

Summary
Aims
Prolonged seizure activity may result in mitochondrial dysfunction and lead to cell death in the hippocampus. Mitochondrial fission may occur in an early stage of neuronal cell death. This study examined the role of the mitochondrial fission protein dynamin‐related protein 1 (Drp1) in the hippocampus following status epilepticus.
Methods
Kainic acid (KA) was microinjected unilaterally into the hippocampal CA3 area in Sprague Dawley rats to induce prolonged seizure activity. Biochemical analysis, electron microscopy, and immunofluorescence staining were performed to evaluate the subsequent molecular and cellular events. The effects of pretreatment with a mitochondrial fission protein inhibitor, Mdivi‐1 (2 nmol), were also evaluated.
Results
Phosphorylation of Drp1 at serine 616 (p‐Drp1(Ser616)) was elevated from 1 to 24 h after the elicited seizure activity. Pretreatment with Mdivi‐1 decreased the Drp1 phosphorylation at Ser616 and limited the mitochondrial fission. Mdivi‐1 rescued the Complex I dysfunction, decreased the levels of oxidized proteins, decreased the activation of cytochrome c/caspase‐3 signaling, and blunted cell death in CA3 neurons.
Conclusion
Our findings suggest that activation of p‐Drp1(Ser616) is related to seizure‐induced neuronal damage. Modulation of p‐Drp1(Ser616) expression is accompanied by decreases in mitochondrial fission, mitochondrial dysfunction, and oxidation, providing a neuroprotective effect against seizure‐induced hippocampal neuronal damage.
Keywords: Dynamin‐related protein 1, Hippocampus, Mdivi‐1, Mitochondrial fission, Neuronal cell death, Status epilepticus
Introduction
Mitochondria are essential organelles in the cell that participate in energy exchange, regulation of signaling cascades, oxidant formation, and cell survival and cell death 1, 2. Recent studies have implicated mitochondria‐dependent neuronal cell death in many neurological diseases, including epileptic seizures 3, 4, 5. Nevertheless, mitochondria are not a static organelle 6. Mitochondria are dynamic and perform their versatile functions by enabling recruitment to critical subcellular compartments, content exchange between mitochondria, communication with the cytosol, and supervision of mitochondria and mitochondrial shape 7, 8. Under stress states, such as in prolonged seizures and ischemia, the perturbed homeostatic condition and inherent production of reactive oxygen species (ROS) by mitochondria in neuronal cells may result in progressive damage and require regulation of the turnover, content, function, morphology, and number of mitochondria 6, 7, 8. A recent step toward understanding the mitochondrial control of apoptosis was the discovery of a dynamic morphological change of this organelle under pathological conditions 9, 10. This process involves a group of conserved, large dynamin‐related GTPases that maintain the critical balance between mitochondrial fission and fusion, allowing mitochondria to maintain their shape or morphology 11, 12, 13. Mitochondrial fission involves the constriction and cleavage of mitochondria by fission proteins, including dynamin‐related protein 1 (Drp1) and mitochondrial fission 1 protein 11, 12, 13. Drp1 is a key mediator of mitochondrial fission 14, 15.
Sustained epileptic seizures will change the redox potential and decrease the ATP content, which may lead to the collapse of energy production and supply in the brain. Emerging evidence has suggested that mitochondrial dysfunction occurs as a consequence of prolonged epileptic seizures and may play a pivotal role in seizure‐induced brain damage and cause subsequent epileptogenesis 3, 16. We have shown previously that dysfunction of Complex I respiratory chain enzymes and mitochondrial ultrastructural damage in the hippocampus are associated with prolonged seizures during experimental status epilepticus 17, 18. Our recent studies 19, 20, 21 demonstrated that increased oxidative and nitrative stress, followed by a reduction in mitochondrial Complex I activity, promotes the release of cytochrome c from the mitochondria to the cytosol, thereby triggering the caspase cascades that lead to apoptotic cell death in the hippocampus. Recently, mitochondrial dynamics has been recognized as a pivotal process in regulating cell survival and death; in particular, mitochondrial fission may occur as an early event in apoptosis and result in neuronal cell death after various cerebral insults 22, 23, 24, 25. Moreover, we recently found that a cerebral ischemia‐induced transient increase in the phosphorylation of Drp1 at serine 616 (p‐Drp1(Ser616)) in hippocampal CA1 neurons 26 may play a crucial role in the hippocampal neuronal damage following ischemia. However, studies regarding the role of Drp1 in prolonged epileptic seizures have been limited 27, 28.
In this study, we hypothesized that seizure‐induced neuronal damage in the hippocampus may relate to the production of p‐Drp1(Ser616) and concomitantly enhance the mitochondrial fragmentation (fission) that leads to hippocampal neuronal apoptosis. The results from this study validated this hypothesis.
Materials and Methods
Animals
All experimental procedures were carried out in compliance with the guidelines for the care and use of experimental animals endorsed by our institutional animal care committee. Specific pathogen‐free adult male Sprague Dawley rats (260–300 g) were purchased from BioLASCO Taiwan Co. Ltd (Taipei, Taiwan) and housed in an environmentally controlled room (temperature 24 ± 1°C; 12 h light/12 h dark cycle) in the Center for Laboratory Animals at Kaohsiung Chang Gung Memorial Hospital. Standard laboratory rat chow and tap water were available ad libitum. All efforts were made to reduce the number of animals used and to minimize animal suffering during the experiment.
Experimental Status Epilepticus
We used an experimental model of status epilepticus that we established previously 17, 18, 19, 20, 21, 29. Briefly, the animal's head was fixed to a stereotaxic headholder (Kopf, Tujunga, CA, USA) after administering 3% of isoflurane via inhalation to induce anesthesia, and the animal's body was placed on a heating pad to maintain the body temperature at 37°C. Kainic acid (KA; 0.5 nmol; Tocris Cookson, Bristol, UK) dissolved in 0.1 M phosphate‐buffered saline (PBS, pH 7.4) was microinjected stereotaxically (3.3–3.6 mm posterior to bregma, 2.4–2.7 mm from the midline, and 3.4–3.8 mm below the cortical surface) into the CA3 subfield of the hippocampus on the left side. This consistently resulted in progressive and concomitant increases in both root mean square and mean power frequency values of bilateral seizurelike hippocampal electroencephalographic (hEEG) activity recorded from the CA3 subfield on the right side 17, 18, 20, 21, 29. According to standard procedures, these experimental manifestations of continuous seizure activity were followed for 60 min, after which they were terminated by the intraperitoneal administration of diazepam (30 mg/kg) 18, 19, 20, 29. The wound was then closed in layers, and sodium penicillin (10,000 IU; YF Chemical Corporation, Taipei, Taiwan) was given intramuscularly to prevent postoperative infection. The animals were returned to the animal room for recovery in individual cages. Animals that received anesthesia and surgical preparations without additional experimental manipulations served as sham controls.
Pharmacological Pretreatments
In experiments that involved pharmacological pretreatments, a Drp1 inhibitor, Mdivi‐1 (2 nmol; Enzo Life Sciences, Farmingdale, NY, USA) 24, 27, was microinjected bilaterally and sequentially into the CA3 subfield of the hippocampus, at a volume of 150 nL on each side. Microinjection of 3% dimethyl sulfoxide (DMSO) served as the vehicle and volume control. To avoid confounding effects of drug interactions, each animal received only a single pharmacological pretreatment, followed 30 min later by microinjection of KA (0.5 nmol) into the left hippocampal CA3.
Collection of Tissue Samples from the Hippocampus
At predetermined time intervals (1, 3, 6, or 24 h; or 7 days) after microinjection of KA or PBS into the hippocampus, rats were again anesthetized with 3% isoflurane and were perfused intracardially with 50 mL of warm (37°C) saline containing heparin (100 U/mL). The brain was rapidly removed under visual inspection and placed on a piece of gauze moistened with ice‐cold 0.9% saline. Using standard procedures, we collected tissues from the ipsilateral (KA injection side) and the contralateral (hEEG recording side) hippocampus. This allowed us to ascertain that the results from those analyses were due directly to prolonged seizures and not indirectly to KA toxicity. Hippocampal samples were stored at −80°C until use in biochemical analyses.
