

Summary
Background
In addition to their original applications for lowering cholesterol, statins display multiple neuroprotective effects. Inflammatory reactions and the PI3K/AKT/caspase 3 pathway are strongly implicated in dopaminergic neuronal death in Parkinson's disease (PD). This study aims to investigate how simvastatin affects 6‐hydroxydopamine‐lesioned PC12 via regulating PI3K/AKT/caspase 3 and modulating inflammatory mediators.
Methods
6‐hydroxydopamine‐treated PC12 cells were used to investigate the neuroprotection of simvastatin, its association with the PI3K/AKT/caspase 3 pathway, and antiinflammatory responses. Dopamine transporters (DAT) and tyrosine hydroxylase (TH) were examined in 6‐hydroxydopamine‐treated PC12 after simvastatin treatment.
Results
Simvastatin‐mediated neuroprotection was associated with a robust reduction in the upregulation induced by 6‐OHDA of inflammatory mediators including IL‐6, COX2, and TNF‐α. The downregulated DAT and TH levels in 6‐OHDA‐lesioned PC12 were restored after simvastatin treatment. Simvastatin reversed 6‐OHDA‐induced downregulation of PI3K/Akt phosphorylation and attenuated 6‐OHDA‐induced upregulation of caspase 3 in PC12. Furthermore, the PI3K inhibitor LY294002 pronouncedly abolished the simvastatin‐mediated attenuation in caspase 3.
Conclusions
Our results demonstrate that simvastatin provides robust neuroprotection against dopaminergic neurodegeneration, partially via antiinflammatory mechanisms and the PI3K/Akt/caspase 3 pathway. These findings contribute to a better understanding of the critical roles of simvastatin in treating PD and might elucidate the molecular mechanisms of simvastatin effects in PD.
Keywords: Apoptosis, Inflammation, Neurodegeneration, Neuroprotection, PI3K/Akt, Simvastatin
Introduction
As hydroxymethylglutaryl‐coenzyme reductase inhibitors, statins have been used clinically to reduce serum low‐density lipoprotein (LDL) cholesterol. Statins reduce the risk of ischemic heart disease events and cerebrovascular stroke and have potential applications in multiple sclerosis, traumatic brain injury, and Alzheimer's disease (AD). Statins impart neuroprotective effects through various mechanisms such as lowering cholesterol; suppressing intracellular adhesion molecule‐1 (ICAM‐1); reducing β‐amyloid production and serum apolipoprotein E (APOE) levels, antithrombotic effects, and antiinflammatory responses; modifying cognition‐related receptors; and augmenting endothelial nitric oxide synthase (eNOS) 1, 2, 3, 4. Antiinflammatory interventions induced by statins were also observed in various neurological diseases, such as AD, PD, and cerebral ischemia 3. Recent attention has been drawn to the effects of statins in PD; however, there is limited knowledge regarding their precise mechanisms.
The PI3‐kinase (PI3K)/Akt/caspase 3 signaling pathway plays an important role in neuronal survival and death 5, 6, 7, 8. Vazquez et al. found that in cerebellar granule neurons, PI3K/Akt inhibitor LY294002 induced apoptosis via a caspase‐dependent and calpain‐independent pathways 9. In human SH‐SY5Y neuroblastoma cells, IFN‐β induced apoptotic cell death via inhibition of the PI3K/Akt pathway and caspase 3 cleavage 10. Decreased neuronal numbers in the cortex were observed in phosphoinositide 3‐kinase enhancer (PIKE)−/− mice, implying important roles of PI3K in neuronal cell death 11. Furthermore, in isolated nuclei, nuclear PI3K and PIKE mediated the antiapoptotic activity of nerve growth factor, demonstrating that PI3K/Akt plays an essential role in cell survival 12. Several lines of evidence indicated that the PI3K/Akt/caspase 3 signaling pathway may cause the pathogenesis of PD; thus, the pathway is a therapeutic target for attenuating some symptoms and motor/nonmotor complications in patients with PD 13.
