

Abstract
The impact of traumatic brain injury (TBI) involves a combination of complex biochemical processes beginning with the initial insult and lasting for days, months and even years post-trauma. These changes range from neuronal integrity losses to neurotransmitter imbalance and metabolite dysregulation, leading to the release of (anti-)apoptotic factors which mediate cell survival or death. Such dynamic processes affecting the brain’s neurochemistry can be monitored using a variety of neuroimaging techniques, whose combined use can be particularly useful for understanding patient-specific clinical trajectories. Here, we describe how TBI changes the metabolism of essential neurochemical compounds, summarize how neuroimaging approaches facilitate the study of such alterations, and highlight promising ways in which neuroimaging can be used to investigate post-TBI changes in brain metabolism.
Keywords: TBI, neuroimaging, metabolism, pathology, neurochemistry
The Injured Human Brain
Traumatic brain injury (TBI) is a physical insult to the brain caused by a blunt mechanical force. TBI demographics are staggering: in the US alone, over 1.4 million cases are recorded every year[1, 2], whereas annual estimates of sports-related concussions range from 1.6 to 3.8 million (males being about twice as likely to be affected as females)[1]. Long-term effects of TBI include an increased vulnerability to psychiatric and neurological disorders. For example, the risk factor for clinical depression is ~1.5 higher in TBI survivors compared to the general population. Furthermore, for Alzheimer’s Disease (AD) this risk increases to 2.3 and 4.5 times higher, depending on whether the TBI is moderate or severe, respectively[3].
Throughout the past few decades, the study of TBI using imaging techniques has garnered an increasing amount of attention from health care professionals, policy makers and the public[4]. This is partly because neuroimaging has led to substantial improvement in our understanding of how TBI can affect brain metabolism even years after injury, particularly in cases where immediate sequelae seem modest, long-term consequences seem unrelated to TBI, or both (see Glossary). Recent advances in the neuroimaging of metabolic compounds have made the study of TBI-related changes in brain function more accessible using noninvasive or minimally-invasive techniques such as magnetic resonance (MR) imaging (MRI), functional MRI (fMRI), magnetic resonance spectroscopy (MRS), diffusion weighted imaging (DWI), diffusion tensor imaging (DTI), single-photon emission computed tomography (SPECT) and positron emission tomography (PET) (Figure 1). The combined use of these techniques has greatly furthered scientific knowledge of post-traumatic changes in neurochemistry and neurophysiology[5]. Nevertheless, substantial additional progress is needed to bridge the gap between the insights which these modalities can deliver regarding human TBI—on the one hand—and the level of sophistication which has been achieved by in vitro and in vivo imaging modalities whose use is currently restricted to animal experiments.
Figure 1. Human neuroimaging, electrophysiology, microdialysis, and microscopy.

In the in vivo study of brain injury, human neuroimaging methods including MRI, PET, and SPECT allow for the examination of brain structural alteration, hemorrhage, edema, connectomic changes, as well as changes in metabolic and bloodflow rates in tissues proximal to lesioned tissue. Likewise, MRS illustrates how the spectra of various brain metabolites are altered in the presence of TBI-related injury and necrosis. Electrophysiological techniques record brain activity at rate much faster than that achieved using MRI or PET. All such methods are often supplemented by microdialysis of intraventricular or spinal CSF to measure neurotransmitter metabolites as well as biomarkers of cell death. Conversely, post-mortem ex vivo examination of cellular disruption, axonal injury, as well as the detection of Tau and Beta-amyloid are generally conducted using microscopy techniques. Accordingly, we have associated each modality with an icon (on the left in each panel) which identifies the locations in the figures which follow where that method has maximal utility for quantifying TBI tissue damage, is measuring a process associated with a particular metabolic pathway, is sensitive to cellular damage, or is capable of providing information about patient outcomes. In this way, we provide a graphical linkage between cellular metabolic alterations in brain injury due to TBI and the neuroimaging as well as accompanying approaches suited for assessing these effects.
The motivation for this survey is to describe how TBI alters the cellular metabolism of essential neurochemical compounds, to outline how studying these alterations has been facilitated by neuroimaging modalities, and to indicate critical goals for neuroimaging researchers who aim to develop new methods for investigating post-traumatic metabolism in greater detail. The ability of macroscale neuroimaging techniques to detect and quantify a given chemical compound is largely predicated upon its concentration; for this reason, the review focuses on the most abundant and/or prominent neurochemical compounds, while also indicating how neuroimaging their metabolism can allow researchers to make valuable inferences concerning the impact of TBI upon the central nervous system (CNS). Though the extent of cell damage and survival is highly dependent upon genetic factors which can either aid recovery or result in the exacerbation of apoptosis, this topic has been discussed extensively elsewhere[6] and is, therefore, outside the scope of this review.
In Figures 2–5 and throughout the course of this review, the notation (X.Y) is used to guide the reader and to provide a parallel between text, table, and figures. Specifically, the first number (X) indicates the number of the figure being referred to, whilst the second number (Y) indicates the step in the physiological pathway illustrated in the figure and discussed in the text. Moreover, Table 1 is included which summarizes the main consequences of damaged brain tissue, the metabolic processes affected, and the neuroimaging modality most suited to its detection and measurement.
Figure 2. Hypoxia and oxidative stress.

Following TBI, blood delivery to brain tissue can be restricted (ischemia) (2.1), which results in neurons being deprived of molecular oxygen (O), i.e. O2 (hypoxia) (2.2). The failure to deliver O2 to brain cells impairs the process of oxidative phosphorylation (2.6), whereby mitochondria process nutrients to generate adenosine triphosphate (ATP) (2.7). Phosphorus (P) is critical to oxidative phosphorylation (2.8), and MRI is useful for identifying brain locations (2.9) where ATP production proceeds at higher or lower than normal rates. Because 31P is a stable phosphorus isotope with non-zero nuclear spin, its presence in tissues is detectable using MRS (2.10). When brain tissues are subjected to TBI-caused environmental stress, O2 is more readily converted into oxidants (e.g. OH−) (2.11) with deleterious effects called reactive oxygen species (ROSs) (2.12). The brain is more sensitive to ROSs than other organs due to the higher concentrations of cholesterol and polyunsaturated fatty acids (FAs) in the CNS, which can be easily broken down to generate free radicals, including FA and hydroxyl radicals (2.13)[132]. The double bonds of unsaturated fatty acids are highly susceptible to ROSs, whereas the brain has relatively low concentrations of ROS scavengers such as glutathione peroxidase. The concentration of ascorbic acid (Asc, an ROS scavenger) is higher in healthy brain tissue than in the presence of injury, and under normal circumstances this compound acts as an ROS scavenger. Following TBI, iron in hemorrhaging blood causes ascorbic acid to aggravate oxidative stress by increasing the production of free radicals (2.14). Ascorbic acid concentrations can be imaged using radioactive carbon (C), specifically using [1-13C]-ascorbic acid as a hyperpolarizing probe in 13C NMR spectroscopy and MRI to study redox state changes in vivo (2.15). Post-TBI, decreases in cerebral blood flow (CBF) (2.16) are followed by mitochondrial damage and to an upregulation of ROS production (see 2.12). Cell membrane damage releases arachidonic acid (AA) into the cytosol (2.17). AA can be oxidized using an alternate lipoxygenase pathway mediated by 5-lipoxygenase, which results in the formation of a modified eicosatetraenoic acid called LTA4 (a leukotriene) (2.20) and further activation of leukotriene receptors (2.21)[18] (the specific role of these compounds is discussed in a later section). Arachidonic acid (AA) is converted into the prostaglandins (PGs) PGD2, PGE2 and PGF2 using PGH2 as a precursor molecule (2.19). Simultaneously, the formation of thromboxane A2 and prostacyclin by the action of the cyclooxygenases (COXs) COX-1 and COX-2 (2.18) causes vasoconstriction, platelet aggregation and vasodilation [133]. Leukotriene-related reactions and phenomena related to ROS activity may be imaged with [3H]hydromethidine (2.22). Hypoxia contributes substantially to the generation of free radicals due to the dissociation of iron (Fe) ions (Fe2+) from the heme group in hemoglobin after TBI, which leads to the formation of hydroxyl radicals (2.23). Formation of these free radicals due to the oxidative degradation of lipids results in the formation of malondialdehyde (MDA) (2.24). MDA molecules diffuse across the mitochondrial membrane and damage mitochondrial DNA (mDNA) by interfering with polymerase function and/or gene expression (2.25)[134]. Under normal circumstances, redox-active iron is maintained at very low levels due to iron transfer and storage proteins present in the cell (such as transferrin and ferritin (2.26), respectively). The ability of these proteins to affect iron concentrations is particularly effective when pH > 6 because iron is less reactive in cellular environments with low acidity[135]. During acute TBI, there is an upregulation in NO levels due to the over-activation of N-Methyl-D-aspartate (NMDA) receptors (NMDARs) (2.36), causing an increase in calcium (Ca) ion (Ca2+) influx (2.37) and binding of Ca2+ to calmodulin (2.38). The latter is involved in the regulation of NO synthase (NOS) levels (39) and in the production of NO from conversion of L-arginine to citrulline by NOS (2.40)[16]. NO also binds to protein disulfide isomerase (a cell surface protein) (2.41) and accumulates inside the ER, where it is involved in mis-folding of newly-synthesized proteins (2.42)[136].
Figure 5. Inflammation, apoptosis and cell survival.