Western Blot Analysis
Western blot analysis for phosphorylated Drp1 (p‐Drp1, cytochrome c, caspase‐3, or β‐actin) was carried out on proteins extracted from total lysate or from mitochondrial or cytosolic fractions of hippocampal samples. The purity of the mitochondrial fraction was verified by the selective expression of a protein specific for the mitochondrial inner membrane, cytochrome c oxidase subunit IV (COX IV). Protein concentration was determined using the BCA protein assay (Pierce, Rockford, IL, USA). The primary antisera used included rabbit polyclonal antisera against Drp1 (Novus Biologicals, Littleton, CO, USA), p‐Drp1(Ser616) (Novus Biologicals), or COX IV (Cell Signaling, Danvers, MA, USA) and mouse monoclonal antisera against cytochrome c (Cell Signaling) or β‐actin (Chemicon, Temecula, CA, USA). This was followed by incubation with secondary antisera including horseradish peroxidase‐conjugated goat anti‐mouse IgG and goat anti‐rabbit IgG (Jackson ImmunoResearch, West Grove, PA, USA) for Drp1, cytochrome c, caspase‐3, and β‐actin. Specific antibody–antigen complexes were detected using an enhanced chemiluminescence western HRP substrate (Merck Millipore, Billerica, MA, USA). The amount of protein was quantified using ImageMaster software (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
Electron Microscopy
At 24 h after status epilepticus, the hippocampal CA3b area was removed and processed for electron microscopy as in our previous reports 17, 30, 31. Tissue samples were diced and submerged in 4% glutaraldehyde (0.1 M sodium cacodylate buffer, pH 7.2). Tissues were postfixed with osmium and en bloc stained with uranyl acetate. After dehydration, each specimen was embedded by infiltration in Spurr's medium. Following trimming of the tissue blocks, sections were cut to a thickness of 90 nm, poststained with uranyl acetate and lead citrate, and viewed on 300 mesh‐coated grids using a JEOL JEM‐1230 (Tokyo, Japan) electron microscope.
Double Immunofluorescence Staining and Laser Confocal Microscopy
Free‐floating sections of the hippocampus (35 μm) were processed for double immunofluorescence staining using procedures we reported previously 18, 19, 20, 21, 29. Double immunofluorescence staining was carried out using a rabbit polyclonal antiserum against p‐Drp1(Ser616) (Novus Biologicals), together with a mouse monoclonal antiserum directed against a specific neuron marker, neuron‐specific nuclear protein (NeuN; Chemicon). The secondary antisera included AlexaFluor 488‐conjugated goat anti‐rabbit IgG, AlexaFluor 568‐conjugated goat anti‐mouse IgG, and AlexaFluor 546‐conjugated goat anti‐rabbit IgG (Molecular Probes, Eugene, OR, USA). The hippocampal CA3b region was viewed under a Fluoview FV10i laser‐scanning confocal microscope (Olympus, Tokyo, Japan).
Immunofluorescent Staining and Quantitative Immunofluorescence Analysis of Apoptotic Neuronal Cells
Immunofluorescence staining was carried out in animals using procedures reported previously 26, 32 with some modifications. In brief, the removed brain tissue was fixed in 4% formaldehyde for 18 h at 4°C and cryoprotected in 30% sucrose solution in PBS. Frozen transverse sections (30 μm) at the level of hippocampus were cut on a cryostat and collected in 0.1 M PBS. Free‐floating sections of the hippocampus were processed for immunoreactivity for NeuN (Chemicon); p‐Drp1(Ser616) (rabbit polyclonal, Novus Biologicals) and activated caspase‐3 (Cell Signaling), as well as a mitochondrial marker, a mouse monoclonal antiserum against ATPB (Anti‐ATPB antibody, Abcam, Cambridge, MA, USA); sections were also stained with 4′,6‐diamidino‐2‐phenylindole (DAPI, Sigma‐Aldrich, St. Louis, MO, USA). Sections were viewed under an Olympus AX‐51 epifluorescence microscope (Olympus, Kyoto, Japan).
Quantitative immunofluorescence analysis of apoptotic neurons in the hippocampal CA3 was performed on representative images from three separate rounds of staining. CellScan (Olympus) was used to assess the images and quantify the relative fluorescent intensity. Each analysis was calculated within a single field (250 μm × 250 μm) in the hippocampus CA3. The numbers of apoptotic cells, defined as those containing condensed chromatin, were manually counted.
Some animals were processed for histopathological analysis of the severity of neuronal death in the hippocampus. For this analysis, 4‐μm paraffin‐embedded brain sections were deparaffinized and stained with cresyl violet.
Assays for Activity of Mitochondrial Respiratory Enzymes
Isolation of rat mitochondria from the hippocampal samples was carried out according to our previous report 17, 18 and modification 33. The activity of Complex I (Nicotinamide adenine dinucleotide (NADH) ubiquinone oxidoreductase) of mitochondrial respiratory enzymes was analyzed via enzyme activity immunocapture assays 33, 34. For this assay, 96‐well plates coated with monoclonal antibodies against the oxidative phosphorylation Complex I (MS141, MitoSciences, Eugene, OR, USA) were used according to manufacturer's instructions. Complex I activity was measured by adding an assay solution, and the oxidation of NADH was monitored by measuring its decrease in absorbance at 450 nm in kinetic mode at 30°C for 2 h. Assays for mitochondrial respiratory enzyme activity were performed using a Multiskan Spectrum reader (Thermo Scientific, Miami, OK, USA). Each tissue sample was evaluated at least in duplicate.
Measurement of Protein Oxidation
Oxidized protein was measured using a protein oxidation detection kit (OxyBlot, Chemicon). Total proteins extracted from the hippocampal CA3 were reacted with 2,4‐dinitrophenylhydrazine and derivatized to 2,4‐dinitrophenylhydrazone (DNP‐hydrazone) 35. The DNP‐derivatized protein samples were separated on a 15% SDS–polyacrylamide gel, followed by Western blotting. The blot was incubated with a rabbit anti‐DNP antibody, followed by incubation with a horseradish peroxidase‐conjugated goat anti‐rabbit IgG according to the manufacturer's instructions.
Qualitative and Quantitative Analyses of DNA Fragmentation
After extraction of total DNA from hippocampal tissues, nucleosomal DNA ladders were amplified by a PCR kit for DNA ladder assays (Maxim Biotech, San Francisco, CA, USA) to enhance the detection sensitivity and were separated by electrophoresis on 1% agarose gels 18, 19, 20, 26, 29. To quantify apoptosis‐related DNA fragmentation, a cell death ELISA (Roche Molecular Biochemicals, Mannheim, Germany) was used to assay the level of histone‐associated DNA fragments in the cytoplasm 36. Proteins from hippocampal samples were used as the antigen source, together with primary antihistone antibody and secondary anti‐DNA antibody coupled with peroxidase. The amount of nucleosomes in the cytoplasm was quantitatively determined using 2,2′‐azino‐di‐[3‐ethylbenzthiazoline] sulfonate as the substrate. Absorbance was measured at 405 nm and referenced at 490 nm using a microtiter plate reader (Anthros Labtec, Salzburg, Austria).
Statistical Analysis
All values are expressed as the mean ± SEM. One‐way analysis of variance (ANOVA) was used, as appropriate, to assess group means, followed by the Scheffé multiple‐range test for post hoc assessment of individual means. The effects of treatments on hEEG signals were assessed using two‐way ANOVA with repeated measures for differences among groups. Values of P < 0.05 were considered to indicate statistical significance.
