

ABSTRACT
The CRISPR/Cas9 system has recently emerged as a highly efficient modality in genetic engineering and has been widely considered for various therapeutic applications. However, since the effector protein, SpCas9, has a bacterial origin, its immunogenicity must be explored in further depth. Here, we found that the intact immune system, in wild-type C57BL/6J and BALB/cL mice, stimulates specific immune response against SpCas9, resulting in the rejection of SpCas9-expressing tumors. However, these tumors effectively grew in syngeneic C57BL/6J immunodeficient, T cell-depleted and Cas9-KI mice. Therefore, these observations suggest that this tumor rejection phenotype is T cell-dependent. The immunological clearance of SpCas9-expressing tumors in the immunocompetent group illustrates the possibility of misinterpreting the impact of CRISPR/Cas9-mediated gene editing on in vivo tumor biology and survival. Thus, these findings have important implications for the use of this exciting approach in in vivo studies, as well as to manipulate cancer cell biology for therapeutic applications.
KEYWORDS: CRISPR-Cas9, SpCas9, CRISPR, IMMUNITY
Introduction
CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats associated nucleus 9) technology has excited the scientific community since its recent discovery, and with good reason. Previously, genome editing used low-efficiency methods of transfection with donor DNA and spontaneous homologous recombination. Later technologies, such as zinc finger nucleases (ZFNs) and transcription-activator-like effector nucleases (TALENs), offered a technique of genome editing with fairly high specificity. However, both required the extremely precise and complex design of a novel protein for each edit.1 Additionally, because ZFN and TALEN function via protein:DNA interactions, their undesired off-target effects are often difficult to predict or prevent.2 The CRISPR-Cas9 system lacks such limitations. This methodology, for the first time, offers a solution of significant ease and efficiency in genome editing, allowing for a simple approach to precise gene addition, edit, or knock-out, rather than knock-down.1,3–6
The effector protein of the system, S. pyogenes Cas9 (SpCas9), guided by single-guide RNA (sgRNA), creates specific double-strand breaks in DNA, which after homology-directed repair (HDR) or nonhomologous end-joining (NHEJ) results in gene replacement.1,7,8 The process is highly specific due to the sgRNA guide and the necessity for a recognized specific protospacer-adjacent-motif (PAM) in the DNA sequence that is compatible with the SpCas9 protein.1 The distinct sequence of the sgRNA and PAM arrangements moreover facilitates the estimation of off-target editing.2
These exciting advantages over prior gene editing techniques have fostered the concept of employing CRISPR-Cas9-mediated genome editing in the research and development of therapeutics. The system has already been successfully employed for in vitro gene knock-in and knock-out studies.9,10 Also, it has been utilized to investigate transcriptional regulation.2 CRISPR-Cas-induced embryo modification has recently led to the development of precisely engineered mice.11 Such animal models represent important additions to the research on the impact of certain genes on disease onset and progression. Furthermore, by harnessing the ability to change a faulty gene itself, the introduction of CRISPR-Cas9 technology could be employed as a therapeutic for hereditary or mutation-based conditions.12
In spite of its promise in human health and in vivo applications, the CRISPR-Cas9 system has some potential pitfalls. Among these is the possibility of a host immune response to the SpCas9 protein. In fact, this enzyme, which is vital for CRISPR-Cas9 functioning, has a bacterial origin. Thus, soon after the development of the CRISPR-Cas9 approach, certain studies have questioned its immunogenicity, and hypothesized that host immunity may restrict its applicability.7,13 A few recent publications have addressed this question. In 2015, while Wang et al. were working on adenovirus-mediated genome editing of Pten by CRISPR-Cas9 technology, they reported SpCas9-specific immune responses in mice. Indeed, they detected elevated serum anti-Cas9 antibodies from two distinct mouse models, C57BL/6 and FVB/NJ, exposed to CRISPR-Cas9.13 Two years later, in 2017, Chew et al. characterized the immunogenicity of SpCas9 in more detail, and showed that SpCas9 can evoke cellular and humoral immune responses. This was validated by the infiltration of myeloid cells and SpCas9-specific active T cells around SpCas9-expressing muscles, and by the induction of SpCas9-specific antibodies, respectively.14 These findings raise the possibility that CRISPR-Cas9 modified tumor cells might have altered immunogenicity.
Here, we show that when mT3-2D pancreatic tumors grow subcutaneously in immunocompetent wild-type (WT) C57BL/6J mice, the intact murine immune system recognizes SpCas9. This immune recognition eventually leads to the complete rejection of the majority of SpCas9-expressing tumor cells. However, SpCas9-expressing tumors successfully grow in syngeneic immunodeficient B6.CB17-Prkdcscid/SzJ (SCID) and T cell-depleted mice. Additionally, some tumors recurred in WT mice with complete SpCas9-expressing tumor rejection. We found that these relapsed WT tumors selectively eliminated the SpCas9 protein, allowing them to escape immune recognition and attack. Furthermore, this phenomenon of SpCas9-expressing tumor rejection is not limited to the mT3-2D model in C57BL/6J mice. SpCas9-expressing 4T1 tumors in immunocompetent BALB/cJ mice also had complete tumor regression, followed by tumor recurrence in one mouse. Collectively, these observations validate the proposed hypothesis that SpCas9 does in fact trigger an efficient adaptive immune response. More importantly, these findings illustrate that SpCas9 immunogenicity may interfere with the applicability of the CRISPR-Cas9 approach for in vivo experiments and therapeutic applications in the context of intact host immunity. Alternatively, this can be overcome by growing SpCas9-expressing mT3=2D tumors in Cas9 knock-in (Cas9-KI+/−) immunocompetent mice, suggesting that the Cas9 transgenic mouse model might serve as an appropriate host for preclinical and biological studies.
Results
Generation of SpCas9-expressing mT3-2D cell lines
To determine the effect of SpCas9 on an in vivo immune response, we infected mT3-2D cells, a murine pancreatic cancer line, with the empty lentiCRISPR (ELC) vector. Then, to isolate a sub-population that highly expressed SpCas9, cells were harvested and sorted to generate single cell colonies using fluorescence-activated cell sorting (FACS). The level of SpCas9 was analyzed in seven SpCas9-expressing mT3-2D clones (mT3-2D-ELC1-7) by Western Blotting (Figure 1a). Based on the relative SpCas9 expression, mT3-2D-ELC1, 3 and 4 clones were selected for further study. Next, we evaluated the in vitro cellular growth of the three SpCas9 expressing mT3-2D cell lines. All three cell lines had similar proliferation rates compared to that of the uninfected lineage (Figure 1b). mT3-2D-ELC4, which expresses SpCas9 and exhibits a near equivalent growth rate to mT3-2D control cells, was used for subsequent in vivo studies.