Parkinson's disease is the second most common neurodegenerative disorder, following AD. PD is characterized by dopaminergic neuronal loss in the substantia nigra, a disturbance of the central dopaminergic system and imbalances in some nondopaminergic systems. Multiple mechanisms, such as inflammatory responses, mitochondrial dysfunction, apoptosis, oxidative stress, and signaling pathways' actions, are involved in dopaminergic neuronal death in PD 4, 13, 14, 15. Our previous studies and several others have reported that in 6‐hydroxydopamine (6‐OHDA)‐lesioned rat model, simvastatin prevented dopaminergic neuronal death by inhibiting inflammatory mediators 4, 16, 17. However, limited knowledge has been obtained as to how simvastatin influences the dopamine neurons in vitro PD models and how it is correlated with the PI3K/Akt/caspase 3 pathway.
To address this issue, we investigated both the neuroprotection of simvastatin in PC12 cells following 6‐OHDA neurotoxicity and its association with neuronal death and antiinflammatory responses. In addition, the association between neuronal apoptosis and the PI3K/Akt/caspase 3 signaling pathway in 6‐OHDA‐incubated PC12 cells with simvastatin treatment has also been investigated.
Materials and Methods
Cell Culture and Treatments
The PC12 cell culture was performed as described in Rodriguez‐Blanco's study 18. Briefly, PC12 cells were routinely maintained in DMEM supplemented with 10% fetal bovine serum, 5% horse serum, 100 U/mL benzyl penicillin, and 100 mg/L streptomycin (Gibco, Grand Island, NY, USA). For all experiments, the cells were seeded on 96‐well plates or 6‐well plates at a density of 1.0 × 105 cells/mL for 24 h. Four groups were treated with either DMEM, simvastatin (0.6 μg/mL), 6‐OHDA (100 μM), or 6‐OHDA (100 μM) + simvastatin (0.6 μg/mL), with three wells in each group. For measurements on Akt and caspase 3, the PI3K inhibitor LY294002 was applied. Incubation with LY294002 was performed at a standard concentration of 10 μM. All of the experiments were repeated three times in different batches of cells.
Cell Counting Kit‐8 Assay and Apoptotic Cell measurement
The cell counting kit‐8 (CCK‐8) assay was performed with modifications according to Park's study 19 to measure the viability of PC12 after 6‐OHDA or 6‐OHDA + simvastatin treatment. Briefly, 10 μL of CCK‐8 kit reagent was added to the cells treated with 6‐OHDA or 6‐OHDA + simvastatin in 96‐well plates and incubated at 37°C for 1 h. Cell viability was assessed at absorbance of 570 nm with the ELISA plate reader. Each treatment group was replicated in three wells. All results were normalized to OD values measured from an identically conditioned well without cell culture. The results were expressed as a percentage of the control group. The PC12 apoptosis was evaluated using flow cytometry with annexin V‐FITC (Abcam, Cambridge, MA, USA): apoptotic cells displayed phosphatidylserine on the outside of the plasma membrane. Changes in phosphatidylserine asymmetry were analyzed by measuring the binding of annexin V to the cell membrane. The cell viability for control group was set as 100%.
Quantitative Real‐Time PCR
Dopamine transporter (DAT), tyrosine hydroxylase (TH), interleukin‐6 (IL‐6), COX2, and tumor necrosis factor‐α (TNF‐α) were measured and modified according to other studies 20, 21. Cells with different treatments were harvested into 1 mL of Ultraspec reagent (Biotecx, Houston, TX, USA). The total RNA was extracted and quantified, and the integrity was tested using gel electrophoresis. From each sample, 1 μg of total RNA was retrotranscripted to cDNA (PrimeScript RT‐PCR Kit; Takara Biotechnology, Dalian, China). Three microliters of each sample was used as a template for amplification reactions conducted with the SYBR® Premix Ex Taq™ II (Takara Biotechnology) following the manufacturer's instructions. The PCR amplifications were conducted in a LightCycler System (Roche Applied Science). For analysis purposes, the amplicon for each of the analyzed genes was cloned, and known amounts of the cloned product were used to generate a standard curve. The primers were designed with the “Primer3Output” program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) (Table 1). The number of copies of the gene of interest in each sample was extrapolated from the corresponding standard curve by the indicated software. For each sample, duplicate determinations were made, and the gene copy number was normalized by the amount of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) in the same sample. The primer selection was performed with the aid of the LightCycler Probe Design Software, version 1.0 (Idaho Technology Inc., Alameda, CA, USA). The mRNA level for the control group was set as 100%.