Results
Temporal Changes in Drp1 Expression in the Hippocampal CA3 Following Experimental Status Epilepticus
We examined whether Drp1 (Ser616) expression in the hippocampal CA3 subfield exhibited changes in expression level after experimental status epilepticus. After unilateral microinjection of KA into the left CA3 region, Western blot analysis revealed a significant increase in p‐Drp1 (Ser616) protein expression from 1 to 24 h in the bilateral hippocampal CA3 subfields (Figure 1A), with no evident change in total Drp1 (Figure 1B). Additionally, compared to the sham control (Figure 1C‐a), there was an increase in p‐Drp1 (Ser616) immunoreactivity in neurons from the hippocampal CA3b subfield on the right side (Figure 1C‐b) at 24 h after KA‐induced status epilepticus.
Figure 1.
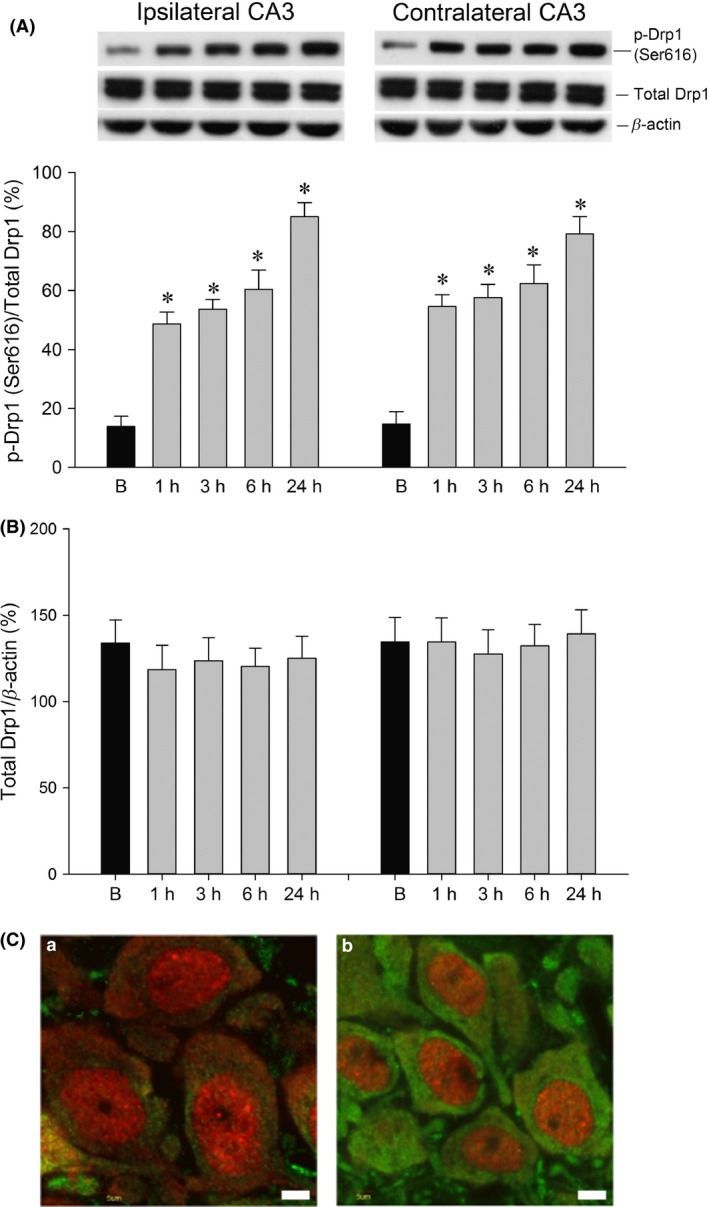
Representative gels (inset) or temporal changes in p‐Drp1 (Ser616) relative to total Drp1 or β‐actin protein (A) and total Drp1 relative to β‐actin protein (B), detected in samples collected from the bilateral hippocampal CA3 at 1, 3, 6, or 24 h after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield. Values are the mean ± SEM of quadruplicate analyses from four animals per experimental group. *P < 0.05 versus the sham‐control group in the Scheffé multiple‐range test. (C). Laser‐scanning confocal microscopic images of the right hippocampal CA3b showing cells that were immunoreactive to a neuronal marker, NeuN (red fluorescence), and additionally stained for p‐Drp1 (Ser616) (green fluorescence), in sham‐control animals (C‐a) or 24 h after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield (C‐b). Compared to the sham control, there was an increase in p‐Drp1 immunoreactivity in neurons from the hippocampal CA3b subregion on the right side 24 h after KA‐induced status epilepticus. Scale bar: 5 μm.
Effects of Mdivi‐1 on Drp1 Expression in Hippocampal CA3 Neurons Following Experimental Status Epilepticus
Bilateral microinjection of the Drp1 agonist Mdivi‐1 (2 nmol) into the hippocampal CA3 region significantly decreased the expression of p‐Drp1 (Ser616) in the CA3 subfield 24 h after the induction of sustained hippocampal seizure discharges (Figure 2A). We also verified the localization of p‐Drp1(Ser616) immunoreactivity in mitochondria by co‐immunofluorescence staining with a mitochondrial marker, Anti‐ATPB, in hippocampal CA3b neurons on the right side (Figure 2B). Compared to the sham control (Figure 2B‐a,b,c and C), p‐Drp1 (Ser616) immunoreactivity increased in neuronal mitochondria on the right‐side CA3 area at 24 h after KA‐induced status epilepticus (Figure 2B‐d,e,f and C). Moreover, pretreatment with Mdivi‐1 (2 nmol) decreased the p‐Drp1 (Ser616) immunoreactivity in the hippocampal CA3 neurons (Figure 2B‐g,h,i and C).
Figure 2.
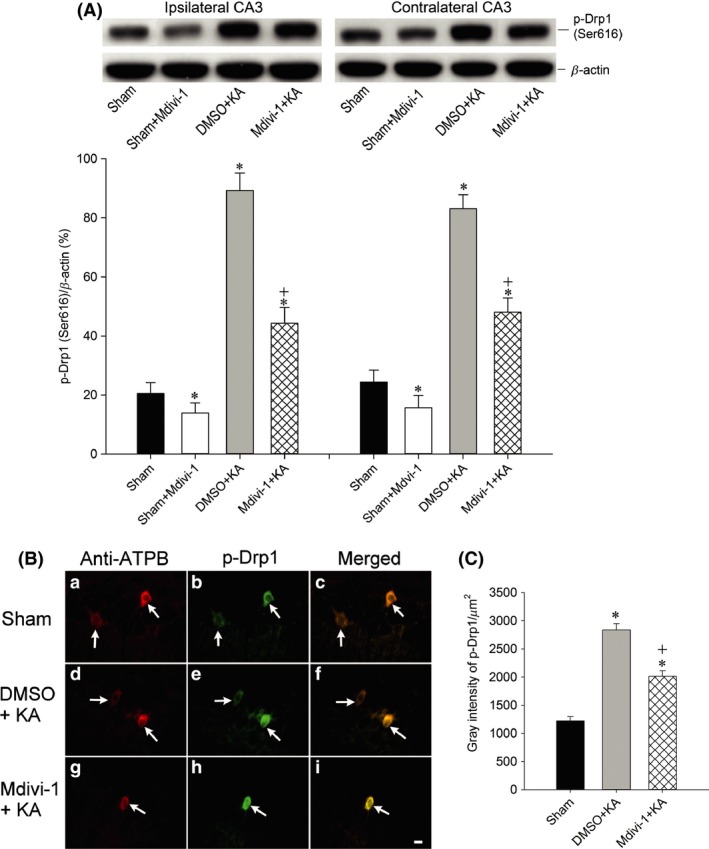
Representative gels (inset) or changes in p‐Drp1 (Ser616) relative to β‐actin protein, detected in samples collected from the bilateral hippocampal CA3 in sham‐control, Mdivi‐1 + sham animals or 24 h after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield in animals that received pretreatment of the bilateral CA3 subfield with 3% DMSO or with Mdivi‐1 (2 nmol) (A). Values are the mean ± SEM of quadruplicate analyses from four to six animals per experimental group. *P < 0.05 versus sham‐control group, and + P < 0.05 versus Mdivi‐1 + KA group in the Scheffé multiple‐range test. Immunofluorescent staining (B) showing p‐Drp1(Ser616) immunoreactivity in mitochondria (arrows) by co‐immunofluorescence staining with a mitochondrial marker, anti‐ATPB antibody, in hippocampal CA3b neurons on the right side, in sham‐control animals (B‐a,b,c), or 24 h after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield in animals that received pretreatment of the bilateral CA3 subfield with 3% DMSO (B‐d,e,f) or with Mdivi‐1 (2 nmol) (B‐g,h,i). Semi‐quantitative data for immunofluorescence intensity of p‐Drp1(Ser616) for Figure 2 B‐b,e,h are shown in (C). *P < 0.05 versus the sham‐control group, and + P < 0.05 versus the Mdivi‐1 + KA group in the Scheffé multiple‐range test.
