g/L in drinking water—equivalent to 60‐80 mg/kg/d in humans and plasma concentrations at ~22 μmol/L) |
Caffeine: ↓ loss of nigral dopamine cell bodies Caffeine at 1 wk or 3 wk: ↓ the loss of nigral cells by 94% and 69%, respectively Caffeine: ↓ microglia activation in the substantia nigra |
Caffeine protects against the loss of nigral dopamine neurons in a chronic progressive rat model of PD. |
| Chen et al., 2001 |
C57BL6 mice n=13 MPTP n=5 saline Male 9 mo |
Caffeine administered 10 min before MPTP administration (5, 10, 20, and 30 mg/kg) |
Caffeine (10 mg/kg): residual dopamine was 40% of control vs 15% of control Caffeine (20 mg/kg): reversed MPTP‐induced dopamine depletion Caffeine at higher dosage caused excessive systemic toxicity. |
There is a potential neural basis for the neuroprotection effects of caffeine in PD. |
| Xu et al., 2006 |
C57BL6 mice n=3‐7 saline treatment n=4‐15 MPTP Females and males Young: 10 wk Old: 6‐9 mo |
10, 20, 40 mg/kg of caffeine treatment Caffeine administration 10 min before MPTP administration |
Caffeine: ↓ MPTP‐induced dopamine loss (38% of the control dopamine levels) in a dose‐dependent manner in young male mice, with maximal effects achieved at 10 mg/kg (78% of the control dopamine levels with caffeine). In old male mice, MPTP‐induced dopamine loss (3% of control without caffeine) ↓ by caffeine pretreatment in a dose‐dependent manner, with 10 mg/kg of caffeine offering the most neuroprotective effect (~30% of control with caffeine). In female mice, caffeine ↓ toxicity only at 40 mg/kg. In ovariectomized mice treated with placebo (striatal dopamine 27% of control), caffeine at 5, 10, 20 mg/kg was neuroprotective. In ovariectomized mice treated with estrogen (striatal dopamine 39% of control), caffeine was neuroprotective only at higher doses (40 mg/kg). In males treated with estrogen, estrogen was neuroprotective, but caffeine in these mice did not attenuate their striatal dopamine loss. |
Estrogen can prevent the neuroprotective effects of caffeine in a model of PD mice. |
| Nakaso et al., 2008 | Human dopaminergic neuroblastoma cell lines | Caffeine concentrations (10 and 100 μmol/L) |
Caffeine ↓cell death and ↓the number of apoptotic nuclei from 13.1% to 9.7% under MPP+‐exposed conditions Caffeine (10 and 100 μmol/L) ↓ caspase 3 in a dose‐dependent manner (21% and 23%, respectively), ↑ Akt phosphorylation (1.96‐fold and 2.96‐fold, respectively). Inhibiting PI3K/Akt pathway abolished caffeine's effects. |
The neuroprotective effects of caffeine involve the PI3K/AKT pathway |
Table 4.
Summary of the human epidemiological studies and the in vivo animal studies investigating the effects of caffeine on amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), and Machado‐Joseph disease (MJD)
| Reference | Participants | Duration | Main results | Conclusion |
|---|---|---|---|---|
| Amyotrophic lateral sclerosis—human meta‐analysis study | ||||
| Fondell et al., 2015 |
1 010 000 participants 494 228 women 60 y 522 775 men 60 y |
18 y At study initiation, participants were not diagnosed with ALS; some developed ALS during study |
Women: consumed 250 mg/d of caffeine Men: consumed 347 mg/d of caffeine Two cups of coffee (274 mg of caffeine): same risk of developing ALS (RR=1.01, CI=0.85‐1.19) vs nonusers. |
Caffeine was found not to be associated with the risk of ALS |
| Huntington's disease—human epidemiological study | ||||
| Simonin et al., 2013 |
80 HD patients Women and men 50 y |
Caffeine consumption over the past 10 y of the patients' lives was assessed, in a self‐survey. | Caffeine consumption (>190 mg/d) → 1.6 y earlier age of onset of HD. | Caffeine consumption at higher dosages may be associated with earlier age of onset for HD. |
| Reference | Subjects | Treatment | Main results | Conclusion |
|---|---|---|---|---|
| Huntington's disease—in vivo animal study | ||||
| Mishra et al., 2014 |
Sprague Dawley rats n=8 for each group Males |
