

Summary
Aim
Tau hyperphosphorylation and amyloid β‐peptide overproduction, caused by altered localization or abnormal activation of glycogen synthase kinase‐3β (GSK‐3β), is a pathogenic mechanism in Alzheimer's disease (AD). Valproic acid (VPA) attenuates senile plaques and neuronal loss. Here, we confirmed that VPA treatment improved spatial memory in amyloid precursor protein (APP)/presenilin 1 (PS 1) double‐transgenic mice and investigated the effect of VPA on synaptic structure and neurite outgrowth.
Methods
We used ultrastructural analysis, immunocytochemistry, immunofluorescence staining, and Western blot analysis to assess the effect of VPA treatment in mice.
Results
VPA treatment thickened the postsynaptic density, increased the number of presynaptic vesicles, and upregulated the expression of synaptic markers PSD‐95 and GAP43. VPA increased neurite length of hippocampal neurons in vivo and in vitro. In VPA‐treated AD mouse brain, inactivated GSK‐3β (pSer9‐GSK‐3β) was markedly increased, while hyperphosphorylation of tau at Ser396 and Ser262 was decreased; total tau levels remained similar. VPA treatment notably improved pSer133‐cAMP response element‐binding protein (CREB) and brain‐derived neurotrophic factor (BDNF) levels, which are associated with synaptic function and neurite outgrowth.
Conclusion
VPA improves behavioral deficits in AD, modifies synaptic structure, and accelerates neurite outgrowth, by inhibiting the activity of GSK‐3β, decreasing hyperphosphorylated tau, enhancing CREB and BDNF expression.
Keywords: Alzheimer's disease, GSK‐3β, Neurite, Synapse, Tau, Valproic acid
Introduction
Alzheimer's disease (AD) is an irreversible neurodegenerative brain disorder that affects a large percentage of the population. It is characterized by progressive cognitive deficits, memory impairment, and behavioral changes 1. In patients with AD, brain regions involved in learning and memory decrease in size due to synaptic and neuronal loss 2. Histopathologically, AD features both extracellular senile plaques (SP), composed chiefly of amyloid β‐peptide (Aβ), and intracellular neurofibrillary tangles (NFTs), mainly composed of abnormally hyperphosphorylated tau 3. Aβ is a proteolytic cleavage product of amyloid precursor protein (APP) and accumulates in AD 4, while hyperphosphorylation of tau reflects the misregulation of several kinases, such as glycogen synthase kinase‐3β (GSK‐3β), and can affect axonal transport 5. In APP/presenilin 1 (PS 1) double‐transgenic mice, Aβ deposition is present from 8 weeks of age and SP are developed as early as 3–4 months, leading to impaired spatial memory and learning 6, 7. Hyperphosphorylated tau appears in the brains of these mice after the onset of Aβ deposition 8, 9.
As AD is complicated and the underlying molecular mechanisms are incompletely understood, effective treatments for AD are still lacking. It is thought that Aβ accumulation is the key factor triggering a cascade of neuropathological events, which in turn lead to alterations in tau, synaptic dysfunction, and neuron death 10. However, how Aβ overproduction leads to cognitive impairment and key AD pathological processes remains unknown. Therefore, identifying the signaling pathways underlying AD pathogenesis may contribute to designing strategies for preventing or curing AD.
It has been proposed that the glycogen synthase kinase‐3β (GSK‐3β) pathway plays a crucial role in AD pathogenesis 11. Altered localization or abnormal activation of GSK‐3β is related to formation of NFTs in AD 12, 13, 14, 15, 16. Besides being the main tau protein kinase, GSK‐3β also mediates APP processing and plays a significant role in synaptic plasticity and memory 17, 18, 19, 20, 21. Furthermore, GSK‐3β inhibits expression of transcription factors, such as β‐catenin and cAMP response element‐binding protein (CREB), ultimately leading to neuronal apoptosis and negatively affecting neuronal plasticity 17, 21. In the light of the evidence linking increased GSK‐3β activity and AD, GSK‐3β inhibition has gained much attention as a promising therapeutic target for AD, and specific and potent GSK‐3β inhibitors are currently under investigation 22.
Valproic acid (VPA) is a widely used mood‐stabilizing and antiepileptic drug 23, which has neuroprotective properties, stimulating neurotrophic factor release, inducing neurogenesis and regulating GSK‐3β activity 24, 25, 26. In our previous study 27, we found that VPA decreased Aβ generation from APP; it also inhibited SP formation. The neuroprotective effect of VPA on AD has also been confirmed by other studies 28, 29, 30. However, the effect of VPA on other key AD processes, such as synaptic alteration and neurite outgrowth, is uncertain.
Here, we treated APP/PS1 double‐transgenic AD model mice with VPA for 4 weeks. Pretreatment with VPA markedly improved memory, rescued hippocampal synaptic structure, and stabilized structural integration of cells. VPA notably decreased hyperphosphorylated tau levels, inhibited the activity of GSK‐3β, and increased levels of CREB and brain‐derived neurotrophic factor (BDNF), which are associated with synaptic function and neurite outgrowth.
Materials and Methods
Transgenic Mice
APP/PS1 double‐transgenic mice, B6C3‐Tg (AβPPswe, PSEN1▵9) (Jackson Laboratories, Bar Harbor, MN, USA), which overexpress mouse/human APP (Mo/Hu AβPP695swe) and mutant human PS1‐▵9, were used in this study. Mice were housed in an air‐conditioned room and maintained on a 12‐h light/dark cycle. At 3 weeks, the genotype of the offspring mice was detected through PCR using DNA extracted from tail tissue 27. All precautions were taken to minimize suffering, and the procedures were undertaken in compliance with institutional guidelines.
Group Design and Drug Treatment
Forty male B6C3‐Tg mice weighing 25–30 g were randomly divided into a control group and a VPA treatment group (n = 20, 3.5–4 months of age). Mice in the VPA treatment group were intraperitoneally injected with 30 mg/kg bodyweight VPA, daily, for 4 weeks, while mice in the control group were injected with the same amount of saline. Fixed injection times and operating personnel were used; the body weight, activities, and food and water consumption of the mice were recorded.