Cellular death signaling initiates apoptosis via two primary pathways. The primary mechanism is initiated by the activation of caspases, which are cysteine proteases whose action is typically regulated by proteins such as Inhibitor of Apoptosis (IAP) to avoid their improper or excessive activation[157]. Caspases are either present in or move to mitochondria, where they induce apoptosis via both intrinsic and extrinsic mechanisms[158, 159]. Intrinsically, mitochondria become permeable and release cytochrome c (5.1), which forms apoptosomes (multi-protein complexes which initiate caspase activation leading to cell death) (5.2). Extrinsically, cell death is induced by factors which activate a death-inducing signaling complex formed by members of the death receptor family of apoptosis-inducing cellular receptors[160]. Caspase activity can be imaged using a fluorescent sensor which consists of two fluorophores (chemical compounds which re-emit photons upon light excitation), namely a monomeric red fluorescent protein (mRFP) and an enhanced green fluorescent protein (eGFP) which are bound to each other by a caspase-sensitive link. As the caspase is activated, the sensor is cleaved, the eGFP translocates to the nucleus and the mRFP remains on the membrane, such that both fluorophores can be imaged using CM[161]. In the secondary apoptotic pathway, caspase-independent apoptosis begins with the release of apoptosis-inducing factors (AIFs) (5.4) and of endogenous G (EndoG) (5.5) enzymes which migrate to the nucleus, where they degrade DNA[16]. This migration is initiated via externalization of phosphatidyl serine (PS), which under normal circumstances lies on the inner membrane. PS externalization (5.3) precedes many apoptotic events, including membrane permeabilization and AIF activation[162]. AIFs initiate their action by binding to chromatin (5.5) and by then causing its peripheral condensation and DNA fragmentation (5.6)[163]. The process also involves overactivation of poly (ADP-ribose) polymerase proteins, which can become highly active in cells with DNA damage (5.7), thereby causing ATP storage depletion quicker than recovery can occur, which results in cell death (5.8)[164]. Estrogen effects upon the injured brain involve (i) estradiol binding to estrogen receptors (5.9), (ii) phosphoinositide-3-kinase (PI-3K) activation (5.10) and (iii) activation of endogenous NO synthase (eNOS) to convert L-arginine into citrulline (5.11). NO is a by-product of this reaction. Estrogen also contributes to controlling apoptosis post-TBI by increasing B-cell lymphoma 2 (Bcl-2) protein expression (5.12)[118]. This prevents the pore forming function of Bcl-2 homology domain 3 (BH-3) proteins (5.13) and the release of other apoptotic factors like bcl-2-like protein (BAX) and Bcl-2 homologous antagonist killer (BAK) (5.14)[119]. BAX/BAK downregulation reduces mitochondrion membrane permeabilization (5.15) and cytochrome c activation (5.16)[120],[121], followed by reduced binding of cytochrome c to apoptotic protease activating factor (APAF) (5.17) and to AIF downregulation (5.18)[122]. Upregulation of estrogen results in an inhibitory effect upon GSK-3β, induced by Wnt signaling, via the removal of beta-catenin from the estrogen-receptor α-GSK-3β Beta-catenin complex (5.19) as well as via the activation of survival pathways in neurons[123, 124]. GSK-3β inhibition causes beta-catenin translocation into the nucleus (5.20), which then coordinates translation via T-cell factors and lymphoid-enhancer binding factors (5.21). Testosterone, conversely, is produced from cholesterol via steriodogenic acute regulatory protein (StAR) (5.22) which is then converted into estradiol through the action of aromatase (5.23)[127], which is a key enzyme involved in the biosynthesis of estrogens. TSPO and StAR participate in homeostatic regulation of steroidogenesis, while enhanced expression of TSPO in microglia and astrocytes reflects inflammatory activation of these cells. However,18-kDa translocator protein (TSPO) contributes to this process.
Table 1:
The main consequences of damage to brain tissue, the metabolic processes affected, and the neuroimaging modality most suited to its detection and measurement are represented. Acute, sub-chronic, and chronic TBI are indicated by open, partially-filled, and filled circles. Colors are included to illustrate consistency with Figures 1–5. The indication of symbols in the rows and columns reflects consistency with what is described in the text and graphically indicated in the figures. While many modalities can, of course, be measured in any stage of TBI (e.g. T1 structural imaging in acute, sub-chronic, or chronic TBI), the presence of the symbols here is intended to illustrate where that modality possesses maximal and/or unique utility for the assessment of alterations in cell structure and metabolism due to traumatic brain injury.
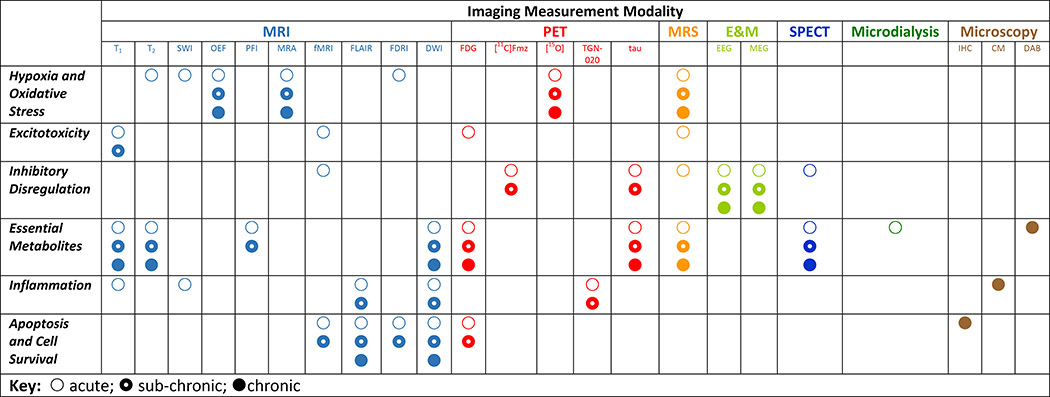 |
Hypoxia and oxidative stress
Hypoxia and oxidative stress are two of the major initiators of cell damage after TBI (Figure 2). Because the failure to deliver O2 to neurons and glia initiates numerous biochemical cascades which lead to secondary injury, mapping O2 usage in the brain is an important neuroimaging goal. One technique which allows researchers to accomplish this task involves 15O PET (2.3), where a radioactive isotope of oxygen is inhaled, ingested or injected, and then important measures of oxygen utilization—e.g. the cerebral metabolic rate for oxygen (CMRO2) (2.4) and the oxygen extraction fraction (OEF) (2.5) of tissues—are obtained[7]. Methods involving the intravenous injection of radioactive ozone molecules (15O-O2) to obtain OEF PET measurements have also been developed for animal models[8], thereby making more sophisticated experiments possible to study oxygen utilization. In humans, 17O2 MRI imaging has been undertaken in both humans and animals[9–11]. The further development of noninvasive approaches suitable for clinical use remains a prominent translational goal.
The ability to image ATP metabolism (2.6–2.7) can greatly aid in studying energy production and/or consumption in the TBI brain. Phosphorus (P) is essential to oxidative phosphorylation (2.8), and MR techniques are well suited for identifying brain locations (2.9) where ATP production proceeds at rates higher or lower than normal. Because 31P is a stable phosphorus isotope with non-zero nuclear spin, its presence in tissues is detectable using MRS (2.10). For example, MRS studies mapping the concentration and spatial distribution of 31P have revealed brain changes in ATP levels using custom-designed 31P/1H MRS head coils[12]. 31P MRI has also been undertaken [13], and although it has not become commonplace due to the low concentration of P in biological tissue compared to H, the use of 31P MRI may increase due to high-field MRI scanners becoming more commonplace. Because O2 delivery and ATP consumption are intimately related to each other, the prospect of using 15O PET in conjunction with 31P MRS/MRI is particularly appealing and may be of interest when attempting to understand how O2 delivery and ATP consumption are affected by TBI. Ironically, after TBI, the presence of iron in hemorrhaging blood causes ascorbic acid to aggravate oxidative stress by increasing the production of free radicals (2.14). Ascorbic acid concentrations can be imaged using radioactive carbon (C), specifically using [1-13C]-ascorbic acid as a hyperpolarizing probe in 13C NMR spectroscopy and MRI to study redox state changes in vivo (2.15)[14] [15].
Subsequent to TBI, there is often a decrease in cerebral blood flow (CBF) (2.16) to affected brain regions followed by damage to mitochondria and to the cellular metabolic machinery, resulting in a shift from aerobic to anaerobic metabolism in neurons (as detailed in a later section) and to an upregulation of ROS production (2.12)[16]. ROS-caused damage involves various pathways—such as chains of reactions for lipid peroxidation—which affect cell membrane integrity. Damage to the cell membrane results in the release of arachidonic acid (AA) into the cytosol (2.17)[17]. AA can be oxidized using an alternate lipoxygenase pathway mediated by 5-lipoxygenase, which results in the formation of a modified eicosatetraenoic acid called LTA4 (a leukotriene) (2.20) and further activation of leukotriene receptors (2.21)[18] (the specific role of these compounds is discussed in a later section). Leukotriene-related reactions as well as other phenomena related to ROS activity can be imaged using [3H]hydromethidine [a free radical trapping radio tracer containing 3H (tritium), which is a radioactive isotope of hydrogen (H)] (2.22). Using ex vivo samples, this tritium radio tracer allows ROS levels to be imaged and quantified using autoradiography (ARG)[19].
Subsequent to injury there is a reduction of pH in the brain which leads to acidosis and to the dissociation of Fe2+ from storage proteins (2.27)[20]. This causes a release of hydroxyl radicals via a Fenton-type reaction where Fe2+ released from hemoglobin decomposes hydrogen peroxide (H2O2) to form methemoglobin (2.28), a hydroxyl free radical and a hydroxyl ion[21]. Changes in free Fe2+ concentrations can be visualized using MRI due to the phenomenon of field-dependent relaxivity increase (FDRI) (2.29)[22]. Acidosis levels and changes in pH can be measured using cerebral microdialysis (2.30), where a probe is inserted into the brain at a location which is typically free of primary injury for the purpose of detecting secondary tissue damage[23]. Methemoglobin created after TBI leads to the generation of hemin (2.31), which at high concentrations has been shown to have a toxic effect upon various cell types[24]. The increase in hydroxyl free radicals results in oxidative stress and in subsequent cell death. Oxidative stress can result in either breakage of deoxyribonucleic acid (DNA) strands (2.32) or in DNA being subjected to excessive electron concentrations, which directly impact its integrity and viability[25, 26]. A later section elaborates on these phenomena.
TBI-related hemorrhage compresses blood vessels and capillaries, which can result in further ischemia, infarction and/or herniation of brain structures. Subarachnoid hemorrhage, in particular, is the most common form of hemorrhage caused by TBI and involves blood accumulation in the cisterns, fissures and sulci of the brain[27]. Gross hemorrhages can be imaged using gradient-recalled echo (GRE) T2*-weighted imaging[28] as well as susceptibility weighted imaging (SWI) (2.33), which relies on iron’s ferromagnetic properties to obviate bleeding in the brain[29],[30]. Blood flow in the presence of hemorrhages can be imaged using magnetic resonance angiography (MRA) (2.34), which can greatly assist in monitoring injury evolution[31].
An important free radical involved in post-TBI pathogenesis is nitrogen (N) monoxide (NO). Though animal studies have imaged this compound in real time using inverted fluorescence microscopy (2.35)[32], there are currently few viable techniques for in vivo imaging of NO brain concentrations in humans due to the side effects of this compound. Novel approaches for addressing this shortcoming of current methods for imaging nitrogen concentrations are thus needed. One ultimate consequence of elevated NO concentrations following TBI is a decrease in BBB integrity. Advanced methods involving contrast-enhanced MRI have allowed scientists to image BBB breakdown in the aging human hippocampus[33] and MRI of N may become more feasible with high-field scanners. The use of manganese (Mn) as an MRI contrast agent may also be of utility [34], in light of its property as a calcium analog. These methods mightbe adopted in TBI research since they would greatly advance our currently-insufficient understanding of BBB dysfunction after trauma.