Effects of Mdivi‐1 on the Mitochondrial Fission in Hippocampal CA3 Neurons Following Experimental Status Epilepticus
As exemplified by a pyramidal neuron in the right hippocampal CA3 area (Figure 3A), electron microscopy showed normal mitochondrial ultrastructure and number in CA3 neurons on both sides at 24 h after microinjection of DMSO into the left CA3 subfield. In contrast, 24 h after microinjection of KA into the CA3 subfield on the left side, neurons in the hippocampal CA3 area showed ultrastructural features of mitochondrial fission including smaller size, altered shape, and increased numbers of mitochondria (Figure 3B). Additionally, pretreatment with Mdivi‐1 (2 nmol) improved the morphology of some mitochondria in the hippocampal CA3 neurons (Figure 3C) that showed a decrease in mitochondrial fission.
Figure 3.
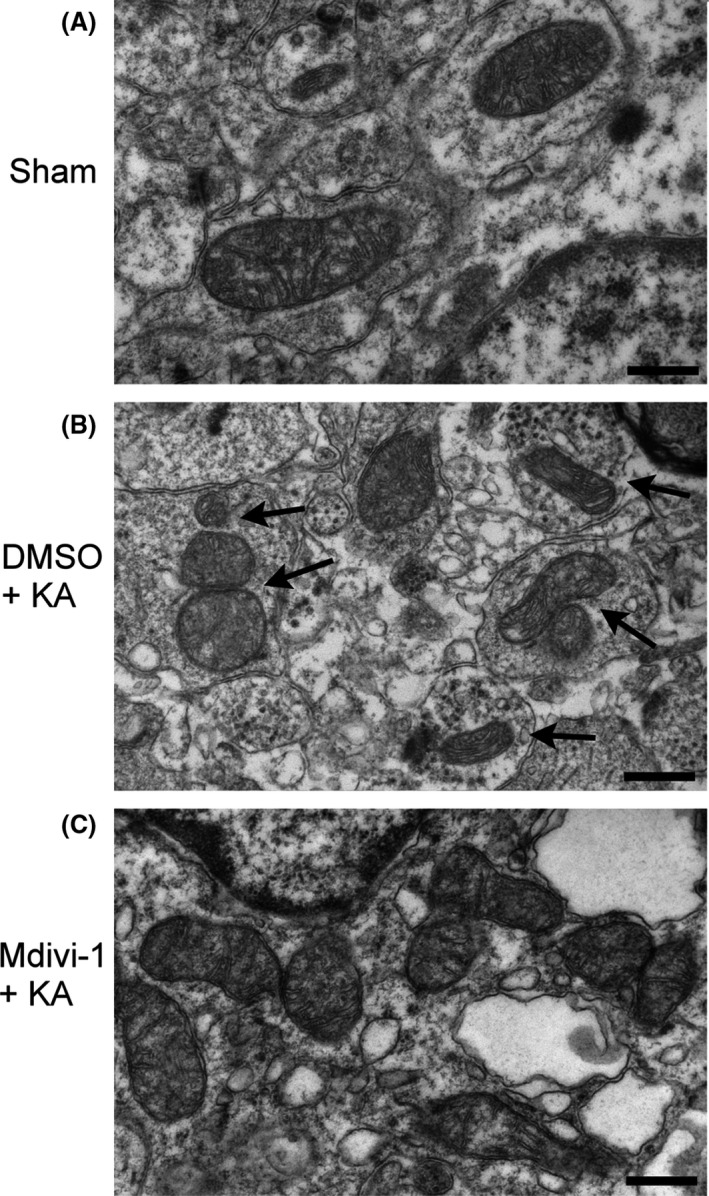
Representative electron photomicrographs of mitochondrial ultrastructure in the hippocampus. (A). Normal mitochondrial ultrastructure and number in right hippocampal CA3 neurons were illustrated from sham‐control rats. (B). Neurons with ultrastructural features of mitochondrial fission were identified in the hippocampal CA3 area 24 h after microinjection of 3% DMSO and KA into the CA3 subfield on the left side, including decreased size, altered shape, and an increased number of mitochondria (arrows). (C). Pretreatment with Mdivi‐1 (2 nmol) improved the morphology of some mitochondria in the hippocampal CA3 neurons, which showed fewer signs of mitochondrial fission 24 h after KA‐induced status epilepticus. (Original magnification, ×50,000; scale bars: 500 nm).
Effects of Mdivi‐1 on the Activity of Mitochondrial Respiratory Enzyme Complex I and Oxidized Protein Expression in the Hippocampal CA3 Subfield Following Experimental Status Epilepticus.
We further examined whether the seizure‐induced mitochondrial Complex I dysfunction and excessive oxidative stress are causally related to Drp1 phosphorylation and mitochondrial fission. We found that the reduction in the activity of mitochondrial respiratory enzyme Complex I in the hippocampus 24 h after local application of KA was significantly blunted by pretreatment with Mdivi‐1 (2 nmol) (Figure 4A). Moreover, bilateral microinjection of Mdivi‐1 (2 nmol) into the hippocampal CA3 region also mitigated the increase in oxidized protein in the CA3 subfield 24 h after KA‐induced status epilepticus (Figure 4B).
Figure 4.