Morris Water Maze Test
Two days after treatment, mice were subjected to the Morris water maze (MWM) test to evaluate spatial learning and memory. The procedures were performed as in Vorhees et al. 31, with the following modifications. The procedures include 1 day of visible platform tests, five consecutive days of hidden platform tests, and a probe trial. In visible platform tests, a 10‐cm platform which was marked with a flag was positioned above a clear water surface. Mice were put into the water from four quadrants and tested for four contiguous trials. In the hidden platform tests, the platform was fixed in the same position, submerged 1 cm below an opaque water surface. The escape latency and path length that mice reached the platform in 60 seconds were analyzed using water maze software (Shanghai Bio‐will Co., Ltd., Shanghai, China). At 24 h after the last hidden platform test, the platform was removed and the probe trial was performed. The number of mice placed into the pool in different quadrant crossing the target platform in 60 second was recorded.
Primary Neuronal Cell Culture
Primary neuronal cell cultures were obtained by dissecting the hippocampus of B6C3‐Tg mice at postnatal day 1 or day 2. Cells were dissociated by enzymatic incubation in trypsin and 0.05% EDTA and repeated pipetting and were resuspended in neurobasal medium (Gibco, Waltham, MA, USA) with 2% B27 (Sigma–Aldrich, St Louis, MO, USA). Cells were then seeded at a density of 1 × 105 cells/well on glass coverslips in 24‐well plates at 37°C in humidified air containing 5% CO2. Primary hippocampal cells, which were cultured for 1 day, were incubated with 5 mm VPA or vehicle solution (saline). The concentration used was chosen based on studies by Qing et al. 27. Cells were photographed after one more day. β‐Tubulin staining was performed on primary cells after VPA treatment for 3 days. The neurite length and number of β‐tubulin‐positive neurons were measured after 3 days of VPA treatment, using Image‐Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA).
Tissue Preparation
Following behavioral testing, the mice were euthanized. Animal handling and tissue harvesting were performed as described by Gao et al. 32. Mice were anesthetized with 10% chloral hydrate and sacrificed by decapitation, and all subsequent procedures were performed on ice. Each mouse brain was divided into two sections: One section was immediately homogenized for protein extraction, and the other was postfixed in fresh 4% PFA in 0.1 m phosphate‐buffered saline (PBS) for 8 h. PFA‐fixed brain tissues were cut into 4‐μm‐thick coronal sections using a microtome (Leica Instruments, Germany). Every 10th slice with the same reference position was mounted onto slides for immunohistochemical (IHC) and immunofluorescent staining.
IHC Staining
Immunohistochemical staining of brain tissue sections was performed as described previously 29. Sections were then immunostained with the following primary antibodies: mouse anti‐β‐tubulin III (1:500; Sigma–Aldrich), rabbit anti‐p‐tau‐Ser262, rabbit anti‐BDNF (both 1:200; Santa Cruz Biotechnology, Dallas, TX, USA), and rabbit anti‐p‐CREB‐Ser133 (1:100; Cell Signaling Technology, Danvers, MA, USA). Incubation was performed at 4°C for overnight. After washing in PBS, sections were incubated with biotinylated goat anti‐mouse and goat anti‐rabbit antibody (1:200; Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature followed by avidin–biotin–peroxidase complex (1:100; Vector Laboratories). The immunostained micrographs were imaged using a digital microscope. In five randomly selected nonoverlapping high‐power fields of the cortex and hippocampus from each slice, the integrated optical density and the number of positive cells were calculated using Image‐Pro Plus 6.0.
Immunofluorescence Staining
Sections were deparaffinized, washed, and blocked in 0.1% Triton and 5% milk in PBS for 1 h. Sections were then immunostained with mouse anti‐NeuN (1:400; Millipore, Billerica, MA, USA), mouse anti‐β‐tubulin (1:500; Sigma–Aldrich), rabbit anti‐p‐CREB‐Ser133, rabbit anti‐BDNF, and rabbit anti‐p‐tau‐Ser262 at 4°C for overnight. The antigens were detected with specific fluorescein‐conjugated secondary antibodies: goat anti‐mouse TRITC and FITC or goat anti‐rabbit TRITC and FITC antibody for 1 h at room temperature; then, nuclei were counterstained with DAPI at room temperature. Images were collected and analyzed with a TCS‐TIV confocal laser scanning microscope (Leica).
Transmission Electron Microscopy
For Transmission Electron Microscopy (TEM) detection, four mice from each group were anesthetized with an overdose of chloral hydrate (i.p.), then transcardially perfused with 0.01 m PBS (pH 7.4) and 2.5% glutaraldehyde/4% paraformaldehyde. The brain was rapidly stripped, and 1 mm3 tissue blocks were cut from the hippocampal CA1 area. Samples were embedded with Epon 812 epoxy resin. Tissue blocks were then cut into 1‐μm‐thick thin sections. After uranyl acetate/lead citrate double staining, neurons and ultrastructures were observed using a Philips EM208S transmission electron microscope (Philips, Amsterdam, the Netherlands). Synapses were identified by the existence of at least three vesicles in the presence of a Postsynaptic density (PSD) and the presynaptic bouton. Using randomly obtained synapse images, the PSD thickness, synaptic cleft width, active zone length, and synaptic interface curvature were measured using the Image‐Pro Plus 6.0 image analysis system, following the approach used by Itarat et al. 33. The synaptic interface curvature (R) was calculated from synaptic interface arc length (a) and chord length (b), viz. R = a/b. The number of presynaptic vesicles within 30 randomly selected synapses was counted.
Western Blot
The cortex and hippocampus of the hemibrain were homogenized in RIPA lyses buffer (Pierce, Rockford, IL, USA). Total proteins from mouse brain samples were measured using a spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Equal amounts of protein were separated by 10% SDS‐PAGE gel; proteins were then transferred onto polyvinylidene fluoride (PVDF) membranes and probed with the following primary antibodies overnight at 4°C: rabbit anti‐growth Associated Protein‐43 (GAP‐43) antibody (1:1000 dilution; Electrochemical Products Inc., New Berlin, WI, USA); mouse anti‐PSD protein‐95 (PSD‐95) antibody, total GSK‐3β antibody, p‐Ser9‐GSK‐3β antibody (1:1000 dilution; Millipore); rabbit anti‐p‐tau‐Ser262, rabbit anti‐p‐tau‐Ser396, rabbit anti‐T‐tau antibody, and rabbit anti‐BDNF (1:1000 dilution; Santa Cruz Biotechnology); and rabbit anti‐total‐CREB antibody and anti‐p‐CREB‐Ser133 antibody (both 1:1000 dilution; Cell Signaling Technology). Subsequently, membranes were incubated with the corresponding horseradish peroxidase‐conjugated secondary antibodies in a 1:5000 dilution (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Immunoreactive bands were visualized by enhanced chemiluminescent detection with exposure to X‐ray film and then quantitated by Quantity One analysis. The initial analysis was carried out by two researchers who were blinded to the treatment status.
