

Summary
Background
Autism spectrum disorders (ASDs) are a heterogeneous group of neurodevelopmental disorders that display complicated behavioral symptoms.
Methods
Using gene expressing profiling and the weighted gene co‐expression network analysis (WGCNA), we studied genes coregulated by similar factors such as genetic variants or environmental effects in the hippocampus in an animal model of autism.
Results
From microarray data, we identified 21,388 robustly expressed genes of which 721 genes were found to be differently expressed in the valproic acid‐treated group compared to the control group. WGCNA identified multiple co‐expression modules known to associate with cognitive function, inflammation, synaptic, and positive regulation of protein kinase activating. Many of these modules, however, have not been previously linked to autism spectrum disorders which included G‐protein signaling, immunity, and neuroactive ligand–receptor interaction pathway. The downregulation of the highly connected (hub) genes Taar7h and Taar7b in neuroactive ligand–receptor interaction pathway was validated by qRT‐PCR. Immunoblotting and immunohistochemistry further showed that TAAR7 expression was downregulated not only in valproic acid‐treated animals, but also BTBR T+tf/J mice.
Conclusions
This study highlights the advantages of gene microarrays to uncover co‐expression modules associated with autism and suggests that Taars and related gene regulation networks may play a significant role in autism.
Keywords: Autism, Gene, Microarray, Neuroactive ligand–receptor interaction pathway, Taar, VPA, WGCNA
Introduction
Autism spectrum disorders (ASDs) are a heterogeneous group of neurodevelopmental disorders that display different levels of interpersonal barriers, language development disorders, narrow interests, and rigid routines that are accompanied by reduced global cognitive function scores 1, 2. Synaptic alterations may underlie the abnormal social and cognitive behaviors observed in ASDs 3.
Gene expression microarrays, combined with network analysis, provide a powerful new tool for studying complex processes such as brain diseases 4, 5, 6, 7. This method is particularly useful for studying the interaction between genes and the environment 8.
The weighted gene co‐expression network analysis (WGCNA) is a commonly used gene network analysis method 8, 9, 10. Rather than calculating the pairwise correlations between gene expression profiles, WGCNA examines groups of genes whose expression profiles are highly correlated across samples and give rise to several gene modules containing subsets of genes with similar biochemical and functional properties as well as anatomical localization 7, 9, 11.
In this study, we used WGCNA to examine gene expression in an animal model of autism; that is, rats prenatally exposed to valproic acid (VPA). We first used differential gene expression analysis to identify differentially expressed genes in VPA‐treated rats and then conducted the weighted gene co‐expression network analysis to gain a system‐level understanding of the correlations between gene expression profiles. GO analysis and pathway analysis were used to further reveal the functional role of the genes. This study highlights the advantages of gene microarrays to uncover co‐expression modules associated with autism and help identify specific molecular networks responsible for different pathological processes in autism.
Materials and Methods
Animals and Prenatal Valproic Acid Treatment
Female Sprague Dawley (SD) rats weighing 200–230 g were housed under climate controlled conditions on a 12‐h light/dark cycle (8:00 AM to 8:00 PM) and provided with standard food and water. An acclimation period of at least 1 week was provided before initiating the experimental protocols. All experimental protocols and animal handling procedures were performed in accordance with the guidelines of the National Institutes of Health (NIH, USA) for the care and use of laboratory animals, and these protocols were approved by the Committees for Animal Experiments of Zhejiang University in China. Valproic acid model was prepared as previously described 12. Briefly, female rats were mated, and treated rats received a single intraperitoneal injection of 600 mg/kg sodium valproic acid (VPA, 2‐propylpentanoic acid sodium salt, Sigma, St. Louis, MO, USA) on E12.5. Dams were housed individually and allowed to raise their own litters. The offspring were weaned on postnatal day (PND) 22, and male and female offspring were then housed separately. The experiments were performed on the male offspring. Additionally, BTBR T+tf/J mice obtained from the Model Animal Research Center of Nanjing University were used as an autism model for validation.
Microarray Data Processing
Tissue samples from rat (50 days old) hippocampus were collected and frozen in liquid nitrogen immediately. Total RNA extracted from these samples was hybridized against a common reference pool on Agilent Rat Gene Expression 4 × 44K Microarray Kit. The arrays were scanned, and raw data were acquired by Agilent Feature Extraction (FE) software (version 9.5.3). Microarray raw data (.txt files) were imported into R × 64 v. 3.0.1 13, then normalized and statistically analyzed with the Bioconductor 14 packages Agi4 × 44PreProcess 15 and samr 16 according to their guidelines. Briefly, after quality check, the microarray probes were filtered and their Processed Signal was log2 transformed and normalized by quantile normalization within and between arrays according to Agi4 × 44Preprocess. Then, the probes were tested for differential expression using unpaired t‐test. Genes with uncorrected P value <0.05 and FC (fold changes) ≥2 were accepted to be differentially expressed. Finally, SAM (significance analysis of microarrays) with FDR (false discovery rate) <5% and FC ≥1.5 was applied to select genes further considered in the network construction.
Weighted Gene Co‐Expression Network Analysis (WGCNA)
Probes filtered by SAM last step (see above) were used in this analysis. With the help of R package WGCNA and its build‐in blockwiseModules function 17, 18, we constructed an unsigned weighted gene co‐expression network. Co‐expression network construction began with the measure of adjacency matrix defined by assigning a power to pairwise Pearson correlation. In our case, we chose the power of 28 to satisfy the scale‐free topology criterion 18. This power adjacency implements “soft” threshold to define connected neighbors of a gene; that is, the network is weighted. From the adjacency matrix, a topological overlap matrix (TOM) is generated to measure the gene dissimilarity and used as input for identifying gene modules based on average linkage hierarchical clustering. Each gene corresponds to a branch of the hierarchical dendrogram, and modules are clustered by cutting branches referring to a cutoff height. In our data, the minimum module size is set to 50 genes. According to WGCNA, the first principal component of each module is denoted as module eigengene (ME), and modules whose eigengenes are more than 0.9 correlated will be remerged. Each module is related to the treatment (control or VPA‐treated) by calculating the correlation between module eigengenes and the treatment. Correlation between gene expression values in each module and its module eigengene is also computed to measure the module membership, kME, which is the basis for hub gene selecting. To visualize module network, the connectivity of genes within each module is calculated (by softConnectivity function in WGCNA package) and the TOM of the top 200 genes is selected. Top 500 of the resulting gene pairs ranked by weighted network edges were plotted using Visant 19.
