

Summary
Aims
This study investigated the neuroprotective properties of icariin (an effective component of traditional Chinese herbal medicine Epimedium) on neuronal function and brain energy metabolism maintenance in a triple‐transgenic mouse model of Alzheimer's disease (3 × Tg‐AD).
Methods
3 × Tg‐AD mice as well as primary neurons were subjected to icariin treatment. Morris water maze assay, magnetic resonance spectroscopy (MRS), Western blotting, ELISA, and immunohistochemistry analysis were used to evaluate the effects of icariin administration.
Results
Icariin significantly improved spatial learning and memory retention in 3 × Tg‐AD mice, promoted neuronal cell activity as identified by the enhancement of brain metabolite N‐acetylaspartate level and ATP production in AD mice, preserved the expressions of mitochondrial key enzymes COX IV, PDHE1α, and synaptic protein PSD95, reduced Aβ plaque deposition in the cortex and hippocampus of AD mice, and inhibited β‐site APP cleavage enzyme 1 (BACE1) expression. Icariin treatment also decreased the levels of extracellular and intracellular Aβ1‐42 in 3 × Tg‐AD primary neurons, modulated the distribution of Aβ along the neurites, and protected against mitochondrial fragmentation in 3 × Tg‐AD neurons.
Conclusions
Icariin shows neuroprotective effects in 3 × Tg‐AD mice and may be a promising multitarget drug in the prevention/protection against AD.
Keywords: Alzheimer's disease, Icariin, Magnetic resonance spectroscopy, Mitochondrial dysfunction, N‐acetylaspartate
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease that is clinically characterized by a gradual and global decline of cognitive functions manifesting as memory loss and changes in behavior and personality. Histopathological investigations of AD brains have revealed multiple cellular changes in the brain, including extracellular amyloid‐beta (Aβ) deposits, intracellular neurofibrillary tangles (NFTs), synaptic loss, mitochondrial abnormalities, and inflammatory responses 1, 2, 3.
Recent studies have demonstrated that mitochondrial deficits occur early in AD pathogenesis, even before plaque formation, indicating that mitochondrial dysfunction is an important factor in the early developmental stages of AD pathology 4, 5, 6, 7. Decreased expression and activity of crucial mitochondrial enzymes cytochrome c oxidase (COX) and pyruvate dehydrogenase (PDH) have been detected in postmortem brain tissue from patients with AD as well as AD animal models 7, 8, 9, 10, 11, 12. The lack of acetyl‐CoA due to PDH defects leads to the reduction in oxidative phosphorylation and a decrease in brain metabolite N‐acetylaspartate (NAA), the synthesis of which is dependent on mitochondrial energy state and the synthesis of acetyl‐CoA 13. NAA is found in relatively high concentrations in the brain, and it displays one of the most prominent peaks in nuclear magnetic resonance (NMR) spectra. 1H magnetic resonance spectroscopy (MRS) is a noninvasive technique that allows the in vivo measure of metabolic changes in the brain and hence has great potential for the diagnosis of brain disorders and monitoring the disease progression 14, 15, 16. NAA could serve as a MRS marker for neuronal viability, and a decrease in NAA level could reflect neuronal dysfunction or neuronal loss 13, 17, 18.
Epimedium is a Chinese herbal medicine that has been traditionally used as an aphrodisiac and antirheumatic remedy in China, Korea, and Japan 19. Icariin (C33H40O15; molecular weight: 676.65, ICA), a natural product isolated from Epimedium herba, is the major pharmacologically active component of this herb. Recent studies have reported that icariin can reduce the production of insoluble Aβ fragments, inhibit abnormal tau protein phosphorylation, and improve spatial learning ability and memory in Alzheimer's disease model rats 20, 21, 22, 23. Only a few studies have prospectively investigated the association between brain metabolite levels and mitochondrial functions in AD. Therefore, the purpose of this study was to assess brain metabolism and mitochondrial key enzyme expression/energy production in an AD mouse model and explore the potential anti‐AD treatment effect of icariin.
Materials and Methods
Drugs and Reagents
Icariin (99% purity) for neuronal cell treatment was purchased from National Institute for the Control of Pharmaceutical and Biological Products in China, and the icariin (98% purity) for drug chow diet (65 mg/kg, which is equivalent to 20 μmol/kg/day for mice administration for 6 months from 2 months of age) preparation was obtained from Nanjing Langze Pharmaceutical Company. All cell culture reagents were purchased from Invitrogen. Protease inhibitor cocktail, penicillin–streptomycin, poly‐D‐lysine, Percoll, Triton X‐100, and DMSO were from Sigma‐Aldrich (St. Louis, MO, USA). Papain was from Worthington. ATP bioluminescence assay kit was from Promega Co. (Madison, WI, USA). Aβ1–42 ELISA kit was from Beijing Airan Technology Co. Ltd. (Beijing, China). The antibodies of Aβ1‐42, BACE1, COX IV, PSD95, synaptophysin, MAP2, β‐tublin, β‐actin, and VDAC were purchased from Abcam. PDHE1α and GAPDH antibodies were purchased from Santa Cruz Biotechnology. All other reagents are of reagent grade.
Animals and Treatment
All animal experiments were complied with Animal Care and Institutional Ethical Guidelines in China. 3 × Tg‐AD mice harboring human transgenes APP (SWE), PS1 (M146V), and Tau (P301L) under the control of Thy1.2 promoters were purchased from Jackson Laboratory and housed under standard laboratory conditions with a 12‐h day/night cycle at room temperature of 22°C. Thirty female transgenic (Tg) mice at 2 months of age were divided into two groups and either administrated with normal diet or with icariin through drug chow diet for 6 months. Fifteen age‐matched female nontransgenic (NTg) mice with the same genome background were administrated with normal diet as normal control.
Behavioral Tasks
Morris Water Maze
The Morris water maze assay 24 was applied on 3 × Tg‐AD mice at 8 months of age to evaluate the learning and memory abilities of the mice for 5 days (d) with continuous icariin treatment. The apparatus and the test procedure were performed as previously described 25. Briefly, the apparatus consisted of a stainless steel circular tank (122 cm in diameter) with a translucent acrylic platform (10 cm in diameter) located in the tank in a fixed position. The tank was virtually divided into four equal quadrants identified as northeast, northwest, southeast, and southwest. The tank was filled with water up to 0.5 cm above the top of the platform and the water was made opaque with milk. A number of visible cues were placed around the room. The mouse was gently released facing the tank wall into the water from one of the four desired starting positions (north, south, east, or west). The swimming path of each mouse was tracked using Watermaze MT‐200 software (MPEG‐4 Image).
Place Navigation Test
Spatial training of the hidden platform in the water maze was performed for 5 consecutive days, with 4 trials per day. The intertrial interval was 2 h. In each trial, each mouse was given 90 second to find the hidden platform. If a mouse failed to find the platform within that time, it was manually placed on the platform and allowed to remain there for 20 second.