Excitotoxicity
An important consequence of abnormal O2/ATP metabolism after TBI is glutamate (Glu) imbalance, which easily leads to excitotoxicity (Figure 3a,b). Glu transporter function is dependent upon the electrochemical transmembrane gradients of ions (primarily Na+ and H+), which implies that membrane depolarization leads to abnormal changes in Glu concentrations within and outside the cell. The malfunction of Glu reuptake transporters prevents Glu removal from the extracellular space, resulting in its accumulation there. Cerebral microdialysis is often used to measure brain Glu levels, though neuroimaging techniques such as 1H MRS may be preferable because microdialysis is an invasive method with comparably poorer regional specificity (3a.4)[35],[36]. Importantly for neuroimaging, neurons can use Glu to generate ATP when deprived of O2 (3a.5), which implies that regional decreases in glucose (Glc) usage revealed by [18F] fluoro-2-deoxy-D-glucose (FDG)-PET can provide insight on Glu metabolism (3a.6). This important relationship between Glu and Glc levels after TBI has been insufficiently explored via neuroimaging, though its study is amenable to the use of FDG-PET and of MRI using natural contrast agents such as D-Glu[37].
Figure 3a,b. Excitotoxicity and inhibitory dysregulation.



In this figure, 3b is a magnified illustration of the synaptic level shown in 3a. In 3a, Glu is the biochemical compound with the highest concentration in the mammalian brain[137]. It is the carboxylate anion of glutamic acid, a non-essential α-amino acid used extensively for protein biosynthesis. Glu is the primary excitatory neurotransmitter in the CNS, where it activates neurons via its dedicated receptors[138]. Within cells, Glu is typically stored in vesicles (3a.1), while extracellular Glu is recycled by plasma membrane transporters at a steady rate to avoid its accumulation in the extracellular space[139]. Since ATP must bind both to the Na+/K+ (sodium/potassium) ATPase (3a.2) and to the plasma membrane Ca2+ ATPase (PMCA) (3a.3) to maintain the electrochemical potential of the cell membrane, TBI-related hypoxia results in ATP production impairment, and thereby to membrane depolarization. In 3b, the accumulation of extracellular Glu results in the opening of an excessive fraction of Ca2+-permeable Glu NMDARs (3b.7) and in the consequent flow of Ca2+ into the neuron (3b.8)[140],[141]. Simultaneously, a membrane potential decrease due to NMDAR activation results in the opening of voltage-gated Ca2+ channels (VLCCs) (3b.9), which leads to further membrane potential decrease. γ-aminobutyric acid (GABA) is produced from Glu by means of the tonically-active glutamic acid decarboxylase (GAD) enzyme (3a.16). In the mammalian CNS, it is the chief neurotransmitter responsible for inhibiting synaptic activity[46, 47], exerting its effect primarily via the activation of ionotropic GABAA receptors. GABA concentrations are relatively higher presynaptically, where its levels are controlled by phasically-active GAD[48]. Synaptically-released GABA is uptaken by glia via GABA transporters (GATs) (3a.18) and converted to Glu in mitochondria (3a.19). Glu is then converted to glutamine (Gln) (3a.20) and transported by system A Gln transporters (SATs) back into neurons (3a.21), where Gln is reconverted to Glu, by the action of glutaminase (GLS-2) (3a.22) and reused to produce GABA[142]. Subsequent to TBI, the number of GATs decreases partly due to the death of GABAergic neurons, where GATs are produced. This lowers the rate at which GABA is reuptaken by glia, such that the extracellular concentration of GABA increases and the rate of its conversion to Glu and then to Gln decreases[143],[144]. The net result of this process is a decrease in the amount of Gln which is exported from glia to the extracellular space and then to GABAergic neurons via SATs[145]. After TBI, GABAergic signaling may be disrupted partly because of modifications to GABAA subunits, including the GABAA α1, α4, γ2 and δ subunits (3b.26). The earliest changes to the α1 subunit occur in the first day after injury and are brought about by disruptions of the pathway involving the Janus Kinase (JaK) and Signal Transducer and Activator of Transcription (STAT) protein (i.e. the JAK/STAT pathway) (3b.25)[146],[147],[148]. Such disruptions result in neuronal membrane depolarization (3b.27) and in weaker downregulation of neuronal excitation (3b.28)[149]. GABAB regulates gene expression by activating protein kinase A (PKA) (3b.30) in the presence of cyclic adenosine monophosphate (cAMP). This second messenger is bound to by cAMP (3b.29) response element-binding (CREB) proteins (3b.31), thereby increasing or decreasing the transcription of downstream genes (3b.32)[150].
Anomalous membrane depolarization after TBI triggers the additional release of Ca2+ from intracellular stores such as the endoplasmic reticulum (ER) (3b.10), resulting in abnormally-high intracellular levels of Ca2+ and in the activation of catabolic cell processes including free radical overproduction, apoptotic signaling activation and increased inflammatory factor release[38], all of which are discussed in later sections. These and related processes involved in post-traumatic Ca metabolism can be studied using both microscopy as well as MRI techniques. In particular, Ca-sensitive fluorescent dyes have been used in rodent models of TBI to study receptors of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and specifically Ca-permeable AMPA receptors (CP-AMPARs) (3b.11), which are becoming increasingly recognized as playing an important role in the pathophysiology of injury[39]. Additionally, calpains - a class of Ca2+-dependent proteases - also play a role in apoptosis by promoting the cleavage of cytoskeletal proteins such as talin and α-actinin, membrane proteins such as EGF receptors, integrins thereby destabilizing the cytoskeletal framework [40, 41]. In animals, imaging experiments which used Ca-sensitive fluorescent dyes have revealed that the selective blockage of CP-AMPARs subsequent to mechanical injury leads to significant reductions in cell death rates 24 hours after injury, to an extent similar to that observed using broad-spectrum NMDAR and AMPAR antagonists. In human studies, neuroimaging of Ca is considerably more challenging and available techniques for mapping NMDAR/AMPAR activity are extremely limited. MR imaging of Ca metabolism has been carried out in bovine brain tissue using Ca-sensitive contrast agents which exploit the interaction between superparamagnetic iron oxide nanoparticles and calmodulin, which binds to Ca2+ ions (3b.12)[42]. In the presence of Ca, calmodulin and polypeptides, these particles form aggregating clusters which can be MR-imaged due to their sufficiently large T1 and T2 relaxivities. There are currently few techniques for neuroimaging Ca metabolism in TBI patients, though promising approaches are being developed[43]. 43Ca is a stable isotope with non-zero nuclear spin, whereas the half-lives of 41Ca, 45Ca and 47Ca are sufficiently long to make MRI of Ca atoms feasible, particularly at high fields.
In addition to Ca, magnesium (Mg) imaging can also be useful to study excitotoxicity. TBI-related mechanical deformations have been found to affect the ability of Mg2+ to close Glu NMDARs (3b.13) and to decrease the desensitization of Glu AMPARs (3b.14)[44]. Because Glu also plays an important role in neurovascular coupling, this makes fMRI suitable for investigating the differential effects of NMDARs and AMPARs upon the generation of blood oxygenation level-dependent (BOLD) signals, upon cerebral blood volume (CBV) as well as upon measures obtained using perfusion-weighted imaging (PWI) from animals (3b.15). Unfortunately, using fMRI to study Mg2+-mediated excitotoxicity in humans is challenging partly because NMDAR and AMPAR activity is difficult to monitor directly without pharmacologically altering Glu levels. In rodents, by contrast, after NMDAR antagonist injections and forepaw stimulation, significant decreases in BOLD responses have been observed, whereas AMPAR antagonist injections have been found to significantly affect somatosensory evoked potentials (SEPs) in addition to BOLD signals[45]. These results have drawn attention to the role of Glu in modulating BOLD responses and CBF, as well as to the differential role of NMDARs and AMPARs in the chain of events which give rise to these fMRI and PWI measures. However, because there are currently no feasible approaches for differentiating between the effects of NMDARs and AMPARs upon human fMRI signals, future imaging research on Mg metabolism is needed, possibly using MRI of 25Mg, which is a stable isotope with non-zero nuclear spin.
Inhibitory dysregulation
In the mammalian CNS, γ-aminobutyric acid (GABA) is the chief neurotransmitter responsible for inhibiting synaptic activity[46, 47], exerting its effect primarily via the activation of ionotropic GABAA receptors. GABA concentrations are relatively higher presynaptically, where its levels are controlled by phasically-active GAD[48]. In TBI, anti-GAD immunofluorescently-labeled antibodies can be imaged in animals using CM (3a.17)[49], whereas in humans this technique cannot be used. [123I]iodotiagabine binds selectively to GABA transporters (GATs) and thereby facilitates its imaging using single-photon emission computed tomography (SPECT) (3a.18) [50].
Abnormal GABAergic signaling plays an important role in the biochemical cascades leading to icto- and epileptogenesis. Sometimes this pathology culminates in post-traumatic epilepsy (PTE)[47], which can be studied using electroencephalography (EEG), magnetoencephalography (MEG), fMRI, as well as simultaneous EEG/fMRI (3b.24)[51]. In particular, anatomically-constrained models of the TBI brain can be used to inversely localize electric potentials (EEG) and magnetic fields (MEG) and thereby to identify the loci of epileptic foci in patients whose PTE is pharmacologically resistant[52].
Changes to GABA inhibitory dynamics (3b.27) can result in substantial functional impairment. After injury, GABAA- and GABAB-mediated tonic inhibition increases due to high GABA concentrations in the extracellular space[53]. Furthermore, increased Glu release causes GABAB inactivation (3b.33) and endocytosis because GABAB is recycled to the plasma from the membrane and subsequently degraded by lysosomes (3b.34)[54]. Glu and GABA signaling are thus intimately coupled, which highlights the importance of studying both neurotransmitters simultaneously in imaging studies. Fortunately, the byproducts of various stages in GABA metabolism can be imaged, thereby providing a mapping of its individual steps. MEGA-PRESS (3b.35) spectroscopy combines MEGA (a frequency-selective editing technique) with PRESS (point-resolved spectroscopy sequence) to increase the sensitivity of single-voxel measurements of both Glu and GABA[55]. GABAA imaging can also be performed using PET tracers such as [11C] flumazenil (a benzodiazepine receptor antagonist) or [11C] Ro 15–4513 (a partial inverse agonist at a benzodiazepine receptor site) (3b.36)[56–58]. Imaging Glu and Gln levels via MRS can also afford an indirect picture of GABA recycling and may additionally help to map its precursors throughout their metabolic cycle[59]. Most notably, [11C]PiB is an imaging agent for fibrillary aggregates of amyloid-beta peptides but not tau proteins. There are several currently available tau PET radioligands, including [18F]AV-1451, [18F]THK5351 and [11C]PBB3, and visualization of tau accumulation by [18F]AV-1451-PET in chronic traumatic encephalopathy has been recently reported [60]. In the future, MRI and EEG techniques should be combined using PET to better understand the relationship between energy consumption and inhibitory dysregulation.