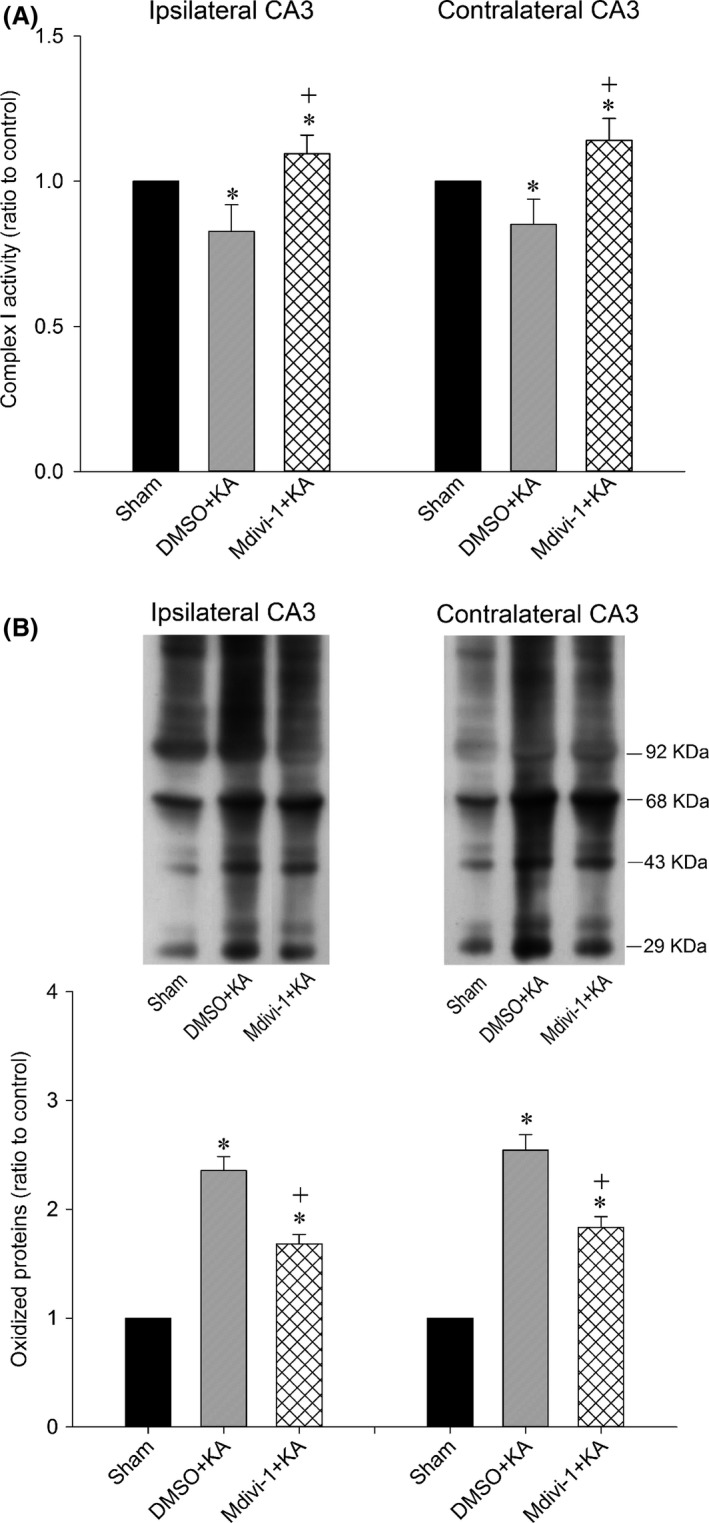
Enzyme assay for the activity of Complex I (A) in mitochondria isolated from the CA3 subfield of hippocampus in sham‐control animals or 24 h after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield in animals in which the bilateral CA3 subfield was pretreated with 3% DMSO or Mdivi‐1 (2 nmol). Values are fold changes with reference to the sham control and are the mean ± SEM of four animals per experimental group. Representative gels (inset) or temporal changes in protein oxidation (B) detected in samples collected from the CA3 subfield of hippocampus in sham‐control animals or 24 h after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield in rats in which the bilateral CA3 subfield was pretreated with 3% DMSO or Mdivi‐1 (2 nmol). Total proteins were extracted from hippocampal CA3 subfield, followed by immunoblot analysis for the extent of protein oxidation. Values in the lower panel are fold changes with reference to the sham control and are the mean ± SEM of four animals per experimental group. *P < 0.05 versus sham‐control group, + P < 0.05 versus the DMSO+KA group in the Scheffé multiple‐range test.
Effects of Mdivi‐1 on the Cytochrome c/caspase‐3 Signaling Cascade in Hippocampal CA3 Neurons Following Experimental Status Epilepticus
The increase in cytosolic cytochrome c (Figure 5A) and the decrease in mitochondrial cytochrome c (Figure 5B) in the bilateral hippocampal CA3 subfields 24 h after the induction of experimental status epilepticus were significantly antagonized by pretreatment with Mdivi‐1 (2 nmol) (Figure 5). Using co‐immunofluorescence staining with NeuN (Figure 6A‐a,e,i; red fluorescence) and activated caspase‐3 (Figure 6A‐b,f,j; green fluorescence), as well as Western blot analysis (Figure 6B), we noted a significant elevation in activated caspase‐3 in the cytosol of the CA3 neurons (Figure 6A‐e,f,g and B) 7 days after the induction of experimental status epilepticus when compared with sham controls (Figure 6A‐a,b,c and Figure 6B). Merged images of NeuN and activated caspase‐3 (Figure 6A‐c,g,k) and Western blot analysis (Figure 6B) also revealed that pretreatment with Mdivi‐1 (2 nmol) also significantly moderated the increase in cytosolic activated caspase‐3 in the hippocampal CA3 neurons.
Figure 5.
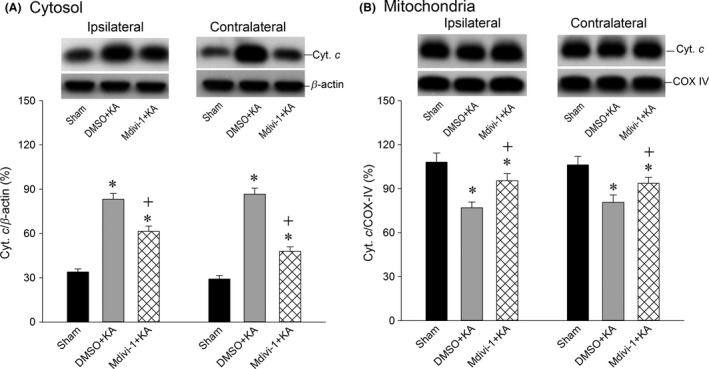
Representative gels (inset) or temporal changes in cytochrome c relative to β‐actin protein detected in the cytosolic fraction (A) or relative to COX IV in the mitochondrial fraction (B) of samples collected from the CA3 subfield of hippocampus in sham‐control animals or 24 h after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield in rats in which the bilateral CA3 subfield was pretreated with 3% DMSO or Mdivi‐1 (2 nmol). Values are the mean ± SEM of quadruplicate analyses from four animals per experimental group. *P < 0.05 versus the sham‐control group, + P < 0.05 versus the DMSO+KA group in the Scheffé multiple‐range test.
Figure 6.
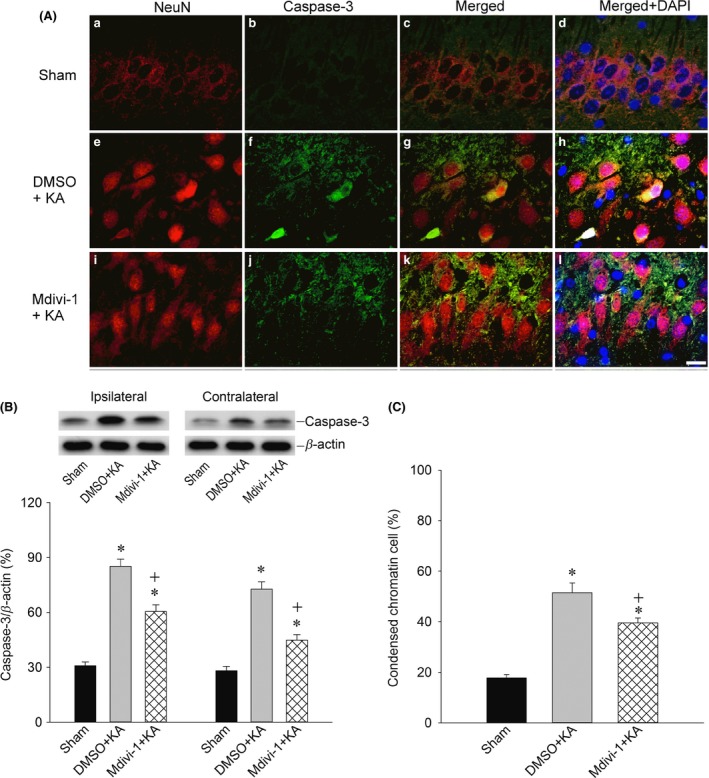
Representative photographs showing double immunofluorescent staining (A) for activated caspase‐3 (green fluorescence) and the neuronal marker NeuN (red fluorescence) in sham‐control animals (A‐a,b,c) or 7 days after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield in rats in which the bilateral CA3 subfield was pretreated with 3% DMSO (A‐e,f,g) or Mdivi‐1 (2 nmol) (A‐i,j,k). Merged images of NeuN and activated caspase‐3 (A‐c,g,k) revealed that pretreatment with Mdivi‐1 (2 nmol) likewise significantly moderated the increase in activated caspase‐3 in the hippocampal CA3 neurons. Representative gels (inset) or changes in activated caspase‐3 relative to β‐actin protein (B), detected in the cytosolic fraction of samples collected from the CA3 subfield of hippocampus in sham‐control animals or 7 days after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield in animals in which the bilateral CA3 subfield was pretreated with 3% DMSO or Mdivi‐1 (2 nmol). Values are the mean ± SEM of quadruplicate analyses from four animals per experimental group. *P < 0.05 versus the sham‐control group, + P < 0.05 versus the DMSO+KA group in the Scheffé multiple‐range test. The nuclei were counterstained with DAPI (A‐d,h,i) (blue fluorescence); pretreatment with Mdivi‐1 (2 nmol) significantly decreased the numbers of apoptotic neurons with condensed chromatin (C). Quantitative analysis of cells containing condensed chromatin and relative fluorescent intensity in the hippocampal CA3 area. Values are the mean ± SEM. *P < 0.05 versus the sham‐control group, + P < 0.05 versus the DMSO+KA group in the Scheffé multiple‐range test.