Probe Trial
To assess short‐term and long‐term memory at the end of learning, two probe trials were given, one at 2 h and another at 24 h after the last training trial (day 5). In each probe trial, the platform was removed and the mice were allowed to swim for 90 second in search of it.
Magnetic Resonance Spectroscopy (MRS)
MRS measurements were taken at the Wuhan Institute of Physics and Mathematics, Chinese Academy of Science, using a Bruker Biospec 7T/30 cm horizontal bore MRS scanner equipped with a ClinScan console (Bruker Biospin, Germany). During the experiments, mice were anesthetized using 1.0–1.5% isoflurane and O2/N2O and positioned in a stereotactic holder to immobilize the head. Body temperature was maintained at 37°C by warm water circulation. Measurements were taken on animals in the drug chow diet/normal diet Tg groups and the NTg group at 8 months of age. VOI (volume of interest) of 1.5 × 2 × 1 mm for 1H MRS spectra was positioned in the hippocampus. Single‐voxel 1H‐MRS was performed with point‐resolved 1H spectroscopy (PRESS) technique with TR = 2500 ms and TE = 20 ms. Quantification of the metabolite concentration was performed using Topspin 2.1 software package. Five brain metabolites that represent the functional metabolic processes were analyzed: N‐acetylaspartate (NAA), choline (Cho), creatine (Cr), myo‐inositol (MI), and lactate (Lac). The ratios of NAA/Cr, Cho/Cr, MI/Cr, and Lac/Cr were calculated. Statistical analysis was performed by SPSS 13.0 software package (SPSS Company, Chicago, IL, USA), with significant differences between means identified using paired two‐tailed t‐test. A P value <0.05 was considered statistically significant.
Upon completion of behavioral test and MRS determination of brain metabolites, mice were sacrificed, and the brains were then removed and bisected in the midsagittal plane. One hemi‐brain was used for ATP extraction, total protein sample preparation, and brain mitochondria isolation; the other hemi‐brain was fixed in 4% paraformaldehyde for immunohistochemistry assay.
Brain Mitochondria Isolation
Brain mitochondria were isolated from the brain tissue following previously established protocol. Briefly, brain cortices were minced and placed in ice‐cold mitochondrial isolation buffer (320 mM sucrose, 1 mM EDTA, 10 mM Tris–HCl, and Protease Inhibitor Mixture Set I from Calbiochem, pH 7.4). The tissue homogenization, separation and density gradient centrifugation were conducted following Yao’ s description 7. The final mitochondrial pellet was resuspended with mitochondrial isolation buffer and stored at −80°C for later protein analysis.
Primary Neuronal Culture
Primary hippocampal neurons were obtained from postnatal (P0‐P1) pups of NTg and 3 × Tg‐AD mice. Briefly, hippocampi were dissected from the brain and digested with 2 mg/mL papain for 30 min at 37°C. Digested tissue was triturated with pipetting in DMEM with 10% FBS. Dissociated cells were cultured in the neurobasal medium with 2% B27 supplement, 0.5 mM l‐glutamine, and 50 U/mL penicillin–streptomycin using poly‐D‐lysine‐coated 6‐well cell culture plates at a density of 0.5 × 106 cells/per well; and for confocal microscope imaging, cells were seeded on 13‐mm glass coverslips precoated with poly‐D‐lysine and incubated in 24‐well cell culture plates at a density of 1.5 × 105 cells/per well. The medium was completely replaced after 4 h, and then, half of the medium was replaced every 3 days. At day 10, neurons were treated with 20 μM icariin (1000 × dilution of 20 mM DMSO dissolved icariin stock solution by culture medium) for 24 h. Neurons from NTg mice or from 3 × Tg‐AD mice treated with the culture medium containing 0.1% DMSO were set up as normal control and AD control, respectively.
Immunohistochemistry/Immunocytochemistry Assay
For immunohistochemistry analysis, brain specimens were paraffin‐embedded, and coronal sections were cut at 6 μm thick each before mounted onto silane‐coated slides. Immunohistochemistry was performed using a standard protocol. Briefly, brain sections were blocked with 5% goat serum and 0.3% Triton X‐100 in PBS before incubation with primary antibody against Aβ1‐42 (1:200) overnight at 4°C, followed by washing and incubation with secondary biotinylated antibody for 1 h at room temperature. Images were taken using an optical microscope (OLYMPUS BX51, Tokyo, Japan). For immunocytochemistry, primary neuronal cultures were administrated with icariin and incubated for 24 h and then fixed in the cell culture plate with 4% paraformaldehyde in PBS. Cells were then permeabilized with 0.2% Triton X‐100 in PBS, and nonspecific binding was blocked with 10% goat serum in PBS. Primary antibodies including Aβ1‐42 (1:200), PDHE1α (1:100), PSD95 (1:1000), and MAP2 (1:8000) were applied followed by incubation with species‐specific secondary antibodies conjugated to Dylight‐488 or Dylight‐594, respectively. The coverslips were then washed with PBS and sealed to slides. Cells were visualized using a confocal microscope (OLYMPUS FV1000). Image J software was used to analyze the density of PDHE1α puncta, and the value was expressed as the percentage of PDHE1α immunoreactivity in Tg and Tg+ICA cells relative to NTg control cells.
Western Blot Assay
In brain tissue‐based assay, brain tissue samples for each group were homogenized with 1 × lysis buffer plus 1 mM PMSF and protease inhibitor cocktail. Brain mitochondrial samples were prepared as described above in Brain mitochondria section. In cell‐based assay, cells were harvested after 24 h 20 μM icariin treatment and then lysed with 1 × lysis buffer. Protein concentrations were determined using BCA protein assay kit. Equal amounts of total protein extract (20 μg per well) for each sample were separated by SDS‐PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. After blocking with 5% fat‐free milk, BACE1, synaptophysin, PSD95, β‐tublin, GAPDH, β‐actin, as well as mitochondrial PDHE1α, COX IV, VDAC were probed by corresponding primary antibodies (1:1000), and followed by the incubation with HRP‐conjugated anti‐rabbit antibody or HRP‐anti‐mouse antibody, respectively. The blots were developed with ECL detection reagents and visualized by KODAK Image Station 4000MM (Carestream Health Inc., New Haven, CT, USA). All band intensities were quantified using Quantity One software.
ELISA for Aβ levels
Aβ levels were determined by ELISA. Primary cultured hippocampal neurons and medium from Tg, Tg+ICA, and NTg groups were collected, respectively, and the levels of extracellular as well as intracellular Aβ1–42 were measured by a sandwich ELISA kit (Beijing Airan Technology Co. Ltd.), in accordance with the manufacturer's instructions.
Determination of ATP Levels
The level of tissue ATP was determined by an ATP bioluminescence assay kit (Promega Co.). Briefly, brain tissues were homogenated with a lysis buffer and mixed for 10 min. Hundred microliters of each sample was mixed with 100 μL of luciferase reagent, and the luminescence was measured using a microplate luminometer. The ATP concentration was normalized to total tissue protein concentration estimated by Bradford protein assay (Bio‐Rad, Hercules, CA, USA).