Essential metabolites
Imaging essential metabolites is fundamental for elucidating TBI metabolism (Figure 4). N-acetylaspartate (NAA) concentrations in the CNS are commonly measured using water-suppressed 1H MRS, which is well-suited for its detection and spatial mapping. NAA level changes relative to creatine (Cre) serve as an indicator of neuronal integrity dynamics and are important biomarkers of injury effects in the brain[61]. In the first 24 hours post-injury, there is a decrease in the NAA/Cre ratio, with substantial decreases often being observed during the first 4 hours post-injury[62]. Decreases in NAA concentrations (4.1) have been found to correlate with neuronal loss of ATP, acetyl-CoA and of associated metabolites involved in energy production[63]. MRS studies have found such decreases to occur as early as in the first 10 minutes after injury, with only partial recovery in the sub-acute and chronic stages of TBI[64].
Figure 4. Essential metabolites.

N-acetylaspartate (NAA), also one of the most abundant molecules in the mammalian brain, is a metabolite specific to the nervous system which is synthesized in neuronal mitochondria from aspartate (Asp) and acetylcoenzyme A (CoA) (4.1)[61]. This synthesis is mediated by the presence of Asp N-acetyltransferase (ASP-NAT) and differs from that in other tissues in that brain mitochondria synthesize relatively higher NAA amounts[151]. In the brain, NAA is critically involved in lipid and myelin synthesis, in energy production and in the synthesis of N-Acetylaspartylglutamate (NAAG), which is the third-most-prevalent neurotransmitter in the CNS. The regulatory pathway of NAA involves biosynthesis with the participation of ASP-NAT followed by neuronal release, uptake by oligodendrocytes and subsequent degradation by aspartocylase (ASPA) (4.3–4.4)[152]. NAA is transported across the neuronal membrane by Na+-dependent high-affinity dicarboxylate transporters (NaDC3) which maintain the osmotic balance of NAA within the neuron (4.2)[153]. During the synthesis of acetyl-CoA, NAA is broken down into acetate via ASPA, which is further converted into acetyl-CoA and aspartate using the cytosolic/nuclear enzyme acetyl-CoA synthatase 1 (4.6)[154]. Histone acetyltransferases (HATs) transfer an acetyl group from acetyl-CoA to the N-terminal of histones and thereby turn gene expression on or off (4.7–4.9)[155]. Acetyl-CoA also serves as a precursor to acetylcholine (Ach) because the former combines with choline (Cho) to form Ach (4.10). Metabolic changes due to TBI lead to alterations in biomolecule concentrations which are indicative of hypoxia and/or mitochondrial dysfunction. One such biomolecule is lactate (Lac); under normal conditions, energy production relies on glucose (Glc) as the primary energy source due to the ease of its transportation across the BBB. Lac is produced as a byproduct of Glc metabolism via the Embden-Meyerhof (EM) pathway, which involves conversion of Glc to pyruvate (Pyr) and then to Lac by Lac dehydrogenase (4.11–4.12)[156],[80]. Lac is also metabolized along the pentose phosphate pathway (PPP), but the brain’s dependence upon this pathway is more limited than in the case of the EM pathway. The initial steps of the PPP pathway lead to the formation of nicotinamide adenine dinucleotide phosphate (NADPH), involved in reductive biosynthesis of lipids and steroids (4.13). NADPH contributes to the production of reduced forms of glutathione (GSH) and thioredoxin (Trx) - cofactors of glutathione peroxide enzymes (GPx) and of peroxiredoxins (Prx), respectively (4.14–4.15). A protective effect induced by Trx involves the conversion of deoxyribonucleotide from ribonucleotide (4.16), which contributes to DNA synthesis and balances apoptotic factors. MRS is ideally suited for detecting changes in the levels of both neurometabolites (NAA, Cho, Cr, Lac) and neurotransmitters (Glu, GABA). Furthermore, the concentration gradient following TBI may be insufficient to allow Lac movement across the BBB. So Lac uptake is assisted by monocarboxylate transporters (MCTs) (4.17). Along with a high Lac concentration, an increase in the Lac/Pyr ratio (LPR) may be present in extracellular fluid and associated with hypoxia and mitochondrial dysfunction (4.18–4.20). TBI also raises brain tissue acidosis (4.21), causing binding of Op18/stathmin (STMN1; 4.22) to tubulin and slowing microtubule growth (4.23).
The complex ways in which NAA levels affect neuromodulatory pathways are important to understand because changes in NAA and in its associated neurometabolites are marked by neuronal integrity loss and by changes in neurotransmitter levels. These compounds can serve as important biomarkers of metabolic dysfunction when studying the initial stages of injury. Importantly, NAA is an acetate carrier for the synthesis of acetyl-CoA, which is required for myelin synthesis (4.5)[65]. Demyelination and reductions in myelin density can be imaged using T1 and T2-weighted imaging (after the administration of gadolinium-based contrast agents) or via macromolecular proton fraction imaging (PFI), which involves the calculation of the magnetization transfer ratio to infer the relative reduction in signal intensity due to off-resonance frequency saturation of macromolecular protons[66],[67]. The synthesis of acetyl-CoA is catalyzed by acetyl-CoA sythatases (AceCS1 and AceCS2; 4.6), which play a critical role in reactions between acetate and CoA[68]. Reductions in the concentrations of the former two compounds are correlated with NAA level changes in TBI models[69, 70]. Since it is an Ach precursor, NAA also modulates Ach levels (4.10) and NAA decreases are thus often paralleled by Ach decreases. This can be quantified using 123I-3-[2(S)-2-azetidinylmethoxy]pyridine (123I-5-IA) SPECT[71] because the decay of the radioactive isotope 123I of iodine allows for the spatial distribution of this element in the brain to be resolved. Additionally, [123I]5IA is a radioligand having specificity for nicotinic Ach receptors, while an analogous PET tracer, [18F]2FA, has recently become available [72]. Both might prove useful in TBI.
Following TBI, there is a shift in this dynamic process where lactate (Lac) is produced more often via the pentose phosphate pathway (PPP) than in the healthy brain. The shift to the PPP is partially due to the neuroprotective properties of the PPP, which guards against oxidative stress and generates biomolecules which may be used for cell repair[73]. The initial steps of the PPP pathway lead to the formation of nicotinamide adenine dinucleotide phosphate (NADPH), which is involved in reductive biosynthesis of lipids and steroids (4.13). NADPH also participates in the production of reduced forms of glutathione (GSH) and thioredoxin (Trx), which are cofactors of glutathione peroxide enzymes (GPx) and of peroxiredoxins (Prx), respectively (4.14–4.15). Both of these molecules scavenge hydroperoxides, thereby helping to minimize oxidative stress[74]. Another protective effect induced by Trx involves the conversion of deoxyribonucleotide from ribonucleotide (4.16), which contributes to DNA synthesis and balances the apoptotic factors being produced as a result of injury[75]. MRS is ideally suited for detecting changes in the levels of both neurometabolites (NAA, Cho, Cr, Lac) and neurotransmitters (Glu, GABA). After TBI, the concentration of Cho (a biomarker of inflammation) increases, leading to a decrease in the NAA/Cho ratio. Simultaneously, levels of NAA (a biomarker of neuronal integrity) decrease[76]. The Cho/Cre ratio increases due to cell membrane degradation, whereas the NAA/Cre ratio decreases due to neuronal integrity loss as well as to increased energy production in neurons attempting to compensate for neighboring cell death[77].
Typically, the brain’s demand for Lac is compensated by the uptake of Lac circulating in the body, which easily crosses the BBB due to its gradient difference across the barrier. After TBI, this concentration gradient is insufficiently large and may not allow Lac movement across the BBB, so Lac uptake is assisted by monocarboxylate transporters (MCTs) (4.17). These co-transporters use the proton gradient between brain tissue and blood to facilitate Lac delivery[78]. Lac uptake increases are visible as early as 3 days after injury and can be observed using [13C]Lac MRS[73],[79]. Along with a high Lac concentration, an increase in the Lac/Pyr ratio (LPR) has also been observed in the extracellular fluid of the brain and has been associated with hypoxia and with mitochondrial dysfunction (4.18–4.20)[80],[81],[82]. This phenomenon has been described as Type I Lac/Cre ratio elevation, whereas Type II Lac/Cre ratio elevation is associated with reduced Pyr concentrations. This could be due to dysfunction of the glycolytic pathway, to inadequate Glc amount entering the glycolytic pathway or to a diversion of Glc to other pathways such as the PPP[83]. Apart from Lac, the contribution of Pyr to the LPR can be imaged using [13C]Pyr MRS[84]. The LPR is helpful in estimating the ratio of oxidized to reduced nicotinamide adenine dinucleotide (NAD and NADH, respectively), which is important for maintaining cellular redox homeostasis and hence constitutes an important indicator of the cellular metabolic rate[85].
The effects of deficiencies in NAA metabolism can be quantified not only at the microscale but also at the macroscale. Mechanical stretching and compression of axons can result in their distortion and breakage—referred to as traumatic axonal injury (TAI)—and to the activation of microtubule depolymerisation mechanisms around the sites of micro-injury[86]. This phenomenon debilitates axonal transport along affected sites[87], causes the accumulation of neurometabolites in these regions and triggers swelling along axons[88]. In fact, histological studies using electron microscopy have determined that axonal damage is associated with mitochondrial swelling, vesicular transport disruption and cytoskeleton breakdown[89]. Other imaging studies involving sensitivity markers of axonal damage as well as Immunohistochemistry for beta-amyloid precursor protein (βAPP) have determined that TBI-affected axons are, quite often, βAPP positive[90]. Additionally, imaging using immunolabeling for dephosphorylated neurofilaments indicates collapse of neurofilament side arms responsible for the maintenance of axon diameters[91].
Despite being a hallmark of TBI, TAI remains poorly understood and additional imaging research is necessary to identify and characterize its full consequences, particularly upon the human connectome[92]. Since TAI-related changes impact the extent and direction of water diffusion along axonal paths, DTI measures such as fractional anisotropy (FA, which conveys the extent to which water diffuses preferentially in certain directions), can be used to quantify TAI. Novel methods for microscale ex vivo imaging, such as CLARITY[93], could be particularly useful for studying TAI. As previously explained, TBI is associated with increases in brain tissue acidosis (4.21), which results in binding of Op18/stathmin (STMN1; 4.22) to tubulin and in a slowdown of microtubule growth (4.23) [94],[95]. Currently, stathmin can only be imaged in vitro using pathology slide scanners and immunofluorescent anti-stathmin antibodies in conjunction with 3,3′-diaminobenzidine (DAB) staining of brain cells[96]. Thus, novel imaging methods should be developed to facilitate the study of this and of related compounds in human TBI patients. Another insufficiently understood consequence of injury-related microtubule deformation is the accumulation of tau protein; specifically, the proteolysis of neuronally-localized, intracellular microtubule-associated protein (MAP-tau) produces cleaved tau, whose accumulation is crucially implicated in AD pathology[97]. Using [11C] Pittsburgh compound-B (PiB) PET imaging, regions with high accumulations of tau can be identified as areas with increased PiB concentrations[2]. The buildup of this and of other neurometabolites is associated with complications and with the development of secondary brain injury, and current studies are attempting to explore the similarities and differences between AD pathology and TBI in this respect.