Figure 1.
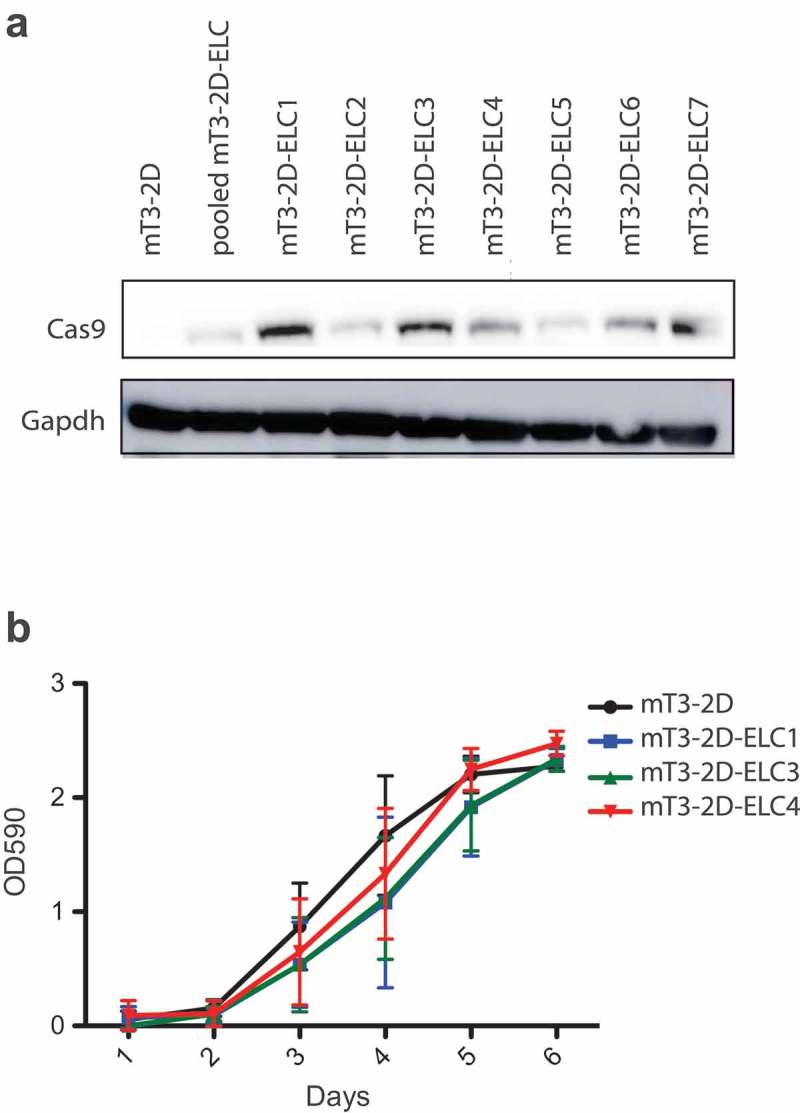
Generation of SpCas9-expressing mT3-2D cell lines. (a) Validation of SpCas9 protein expression in SpCas9-expressing mT3-2D single-cell clones by western blot. (b) The effect of SpCas9 introduction on in vitro cellular proliferation rate.
SpCas9-expressing mT3-2D tumors are rejected in immunocompetent but not in immunodeficient C57BL/6 mice
Because in vivo tumor growth can indirectly predict the immunogenicity of SpCas9, mT3-2D and mT3-2D-ELC4 cell lines were inoculated subcutaneously into immunocompetent WT C5BL/6 mice. Interestingly, SpCas9-expressing mT3-2D tumors were initially established and grew until approximately day 13. However, all these tumors were eventually completely rejected (Figure 2a). This observation suggested the induction of an in vivo adaptive host immune response against SpCas9. To directly determine the stimulation of immune response against SpCas9, the levels of IgG1 were measured from the serum of these WT mice. Serum SpCas9-specific IgG1 was detected in 9/10 mice that experienced complete tumor rejection (Figure 2b), though SpCas9-specific IgG2a and IgG2b concentrations were negative or below the detection limit (data not shown). These data are consistent with the interpretation that SpCas9 stimulates host immunity that leads to in vivo tumor rejection.
Figure 2.
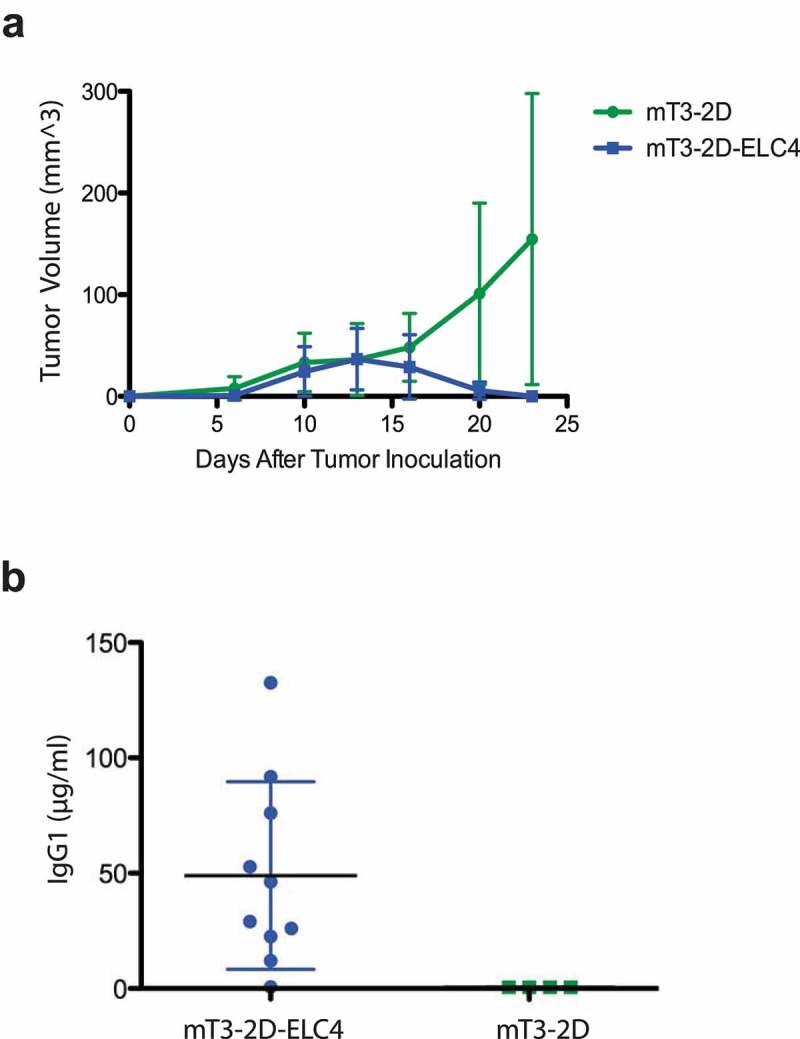
SpCas9 stimulates immune response in C57BL/6J immunocompetent mice. (a) Growth curves depicting average tumor volumes (±SD) of mT3-2D (n = 10) and mT3-2D-ELC4 (n = 10) grown in immunocompetent WT mice. (b) Detection of anti-SpCas9 IgG1 levels in serum of immunocompetent mice.