Statistical Analysis
Data were analyzed using SPSS 17.0 software. All values were expressed as mean ± standard deviation (SD). Statistical comparisons between control and VPA treatment groups were determined using analysis of variance (ANOVA) followed by Student's t‐test. P values <0.05 were considered significant.
Results
Long‐Term Treatment With VPA Improves Spatial Learning and Memory in APP/PS1 Transgenic Mice
In the visible platform test, VPA‐ and vehicle‐treated AD mice had similar escape latencies (t = 0.084, P > 0.05, n = 20, Figure 1A) and path lengths (t = 0.073, P > 0.05, n = 20, Figure 1B), indicating that VPA treatment did not affect mouse motility or vision. In the hidden platform test, VPA‐treated AD model mice had significantly improved escape latency (F = 12.89, P < 0.001) and path length (F = 3.92, P = 0.006) compared with saline‐treated controls. A significant decrease in escape latency (P < 0.05, Figure 1C) and path length (P < 0.05, Figure 1D) in finding the platform was observed in the VPA‐treated mice on days 3, 4, and 5, as compared to controls. In probe trials, VPA‐treated mice crossed the platform in the target quadrant significantly more often than vehicle‐treated controls (t = 2.578, P < 0.028, n = 20, Figure 1E). These data indicated that VPA treatment significantly improved spatial learning and memory in APP/PS1 transgenic AD model mice.
Figure 1.
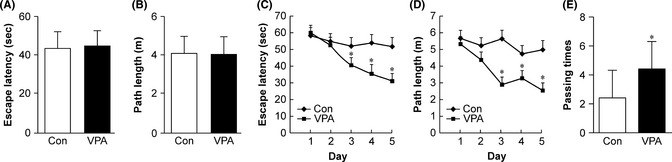
The Morris water maze test analysis of behavioral changes in control and valproic acid (VPA) treatment in amyloid precursor protein (APP)/PS1 transgenic mice (n = 10 mice). (A) During the first day of visible platform testing, the VPA‐treated (VPA) and saline‐treated control (Con) Alzheimer's disease (AD) mice exhibited a similar latency for escape onto the visible platform (P > 0.05). (B) The VPA‐treated and control group covered similar swimming distances before escaping onto the visible platform (P > 0.05). (C) In the hidden platform test, mice were trained with four trails per day for 5 days. VPA‐treated AD mice showed a shorter latency for escape onto the hidden platform on the third, fourth, and fifth day (*P < 0.01). (D) The VPA‐treated AD mice demonstrated a shorter swimming distance before escaping onto the hidden platform on the third, fourth, and fifth day than did the controls (*P < 0.01). (E) In the probe trial, the VPA‐treated AD mice traveled across the target platform significantly more often than did the controls (*P < 0.05).
VPA Modifies Synaptic Structure and Upregulates the Expression of Synapse Plasticity‐Related Protein
Learning and memory are related to biochemical and morphological changes at the synaptic level. TEM analysis showed that the number of synapses in the hippocampus of VPA‐treated mice (3.21 ± 0.88) was higher than that of vehicle‐treated group (2.74 ± 1.02), although not significantly so (t = 1.047, P = 0.322, n = 10, Figure 2A,B). However, as compared with saline‐treated controls (Figure 2Ab), the number and density of synaptic vesicles in the presynapses of 4‐week VPA‐treated mice (Figure 2Ad) were markedly increased.
Figure 2.
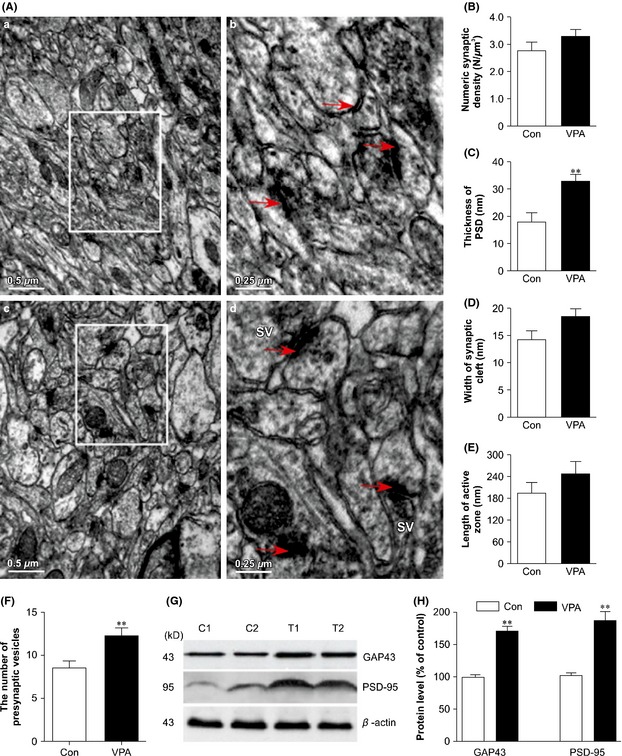
Changes in the synapses in valproic acid (VPA)‐treated and saline‐treated (Con) amyloid precursor protein (APP)/PS1 transgenic mice. (A) Electron micrographs showing the ultrastructural features of synapses in the hippocampal CA1 area of mice (scale bar = 0.5 μm). SV: Synaptic vesicles. (B) Effects of VPA (30 mg/kg, i.p. daily for 4 weeks) on numeric synaptic density (N/μm3) in mouse brains. The results are expressed as mean ± SEM, n = 30 synapses. No statistical differences in numeric synaptic density were detected by one‐way ANOVA. (C–F) Effects of VPA (30 mg/kg, i.p. daily for 4 weeks) on the structural parameters of synaptic interfaces in mice. (C) The width of the synaptic cleft (**P < 0.01 vs. control mice); (D) the thickness of the postsynaptic density (PSD); (E) the length of the synaptic active zone; (F) the number of presynaptic vesicles; n = 30 synapses (**P < 0.01 vs. control mice). (G) Western blot showed the expression of GAP43 and PSD‐95 in VPA‐treated and control mice (two representative images are shown, n = 4). (H) Quantification showing the significant elevation of GAP43 and PSD‐95 expression in VPA‐treated mice compared with controls (**P < 0.01 vs. control mice).
