

Summary
Aim
Anxiety is one of common mood disorders, in which the deficit of serotonergic and GABAergic synaptic functions in the amygdala and prefrontal cortex is believed to be involved. The pathological changes at the glutamatergic synapses and neurons in these brain regions as well as their underlying mechanisms remain elusive, which we aim to investigate.
Methods
An agonist of kainate‐type glutamate receptors, kainic acid, was applied to induce anxiety‐related behaviors. The morphology and functions of glutamatergic synapses in the prelimbic region of mouse prefrontal cortex were analyzed using cellular imaging and electrophysiology.
Results
After kainate‐induced anxiety is onset, the signal transmission at the glutamatergic synapses is upregulated, and the dendritic spine heads are enlarged. In terms of the molecular mechanisms, the upregulated synaptic plasticity is associated with the expression of more protein kinase C (PKC) in the dendritic spines. Chelerythrine, a PKC inhibitor, reverses kainate‐induced anxiety and anxiety‐related glutamatergic synapse upregulation.
Conclusion
The activation of glutamatergic kainate‐type receptors leads to anxiety‐related behaviors and glutamatergic synapse upregulation through protein kinase C in the prelimbic region of the mouse prefrontal cortex.
Keywords: Anxiety, Glutamate, Prefrontal cortex, Protein kinase C, Synapse
Introduction
Anxiety, featured as unstable mood, negative interpretation, and social phobia, is one of prevalent psychological disorders 1, 2. The malfunction of amygdala and prefrontal cortex has been shown as a major origin of anxiety pathogenesis 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22. In terms of anxiety‐associated cellular pathology, GABAergic neurons and synapses appear deficit in these brain areas 23, 24, 25, 26, 27, 28, 29, 30, 31, 32. However, anxiety‐associated pathological changes at the glutamatergic synapses and neurons in these brain areas remain elusive 33, 34.
A long‐time belief is that the deficits of serotonergic and GABAergic synaptic transmission are associated with anxiety 1, 2, 35, 36. Selective serotonin reuptake inhibitors and GABA receptor enhancers are used to treat anxiety disorders 37, 38. Their effectiveness needs to be reevaluated due to unfavorable side effects 39, 40, 41, 42. Recent reports show that glutamate is associated with anxiety 43 and the antagonists of ionotropic glutamate receptor (iGluR) are potential reagents for anxiety therapy 44, 45, 46. In this regard, iGluR agonists hypothetically induce anxiety‐related behavior. We aim to examine an anxiogenic role of iGluR agonist in mood behavior and its mechanism. This study should shed light on the correlation between glutamate and anxiety.
In our study, an agonist of iGluR, kainic acid, was given to induce anxiety‐related behaviors. To analyze anxiety‐associated pathology at the glutamatergic synapses, cerebral excitatory neurons were genetically labeled with the yellow fluorescent protein for their morphological identification under a fluorescent microscope. The roles of protein kinase C (PKC) in kainate‐induced anxiety and cellular pathology were examined. Our results indicate that an activation of kainate receptors leads to anxiety‐related behavior as well as glutamatergic synapse upregulation through PKC in the prelimbic area of the prefrontal cortex.
Materials and methods
The entire procedures in our experiments have been approved by Institutional Animal Care Unit Committee in the Administration Office of Laboratory Animals, Beijing, China (B10831).
The Mice Model of Anxiety‐Related Behaviors
The mice applied in our study were cross‐matched from strains of C57(Thy1YFP)BL/6N and FVB‐Tg(GADGFP)4570Swn/J (Jackson Lab, Bar Harbor, ME, USA). The glutamatergic neurons in these mice were genetically labeled by yellow fluorescent protein (YFP), in which the promoter was Thy1 on the upstream of YFP. The GABAergic neurons in FVB mice were labeled by green fluorescent protein (GFP), in which a promoter was GAD on GFP upstream 47. The mice in postnatal day (PND) 30–32 were divided into two groups, control and kainate treatments, respectively. Saline or kainic acid (9 mM or 24 mg/kg) in saline was treated to either of two groups of mice by the intraperitoneal injection. Shortly after the injection of kainic acid, the mice demonstrated paniclike reactions, such as spontaneous muscle contraction in their heads and forearms. However, these mice did not express seizure like behaviors 1 month (n = 15) and 24 h after the kainate injection by grading motor seizure 48, in which none of these kainate‐injected mice showed motor seizure above grade two. In this regard, we assume that the mice in our studies are not seizure model, which may be due to a possibility that the mice from strains C57 and FVB are not sensitive to kainic acid 49.
After these treatments for 24 h, the mice were used to conduct the experiments of behavioral tasks by elevated plus‐maze, morphology by confocal microscope and electrophysiology by whole‐cell voltage‐clamp recording. The elevated plus‐maze was used to evaluate anxiety‐related behaviors. Patch‐clamp was used to record the transmission of glutamatergic synapses at pyramidal neurons in cortical slices. A laser scanning confocal microscopy was applied to image the subcellular structure of pyramidal neurons.
Behavioral Study
Anxiety‐related behaviors in the two groups of mice were evaluated by an elevated plus‐maze (EPM), which was described as the validated method to assess the level of anxiety in the rodents 32, 50, 51. The EPM consisted of two open arms (30 × 5 cm) opposite to two closed arms (30 × 5×15.25 cm). The arms were extended from a central platform (5 × 5 cm). The EPM was located 40 cm above the floor (Figure 1A). The mice were housed in plastic cages with food/water availability ad libitum and a schedule of alternative light and dark (12 h for each condition), in which the light was on 19:00. All experiments were performed between 10:00 and 16:00. The mice were at the age about 1 month when the tests were conducted.
Figure 1.
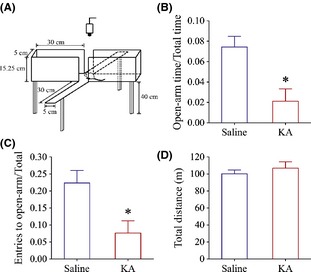
Kainate induces anxiety‐related behaviors in the mice. An elevated plus‐maze was used to evaluate anxiety‐related behavior. (A) The diagram shows experimental methods to examine a mouse staying in the open arms versus closed ones in an elevated plus‐maze. (B) illustrates a comparison in the time for mice staying in the open arms jr total arms (open‐arm time/total‐arm time) in the mice with the intraperitoneal injections of kainic acid (red bar, n = 11) and saline (blue, n = 6), respectively (*P < 0.05). (C) illustrates a comparison in the entry times of mice into the open arms versus total arms (entry to open arm/total arm) in the mice with the peritoneal injections of kainic acid (red bar, n = 11) and saline (blue, n = 6), respectively (*P < 0.05). The duration for staying in the open arms is shorter, and the entry times are lower significantly in the intraperitoneal injection of kainic acid, implying kainate‐induced anxiety. (D) illustrates the total distances traveled in the arms from control and anxiety‐related behavior mice, indicating their normal locomotion.
Mice naturally avoid the open field. On the other hand, they intend to explore a new environment for the food. In this regard, the measurement for mice avoiding the open field was the duration when mice stayed in the open arms, that is, the duration in the open arms versus total experimental time, whereas the measurement for mice exploring the new environment was their entries into open arms. Therefore, the entry times into the open arms and the duration in the open arms were used to evaluate the level of anxiety, which were recorded by an automatic video‐tracking system for 5 min. The mice were placed at the open field of an elevated plus‐maze with facing to a closed arm at the beginning of experiments. High anxiety‐related behaviors are described as mice spending more time in the closed arms as well as having lower entry times into the open arms.