Statistical Analysis
Statistical data were expressed as mean ± standard deviation (SD). Student's t‐test and analysis of variance (ANOVA) were used to determine statistical significance. P value of <0.05 was considered to be statistically significant.
Results
Icariin Improved Spatial Learning and Memory Retention in 3 × Tg‐AD Mice
We have previously reported cognitive impairment in 3 × Tg‐AD mice 26, which is consistent with findings from other studies 27, 28. Here, we sought to explore the anti‐AD effect of icariin; therefore, we first performed the Morris water maze assay to examine the potential of icariin in the amelioration of spatial learning and memory deficits in 3 × Tg‐AD mice. In the place navigation test, the escape latencies to find the platform for the 3 × Tg‐AD mice were decreased across the 5‐day training period by icariin administration, with a significant effect from day 3 onward (Figure 1A). These data suggest that icariin was effective in improving spatial learning ability in 3 × Tg‐AD mice. Next, two probe trials were administered to assess short‐ and long‐term memory at 2 h and 24 h after training, respectively (Figure 1C,D). Icariin‐treated transgenic AD mice crossed over the hidden location of the platform more frequently, compared with the normal‐diet transgenic mice in both the 2‐h short‐term and 24‐h long‐term memory tests, indicating that icariin treatment could ameliorate spatial memory deficits in 3xTg‐AD mice. There was no difference in swimming speed between the two groups of mice across the 5 training days, excluding the possibility that the administration of icariin induced changes in body (Figure 1B).
Figure 1.

Icariin treatment improved spatial learning and memory deficits in 3 × Tg‐AD mice. Morris water maze test was performed to evaluate spatial memory in Tg mice. (A) In 5‐day training trials, the escaping latencies of mice were measured to assess the mouse memory ability. Icariin‐treated Tg mice showed significant improvement in escape latency with successive days of training as compared with vehicle control Tg mice. (B) No significant differences were observed in the swimming speed of the icariin‐treated Tg mice versus Tg controls. (C) In the probe trial, the swimming distance in all quadrants was analyzed. Compared with the vehicle‐treated Tg mice, icariin treatment significantly increased the chances of Tg mice searching for the hidden platform in the target quadrant in both 2‐h and 24‐h memory tests. (D) In the probe trial, the times for mice crossing the area where the submerged platform was located in training trials were recorded. Icarrin‐treated Tg mice crossed over the platform site more frequently compared with vehicle control Tg mice. Data are shown as mean ± SD, *P < 0.05, n = 15.
Icariin Modulated the Levels of Neurometabolites in 3×Tg‐AD Mice
Proton magnetic resonance spectroscopy (1H MRS) was applied to evaluate the efficacy of icariin in the regulation of neurometabolites in the hippocampus of AD mice. The spectra shown in Figure 2 are representative of the quality consistently achieved in this study. These spectra were analyzed using Topspin 2.1 software package to obtain ratios of concentrations of the 4 metabolites with respect to Cr. At the age of 8 months, 3 × Tg‐AD mice showed a significantly lower ratio of NAA/Cr than did NTg controls (P < 0.05), implying decreased neuronal health with or without increased neurodegeneration in Tg mice (Table 1). Upon icariin treatment, the ratio of NAA/Cr was significantly elevated (P < 0.05), to a level that was similar to that in NTg control mice, suggesting that icariin could promote neuronal cell activity. At the same time, the MI/Cr ratio was significantly elevated with icariin administration (P < 0.05). The ratio of Cho/Cr was decreased in 3 × Tg‐AD mice, relative to NTg controls, and it was enhanced in the icariin treatment group; however, the differences were not statistically significant. Despite this, a much higher ratio of Lac/Cr was observed in 3 × Tg‐AD mice, compared with NTg controls (P < 0.05), indicating a shift from oxidative phosphorylation to anaerobic glycolysis. Low NAA/Cr ratios and high Lac/Cr ratios are generally indicative of poor energetic metabolism 13, 17, 29. In the brain, NAA is produced and exported from mitochondria, and its synthesis requires aspartate and acetyl‐CoA. Thus, the reduction in NAA levels in Tg mice may also imply impairment of expression/activity of key regulatory enzymes involved in energetic metabolism, including acetyl‐CoA production, oxaloacetate–glutamate transamination, and oxidative phosphorylation. After icariin administration, the Lac/Cr ratio was significantly decreased in 3 × Tg‐AD mice (P < 0.05), suggesting that icariin may sustain aerobic metabolism and hence cause a reduction in lactic acid‐producing glycolysis in AD brains.
Figure 2.
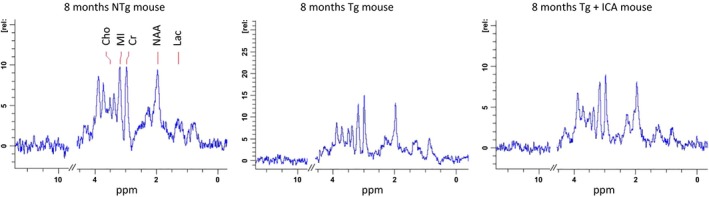
Representative 1H‐MRS spectra from different groups obtained after 6‐month‐long treatment. The spectra are shown with similar line widths and with amplitude adjusted using creatine peak at 3.03 ppm. The areas corresponding to NAA, MI, Cr, Lac, and Cho have been labeled.
Table 1.
1H‐MRS mean metabolite ratios in NTg, Tg, and Tg+ICA groups (TE = 20 ms)
| Metabolite | Ratio to Cr (mean ± SD) | ||
|---|---|---|---|
| NTg | Tg | Tg+ICA | |
| Creatine (Cr) | 1.000 | ||
| N ‐acetyl –aspartate (NAA) | 1.239 | 1.02 + 0.26* | 1.41 + 0.21# |
| Myo‐inositol (MI) | 1.073 | 1.06 + 0.07 | 1.30 + 0.25# |
| Choline (Cho) | 0.389 | 0.29 + 0.06 | 0.32 + 0.06 |
| Lactic acid (Lac) | 0.208 | 0.25 + 0.11* | 0.15 + 0.06# |
*P < 0.05, relative to NTg group. #P < 0.05, relative to Tg group. n = 4.
Icariin Preserved the Expression of Mitochondrial Key Enzymes and Synaptic Proteins
Given that NAA reductions might be related to neuronal energy impairment and dysfunction, we next investigated the expression of mitochondrial proteins PDH (PDHE1α subunit) and COX (COX subunit IV), which are crucial enzymes involved in the tricarboxylic acid cycle and respiratory chain, respectively. As shown by Western blot analysis in Figure 3A, PDHE1α and COX IV protein levels in the 3 × Tg‐AD mouse brain were much lower than that in the control NTg mouse brain (P < 0.01), which is consistent with previous publications 7. Icariin administration effectively reversed the reduced levels of PDHE1α and COX IV in Tg mice (P < 0.01), indicating that icariin had protective effects in maintaining expression of certain mitochondrial proteins. Correlatively, our data revealed a significant reduction in brain ATP levels in 3 × Tg‐AD mice, compared with NTg controls (P < 0.01), while icariin demonstrated significant treatment effects (P < 0.01) (Figure 3C).