To further confirm that the triggered adaptive immune response against the SpCas9 protein was responsible for this tumor rejection and that these cells do not lack the ability to grow in vivo, mT3-2D-ELC4 cells were inoculated into both immunocompetent WT and syngeneic immunodeficient SCID C5BL/6 mice. While 9/10 mT3-2D-ELC4 tumors were rejected in the WT mice, all mT3-2D-ELC4 tumors successfully grew in the SCID mice (Figure 3a). SpCas9 levels were detected by WB (Figure 3b) in mT3-2D-ELC4 SCID tumors, demonstrating the persistent expression of SpCas9 in these tumors. Collectively, these data indicate that the adaptive immune response is key to SpCas9 recognition and subsequent tumor rejection.
Figure 3.
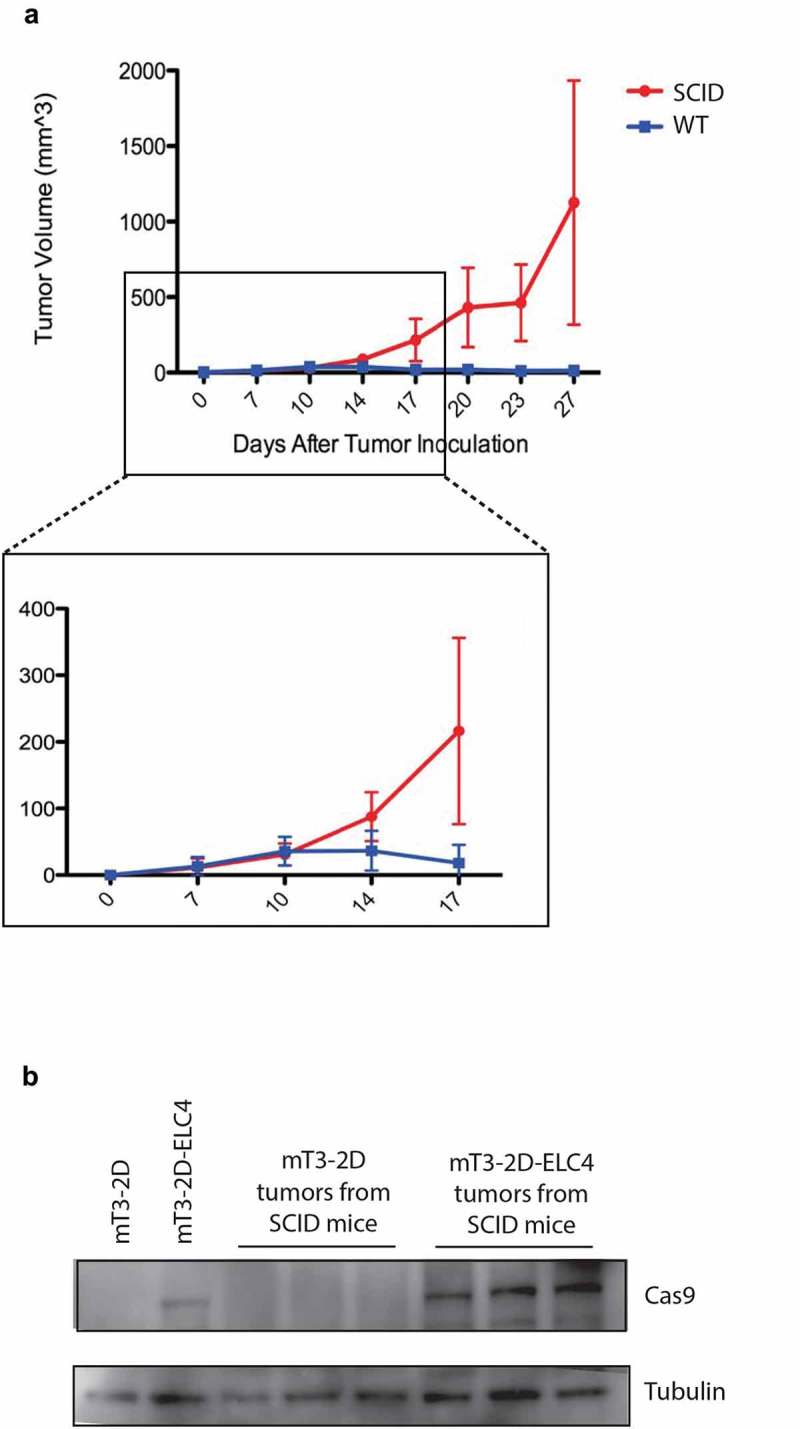
SpCas9-expressing tumors grow in immunodeficient mice. (a) Growth curves depicting average mT3-2D-ELC4 tumor volumes (±SD) in immunocompetent WT mice (n = 10) and in immunodeficient SCID mice (n = 10). (b) Validation of SpCas9 protein expression in mT3-2D-ELC4 SCID tumors (n = 3) compared to mT3-2D SCID tumors (n = 3) and the cell lines.
The rejection of SpCas9-expressing tumors is T cell-dependent
Since the major difference between the WT and SCID mice is the presence of T cells and B cells, we hypothesized that T cell immunity is required for tumor rejection. To examine the role of T cells in mT3-2D-ELC4 tumor immunological clearance, we depleted CD4 and CD8 T cells simultaneously in vivo. While SpCas9-expressing tumors were rejected in the untreated WT mice as seen previously, these tumors grew rapidly in the T cell-depleted mice (Figure 4), suggesting that impaired mT3-2D-ELC4 tumor growth in WT mice is a T cell-dependent phenomenon. To validate that we had successfully depleted T cells in vivo, splenic lymphocytes were analyzed using flow cytometry analysis, which confirmed the reduction of CD4 and CD8 T cells to 1.02% and 1.15%, respectively (Supplementary Figure 1).
Figure 4.