In the test of locomotion behavior, each of mice was placed in the center of an activity monitor chamber (40 × 40 × 35 centimeters; Ecgonline, China). The mice were allowed to stay in the chamber for 5 min, and their activity was recorded by an automatic video‐tracking system. The total distance was used to evaluate the locomotion of the mice. These behavioral data were presented as means ± SE and statistically analyzed by one‐way ANOVA (Origin Lab, Northampton, MA, USA).
Electrophysiology Study
The brain slices in the coronal section including the prefrontal cortex and other subcortical areas (350 μm) were prepared from two groups of the mice. These mice were anesthetized by injecting chloral hydrate (300 mg/kg) and decapitated by guillotine. The slices were sectioned by a vibratome in the modified and oxygenized (95% O2/5% CO2) artificial cerebrospinal fluid (mM: 124 NaCl, 3 KCl, 1.2 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgSO4, 10 dextrose, and 5 HEPES; pH 7.35) at 4°C, and then were held in the normal oxygenated ACSF (mM: 124 NaCl, 3 KCl, 1.2 NaH2PO4, 26 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, 10 dextrose, and 5 HEPES; pH 7.35) 25°C for 1–2 h. A slice was transferred into a submersion chamber (Warner RC‐26G) that was perfused by normal ACSF at 31°C for electrophysiological experiments 52, 53, 54, 55.
YFP‐labeled pyramidal neurons in the prelimbic region of the prefrontal cortex were recorded by whole‐cell clamp under a visualized condition (DIC/FN‐E600, Nikon, Japan). Spontaneous excitatory postsynaptic currents (sEPSC) from the glutamatergic synapses were recorded under a voltage‐clamp model (MultiClamp 700B and pClamp 10; Axon Instrument, Foster, CA, USA). The standard pipette solution contained (mM) 150 K‐gluconate, 5 NaCl, 0.4 EGTA, 4 Mg‐ATP, 0.5 Tris‐GTP, 4 Na‐phosphocreatine, and 10 HEPES (pH 7.4 adjusted by 2M KOH). Pipette solution osmolarity was 295–305 mOsmol, and pipette resistance was 6–8 MΩ. Series and input resistances for all neurons were monitored using hyperpolarization pulses (5 mV/50 ms) in each experiment and calculated by voltage pulses versus instantaneous and steady‐state currents. Bicuculline (10 μM) was present in ACSF to block GABAA receptor‐channels in GABAergic synapses and isolate glutamatergic sEPSCs 56, 57. At the end of experiments, 6‐Cyano‐7‐nitroquinoxaline ‐2,3‐(1H, 4H)‐dione (10 μM) and D‐amino‐5‐phosphonovanolenic acid (40 μM) were added in ACSF to test whether sEPSCs were glutamatergic. They did block synaptic currents recorded in our studies.
Data were analyzed if the recorded neurons had resting membrane potentials negatively more than −70 mV. The criteria for the acceptation of each experiment also included <5% changes in the resting membrane potential, spike magnitudes, and input/seal resistance 58, 59, 60, 61, 62. The values of sEPSCs are presented as mean ± SE. The comparisons of the data from behavioral tasks, electrophysiology as well as cell imaging between groups are statistically performed by one‐way ANOVA in the software Origin (version, 7.0, Northampton, MA, USA).
Studies in Morphology and Immunocytochemistry
After the treatments for 24 h and anxiety‐related behavior evaluation, the mice in groups of control and kainate treatment were anesthetized by the intraperitoneal injection of sodium pentobarbital and were perfused with 4% paraformaldehyde in 0.1 M phosphate buffer solution (PBS) from left ventricle/aorta until their bodies were rigid. The brains were quickly isolated and fixed in 4% paraformaldehyde PBS for additional 24 h. Cortical tissues were sliced in the cross‐section including the prefrontal cortices at 100 μm by vibratome. The sections were washed by PBS for three times air‐dried and cover‐slipped before cellular imaging under a confocal microscope (Olympus FV‐1000, Tokyo, Japan). The structure of the dendritic spines on YFP‐labeled glutamatergic neurons in the prelimbic region of the prefrontal cortex was analyzed.
The analyses to the structures in the spines of pyramidal neurons were carried out by an open‐accessed software Fiji Image‐J (ver. 1.48F, NIH, Bethesda, MA, USA). As the brain tissues were sliced in series sections, the counting and analysis in cell structures were able to be carried out at least from two sections for each of sections. The analyzed sections were chosen in a manner of one section from every two to prevent the influence of cells that crossed the neighboring sections on the analysis. In the analyses of dendritic spines, their density and size from primary processes (branches from somata) of pyramidal neurons were measured 63. The spines were the protrusion extended from the dendrites, which were counted as spines per μm in their number and were measured as spine width in their head size.
In immunocytochemistry study of PKC expression, the cortical tissues were sliced by vibratome for 20 μm. The sections were incubated in a polyclonal anti‐PKC (1:100) antibody (Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA) at 4°C with shaking for 18 h and then were incubated in red‐fluorescent‐conjugated anti‐rabbit (1:1000) antibody 64. The images of YFP‐labeled spines (yellow) and PKC (red)‐stained neurons in the prelimbic region of the prefrontal cortex were taken by laser scanning confocal microscope, in which the parameters of laser beam and PMT were fixed for all of the experiments. As the size of the spines is variable, the quantity of PKC expression in the spines was calculated by PKC areas versus spine areas.
The effects of PKC on anxiety‐related behavior and synaptic plasticity were examined using its selective and potent inhibitor of PKC activity, chelerythrine chloride (CHE; IC50 = 0.6 μM; 65, in which CHE was dissolved in dimethyl sulfoxide (DMSO) with final concentration at 2 mM. Although this concentration was fairly higher than its IC50, the realistic concentration in the brain was low due to the presences of the blood‐brain barrier, the metabolism by the liver and the elimination by the kidneys. It is noteworthy that this concentration of CHE alone does not affect the mouse anxiety‐related behavior, spine morphology, and PKC expression in the spine (Figures S1–S3). One could argue why other PKC inhibitors were not selected for our studies. Based on the literatures, CHE is thought to be one of PKC inhibitors that effectively penetrates across the blood‐brain barrier.
Results
To reveal the relationship between anxiety and glutamate as well as the anxiety‐related pathology of the glutamatergic synapses in the prelimbic area of the prefrontal cortex, we set up a mouse model of kainite‐induced anxiety. After this anxiety was onset, the signal transmission at the glutamatergic synapses was analyzed by recording spontaneous excitatory postsynaptic current (sEPSC) in the YFP‐labeled glutamatergic neurons from the prefrontal cortical slice. The morphology of the glutamatergic synapses was analyzed by measuring the size and number of the dendritic spines. The role of protein kinase C in the anxiety‐associated change of the glutamatergic synapses was studied using protein kinase C inhibitor and immunocytochemistry.