Quinolinic acid (QA) (200 nmol/2 μL saline) administered, followed by chronic caffeine (10, 20, 40 mg/kg) administration Caffeine and QA administration for 21 d |
Caffeine at 40 mg/kg for 7, 14, and 21 d completely ↑ motor function, and body weight Caffeine at 40 mg/kg for 21 d ↓ MDA, ↓ nitrite and ↑ SOD to healthy control levels Caffeine at 10 and 20 mg/kg ↓ MDA (15% and 42%, respectively) and nitrite (5% and 11%, respectively), and ↑ SOD (19% and 62%, respectively) and catalase (19% and 65%, respectively). QA ↓ complex I, II, and III in the mitochondria, but caffeine (40 mg/kg) ↑ these levels. |
Caffeine significantly reduced the oxidative stress in QA‐treated rats by increasing endogenous antioxidant capacity and decreasing oxidative damage, in a dose‐dependent manner. |
| Machado‐Joseph disease—in vivo animal study | ||||
| Gonçalves et al., 2013 |
C57BL6 mice n=6 caffeine n=5 water Males 7 wk |
Caffeine (1 g/L) through drinking water Caffeine administration for 2‐12 wk |
Chronic caffeine and genetic A2AR deletion: ↓ progressive degeneration and neuronal death, neuronal dysfunction and reactive gliosis, sequestered noxious ataxin‐3 and prevented ataxin‐3‐induced synaptotoxicity | Pharmacological and genetic manipulation of A2AR can delay MJD‐associated striatal pathology. |
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
There was no funding for this review.
Kolahdouzan M, Hamadeh MJ. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci Ther. 2017;23:272–290. 10.1111/cns.12684
References
- 1. Xu F, Liu P, Pekar JJ, Lu H. Does acute caffeine ingestion alter brain metabolism in young adults? NeuroImage. 2015;110C:39‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ullrich S, de Vries Y, Kühn S, Repantis D, Dresler M, Ohla K. Feeling smart: effects of caffeine and glucose on cognition, mood and self‐judgment. Physiol Behav. 2015;151:629‐637. [DOI] [PubMed] [Google Scholar]
- 3. Pong NY, Or TCT, Ip NY. Plant alkaloids as drug leads for Alzheimer's disease. Neurochem Int. 2015;89:260‐270. [DOI] [PubMed] [Google Scholar]
- 4. McCusker RR, Goldberger BA, Cone EJ. Caffeine content of specialty coffees. J Anal Toxicol. 2003;27:520‐522. [DOI] [PubMed] [Google Scholar]
- 5. Fox GP, Wu A, Yiran L, Force L. Variation in caffeine concentration in single coffee beans. J Agric Food Chem. 2013;61:10772‐10778. [DOI] [PubMed] [Google Scholar]
- 6. Caffeine . Centre of Addiction and Mental Health. 2013. p. 1.
- 7. Arendash GW, Schleif W, Rezai‐Zadeh K, et al. Caffeine protects Alzheimer's mice against cognitive impairment and reduces brain Beta‐amyloid production. Neuroscience. 2006;142:941‐952. [DOI] [PubMed] [Google Scholar]
- 8. Smith A. Effects of caffeine on human behavior. Food Chem Toxicol. 2002;40:1243‐1255. [DOI] [PubMed] [Google Scholar]
- 9. Ritchie K, Carriere I, de Mendonca A, et al. The neuroprotective effects of caffeine: a prospective population study (the Three City Study). Neurology. 2007;69:536‐545.17679672 [Google Scholar]
- 10. van Gelder BM, Buijsse B, Tijhuis M, et al. Coffee consumption is inversely associated with cognitive decline in elderly European men: the FINE Study. Eur J Clin Nutr. 2007;61:226‐232. [DOI] [PubMed] [Google Scholar]
- 11. Eskelinen MH, Ngandu T, Tuomilehto J, Soininen H, Kivipelto M. Midlife coffee and tea drinking and the risk of late‐life dementia: a population‐based CAIDE study. J Alzheimers Dis. 2009;16:85‐91. [DOI] [PubMed] [Google Scholar]
- 12. Devasagayam TP, Kamat JP, Mohan H, Kesavan PC. Caffeine as an antioxidant: inhibition of lipid peroxidation induced by reactive oxygen species. Biochim Biophys Acta. 1996;1282:63‐70. [DOI] [PubMed] [Google Scholar]
- 13. Mishra J, Kumar A. Improvement of mitochondrial NAD(+)/FAD(+)‐linked state‐3 respiration by caffeine attenuates quinolinic acid induced motor impairment in rats: implications in Huntington's disease. Pharmacol Rep. 2014;66:1148‐1155. [DOI] [PubMed] [Google Scholar]