Table 1.
Primers used for real‐time RT‐PCR
| Target gene | Primer sequences | Amplicon (bp) | |
|---|---|---|---|
| TNF‐α | FP | 5′‐ATGAGCACGGAAAGCATG‐3′ | 276 |
| RP | 5′‐TACGGGCTTGTCACTCGAGTT‐3′ | ||
| TH | FP | 5′‐ACCGAGAGGACAGGATCCCACA‐3′ | 105 |
| RP | 5′‐AATCACGGGCGGACAGTAGACC‐3′ | ||
| IL‐6 | FP | 5′‐GACAAAGCCAGAGTCATTCA‐3′ | 229 |
| RP | 5′ ‐ACTAGGTTTGCCGAGTAGAC‐3′ | ||
| DAT | FP | 5′ ‐TGGCATCAGAGCATACCTAC‐3′ | 114 |
| RP | 5′‐ ATCAGCACTCCAAACCCAAC‐3′ | ||
| COX2 | FP | 5′ ‐GAACAACATTCCCTTCCTTCGG‐3′ | 83 |
| RP | 5′‐AAGTTGGTGGGCTGTCAATCA‐3′ | ||
| GAPDH | FP | 5′ ‐AAC CTG CCA AGT ATG ATG‐3′ | 119 |
| RP | 5′‐GGA GTT GCT GTT GAA GTC‐3′ | ||
FP, forward primer; RP, reverse primer; DAT, dopamine transporter; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
Immunocytochemistry
Immunocytochemistry was performed and modified according to Iida's study 22. Briefly, PC12 cells were fixed with PBS containing 4% (wt/vol) paraformaldehyde and 4% (wt/vol) sucrose for 15 min or with cold methanol for 10 min at −20°C and permeabilized with 0.25% (wt/vol) Triton X‐100 in PBS for 5 min. For the immunocytostaining with anti‐TH (1:200; Cell Signaling Technology, Inc., Danvers, MA, USA) and anti‐DAT (1:100; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) antibodies, methanol fixation was used. After the nonspecific reaction was blocked with PBS containing 10% (wt/vol) bovine serum albumin (BSA), the cells were incubated with the primary antibody in PBS containing 3% (wt/vol) BSA overnight, washed with PBS, and incubated with the second antibody (1:500) in PBS containing 3% (wt/vol) BSA for 1 h. After the samples were washed with PBS three times, they were embedded in 200 μL of Hoechst 33342 (10 μg/mL) for 5 min. The images were obtained using a Leica DMI 4000B microscope (Leica Corp., Lasertechnik, Heidelberg, Germany). Mean fluorescence intensity was calculated by Image Pro (Media Cybernetics, Silver Spring, MD, USA).