Inflammation
Brain injury results in a cascade of neurochemical reactions which release inflammatory molecules and which can lead to a substantial imbalance of metabolites (Figure 5). Essential to this process are a family of compounds called leukotrienes, which are molecules produced by leukocytes, by mastocytoma cells and by various other cells subsequent to immunological or non-immunological triggers. They contribute primarily to the stimulation of bronchial muscle contractions, to vascular permeability and to the activation of leukocytes[98]. During TBI, there is an upregulation of leukotrienes and of their receptors in the brain. This occurs due to the interaction of cytosolic phospholipase A2 with phospholipids on the nuclear envelope, which results in the formation of arachidonic acid. The latter binds to 5-lipoxygenase binding protein and reacts with 5-lipoxygenase to form LTA4[18], which is further converted into several other types of leukotrienes by various synthases and hydrolases[99]; this contributes to the onset of edema[100]. An MRI sequence type called fluid-attenuated inversion recovery (FLAIR) is ideal for imaging edema due to the fact that brain parenchyma perfused by cerebrospinal fluid (CSF) after injury appears hyperintense in FLAIR images[30].
As mentioned in a previous section, leukotrienes exert an effect upon various associated receptors to enact a variety of inflammatory responses in the brain. One such receptor, cys-LT1, has been shown to result in an increase in BBB permeability, in brain edema and in astrocyte proliferation. By contrast, reactions involving the cys-LT2 receptor result in the onset of cytotoxic brain edema after ischemic injury, which occurs because of upregulation of aquaporin 4 (AQP4) expression[101]. Incidentally, DWI has been proposed as one of the few imaging methods suitable for differentiating between cytotoxic and vasogenic edema[102],[30]. AQP4 is involved in maintenance of water homeostasis and is heavily expressed after TBI, resulting in brain edema and hydrocephalus[103]. The effects elicited by AQP4 can be imaged using a 11C-labelled analogue of 2-nicotinamido-1,3,4-thiadiazole (TGN-020) PET, which is an AQP4 ligand[104]. Although many of the compounds associated with leukotriene action cannot be individually imaged, their combined effect in the form of inflammation can be quantified using contrast dyes to reveal the extent of inflammatory progression. An example of this is the use of a radiolabeled peptide (IELLQAR) as an MRI contrast agent to target E-selectin, an important intercellular adhesion molecule involved in leukocyte rolling and recruitment[105]. Future studies should aim at developing additional techniques for imaging this important class of inflammatory agents, particularly for bedside use.
Apoptosis and cell survival
Cellular death signaling which leads to apoptosis (Figure 5) involves the release of apoptosis-inducing factors (AIFs). Imaging studies of intranuclear AIF localization in TBI-affected rodents used immunoelectromicroscopy to study DNA fragmentation in cortex and hippocampus between 2 and 72 hours after injury under conditions of oxidative/nitrosative stress[106]. Rabbit anti-AIF and anti-EndoG antibodies have also been imaged using fluorescent microscopy (FM)[107],[108]. In cryo-lesioned mouse TBI models, near-infrared molecular probes and tracers have been used to image the extent of cell death during noninvasive whole-body fluorescent imaging[109]. For example, lysotracer red dye (which concentrates in areas of high lysosomal and phagocyctic activity) can facilitate three-dimensional (3D) imaging of apoptotic cells using laser CM[110]. Despite these encouraging results from experiments in animal models, the available methods for imaging apoptotic processes in the human brain are comparatively crude and future efforts should be dedicated to bridging this technological and translational gap.
Apoptosis and cell survival are both influenced to a large extent by hormones, particularly estrogen and testosterone. Estrogen is a sex hormone which plays an important role in neuronal activity and synaptogenesis. Estradiol, a member of the estrogen family, is extensively involved in neural network formation, plays an important role in brain development—including neuron morphology, apoptosis and synaptogenesis[111]—and has also been shown to reduce apoptosis and BBB permeability[112]. TBI affects sex hormone levels, with significant decreases in testosterone levels (males) and in estrogen levels (females) in the first 24 hours after injury[113]. One other consequence of TBI can be hypopituitarism, whose symptoms range from fluctuations in blood pituitary hormone concentration immediately after TBI to alterations in pituitary hormone secretion, which may occur much later[114].
Following brain injury, one of the main causes of brain cell apoptosis is an inadequate blood supply to brain areas affected by primary injuries. Estrogen modulates this process by increasing vascular permeability and NO production in affected vascularized areas, thereby improving blood flow in affected regions. FDG-PET and DTI studies investigating the protective effects of estrogen upon the TBI brain have found that estrogen upregulation can result in cell activity increases and in reduced edema[115]. One key mechanism upon which estrogen effects upon the injured brain are predicated involves (i) the binding of estradiol to estrogen receptors (5.9), (ii) the activation of phosphoinositide-3-kinase (PI-3K) (5.10) and (iii) further activation of endogenous NO synthase (eNOS) to convert L-arginine into citrulline (5.11). NO is a by-product of this reaction, which increases CBF and can be studied by monitoring BOLD fMRI signals[116]. Other means of production involve the modulatory action of estrogen receptors upon estrogen-encoding segments of DNA subsequent to estrogen binding. Furthermore, the binding of estradiol to G-protein-coupled estrogen receptor 1 results in the activation of transcription factors (such as cAMP) whose action leads to additional eNOS expression[117]. The consequences of this process can be clearly visualized using DTI after administration of estrogen, which increases microcirculation and facilitates the transport of water molecules in TBI-affected regions[115].
Estrogen plays an important role in controlling apoptosis post-TBI by increasing the expression of B-cell lymphoma 2 (Bcl-2) proteins (5.12), a family of anti-apoptotic proteins[118]. This prevents the pore forming function of Bcl-2 homology domain 3 (BH-3) proteins (5.13) and the release of other apoptotic factors like bcl-2-like protein (BAX) and Bcl-2 homologous antagonist killer (BAK) (5.14)[119]. BAX/BAK downregulation reduces mitochondrion membrane permeabilization (5.15) and cytochrome c activation (5.16)[120],[121], followed by reduced binding of cytochrome c to apoptotic protease activating factor (APAF) (5.17) and to AIF downregulation (5.18)[122].
Changes in estrogen levels have also been associated with changes in the levels of beta-catenin—a pro-survival cell adhesion molecule—after injury. Upregulation of estrogen results in an inhibitory effect upon GSK-3β, induced by Wnt signaling, via the removal of beta-catenin from the estrogen-receptor α-GSK-3β Beta-catenin complex (5.19) as well as via the activation of survival pathways in neurons[123, 124]. The inhibition of GSK-3β causes the translocation of beta-catenin into the nucleus (5.20), which then coordinates translation via T-cell factors and lymphoid-enhancer binding factors (5.21). This results in the upregulation of pro-survival molecules[125] and in increased Glu metabolism; these consequence of GSK-3β level decreases have been investigated using FDG-PET[126].
Effects which are analogous to those observed in estrogen metabolism are also observed when testosterone levels change after TBI. Testosterone is produced in the brain from cholesterol via steriodogenic acute regulatory protein (StAR) (5.22) and is converted into estradiol through the action of aromatase (5.23)[127], which is a key enzyme involved in the biosynthesis of estrogens. In the first 24 hours post-injury, there is a significant drop in testosterone levels in males and this reduction can be significant 6 hours after injury and can remain so for up to 24 hours[128, 129]. It takes ~3–6 months for testosterone level to reach pre-injury levels[129], which illustrates the longer time range of anti-apoptotic processes. However, StAR may not be the only element mediating the transfer of cholesterol to the inner matrix of mitochondria for its conversion to steroids, but 18-kDa translocator protein (TSPO) is also responsible for this process. In addition, TSPO can be visualized in TBI patients and animal models with specific PET ligands [130, 131]. TSPO and StAR participate in homeostatic regulation of steroidogenesis, while enhanced expression of TSPO in microglia and astrocytes reflects inflammatory activation of these cells.
Concluding Remarks and Future Directions
The first few hours post-TBI are critical for understanding the trajectory of injury evolution, though neuroimaging patients within this time frame remains challenging due to their potentially unstable condition as well as to the difficulties associated with the presence of motion artifact in acquired imaging volumes. Though pharmacological agents are routinely administered to minimize brain damage as well as patient discomfort, the effects of such medications upon injury progression remain insufficiently understood and could greatly benefit from the development of novel neuroimaging methods. Animal models are only partly adequate for investigating the pathogenesis of secondary injury due to their differences from humans in brain structure, immune responses and metabolism. For this reason, noninvasive or minimally-invasive human neuroimaging methods and translational strategies should be designed so that clinicians can perform sophisticated monitoring of post-TBI metabolism in human patients. Due to the increased use of high-field MRI, there are now more opportunities than ever before for improving the MR imaging of Ca, Mg, C, P and of other elements involved in post-traumatic metabolism. This implies that the next decade is likely to witness a proliferation of MR protocols for the sophisticated imaging of nuclei whose MR signal at low fields is too weak for accurate brain mapping. Imaging the effects of injury severity upon the regulation of apoptotic pathways using such methods (which are currently inexistent or very limited in their capabilities) would provide insight on how to optimize clinical intervention and thereby minimize brain damage. In addition, increased efforts should be dedicated to validating the techniques we have highlighted (many of which are now in early translational stages) against post-mortem histological examinations of human brain tissue and against animal models. This would turn functional and structural imaging of TBI at the micro-, meso- and macroscale into substantially more promising approaches for delivering insights on metabolic responses to injury and on how these can be altered clinically. Improvements in functional techniques would be particularly welcome because of these methods’ ability to monitor brain activity at relatively high temporal resolution, which is critical in a condition as dynamic as TBI. The availability of multimodal, multi-scale neuroimaging of metabolism and neurotransmitter levels in hospital settings would also greatly advance the goals of personalized medicine and could greatly accelerate the process of incorporating genetic profiling and gene therapy into clinical care protocols. Collectively, these improvements in the state of the art would not only optimize individual patient outcome, but also alleviate the epidemiological burden of what continues to be an extremely challenging condition to understand mechanistically and treat adequately.