Morphological changes were observed in the brains of the two groups. In the hippocampus of vehicle‐treated mice, the majority of synapses were II‐type synapses with a straight synaptic interface, and the minority were asymmetrical synapses with slightly curved presynaptic or postsynaptic membranes, while in the brain of VPA‐treated mice, there were more concave or convex asymmetrical synapses. Synaptic morphological image analysis showed that some of the structural parameters of the synaptic interface were altered in these brains. VPA treatment for 4 weeks significantly increased the thickness of the PSD (t = 4.797, P = 0.020, Figure 2C). However, other synaptic interface parameters, such as the width of the synaptic cleft (t = 2.077, P = 0.807, Figure 2D) and synaptic active zone length (t = 1.190, P = 0.288, Figure 2E), were not statistically significantly different between vehicle‐treated and VPA‐treated mouse brains. VPA treatment did, however, significantly increase the number of presynaptic vesicles (t = 4.797, P = 0.020, Figure 2F).
Synaptic function is associated with cognition and depends on the normal integration of glutamate receptors at the PSD. Expression of PSD‐95 protein in the cortex and hippocampus of VPA‐treated mice was significantly higher than that in the control group (t = 6.570, P = 0.002, n = 10). GAP‐43 expression was also significantly higher in the VPA‐treated AD model mouse brains than in the vehicle‐treated group (t = 6.103, P = 0.0017, n = 10).
VPA Accelerates Neurite Outgrowth
A pronounced loss of neurites in the brain is one of the functionally most relevant histopathological lesions in AD; hence, we investigated the potential effects of VPA on neurites. Numerous dystrophic neurites were observed in control mice using β‐tubulin staining, while in VPA groups, the neurites grew well and were orderly arranged, with notably fewer dystrophic neurites (Figure 3A). One day after 5 mm VPA or saline treatment, growing cells were photographed under a microscope (Figure 3Ba,b), and the processes of VPA‐treated primary cells were more numerous and longer than those of vehicle‐treated cells. Three days after treatment, β‐tubulin staining also showed that VPA treatment increased the processes in primary hippocampal cells (Figure 3Bc,d). More specifically, both the number of neurites per cell (t = 4.293, P = 0.002, n = 20, Figure 3C) and the length of the longest neurite per cell (t = 4.031, P = 0.003, n = 20, Figure 3D) were increased in VPA‐treated cells in comparison with vehicle‐treated cells.
Figure 3.

Morphological analysis of neurite outgrowth in valproic acid (VPA)‐treated and vehicle‐treated (Con) amyloid precursor protein (APP)/PS1 transgenic mice and primary hippocampal cells. (A) β‐Tubulin staining showing the neurite outgrowth of β‐tubulin III + cells in the hippocampus of VPA‐treated and control mice (scale bar = 50 μm): (a–c) hippocampus of the control mice and (d–f) hippocampus of the VPA‐treated mice. (B) Changes in neurite outgrowth in VPA‐treated and vehicle‐treated primary hippocampal cells. Hippocampal cells were cultured for 1 day and then treated with vehicle solution (saline) and 5 mm VPA (scale bar = 20 μm). (a, b) Photographs of 1 day VPA‐ and saline‐treated primary cells; (c, d) β‐tubulin staining showing 3 days VPA‐ and saline‐treated primary cells. (C) Quantification showing the significantly increased number of neurites per cell in VPA‐treated primary cells in relative to controls. (D) Quantification showing the significantly increased length of the longest neurite in VPA‐treated cells in comparison with saline‐treated cells (**P < 0.01 vs. control mice).
VPA Decreases the Abnormal Hyperphosphorylated Tau
We then examined whether the reduction of dystrophic neurites in VPA‐treated mice was caused by a decrease of tau hyperphosphorylation. IHC and immunofluorescence staining showed that the p‐tau‐Ser262‐positive cells were distributed in various brain regions, such as the cerebral cortex, hippocampus, cerebellum, and olfactory bulb. The optical density of p‐tau‐Ser262‐positive cells in the cortex and hippocampus of VPA‐treated mice was markedly less than that in the control group (cortex: t = 4.13, P < 0.05; hippocampus: t = 3.73, P < 0.05, Figure 4A–C). There was no significant difference in the levels of total (t) tau between VPA‐treated and control brains (t = 0.000, P > 0.05, n = 8), but phospho‐tau levels were significantly reduced to 78.4% (Ser262, t = 4.425, P = 0.001) and 37.8% (Ser396, t = 6.942, P < 0.005) in VPA‐treated transgenic mouse brains compared with those of the saline‐treated group (Figure 4D,E). Thus, VPA treatment significantly attenuated tau hyperphosphorylation in the APP/PS1 mice brain.
Figure 4.