Protein Extraction, Subcellular Fractionation, and Western Blotting Analysis
After the 6‐OHDA or 6‐OHDA + simvastatin treatment, the cells were harvested using cell scrapers and washing in ice‐cold PBS, and then, they were lysed with two different ice‐cold lysis buffers 18. The supernatants were collected for protein determination using a BCA assay (Pierce, Inc., Rockford, IL, USA), and the protein was run in NuPage Bis‐Tris 10% gels (Invitrogen, Carlsbad, CA, USA) and transferred to PVDF membranes (Amersham Bioscience, Ltd., Buckinghamshire, UK). The membranes were blocked in 5% skim milk, 0.05% Tween 20, and Tris‐buffered saline (TBS) for 1 h. The PVDF membranes were incubated with primary antibodies: rabbit polyclonal Akt antibody (1:500), rabbit polyclonal phospho‐Akt antibody (1:500), rabbit polyclonal PI3K antibody (1:500), rabbit polyclonal phospho‐PI3K antibody (1:500), rabbit polyclonal caspase 3 antibody (1:500), and rabbit anti‐β‐actin (1:500) (all from Cell Signaling Technology, Inc.) overnight at 4°C. The next day, horseradish peroxidase‐conjugated secondary antibodies (Calbiochem, San Diego, CA, USA) were applied. Peroxidase‐conjugated streptavidin and substrate were used for detection. Negative controls were performed by omitting the primary antibodies. The images were analyzed using the NIH Image J software (Bethesda, MD, USA), and the mean optical density of control group was set as 100%.
Statistical Analysis
The data were expressed as the mean ± SEM. The data related to DAT, TH immunocytochemistry staining, real‐time PCR, and protein quantification with Western blot were analyzed using a one‐way ANOVA followed by Tukey's post hoc analysis (SPSS 15.0 program; SPSS, Chicago, IL, USA). Student's t‐test was employed to determine the statistical significance of apoptosis and cell viability. P‐values of <0.05 were regarded as statistically significant.
Results
Effect of 6‐OHDA and Simvastatin on the Levels of IL‐6, TNF‐α, and COX2
Real‐time PCR analysis showed that the mRNAs for proinflammatory cytokines IL‐6, COX2, and TNF‐α were induced 24 h after the 6‐OHDA treatment (Figure 1). The levels of IL‐6 increased 331% with respect to the control (**P < 0.001, 6‐OHDA vs. controls, n = 6–9, Figure 1A); this increase was prevented by simvastatin incubation (137% of control values; ††P < 0.01, 6‐OHDA vs. 6‐OHDA + sim, n = 6–9, Figure 1A). The changes in the levels of COX2 and TNF‐α were similar 24 h after the 6‐OHDA treatment, with the increases of 153% and 508%, respectively (**P < 0.001, 6‐OHDA vs. controls, n = 6–9, Figure 1B,C); this increase was also prevented by simvastatin (††P < 0.01, 6‐OHDA vs. 6‐OHDA + sim, n = 6–9, Figure 1B,C). However, the simvastatin + 6‐OHDA treatment produced significant increases in the expressions of IL‐6, TNF‐α, and COX2 (##P < 0.01, 6‐OHDA + sim vs. controls, n = 6–9, Figure 1A–C) compared with the controls. The treatment with simvastatin did not alter the expression of these cytokines IL‐6, COX2, and TNF‐α in PC12 cells.
Figure 1.
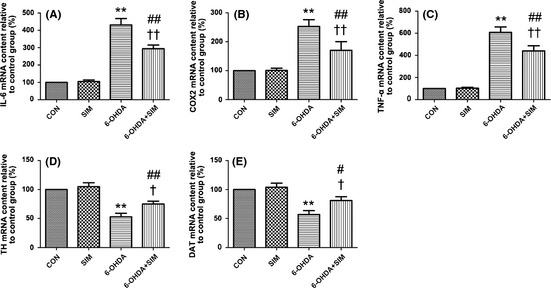
(A–C) Simvastatin reduced the 6‐OHDA‐mediated elevation in the expressions of IL‐6, TNF‐α, and COX2. 6‐OHDA pronouncedly increased the expression of IL‐6, TNF‐α, and COX2 compared with the controls (**P < 0.001, A–C). Simvastatin significantly abolished the elevation (††P < 0.01, A–C). The simvastatin + 6‐OHDA treatment produced significant increases in the expressions of IL‐6, TNF‐α, and COX2 (##P < 0.01, A–C) compared with the controls. D–E. Simvastatin attenuated the 6‐OHDA‐mediated decrease in TH and dopamine transporter (DAT) mRNA levels. Compared with the controls, the 6‐OHDA incubation pronouncedly decreased the TH and DAT mRNA (**P < 0.01, D–E); this decrease was significantly prevented following simvastatin treatment (†P < 0.05, D–E). All of the results are expressed as the mean ± SEM.