Gene Ontology and Pathway Enrichment Analysis
We performed a gene ontology (GO) enrichment analysis for network modules using the DAVID database (http://david.abcc.ncifcrf.gov) 20. For all analysis, the background was set to Rattus norvegicus, and the enrichment threshold was P < 0.05 (modified Fisher Exact P value).
Semi‐Quantitative PCR
Total RNA (2.5 μg) extracted from eight rats' hippocampus (four controls and four VPA‐treated autism rats) was reverse‐transcribed using PrimeScript Reverse Transcriptase (Takara, Shiga, Japan) and random hexa nucleotide primers (Takara), followed by the cDNA (50 ng) polymerase chain reaction (PCR). Then, 1% agarose gel stained with Gel‐red (Biotium) was applied to separate PCR products. The details of used primers are given in Table S11.
Quantitative RT‐PCR
qRT‐PCR was performed in 96‐well plates using a Mastercycler® ep realplex machine (Eppendorf, Hamburger, Germany). 0.5 μg real‐time PCR was conducted in 10 μL volume containing SYBR® Premix Ex Taq™ (Takara) and primers at a concentration of 0.5 mM each. β‐actin levels were used as an internal control. PCR condition was 95°C for 2 min, 40 cycles of each 95°C for 15 second, 55°C for 15 second, and 68°C for 25 second. Melting curve analysis was performed with the default settings on the instrument from 50 to 85°C. The Ct values were automatically calculated using commercial software with the Mastercycler® ep realplex. The details of primer sequences are given in Table S11.
Western Blotting Analysis
Tissue samples of hippocampus from rats (P14) were collected, and membrane fractionation was performed according to reported protocol 21. Briefly, tissues were homogenized, and cytosolic fraction was first precipitated by centrifugation (10 min, 1000 × g, 4°C) and then re‐suspended in extraction buffer (0.05 mol/L Tris‐HCl pH 7.4, 2% Triton X‐100, 100 mM sodium chloride, and protease/phosphatase inhibitor). Membrane parts were obtained by centrifugation (13,000 × g, 10 min) after 30 min on ice. The extracted membrane proteins were probed with the TAAR7 antibody (NB110‐74882, Novus Biologicals, Littleton, CO, USA), TAAR7D antibody (NB110‐74883, Novus Biologicals), and caveolin‐1 monoclonal antibody (Sigma, St. Louis, MO, USA).
Immunohistochemistry
Brain slices from BTBR T+tf/J mice were fixed in 4% formaldehyde/PBS as previously described 22. Slices were treated without Triton X‐100 and labeled with TAAR7 antibody (NB110‐74882, Novus Biologicals). The 3D surface visualization of grayscale intensity was plotted through ImageJ v1.50H and its “Interactive 3D Surface Plot” plug‐in.
Statistical Analysis
Data are represented as the means ± SEMs. Statistical significance was determined with two‐tailed t‐test. The results were considered significant when P < 0.05. All gene sets overlap were analyzed in Matlab (Mathworks Inc., Natick, MA, USA).
Results
Gene Expression in the Hippocampus of VPA‐Treated Rats
Lower global cognitive function scores are a common symptom of autism spectrum disorders which is closely associated with hippocampal dysfunction (Figure 1A) 12. Compared to control animals, VPA‐treated rats showed an abnormal hippocampal protein expression, which may be responsible for their deviant behavioral phenotype 12. Hierarchical clustering dendrogram was used to cluster animal samples. Each sample stands for a single tight‐clustering branch of the dendrogram. Total RNA expression in hippocampus of both VPA‐treated and control rats was classified into distinct two clusters (Figure 1B), indicating that there is no outlier of arrays. Principal component analysis (PCA) was also performed to identify potential outliers and further proved that there is no outlier among samples (Figure 1C).
Figure 1.