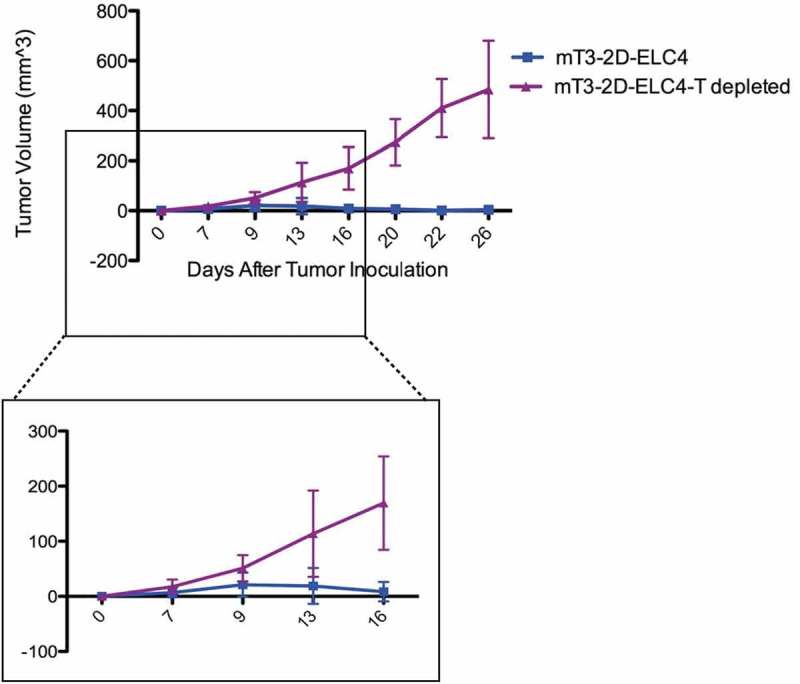
SpCas9-expressing tumors grow in CD4/CD8 T cell-depleted mice. Growth curves depicting average mT3-2D-ELC4 tumor volumes (±SD) in immunocompetent WT mice (n = 7) and in T cell-depleted mice (n = 7).
Selective elimination of SpCas9 protein results in tumor recurrence in immunocompetent mice
We injected 5 WT mice with mT3-2D-ELC4 cells and monitored the mice over time. About 2 weeks after tumor inoculation, one mouse had nearly complete tumor regression, and four mice had complete tumor rejection. Interestingly, among the four tumor-free mice, three developed late tumor recurrence (Figure 5a). We hypothesized that the anti-SpCas9 immune response had selectively eliminated cells expressing this immunogenic protein in vivo. Indeed, as assessed by Western blot, we found that the level of SpCas9 was reduced in mT3-2D-ELC4 relapsed WT tumors compared to the SCID tumors injected with the same cell line. In order to exclude the possibility that the mT3-2D relapsed WT tumors have lower epithelial/stroma ratio compared to the SCID tumors, we evaluated the relative pan-keratin levels and found no reduction of the epithelial content but rather selective elimination of SpCas9 (Figure 5b).
Figure 5.
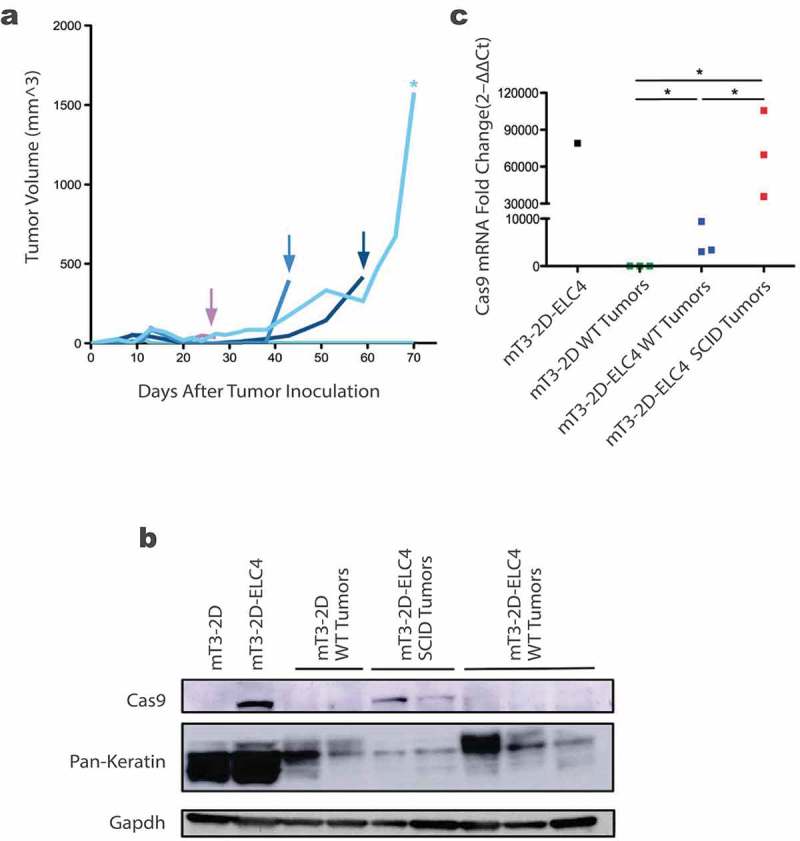
Selective elimination of SpCas9 protein in SpCas9-expressing relapsed WT tumors. (a) Growth curves of individual mT3-2D-ELC4 tumors in immunocompetent WT mice (n = 5), the arrows indicate the relapsed tumors after complete rejection and the asterisk indicates the slowly progressed tumor. (b) Evaluation of SpCas9 and Pan-keratin protein levels in mT3-2D-ELC4 relapsed WT tumors (n = 3) compared to mT3-2D-ELC4 SCID tumors (n = 2), mT3-2D WT tumors (n = 2), and the cell lines. (c) Evaluation of SpCas9 mRNA levels in mT3-2D-ELC4 relapsed WT tumors (n = 3) compared to mT3-2D-ELC4 SCID tumors (n = 3), mT3-2D WT tumors (n = 3), and mT3-2D-ELC4 cell line by RT-qPCR. Statistical analysis was performed using the Mann Whitney test, one-tailed. One asterisk (*) indicates p value p < 0.05.
To further investigate this observation, and to determine whether this is a result of a selective loss of the SpCas9 expression or there was an epigenetic silencing of the SpCas9 gene, we performed RT-qPCR on the WT relapsed tumor samples. As indicated in Figure 5c, we found that changes in SpCas9 protein levels parallel the mRNA Cas9 gene expression. The selective loss of SpCas9 expression further illustrates the immunogenicity of this bacterial protein.