Effect of 6‐OHDA and Simvastatin on the Levels of TH and DAT
In this study, we employed relative quantitative method “real‐time PCR” to evaluate the TH and DAT mRNA levels, and the mRNA level of control group was considered as 100%. Real‐time PCR analysis showed that the expression of mRNAs for TH and DAT decreased 24 h after the 6‐OHDA treatment (Figure 1D,E). TH and DAT mRNA levels for different treated groups were compared with the controls and calculated as the percentage of the controls. The mRNA expression of TH was 53% (decreased 47%) and DAT 57% (decreased 43%) of the controls, respectively (**P < 0.01, 6‐OHDA vs. controls, n = 6–9, Figure 1D,E). These decreases were attenuated by simvastatin incubation (22% and 24% of control values for TH and DAT, †P < 0.05, 6‐OHDA vs. 6‐OHDA + sim, n = 6–9, Figure 1D,E). However, this reversal in the expression of TH and DAT mRNA was not restored to the control levels (##P < 0.01 for TH, 6‐OHDA + sim vs. controls; #P < 0.05 for DAT, 6‐OHDA + sim vs. controls, Figure 1D,E). Similarly, the pattern of alteration for DAT and TH immunocytochemistry staining is consistent with the real‐time PCR results. Immunocytochemistry analysis showed that the protein levels for TH and DAT decreased 51% and 34.1%, respectively, with respect to the control 24 h after the 6‐OHDA treatment (Figure 2); these decreases were attenuated by simvastatin incubation (66% and 80.2% of control values, respectively). Decreasing levels of DAT expression are associated with decreasing levels of TH and imply a reduction in dopamine reuptake. This reduction is most likely underlying a compensatory mechanism for the decreases in dopamine release caused by 6‐OHDA neurotoxicity in the PC12, which is consistent with previous studies 23, 24.
Figure 2.
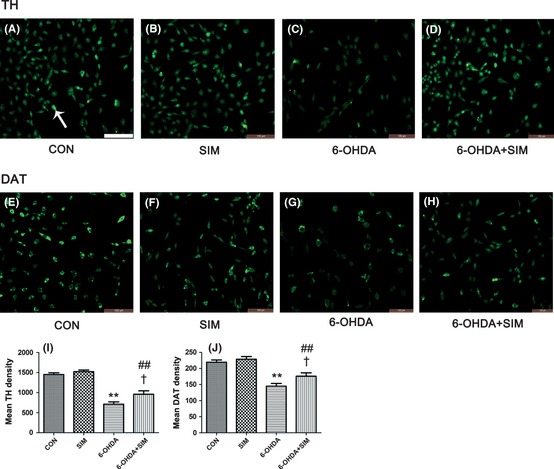
Effects of 6‐OHDA and simvastatin on TH and dopamine transporter (DAT) immunocytochemistry staining in PC12 cells. The arrow indicates a positive cell. (A–D) show the TH staining in the PC12 cells of the control, simvastatin, 6‐OHDA, and 6‐OHDA + simvastatin groups, respectively. (E–H) show the DAT staining in the control, simvastatin, 6‐OHDA, and 6‐OHDA + simvastatin groups, respectively. Bar = 100 μm. (I) represents the average number of TH‐positive PC12 cells of the control, simvastatin, 6‐OHDA, and 6‐OHDA + simvastatin groups, respectively. (J) represents the average number of DAT‐positive PC12 cells of each group. The pattern of alteration for DAT and TH immunocytochemistry staining is consistent with the real‐time PCR results.