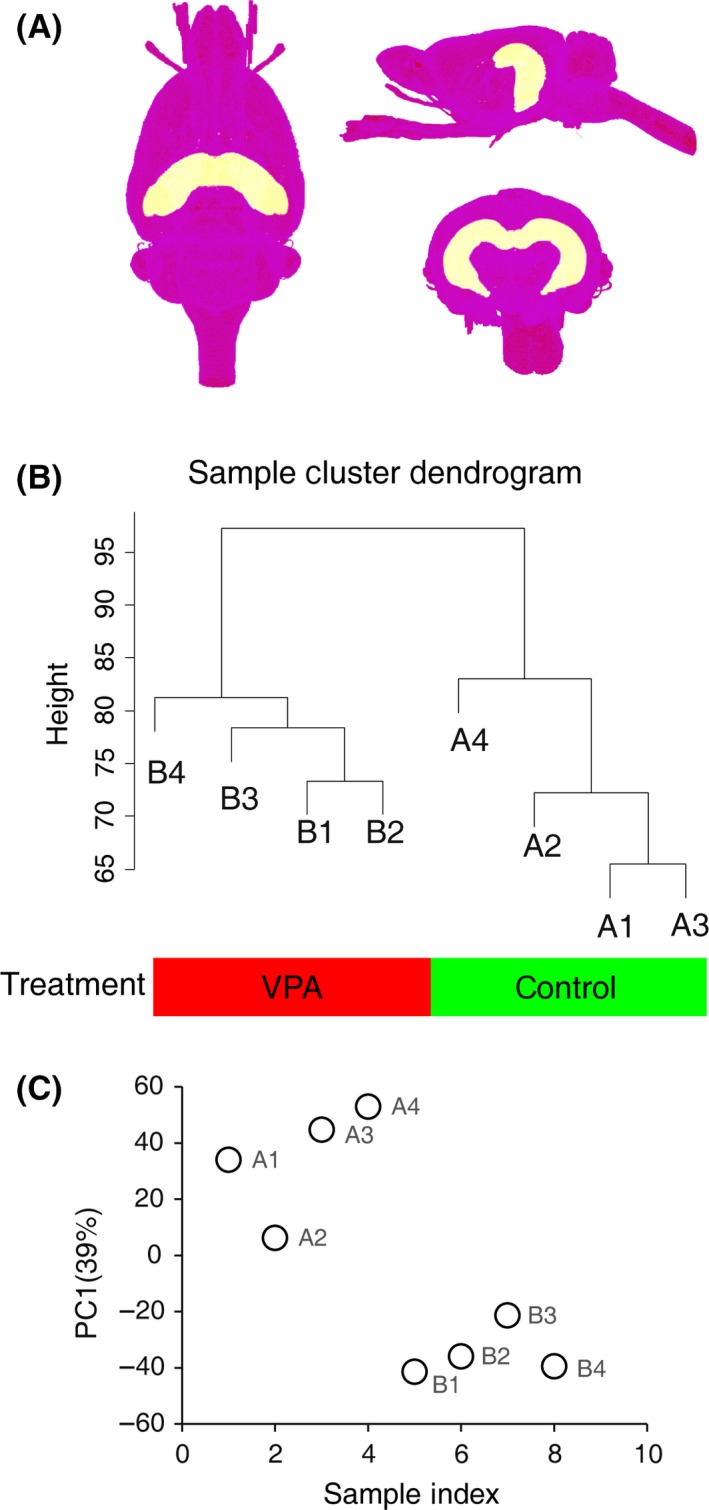
Gene expression data in hippocampus of VPA‐treated rat. (A) 3D views of hippocampus in the brain. The atlas data are from 47. (B) Hierarchical clustering of samples on all genes in rats' hippocampus. (C) Principal component analysis (PCA) of gene expression profiles of all samples.
Identification of Gene Expression Changes in the Hippocampus of VPA‐Treated Animals Using Classic Fold Change Analysis
We initially identified 21,388 robustly expressed genes from microarray data (Table S1), and 721 genes were found to be differently expressed between the VPA‐treated and control group when threshold was set to fold change ≥2 and t‐test P value ≤0.05 (Figure 2A,B; Table S2), most of which were downregulated. These data demonstrated a large number of genes related or potentially related to autism.
Figure 2.
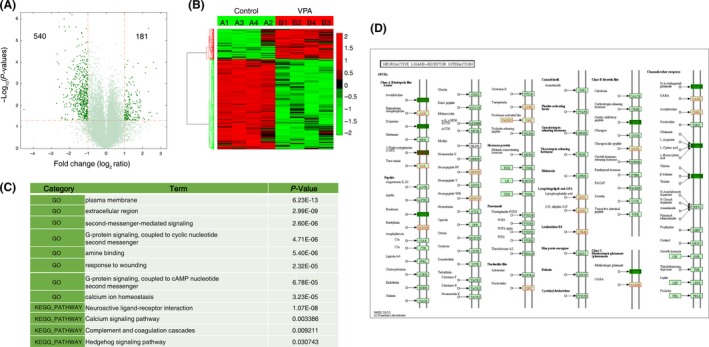
Gene expression changes in the hippocampus of VPA‐treated rats. (A) Volcano plots depicting fold change versus P‐values for gene expression between VPA‐treated and control rats. (B) Heatmap of all differentially expressed genes between VPA‐treated and control rats. The threshold was set to fold change ≥2 and t‐test P value ≤0.05 as illustrated in A. Data were standardized along the rows. (C) Relevant gene ontology categories of differentially expressed genes. (D) Upregulated (green) or downregulated (red) genes in neuroactive ligand–receptor interaction pathway.
To characterize biological themes present in these differentially expressed genes, we employed gene ontology analysis using David (http://david.abcc.ncifcrf.gov/) to determine enrichment of GO categories (Table S3). Several potentially important observations emerged (Figure 2C), and the enriched terms include plasma membrane, G‐protein signaling, amine binding, and calcium signaling.
Notably, one KEGG signaling pathway, the neuroactive ligand–receptor interaction pathway, is highlighted. Its members including Taar7h, Taar7b, Taar5, Taar8c, and Gpr156 show significant alterations in expression (Figure 2C,D; Table S4). Another analysis through WebGestalt (WEB‐based Gene SeT AnaLysis Toolkit) showed similar results (Figure S1).
Gene Subset Established by SAM for WGCNA
Although the differentially expression analysis points out several potential key alterations in biological and molecular functions coupled with VPA‐treated rats, further system‐level framework study is essential to reveal the underlying organization of gene co‐expression network. We chose WGCNA to conduct this analysis.
WGCNA examines groups of genes whose expression profiles are highly correlated across samples and give rise to several gene modules containing subsets of genes with similar biochemical and functional properties as well as anatomical localization 7, 9, 11. A gene subset containing 2338 genes was first identified by SAM (FDR < 5%, FC ≥ 1.5; Table S5, Figure S2A). These genes included all differently expressed genes identified above (Figure 3A). We also compared our dataset to genes listed in SFARI (https://gene.sfari.org/autdb/Welcome.do), a database for autism research. We identified 723 genes from SFARI that may be associated with autism (up to June 16, 2015, Table S6), 603 of which were included in our microarray probes. This comparison returned an enrichment in autism‐related genes for the gene subset selected by SAM (Figure S2B,C; Fisher's exact test, P < 0.001).