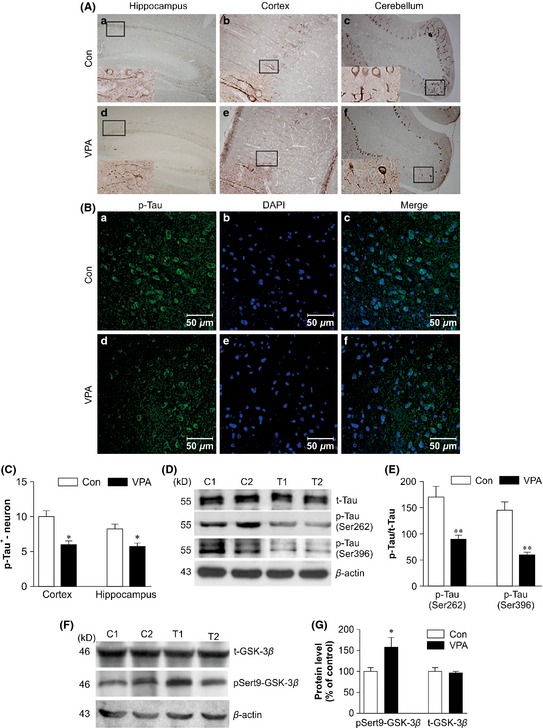
Immunohistochemical (IHC), immunofluorescent, and Western blot analysis of the phosphorylated tau levels in valproic acid (VPA)‐treated and saline‐treated (Con) amyloid precursor protein (APP)/PS1 transgenic mice and Western blot analysis of the expression of pSer9‐GSK‐3β and total GSK‐3β in VPA‐treated and saline‐treated (Con) APP/PS1 transgenic mice. (A) IHC analysis of p‐tau‐Ser262‐positive cells in VPA‐treated and control mice: (a, e) hippocampus CA1; (b, d) cortex; (c, f) cerebellum (a–f: magnification 100×). (B) Immunofluorescent analysis of p‐tau‐Ser262+ cells in the cerebral cortex of VPA‐treated and control mice: (a–c) cerebral cortex of the control mice and (d–f) cerebral cortex of the VPA‐treated mice (a–f: scale bar = 50 μm). (C) Quantification showing the significantly decreased number of p‐tau‐Ser262+ cells in VPA‐treated mice relative to controls (n = 8, *P < 0.05 vs. control mice). (D) Western blot showing the expression of total tau, p‐tau‐Ser262, and p‐tau‐Ser396 in VPA‐treated and control mice (two representative images are shown, n = 4). (E) Quantification showing the significantly decreased p‐tau‐Ser262 and p‐tau‐Ser396 expression in VPA‐treated mice compared with controls (**P < 0.01 vs. control mice). (F) Western blot showing the expression of pSer9‐GSK‐3β and total GSK‐3β (two representative images are shown, n = 4). (G) Quantification showing the significant elevation of pSer9‐GSK‐3β expression in VPA‐treated mice in relative to controls (*P < 0.05 vs. control mice).
VPA Inhibits GSK‐3β Activity
Levels of total GSK‐3β (t‐GSK‐3β) and p‐Ser9‐GSK‐3β were determined in mouse brain homogenates. VPA‐treated AD model mice displayed significantly increased levels of pSer9‐GSK‐3β, as compared with controls (t = 2.75, P < 0.05, n = 10, Figure 4F,G), although levels of t‐GSK‐3β did not differ significantly between VPA‐treated and control mice (t = 1.340, P = 0.210, n = 10). Therefore, VPA treatment significantly inhibited the activity of GSK‐3β in the APP/PS1 mouse brain.
VPA Upregulates CREB Phosphorylation and BDNF Expression in APP/PS1 Transgenic Mice
One of the transcriptional factors that may be regulated by GSK‐3β is CREB. IHC staining showed that the p‐Ser133‐CREB‐positive cells were distributed in all hippocampal and cortical fields (Figure 5A). The optical density of p‐Ser133‐CREB‐positive cells in VPA‐treated mice was markedly higher than that in the control group (t = 4.265, P = 0.0021, Figure 5D). There was a significantly increased number of p‐Ser133‐CREB‐positive cells in VPA‐treated mice compared with controls (t = 4.103, P = 0.003, Figure 5E). Western blot analysis confirmed that the level of p‐Ser133‐CREB protein in VPA‐treated mice was notably increased compared with that of vehicle‐treated mice (t = 7.664, P = 0.001, n = 10), while the total CREB level was not significantly different between the two groups (t = 4.700, P = 0.001, Figure 5B,C).
Figure 5.
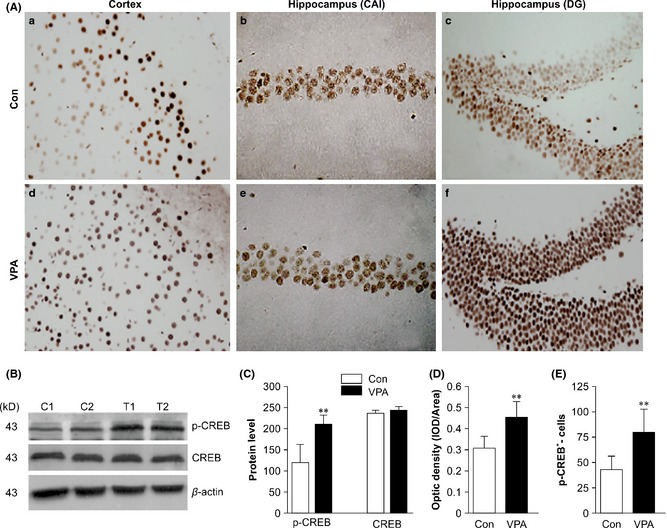
Immunohistochemical (IHC) and Western blot analysis of the phosphorylated cAMP response element‐binding protein (CREB) in valproic acid (VPA)‐treated and saline‐treated (Con) amyloid precursor protein (APP)/PS1 transgenic mice. (A) IHC analysis of p‐Ser133‐CREB‐positive cells in VPA‐treated and control mice: (a, e) Cerebral cortex, (b, d) hippocampal CA1, (c, f) hippocampal dentate gyrus (a–f: magnification 400×). (B) Western blot showing the expression of p‐Ser133‐CREB and total CREB in VPA‐treated and control mice (two representative images are shown, n = 4). (C) Quantification showing the significant elevation of p‐Ser133‐CREB expression in VPA‐treated mice relative to controls (**P < 0.01 vs. control mice); there was no significant change in total CREB expression (P > 0.05 vs. control mice). (D) Quantitative analysis of the optical density of p‐CREB + neurons (n = 8, **P < 0.01 vs. control mice). (E) Quantitative analysis of the number of p‐Ser133‐CREB + neurons (n = 8, **P < 0.01 vs. control mice).