Simvastatin Reversed the Reduced PI3K/Akt Phosphorylation and Abolished the Increased Caspase 3 Induced by 6‐OHDA
The protein levels of phosphorylated PI3K/Akt and caspase 3 were determined by Western blot analysis. The simvastatin treatment pronouncedly increased the levels of phosphorylated PI3K and Akt in PC12 compared with the controls (oo p<0.01, simvastatin vs. controls, n = 5, Figure 3A,B). The 6‐OHDA incubation decreased the protein levels of phosphorylated PI3K and Akt compared with the controls (**P < 0.01, 6‐OHDA vs. controls, n = 5, Figure 3A,B), but this reduction was significantly reversed following simvastatin treatment (††P < 0.01, 6‐OHDA vs. 6‐OHDA + sim, n = 5, Figure 3A,B). Although simvastatin treatment reversed the levels of phosphorylated PI3K/Akt, the protein levels did not reach the control levels. The phosphorylated PI3K/Akt levels in the simvastatin treated group were higher than the controls (#P < 0.05 for PI3K, ##P < 0.01 for AKT, 6‐OHDA + sim vs. controls, n = 5, Figure 3A,B).
Figure 3.

Effect of 6‐OHDA and simvastatin on the levels of PI3K, Akt, and caspase 3. Simvastatin significantly increased the protein levels of phosphorylated PI3K and Akt compared with the controls (oo p<0.01, A, B). 6‐OHDA incubation significantly decreased the levels of phosphorylated PI3K and Akt compared with the controls (**P < 0.01, A, B), but this reduction was reversed by simvastatin treatment (††P < 0.01, A, B). 6‐OHDA pronouncedly increased the protein levels of caspase 3 (**P < 0.01, C). This increase was partially inhibited by simvastatin treatment (††P < 0.01, C). However, coincubation with simvastatin and LY294002 partially attenuated the effect of simvastatin alone (&&P < 0.01, C). All of the results are expressed as the mean ± SEM.
Simvastatin had no effect on the protein levels of caspase 3 in PC12 cells without 6‐OHDA treatment. The 6‐OHDA incubation considerably increased the protein levels of caspase 3 (**P < 0.01, 6‐OHDA vs. controls, n = 5, Figure 3C), but this increase was significantly abolished after the simvastatin treatment (††P < 0.01, 6‐OHDA vs. 6‐OHDA + sim, n = 5, Figure 3C). However, compared with the controls, the protein levels of caspase 3 in the simvastatin treated group was higher (#P < 0.05, 6‐OHDA + sim vs. controls, n = 5, Figure 3C). Interestingly, when the 6‐OHDA‐treated PC12 cells were coincubated with simvastatin and LY294002, the expression of caspase 3 significantly increased compared with the group only treated with simvastatin (&&P < 0.01, 6‐OHDA + sim + LY vs. sim, n = 5, Figure 3C).
Effects of 6‐OHDA and Simvastatin on PC12 Cell Viability and Apoptosis
The CCK‐8 value in the 6‐OHDA‐treated group was significantly reduced compared with the controls (**P < 0.01, 6‐OHDA vs. controls, n = 9; Figure 4A), but simvastatin attenuated this reduction (††P < 0.01, 6‐OHDA vs. 6‐OHDA + sim; ##P < 0.01, 6‐OHDA + sim vs. controls, n = 9; Figure 4A). We examined the cultures exposed to 6‐OHDA for the presence of apoptotic nuclei in PC12 cells using flow cytometry. The apoptosis in the cells was further verified using flow cytometry analysis after being labeled with annexin V. The red dots in the lower left quarter of Figure 4C–F represent the normal cells; those in lower right quarter represent early apoptosis cells; those in higher right quarter represent later apoptosis cells; and those in higher left quarter represent necrosis cells. The percentage of apoptosis rate was calculated by the numbers of red dots in the higher right quarter divided by those in the lower left quarter. The result showed that 6‐OHDA induced significant apoptosis (4.22 ± 0.28% vs. 16.58 ± 1.53%, controls vs. 6‐OHDA, **P < 0.01, n = 5; Figure 4B,C,E), but simvastatin incubation attenuated this apoptotic cell death (16.58 ± 1.53% vs. 11.05 ± 1.33%, 6‐OHDA vs. 6‐OHDA + sim, ††P < 0.01, ##P < 0.01, 6‐OHDA + sim vs. controls, n = 5; Figure 4B,E,F).