- 14. Ullah F, Ali T, Kim MO. Caffeine prevents D‐galactose‐induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem Int. 2015;90:114‐124. [DOI] [PubMed] [Google Scholar]
- 15. Carelli‐Alinovi C, Ficarra S, Russo AM, et al. Involvement of acetylcholinesterase and protein kinase C in the protective effect of caffeine against β‐amyloid‐induced alterations in red blood cells. Biochimie. 2016;121:52‐59. [DOI] [PubMed] [Google Scholar]
- 16. Spiff AI, Uwakwe AA. Activation of human erythrocyte glutathione‐s‐ transferase (EC. 2.5.1.18) by caffeine (1,3,7‐ trimethylxanthine). J Appl Sci Environ Manag. 2003;7:45‐48. [Google Scholar]
- 17. Chee HK, Oh SJ. Molecular vibration‐activity relationship in the agonism of adenosine receptors. Genomics Inform. 2013;11:282‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moro S, Deflorian F, Spalluto G, et al. Demystifying the three dimensional structure of G protein‐coupled receptors (GPCRs) with the aid of molecular modeling. Chem Commun (Camb). 2003;24:2949‐2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pedata F, Dettori I, Coppi E, et al. Purinergic signalling in brain ischemia. Neuropharmacology. 2016;104:105‐130. [DOI] [PubMed] [Google Scholar]
- 20. Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastião AM. Adenosine: setting the stage for plasticity. Trends Neurosci. 2013;36:248‐257. [DOI] [PubMed] [Google Scholar]
- 21. Kamp TJ, Hell JW. Regulation of cardiac L‐type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095‐1102. [DOI] [PubMed] [Google Scholar]
- 22. Fredholm BB, Abbracchio MP, Burstock G, et al. Nomenclature and classification of purinoceptors. Pharmacol Rev. 1994;46:143‐156. [PMC free article] [PubMed] [Google Scholar]
- 23. Halldner L, Adén U, Dahlberg V, Johansson B, Ledent C, Fredholm BB. The adenosine A1 receptor contributes to the stimulatory, but not the inhibitory effect of caffeine on locomotion: a study in mice lacking adenosine A1 and/or A2A receptors. Neuropharmacology. 2004;46:1008‐1017. [DOI] [PubMed] [Google Scholar]
- 24. DallIgna OP, Porciúncula LO, Souza DO, Cunha RA, Lara DR. Neuroprotection by caffeine and adenosine A2A receptor blockade of beta‐amyloid neurotoxicity. Br J Pharmacol. 2003;138:1207‐1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shi D, Nikodijevic O, Jacobson KA, Daly JW. Chronic caffeine alters the density of adenosine, adrenergic, cholinergic, GABA, and serotonin receptors and calcium channels in mouse brain. Cell Mol Neurobiol. 1993;13:247‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsutsui S, Schnermann J, Noorbakhsh F, et al. A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. Neurobiol Dis. 2004;24:1521‐1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giunta S, Andriolo V, Castorina A. Dual blockade of the A1 and A2A adenosine receptor prevents amyloid beta toxicity in neuroblastoma cells exposed to aluminum chloride. Int J Biochem Cell Biol. 2014;54C:122‐136. [DOI] [PubMed] [Google Scholar]
- 28. Mohamed RA, Agha AM, Nassar NN. SCH58261 the selective adenosine A2A receptor blocker modulates ischemia reperfusion injury following bilateral carotid occlusion: role of inflammatory mediators. Neurochem Res. 2012;37:538‐547. [DOI] [PubMed] [Google Scholar]
- 29. Sawynok J. Adenosine receptor targets for pain. Neuroscience. 2016;338:1‐18. [DOI] [PubMed] [Google Scholar]
- 30. Mohamed R, Agha A, Abdel‐Rahman A, Nassar N. Role of adenosine A2A receptor in ischemia reperfusion injury: signaling to extracellular signal‐regulated protein kinase. Neuroscience. 2016;314:145‐159. [DOI] [PubMed] [Google Scholar]