Kainic Acid Induces Anxiety‐Related Behaviors
The mice were divided into two groups, controls (the peritoneal injection of saline) and kainate treatments (the peritoneal injection of kainic acid). The treatment of kainic acid was based on the fact that kainic acid given to the brain led to the generalized anxiety and panic‐related defensive responses 66, 67, 68. After this treatment for 24 h, anxiety‐related behaviors in these mice were examined on an elevated plus‐maze (Figure 1A). The typical feature of anxiety‐related behaviors in the mice is their avoidance, so that they spend more time in the closed arms and have less entry times into the open arms. Based on these analyses, the duration of staying in the open arms versus total arms (open‐arm time/total time) is shorter in kainite‐induced anxiety mice (red bar, n = 11, Figure 1B) than controls (blue; n = 6; P < 0.05). Their entry times into open arms (entries into open arm/total arms) appear to be lower in kainate‐induced anxiety mice (red bar, n = 11, Figure 1C) than controls (blue; n = 6; P < 0.05). It is noteworthy that there is no difference in the total traveled distances in two groups of the mice (Figure 1D), showing their normal locomotion. Therefore, kainate evokes anxiety‐related behaviors in the mice.
We subsequently investigated the anxiety‐associated changes of the glutamatergic neurons in the prelimbic areas of the prefrontal cortices in terms of the morphology and functions of glutamatergic synapses.
The Glutamatergic Synapses in Prefrontal Cortex are Upregulated in Kainate‐Induced Anxiety
To reveal the anxiety‐associated changes in the glutamatergic synapses on the pyramidal neurons of prelimbic prefrontal cortex, we analyzed their dendritic spines by confocal microscope as well as their excitatory synaptic transmission by recording sEPSCs in kainate‐induced anxiety and control mice.
Figure 2 illustrates the functional alternation of glutamatergic synapses in the cortical slices from kainate‐induced anxiety mice. sEPSCs were recorded using whole‐cell voltage‐clamp at prelimbic cortical pyramidal neurons in the presence of 10 μM bicuculline. sEPSC amplitude and frequency appear to be higher in kainate‐induced anxiety mice (red in right panel of Figure 2A) than in controls (blue in 2A left panel). Figure 2B,C shows cumulative probability versus sEPSC amplitudes (2B) or inter‐sEPSC intervals (2C) in kainate‐induced anxiety mice (red symbols) and controls (blues). sEPSC amplitudes at cumulative probability to 67% (CP67) that merit ionotropic glutamate receptor responsiveness are 5.8 ± 0.28 pA in controls (blue bar in Figure 2D; n = 11) and 6.85 ± 0.38 pA in kainate‐induced anxiety mice (red in 2D, n = 7, P < 0.05). Inter‐sEPSC intervals at CP67 that merit glutamatergic synaptic active frequency are 541 ± 34 ms in controls (blue bar in Figure 2E; n = 11) and 386 ± 23 ms in kainate‐induced anxiety mice (red, n = 7, P < 0.05). Therefore, the upregulation of glutamatergic synaptic transmission in the prelimbic cortex is associated with anxiety‐related behaviors.
Figure 2.
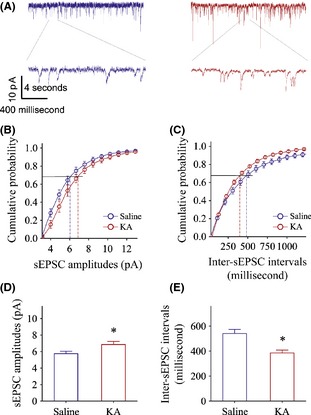
Excitatory synaptic transmission is upregulated in the pyramidal neurons of the prelimbic cortices from the kainate‐induced anxiety mice. Spontaneous excitatory postsynaptic currents (sEPSC) were recorded by whole‐cell voltage‐clamp at the prefrontal cortical pyramidal neurons of brain slices from kainate‐induced anxiety and control mice. (A) Left panels show sEPSCs recorded from a control mouse (blue trace) and right panels shows sEPSCs recorded from kainate‐induced mouse (red traces). (B) shows cumulative probability versus sEPSCs amplitudes from kainate‐induced anxiety mice (red symbols) and control (blue ones). (C) illustrates cumulative probability versus inter‐sEPSC intervals from kainate‐induced anxiety mice (red symbols) and control (blues). (D) shows sEPSC amplitudes at the cumulative probability to 67% (CP67) in the controls (blue bar; n = 11) and in kainate‐induced anxiety mice (red; n = 7, *P < 0.05). (E) shows inter‐EPSC intervals at 67% cumulative probability in the controls (blue bar; n = 11) and in kainate‐induced anxiety mice (red bar, n = 7, *P < 0.05).
Figure 3 shows the morphological features of the dendritic spines in prelimbic cortical pyramidal neurons from kainate‐induced anxiety mice. We analyzed the head size and the number of dendritic spines. The spine head appears to be enlarged and the number appears to be reduced in anxiety mice versus control (Figure 3A), which is supported by cumulative changes in spine number versus spine size (Figure 3B). Figure 3C,D shows the averaged spine width and spine density (spines per micrometer) in control (blue line and symbols) and anxiety mice (reds). The values of spine width are 0.64 ± 0.006 μm in controls (blue bar in Figure 3C, n = 1538 spines) and 0.75 ± 0.007 μm in anxiety (red bar, n = 1345, P < 0.05), respectively. The values of spine density are 1.06 ± 0.02 μm in control (blue bar in Figure 3D, n = 6 neurons) and 0.94 ± 0.04 μm in anxiety (red, n = 7, P < 0.05), respectively. These results indicate that the upregulation of excitatory synapses in the prelimbic cortical pyramidal neurons is associated with kainate‐induced anxiety, which is consistent to their functional study in Figure 2. It is pointed out that the upregulation of spine size and the downregulation of spine number indicate a homeostatic change in the excitatory synapses.
Figure 3.

Kainate‐induced anxiety is associated with the upregulation of dendritic spine head and the downregulation of spine number in YFP‐labeled pyramidal neurons of prelimbic cortices. (A) shows the spine head appears to be enlarged and the spine number is reduced in kainate‐induced anxiety mice versus controls. (B) illustrates cumulative changes in spine number versus spine size in kainate‐induced anxiety mice (red symbols) versus controls (blues). A shift of such cumulative curves toward right in kainate‐induced anxiety mice indicates the rise of their spine heads. (C) shows the averaged spine width in controls (blue bar; n = 1538 spines) and kainate‐induced anxiety mice (red bar, n = 1345, *P < 0.05), respectively. (D) shows spine density (spines per micrometer) in the controls (blue bar) and in kainate‐induced anxiety (red bar), respectively. The upregulation of the individual excitatory synapses is associated with kainate‐induced anxiety. The upregulation of spine size and the downregulation of spine number indicate the homeostatic change in excitatory synapses.
In terms of molecular mechanism underlying kainate‐induced anxiety and glutamatergic synapse upregulation, we studied the role of protein kinase C (PKC) in these events by immunocytochemistry and its inhibitor. The rationale to examine PKC effect is based on the facts that PKC plays critical role in cognitive behaviors, such as learning and memory 69, 70, 71, 72.
The Role of Protein Kinase C in Kainate‐Induced Anxiety and Glutamatergic Synapse Upregulation
To examine the essential role of PKC in the kainate‐induced anxiety and glutamatergic synapse upregulation, we used the following strategies. If PKC is involved in these events, we should see the expression of more protein kinase C in dendritic spines by immunocytochemistry. If PKC is required for these events, we should see that PKC inhibitor reverses kainate‐induced changes.