Figure 3.
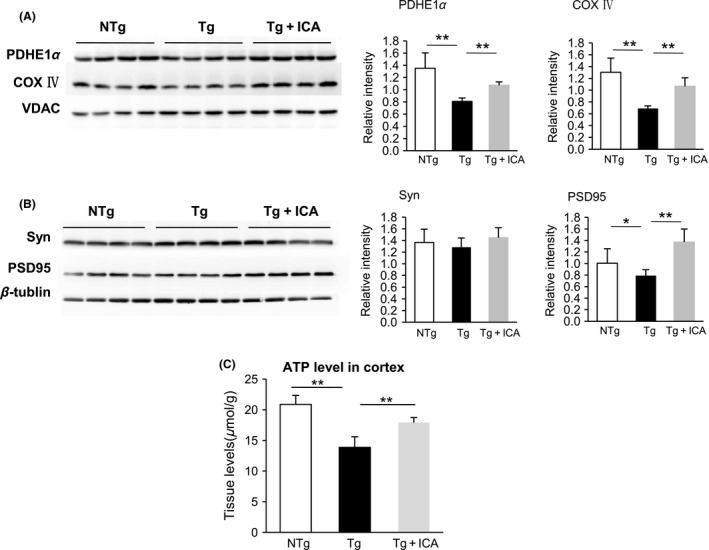
Icariin preserved mitochondrial and synaptic protein expression in AD mouse brain. (A&B) Western blot assay demonstrated that icariin enhanced the protein levels of PDHE1α and COX IV (A) and PSD95 (B) in the brains of Tg mice, whereas the difference in synaptophysin expression was not statistically significant among the groups. (C) ATP level was increased in icariin‐treated Tg mice compared with Tg controls. Bars graph (means ± SD)represented three independent experiments. *P < 0.05, **P < 0.01, n = 10.
Because NAA is synthesized in neurons and its depletion reflects neuronal loss or dysfunction, we further examined synaptic protein expression in the 3 × Tg‐AD mice. Our results showed that the expression of postsynaptic protein PSD95 was decreased in 3 × Tg‐AD mice, compared with that in NTg control mice (P < 0.05). Icariin treatment significantly enhanced the protein level of PSD95 (P < 0.01), indicating that icariin could partially reverse synaptic protein deficits in 3 × Tg‐AD mice (Figure 3B). We did not find a significant alteration in the expression of presynaptic protein synaptophysin in 3 × Tg‐AD mice, compared with NTg controls (Figure 3B), which is consistent with previous reports on 3 × Tg‐AD mice and other AD animal models 30, 31, 32. We repeated the experiments more than three times to ensure the accuracy and credibility of the results. Student's t‐test and analysis of variance (ANOVA) were used to determine statistical significance.
Icariin Reduced Amyloid Plaque Deposition in 3×Tg‐AD Mice
Given that Aβ is a major pathological component in AD development, we next performed immunohistochemistry analysis on the brain slices to examine the potential of icariin in the amelioration of amyloid protein deposition. As detected by Aβ immunolabeling with an anti‐Aβ1‐42 antibody, Aβ plaque load in the cortex and hippocampus of Tg mice was remarkably increased relative to that in NTg mice, while treatment with icariin for 6 months clearly decreased Aβ plaque deposition in comparison with the vehicle‐administered Tg mice (Figure 4A). Because Aβ is generated from a sequential cleavage of amyloid precursor protein (APP) by β‐site APP cleavage enzyme 1 (BACE1) and γ‐secretase 33, we further investigated the potential influence of icariin on BACE1 expression, using Western blot assay. Our results indicated significantly higher levels of BACE1 expression in Tg mice, compared with those in NTg controls (P < 0.05). The high levels of BACE1 expression in Tg mice were apparently inhibited by icariin treatment (P < 0.05), suggesting that the decreased accumulation of Aβ might be attributed to the inhibitory effect of icariin on BACE1 (Figure 4B).
Figure 4.
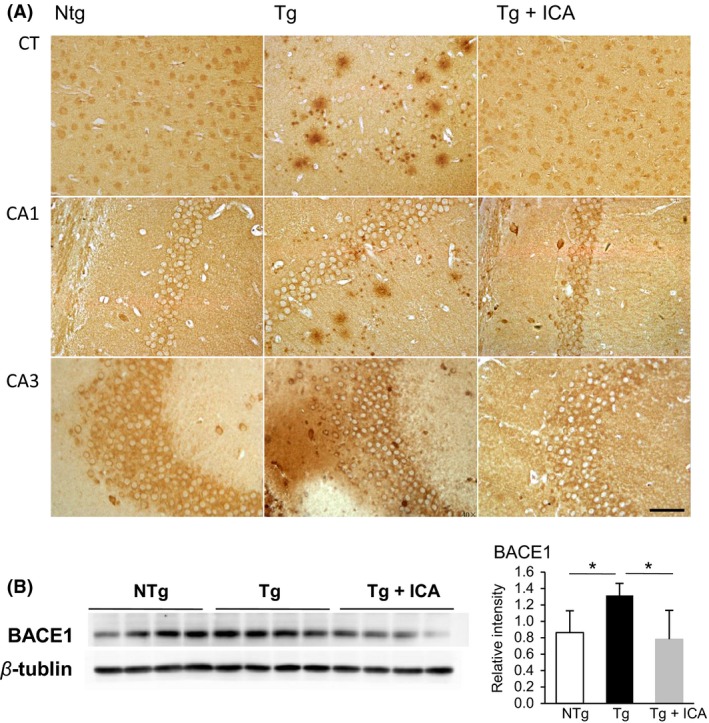
Icariin attenuated Aβ expression in the cortex and hippocampus of 3 × Tg‐AD mice. (A) Immunohistochemistry staining of Aβ1‐42 in the cortex and hippocampus of NTg, Tg, and icariin‐treated Tg mice, respectively. Aβ plaque deposition was significantly decreased in the cortex (CT) and the CA1, CA3 areas of hippocampus of icariin‐treated Tg mice compared with Tg controls. (B) Western blot assay demonstrated that icariin decreased BACE1 protein level in the brains of Tg mice. Bars graph (means ± SD)represented three independent experiments. *P < 0.05, n = 10. Scale bar in (A) = 50 μm.