Figure 3.
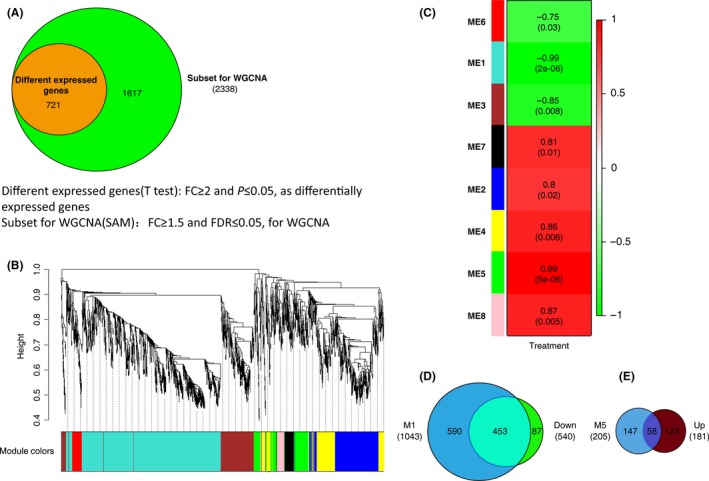
Weighted gene co‐expression network analysis (WGCNA). (A) Gene subset containing 2338 genes is selected for WGCNA analysis (SAM, FC ≥ 1.5 and FDR ≤ 0.05), which totally includes differently expressed genes (FC ≥ 2 and t‐test P ≤ 0.05). (B) Gene dendrogram and clustered modules coded by different colors. (C) Relationship between modules and treatment. Eight modules were clustered by WGCNA (ME1–ME8). ME, module eigengene, the 1st principal component of each module. Numbers in each cell represent the correlations between the corresponding ME and the treatment, with P‐values in round brackets. (D) M1, the module 1, contains 1043 genes and has an overlap of 453 genes with downregulated genes in Figure 2A. (E) M5, the module 5 contains 205 genes and has an overlap of 58 genes with upregulated genes in Figure 2A.
WGCNA Identifies Multiple Co‐Expression Modules Which Enriched in G‐Protein Signaling, Synaptic, Immunity, and Neuroactive Ligand–Receptor Interaction Pathway
We constructed a co‐expression network using the dataset generated by SAM and compared data from VPA‐treated and control rats. Gene expression profiles were classified into eight modules (ME1‐8) with the expression levels of each module summarized by the first principle component (the module eigengene, ME) 9 (Figure 3B,C; Figure S2D–K), most of which were significantly correlated with VPA‐induced autism (correlation > 0.85, P < 0.05). Two of these modules were of particular interest: module 1 (ME1, denoted as M1) and module 5 (ME5, denoted as M5) whose module eigengenes are most correlated with VPA treatment (Figure 3C).
M1 contains 1043 genes with a high correlation between its module membership and gene significance for VPA treatment (Figure 4A,B; Table S5). The M1 eigengene was down‐expressed in VPA‐treated autism cases, indicating that genes in this module were downregulated in the autistic hippocampus. Genes in M1 also highly overlapped most downregulated genes in differently expressed genes (Figures 2A,B and 3D). GO and pathway analysis of this module demonstrated that it was enriched in membrane signaling transductions, especially the neuroactive ligand–receptor interaction pathway (Figure 4C, Table S7, Figure S4). An advantage of network analysis over standard analysis for differential expressed genes is that it allows one to infer the functional relevance of genes based on their network position. Notable highly connected genes (hub) in this module, including Taar7h, Taars7b, and Cyp27b1, may be the key genes that could represent response to VPA‐induced autism at transcriptional level (Figure 4D). Cyp27b1 is one of the cytochrome P450 enzyme families, and it is linked to autism because of its essential role in vitamin D metabolism 23. Arntl2, another hub, is recently reported to affect sleep in patients with ASD 24. These findings verify the network analysis to some extent.
Figure 4.
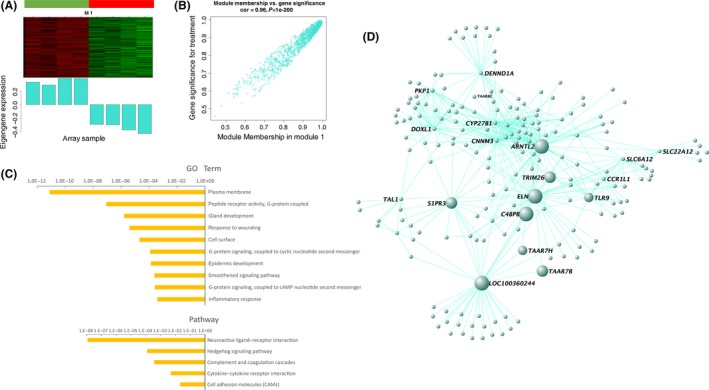
Gene expression network analysis in M1. (A) Heatmap of gene expression profiles of M1 (up panel) and corresponding eigengene was down‐expressed in VPA‐treated autism cases (bottom panel). (B) Gene significance (y axis) versus module membership of genes in M1 (x axis). Gene significance measures the correlation between genes and VPA‐treated autism cases. Module membership measures the correlation between genes and module eigengenes. (C) GO and pathway analysis of M1. The results showed enrichment in membrane signaling transductions and neuroactive ligand–receptor interaction pathway. (D) Co‐expression network of top 500 gene–gene interactions among genes in M1. The hub genes, which has highest correlation with the module eigengene, appear in large node size. Genes are connected by an edge if the correlation between their expression profiles is significant. Taar7h and Taars7b, belonging to members of neuroactive ligand–receptor interaction pathway, are highlighted.