Figure 4 shows PKC expression in the dendritic spines, in which its spot size (PKC area vs. spine area) is used to merit PKC quantity. The spot size of PKC appears larger in the dendritic spines from kainate‐induced anxiety mice than controls (Figure 4A). The ratios of PKC areas to spine areas are 0.224 ± 0.025 in anxiety mice (n = 78 spines, red bar in Figure 4B) and 0.145 ± 0.02 in control (n = 82, blue bar in 4B), respectively (P < 0.05). The upregulated PKC expression in the dendritic spines from kainate‐induced anxiety mice indicates the involvements of PKC in the anxiety‐related behaviors and glutamatergic synapse upregulation. We subsequently investigated the requirement of PKC for these processes using its inhibitor.
Figure 4.
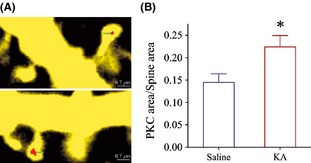
Protein kinase C is over‐expressed in the dendritic spines of the prelimbic cortical pyramidal neurons from kainate‐induced anxiety mice. Pyramidal neurons are genetically labeled by YFP, and protein kinase C expression is showed as red patches. (A) The areas of protein kinase C appear to be larger in the spines from kainate‐induced anxiety mice (bottom panel) than in controls (top panel). (B) illustrates a comparison of PKC area versus spine size in kainate‐induced anxiety mice (red bar, n = 78 spines) and controls (blue bar; n = 82, *P < 0.05).
Chelerythrine chloride (CHE), a selective and potent inhibitor of PKC 65, was used to lower PKC activity 73, 74, 75, 76. CHE was dissolved in dimethyl sulfoxide (DMSO) with the concentration at 2 mM (IC50 = 0.6 μM), in which final DMSO concentration was 8%. Our experiments were performed in three groups of the mice, DMSO control, kainate plus DMSO as well as the mix of CHE, kainate and DMSO, which were given by the intraperitoneal injection. CHE in DMSO was injected 30 min before kainate application.
Figure 5 illustrates the effect of PKC inhibitor on kainate‐induced anxiety. The ratios of the time staying in open arms to the time in entire maze (open‐arm time/total time) are 0.141 ± 0.03 in DMSO control (blue bar in Figure 5A, n = 8), 0.028 ± 0.02 in kainate plus DMSO (red, n = 8), and 0.142 ± 0.06 in CHE/kainate/DMSO (green; n = 8; asterisk, P < 0.05). The ratios of entry times into open arms to times into all arms (entries to open‐arm/total arms) are 0.23 ± 0.036 in DMSO control (blue bar in Figure 5B, n = 8), 0.05 ± 0.024 in kainate plus DMSO (red bar, n = 8), and 0.24 ± 0.04 in CHE/kainate/DMSO (green; n = 8; asterisk, P < 0.05). It is noteworthy that there are no differences in total traveled distances among three groups of mice (Figure 5C), indicating their normal locomotion. As CHE alone does not change anxiety‐related behavior (Figure S1), PKC inhibition reverses kainate‐induced anxiety.
Figure 5.
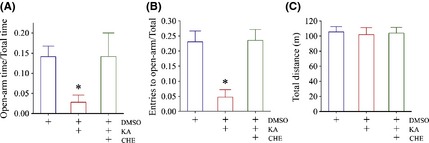
PKC inhibition reverses kainate‐induced anxiety‐related behaviors. Chelerythrine (CHE), a selective and potent inhibitor of PKC, was dissolved in DMSO for intraperitoneal injection to kainate‐induced anxiety mice. (A) illustrates open‐arm time versus total time in DMSO control (blue bar, n = 8), kainite‐induced anxiety mice (red bar, n = 8), and the mix of CHE, kainate, and DMSO (green; n = 8; asterisk, P < 0.05). (B) shows entries to open arm/total arms in DMSO control (blue bar, n = 8), kainite‐induced anxiety mice (red bar, n = 8), and the mix of CHE, kainate, and DMSO (green; n = 8; asterisk, P < 0.05). (C) There is no difference in total traveled distances among three groups of mice, indicating their normal locomotion.
Figure 6 illustrates an influence of PKC inhibitor CHE on kainate‐induced glutamatergic synapse upregulation. CHE appears to reverse kainate‐induced sEPSC upregulation in Figure 6A from DMSO control (blue trace), kainate plus DMSO (red), and a mix of CHE, kainate, and DMSO (green). Figure 6B,C illustrates cumulative probability versus sEPSC amplitudes (6B) or inter‐sEPSC intervals (6C) in DMSO (blue symbols), kainate/DMSO (red), and CHE/kainate/DMSO (greens). sEPSC amplitudes at 67% cumulative probability are 6.94 ± 0.46 pA in DMSO (blue bar in Figure 6D, n = 12), 9.35 ± 0.92 pA in kainite/DMSO (red, n = 6), and 7.1 ± 0.51 pA in CHE/kainate/DMSO (green; n = 12; an asterisk, P < 0.05). Inter‐sEPSC intervals at 67% cumulative probability are 353 ± 40 ms in DMSO (blue bar in Figure 6E; n = 12), 237 ± 15 ms in kainate/DMSO (red, n = 6), and 349 ± 30 ms in CHE/kainate/DMSO (green; n = 12; P < 0.05). PKC inhibition reverses the kainate‐induced upregulation of the glutamatergic synaptic transmission.
Figure 6.
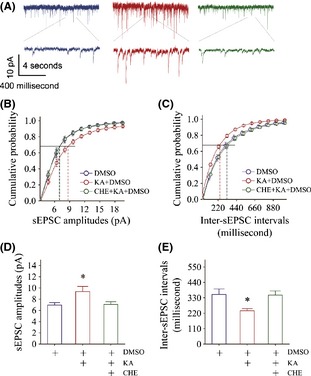
PKC inhibition reverses kainate‐induced upregulation of excitatory synaptic transmission at prelimbic cortical pyramidal neurons. (A) shows sEPSCs recorded from DMSO (blue trace), kainate plus DMSO (red), and the mix of CHE, kainate, and DMSO (green). (B) shows cumulative probability versus sEPSC amplitudes in DMSO control (blue symbols), kainate‐induced anxiety (reds), and the mixing of CHE, DMSO, and kainate (greens). (C) shows cumulative probability versus inter‐sEPSC intervals in DMSO control (blue symbols), kainate‐induced anxiety (reds), and the mixing of CHE, DMSO, and kainate (greens). (D) shows sEPSC amplitudes at 67% cumulative probability that merit ionotropic glutamate receptor responsiveness in DMSO control (blue bar, n = 12), kainite‐induced anxiety (red, n = 6), and in the mix of CHE, kainate, and DMSO (green; n = 12; asterisk, P < 0.05). (E) illustrates inter‐sEPSC intervals at 67% cumulative probability that merit the activity frequency of glutamatergic synapses in DMSO control (blue bar; n = 12), kainate‐induced anxiety (red bar, n = 6), and the mix of CHE, kainate, and DMSO (green; n = 12; asterisk, P < 0.05).