Effects of Mdivi‐1 on Apoptosis in the Hippocampal CA3 Subfield Following Experimental Status Epilepticus
Our next series of experiments explored whether the Drp1‐related mitochondrial fission pathway plays a significant role in neuronal apoptosis. We found that pretreatment with Mdivi‐1 (2 nmol) significantly decreased the numbers of condensed chromatin apoptotic neuronal cells (Figure 6A‐d,h,l and C). Additionally, pretreatment with Mdivi‐1 (2 nmol) significantly reduced the qualitative (Figure 7A) and quantitative (Figure 7B) values of DNA fragmentation as another index for apoptosis, 7 days after the induction of status epilepticus. Histological analysis demonstrated that prominent neuronal cell loss occurred in the bilateral hippocampal CA3 7 days after the induction of status epilepticus. Those neuronal cell losses in CA3 neurons were markedly reduced in rats pretreated with Mdivi‐1 (2 nmol) (Figure 7C,D).
Figure 7.
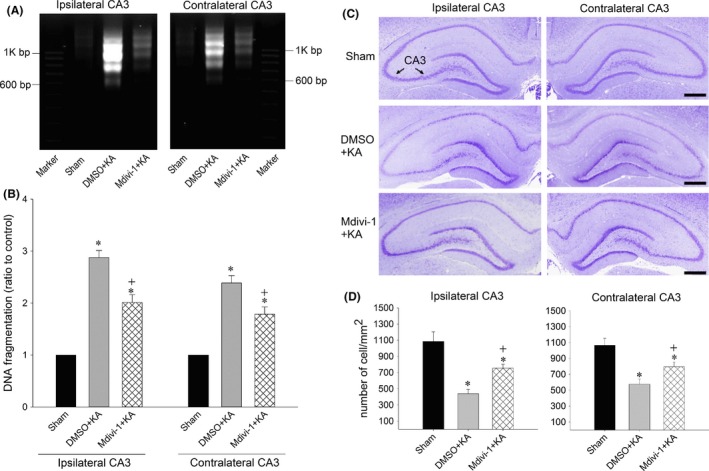
Qualitative (A) or quantitative (B) analysis of DNA fragmentation detected in samples collected from the CA3 subfield of hippocampus in sham‐control animals or 7 days after microinjection of KA (0.5 nmol) into the left hippocampal CA3 subfield in animals in which the bilateral CA3 subfield was pretreated with 3% DMSO or Mdivi‐1 (2 nmol). Values are the mean ± SEM of quadruplicate analyses from four to six animals per experimental group. *P < 0.05 versus sham‐control group, + P < 0.05 versus the DMSO+KA group in the Scheffé multiple‐range test. (C and D): Brain sections stained with cresyl violet show the neuronal cell loss in the bilateral hippocampal CA3 area 7 days after microinjection of KA (0.5 nmol) into the left hippocampal CA3 in animals in which the bilateral CA3 subfield was pretreated with 3% DMSO or Mdivi‐1 (2 nmol). These results were obtained from four animals from each experimental group. *P < 0.05 versus the sham‐control group, + P < 0.05 versus the DMSO+KA group in the Scheffé multiple‐range test. Scale bar, 5 mm.
Effect of Mdivi‐1 on hEEG Signals in the Hippocampal CA3 Subfield Following Experimental Status Epilepticus
Our final experiments examined whether Mdivi‐1 affects KA‐induced seizure activity. Unilateral microinjection of KA into the CA3 subfield of the hippocampus resulted in a progressive buildup of seizure activity. Pretreatment with Mdivi‐1 (2 nmol) did not affect the increases in either MPF or RMS values observed during experimental status epilepticus (Figure 8).
Figure 8.
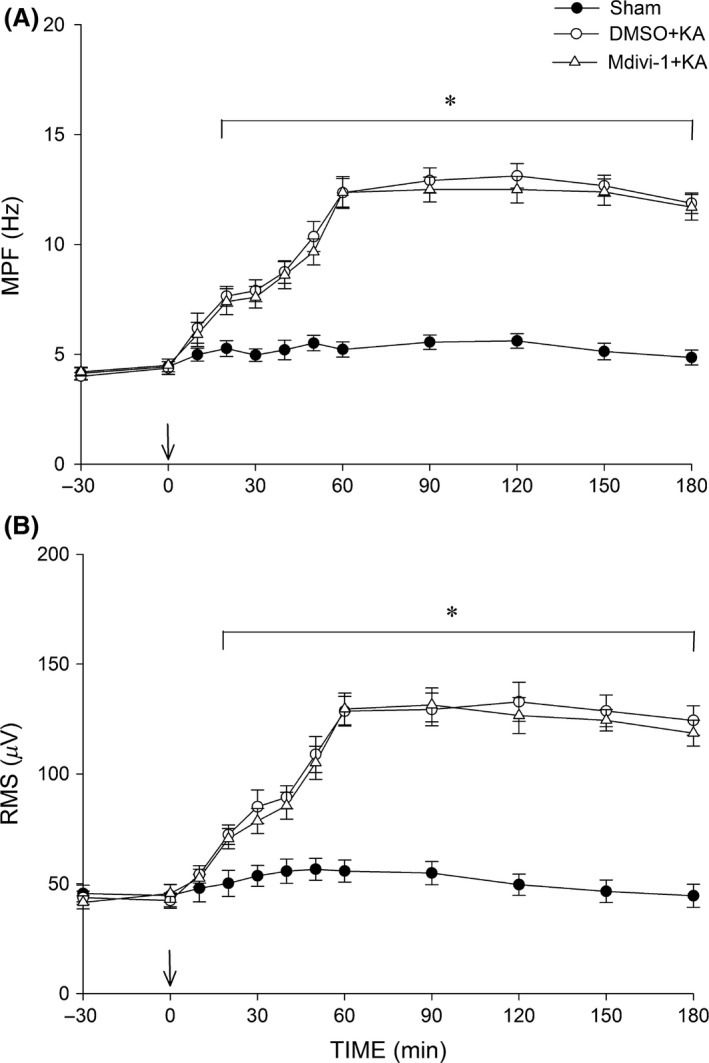
Time‐course alternations in root mean square (RMS) (A) and mean power frequency (MPF) (B) values of hEEG signals before and after microinjection of KA (0.5 nmol) or PBS into the left hippocampal CA3 in rats in which the bilateral CA3 subfield (at arrow) was pretreated with 3% DMSO or Mdivi‐1 (2 nmol). Values are the mean ± SEM from four animals per group. *P < 0.05 versus the sham control and DMSO+KA groups during the indicated time points in two‐way ANOVA.
Discussion
Using a clinically relevant animal model, the present study provides novel evidence to support the changes in mitochondrial dynamics and the mitochondrial dysfunction observed in prolonged seizures. Specifically, our results revealed that p‐Drp1(Ser616), rather than total Drp1, plays an important role in mitochondrial fission during experimental status epilepticus. Sustained epileptic seizures upregulated Drp1 phosphorylation at Ser616, which was accompanied by mitochondrial dysfunction and oxidative stress, and it further triggered the cytochrome c/caspase‐3 signaling cascade and contributed to neuronal cell death in the hippocampal CA3 area. Our results also demonstrated that a Drp1 inhibitor can partially reverse these detrimental effects and lessen neuronal injury in this experimental model of status epilepticus.