Icariin Exerted Neuroprotective Effects in Primary Neuronal Culture
To validate our in vivo studies, we performed in vitro studies to determine whether icariin treatment could affect Aβ expression and preserve mitochondrial key enzymes and synaptic proteins in 3 × Tg‐AD primary neurons. First, we examined the effect of icariin on the regulation of Aβ production in neurons using ELISA to measure the levels of extracellular and intracellular Aβ. The level of extracellular Aβ1‐42 was found to be significantly increased in 3 × Tg‐AD neurons, compared with NTg controls (P < 0.05), which was markedly reduced after 24 h of treatment with icariin (P < 0.01). These results indicate that the secretion of Aβ1‐42 could be attenuated by icariin. In addition, the level of intracellular Aβ1‐42 was significantly decreased by treatment with icariin (P < 0.01), which suggests that icariin probably exerts its neuroprotective effect through the regulation of Aβ1‐42 production (Figure 5A).
Figure 5.
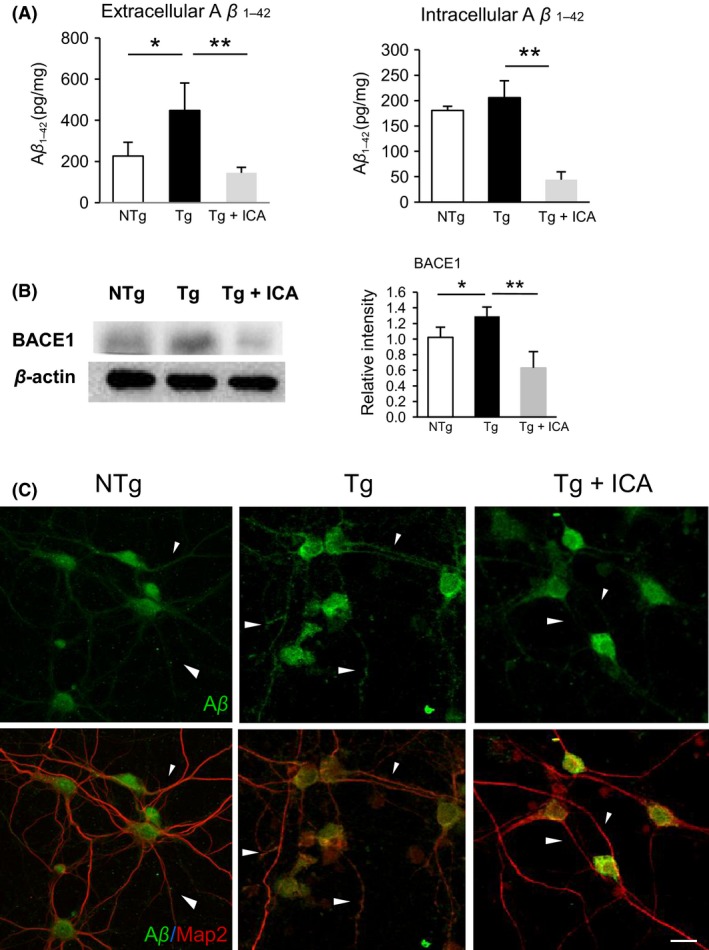
Icariin attenuated Aβ expression in 3 × Tg‐AD primary neurons. (A) ELISA demonstrated that icariin reduced extracellular and intracellular Aβ levels in Tg neurons. (B) Western blot assay demonstrated that icariin decreased BACE1 protein level in Tg neurons. (C) Double‐labeling analysis of Aβ with neuronal marker MAP2 in neurons from NTg, Tg, and Tg+ICA groups. Tg cells showed an overexpression of Aβ in the soma and neurites (arrow) as compared to NTg group, which was partially reduced in the neurites and restricted to the soma after icariin treatment. Bars graph (means ± SD)represented three independent experiments. *P < 0.05, **P < 0.01. Scale bar in (C) = 20 μm.
Next, we investigated whether icariin treatment decreased BACE1 levels in 3 × Tg‐AD primary neurons, using Western blot assay. The results revealed that icariin significantly decreased BACE1 level in transgenic neurons (P < 0.05), implying that icariin possibly inhibits Aβ production by decreasing BACE1 protein level (Figure 5B). To further evaluate the potential effect of icariin on Aβ distribution in neurons, immunofluorescence analysis of Aβ and neuronal marker MAP2 double staining in primary neurons was performed (Figure 5C). Localization of Aβ in NTg neurons was limited to the soma, with moderate staining along the neurites, whereas in Tg neurons, an overexpression of Aβ was seen throughout the soma and neuronal processes. After icariin treatment, Aβ expression was diminished along the neurites and restricted to the soma of the neurons, suggesting that icariin might inhibit the distribution of Aβ along the neurites.
Next, we investigated the potential of icariin in preserving mitochondrial functional protein PDH (PDHE1α) that naturally localizes in the mitochondrial matrix and adjacent to the internal membrane and, hence, may be used as a mitochondrial indicator. As shown by double‐labeling analysis of PDHE1α and MAP2 antibodies, PDHE1α expression in NTg neurons was abundant in the cytoplasm and was evenly distributed along the neuronal processes (Figure 6A). However, we found decreased and fragmented immunoreactivity of PDHE1α in neurites from 3 × Tg‐AD mouse neurons, compared with that in NTg controls (NTg, 100 ± 11.4%; Tg, 76.3 ± 10.2%, P < 0.01, n = 10, Figure 6B), suggestive of increased mitochondrial fragmentation in the diseased neurons. Upon icariin treatment, PDHE1α expression was enhanced and more evenly distributed along the neurites (98.3 ± 15.8%, P < 0.05, compared with Tg cells). Further examination using Western blot assay showed that icariin effectively elevated PDHE1α level in neurons from Tg mice (P < 0.01) (Figure 6C). These results indicate that icariin could regulate the expression of PDHE1α and protect against mitochondrial fragmentation in 3 × Tg‐AD neurons.
Figure 6.