We further examined the influence of PKC inhibitor on kainate‐induced upregulation of dendritic spines (Figure 7). CHE appears to reverse kainate‐induced spine enlargement, as showed in Figure 7A from DMSO control (top panel), kainate plus DMSO (middle), and a mix of CHE, kainate, and DMSO (bottom), which is granted by cumulative change in spine size versus spine number (Figure 7B). The values of spine width are 0.58 ± 0.005 μm in DMSO control (blue bar in Figure 7C, n = 1520 spines), 0.63 ± 0.006 μm in kainite plus DMSO (red, n = 972), and 0.59 ± 0.005 μm in CHE/kainate/DMSO (green; n = 1211; an asterisk, P < 0.05). The values of spine density (spines per micrometer) are 1.13 ± 0.044 in DMSO (blue bar in Figure 7D, n = 10 neurons), 0.92 ± 0.055 in kainite plus DMSO (red bar, n = 8), and 1.24 ± 0.068 in CHE/kainate/DMSO (green; n = 10; an asterisk, P < 0.05). As CHE alone does not change spine morphology (Figure S2), the inhibition of PKC activity reverses a kainate‐induced upregulation of glutamatergic spines.
Figure 7.
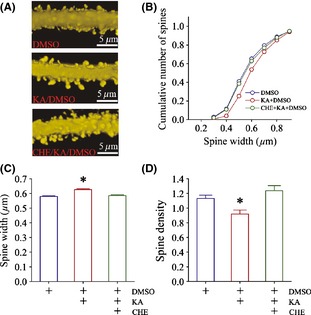
PKC inhibition reverses the upregulation of dendritic spines at prelimbic cortical pyramidal neurons. (A) illustrates the dendritic spines from DMSO (top panel), kainate plus DMSO (middle), and the mix of CHE, kainate, and DMSO (bottom). (B) shows cumulative changes in spine number versus spine size in DMSO (blue symbols), kainate plus DMSO (red symbols), and the mix of CHE, kainate, and DMSO (green symbols). (C) shows the values of spine width in DMSO control (blue bar, n = 1520 spines), kainite‐induced anxiety mice (red, n = 972), and the mixing of CHE, kainate, and DMSO (green; n = 1211; asterisk, P < 0.05). (D) shows spine density (spines per micrometer) in DMSO control (blue bar, n = 10 neurons), kainite‐induced anxiety mice (red, n = 8), and the mix of CHE, kainate, and DMSO (green; n = 10; asterisk, P < 0.05).
Conclusion
In summary, kainic acid induces anxiety‐related behaviors in the mice (Figure 1) and upregulates their glutamatergic synapses on the pyramidal neurons in the prelimbic area of the prefrontal cortices (Figures 2, 3). These kainate‐induced anxiety and glutamatergic synapse upregulations are essentially mediated by protein kinase C (Figures 4, 5, 6, 7). Our results provide the evidences for a hypothesis about the correlation between glutamate and anxiety as well as for a viewpoint about the anxiety therapy by applying the antagonists of ionotropic glutamate receptors. Our studies also present a new finding that protein kinase C plays a critical role in anxiety disorders.
In terms of the subcellular targets of glutamatergic synapses acted by kainate, our study indicates that kainic acid makes dendritic spines to be enlarged (Figure 3) and sEPSC amplitude to be increased (Figure 2), which are the indices for showing postsynaptic change 77. In addition, kainic acid enhances sEPSC frequency (Figure 2), that is, an increase of presynaptic transmitter release 78. Therefore, the changes in both presynaptic and postsynaptic compartments are involved in kainate‐induced anxiety. It is noteworthy that kainate upregulates spine volumes and downregulates spine densities, which appears a homeostatic process 59. As larger spines function for synapse formation 79, this homeostasis between spine volume and density turns dendritic spines to be either function or extinction, and this refinement of dendritic spines is structural buildup‐efficiency.
Our studies indicate the intracellular signaling mechanism underlying kainate‐induced anxiety, in which protein kinase C plays essential roles in anxiety‐related behaviors as well as anxiety‐associated upregulation in the function and morphology of the glutamatergic synapses (Figures 5, 6, 7). The action target of protein kinase C is located at the dendritic spines of the prelimbic cortical excitatory neurons (Figure 4). This is the first time to reveal the role of protein kinase C in anxiety disorder pathogenesis, which presents a possibility that anxiety disorders may be treated by applying the PKC inhibitors. It is noteworthy that our study provides new information about a role of PKC in anxiety disorder; however, other molecules have been found to be involved in anxiety, such as CREB, CRHR1, FKBP5, Egr‐1, Glo1, Gsr, AC8, CaMKIV, dystrophin, HTTLPR, and COMT Met158 80, 81. Such molecules either include the motifs of protein kinases or phosphorylate other proteins, which lead to anxiety. How these molecules interact each other to be responsible for anxiety disorders remains to be investigated.
Many brain areas have been found to be related to anxiety pathogenesis, such as prefrontal cortex, amygdala, cingulate, hypothalamus, and hippocampus 1, 7, 10, 14, 29. These areas are physically connected to constitute neural circuits for encoding emotional processes and cognitive behaviors 82, 83, 84. The prefrontal cortex sends glutamatergic axon projections to the amygdala and hippocampus, and in turn the nucleus accumbens for modulating the emotional and mnemonic functions. Both physiological and pathological alterations in the prefrontal cortex influence the activities of these limbic regions 82, 83, 84. In this regard, the prefrontal cortex may be an origin to trigger pathologically emotional behaviors, such as anxiety, which is supported by our result that the upregulated activity of pyramidal neurons in the prelimbic prefrontal cortex (Figures 2, 3) is associated with anxiety disorder. It is noteworthy that we focus on studying the involvement of the prelimbic cortex in kainate‐induced anxiety‐related behaviors. In fact, the intraperitoneal injections of kainic acid may affect other brain areas. How other brain areas are influenced by kainate and involved in anxiety‐related behaviors will be examined using stereotactic intracerebral injection, which is challenge because these cerebral areas are small in their size.
Previous studies indicate that the deficit of GABAergic synaptic transmission is associated with anxiety disorders 1, 2, 32. GABAAR enhancers are used to be anxiolytic reagents 37, 38, which remain to be reevaluated due to the unfavorable side effects 39, 42. Recently, glutamate is presumably associated to anxiety 43, and the antagonists of ionotropic glutamate receptors are thought to be potential anxiolytic reagents 44, 45, 46. Our studies in kainate‐induced anxiety strengthen these thoughts, in which one of cellular targets acted by kainate is the glutamate synapses in the excitatory neurons of the prelimbic cortex. As the balance between glutamate and GABA signal transmission on the excitatory neurons is critical for a functional homeostasis in the cerebral cortices, both the upregulation of glutamatergic synapses and the deficit of GABAergic synapses are involved in anxiety pathogenesis. The medications to anxiety disorders may be beneficial from a combined use of ionotropic glutamate receptor antagonists and GABAAR enhancers with low dosages, which would reduce their side effects.
In addition to GABA and glutamate, the dysfunctional interactions in ligands and receptors, such as serotonin, norepinephrine, dopamine, and hormones, are associated with anxiety disorder 38, 85, 86, 87. Chemical reagents strengthening the efficacy of their interactions have been applied for the psychotropic medications of anxiety disorders, such as selective serotonin reuptake inhibitors, monoamine oxidase inhibitors, and tricyclic antidepressants 42. In spite of the notable advances, numerous patients suffering from anxiety disorder fail to adequately and timely respond to these pharmacologic reagents. The effort to combine these medications should be worthy in the future.