Neurodegenerative diseases are a large group of disabling disorders of the nervous system, and they are characterized by the relatively selective death of neuronal subtypes. In most neurological diseases, there is overwhelming evidence of impaired mitochondrial function as a causative factor 37, 38. More recently, evidence has emerged indicating that impairment of mitochondrial dynamics, including shape, size, fission‐fusion, distribution, and movement, contributes to neurodegenerative diseases such as Parkinson's disease, Huntington's disease, amyotrophic lateral sclerosis, and Alzheimer's disease 37, 38, 39, 40. In our recent study, cerebral ischemia increased the level of p‐Drp1(Ser616), with milder effects on total Drp1 and p‐Drp1(Ser637), and augmented p‐Drp1(Ser616) expression could heighten DNA oxidation and enhance neuronal damage in the hippocampal CA1 subfield 26. These results indicated a pivotal role for Drp1 in cerebral ischemia and suggest that attenuation of p‐Drp1 (Ser616) levels may be neuroprotective against ischemia/reperfusion injury. The role of mitochondrial dynamics in epileptic seizure is only now emerging 27, 28. Drp1 is required for normal spatial memory and synaptic function, and loss of Drp1 may compromise the intrinsic bioenergetic function of axonal mitochondria 41. However, accumulating evidence indicates that aberrant mitochondrial fission is an early event following acute brain injury and is a key player in ensuring neuronal apoptosis and that the Drp1‐mediated fission pathway may precede the initiation of cell death signaling 22, 42, 43. It has been reported that inhibition of Drp1 provides neuroprotection and that Drp1 inhibitors protected primary neurons against glutamate excitotoxicity and oxygen glucose deprivation and reduced the infarct volume in a mouse model of transient focal ischemia 23. The important role of p‐Drp1(Ser616) rather than total Drp1 expression is well noted in other models of acute stress 26, 44, 45. A recent study 27 using pilocarpine to elicit seizures in rats showed that mitochondrial fission was increased after seizures and that the inhibition of mitochondrial fission by Mdivi‐1 significantly attenuated the postseizure oxidative stress and neuronal loss. To further characterize the pivotal role of Drp1 under prolonged seizure conditions, we used an experimental status epilepticus model. We demonstrated here that under prolonged seizure, p‐Drp1(Ser616) increased from 1 to 24 h after microinjection of KA to the hippocampal CA3 region, with no evident change in total Drp1.
Sustained epileptic seizures will change the redox potential and decrease the ATP content, which may lead to the collapse of energy production and supply in the brain 3, 32. Increasing evidence indicates that mitochondrial dysfunction is associated with epilepsy and status epilepticus both in animal models and in human samples 3, 46, 47. We and others 17, 18, 47, 48, 49 have found that prolonged seizures were associated with the dysfunction of Complex I in the mitochondrial electron transport chain, increased oxidative stress, and mitochondrial ultrastructural injury in the hippocampus. These results indicate that seizure‐induced mitochondrial dysfunction and oxidative stress play crucial roles in inducing neuronal apoptosis in the hippocampus after experimental status epilepticus. The role of mitochondrial fission in epileptic seizures still requires elucidation. In the present study, we showed that pretreatment with a mitochondrial fission inhibitor, Mdivi‐1, significantly decreased Drp1 phosphorylation at Ser616 (Figure 2) as well as reducing the ultrastructural changes in mitochondrial fission, including smaller size, altered shape, and increased number of mitochondria (Figure 3), in the hippocampal CA3 area after KA‐induced prolonged seizure. A recent study 27 showed that Mdivi‐1 decreased the 8‐hydroxy‐deoxyguanosine (8‐oHdG) content, increased the superoxide dismutase activity, decreased the expression of cytochrome c and caspase‐3, and increased the neuronal survival in the hippocampus in a pilocarpine‐induced seizure model.
In our present study, we demonstrate that decreased expression of p‐Drp1(Ser616) may be accompanied by reducing mitochondrial fragmentation. This may rescue the decrease in mitochondrial Complex I enzyme activity as well as lessening the expression of oxidized proteins in the hippocampus in our experimental model of temporal lobe status epilepticus. It is well known that mitochondrial dysfunction and oxidative stress play crucial roles in inducing neuronal apoptosis in the hippocampus after experimental status epilepticus 3, 4, 48. Our present study suggested that the aberrant mitochondrial fission may impair the function of mitochondrial respiratory chain enzymes, increase ROS production and trigger a cytochrome c/caspase‐3 signaling cascade that results in apoptotic cell death in hippocampal CA3 neurons after experimental status epilepticus. Consistent with previous studies showing that inhibition of Drp1 provides neuroprotection and that Drp1 inhibitors can protect against various insults, including glutamate excitotoxicity, oxygen glucose deprivation, and focal ischemia 23, the biochemical, electron microscopic, and immunofluorescence experiments in the present study show that the phenomena of mitochondrial fission and the detrimental effects on neuronal cells were partially rescued by pretreatment with Mdivi‐1. These results indicate a potential role for p‐Drp1(Ser616) in contributing to the hippocampal neuronal cell death following experimental status epilepticus.
We are aware that pretreatment with Mdivi‐1 did not affect the seizure activity and suggest that the mitochondrial fission pathway is not likely to be involved in the mechanism of anti‐epileptogenesis. Blockade of p‐Drp1 expression antagonized the seizure‐induced neuronal cell death in the hippocampus, supporting the idea that these cellular events are mainly involved in the mitochondrial fission‐dependent signaling pathway rather than in the mechanism of anti‐epileptogenesis.
In conclusion, the present study demonstrated that activation of p‐Drp1(Ser616) is related to seizure‐induced neuronal cell damage in the hippocampus and that inhibition of p‐Drp1(Ser616) expression is accompanied by decreases in mitochondrial fragmentation, mitochondrial dysfunction, and oxidation, as well as the resulting apoptosis and neural damage. Thus, modulation of the p‐Drp1(Ser616) signaling pathway may offer a therapeutic strategy against seizure‐induced hippocampal neuronal cell damage following status epilepticus and may offer an avenue to counteract the vicious cycle related to mitochondrial dysfunction and the mitochondria‐related apoptotic pathway.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported in part by research grants NSC102‐2314‐B‐182A‐090‐MY3 to Y.C.C. from the National Science Council, and CMRPG8B1031 and CMRPG8E0791G to Y.C.C. from Chang Gung Memorial Hospital‐Kaohsiung, Taiwan.