- 31. Kaster MP, Machado NJ, Silva HB, et al. Caffeine acts through neuronal adenosine A 2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc Natl Acad Sci. 2015;112:201423088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pedata F, Pugliese AM, Coppi E, et al. Adenosine A2A receptors modulate acute injury and neuroinflammation in brain ischemia. Mediators Inflamm. 2014;2014:805198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dobson KL, Jackson C, Balakrishnan S, Bellamy TC. Caffeine modulates vesicle release and recovery at cerebellar parallel fibre terminals, independently of calcium and cyclic AMP signalling. PLoS One. 2015;10:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu J, Supnet C, Sun S, et al. The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels. 2014;8:230‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fill M, Copell JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893‐922. [DOI] [PubMed] [Google Scholar]
- 36. Yoshimura H. The potential of caffeine for functional modification from cortical synapses to neuron networks in the brain. Curr Neuropharmacol. 2005;3:309‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kong H, Jones PP, Koop A, et al. Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem J. 2009;414:441‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fredholm BB, Bättig K, Holmén J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. 1999;51:83‐133. [PubMed] [Google Scholar]
- 39. Garrett BE, Griffiths RR. The role of dopamine in the behavioral effects of caffeine in animals and humans. Pharmacol Biochem Behav. 1997;57:533‐541. [DOI] [PubMed] [Google Scholar]
- 40. Boswell‐Smith V, Spina D, Page CP. Phosphodiesterase inhibitors. Br J Pharmacol. 2009;147(S1):S252‐S257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simonin C, Duru C, Salleron J, et al. Association between caffeine intake and age at onset in Huntington's disease. Neurobiol Dis. 2013;58:179‐182. [DOI] [PubMed] [Google Scholar]
- 42. LaFerla FM. Calcium dyshomeostasis and intracellular signalling in alzheimer's disease. Nat Rev. 2002;3:862‐872. [DOI] [PubMed] [Google Scholar]
- 43. Basurto‐Islas G, Blanchard J, Tung YC, et al. Therapeutic benefits of a component of coffee in a rat model of Alzheimer disease. Neurobiol Aging. 2014;35:2701‐2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer's disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol. 2002;156:445‐453. [DOI] [PubMed] [Google Scholar]
- 45. Maia L, de Mendonca A. Does caffeine intake protect from Alzheimer ‘s disease ? Eur J Neurol. 2002;9:377‐382. [DOI] [PubMed] [Google Scholar]
- 46. Kim Y, Mi S, Myung S. Caffeine intake from coffee or tea and cognitive disorders : a meta‐analysis of observational studies. Neuroepidemiology. 2015;769:51‐63. [DOI] [PubMed] [Google Scholar]
- 47. Popoli P, Pintor A, Domenici MR, et al. Blockade of striatal adenosine A2A receptor reduces, through a presynaptic mechanism, quinolinic acid‐induced excitotoxicity: possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum. J Neurosci. 2002;22:1967‐1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Laurent C, Burnouf S, Ferry B, et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol Psychiatry. 2014;2016:97‐107. [DOI] [PubMed] [Google Scholar]
- 49. Chu YF, Chang WH, Black RM, et al. Crude caffeine reduces memory impairment and amyloid β1‐42 levels in an Alzheimer's mouse model. Food Chem. 2012;135:2095‐2102. [DOI] [PubMed] [Google Scholar]
- 50. Arendash GW, Mori T, Cao C, et al. Caffeine reverses cognitive impairment and decreases brain amyloid levels in aged alzheimer's disease mice. J Alzheimers Dis. 2009;17:661‐680. [DOI] [PubMed] [Google Scholar]
- 51. Rajendran P, Bhatt A, Manthuruthil S, Pericherla S. Caffeine and Alzheimer's disease. Int J Biol Med Res. 2013;3:3513‐3514. [Google Scholar]