It is noteworthy that the dendritic spines include both ionotropic glutamate receptors (iGluR) and metabotropic glutamate receptors (mGluR). The attention has been paid to reveal the roles of mGluR reagents in anxiety disorders 88, 89, 90. For instance, a current report demonstrates that the activation of mGluR1,5 can improve anxiety‐related behaviors, synchronizes neuronal activity, and enhances GABAergic synaptic transmission in anxiety‐behavior mice 32. mGluR agonists may be potentially applied for the therapy of anxiety disorders.
Our studies were conducted at the glutamatergic synapses and neurons in the prelimbic cortex, in which the glutamatergic neurons were genetically labeled by yellow fluorescent protein. This labeling confers our study to be neuron‐type specific 47, such that the synaptic spines in morphological analysis and the synaptic transmission in electrophysiological recording can be clearly identified. The identification of cellular and subcellular origins in the study of anxiety pathogenesis will provide clues for developing pharmacological reagents with specific subcellular targets.
Disclosure
None.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. PKC inhibitor, chelerythrine (CHE), does not affect anxiety‐related behaviors in the mice. The mice were treated by the intraperitoneal injection of either DMSO or CHE/DMSO. An elevated plus‐maze was used to evaluate anxiety‐related behavior.
Figure S2. Chelerythrine (CHE), a PKC inhibitor, does not affect dendritic spines on the YFP‐labeled pyramidal neurons of prelimbic cortices.
Figure S3. The expression of protein kinase C in the dendritic spines of prelimbic cortical pyramidal neurons is not influenced by chelerythrine (CHE). Pyramidal neurons are genetically labeled by YFP, and protein kinase C expression is showed as red patches.
Acknowledgments
This study is supported by National Basic Research Program (2013CB531304 and 2011CB504405) and the Natural Science Foundation China (30990261 and 81171033) to JHW.
References
- 1. Bishop SJ. Neurocongnitive mechanisms of anxiety: an integrative account. Trends Cogn Sci 2007;11:307–316. [DOI] [PubMed] [Google Scholar]
- 2. Rickeles K, Rynn M. Overview and clinical presentation of generalized anxiety disorder. Psychiatr Clin North Am 2001;24:1–17. [DOI] [PubMed] [Google Scholar]
- 3. Anand A, Shekhar A. Brain imaging studies in mood and anxiety disorders: Special emphasis on the amygdala. Ann N Y Acad Sci 2003;985:370–388. [DOI] [PubMed] [Google Scholar]
- 4. Adams JD Jr. Chemical interactions with pyramidal neurons in layer 5 of the cerebral cortex: Control of pain and anxiety. Curr Med Chem 2009;16:3476–3479. [DOI] [PubMed] [Google Scholar]
- 5. Bondi CO, Rodriguez G, Gould GG, Frazer A, Morilak DA. Chronic unpredictable stress induces a cognitive deficit and anxiety‐like behavior in rats that is prevented by chronic antidepressant drug treatment. Neuropsychopharmacology 2008;33:320–331. [DOI] [PubMed] [Google Scholar]
- 6. Bremner JD. Brain imaging in anxiety disorders. Exp Rev Neurother 2004;4:275–284. [DOI] [PubMed] [Google Scholar]
- 7. Craske MG, Rauch SL, Ursano R, Prenoveau JP, D.S., Zinbarg RE. What is an anxiety disorder? Depress Anx 2009;26:1066–1085. [DOI] [PubMed] [Google Scholar]
- 8. Damsa C, Kosel M, Moussally J. Current status of brain imaging in anxiety disorders. Curr Opin Psychiatry 2009;22:96–110. [DOI] [PubMed] [Google Scholar]
- 9. Davidson RJ. Anxiety and affective style: One of prefrontal cortex and amygdala. Biol Psychiatry 2002;51:68–80. [DOI] [PubMed] [Google Scholar]
- 10. Davis M, Walker DL, Miles L, Grillon C. Phasic versus sustained fear in rats and humans: Role of the extended amygdala in fear versus anxiety. Neuropsychopharmacology 2010;35:105–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freitas‐Feerrari MC, Hallak JE, Trzesniak C, Filho AS et al. Neuroimaging in social anxiety disorder: A systematic review of the literature. Prog Neuropyschopharmacol Biol Psychiatry 2010;34:565–580. [DOI] [PubMed] [Google Scholar]
- 12. Gross C, Hen R. The developmental origins of anxiety. Nat Rev Neurosci 2004;5:545–552. [DOI] [PubMed] [Google Scholar]
- 13. Garrett A, Chang K. The role of the amygdala in bipolar disorder development. Dev Psychopathol 2008;20:1285–1296. [DOI] [PubMed] [Google Scholar]
- 14. LeDoux J. Emotion circuits in the brain. Annu Rev Neurosci 2000;23:155–184. [DOI] [PubMed] [Google Scholar]
- 15. Milad MR, Rauch SL. The role of the orbitofrontal cortex in anxiety disorders. Ann N Y Acad Sci 2007;1121:546–561. [DOI] [PubMed] [Google Scholar]
- 16. Mennes M, Stiers P, Lagae L, Van den Bergh B. Long‐term cognitive sequelae of antenatal maternal anxiety: Involvement of the orbitofrontal cortex. Neurosci Biobehav Rev 2006;30:1078–1086. [DOI] [PubMed] [Google Scholar]
- 17. Myers‐Schulz B, Koenigs M. Functional anatomy of ventromedial prefrontal cortex: Implications for mood and anxiety disorders. Mol Psychiatry 2011;17:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist 2004;10:221–234. [DOI] [PubMed] [Google Scholar]
- 19. Rauch SL, Shin LM, Wright CI. Neuroimaging studies of amygdala function in anxiety disorders. Ann N Y Acad Sci 2003;985:389–410. [DOI] [PubMed] [Google Scholar]
- 20. Roozendaal B, McEwen BS, Chattarji S. Stress, memory and amygdala. Nat Rev Neurosci 2009;10:423–433. [DOI] [PubMed] [Google Scholar]
- 21. Stein MB, Stein DJ. Social anxiety disorders. Lancet 2008;371:1115–1125. [DOI] [PubMed] [Google Scholar]
- 22. Yu T, Guo M, Garza J, Rendon S, Sun XL, Zhang W, Lu XY. Cognitive and neural correlates of depression‐like behaviour in socially defeated mice: An animal model of depression with cognitive dysfunction. Int J Neuropsychopharmacol 2010;14:303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amano T, Unal CT, Pare D. Synaptic correlates of fear extinction in the amygdala. Nat Neurosci 2010;13:489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crestani F, Lorez M, Bear K. Decreased GABAA‐receptor clustering results in enhanced anxiety and a bias for threat cue. Nat Neurosci 1999;2:833–839. [DOI] [PubMed] [Google Scholar]
- 25. Delgado MR, Olsson A, Phelps EA. Extending animal models of fear conditioning to humans. Biol Psychol 2006;73:39–48. [DOI] [PubMed] [Google Scholar]
- 26. DuBios DW, A.P, Floyd DW, Weiner JL, McCool BA. et al. Distinct functional characteristics of the lateral/basolateral amygdala GABAergic system in C57BL/6J and DBA/2J mice. J Pharmacol Exp Ther 2006;318:629–640. [DOI] [PubMed] [Google Scholar]
- 27. Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron 2009;62:757–771. [DOI] [PubMed] [Google Scholar]