CREB binds to the promoter region of many genes, including BDNF, which is associated with memory and synaptic plasticity. IHC and immunofluorescence analyses showed that VPA treatment significantly increased the average optical density of the BDNF+ cells in the APP/PS1 mice (Figure 6A,B). The optical density of BDNF+ cells was much higher in VPA‐treated mouse cortex and hippocampus fields than in those of the control group (cortex: t = 6.29, P = 0.000; hippocampus: t = 5.90, P = 0.004, Figure 6C). BDNF protein levels were also significantly increased in VPA‐treated AD mice compared with those of vehicle‐treated mice (t = 4.303, P = 0.001, Figure 6D,E). These data strongly suggested that VPA alleviated synaptic dysfunction by triggering the GSK‐3β–CREB–BDNF signaling pathway.
Figure 6.
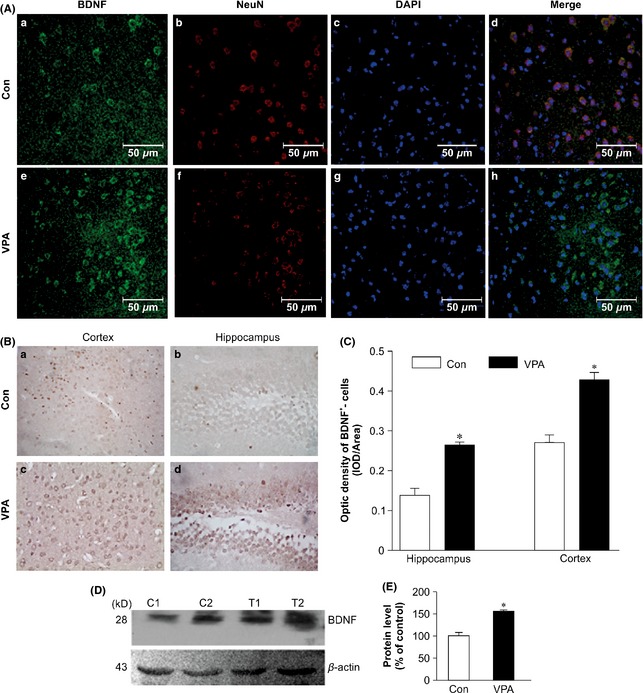
Immunohistochemical (IHC), immunofluorescent, and Western blot analysis of BDNF expression in valproic acid (VPA)‐treated and saline‐treated (Con) amyloid precursor protein (APP)/PS1 transgenic mice. (A) Immunofluorescent‐stained sections showing the increased expression of BDNF in the cerebral cortex of VPA‐treated mice: (a, e) immunostained with BDNF; (b, f) immunostained with NeuN; (c, g) stained nuclei with DAPI; (d, h) BDNF, NEUN, and DAPI merged (a–h: scale bar = 50 μm). (B) IHC analysis showing the increased expression of BDNF: (a, c) cortex of mice and (b, d) hippocampal dentate gyrus of the mice (a–d: magnification 400×). (C) Quantitative analysis of the optical density of BDNF + neurons (n = 8, *P < 0.01 vs. control mice). (D) Western blot showing the expression of BDNF in VPA‐treated and control mice (two representatives are shown, n = 4). (E) Quantifications showing the significant elevation of BDNF expression in VPA‐treated mice in relative to controls (*P < 0.01 vs. control mice).
Discussion
AD, a multifactorial neurodegenerative disease accompanied by severe cognitive impairment, is also one of the earliest disorders to be linked to GSK‐3β dysfunction 34, 35. GSK‐3β hyperactivity or overexpression is increasingly shown to be closely related to Aβ generation, tau hyperphosphorylation, and neuronal loss 36. Therefore, GSK‐3β inhibitors have attracted great attention in AD research. VPA, a inhibitor of histone deacetylase (HDACs), has also been shown to inhibit GSK‐3 recently 27. So, VPA could be used as a potential therapeutic agent against AD.
In this study, the data showed that VPA‐treated mice exhibited significantly increased spatial learning and memory performance, consistent with previous findings 27, 37, 38, indicating that VPA remediates cognitive deficits in AD transgenic mice. The changes underlying progressive cognitive decline and memory loss are strongly correlated with exhausted neural plasticity and synaptic loss 39, 40, and the latter is an early event in AD progression 41. The modification of synaptic connectivity involves a change in synapse number and density, which contributes to neural system dysfunction, and causes cognitive deficits 42. Here, TEM was employed to examine the effect of VPA on mouse hippocampal synapses. It revealed 4‐week VPA treatment thickened the PSD compared with the control group. The PSD is an electron‐dense structure located beneath the postsynaptic membrane in register with the active zone of the presynaptic terminal and is a structural and functional multiprotein crossroad 43. Additionally, the number and density of presynaptic vesicles were markedly increased after VPA treatment. VPA led to upregulation of expression of the synaptic protein markers, GAP43 and PSD‐95, which are distributed on presynaptic and postsynaptic membranes, and plays a role in axonal remodeling and regeneration in adults and regulates synaptic strength, respectively. Thus, VPA treatment can significantly rescue early synaptic structural and functional deficits in AD model mice.
The positive effects of increased PSD‐95 and GAP43 expression after VPA treatment may relate to adjusting new synaptic connections and axonal growth during neuronal growth cone formation. We examined the effect of VPA on neurite outgrowths of hippocampal neurons both in vivo and in vitro and showed rescue of dystrophic neurites and promotion of neurite outgrowth. Moreover, VPA significantly increased the number and length of β‐tubulin III+ cellular processes. Microtubule stability, involved in structure and network formation for axonal transport, is crucial for neuronal functioning. Compelling evidence showed that VPA could promote neurite outgrowth and stabilize cell structural integrity in the brains of APP/PS1 transgenic mice.
Microtubule assembly and axonal transport are regulated by the microtubule‐associated protein tau under normal physiological conditions. Tau hyperphosphorylation causes its detachment from microtubules, impairing their function, axonal transport, synaptic dysfunction, and neurodegeneration 44. Our data showed that 4‐week VPA treatment significantly decreased p‐tau‐Ser262 and p‐tau‐Ser396 expression, which consistent with previous data 30. Ser262 is close to the region involved in the formation of microtubules; abnormal phosphorylation at Ser262 enhances tau separation from microtubules and NFT formation, leading to disruption of normal cell structure and loss of transportation function 45.