M5 contains 205 genes, 58 of which overlap upregulated genes in differently expression analysis (Figures 2A,B, 3E and 5A,B). Although it has a high correlation with VPA‐induced autism, M5 has only eight genes overlapped with SFARI genes (Figure 3C; Table S8). Genes in this module are more likely to be related to vesicle, immunity, endopeptidase activity, and cation binding (Figure 5C; Table S9; Figure S5). The pathway analysis revealed that genes in this module are enriched mainly in cancer pathway (Figure 5C,D). Serpine1, one of the hub genes in this module (Figure 5D),has also been reported correlation with autism 25.
Figure 5.
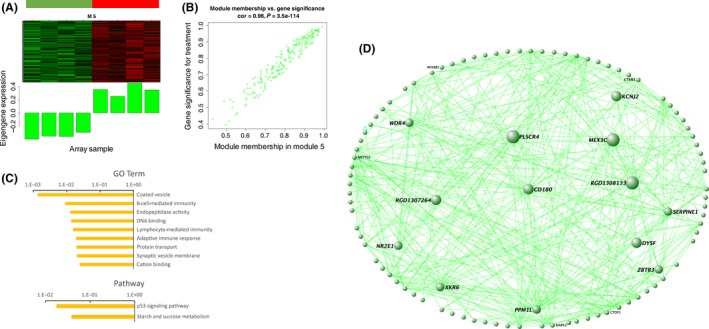
Gene expression network analysis in M5. (A) Heatmap of gene expression profiles of M5 (up panel) and corresponding eigengene was upregulated in VPA‐treated autism cases (bottom panel). (B) Gene significance (y axis) versus module membership of genes in M5 (x axis) with a correlation coefficient 0.96 to each other. (C) GO and pathway analysis of M5. It was enriched in plasma membrane, secretion, kinase activity, and synapse. (D) Co‐expression network of gene–gene interactions among genes in M5.
Module 2 (M2) is also a high correlated module including 357 genes (Figure 3C; Figures S3 and S6A,B). It has a relatively high overlap with upregulated genes in VPA‐treated rats (Figure 2A,B; Figure S3) and the highest overlap with the SFARI autism‐related genes (Table S8). These upregulated genes are enriched in terms of plasma membrane, secretion, kinase activity, and synapse (Figures S6C and S7; Table S10). Similar to M1, M2 suggests the importance of the neuroactive ligand–receptor interaction pathway in VPA‐treated rats (Figure S6C; Table S10). The hubs, Gabra3, Cadm1, Bcl2, and Magel2, are genes included in the SFARI (Figure S6D; Table S6). Other hubs, like Ndn and Nos1, are also related to autism 26, 27.
Validation of TAARs at the Gene and Protein Levels
Neuroactive ligand–receptor interaction pathway stands out in both M1 and M2. Gabra3, one of the hubs in M2, belongs to neuroactive ligand–receptor interaction pathway, and it is known as autism‐related genes. In M1, Taars7b is the outstanding one belonging to neuroactive ligand–receptor interaction pathway, and another gene of Taars family, Taars7h, is also one of the hubs. To confirm the expression level of these two Taars genes, we employed PCR and qRT‐PCR to validate the results of microarray analysis. Taar7h and Taars7b showed significant low expression in VPA‐treated hippocampus, which was in accordance with the microarray results (Figure 6A,B). Two other genes in Taars family, Taar1 and Taar8a, showed no difference between control and VPA‐treated rats (Figure S8A).
Figure 6.
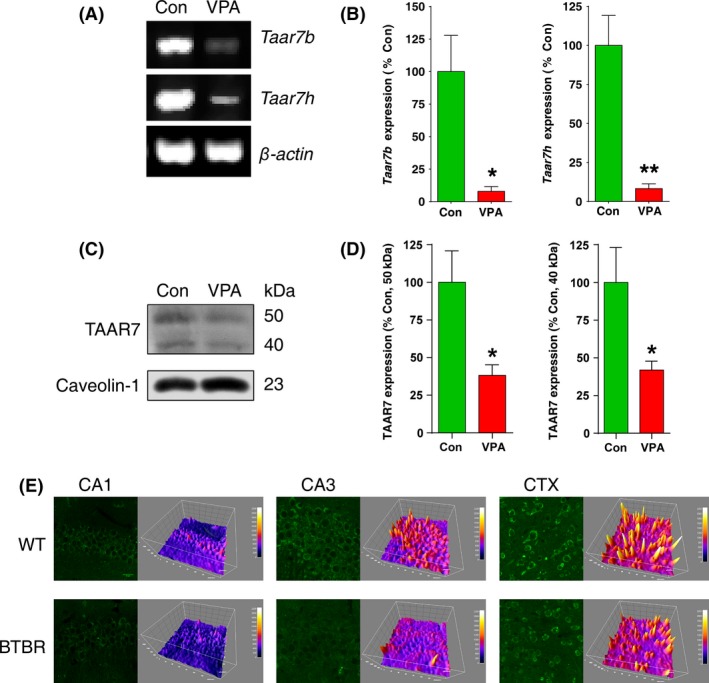
Validation for microarray analysis. Taars genes expression in control and VPA‐treated autism rats (P50) by PCR (A) and qRT‐PCR (B) *P < 0.05, **P < 0.01, versus control rats, n = 4. (C) TAAR7 protein expression in control and VPA‐treated rats (P14). (D) Quantification of (C) was performed by densitometry. The data are expressed as the densitometry ratio of the control (mean ± SEM). *P < 0.05, versus control rats, n = 6. (E) Fluorescent immunohistochemical staining of TAAR7 in hippocampus and brain cortex of BTBR T+tf/J mice. Scale bars: 20 μm.