- 28. Liu J, Garza JC, Truong HV, Henschel J, Zhang W, Lu XY. The melanocortinergic pathway is rapidly recruited by emotional stress and contributes to stress‐induced anorexia and anxiety‐like behavior. Endocrinology 2007;148:5531–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression and extinction of conditioned fear. Physiol Rev 2010;90:419–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shin LM, Liberzon I. The neurocircuitry of fear, stress and anxiety disorders. Neuropsychopharmacology 2010;35:169–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shumyatsky GP, Tsvetkov E, Malleret G et al. Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell 2002;111:905–918. [DOI] [PubMed] [Google Scholar]
- 32. Zhang F, Liu B, Lei Z, Wang J. mGluR1,5 activation improves network asynchrony and GABAergic synapse attenuation in the amygdala: Implication for anxiety‐like behavior in DBA/2 mice. Mol Brain 2012;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leuner B, Shors TJ. Stress, anxiety, and dendritic spines: What are the connections? Neuroscience 2012;251:108–119. [DOI] [PubMed] [Google Scholar]
- 34. Wu LJ, Ko SW, Toyoda H et al. Increased anxiety‐like behavior and enhanced synaptic efficacy in the amygdala of GluR5 knockout mice. PLoS ONE 2007;2:e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Furmaga H, Shah A, Frazer A. Serotonergic and noradrenergic pathways are required for the anxiolytic‐like and antidepressant‐like behavioral effects of repeated vagal nerve stimulation in rats. Biol Psychiatry 2011;70:937–945. [DOI] [PubMed] [Google Scholar]
- 36. Graeff FG, Guimaraes FS, De Andrade TG, Deakin JF. Role of 5‐HT in stress, anxiety, and depression. Pharmacol Biochem Behav 1996;54:129–141. [DOI] [PubMed] [Google Scholar]
- 37. Bourin M, Hascoet M. Drug mechanisms in anxiety. Curr Opin Investig Drugs 2001;2:259–265. [PubMed] [Google Scholar]
- 38. Dell'Osso B, Buoli M, Baldwin DS, Altamura AC. Serotonin norepinephrine reuptake inhibitors (SNRIs) in anxiety disorders: A comprehensive review of their clinical efficacy. Hum Psychopharmacol 2010;25:17–29. [DOI] [PubMed] [Google Scholar]
- 39. Amiel JM, Mathew SJ. Glutamate and anxiety disorders. Curr Psychiatry Rep 2007;9:278–283. [DOI] [PubMed] [Google Scholar]
- 40. Bowden CL, Perlis RH, Thase ME et al. Aims and results of the NIMH systematic treatment enhancement program for bipolar disorder (STEP‐BD). CNS Neurosci Ther 2012;18:243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vilhauer JS, Young S, Kealoha C et al. Treating major depression by creating positive expectations for the future: A pilot study for the effectiveness of future‐directed therapy (FDT) on symptom severity and quality of life. CNS Neurosci Ther 2012;18:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ravindran LN, Stein MB. The pharmacologic treatment of anxiety disorders: A review of progress. J Clin Psychiatry 2010;71:839–854. [DOI] [PubMed] [Google Scholar]
- 43. Bergink V, van Megen HJ, Westenberg HG. Glutamate and anxiety. Eur Neuropsychopharmacol 2004;14:175–183. [DOI] [PubMed] [Google Scholar]
- 44. Chaki S, Okubo T, Sekiguchi Y. Non‐monoamine‐based approach for the treatment of depression and anxiety disorders. Recent Pat CNS Drug Discov 2006;1:1–27. [DOI] [PubMed] [Google Scholar]
- 45. Cortese BM, Phan KL. The role of glutamate in anxiety and related disorders. CNS Spectr 2005;10:820–830. [DOI] [PubMed] [Google Scholar]
- 46. Simon AB, Gorman JM. Advances in the treatment of anxiety: Targeting glutamate. NeuroRx 2006;3:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang G, Gao Z, Guan S, Zhu Y, Wang JH. Upregulation of excitatory neurons and downregulation of inhibitory neurons in barrel cortex are associated with loss of whisker inputs. Mol Brain 2013;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972;32:281–294. [DOI] [PubMed] [Google Scholar]
- 49. McLin JP, Steward O. Comparison of seizure phenotype and neurodegeneration induced by systemic kainic acid in inbred, outbred, and hybrid mouse strains. Eur J Neurosci 2006;24:2191–2202. [DOI] [PubMed] [Google Scholar]
- 50. Pellow S, Chopin P. Validation of open: Closed arm entries in an elevated plus‐maze as a measure of anxiety in the rats. J Neurosci Methods 1985;14:149–167. [DOI] [PubMed] [Google Scholar]
- 51. Walf AA, Frye CA. The use of elevated plus maze as an assay of anxiety‐related behavior in rodents. Nat Protoc 2007;2:322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ge R, Qian H, Wang JH. Physiological synaptic signals initiate sequential spikes at soma of cortical pyramidal neurons. Mol Brain 2011;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang J‐H. Short‐term cerebral ischemia causes the dysfunction of interneurons and more excitation of pyramidal neurons. Brain Res Bull 2003;60:53–58. [DOI] [PubMed] [Google Scholar]
- 54. Yu J, Qian H, Chen N, Wang JH. Quantal glutamate release is essential for reliable neuronal encodings in cerebral networks. PLoS ONE 2011;6:e25219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang Z, Gu E, Lu X, Wang JH. Essential role of axonal VGSC inactivation in time‐dependent deceleration and unreliability of spike propagation at cerebellar Purkinje cells. Mol Brain 2014;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang J‐H, Stelzer A. Shared calcium signaling pathways in the induction of long‐term potentiation and synaptic disinhibition in CA1 pyramidal cell dendrites. J Neurophysiol 1996;75:1687–1702. [DOI] [PubMed] [Google Scholar]
- 57. Wei J, Zhang M, Zhu Y, Wang JH. Ca2+‐calmodulin signalling pathway upregulates GABA synaptic transmission through cytoskeleton‐mediated mechanisms. Neuroscience 2004;127:637–647. [DOI] [PubMed] [Google Scholar]
- 58. Chen N, Yu J, Qian H, Ge R, Wang JH. Axons amplify somatic incomplete spikes into uniform amplitudes in mouse cortical pyramidal neurons. PLoS ONE 2010;5:e11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen N, Chen X, Wang J‐H. Homeostasis established by coordination of subcellular compartment plasticity improves spike encoding. J Cell Sci 2008;121:2961–2971. [DOI] [PubMed] [Google Scholar]
- 60. Ge R, Qian H, Chen N, Wang JH. Input‐dependent subcellular localization of spike initiation between soma and axon at cortical pyramidal neurons. Mol Brain 2014;7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lu W, Wen B, Zhang F, Wang JH. Voltage‐independent sodium channels emerge for an expression of activity‐induced spontaneous spikes in GABAergic neurons. Mol Brain 2014;7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang JH, Wei J, Chen X, Yu J, Chen N, Shi J. The gain and fidelity of transmission patterns at cortical excitatory unitary synapses improve spike encoding. J Cell Sci 2008;121:2951–2960. [DOI] [PubMed] [Google Scholar]