GSK‐3β is activated by phosphorylation at Tyr216 and is inhibited by phosphorylation at Ser9 46. Zhu et al. 47 reported that GSK‐3 upregulation impairs synaptic plasticity both structurally and functionally, which may underlie the GSK‐3‐mediated memory deficits. We showed that VPA treatment significantly increased p‐Ser9‐GSK‐3β levels in AD model mice. GSK‐3β may also regulate tau, as inhibition of GSK‐3β by VPA treatment caused a sharp decrease in hyperphosphorylated tau levels.
GSK‐3β also regulates CREB, which modulates plasticity and memory 48. CREB activation is regulated by phosphorylation at Ser133, which controls the induction of many genes, including BDNF 49. Here, p‐CREB‐Ser133 levels in VPA‐treated mice were increased markedly, but not total CREB levels. Several reports suggested that CREB improves synaptic plasticity through upregulating BDNF which is well known to promote hippocampal synaptogenesis, as well as learning and memory in AD model mice 50, 51. After 4‐week VPA treatment, more BDNF+ cells were distributed in the cortex and hippocampus of VPA‐treated mice, and the increased levels were confirmed by Western blot analysis; thus, VPA could promote CREB activity and increase BDNF expression levels.
Taken together, our data suggest that VPA could markedly improve memory, rescue hippocampal synaptic structure, stabilize structural integration of cells, and accelerate neurite outgrowth. VPA could inhibit GSK‐3β by phosphorylation of GSK‐3β at Ser9 and activate CREB by phosphorylation at Ser133, which leads to an increase in downstream protective factors, such as BDNF. Inhibition of GSK‐3β reduces abnormal tau hyperphosphorylation. Consequently, VPA‐derived BDNF and CREB‐Ser133 enhance axonal outgrowth, markedly increase hippocampal synaptic protein levels, and reduce disruption of neuronal filaments. Taken together, these findings strongly suggest that VPA modifies synaptic structure and accelerates neurite outgrowth via the GSK‐3β signaling pathway, improving behavioral deficits in AD mouse models.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
All authors have contributed to the work and agree with the presented findings, and the work has neither been published before nor is it being considered for publication in another journal. This work was supported by National Natural Science Foundation of China (81371221, 30970986), Program for New Century Excellent Talents in University (NCET‐11‐1084).
The first two authors contributed equally to this work.
References
- 1. Smith DL, Pozueta J, Gong B, et al. Reversal of long‐term dendritic spine alterations in Alzheimer disease models. Proc Natl Acad Sci 2009;106:16877–16882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wolfe MS, Xia W, Ostaszewsk BL, et al. Two transmembrane aspartates in presenilin‐1 required for presenilin endoproteolysis and gamma‐secretase activity. Nature 2009;398:513–517. [DOI] [PubMed] [Google Scholar]
- 3. Hernandez F, Avila J. The role of glycogen synthase kinase 3 in the early stages of Alzheimer's disease. FEBS Lett 2008;582:3848–3854. [DOI] [PubMed] [Google Scholar]
- 4. Mattson MP. Pathways towards and away from Alzheimer's disease. Nature 2004;430:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. LaPointe NE, Morfini G, Pigino G, et al. The amino terminus of tau inhibits kinesin‐dependent axonal transport: Implications for filament toxicity. J Neurosci Res 2009;87:440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wengenack TM, Whelan S, Curran GL, et al. Quantitative histological analysis of amyloid deposition in Alzheimer's double transgenic mouse brain. Neuroscience 2000;101:939–944. [DOI] [PubMed] [Google Scholar]
- 7. Holcomb L, Gordon MN, McGowan E, et al. Accelerated Alzheimer‐type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 1998;4:97–100. [DOI] [PubMed] [Google Scholar]
- 8. Kurt MA, Davies DC, Kidd M, et al. Hyperphosphorylated tau and paired helical filament‐like structures in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin‐1 transgenes. Neurobiol Dis 2003;14:89–97. [DOI] [PubMed] [Google Scholar]
- 9. Gong B, Vitolo OV, Trinchese F, et al. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 2004;114:1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hardy J. A hundred years of Alzheimer's disease research. Neuron 2006;52:3–13. [DOI] [PubMed] [Google Scholar]
- 11. Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem 2008;104:1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li X, Lv B, Xie J, et al. AGEs induce Alzheimer‐like tau pathology and memory deficit via RAGE‐mediated GSK‐3 activation. Neurobiol Aging 2012;33:1400–1410. [DOI] [PubMed] [Google Scholar]
- 13. Lucas JJ, Hernandez F, Gomez‐Ramos P, et al. Decreased nuclear beta‐catenin, tau hyperphosphorylation and neurodegeneration in GSK‐3beta conditional transgenic mice. EMBO J 2001;20:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Munoz‐Montano JR, Moreno FJ, Avila J, et al. Lithium inhibits Alzheimer's disease‐like tau protein phosphorylation in neurons. FEBS Lett 1997;411:183–188. [DOI] [PubMed] [Google Scholar]
- 15. Hanger DP, Hughes K, Woodgett JR, et al. Glycogen synthase kinase‐3 induces Alzheimer's disease‐like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 1992;147:58–62. [DOI] [PubMed] [Google Scholar]
- 16. Zhao R, Zhang Z, Song Y, et al. Implication of phosphatidylinositol‐3 kinase/Akt/glycogen synthase kinase‐3beta pathway in ginsenoside Rb1's attenuation of beta‐amyloid‐induced neurotoxicity and tau phosphorylation. J Ethnopharmacol 2011;133:1109–1116. [DOI] [PubMed] [Google Scholar]
- 17. DaRocha‐Souto B, Coma M, Perez‐Nievas BG, et al. Activation of glycogen synthase kinase‐3 beta mediates beta‐amyloid induced neuritic damage in Alzheimer's disease. Neurobiol Dis 2012;45:425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muyllaert D, Terwel D, Borghgraef P, et al. Transgenic mouse models for Alzheimer's disease: The role of GSK‐3β in combined amyloid and tau‐pathology. Rev Neurol 2006;162:903–907. [DOI] [PubMed] [Google Scholar]