- 52. Qosa H, Abuznait AH, Hill RA, Kaddoumi A. Enhanced brain amyloid‐β clearance by Rifampicin and caffeine as a possible protection mechanism against Alzheimer's disease. J Alzheimers Dis. 2012;31:151‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Han K, Jia N, Li J, Yang L, Min LQ. Chronic caffeine treatment reverses memory impairment and the expression of brain BNDF and TrkB in the PS1/APP double transgenic mouse model of Alzheimer's disease. Mol Med Rep. 2013;8:737‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghoneim FM, Khalaf HA, Elsamanoudy AZ, El‐khair SMA, Helaly AMN. Protective effect of chronic caffeine intake on gene expression of brain derived neurotrophic factor signaling and the immunoreactivity of glial fibrillary acidic protein and Ki‐67 in Alzheimer's disease. Int J Clin Exp Pathol. 2015;8:7710‐7728. [PMC free article] [PubMed] [Google Scholar]
- 55. Zeitlin R, Patel S, Burgess S, Arendash GW, Echeverria V. Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer's transgenic mice. Brain Res. 2011;1417:127‐136. [DOI] [PubMed] [Google Scholar]
- 56. Dragicevic N, Delic V, Cao C, et al. Caffeine increases mitochondrial function and blocks melatonin signaling to mitochondria in Alzheimer's mice and cells. Neuropharmacology. 2012;63:1368‐1379. [DOI] [PubMed] [Google Scholar]
- 57. Sato A, Terata K, Miura H, et al. Mechanism of vasodilation to adenosine in coronary arterioles from patients with heart disease. Am J Physiol Heart Circ Physiol. 2005;288:H1633‐H1640. [DOI] [PubMed] [Google Scholar]
- 58. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921‐923. [DOI] [PubMed] [Google Scholar]
- 59. Prasanthi JRP, Bhanu D, Marwarha G, et al. Caffeine protects against oxidative stress and Alzheimer's disease‐like pathology in rabbit hippocampus induced by cholesterol‐enriched diet. Free Radic Biol Med. 2010;49:1212‐1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boerwinkle E, Utermannt G. Simultaneous effects of the apolipoprotein E polymorphism on apolipoprotein E, apoprotein E and cholesterol metabolism. Am J Hum Genet. 1988;42:104‐112. [PMC free article] [PubMed] [Google Scholar]
- 61. Hall K, Murrell J, Ogunniyi A, et al. Cholesterol, APOE genotype, and Alzheimer disease: an epidemiologic study of Nigerian Yoruba. Neurology. 2006;66:223‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kumar PM, Paing SST, Li H, et al. Differential effect of caffeine intake in subjects with genetic susceptibility to Parkinson's Disease. Sci Rep. 2015;5:15492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bagga P, Chugani AN, Patel AB. Neuroprotective effects of caffeine in MPTP model of Parkinson's disease: a 13C NMR study. Neurochem Int. 2016;92:24‐35. [DOI] [PubMed] [Google Scholar]
- 64. Prediger RDS. Effects of caffeine in Parkinson's disease: from neuroprotection to the management of motor and non‐motor symptoms. J Alzheimers Dis. 2010;20:s205‐s220. [DOI] [PubMed] [Google Scholar]
- 65. Hayashi A, Matsunaga N, Okazaki H, et al. A disruption mechanism of the molecular clock in a MPTP mouse model of parkinson's disease. Neuromolecular Med. 2013;15:238‐251. [DOI] [PubMed] [Google Scholar]
- 66. Postuma RB, Lang AE, Munhoz RP, et al. Caffeine for treatment of Parkinson disease: a randomized controlled trial. Neurology. 2012;79:651‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ross GW, Abbott RD, Petrovitch H, et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA. 2000;283:2674‐2679. [DOI] [PubMed] [Google Scholar]
- 68. Liu R, Guo X, Park Y, et al. Caffeine intake, smoking, and risk of Parkinson disease in men and women. Am J Epidemiol. 2012;175:1200‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Qi H, Li S. Dose‐response meta‐analysis on coffee, tea and caffeine consumption with risk of Parkinson's disease. Geriatr Gerontol Int. 2014;14:430‐439. [DOI] [PubMed] [Google Scholar]
- 70. Palacios N, Gao X, McCullough ML, et al. Caffeine and risk of Parkinson's disease in a large cohort of men and women. Mov Disord. 2012;27:1276‐1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hu G, Bidel S, Jousilahti P, Antikainen R, Tuomilehto J. Coffee and tea consumption and the risk of Parkinson's disease. Mov Disord. 2007;22:2242‐2248. [DOI] [PubMed] [Google Scholar]
- 72. Ascherio A, Chen H, Schwarzschild M, Zhang S, Colditz G, Speizer F. Caffeine, postmenopausal estrogen, and risk of Parkinson's disease. Neurology. 2003;60:790‐795. [DOI] [PubMed] [Google Scholar]