We next investigated the TAAR7s protein expression in P14 of VPA‐treated rats. The TAAR7 antibody used here recognizes rat TAAR7A, B, C, E, G, H containing proteins coded by the two hubs of genes. As shown in Figure 6C,D, TAAR7s protein levels, which was detected in the membrane fraction, were significantly decreased in VPA‐treated rats in comparison with control animals (Figure 6C,D). However, TAAR7D was not detectable in either VPA‐treated rats or control animals (Figure S8B). The BTBR T+tf/J mouse is an animal model of autism spectrum disorders with abnormal behaviors that resemble the autism‐relevant behaviors 12, 28. Immunohistochemical staining showed distributions of TAAR7 in the soma of pyramidal neurons in the hippocampal CA1, CA3, and cortex region and a decrease in intensity in the hippocampus and cortex in BTBR T+tf/J mice (Figure 6E).
Discussion
Autism spectrum disorders are a common and heritable neurodevelopmental condition with marked genetic heterogeneity 29, 30. Microarray approaches have been used to explore differences in gene expression between different divergent patient populations 31, 32. The present study used DNA microarray analysis to investigate differential gene expression and to uncover co‐expression modules potentially associated with autism.
Others and we previously reported that disorders of intracellular biochemical events in the hippocampus may underlie ASD‐mediated cognitive deficits 12, 33, 34. However, the general mapping of intercellular signaling is still largely lacking. Using VPA‐treated rat model, we profiled gene expression in the hippocampus and found significant changes; compared to the control group, at least 721 genes were affected in VPA‐exposed rats.
We used GO and pathway analysis to understand the functional role of these alterations and found that most affected genes were related to plasma membrane, G‐protein signaling, and neuroactive ligand–receptor interaction pathway. Kitagishi et al. 35 reported the importance of several protein kinases that regulate the membrane trafficking involved in autism and ADHD, suggesting new targets for therapeutic intervention. In addition, children with autism exhibit a dysregulation in alterations in stimulatory G proteins signaling pathways and oxytocin levels 36.
Weighted gene co‐expression network analysis was used to reveal the underlying functional interaction between affected genes 37, 38, 39. We constructed a co‐expression network using the dataset screened by SAM and obtained eight modules (denoted as M1 to M8) affected by the VPA treatment. All the eight modules identified by WGCNA were significantly correlated with VPA treatment, of which M5 and M1 showed the highest correlation. M1 contains 1043 genes with a high correlation between its module membership and gene significance for VPA treatment. M5 contains 205 genes, eight of which overlapped with SFARI genes. Genes in M5 are more likely to be related to vesicle, immunity, endopeptidase activity, and cation binding. This result suggests that the identified autism‐related gene groups might be helpful to elucidate the mechanisms of dysfunction of the core phenotype during pathological process of autism.
Neuroactive ligand influences neuronal function by binding to intracellular receptors that can act as transcription factors and regulate gene expression 40, 41. Pharmacological intervention at the neuroactive ligand–receptor interaction gene set leads to significant reduction in aversive memory 42. In the present study, most significant enrichment showed neuroactive ligand–receptor interaction pathway. Tarrs are the key drivers, hub genes of functional modules belonging to neuroactive ligand–receptor interaction pathway. We employed PCR and qRT‐PCR to validate the results of microarray, as well as immunoblotting and immunostaining. It further demonstrated the reliability of the gene microarray analysis. Indeed, trace amine‐associated receptors (Taars) are vertebrate olfactory receptors. However, as most are “orphan receptors,” Taars are still poorly understood 43.
Our results highlight several advantages of different methods for gene microarray analysis. The statistical power of the original analysis was limited owing to the need to correct for multiple comparisons 44. In contrast, the network analysis changes the experimental unit to modules, thus reduces the dimensionality of the data, and therefore significantly increased our ability to screen out subsets with homologous expression among the multiple genes 45. More importantly, network analysis provides information on the relationships between genes. Specifically, we identified a set of genes that share similar functions and are connected to one another, some of which have already been shown to play an important role in autism. Several genomewide association studies (GWASs) of ASDs have been published, and three signaling networks, regulating steroidogenesis, neurite outgrowth, and (glutamatergic) synaptic function, respectively, have been identified 46.
This study identified a number of candidate genes potentially responsible for autism including Taar7h and Taars7b. Some of the identified genes, such as those in Module 1, are novel with respect to autism. Our results may have important implications in diagnosis, intervention, and prevention of ASD.
Conclusions
In conclusion, we have used differential gene expression analysis to identify differently expressed genes in VPA‐treated autism rats and weighted gene co‐expression network analysis to elucidate modules and genes that may play an important role in autism. This analysis highlights the advantages of using large‐scale gene expression analysis to investigate the molecular basis of autism and suggests that Taars and related gene regulation networks may play a significant role in autism.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Differential gene expression analysis using WebGestalt (WEB‐based Gene SeT AnaLysis Toolkit).
Figure S2. Weighted Gene Co‐Expression Network Analysis (WGCNA).
Figure S3. Venn diagram, depicting the overlap between genes in each module and differently expressed genes (Eigengenes in M1, M3 and M6 are down‐regulated, they overlap with down‐regulated genes in differentially expressed genes selected by FC ≥ 5 and t test P value ≤0.05, as shown in figure 2. Left modules overlap with the up regulated genes).
Figure S4. Analysis of M1 genes through WebGestalt with GO categories (A) and pathway enrichment (B). The result is similar to the analysis through DAVID database.
Figure S5. Analysis of M5 genes through WebGestalt with GO categories (A) and pathway enrichment (B). The result is similar to the analysis through DAVID database.
Figure S6. Gene expression network analysis in M2.
Figure S7. Functional analysis of M2 through WebGestalt with GO categories (A) and pathway enrichment (B). The result is similar to DAVID database.
Figure S8. Validation of TAARs genes and protein expression.
Table S1. All gene expression profiles of our microarray data.
Table S2. Differentially expressed genes identified by fold change ≥2 and t test P value ≤0.05.
Table S3. GO and pathway analysis for differentially expressed genes using David.
Table S4. Differentially expressed genes included in neuroactive ligand‐receptor interaction pathway.
Table S5. Genes selected by SAM (FDR < 5%, FC ≥ 1.5) with module label attached to each gene clustered by WGCNA.