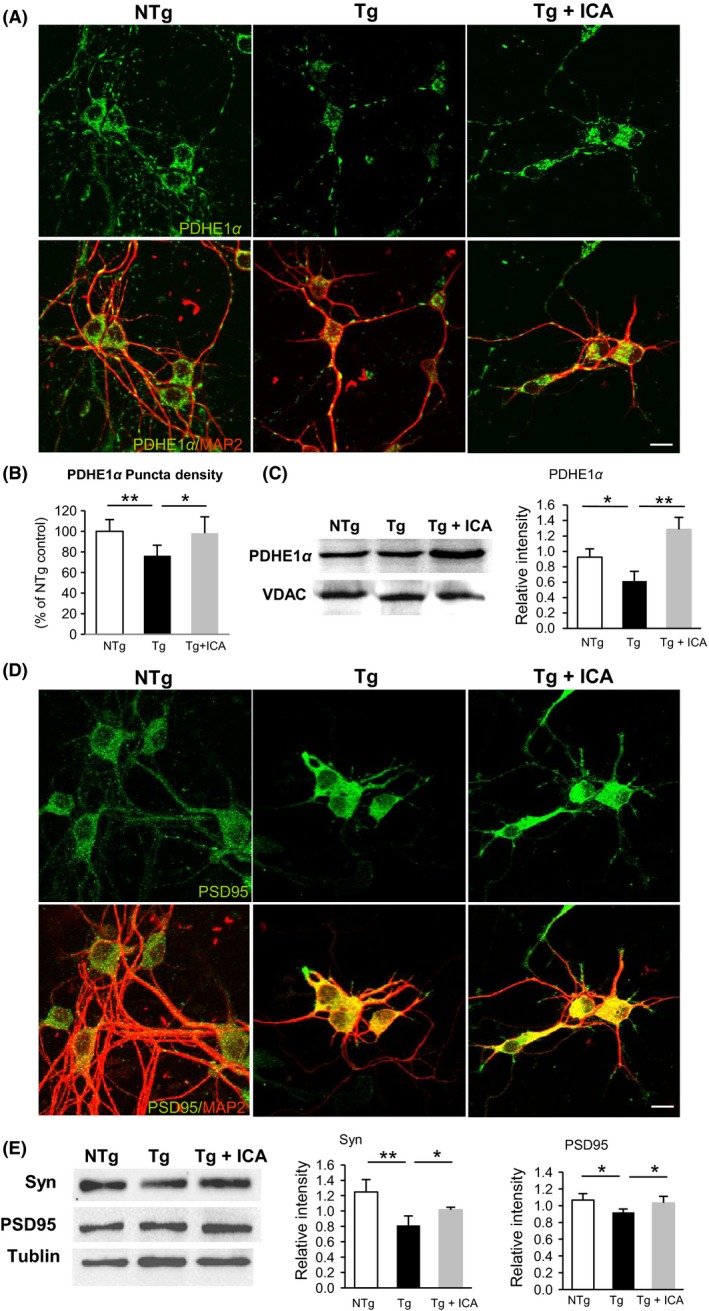
Icariin preserved mitochondrial and synaptic functional proteins in 3 × Tg‐AD primary neurons. (A&D) Double‐labeling analysis of neuronal marker MAP2 with PDHE1α (A) and PSD95 (D) in neurons from NTg, Tg, and Tg+ICA groups. Tg cells showed fragmented expression of PDHE1α, whereas icariin‐treated cells showed more evenly distributed PDHE1α along the neurites (A). PSD95 expression was reduced in the neurites of Tg cells and partially increased in icariin‐treated cells (D). (B) Quantification of PDHE1α immunoreactivity showed a significant increase of PDHE1α expression in Tg+ICA neurons relative to Tg cells. (C&E) Western blot assay demonstrated that icariin enhanced the protein levels of PDHE1α (C) and synaptophysin and PSD95 (E) in Tg neurons. Bars graph (means ± SD)represented three independent experiments. *P < 0.05, **P < 0.01. Scale bars in (A) & (D) = 20 μm.
To determine the effect of icariin treatment on synaptic functional protein expression, immunostaining analysis was performed in primary neurons using synaptic marker PSD95 and MAP2. As shown in Figure 6D, PSD95 was distributed evenly in the soma as well as along the neurites in NTg neurons, whereas in Tg cases, the expression of PSD95 was diminished along the neurites and restricted to the soma. After icariin treatment, PSD95 expression was partially recovered in the neurites of Tg neurons. In addition, Western blot assay revealed significantly reduced levels of synaptophysin and PSD95 in Tg neurons, compared with that in NTg neurons, which could be reversed by treatment with icariin (P < 0.05) (Figure 6E). These data, together with the in vivo results mentioned above, indicate that icariin could abolish Aβ generation while preserving key mitochondrial enzymes and synaptic functional proteins. Furthermore, we performed these experiments more than three times in order to ensure the accuracy and credibility of the results. Student's t‐test and analysis of variance (ANOVA) were used to determine statistical significance.
Discussion
In the present study, we reported the first evidence that icariin treatment modulated brain metabolism and maintained mitochondrial functions in 3 × Tg‐AD mice. Our results revealed a reduction in NAA levels accompanied with a decrease in the expression of mitochondrial PDH and COX enzymes and ATP production in AD mice, revealing a biochemical coupling between NAA synthesis and ATP generation, as proposed in previous publications 34. Despite complicating issues, many in vivo MRS studies support NAA as a surrogate for both neuronal loss and mitochondria metabolism dysfunction 29, 35, 36. Clinically, reduction in NAA has been used as an indicator of the disease progression in AD, and to identify patients with mild cognitive impairment (MCI), mild dementia, or AD 14, 15, 16, 37, 38. Additionally, we observed an elevation in the MRS metabolite ratio of Lac/Cr in the 3xTg‐AD mice, indicating that glycolysis had been initiated in an oxygen‐deficient environment. Previous studies also detected a shift from oxidative phosphorylation to anaerobic glycolysis, as indicated by a decrease in the oxygen consumption rates (OCR) and an increase in the extracellular acidification rates in primary neurons from 3 × Tg‐AD mice 7. Thus, change in lactate concentrations as measured by MRS could indicate mitochondrial disorders. In this study, MRS analysis also detected a treatment effect of icariin in 3 × Tg‐AD mice, suggesting that this method can be used as a noninvasive tool to evaluate anti‐AD therapeutic efficacy and accelerate drug discovery.
The mechanism underlying the anti‐Aβ action of icariin has been intensively investigated in previous publications. Zhang et al. suggested that icariin decreases Aβ contents by reducing the expression of APP and BACE‐1 23. Nie et al. reported that icariin could reduce the mRNA level of BACE‐1 in a dose‐dependent manner and increase the level of SOD‐2 in AD mouse brains, suggesting that the inhibitory effect of icariin on β‐secretase might be partially attributed to its anti‐oxidative activity 20. Our studies also showed that icariin treatment could reduce Aβ production, plaque deposition, and BACE1 levels in AD animal models. We also demonstrated that icariin administration significantly recovered the learning and memory deficits in 3 × Tg‐AD mice, which is consistent with results of studies in other AD animal models 20, 23, 39. As reported in previous publications, 3 × Tg‐AD mice did not display deficits in spatial learning memory before 3 months of age 28; thus, the Tg mice in our study were not assumed to have developed spatial learning impairment at the time when they began to receive icariin treatment at 2 months of age. Therefore, we speculate that the beneficial effects of icariin on improving spatial learning and memory abilities may be attributable to the prevention or delay of cognitive decline in AD mice rather than a reversal of preexisting cognitive impairment.