- 73. Greene N, Lassen CF, Rugbjerg K, Ritz B. Reproductive factors and Parkinson's disease risk in Danish women. Eur J Neurol. 2014;21:1168, e68. [DOI] [PubMed] [Google Scholar]
- 74. Checkoway H, Powers K, Smith‐Weller T, Franklin GM, Longstreth WT, Swanson PD. Parkinson's disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am J Epidemiol. 2002;155:732‐738. [DOI] [PubMed] [Google Scholar]
- 75. Xu K, Xu Y, Brown‐Jermyn D, et al. Estrogen prevents neuroprotection by caffeine in the mouse 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine model of Parkinson's disease. J Neurosci. 2006;26:535‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sonsalla PK, Wong L‐Y, Harris SL, et al. Delayed caffeine treatment nigral dopamine neuron loss in a progressive rat model of Parkinson's disease. Exp Neurol. 2012;234:482‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen JF, Xu K, Petzer JP, et al. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson's disease. J Neurosci. 2001;21:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nakaso K, Ito S, Nakashima K. Caffeine activates the PI3K/Akt pathway and prevents apoptotic cell death in a Parkinson's disease model of SH‐SY5Y cells. Neurosci Lett. 2008;432:146‐150. [DOI] [PubMed] [Google Scholar]
- 79. Arita R, Yanagi Y, Honda N, et al. Caffeine increases tear volume depending on polymorphisms within the adenosine A2a receptor gene and cytochrome P450 1A2. Ophthalmology. 2012;119:972‐978. [DOI] [PubMed] [Google Scholar]
- 80. El‐Sohemy A, Cornelis MC, Kabagambe EK, Campos H. Coffee, CYP1A2 genotype and risk of myocardial infarction. Genes Nutr. 2007;2:155‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tassaneeyakul W, Birkett DJ, McManus ME, et al. Caffeine metabolism by human hepatic cytochromes P450: contributions of 1A2, 2E1 and 3A isoforms. Biochem Pharmacol. 1994;47:1767‐1776. [DOI] [PubMed] [Google Scholar]
- 82. Pollock BG, Wylie M, Stack JA, et al. Inhibition of caffeine metabolism by estrogen replacement therapy in postmenopausal women. J Clin Pharmacol. 1999;39:936‐940. [DOI] [PubMed] [Google Scholar]
- 83. Ghotbi R, Christensen M, Roh HK, Ingelman‐Sundberg M, Aklillu E, Bertilsson L. Comparisons of CYP1A2 genetic polymorphisms, enzyme activity and the genotype‐phenotype relationship in Swedes and Koreans. Eur J Clin Pharmacol. 2007;63:537‐546. [DOI] [PubMed] [Google Scholar]
- 84. Josse AR, Da Costa LA, Campos H, El‐Sohemy A. Associations between polymorphisms in the AHR and CYP1A1‐CYP1A2 gene regions and habitual caffeine consumption. Am J Clin Nutr. 2012;96:665‐671. [DOI] [PubMed] [Google Scholar]
- 85. Begas E, Kouvaras E, Tsakalof A, Papakosta S, Asprodini E. In vivo evaluation of CYP1A2, CYP2A6, NAT‐2 and xanthine oxidase activities in a Greek population sample by the RP‐HPLC monitoring of caffeine metabolic ratios. Biomed Chromatogr. 2007;21:190‐200. [DOI] [PubMed] [Google Scholar]
- 86. Cornelis MC, Byrne EM, Esko T, et al. Genome‐wide meta‐analysis identifies six novel loci associated with habitual coffee consumption. Mol Psychiatry. 2015;20:647‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Xu K, Xu YH, Chen JF, Schwarzschild MA. Neuroprotection by caffeine: time course and role of its metabolites in the MPTP model of Parkinson's disease. Neuroscience. 2010;167:475‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ng SK, Higashimori H, Tolman M, Yang Y. Suppression of adenosine 2a receptor (A2aR)‐mediated adenosine signaling improves disease phenotypes in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2015;267:115‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nascimento F, Pousinha PA, Correia AM, Gomes R, Sebastião AM, Ribeiro JA. Adenosine A2A receptors activation facilitates neuromuscular transmission in the pre‐symptomatic phase of the SOD1(G93A) ALS mice, but not in the symptomatic phase. PLoS One. 2014;9:e104081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Moghimi E, Solomon JA, Gianforcaro A, Hamadeh MJ. Dietary vitamin D3 restriction exacerbates disease pathophysiology in the spinal cord of the G93A mouse model of amyotrophic lateral sclerosis. PLoS One. 2015;10:e0126355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39‐59. [DOI] [PubMed] [Google Scholar]