Table S6. Genes listing in SFARI (up to June 16, 2015). Excluding a small part of genes, 603 of them can be found in all our microarray data.
Table S7. GO and pathway analysis for M1.
Table S8. Gene overlap between each module and SFARI.
Table S9. GO and pathway analysis for M5.
Table S10. GO and pathway analysis for M2.
Table S11. Primer information for PCR and qRT‐PCR.
Acknowledgments
This work was supported in part by National Natural Science Foundations of China (81402908, 81403024). Zhejiang Provincial Natural Science Foundation of China (LQ13H310001).
The first two authors contributed equally to this work.
Contributor Information
Feng Han, Email: changhuahan@zju.edu.cn.
Ying‐Mei Lu, Email: lufx@zju.edu.cn.
References
- 1. Lord C, Cook EH, Leventhal BL, Amaral DG. Autism spectrum disorders. Neuron 2000;28:355–363. [DOI] [PubMed] [Google Scholar]
- 2. Oswald DP, Haworth SM. Autism spectrum disorders In: Alfiee MB, Cheryl SA, Nirbhay NS, editors. Handbook of mental health in African American youth. New York: Springer, 2016;271–285. [Google Scholar]
- 3. Codagnone MG, Podesta MF, Uccelli NA, Reines A. Differential local connectivity and neuroinflammation profiles in the medial prefrontal cortex and hippocampus in the valproic acid rat model of autism. Dev Neurosci 2015;37:215–231. [DOI] [PubMed] [Google Scholar]
- 4. Mirnics K, Middleton FA, Lewis DA, Levitt P. Analysis of complex brain disorders with gene expression microarrays: schizophrenia as a disease of the synapse. Trends Neurosci 2001;24:479–486. [DOI] [PubMed] [Google Scholar]
- 5. Miklos GL, Maleszka R. Microarray reality checks in the context of a complex disease. Nat Biotechnol 2004;22:615–621. [DOI] [PubMed] [Google Scholar]
- 6. Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci 2012;32:1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011;474:380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Farber CR. Identification of a gene module associated with BMD through the integration of network analysis and genome‐wide association data. J Bone Miner Res 2010;25:2359–2367. [DOI] [PubMed] [Google Scholar]
- 9. Zhao YT, Goffin D, Johnson BS, Zhou Z. Loss of MeCP2 function is associated with distinct gene expression changes in the striatum. Neurobiol Dis 2013;59:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DiLeo MV, Strahan GD, den Bakker M, Hoekenga OA. Weighted correlation network analysis (WGCNA) applied to the tomato fruit metabolome. PLoS One 2011;6:e26683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malki K, Tosto MG, Jumabhoy I, et al. Integrative mouse and human mRNA studies using WGCNA nominates novel candidate genes involved in the pathogenesis of major depressive disorder. Pharmacogenomics 2013;14:1979–1990. [DOI] [PubMed] [Google Scholar]
- 12. Tian Y, Yabuki Y, Moriguchi S, et al. Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J Pineal Res 2014;56:1–11. [DOI] [PubMed] [Google Scholar]
- 13. R Core Team . R: A language and environment for statistical computing. Version 3.0.1 ed; 2013.
- 14. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lopez‐Romero P. Agi4x44PreProcess: PreProcessing of Agilent 4x44 array data. R package version 1.20.0 ed; 2013.
- 16. Tibshirani R, Chu G, Narasimhan B, Li J. samr: SAM: Significance Analysis of Microarrays. R package version 2.0. ed; 2011.
- 17. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang B, Horvath S. A general framework for weighted gene co‐expression network analysis. Stat Appl Genet Mol Biol 2005;4:e17. [DOI] [PubMed] [Google Scholar]
- 19. Hu Z, Mellor J, Wu J, Yamada T, Holloway D, Delisi C. VisANT: data‐integrating visual framework for biological networks and modules. Nucleic Acids Res 2005;33:W352–W357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dennis GJ, Sherman BT, Hosack DA, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 2003;4:P3. [PubMed] [Google Scholar]
- 21. Lu YM, Gao YP, Tao RR, et al. Calpain‐dependent ErbB4 cleavage is involved in brain ischemia‐induced neuronal death. Mol Neurobiol 2016;53:2600–2609. [DOI] [PubMed] [Google Scholar]
- 22. Han F, Chen YX, Lu YM, et al. Regulation of the ischemia‐induced autophagy‐lysosome processes by nitrosative stress in endothelial cells. J Pineal Res 2011;51:124–135. [DOI] [PubMed] [Google Scholar]
- 23. Currenti SA. Understanding and determining the etiology of autism. Cell Mol Neurobiol 2010;30:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang Z, Matsumoto A, Nakayama K, et al. Circadian‐relevant genes are highly polymorphic in autism spectrum disorder patients. Brain Dev 2016;38:91–99. [DOI] [PubMed] [Google Scholar]
- 25. Campbell DB, Li C, Sutcliffe JS, Persico AM, Levitt P. Genetic evidence implicating multiple genes in the MET receptor tyrosine kinase pathway in autism spectrum disorder. Autism Res 2008;1:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Connor CM, Crawford BC, Akbarian S. White matter neuron alterations in schizophrenia and related disorders. Int J Dev Neurosci 2011;29:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakatani J, Tamada K, Hatanaka F, et al. Abnormal behavior in a chromosome‐engineered mouse model for human 15q11‐13 duplication seen in autism. Cell 2009;137:1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mercier F, Kwon YC, Douet V. Hippocampus/amygdala alterations, loss of heparan sulfates, fractones and ventricle wall reduction in adult BTBR T+ tf/J mice, animal model for autism. Neurosci Lett 2012;506:208–213. [DOI] [PubMed] [Google Scholar]
- 29. Dell'Osso L, Dalle LR, Cerliani C, Bertelloni CA, Gesi C, Carmassi C. Unexpected subthreshold autism spectrum in a 25‐year‐old male stalker hospitalized for delusional disorder: a case report. Compr Psychiatry 2015;61:10–14. [DOI] [PubMed] [Google Scholar]