SpCas9-expressing mT3-2D tumors were not rejected in Cas9-KI+/− immunocompetent mice
To overcome the immunogenicity of SpCas9, we hypothesized that Cas9 transgenic mice can recognize SpCas9 as self. Thus, these mice will not immunologically respond to the SpCas9-expressing tumor cells, making this model a good alternative strategy to test the impact of specific gene knockouts in immunocompetent murine models. To test this hypothesis and evaluate the applicability of this model, we crossed Rosa26-LSL-Cas9 knock-in (Cas9-KI+/+) male mice, which have C57BL/6J backgrounds,15 to WT C57BL/6J female mice. After the generation of these heterozygous Cas9 transgenic mice (Cas9-KI+/−), we injected these mice subcutaneously with mT3-2D and mT3-2D-ELC4 cells, and tumor growth was monitored over time. SpCas9-expressing cancer cells grew successfully in the Rosa26-LSL-Cas9 knock-in(+/−) mice, with no significant differences in the average tumor growth rates between the mT3-2D and mT3-2D-ELC4 groups (Figure 6). To evaluate the immune phenotype of the naïve heterozygous Cas9KI mice, we performed multi-color flow cytometry. The analysis revealed that there is no significant difference in the splenic immune cell populations between naïve Cas9 transgenic mice and naïve WT C57BL/6J mice, including total T cells, CD4 and CD8 T cells, B cells, NK cells, MHCII− monocytes/macrophages, MHCII+ monocytes/macrophages conventional DCs (cDCs) and neutrophils (Supplementary Figure 2). These proof of concept data suggest that SpCas9 is immunogenic and that this Cas9 transgenic mouse model is a useful alternative approach for studies in which immunogenicity of the SpCas9 protein could be challenging.
Figure 6.
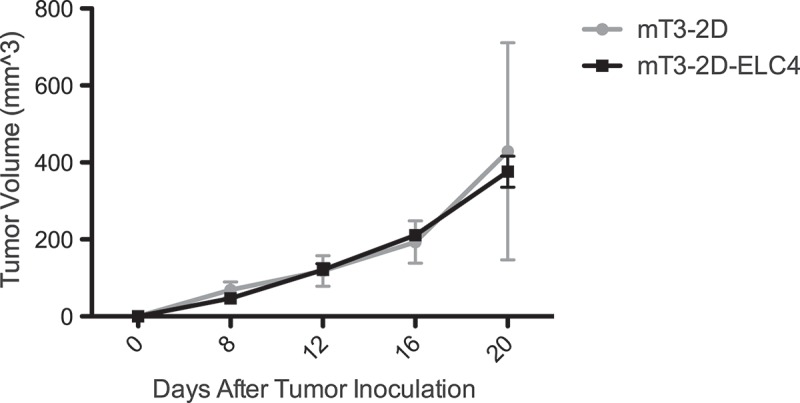
SpCas9-expressing tumors grow in Cas9-KI immunocompetent mice. Growth curves depicting average tumor volume s (±SD) of mT3-2D (n = 5) and mT3-2D-ELC4 (n = 5) in Cas9-KI mice.
SpCas9-expressing 4T1 tumors are rejected in immunocompetent BALB/cJ mice
To investigate whether the phenomenon of immunological clearance of SpCas9-expressing tumors is generalizable, we conducted similar experiments using the 4T1 model, a murine breast cancer cell line. First, we generated three SpCas9-expressing 4T1 clones (4T1-ELC16, 23 and 25). Based on the relative SpCas9 expression (Figure 7a), and the similar proliferation rate of the 4T1-ELC16 clone compared to the uninfected lineage (Figure 7b), the 4T1-ELC16 cell line was selected for the in vivo study. In this experiment, the 4T1 and 4T1-ELC16 cell lines were inoculated subcutaneously into immunocompetent WT BALB/cJ mice. Interestingly, 5/10 SpCas9-expressing 4T1 tumors were initially established but then all of them were eventually completely rejected. Additionally, similar to the mT3-2D-ELC4 model, one mouse inoculated with 4T1-ELC16 had tumor recurrence after complete tumor disappearance (Figure 7c). These observations further support our hypothesis that SpCas9 is immunogenic. Also, it demonstrates that the effect of SpCas9 immunogenicity on tumor growth is not limited either to the mT3-2D lineage nor to C57BL/6J mouse strain but generalizable to other in vivo models.
Figure 7.
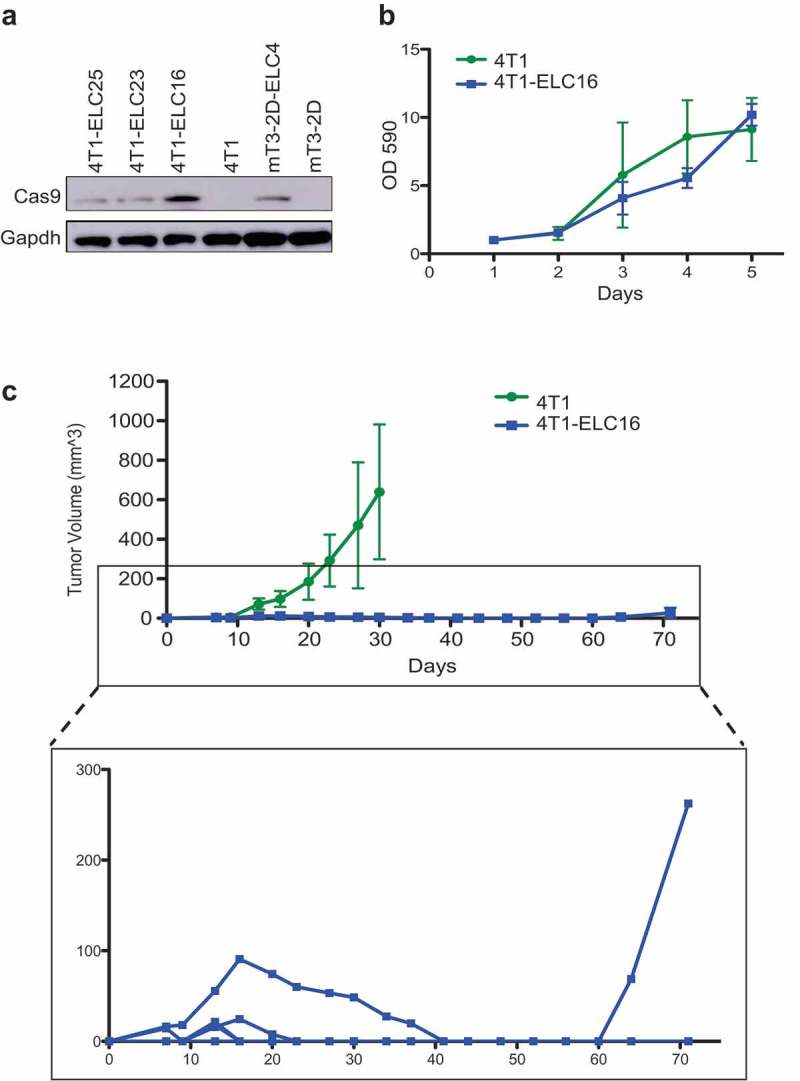
SpCas9-expressing tumors are rejected in immunocompetent BALB/cJ mice. (a) Validation of SpCas9 protein expression in SpCas9-expressing 4T1 single-cell clones by western blot. (b) The effect of SpCas9 introduction on in vitro 4T1-ELC16 cellular proliferation rate. (c) Growth curves depicting average tumor volumes (±SD) of 4T1 (n = 10) and 4T1-ELC16 (n = 10) grown in immunocompetent WT mice.