The accumulation of Aβ in subcellular compartments such as mitochondria and synapses is associated with mitochondrial impairment and neuronal dysfunction 40, 41, 42, 43, 44, 45, 46, 47, 48, 49. Extracellular deposits of Aβ may access mitochondria through permeabilization of the cellular membranes. The interaction between Aβ and the mitochondrial protein Aβ‐binding alcohol dehydrogenase (ABAD) results in a decreased activity of a key mitochondrial enzyme, COX, and induces oxidative stress 44, 45, 48. The increased levels of reactive oxygen species and impairment in mitochondrial functions further trigger the production of Aβ, resulting in a vicious cycle of Aβ accumulation and mitochondrial dysfunction that leads to neurodegeneration 46, 47, 49. Icariin, the major component of Epimedium herba, acts as a phytoestrogen and exhibits estrogenic activity in vivo 50. Estrogen has been shown to have beneficial effects on brain bioenergetics, including facilitating glucose transport into cells, promoting neuronal aerobic glycolysis and oxidative phosphorylation, and sustaining ATP generation 46. In addition, it has been demonstrated that estrogen action can ameliorate Aβ elevation in mitochondria and regulate the activity and expression of key mitochondrial enzymes such as PDH, COX IV, and ATP synthase, while also protecting the electron transport chain and increasing mitochondrial respiration in neurons 51, 52. The mechanism by which estrogen promotes the degradation and clearance of Aβ may be related to the upregulation of insulin‐degrading enzyme and neprilysin gene expression 53, 54. Based on these observations, we extrapolate our findings from 3 × Tg‐AD mice to indicate a therapeutic effect of icariin related to its estrogenic activities. We speculate that icariin administration could ameliorate Aβ production and accumulation, while simultaneously sustaining mitochondrial functions and energy generation in the 3 × Tg‐AD mouse brain. Because neuronal mitochondria are essential in the regulation of synaptogenesis and provide ATP to support the release and recycling of neurotransmitters, icariin might preserve synaptic protein expressions/functions through modulation of mitochondrial functions, thus improving learning and memory abilities. In our present study, there were no abnormal changes in the general health of the 8‐month‐old 3 × Tg‐AD mice that had been administered icariin for 6 months beginning at 2 months of age. Some studies also showed no significant alteration of neuron morphology, full‐length APP, or Aβ expression in the brain of normal mice after icariin treatment 23, 39, 55. These data indicate that as an effective component of traditional Chinese medicine, icariin exhibits no significant toxicity in mice, and we speculate that the early administration of icariin could defeat AD at an early stage. Some limitations of this study are that it remains to be clarified how icariin modulates mitochondrial dynamics and synaptic activity in the AD‐affected neurons. Further investigation of the underlying cellular and molecular mechanisms needs to be conducted in future.
In conclusion, our results demonstrated that icariin could modulate brain metabolism to promote neuronal cell activity, preserve mitochondrial and synaptic functional proteins, inhibit Aβ expression, and improve cognitive functions in AD mice. These results strongly suggest icariin as a promising multitarget drug in the prevention/protection against AD.
Disclosure
This work has not been published or submitted elsewhere. The authors declare no financial or other conflict of interests.
Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (Grant 21271131), and the China Postdoctoral Science Foundation Funded Project (Grant 2014M552236). The MRS measurements and data analysis in this work were supported by the State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Center for Magnetic Resonance, Wuhan institute of Physics and Mathematics, Chinese Academy of Science with funding from the Institute Grants Committee (Grant T151006).
The first two authors contributed equally to this work.
References
- 1. Oddo S, Caccamo A, Shepherd JD, et al. Triple‐transgenic model of Alzheimer's disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003;39:409–421. [DOI] [PubMed] [Google Scholar]
- 2. Perez‐Cruz C, Nolte MW, van Gaalen MM, et al. Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer's disease. J Neurosci 2011;31:3926–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Selkoe DJ. Alzheimer's disease: Genes, proteins, and therapy. Physiol Rev 2001;81:741–766. [DOI] [PubMed] [Google Scholar]
- 4. Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A 2010;107:18670–18675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim Biophys Acta 2014;1842:1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci 2009;29:9090–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2009;106:14670–14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A 2004;101:2173–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blass JP, Sheu RK, Gibson GE. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann N Y Acad Sci 2000;903:204–221. [DOI] [PubMed] [Google Scholar]
- 10. Cottrell DA, Blakely EL, Johnson MA, Ince PG, Turnbull DM. Mitochondrial enzyme‐deficient hippocampal neurons and choroidal cells in AD. Neurology 2001;57:260–264. [DOI] [PubMed] [Google Scholar]
- 11. Perry EK, Perry RH, Tomlinson BE, Blessed G, Gibson PH. Coenzyme A‐acetylating enzymes in Alzheimer's disease: Possible cholinergic ‘compartment’ of pyruvate dehydrogenase. Neurosci Lett 1980;18:105–110. [DOI] [PubMed] [Google Scholar]
- 12. Valla J, Yaari R, Wolf AB, et al. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late‐onset Alzheimer's susceptibility gene. J Alzheimers Dis 2010;22:307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AM. N‐Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog Neurobiol 2007;81:89–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kantarci K, Weigand SD, Petersen RC, et al. Longitudinal 1H MRS changes in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging 2007;28:1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Targosz‐Gajniak MG, Siuda JS, Wicher MM, et al. Magnetic resonance spectroscopy as a predictor of conversion of mild cognitive impairment to dementia. J Neurol Sci 2013;335:58–63. [DOI] [PubMed] [Google Scholar]
- 16. Zimny A, Szewczyk P, Trypka E, et al. Multimodal imaging in diagnosis of Alzheimer's disease and amnestic mild cognitive impairment: Value of magnetic resonance spectroscopy, perfusion, and diffusion tensor imaging of the posterior cingulate region. J Alzheimers Dis 2011;27:591–601. [DOI] [PubMed] [Google Scholar]
- 17. Gasparovic C, Arfai N, Smid N, Feeney DM. Decrease and recovery of N‐acetylaspartate/creatine in rat brain remote from focal injury. J Neurotrauma 2001;18:241–246. [DOI] [PubMed] [Google Scholar]
- 18. Narayanan S, De Stefano N, Francis GS, et al. Axonal metabolic recovery in multiple sclerosis patients treated with interferon beta‐1b. J Neurol 2001;248:979–986. [DOI] [PubMed] [Google Scholar]
- 19. Ma H, He X, Yang Y, Li M, Hao D, Jia Z. The genus Epimedium: An ethnopharmacological and phytochemical review. J Ethnopharmacol 2011;134:519–541. [DOI] [PubMed] [Google Scholar]