- 30. Brown AB, Elder JH. Communication in autism spectrum disorder: a guide for pediatric nurses. Pediatr Nurs 2014;40:219–225. [PubMed] [Google Scholar]
- 31. Myers EM, Bartlett CW, Machiraju R, Bohland JW. An integrative analysis of regional gene expression profiles in the human brain. Methods 2015;73:54–70. [DOI] [PubMed] [Google Scholar]
- 32. Hu H, Dai Y. A model‐based approach to transcription regulatory network reconstruction from time‐course gene expression data. Conf Proc IEEE Eng Med Biol Soc 2014;2014:4767–4770. [DOI] [PubMed] [Google Scholar]
- 33. Codagnone MG, Podesta MF, Uccelli NA, Reines A. Differential local connectivity and neuroinflammation profiles in the medial prefrontal cortex and hippocampus in the valproic acid rat model of autism. Dev Neurosci 2015;37:215–231. [DOI] [PubMed] [Google Scholar]
- 34. Provenzano G, Pangrazzi L, Poli A, Sgado P, Berardi N, Bozzi Y. Reduced phosphorylation of synapsin I in the hippocampus of Engrailed‐2 knockout mice, a model for autism spectrum disorders. Neuroscience 2015;286:122–130. [DOI] [PubMed] [Google Scholar]
- 35. Kitagishi Y, Minami A, Nakanishi A, Ogura Y, Matsuda S. Neuron membrane trafficking and protein kinases involved in autism and ADHD. Int J Mol Sci 2015;16:3095–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jacobson JD, Ellerbeck KA, Kelly KA, et al. Evidence for alterations in stimulatory G proteins and oxytocin levels in children with autism. Psychoneuroendocrinology 2014;40:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holtman IR, Raj DD, Miller JA, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co‐expression meta‐analysis. Acta Neuropathol Commun 2015;3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller JA, Woltjer RL, Goodenbour JM, Horvath S, Geschwind DH. Genes and pathways underlying regional and cell type changes in Alzheimer's disease. Genome Med 2013;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Allen JD, Xie Y, Chen M, Girard L, Xiao G. Comparing statistical methods for constructing large scale gene networks. PLoS One 2012;7:e29348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rupprecht R, Koch M, Montkowski A, et al. Assessment of neuroleptic‐like properties of progesterone. Psychopharmacology 1999;143:29–38. [DOI] [PubMed] [Google Scholar]
- 41. Xu LM, Li JR, Huang Y, Zhao M, Tang X, Wei L. AutismKB: an evidence‐based knowledgebase of autism genetics. Nucleic Acids Res 2012;40:D1016–D1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Papassotiropoulos A, de Quervain DJ. Failed drug discovery in psychiatry: time for human genome‐guided solutions. Trends Cogn Sci 2015;19:183–187. [DOI] [PubMed] [Google Scholar]
- 43. Ferrero DM, Wacker D, Roque MA, Baldwin MW, Stevens RC, Liberles SD. Agonists for 13 trace amine‐associated receptors provide insight into the molecular basis of odor selectivity. ACS Chem Biol 2012;7:1184–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu Y, Huggins P, Bar‐Joseph Z. Cross species analysis of microarray expression data. Bioinformatics 2009;25:1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Satterthwaite TD, Vandekar SN, Wolf DH, et al. Connectome‐wide network analysis of youth with Psychosis‐Spectrum symptoms. Mol Psychiatry 2015;20:1508–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poelmans G, Franke B, Pauls DL, Glennon JC, Buitelaar JK. AKAPs integrate genetic findings for autism spectrum disorders. Transl Psychiatry 2013;3:e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Papp EA, Leergaard TB, Calabrese E, Johnson GA, Bjaalie JG. Waxholm Space atlas of the Sprague Dawley rat brain. Neuroimage 2014;97:374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Differential gene expression analysis using WebGestalt (WEB‐based Gene SeT AnaLysis Toolkit).
Figure S2. Weighted Gene Co‐Expression Network Analysis (WGCNA).
Figure S3. Venn diagram, depicting the overlap between genes in each module and differently expressed genes (Eigengenes in M1, M3 and M6 are down‐regulated, they overlap with down‐regulated genes in differentially expressed genes selected by FC ≥ 5 and t test P value ≤0.05, as shown in figure 2. Left modules overlap with the up regulated genes).
Figure S4. Analysis of M1 genes through WebGestalt with GO categories (A) and pathway enrichment (B). The result is similar to the analysis through DAVID database.
Figure S5. Analysis of M5 genes through WebGestalt with GO categories (A) and pathway enrichment (B). The result is similar to the analysis through DAVID database.
Figure S6. Gene expression network analysis in M2.
Figure S7. Functional analysis of M2 through WebGestalt with GO categories (A) and pathway enrichment (B). The result is similar to DAVID database.
Figure S8. Validation of TAARs genes and protein expression.
Table S1. All gene expression profiles of our microarray data.
Table S2. Differentially expressed genes identified by fold change ≥2 and t test P value ≤0.05.
Table S3. GO and pathway analysis for differentially expressed genes using David.
Table S4. Differentially expressed genes included in neuroactive ligand‐receptor interaction pathway.
Table S5. Genes selected by SAM (FDR < 5%, FC ≥ 1.5) with module label attached to each gene clustered by WGCNA.
Table S6. Genes listing in SFARI (up to June 16, 2015). Excluding a small part of genes, 603 of them can be found in all our microarray data.
Table S7. GO and pathway analysis for M1.
Table S8. Gene overlap between each module and SFARI.
Table S9. GO and pathway analysis for M5.
Table S10. GO and pathway analysis for M2.
Table S11. Primer information for PCR and qRT‐PCR.