Discussion
Due to the diverse and widespread applicability of the CRISPR-Cas9 approach, research relating to this system has grown exponentially since its emergence in 2012. Because the CRISPR effector protein, SpCas9, has a bacterial origin, several studies have postulated its immunogenicity.13 Validating the immunogenicity of SpCas9 may limit the clinical, translational and biomedical applicability of the CRISPR-Cas9 approach, and thus illustrates the importance of employing alternative technologies and modifications. Accordingly, we generated SpCas9-expressing mT3-2D murine pancreatic cancer cells and studied the immunogenicity of these cells using four C57BL/6J mouse models (WT, SCID, T cell-depleted and Cas9-KI+/− mice). Here, we demonstrate that immunocompetent mice with intact immunity reject syngeneic pancreatic tumors bearing a CRISPR-Cas9 vector. However, corresponding T cell-depleted, SCID and immunocompetent Cas9 transgenic mice support tumor growth, suggesting that SpCas9 is immunogenic. Additionally, using the 4T1 BALB/cJ murine breast cancer model, we demonstrated that the immunological clearance of SpCas9-expressing tumors is not limited to the mT3-2D C57BL/6J mouse model but rather is a generalizable phenomenon. These findings have important implications for the use of this exciting and potentially important approach to manipulating cancer cell biology and serve as a caveat for such efforts.
Our findings suggest that SpCas9 triggers an efficient adaptive immune response. More specifically, by simultaneously depleting CD4 and CD8 T cells, we were able to demonstrate that SpCas9 tumor rejection is mainly T cell-dependent. Also, we identified SpCas9-specific IgG1 antibodies in the sera of WT mice bearing CRISPR-Cas9-modified tumors, consistent with associated T cell engagement. Although our experimental design does not consider the role of each T cell subtype individually, Chew et al. associated the SpCas9 host response with a marked elevation in both CD4 and CD8 T cells,14 suggesting that both T cell subsets may play a role in the immunological clearance of the SpCas9-expressing tumors.
It is worth noting that our results do not completely rule out the possibility that the innate immune response is involved in SpCas9 immunogenicity. Nonetheless, since SCID and T cell-depleted mice possess the intact activity of natural killer (NK) cells, granulocytes, macrophages, and complement proteins, the likelihood that the innate immunity can play a significant role in tumor rejection is quite slim.16 In line with that, the use of pooled CRISPR library approach has been successful in several previous studies that either used the CRISPR-Cas9 system in vitro17 or injected the SpCas9-expressing cells into immunodeficient mice.18
In addition, reduction in tumor immunogenicity has been reported in multiple cancer types, including pancreatic cancer, in both mouse models as well as clinical samples.19–21 As a result, the loss of SpCas9 expression is an expected immune escape mechanism to be employed by mT3-2D-ELC4 tumor cells allowing relapse in WT mice. However, any immune escape mechanism, such as the down-regulation of the major histocompatibility complex (MHC) molecules, the induction of immune checkpoints and the stimulation of immunosuppressive cells, cytokines, and chemokines, could be relevant to the immune response in other SpCas9-expressing cancer models. Moreover, while we were able to show that there was a significant loss of SpCas9 gene expression in relapsed mT3-2D-ELC4 WT tumors, our studies do not determine if there was selective loss of the SpCas9 protein in the mT3-2D-ELC4 tumor cells, or there was in vivo selection of less immunogenic mT3-2D-ELC4 clones. Nonetheless, whether the SpCas9 elimination occurred at the protein level or the cellular level, this does not change the finding that there was selective loss of SpCas9 expression, which strongly supports our hypothesis.
We did not employ a lentiviral vector as a negative control in our study and thus cannot exclude its role in tumor rejection. Nevertheless, the majority of the mice had detectable levels of SpCas9-specific IgG1. Also, SpCas9-expressing tumors were not rejected in Cas9-KI+/− mice. In our view, these data confirm both SpCas9 and the impact of this immunogenicity on in vivo tumor biology in immunocompetent murine models.
Furthermore, despite the fact that not all the BALB/cJ mice subcutaneously injected with SpCas9-expressing 4T1 cells had detectable tumors, tumors that successfully grew within the first 2 weeks showed complete immunological clearance. In line with that, other groups have reported similar observations of impaired SpCas9-expressing tumor growth using other cell lines and different mice strains.13,14 Collectively, these data demonstrate that tumor biology-relevant SpCas9 immunogenicity is generalizable to multiple in vivo models, and not specific to the mT3-2D and 4T1 models.
Since genome editing techniques have broad and useful applications in vivo, it is crucial to create alternative approaches of stable CRISPR-Cas9 transfection. The Cas9-KI mice provide a strong model for such investigation. This mouse model has been reported to be impervious to morphological abnormalities, changes in DNA damage, variations in membrane function and integrity, and elevation in apoptosis. However, SpCas9 functions by generating targeted DNA double-stranded breaks. Therefore, uncertainty regarding the potential adverse effects of this protein in the Cas9 transgenic mice cannot be eliminated with complete confidence. For example, Germano et al. have shown that executing CRISPR-Cas9 transient transfection is a useful strategy to overcome the challenge of SpCas9-expressing tumor rejection in non-Cas9-KI immunocompetent mice.22 Indeed, this approach relies on the expectation that the SpCas9 will rapidly dissipate, leaving behind the desired targeted gene knock-out. Due to the immunogenicity of SpCas9 protein and despite the advantages of the CRISPR-Cas9 system, other genetic engineering techniques might be more useful for in vivo applications.
In conclusion, our data further support the proposed hypothesis that SpCas9 is immunogenic. Also, it suggests that T cell immunity modulates the observed host response and tumor rejection. This immune-mediated tumor rejection in WT mice may confound the interpretation of the impact of gene editing on in vivo tumor biology and survival. Moreover, in this study, we show that the SpCas9 transgenic mouse model might serve as a useful alternative approach for preclinical studies. Collectively, these findings have important implications for the use of this exciting approach in in vivo studies. Also, it highlights the need to improve the applicability of the CRISPR-Cas9 system for clinical applications.