References
- 1. Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 2009;109(Suppl 1):133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liou AKF, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: A review on the stress‐activated signaling pathways and apoptotic pathways. Prog Neurobiol 2003;69:103–142. [DOI] [PubMed] [Google Scholar]
- 3. Chen SD, Chang AYW, Chuang YC. The potential role of mitochondrial dysfunction in seizure‐associated cell death in the hippocampus and epileptogenesis. J Bioenerg Biomembr 2010;42:461–465. [DOI] [PubMed] [Google Scholar]
- 4. Henshall DC, Simon RP. Epilepsy and apoptosis pathways. J Cereb Blood Flow Metab 2005;25:1557–1572. [DOI] [PubMed] [Google Scholar]
- 5. Patel M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic Biol Med 2004;37:1951–1962. [DOI] [PubMed] [Google Scholar]
- 6. Diaz F, Moraes CT. Mitochondrial biogenesis and turnover. Cell Calcium 2008;44:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet 2009;18:R169–R176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol 2014;2:702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev 2008;22:1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Herzig S, Martinou JC. Mitochondrial dynamics: To be in good shape to survive. Curr Mol Med 2008;8:131–137. [DOI] [PubMed] [Google Scholar]
- 11. Mihara K, Ishihara N, Otera H, Oka T. Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid Redox Signal 2013;19:389–399. [DOI] [PubMed] [Google Scholar]
- 12. Westermann B. Molecular machinery of mitochondrial fusion and fission. J Biol Chem 2008;283:13501–13505. [DOI] [PubMed] [Google Scholar]
- 13. Zorzano A, Sebastian D, Segales J, Palacin M. The molecular machinery of mitochondrial fusion and fission: An opportunity for drug discovery? Curr Opin Drug Discov Devel 2009;12:597–606. [PubMed] [Google Scholar]
- 14. Berman SB, Pineda FJ, Hardwick JM. Mitochondrial fission and fusion dynamics: The long and short of it. Cell Death Differ 2008;15:1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 2010;10:640–648. [DOI] [PubMed] [Google Scholar]
- 16. Waldbaum S, Patel M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res 2010;88:23–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chuang YC, Chang AYW, Lin JW, Hsu SP, Chan SHH. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid‐induced status epilepticus in the rat. Epilepsia 2004;45:1202–1209. [DOI] [PubMed] [Google Scholar]
- 18. Chuang YC, Lin TK, Huang HY, et al. Peroxisome proliferator‐activated receptors gamma/mitochondrial uncoupling protein 2 signaling protects against seizure‐induced neuronal cell death in the hippocampus following experimental status epilepticus. J Neuroinflammation 2012;9:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chuang YC, Chen SD, Lin TK, et al. Transcriptional upregulation of nitric oxide synthase II by nuclear factor‐kappaB promotes apoptotic neuronal cell death in the hippocampus following experimental status epilepticus. J Neurosci Res 2010;88:1898–1907. [DOI] [PubMed] [Google Scholar]
- 20. Chuang YC, Chen SD, Liou CW, et al. Contribution of nitric oxide, superoxide anion, and peroxynitrite to activation of mitochondrial apoptotic signaling in hippocampal CA3 subfield following experimental temporal lobe status epilepticus. Epilepsia 2009;50:731–746. [DOI] [PubMed] [Google Scholar]
- 21. Chuang YC, Chen SD, Lin TK, et al. Upregulation of nitric oxide synthase II contributes to apoptotic cell death in the hippocampal CA3 subfield via a cytochrome c/caspase‐3 signaling cascade following induction of experimental temporal lobe status epilepticus in the rat. Neuropharmacology 2007;52:1263–1273. [DOI] [PubMed] [Google Scholar]
- 22. Barsoum MJ, Yuan H, Gerencser AA, et al. Nitric oxide‐induced mitochondrial fission is regulated by dynamin‐related GTPases in neurons. EMBO J 2006;25:3900–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grohm J, Kim SW, Mamrak U, et al. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ 2012;19:1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao YX, Cui M, Chen SF, Dong Q, Liu XY. Amelioration of ischemic mitochondrial injury and Bax‐dependent outer membrane permeabilization by Mdivi‐1. CNS Neurosci Ther 2014;20:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci 2009;29:9090–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen SD, Lin TK, Yang DI, et al. Roles of PTEN‐induced putative kinase 1 and dynamin‐related protein 1 in transient global ischemia‐induced hippocampal neuronal injury. Biochem Biophys Res Commun 2015;460:397–403. [DOI] [PubMed] [Google Scholar]
- 27. Qiu X, Cao L, Yang X, et al. Role of mitochondrial fission in neuronal injury in pilocarpine‐induced epileptic rats. Neuroscience 2013;245:157–165. [DOI] [PubMed] [Google Scholar]
- 28. Xie N, Wang C, Lian Y, Zhang H, Wu C, Zhang Q. A selective inhibitor of Drp1, mdivi‐1, protects against cell death of hippocampal neurons in pilocarpine‐induced seizures in rats. Neurosci Lett 2013;545:64–68. [DOI] [PubMed] [Google Scholar]
- 29. Chang CC, Chen SD, Lin TK, et al. Heat shock protein 70 protects against seizure‐induced neuronal cell death in the hippocampus following experimental status epilepticus via inhibition of nuclear factor‐kappaB activation‐induced nitric oxide synthase II expression. Neurobiol Dis 2014;62:241–249. [DOI] [PubMed] [Google Scholar]
- 30. Chen SD, Lin TK, Lin JW, et al. Activation of calcium/calmodulin‐dependent protein kinase IV and peroxisome proliferator‐activated receptor gamma coactivator‐1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J Neurosci Res 2010;88:3144–3154. [DOI] [PubMed] [Google Scholar]
- 31. Lin TK, Cheng CH, Chen SD, Liou CW, Huang CR, Chuang YC. Mitochondrial dysfunction and oxidative stress promote apoptotic cell death in the striatum via cytochrome c/Caspase‐3 signaling cascade following chronic rotenone intoxication in rats. Int J Mol Sci 2012;13:8722–8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chuang YC, Lin JW, Chen SD, et al. Preservation of mitochondrial integrity and energy metabolism during experimental status epilepticus leads to neuronal apoptotic cell death in the hippocampus of the rat. Seizure 2009;18:420–428. [DOI] [PubMed] [Google Scholar]
- 33. Nadanaciva S, Dykens JA, Bernal A, Capaldi RA, Will Y. Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration. Toxicol Appl Pharmacol 2007;223:277–287. [DOI] [PubMed] [Google Scholar]
- 34. Willis JH, Capaldi RA, Huigsloot M, Rodenburg RJ, Smeitink J, Marusich MF. Isolated deficiencies of OXPHOS complexes I and IV are identified accurately and quickly by simple enzyme activity immunocapture assays. Biochim Biophys Acta 2009;1787:533–538. [DOI] [PubMed] [Google Scholar]
- 35. Smith CD, Carney JM, Starke‐Reed PE, et al. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A 1991;88:10540–10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saito A, Narasimhan P, Hayashi T, Okuno S, Ferrand‐Drake M, Chan PH. Neuroprotective role of a proline‐rich Akt substrate in apoptotic neuronal cell death after stroke: Relationships with nerve growth factor. J Neurosci 2004;24:1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakamura T, Cho DH, Lipton SA. Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp Neurol 2012;238:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 2012;342:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Knott AB, Bossy‐Wetzel E. Impairing the mitochondrial fission and fusion balance: A new mechanism of neurodegeneration. Ann N Y Acad Sci 2008;1147:283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Santos RX, Correia SC, Wang X, et al. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer's disease. J Alzheimers Dis 2010;20(Suppl 2):S401–S412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shields LY, Kim H, Zhu L, et al. Dynamin‐related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis 2015;6:e1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Almeida A, Delgado‐Esteban M, Bolanos JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem 2002;81:207–217. [DOI] [PubMed] [Google Scholar]
- 43. Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci 1996;16:6125–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ribeiro M, Rosenstock TR, Oliveira AM, Oliveira CR, Rego AC. Insulin and IGF‐1 improve mitochondrial function in a PI‐3K/Akt‐dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington's disease knock‐in striatal cells. Free Radic Biol Med 2014;74:129–144. [DOI] [PubMed] [Google Scholar]
- 45. Cho MH, Kim DH, Choi JE, Chang EJ, Seung Y. Increased phosphorylation of dynamin‐related protein 1 and mitochondrial fission in okadaic acid‐treated neurons. Brain Res 2012;1454:100–110. [DOI] [PubMed] [Google Scholar]
- 46. Kudin AP, Zsurka G, Elger CE, Kunz WS. Mitochondrial involvement in temporal lobe epilepsy. Exp Neurol 2009;218:326–332. [DOI] [PubMed] [Google Scholar]
- 47. Kunz WS, Kudin AP, Vielhaber S, et al. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann Neurol 2000;48:766–773. [PubMed] [Google Scholar]
- 48. Folbergrova J, Jesina P, Haugvicova R, Lisy V, Houstek J. Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem Int 2010;56:394–403. [DOI] [PubMed] [Google Scholar]
- 49. Folbergrova J, Jesina P, Drahota Z, et al. Mitochondrial complex I inhibition in cerebral cortex of immature rats following homocysteic acid‐induced seizures. Exp Neurol 2007;204:597–609. [DOI] [PubMed] [Google Scholar]