- 19. Lim HJ, Cho JS, Oh JH, et al. NSE‐controlled carboxyl‐terminus of APP gene over‐expressing in transgenic mice induces altered expressions in behavior, Abeta‐42, and GSK3beta binding proteins. Cell Mol Neurobiol 2005;25:833–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liang MH, Chuang DM. Regulation and function of glycogen synthase kinase‐3 isoforms in neuronal survival. J Biol Chem 2007;282:3904–3917. [DOI] [PubMed] [Google Scholar]
- 21. Beurel E, Jope RS. The paradoxical pro‐ and anti‐apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol 2006;79:173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z, Zhao R, Qi J, et al. Inhibition of glycogen synthase kinase‐3beta by Angelica sinensis extract decreases beta‐amyloid‐induced neurotoxicity and tau phosphorylation in cultured cortical neurons. J Neurosci Res 2011;89:437–447. [DOI] [PubMed] [Google Scholar]
- 23. Phiel CJ, Zhang F, Huang EY, et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001;276:36734–36741. [DOI] [PubMed] [Google Scholar]
- 24. Chen PS, Peng GS, Li G, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry 2006;11:1116–1125. [DOI] [PubMed] [Google Scholar]
- 25. Kim HJ, Rowe M, Ren M, et al. Histone deacetylase inhibitors exhibit anti‐inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J Pharmacol Exp Ther 2007;321:892–901. [DOI] [PubMed] [Google Scholar]
- 26. Williams RS, Cheng L, Mudge AW, et al. A common mechanism of action for three mood‐stabilizing drugs. Nature 2002;417:292–295. [DOI] [PubMed] [Google Scholar]
- 27. Qing H, He G, Ly PTT, et al. Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J Exp Med 2008;205:2781–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z, Zhang XJ, Li T, et al. Valproic acid reduces neuritic plaque formation and improves learning deficits in APP(Swe)/PS1(A246E) transgenic mice via preventing the prenatal hypoxia‐induced down‐regulation of neprilysin. CNS Neurosci Ther 2014;20:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Long Z, Zheng M, Zhao L, et al. Valproic acid attenuates neuronal loss in the brain of APP/PS1 double transgenic Alzheimer's disease mice model. Curr Alzheimer Res 2013;10:261–269. [DOI] [PubMed] [Google Scholar]
- 30. Hu JP, Xie JW, Wang CY, et al. Valproate reduces tau phosphorylation via cyclin‐dependent kinase 5 and glycogen synthase kinase 3 signaling pathways. Brain Res Bull 2011;85:194–200. [DOI] [PubMed] [Google Scholar]
- 31. Vorhees CV, Williams MT. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat Protoc 2006;1:848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao B, Long Z, Zhao L, et al. Effect of normobaric hyperoxia on behavioral deficits and neuropathology in Alzheimer's disease mouse model. J Alzheimers Dis 2011;27:317–326. [DOI] [PubMed] [Google Scholar]
- 33. Itarat W, Jones DG. Morphological characteristics of perforated synapses in the latter stages of synaptogenesis in rat neocortex: Stereological and three‐dimensional approaches. J Neurocytol 1993;22:753–764. [DOI] [PubMed] [Google Scholar]
- 34. Gandy JC, Melendez‐Ferro M, Bijur GN, et al. Glycogen synthase kinase‐3β (GSK3β) expression in a mouse model of Alzheimer's disease: A light and electron microscopy study. Synapse 2013;67:313–327. [DOI] [PubMed] [Google Scholar]
- 35. Blalock EM, Geddes JW, Chen KC, et al. Incipient Alzheimer's disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA 2004;101:2173–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Medina M, Avila J. Glycogen synthase kinase‐3 (GSK‐3) inhibitors for the treatment of Alzheimer's disease. Curr Pharm Des 2010;16:2790–2798. [DOI] [PubMed] [Google Scholar]
- 37. Yao Z, Liang L, Liu Y, et al. Valproate improves memory deficits in an Alzheimer's disease mouse model: Investigation of possible mechanisms of action. Cell Mol Neurobiol 2014;34:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xuan A, Pan X, Wei P, et al. Valproic acid alleviates memory deficits and attenuates amyloid‐β deposition in transgenic mouse model of Alzheimer's disease. Mol Neurobiol 2015;51:300–312. [DOI] [PubMed] [Google Scholar]
- 39. D'Amelio M, Cavallucci V, Middei S, et al. Caspase‐3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci 2011;14:69–76. [DOI] [PubMed] [Google Scholar]
- 40. de Calignon A, Fox LM, Pitstick R, et al. Caspase activation precedes and leads to tangles. Nature 2010;464:1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheff SW, Price DA, Schmitt FA, et al. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007;68:1501–1508. [DOI] [PubMed] [Google Scholar]
- 42. Citri A, Malenka RC. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008;33:18–41. [DOI] [PubMed] [Google Scholar]
- 43. de Bartolomeis A, Latte G, Tomasetti C, et al. Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: Relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches. Mol Neurobiol 2014;49:484–511. [DOI] [PubMed] [Google Scholar]
- 44. Avila J, Lucas JJ, Perez M, et al. Role of tau protein in both physiological and pathological conditions. Physiol Rev 2004;84:361–384. [DOI] [PubMed] [Google Scholar]
- 45. Buee L, Bussiere T, Buee‐Scherrer V, et al. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 2000;33:95–130. [DOI] [PubMed] [Google Scholar]
- 46. Jope RS, Roh MS. Glycogen synthase kinase 3 (GSK‐3) in psychiatric disease and therapeutic interventions. Curr Drug Targets 2006;7:1421–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu LQ, Wang SH, Liu D, et al. Activation of glycogen synthase kinase‐3 inhibits long term potentiation with synapse‐associated impairments. J Neurosci 2007;27:12211–12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Banks PJ, Warburton EC, Brown MW, et al. Mechanisms of synaptic plasticity and recognition memory in the perirhinal cortex. Prog Mol Biol Transl Sci 2014;122:193–209. [DOI] [PubMed] [Google Scholar]
- 49. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002;35:605–623. [DOI] [PubMed] [Google Scholar]
- 50. Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog Neurobiol 2005;76:99–125. [DOI] [PubMed] [Google Scholar]
- 51. Ji Y, Lu Y, Yang F, et al. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat Neurosci 2010;13:302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]