- 92. Fondell E, ÉiJ O, Fitzgerald KC, et al. Intakes of caffeine, coffee and tea and risk of amyotrophic lateral sclerosis: results from five cohort studies. Amyotroph Lateral Scler Frontemporal Degener. 2014;2015:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nascimento F, Sebastião AM, Ribeiro JA. Presymptomatic and symptomatic ALS SOD1 (G93A) mice differ in adenosine A1 and A2A receptor‐mediated tonic modulation of neuromuscular transmission. Purinergic Signal. 2015;11:471‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Van Kan HJM, Groeneveld CJ, Kalmijn S, Spieksma M, Van Den Berg LH, Guchelaar HJ. Association between CYP1A2 activity and riluzole clearance in patients with amyotrophic lateral sclerosis. Br J Clin Pharmacol. 2005;59:310‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sanderink G‐J, Bournique B, Stevens J, Petry M, Martinet M. Involvement of human CYP1A isoenzymes in the metabolism and drug interactions of riluzole in vitro. J Pharmacol Exp Ther. 1997;282:1465‐1472. [PubMed] [Google Scholar]
- 96. Mievis S, Blum D, Ledent C. A2A receptor knockout worsens survival and motor behaviour in a transgenic mouse model of Huntington's disease. Neurobiol Dis. 2011;41:570‐576. [DOI] [PubMed] [Google Scholar]
- 97. Blum D, Hourez R, Galas M, Popoli P, Schiffmann SN. Adenosine receptors and Huntington's disease: implications for pathogenesis and therapeutics. Lancet Neurol. 2003;2:366‐374. [DOI] [PubMed] [Google Scholar]
- 98. Alfinito PD, Wang S‐P, Manzino L, Rijhsinghani S, Zeevalk GD, Sonsalla PK. Adenosinergic protection of dopaminergic and GABAergic neurons against mitochondrial inhibition through receptors located in the substantia nigra and striatum, respectively. J Neurosci. 2003;23:10982‐10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chen X, Wu J, Lvovskaya S, Herndon E, Supnet C, Bezprozvanny I. Dantrolene is neuroprotective in Huntington's disease transgenic mouse model. Mol Neurodegener. 2011;6:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li X, Liu H, Fischhaber PL, Tang T‐S. Toward therapeutic targets for SCA3: insight into the role of Machado‐Joseph disease protein ataxin‐3 in misfolded proteins clearance. Prog Neurobiol. 2015;132:34‐58. [DOI] [PubMed] [Google Scholar]
- 101. Koch P, Breuer P, Peitz M, et al. Excitation‐induced ataxin‐3 aggregation in neurons from patients with Machado‐Joseph disease. Nature. 2011;480:543‐548. [DOI] [PubMed] [Google Scholar]
- 102. Gonçalves N, Simões AT, Cunha RA, De Almeida LP. Caffeine and adenosine A2A receptor inactivation decrease striatal neuropathology in a lentiviral‐based model of Machado‐Joseph disease. Ann Neurol. 2013;73:655‐666. [DOI] [PubMed] [Google Scholar]
- 103. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer's disease. J Alzheimers Dis. 2010;20(Suppl. 1):117‐126. [DOI] [PubMed] [Google Scholar]
- 104. Freedman ND, Park Y, Abnet CC, Hollenbeck A, Sinha R. Association of Coffee Drinking with total and all cause mortality. N Engl J Med. 2012;366:1891‐1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ford B. Pain in Parkinson's disease. Mov Disord. 2012;27:485‐491. [DOI] [PubMed] [Google Scholar]
- 106. Handy CR, Krudy C, Boulis N, Federici T. Pain in amyotrophic lateral sclerosis: a Neglected aspect of disease. Neurol Res Int. 2011;2011:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bell DG, McLellan TM. Exercise endurance 1, 3, and 6 h after caffeine ingestion in caffeine users and nonusers. J Appl Physiol. 2002;93:1227‐1234. [DOI] [PubMed] [Google Scholar]