Acknowledgements
The authors wish to express our sincere gratitude to Ms. Caroline O’Driscoll for her artistic contributions as well as to thank the staff of the USC Mark and Mary Stevens Neuroimaging and Informatics Institute. This work was support by NIH grant R44 NS081792-03A1 to J.D.V.H.
Glossary
- Blood–Brain Barrier (BBB)
The highly selective permeability barrier which separates the circulating blood from the brain extracellular fluid in the central nervous system (CNS).
- Diffuse Axonal Injury (DAI)
Diffuse (or traumatic) axonal injury represents damage in the form of extensive lesions in white matter tracts occurs over a widespread area. DAI represents one of the most common and devastating classes of traumatic brain injury, and is a major cause of unconsciousness and persistent vegetative state after severe head trauma.
- Electrophysiology
Use of scalp electrode systems to record brain electrical activity at multiple sites over the cortex.
- Endocytosis
A process in which cell takes in materials from the extra-cellular space by surrounding and fusing them with its plasma membrane.
- Epileptogenesis
The systematic process by which a normal brain develops epilepsy. Epilepsy is a chronic condition in which seizures occur. These changes to the brain occasionally cause neurons to fire in a hyper-synchronous manner. This hyper-synchronous firing of neurons is called a seizure (ictogenesis).
- Evoked Potentials
An evoked potential or evoked response is an electrical potential recorded from the nervous system of a human or other animal following presentation of a stimulus, as distinct from spontaneous potentials as detected by electroencephalography (EEG), electromyography (EMG), or other electrophysiological recording method.
- Excitotoxicity
The pathological process by which nerve cells are damaged or killed by excessive stimulation from neurotransmitters such as glutamate or similar substances.
- Human Connectome
The comprehensive map of all neural connections in the brain.
- Hypoxia
A deficiency in the amount of oxygen reaching neural tissues.
- Inflammation
A component of the complex biological response of body tissues to harmful stimuli, such as cellular injury, damage, or irritation, and is considered as a protective response involving immune cells, blood vessels, and molecular mediators.
- Metabolism
The biochemical processes which occur within a living organism or cellular system in order to maintain life.
- Microdialysis
A process used to measure neurotransmitter metabolites as well as biomarkers of cell death from cerebrospinal fluid.
- Neuroimaging
The collection of medical imaging methods, including Magnetic Resonance Imaging (MRI), Positron Emission Tomography (PET), and Single Photon Emission Computed Tomography (SPECT), allowing for the examination of brain structural alteration, hemorrhage, edema, connectomic changes, as well as changes in metabolic and blood-flow rates in tissues proximal to lesioned tissue. Magnetic Resonance Spectroscopy (MRS), for instance, illustrates how the spectra of various brain metabolites are altered in the presence of TBI-related injury and necrosis.
- Oxidative Stress
Reflecting an imbalance between the systemic manifestation of reactive oxygen species and a biological system’s ability to readily reclaim the reactive intermediates or to repair the resulting damage.
- Traumatic Brain Injury (TBI)
Occurs as a consequence of a focal impact upon the head, by a sudden acceleration/deceleration within the cranium, or by a complex combination of both movement and abrupt head impact.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Langlois JA et al. (2006) The epidemiology and impact of traumatic brain injury - A brief overview. Journal of Head Trauma Rehabilitation 21 (5), 375–378. [DOI] [PubMed] [Google Scholar]
- 2.Maruyama M et al. (2013) Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 79 (6), 1094–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holsinger T et al. (2002) Head injury in early adulthood and the lifetime risk of depression. Archives of General Psychiatry 59 (1), 17–22. [DOI] [PubMed] [Google Scholar]
- 4.Irimia A et al. (2012) Neuroimaging of structural pathology and connectomics in traumatic brain injury: Toward personalized outcome prediction. Neuroimage Clin 1 (1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin AP et al. (2012) Metabolic imaging of mild traumatic brain injury. Brain Imaging Behav 6 (2), 208–23. [DOI] [PubMed] [Google Scholar]
- 6.Jordan BD (2007) Genetic influences on outcome following traumatic brain injury. Neurochem Res 32 (4–5), 905–15. [DOI] [PubMed] [Google Scholar]
- 7.Mintun MA et al. (1984) Brain oxygen utilization measured with O-15 radiotracers and positron emission tomography. J Nucl Med 25 (2), 177–87. [PubMed] [Google Scholar]
- 8.Temma T (2008) In-vivo positron emission tomography (PET) measurement of cerebral oxygen metabolism in small animals. Yakugaku Zasshi 128 (9), 1267–73. [DOI] [PubMed] [Google Scholar]
- 9.Tailor DR et al. (2004) Proton MRI of metabolically produced H2 17O using an efficient 17O2 delivery system. Neuroimage 22 (2), 611–8. [DOI] [PubMed] [Google Scholar]
- 10.Gordji-Nejad A et al. (2014) Characterizing cerebral oxygen metabolism employing oxygen-17 MRI/MRS at high fields. MAGMA 27 (1), 81–93. [DOI] [PubMed] [Google Scholar]
- 11.Lu H et al. (2013) Noninvasive functional imaging of cerebral blood volume with vascular-space-occupancy (VASO) MRI. NMR Biomed 26 (8), 932–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novak J et al. (2014) Clinical protocols for (3)(1)P MRS of the brain and their use in evaluating optic pathway gliomas in children. Eur J Radiol 83 (2), e106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frey MA et al. (2012) Phosphorus-31 MRI of hard and soft solids using quadratic echo line-narrowing. Proc Natl Acad Sci U S A 109 (14), 5190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohndiek SE et al. (2011) Hyperpolarized [1–13C]-ascorbic and dehydroascorbic acid: vitamin C as a probe for imaging redox status in vivo. J Am Chem Soc 133 (30), 11795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golman K et al. (2008) Cardiac metabolism measured noninvasively by hyperpolarized 13C MRI. Magn Reson Med 59 (5), 1005–13. [DOI] [PubMed] [Google Scholar]
- 16.Slemmer JE et al. (2008) Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr Med Chem 15 (4), 404–14. [DOI] [PubMed] [Google Scholar]
- 17.Novack TA et al. (1996) Neurochemical mechanisms in brain injury and treatment: a review. J Clin Exp Neuropsychol 18 (5), 685–706. [DOI] [PubMed] [Google Scholar]
- 18.Boyce JA (2007) Mast cells and eicosanoid mediators: a system of reciprocal paracrine and autocrine regulation. Immunol Rev 217, 168–85. [DOI] [PubMed] [Google Scholar]
- 19.Abe K et al. (2015) Imaging of reactive oxygen species in focal ischemic mouse brain using a radical trapping tracer [(3)H]hydromethidine. EJNMMI Res 5 (1), 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall ED et al. (2010) Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7 (1), 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadrzadeh SM et al. (1984) Hemoglobin. A biologic fenton reagent. J Biol Chem 259 (23), 14354–6. [PubMed] [Google Scholar]
- 22.Pfefferbaum A et al. (2009) MRI estimates of brain iron concentration in normal aging: comparison of field-dependent (FDRI) and phase (SWI) methods. Neuroimage 47 (2), 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clausen T et al. (2005) Association between elevated brain tissue glycerol levels and poor outcome following severe traumatic brain injury. J Neurosurg 103 (2), 233–8. [DOI] [PubMed] [Google Scholar]
- 24.Balla G et al. (1992) Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem 267 (25), 18148–53. [PubMed] [Google Scholar]
- 25.Clark RSB et al. (2001) Detection of single- and double-strand DNA breaks after traumatic brain injury in rats: comparison of in situ labeling techniques using DNA polymerase I, the Klenow fragment of DNA polymerase I, and terminal deoxynucleotidyl transferase. J Neurotrauma 18 (7), 675–89. [DOI] [PubMed] [Google Scholar]
- 26.Liu PK et al. (2002) The association between neuronal nitric oxide synthase and neuronal sensitivity in the brain after brain injury. Ann N Y Acad Sci 962, 226–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee B and Newberg A (2005) Neuroimaging in traumatic brain imaging. NeuroRx 2 (2), 372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chavhan GB et al. (2009) Principles, techniques, and applications of T2*-based MR imaging and its special applications. Radiographics 29 (5), 1433–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haacke EM et al. (2004) Susceptibility weighted imaging (SWI). Magn Reson Med 52 (3), 612–8. [DOI] [PubMed] [Google Scholar]
- 30.Irimia A et al. (2011) Comparison of acute and chronic traumatic brain injury using semi-automatic multimodal segmentation of MR volumes. J Neurotrauma 28 (11), 2287–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bryant DJ et al. (1984) Measurement of flow with NMR imaging using a gradient pulse and phase difference technique. J Comput Assist Tomogr 8 (4), 588–93. [DOI] [PubMed] [Google Scholar]
- 32.Huang KJ et al. (2007) Real-time imaging of nitric oxide production in living cells with 1,3,5,7-tetramethyl-2,6-dicarbethoxy-8-(3’,4’-diaminophenyl)-difluoroboradiaza-s- indacence by invert fluorescence microscope. Nitric Oxide 16 (1), 36–43. [DOI] [PubMed] [Google Scholar]
- 33.Montagne A et al. (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85 (2), 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talley Watts L et al. (2015) Manganese-Enhanced Magnetic Resonance Imaging of Traumatic Brain Injury. J Neurotrauma 32 (13), 1001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benveniste H et al. (1984) Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 43 (5), 1369–74. [DOI] [PubMed] [Google Scholar]
- 36.Prost RW et al. (1997) Detection of glutamate/glutamine resonances by 1H magnetic resonance spectroscopy at 0.5 tesla. Magn Reson Med 37 (4), 615–8. [DOI] [PubMed] [Google Scholar]
- 37.Xu X et al. (2015) Dynamic Glucose-Enhanced (DGE) MRI: Translation to Human Scanning and First Results in Glioma Patients. Tomography 1 (2), 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng G et al. (2012) Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol 167 (4), 699–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spaethling JM et al. (2008) Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma 25 (10), 1207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glading A et al. (2000) Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J Biol Chem 275 (4), 2390–8. [DOI] [PubMed] [Google Scholar]
- 41.Yan B et al. (2001) Calpain cleavage promotes talin binding to the beta 3 integrin cytoplasmic domain. J Biol Chem 276 (30), 28164–70. [DOI] [PubMed] [Google Scholar]