- 63. Ni H, Huang L, Chen N et al. Upregulation of barrel GABAergic neurons is associated with cross‐modal plasticity in olfactory deficit. PLoS ONE 2010;5:e13736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Qi Y, Huang L, Ni H et al. Intracellular Ca2+ regulates spike encoding at cortical GABAergic neurons and cerebellar Purkinje cells differently. Biochem Biophys Res Commun 2009;381:129–133. [DOI] [PubMed] [Google Scholar]
- 65. Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun 1990;172:993–999. [DOI] [PubMed] [Google Scholar]
- 66. Alt A, Weiss B, Ogden AM et al. In vitro and in vivo studies in rats with LY293558 suggest AMPA/kainate receptor blockade as a novel potential mechanism for the therapeutic treatment of anxiety disorders. Psychopharmacology 2006;185:240–247. [DOI] [PubMed] [Google Scholar]
- 67. Dos Santos L, de Andrade TG, Zangrossi H Jr. Serotonergic neurons in the median raphe nucleus regulate inhibitory avoidance but not escape behavior in the rat elevated T‐maze test of anxiety. Psychopharmacology 2005;179:733–741. [DOI] [PubMed] [Google Scholar]
- 68. Pobbe RL, Zangrossi H Jr. Involvement of the lateral habenula in the regulation of generalized anxiety‐ and panic‐related defensive responses in rats. Life Sci 2008;82:1256–1261. [DOI] [PubMed] [Google Scholar]
- 69. Govoni S, Amadio M, Battaini F, Pascale A. Senescence of the brain: Focus on cognitive kinases. Curr Pharm Des 2010;16:660–671. [DOI] [PubMed] [Google Scholar]
- 70. Sun MK, Alkon DL. Protein kinase C pharmacology: Perspectives on therapeutic potentials as antidementic and cognitive agents. Recent Pat CNS Drug Discov 2006;1:147–156. [DOI] [PubMed] [Google Scholar]
- 71. Van Kolen K, Pullan S, Neefs JM, Dautzenberg FM. Nociceptive and behavioural sensitisation by protein kinase Cepsilon signalling in the CNS. J Neurochem 2008;104:1–13. [DOI] [PubMed] [Google Scholar]
- 72. Wang JH, Feng DP. Postsynaptic protein kinase C essential to induction and maintenance of long‐term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci USA 1992;89:2576–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Begon S, Pickering G, Eschalier A, Mazur A, Rayssiguier Y, Dubray C. Role of spinal NMDA receptors, protein kinase C and nitric oxide synthase in the hyperalgesia induced by magnesium deficiency in rats. Br J Pharmacol 2001;134:1227–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Keenan C, Goode N, Pears C. Isoform specificity of activators and inhibitors of protein kinase C gamma and delta. FEBS Lett 1997;415:101–108. [DOI] [PubMed] [Google Scholar]
- 75. Kukreja RC, Qian YZ, Okubo S, Flaherty EE. Role of protein kinase C and 72 kDa heat shock protein in ischemic tolerance following heat stress in the rat heart. Mol Cell Biochem 1999;195:123–131. [DOI] [PubMed] [Google Scholar]
- 76. Serrano PA, Rodriguez WA, Pope B, Bennett EL, Rosenzweig MR. Protein kinase C inhibitor chelerythrine disrupts memory formation in chicks. Behav Neurosci 1995;109:278–284. [DOI] [PubMed] [Google Scholar]
- 77. Wang JH, Ko G, Kelly PT. Cellular and molecular bases of memory: Synaptic and neuronal plasticity. J Clin Neurophysiol 1997;14:264–293. [DOI] [PubMed] [Google Scholar]
- 78. Zucker RS, Regehr WG. Short‐term synaptic plasticity. Ann Rev Physiol 2002;25:355–405. [DOI] [PubMed] [Google Scholar]
- 79. Kasai H, Fukuda M, Watanabe S, Hayashi‐Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci 2010;33:121–129. [DOI] [PubMed] [Google Scholar]
- 80. Wu L‐J, Kim SS, Zhuo M. Molecular targets of anxiety: From membrane to nucleus. Neurochem Res 2008;33:1925–1932. [DOI] [PubMed] [Google Scholar]
- 81. Xia K, Xiong H, Shin Y et al. Roles of KChIP1 in the regulation of GABA‐mediated transmission and behavioral anxiety. Mol Brain 2010;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Del Arco A, Mora F. Neurotransmitters and prefrontal cortex‐limbic system interactions: Implications for plasticity and psychiatric disorders. J Neural Transm 2009;116:941–952. [DOI] [PubMed] [Google Scholar]
- 83. Mega MS, Cummings JL, Salloway S, Malloy P. The limbic system: An anatomic, phylogenetic, and clinical perspective. J Neuropsychiatry Clin Neurosci 1997;9:315–330. [DOI] [PubMed] [Google Scholar]
- 84. Stathis P, Panourias IG, Themistocleous MS, Sakas DE. Connections of the basal ganglia with the limbic system: Implications for neuromodulation therapies of anxiety and affective disorders. Acta Neurochir Suppl 2007;97:575–586. [DOI] [PubMed] [Google Scholar]
- 85. Charney DS. Neuroanatomical circuits modulating fear and anxiety behavior. Acta Psychiatry Scand Suppl 2003;417:38–50. [DOI] [PubMed] [Google Scholar]
- 86. Mathew SJ, Price RB, Charney DS. Recent advances in the neurobiology of anxiety disorders: Implications for novel therapeutics. Am J Med Genet C Semin Med Genet 2008;148C:89–98. [DOI] [PubMed] [Google Scholar]
- 87. Siever LJ, Weinstein LN. The neurobiology of personality disorders: Implications for psychoanalysis. J Am Psychoanal Assoc 2009;57:361–398. [DOI] [PubMed] [Google Scholar]
- 88. Krystal JH, Mathew SJ, D'Souza DC, Garakani A, Gunduz‐Bruce H, Charney DS. Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs 2010;24:669–693. [DOI] [PubMed] [Google Scholar]
- 89. Lima VC, Molchanov ML, Aguiar DC, Campos AC, Guimaraes FS. Modulation of defensive responses and anxiety‐like behaviors by group I metabotropic glutamate receptors located in the dorsolateral periaqueductal gray. Prog Neuropsychopharmacol Biol Psychiatry 2008;32:178–185. [DOI] [PubMed] [Google Scholar]
- 90. Muly EC, Mania I, Guo JD, Rainnie DG. Group II metabotropic glutamate receptors in anxiety circuitry: Correspondence of physiological response and subcellular distribution. J Comp Neurol 2007;505:682–700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. PKC inhibitor, chelerythrine (CHE), does not affect anxiety‐related behaviors in the mice. The mice were treated by the intraperitoneal injection of either DMSO or CHE/DMSO. An elevated plus‐maze was used to evaluate anxiety‐related behavior.
Figure S2. Chelerythrine (CHE), a PKC inhibitor, does not affect dendritic spines on the YFP‐labeled pyramidal neurons of prelimbic cortices.
Figure S3. The expression of protein kinase C in the dendritic spines of prelimbic cortical pyramidal neurons is not influenced by chelerythrine (CHE). Pyramidal neurons are genetically labeled by YFP, and protein kinase C expression is showed as red patches.