Materials & methods
Cell lines: mT3-2D, a murine pancreatic cancer cells that are syngeneic in C57Bl/6 mice (gift from David Tuveson; Cold Spring Harbor Laboratory; Laurel Hollow, NY),23 as well as 4T1 and HEK293T cells (Georgetown Tissue Culture Shared Resources (TCSR); Georgetown University; Washington, DC) were grown in standard conditions and maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 5% 2mM L-glutamine. All cell lines were tested and determined to be free of Mycoplasma and other rodent pathogens; no other authentication assay
was performed.
Generation of CRISPR-Cas9 Transfected Cell Lines: Bacterial DNA carrying empty lentiCRISPRv2 vectors (addgene #52961) was isolated and kept at −80°C. This lentiCRISPRv2 plasmid (one vector system) contains the S. pyogenes CRISPR-Cas9 gene. In Opti-Mem media, isolated DNA was combined with VSV-G (gift from Dr. Todd Waldman; Georgetown University; Washington, DC) and psPAX (Addgene plasmid 12260 submitted by Dr. Didier Trono) reagents at a 2:1:1.5 ratio (10.8 μg, 5.4 μg, 8.1 μg). Lipofectamine 2000 (40 μl, Thermofisher) was administered for transfection. After incubation, Opti-MEM containing each vector was added drop-wise to HEK293 cells in order to raise virions. After 48-72 hours of further incubation, media was collected from HEK293 cells, centrifuged, and filtered with a 0.22 micron syringe filter to be immediately used as virus to transfect 50% confluent plates of mT3-2D or 4T1 murine cell lines. After exposure to 3mL of virus containing media for 3 hours at 37 degrees, 7mL of DMEM complete medium was added. Twenty-four hours following the addition of DMEM, selection for puromycin began and media was changed every 48–72 hours for two weeks. Generated cell lines were harvested and then sorted based on cell size to generate single cell colonies using the fluorescence-activated cell sorting (FACS) machine BD FACSAria IIu.
Crystal Violet Staining: 1 × 103 cells per well were cultured in 96-well plates. At an indicated time points, cells were subjected to 0.52% Crystal Violet in 25% methanol. For quantitative analysis, cells were dissolved in 100mM Sodium Citrate with 50% ethanol. Then, the absorbance of plates was measured at 570nm.
Animal xenograft studies: 1 × 105 of mT3-2D and mT3-2D-ELC4 cells were subcutaneously injected in the right flank of C57Bl/6J, B6.CB17-Prkdcscid/SzJ, and Cas9-KI+/− mice. 5 × 103 4T1 and 4T1-ELC16 cells were subcutaneously injected in the right flank of BALB/cJ mice. All mice used in the study were 6–8 weeks of age. C57Bl/6J, B6.CB17-Prkdcscid/SzJ and BALB/cJ mice were purchased from The Jackson Laboratory15. Homozygous B6J.129(Cg)Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J male mice were purchased from The Jackson Laboratory and they were crossed to C57BL/6J female mice. Generated Cas9-KI+/− heterozygous mice were then used for this study. Tumors were measured twice-weekly using calipers. Volume was calculated as (length × width2)/2. When tumors reached 1 cm3 or when mice showed signs of pain or distress, mice were euthanized using CO2 inhalation, and the tumors were excised. After euthanizing the mice, blood was collected for ELISA analysis. All studies involving animals were reviewed and approved by the Georgetown University Institutional Animal Care and Use Committee (GU IACUC).
In vivo T cell depletion animal experiment: 1 × 105 of mT3-2D and mT3-2D-ELC4 cells were subcutaneously injected in the right flank of heterozyogous Rosa26-LSL-Cas9 knock-in(+/−) mice. For depletion of CD4+ and CD8+ T cells, each mouse was given 200 μg of anti-mouse CD4 antibody (GK1.5, rat IgG2b; BioXCell) + 200 μg CD8 antibody (2.43, rat IgG2b; BioXCell) i.p. on days −5 (5 days before tumor inoculation), day −2, day 1, and the treatment continued twice weekly until sacrificing the mice. Tumors were measured twice-weekly using calipers. Volume was calculated as (length × width2)/2. When tumors reached 1 cm3 or when mice showed signs of pain or distress, mice were euthanized using CO2 inhalation, and the tumors were excised. After euthanizing the mice, splenocytes were collected to evaluate T cell depletion by flow cytometry. All studies with animals were approved by the Georgetown University Institutional Animal Care and Use Committee (GU IACUC).
Flow cytometry analysis of splenocytes from T cell-depleted mice: Two million splenocytes from sacrificed WT and T cell-depleted mice were labeled with Live/Dead (Invitrogen, Thermofisher, Ref: L34964). Subsequently, cells were washed and incubated for 10 minutes with anti-mouse CD16/CD32 (Biolegend, clone 93; Ref: 101320) to block Fc receptors before adding a cocktail of mAbs: CD45 (Biolegend, clone 30–F11; Ref: 103116), CD3e (Biolegend, clone 145–2C11; Ref: 100312), CD4 (eBioscience, clone RM4–5, Ref: 11–004–2–85), CD8a (BD Bioscienceclone 53–6.7, Ref: 552877). After staining cells were acquired with BD LSRFortessa Cell Analyzer, BD Biosciences and analyzed with FlowJo.