- 42.Atanasijevic T et al. (2006) Calcium-sensitive MRI contrast agents based on superparamagnetic iron oxide nanoparticles and calmodulin. Proc Natl Acad Sci U S A 103 (40), 14707–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jasanoff A (2007) MRI contrast agents for functional molecular imaging of brain activity. Curr Opin Neurobiol 17 (5), 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goforth PB et al. (1999) Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J Neurosci 19 (17), 7367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gsell W et al. (2006) Differential effects of NMDA and AMPA glutamate receptors on functional magnetic resonance imaging signals and evoked neuronal activity during forepaw stimulation of the rat. J Neurosci 26 (33), 8409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whiting PJ (2003) GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discov Today 8 (10), 445–50. [DOI] [PubMed] [Google Scholar]
- 47.Wu C and Sun D (2015) GABA receptors in brain development, function, and injury. Metab Brain Dis 30 (2), 367–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stagg CJ et al. (2011) What are we measuring with GABA magnetic resonance spectroscopy? Commun Integr Biol 4 (5), 573–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X et al. (2014) Immunofluorescently labeling glutamic acid decarboxylase 65 coupled with confocal imaging for identifying GABAergic somata in the rat dentate gyrus-A comparison with labeling glutamic acid decarboxylase 67. J Chem Neuroanat 61–62, 51–63. [DOI] [PubMed] [Google Scholar]
- 50.Schijns O et al. (2013) Development and characterization of [123I]iodotiagabine for in-vivo GABA-transporter imaging. Nucl Med Commun 34 (2), 175–9. [DOI] [PubMed] [Google Scholar]
- 51.Storti SF et al. (2012) A multimodal imaging approach to the evaluation of post-traumatic epilepsy. Magma 25 (5), 345–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Irimia A and Van Horn JD (2015) Epileptogenic focus localization in treatment-resistant post-traumatic epilepsy. J Clin Neurosci 22 (4), 627–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mtchedlishvili Z et al. (2010) Increase of GABAA receptor-mediated tonic inhibition in dentate granule cells after traumatic brain injury. Neurobiol Dis 38 (3), 464–75. [DOI] [PubMed] [Google Scholar]
- 54.Maier PJ et al. (2010) Sustained glutamate receptor activation down-regulates GABAB receptors by shifting the balance from recycling to lysosomal degradation. J Biol Chem 285 (46), 35606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edden RA and Barker PB (2007) Spatial effects in the detection of gamma-aminobutyric acid: improved sensitivity at high fields using inner volume saturation. Magn Reson Med 58 (6), 1276–82. [DOI] [PubMed] [Google Scholar]
- 56.Mullins PG et al. (2014) Current practice in the use of MEGA-PRESS spectroscopy for the detection of GABA. Neuroimage 86, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asahina N et al. (2008) [(11)C]flumazenil positron emission tomography analyses of brain gamma-aminobutyric acid type A receptors in Angelman syndrome. J Pediatr 152 (4), 546–9, 549 e1–3. [DOI] [PubMed] [Google Scholar]
- 58.Lingford-Hughes A et al. (2002) Imaging the GABA-benzodiazepine receptor subtype containing the alpha5-subunit in vivo with [11C]Ro15 4513 positron emission tomography. J Cereb Blood Flow Metab 22 (7), 878–89. [DOI] [PubMed] [Google Scholar]
- 59.Ramadan S et al. (2013) Glutamate and glutamine: a review of in vivo MRS in the human brain. NMR Biomed 26 (12), 1630–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dickstein DL et al. (2016) Cerebral [18 F]T807/AV1451 retention pattern in clinically probable CTE resembles pathognomonic distribution of CTE tauopathy. Transl Psychiatry 6 (9), e900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moffett JR et al. (2007) N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol 81 (2), 89–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu S et al. (2011) Early microstructural and metabolic changes following controlled cortical impact injury in rat: a magnetic resonance imaging and spectroscopy study. J Neurotrauma 28 (10), 2091–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vagnozzi R et al. (2007) Temporal window of metabolic brain vulnerability to concussions: mitochondrial-related impairment--part I. Neurosurgery 61 (2), 379–88; discussion 388–9. [DOI] [PubMed] [Google Scholar]
- 64.Signoretti S et al. (2001) N-Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J Neurotrauma 18 (10), 977–91. [DOI] [PubMed] [Google Scholar]
- 65.Moffett JR and Namboodiri MA (1995) Differential distribution of N-acetylaspartylglutamate and N-acetylaspartate immunoreactivities in rat forebrain. J Neurocytol 24 (6), 409–33. [DOI] [PubMed] [Google Scholar]
- 66.Fox RJ et al. (2011) Measuring myelin repair and axonal loss with diffusion tensor imaging. AJNR Am J Neuroradiol 32 (1), 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yarnykh VL et al. (2015) Fast whole-brain three-dimensional macromolecular proton fraction mapping in multiple sclerosis. Radiology 274 (1), 210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ariyannur PS et al. (2010) Nuclear-cytoplasmic localization of acetyl coenzyme a synthetase-1 in the rat brain. J Comp Neurol 518 (15), 2952–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szutowicz A et al. (2013) Acetyl-CoA the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. Neurochem Res 38 (8), 1523–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dixon CE et al. (1997) Reduced evoked release of acetylcholine in the rodent neocortex following traumatic brain injury. Brain Res 749 (1), 127–30. [DOI] [PubMed] [Google Scholar]
- 71.Esterlis I et al. (2013) Imaging changes in synaptic acetylcholine availability in living human subjects. J Nucl Med 54 (1), 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kimes AS et al. (2003) 2-[18F]F-A-85380: PET imaging of brain nicotinic acetylcholine receptors and whole body distribution in humans. Faseb J 17 (10), 1331–3. [DOI] [PubMed] [Google Scholar]
- 73.Jalloh I et al. (2013) Lactate uptake by the injured human brain: evidence from an arteriovenous gradient and cerebral microdialysis study. J Neurotrauma 30 (24), 2031–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jalloh I et al. (2015) Glycolysis and the pentose phosphate pathway after human traumatic brain injury: microdialysis studies using 1,2-(13)C2 glucose. J Cereb Blood Flow Metab 35 (1), 111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Herrick J and Sclavi B (2007) Ribonucleotide reductase and the regulation of DNA replication: an old story and an ancient heritage. Mol Microbiol 63 (1), 22–34. [DOI] [PubMed] [Google Scholar]
- 76.Kubas B et al. (2010) Proton MR spectroscopy in mild traumatic brain injury. Pol J Radiol 75 (4), 7–10. [PMC free article] [PubMed] [Google Scholar]
- 77.Garnett MR et al. (2000) Evidence for cellular damage in normal-appearing white matter correlates with injury severity in patients following traumatic brain injury: A magnetic resonance spectroscopy study. Brain 123 ( Pt 7), 1403–9. [DOI] [PubMed] [Google Scholar]
- 78.Poole RC and Halestrap AP (1993) Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am J Physiol 264 (4 Pt 1), C761–82. [DOI] [PubMed] [Google Scholar]
- 79.Qu H et al. (2000) (13)C MR spectroscopy study of lactate as substrate for rat brain. Dev Neurosci 22 (5–6), 429–36. [DOI] [PubMed] [Google Scholar]
- 80.Carpenter KL et al. (2015) Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci 9, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Timofeev I et al. (2011) Cerebral extracellular chemistry and outcome following traumatic brain injury: a microdialysis study of 223 patients. Brain 134 (Pt 2), 484–94. [DOI] [PubMed] [Google Scholar]
- 82.Hillered L et al. (2005) Translational neurochemical research in acute human brain injury: the current status and potential future for cerebral microdialysis. J Neurotrauma 22 (1), 3–41. [DOI] [PubMed] [Google Scholar]
- 83.Bartnik BL et al. (2005) Upregulation of pentose phosphate pathway and preservation of tricarboxylic acid cycle flux after experimental brain injury. J Neurotrauma 22 (10), 1052–65. [DOI] [PubMed] [Google Scholar]
- 84.Hurd RE et al. (2010) Metabolic imaging in the anesthetized rat brain using hyperpolarized [1–13C] pyruvate and [1–13C] ethyl pyruvate. Magn Reson Med 63 (5), 1137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sun F et al. (2012) Biochemical issues in estimation of cytosolic free NAD/NADH ratio. PLoS One 7 (5), e34525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tang-Schomer MD et al. (2010) Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J 24 (5), 1401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kuznetsov IA and Kuznetsov AV (2015) Modelling organelle transport after traumatic axonal injury. Comput Methods Biomech Biomed Engin 18 (6), 583–91. [DOI] [PubMed] [Google Scholar]
- 88.Tang-Schomer MD et al. (2012) Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol 233 (1), 364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Armstrong RC et al. (2016) White matter involvement after TBI: Clues to axon and myelin repair capacity. Exp Neurol 275 Pt 3, 328–33. [DOI] [PubMed] [Google Scholar]
- 90.Geddes JF et al. (2001) Neuropathology of inflicted head injury in children. II. Microscopic brain injury in infants. Brain 124 (Pt 7), 1299–306. [DOI] [PubMed] [Google Scholar]
- 91.Stone JR et al. (2000) Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res 871 (2), 288–302. [DOI] [PubMed] [Google Scholar]
- 92.Irimia A et al. (2012) Patient-tailored connectomics visualization for the assessment of white matter atrophy in traumatic brain injury. Front Neurol 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chung K and Deisseroth K (2013) CLARITY for mapping the nervous system. Nat Methods 10 (6), 508–13. [DOI] [PubMed] [Google Scholar]
- 94.Clausen T et al. (2005) Cerebral acid-base homeostasis after severe traumatic brain injury. J Neurosurg 103 (4), 597–607. [DOI] [PubMed] [Google Scholar]
- 95.Belmont LD and Mitchison TJ (1996) Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 84 (4), 623–31. [DOI] [PubMed] [Google Scholar]
- 96.Schmitt S et al. (2013) Stathmin regulates keratinocyte proliferation and migration during cutaneous regeneration. PLoS One 8 (9), e75075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gabbita SP et al. (2005) Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J Neurotrauma 22 (1), 83–94. [DOI] [PubMed] [Google Scholar]
- 98.Hammarstrom S (1983) Leukotrienes. Annu Rev Biochem 52, 355–77. [DOI] [PubMed] [Google Scholar]
- 99.Singh RK et al. (2010) Cysteinyl leukotrienes and their receptors: molecular and functional characteristics. Pharmacology 85 (6), 336–49. [DOI] [PubMed] [Google Scholar]
- 100.Rao AM et al. (1999) Arachidonic acid and leukotriene C4: role in transient cerebral ischemia of gerbils. Neurochem Res 24 (10), 1225–32. [DOI] [PubMed] [Google Scholar]
- 101.Wang ML et al. (2006) Leukotriene D4 induces brain edema and enhances CysLT2 receptor-mediated aquaporin 4 expression. Biochem Biophys Res Commun 350 (2), 399–404. [DOI] [PubMed] [Google Scholar]
- 102.Huisman TA et al. (2003) Diffusion-weighted imaging for the evaluation of diffuse axonal injury in closed head injury. J Comput Assist Tomogr 27 (1), 5–11. [DOI] [PubMed] [Google Scholar]