- 20. Nie J, Luo Y, Huang XN, Gong QH, Wu Q, Shi JS. Icariin inhibits beta‐amyloid peptide segment 25‐35 induced expression of beta‐secretase in rat hippocampus. Eur J Pharmacol 2010;626:213–218. [DOI] [PubMed] [Google Scholar]
- 21. Urano T, Tohda C. Icariin improves memory impairment in Alzheimer's disease model mice (5xFAD) and attenuates amyloid beta‐induced neurite atrophy. Phytother Res 2010;24:1658–1663. [DOI] [PubMed] [Google Scholar]
- 22. Zeng KW, Ko H, Yang HO, Wang XM. Icariin attenuates beta‐amyloid‐induced neurotoxicity by inhibition of tau protein hyperphosphorylation in PC12 cells. Neuropharmacology 2010;59:542–550. [DOI] [PubMed] [Google Scholar]
- 23. Zhang L, Shen C, Chu J, Zhang R, Li Y, Li L. Icariin decreases the expression of APP and BACE‐1 and reduces the beta‐amyloid burden in an APP transgenic mouse model of Alzheimer's disease. Int J Biol Sci 2014;10:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morris R. Developments of a water‐maze procedure for studying spatial learning in the rat. J Neurosci Methods 1984;11:47–60. [DOI] [PubMed] [Google Scholar]
- 25. Vorhees CV, Williams MT. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat Protoc 2006;1:848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu J, Luo X, Xu H, et al. Identification of the key molecules involved in chronic copper exposure‐aggravated memory impairment in transgenic mice of Alzheimer's disease using proteomic analysis. J Alzheimers Dis 2015;44:455–469. [DOI] [PubMed] [Google Scholar]
- 27. Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease‐related cognitive deficits in transgenic mice. Neuron 2005;45:675–688. [DOI] [PubMed] [Google Scholar]
- 28. Clinton LK, Billings LM, Green KN, et al. Age‐dependent sexual dimorphism in cognition and stress response in the 3xTg‐AD mice. Neurobiol Dis 2007;28:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Signoretti S, Marmarou A, Tavazzi B, Lazzarino G, Beaumont A, Vagnozzi R. N‐Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J Neurotrauma 2001;18:977–991. [DOI] [PubMed] [Google Scholar]
- 30. Arsenault D, Julien C, Tremblay C, Calon F. DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg‐AD mice. PLoS ONE 2011;6:e17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Calon F, Lim GP, Yang F, et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron 2004;43:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. King DL, Arendash GW. Maintained synaptophysin immunoreactivity in Tg2576 transgenic mice during aging: Correlations with cognitive impairment. Brain Res 2002;926:58–68. [DOI] [PubMed] [Google Scholar]
- 33. Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: An emperor in need of clothes. Nat Neurosci 2012;15:349–357. [DOI] [PubMed] [Google Scholar]
- 34. Bates TE, Strangward M, Keelan J, Davey GP, Munro PM, Clark JB. Inhibition of N‐acetylaspartate production: Implications for 1H MRS studies in vivo. NeuroReport 1996;7:1397–1400. [PubMed] [Google Scholar]
- 35. Kantarci K, Reynolds G, Petersen RC, et al. Proton MR spectroscopy in mild cognitive impairment and Alzheimer disease: Comparison of 1.5 and 3 T. AJNR Am J Neuroradiol 2003;24:843–849. [PMC free article] [PubMed] [Google Scholar]
- 36. Signoretti S, Marmarou A, Tavazzi B, et al. The protective effect of cyclosporin A upon N‐acetylaspartate and mitochondrial dysfunction following experimental diffuse traumatic brain injury. J Neurotrauma 2004;21:1154–1167. [DOI] [PubMed] [Google Scholar]
- 37. Jessen F, Gur O, Block W, et al. A multicenter (1)H‐MRS study of the medial temporal lobe in AD and MCI. Neurology 2009;72:1735–1740. [DOI] [PubMed] [Google Scholar]
- 38. Zimny A, Bladowska J, Macioszek A, et al. Evaluation of the posterior cingulate region with FDG‐PET and advanced MR techniques in patients with amnestic mild cognitive impairment: Comparison of the methods. J Alzheimers Dis 2014;44:329–338. [DOI] [PubMed] [Google Scholar]
- 39. Luo Y, Nie J, Gong QH, Lu YF, Wu Q, Shi JS. Protective effects of icariin against learning and memory deficits induced by aluminium in rats. Clin Exp Pharmacol Physiol 2007;34:792–795. [DOI] [PubMed] [Google Scholar]
- 40. Pinho CM, Teixeira PF, Glaser E. Mitochondrial import and degradation of amyloid‐beta peptide. Biochim Biophys Acta 2014;1837:1069–1074. [DOI] [PubMed] [Google Scholar]
- 41. Calkins MJ, Reddy PH. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim Biophys Acta 2011;1812:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosales‐Corral S, Acuna‐Castroviejo D, Tan DX, et al. Accumulation of exogenous amyloid‐beta peptide in hippocampal mitochondria causes their dysfunction: A protective role for melatonin. Oxid Med Cell Longev 2012;2012:843649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xie H, Guan J, Borrelli LA, Xu J, Serrano‐Pozo A, Bacskai BJ. Mitochondrial alterations near amyloid plaques in an Alzheimer's disease mouse model. J Neurosci 2013;33:17042–17051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 2004;304:448–452. [DOI] [PubMed] [Google Scholar]
- 45. Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med 2008;14:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rettberg JR, Yao J, Brinton RD. Estrogen: A master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol 2014;35:8–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis 2010;20(Suppl 2):S265–S279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takuma K, Yao J, Huang J, et al. ABAD enhances Abeta‐induced cell stress via mitochondrial dysfunction. FASEB J 2005;19:597–598. [DOI] [PubMed] [Google Scholar]
- 49. Yao J, Rettberg JR, Klosinski LP, Cadenas E, Brinton RD. Shift in brain metabolism in late onset Alzheimer's disease: Implications for biomarkers and therapeutic interventions. Mol Aspects Med 2011;32:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kang HK, Choi YH, Kwon H, et al. Estrogenic/antiestrogenic activities of a Epimedium koreanum extract and its major components: In vitro and in vivo studies. Food Chem Toxicol 2012;50:2751–2759. [DOI] [PubMed] [Google Scholar]
- 51. Nilsen J, Irwin RW, Gallaher TK, Brinton RD. Estradiol in vivo regulation of brain mitochondrial proteome. J Neurosci 2007;27:14069–14077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yao J, Irwin R, Chen S, Hamilton R, Cadenas E, Brinton RD. Ovarian hormone loss induces bioenergetic deficits and mitochondrial beta‐amyloid. Neurobiol Aging 2012;33:1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jayaraman A, Carroll JC, Morgan TE, et al. 17beta‐estradiol and progesterone regulate expression of beta‐amyloid clearance factors in primary neuron cultures and female rat brain. Endocrinology 2012;153:5467–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao L, Yao J, Mao Z, Chen S, Wang Y, Brinton RD. 17beta‐Estradiol regulates insulin‐degrading enzyme expression via an ERbeta/PI3‐K pathway in hippocampus: Relevance to Alzheimer's prevention. Neurobiol Aging 2011;32:1949–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li F, Gong QH, Wu Q, Lu YF, Shi JS. Icariin isolated from Epimedium brevicornum Maxim attenuates learning and memory deficits induced by d‐galactose in rats. Pharmacol Biochem Behav 2010;96:301–305. [DOI] [PubMed] [Google Scholar]