Multi-color flow cytometry analysis of splenocytes from Cas9-KI mice: Two million splenocytes from sacrificed WT and Cas9-KI mice were labeled with Live/Dead (Invitrogen, Thermofisher, Ref: l-23105). Subsequently, cells were washed and incubated for 10 minutes with CD16/CD32 (clone 2.4G2; 553141) to block Fc receptors before adding a cocktail of mAbs: anti-mouse-CD45 (clone 30–F11; 564590), NK-1.1(clone PK136; 562864), CD3e (clone 145–2C11; 564661), CD4 (clone RM4–5, 563151), CD8a (clone 53–6.7, 564920), B220 (clone RA3–6B2; 563103), all from BD Biosciences. After staining cells were acquired with FACS Symphony, BD Biosciences and analyzed with FlowJo and Graphpad software
ELISA for SpCas9-specific antibody: 96-well plate (Thermo Fisher Scientific, Cat# 12565138) were coated with the SpCas9 protein (0.5 μg/well, PNA Bio, cat # CP01) in 1 × coating buffer diluted from Coating Solution Concentrate Kit (KPL) overnight at 4°C. The plates were washed with 1 × PBS (gibco by life technologies/Thermo Fisher, cat # 70011–044) + 0.05% Tween-20 pH7.4 (Fisher BioReagents, BP337–500) and blocked with 5% BSA (Sigma–Aldrich, cat # A7906–100G) for 1 h at room temperature. Mouse sera were diluted 100-fold with dilution buffer (1% BSA in 1 × PBS) and added to the wells, and incubated for 1 hr at room temperature with shaking. The mouse monoclonal antibody against SpCas9 (Epigentek; clone 7A9, cat # A-9000–100) was serially diluted and used as a standard to quantify IgG1. After washing, each well was incubated with 100 μl of HRP-labeled anti-mouse IgG1 (BD Pharmingen, cat # 559626), IgG2a (abcam, cat # ab11571–100), or IgG2b (Caltag Laboratories, cat # M32507), all diluted 1:4000, for 1 h at room temperature. The wells were washed four times and incubated with 100 μl of ABTS ELISA HRP Substrate (KPL, cat # 50–76–00). Optical density at 410 nm was measured. The general protocol of this assay was obtained from Wang et al.13
Western Blotting: Cell pellets were lysed by heating to 100°C for 10 min in 100 μl of Boiling Buffer (1% SDS, 10 mM Tris, 1 mM Na3NO4, ddH20, and 1 protease inhibitor tablet (Sigma: S8820)). Then, protein concentration was determined by the BCA assay (Bio-Rad). Approximately 40 μg of protein was run on Tris-glycine gels under reducing conditions. After transfer to a nitrocellulose membrane, the membrane was blocked with 5% milk for 1 h (Bio-Rad). Primary blotting for proteins was performed overnight at 4°C using the following antibodies: CRISPR-Cas9 (EpiGentek #A-90000) (Figures 5b and 7a), CRISPR-Cas9 (Abcam #ab204448) (Figures 1a and 3b), Pan-keratin (Cell Signaling #4545), Tubulin (Abcam # ab6160) and GAPDH (D16H11) (Cell Signaling). All antibodies were diluted 1:500–1:1000 in 1 × PBS + 0.1% Tween-20 with 5% milk. Membranes were exposed to a secondary IgG antibodies (horseradish peroxidase (HRP)-labeled anti-rabbit or anti-mouse IgG; GE Healthcare) diluted 1:4000 in 1 × PBS + 0.1% Tween-20 with 5% milk and incubated for 1 h at room temperature. Supersignal West Femto high-sensitivity substrate and Supersignal West Pico (Thermo Scientific) were utilized for visualization of the western blots.
RNA extraction and Reverse Transcriptase Real-Time Quantitative PCR (RT-qPCR): RNA was isolated using an RNA isolation kit (Direct-zol Miniprep #R2050). RNA concentration was identified spectrophotometrically using NanoDrop 1000 (Thermo Scientific, Waltham, MA). The RNA concentration for each sample was 100 ng/μl. According to the manufacturer’s recommendations, the RT-qPCR was performed on StepOne Plus Real-Time PCR system (Applied Biosystems, Carlsbad, CA) using GoTaq 2-Step RT-qPCR kit (Promega, Madison, WI). Analysis was performed by using the ΔΔCT method, with GAPDH as the endogenous loading control and mT3-2D cell line as a baseline, with three technical replicates/sample. The primers sequences are listed in the table below.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | 5`-tcaccaccatggagaaggc-3` | 5`-gctaagcagttggtggtgca-3` |
| Cas9 | 5`-ggactcccggatgaacacta-3` | 5`-tcgctttccagcttagggta-3` |
Funding Statement
This work was supported by the National Cancer Institute [NCI P30 CA51008 (LMW)];
Acknowledgments
This research was supported by NCI R01 CA50633 (LMW) and by the following Shared Resources at Lombardi Comprehensive Cancer Center: the Flow Cytometry and Cell Sorting Shared Resource, Division of Comparative Medicine and the Tissue Culture Shared Resource. All Lombardi Comprehensive Cancer Center Shared Resources are partially supported by National Institutes of Health (NIH)/National Cancer Institute (NCI) Grant P30-CA051008. Financial support for Reham Ajina by the Saudi Arabian Cultural Mission (SACM) and King Saud bin Abdulaziz University for Health Sciences (KSAU-HS). Danielle Zamalin received the Discovery Student Research Grant offered by the Georgetown University Department of Human Science of the School of Nursing and Health Studies. The authors are grateful to Anton Wellstein (Georgetown University) for helpful discussions.
Supplemental Material
Supplemental data for this article can be accessed on the publisher’s website.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Komor AC, Badran AH, Liu DR.. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168:20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun L, Lutz BM, Tao Y-X.. The CRISPR/Cas9 system for gene editing and its potential application in pain research. Transl Perioperative Pain Med. 2016;1:22–33. [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Z, Mao Y, Ha S, Liu W, Botella JR, Zhu J-K. A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis. Plant Cell Rep. 2016;35:1519–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013;8:2180–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science (New York, N Y). 2013;339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai W-J, Zhu L-Y, Yan Z-Y, Xu Y, Wang Q-L, Lu X-J. CRISPR-Cas9 for in vivo gene therapy: promise and hurdles. Mol Ther Nucleic Acids. 2016;5:e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song H-Y, Chiang H-C, Tseng W-L, Wu P, Chien C-S, Leu H-B, Yang Y-P, Wang M-L, Jong Y-J, Chen C-H, et al. Using CRISPR/cas9-mediated GLA gene knockout as an in vitro drug screening model for fabry disease. Int J Mol Sci. 2016;17(12):2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park K-E, Powell A, Sandmaier SES, Kim C-M, Mileham A, Donovan DM, Telugu BP. Targeted gene knock-in by CRISPR/Cas ribonucleoproteins in porcine zygotes. Sci Rep. 2017;7:42458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin L-F, Li J-S. Generation of genetically modified mice using CRISPR/Cas9 and haploid embryonic stem cell systems. Dong wu xue yan jiu = Zool Res. 2016;37:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma H, Marti-Gutierrez N, Park S-W, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548(7668):413–419. [DOI] [PubMed] [Google Scholar]
- 13.Wang D, Mou H, Li S, Li Y, Hough S, Tran K, Li J, Yin H, Anderson DG, Sontheimer EJ, et al. Adenovirus-mediated somatic genome editing of pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum Gene Ther. 2015;26(7):432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chew WL, Tabebordbar M, Cheng JKW, Mali P, Wu EY, Ng AHM, Zhu K, Wagers AJ, Church GM. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods. 2016;13:868–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159(2):440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Jackson Laboratory B6.CB17-Prkdcscid/SzJ [accessed 2018. March 11]. https://www.jax.org/strain/001913
- 17.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160(6):1246–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, Bajor DL, Byrne KT, Stanger BZ, Riley JL, et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight. 2016;1:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552(7683):116–120. [DOI] [PubMed] [Google Scholar]
- 23.Boj SF, Hwang C-I, Baker LA, Chio IIC, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.