- 103.Lopez-Rodriguez AB et al. (2015) Changes in cannabinoid receptors, aquaporin 4 and vimentin expression after traumatic brain injury in adolescent male mice. Association with edema and neurological deficit. PLoS One 10 (6), e0128782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Suzuki Y et al. (2013) Aquaporin-4 positron emission tomography imaging of the human brain: first report. J Neuroimaging 23 (2), 219–23. [DOI] [PubMed] [Google Scholar]
- 105.Chapon C et al. (2009) Imaging E-selectin expression following traumatic brain injury in the rat using a targeted USPIO contrast agent. MAGMA 22 (3), 167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang X et al. (2002) Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem 82 (1), 181–91. [DOI] [PubMed] [Google Scholar]
- 107.Varecha M et al. (2009) Prediction of localization and interactions of apoptotic proteins. J Biomed Sci 16, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Siu PM et al. (2007) Response of caspase-independent apoptotic factors to high salt diet-induced heart failure. J Mol Cell Cardiol 42 (3), 678–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smith BA et al. (2012) Multicolor fluorescence imaging of traumatic brain injury in a cryolesion mouse model. ACS Chem Neurosci 3 (7), 530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zucker RM et al. (2000) Confocal laser scanning microscopy of rat follicle development. J Histochem Cytochem 48 (6), 781–91. [DOI] [PubMed] [Google Scholar]
- 111.McCarthy MM (2008) Estradiol and the developing brain. Physiol Rev 88 (1), 91–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Day NL et al. (2013) 17beta-estradiol confers protection after traumatic brain injury in the rat and involves activation of G protein-coupled estrogen receptor 1. J Neurotrauma 30 (17), 1531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bondanelli M et al. (2005) Hypopituitarism after traumatic brain injury. Eur J Endocrinol 152 (5), 679–91. [DOI] [PubMed] [Google Scholar]
- 114.Fernandez-Rodriguez E et al. (2011) Hypopituitarism following traumatic brain injury: determining factors for diagnosis. Front Endocrinol (Lausanne) 2, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim H et al. (2015) Salutary Effects of Estrogen Sulfate for Traumatic Brain Injury. J Neurotrauma 32 (16), 1210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.D’Esposito M et al. (2003) Alterations in the BOLD fMRI signal with ageing and disease: a challenge for neuroimaging. Nat Rev Neurosci 4 (11), 863–72. [DOI] [PubMed] [Google Scholar]
- 117.Novella S et al. (2012) Vascular Aging in Women: is Estrogen the Fountain of Youth? Front Physiol 3, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Raghupathi R et al. (2003) Temporal alterations in cellular Bax:Bcl-2 ratio following traumatic brain injury in the rat. J Neurotrauma 20 (5), 421–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kachadroka S et al. (2010) Effect of endogenous androgens on 17beta-estradiol-mediated protection after spinal cord injury in male rats. J Neurotrauma 27 (3), 611–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Waterhouse NJ et al. (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol 153 (2), 319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lalier L et al. (2007) Bax activation and mitochondrial insertion during apoptosis. Apoptosis 12 (5), 887–96. [DOI] [PubMed] [Google Scholar]
- 122.Fang X et al. (2015) Berberine induces cell apoptosis through cytochrome C/apoptotic protease-activating factor 1/caspase-3 and apoptosis inducing factor pathway in mouse insulinoma cells. Chin J Integr Med. [DOI] [PubMed] [Google Scholar]
- 123.Cross DA et al. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378 (6559), 785–9. [DOI] [PubMed] [Google Scholar]
- 124.Cardona-Gomez P et al. (2004) Estradiol inhibits GSK3 and regulates interaction of estrogen receptors, GSK3, and beta-catenin in the hippocampus. Mol Cell Neurosci 25 (3), 363–73. [DOI] [PubMed] [Google Scholar]
- 125.Arevalo MA et al. (2015) The neuroprotective actions of oestradiol and oestrogen receptors. Nat Rev Neurosci 16 (1), 17–29. [DOI] [PubMed] [Google Scholar]
- 126.de Cristobal J et al. (2014) A longitudinal FDG-PET study of transgenic mice overexpressing GSK- 3beta in the brain. Curr Alzheimer Res 11 (2), 175–81. [DOI] [PubMed] [Google Scholar]
- 127.Seol HJ et al. (2009) The pattern of gene expression and possible relation of steroidogenic genes in oligodendroglial tumors. Int J Oncol 34 (1), 181–90. [PubMed] [Google Scholar]
- 128.Meffre D et al. (2007) Steroid profiling in brain and plasma of male and pseudopregnant female rats after traumatic brain injury: analysis by gas chromatography/mass spectrometry. Endocrinology 148 (5), 2505–17. [DOI] [PubMed] [Google Scholar]
- 129.Iglesias P et al. (1996) Spontaneous recovery from post-traumatic hypopituitarism. J Endocrinol Invest 19 (5), 320–3. [DOI] [PubMed] [Google Scholar]
- 130.Vivash L and O’Brien TJ (2016) Imaging Microglial Activation with TSPO PET: Lighting Up Neurologic Diseases? J Nucl Med 57 (2), 165–8. [DOI] [PubMed] [Google Scholar]
- 131.Yu I et al. (2010) Glial cell-mediated deterioration and repair of the nervous system after traumatic brain injury in a rat model as assessed by positron emission tomography. J Neurotrauma 27 (8), 1463–75. [DOI] [PubMed] [Google Scholar]
- 132.Ikeda Y and Long DM (1990) The molecular basis of brain injury and brain edema: the role of oxygen free radicals. Neurosurgery 27 (1), 1–11. [DOI] [PubMed] [Google Scholar]
- 133.Taiwo YO and Levine JD (1990) Effects of cyclooxygenase products of arachidonic acid metabolism on cutaneous nociceptive threshold in the rat. Brain Res 537 (1–2), 372–4. [DOI] [PubMed] [Google Scholar]
- 134.Cline SD (2012) Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim Biophys Acta 1819 (9–10), 979–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Halliwell B and Chirico S (1993) Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr 57 (5 Suppl), 715S–724S; discussion 724S-725S. [DOI] [PubMed] [Google Scholar]
- 136.Zai A et al. (1999) Cell-surface protein disulfide isomerase catalyzes transnitrosation and regulates intracellular transfer of nitric oxide. J Clin Invest 103 (3), 393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Meldrum BS (2000) Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 130 (4S Suppl), 1007S–15S. [DOI] [PubMed] [Google Scholar]
- 138.Choi SW et al. (2004) Vitamins C and E: acute interactive effects on biomarkers of antioxidant defence and oxidative stress. Mutat Res 551 (1–2), 109–17. [DOI] [PubMed] [Google Scholar]
- 139.Greenwood SM and Connolly CN (2007) Dendritic and mitochondrial changes during glutamate excitotoxicity. Neuropharmacology 53 (8), 891–8. [DOI] [PubMed] [Google Scholar]
- 140.Cross JL et al. (2010) Modes of Neuronal Calcium Entry and Homeostasis following Cerebral Ischemia. Stroke Res Treat 2010, 316862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shirakawa T et al. (2013) [18F]FDG-PET as an imaging biomarker to NMDA receptor antagonist-induced neurotoxicity. Toxicol Sci 133 (1), 13–21. [DOI] [PubMed] [Google Scholar]
- 142.Hyland NP and Cryan JF (2010) A Gut Feeling about GABA: Focus on GABA(B) Receptors. Front Pharmacol 1, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Palmer AM et al. (1994) Increased transmitter amino acid concentration in human ventricular CSF after brain trauma. Neuroreport 6 (1), 153–6. [DOI] [PubMed] [Google Scholar]
- 144.Nilsson P et al. (1990) Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J Cereb Blood Flow Metab 10 (5), 631–7. [DOI] [PubMed] [Google Scholar]
- 145.Li Y et al. (2010) Increased GAD expression in the striatum after transient cerebral ischemia. Mol Cell Neurosci 45 (4), 370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oliva AA Jr. et al. (2012) STAT3 signaling after traumatic brain injury. J Neurochem 120 (5), 710–20. [DOI] [PubMed] [Google Scholar]
- 147.Brooks-Kayal AR et al. (1998) Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med 4 (10), 1166–72. [DOI] [PubMed] [Google Scholar]
- 148.Raible DJ et al. (2012) GABA(A) receptor regulation after experimental traumatic brain injury. J Neurotrauma 29 (16), 2548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Harvey RJ and Yee BK (2013) Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat Rev Drug Discov 12 (11), 866–85. [DOI] [PubMed] [Google Scholar]
- 150.Hu Y et al. (2008) Surface expression of GABAA receptors is transcriptionally controlled by the interplay of cAMP-response element-binding protein and its binding partner inducible cAMP early repressor. J Biol Chem 283 (14), 9328–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Patel TB and Clark JB (1979) Synthesis of N-acetyl-L-aspartate by rat brain mitochondria and its involvement in mitochondrial/cytosolic carbon transport. Biochem J 184 (3), 539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Di Pietro V et al. (2014) The molecular mechanisms affecting N-acetylaspartate homeostasis following experimental graded traumatic brain injury. Mol Med 20, 147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huang W et al. (2000) Transport of N-acetylaspartate by the Na(+)-dependent high-affinity dicarboxylate transporter NaDC3 and its relevance to the expression of the transporter in the brain. J Pharmacol Exp Ther 295 (1), 392–403. [PubMed] [Google Scholar]
- 154.Goldberg RP and Brunengraber H (1980) Contributions of cytosolic and mitochondrial acetyl-CoA syntheses to the activation of lipogenic acetate in rat liver. Adv Exp Med Biol 132, 413–8. [DOI] [PubMed] [Google Scholar]
- 155.Verdone L et al. (2005) Role of histone acetylation in the control of gene expression. Biochem Cell Biol 83 (3), 344–53. [DOI] [PubMed] [Google Scholar]
- 156.Nordstrom CH (2010) Cerebral energy metabolism and microdialysis in neurocritical care. Childs Nerv Syst 26 (4), 465–72. [DOI] [PubMed] [Google Scholar]
- 157.Verhagen AM et al. (2001) Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol 2 (7), REVIEWS3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Degterev A et al. (2003) A decade of caspases. Oncogene 22 (53), 8543–67. [DOI] [PubMed] [Google Scholar]
- 159.Lemasters JJ (2005) Dying a thousand deaths: redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology 129 (1), 351–60. [DOI] [PubMed] [Google Scholar]
- 160.Kischkel FC et al. (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14 (22), 5579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bardet PL et al. (2008) A fluorescent reporter of caspase activity for live imaging. Proc Natl Acad Sci U S A 105 (37), 13901–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Denecker G et al. (2000) Phosphatidyl serine exposure during apoptosis precedes release of cytochrome c and decrease in mitochondrial transmembrane potential. FEBS Lett 465 (1), 47–52. [DOI] [PubMed] [Google Scholar]
- 163.Stoica BA and Faden AI (2010) Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 7 (1), 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chaitanya GV et al. (2010) PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]