Figure 4.
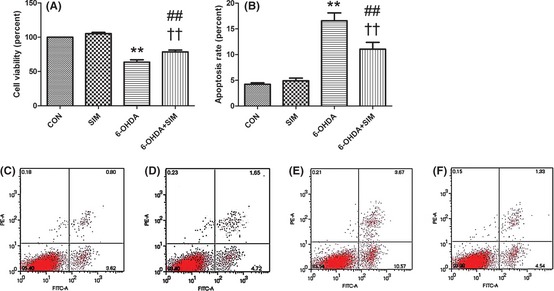
Simvastatin protected PC12 cells against 6‐OHDA neurotoxicity. (A) The CCK‐8 value in the 6‐OHDA‐treated group was significantly reduced compared with the controls (**P < 0.01), but simvastatin attenuated this reduction (††P < 0.01). (B–F) Simvastatin protected PC12 cells against 6‐OHDA‐induced apoptosis. (B) represents the apoptosis rate for different groups. (C–F) represent controls, simvastatin, 6‐OHDA‐treated, and 6‐OHDA + sim‐treated group, respectively. 6‐OHDA induced significant apoptosis (**P < 0.01, controls vs. 6‐OHDA; B–F), but simvastatin incubation attenuated this apoptotic cell death (6‐OHDA vs. 6‐OHDA + sim, ††P < 0.01; 6‐OHDA + sim vs. controls, ##P < 0.01, B–F). All of the results are expressed as the mean ± SEM.
Discussion
PC12, a cell line derived from a pheochromocytoma of the rat adrenal medulla, is a dopaminergic neuronal cell and can synthesize, store, secrete, and take up dopamine. It has been widely used for a cellular model system relevant to PD 25. In this study, we investigated whether simvastatin attenuates PC12 cell death using a 6‐OHDA‐lesioned PC12 model. The results showed that simvastatin reduced neuronal death and cell apoptosis, as measured by cell viability and flow cytometry analysis. Second, simvastatin restored the downregulation of TH and DAT levels in 6‐OHDA‐incubated PC12. In addition, simvastatin reduced inflammatory reactions as measured using the levels of IL‐6, COX2, and TNF‐α determined with real‐time PCR measurement. Finally, we found evidence that simvastatin reversed the downregulation of PI3K and Akt expression and attenuated the upregulation of caspase 3 after applying the neurotoxin, 6‐OHDA. To our knowledge, these findings are the first report that simvastatin exerts neuroprotection via antiapoptotic and antiinflammatory effects and the PI3K/Akt/caspase 3 pathway in PC12 cells, which are dopaminergic neurons.
To explore whether inflammatory mediators in PC12 cells changed after the 6‐OHDA and simvastatin treatments, we measured the expression of COX2, IL‐6, and TNF‐α. Our study showed increased expression of COX2, IL‐6, and TNF‐α in 6‐OHDA‐induced PC12 cells (Figure 1); simvastatin obviously prevented these inflammatory responses. These results are in agreement with previous studies, including our own 3, 4, 26, 27, 28, indicating that simvastatin‐mediated neuroprotection in PD occurs partially via the modulation of inflammatory actions and the reduction of oxidative damage. TH and DAT levels significantly decrease in PD patients, indicating an obvious dopaminergic neurodegeneration 29, 30, 31. To verify whether the increased inflammatory responses cause impaired dopaminergic activation, we measured the levels of TH and DAT in PC12 following 6‐OHDA incubation with and without simvastatin treatment. In this study, pronounced reductions of TH and DAT, at both the transcriptional and translational levels, were observed in 6‐OHDA‐incubated PC12, demonstrating an obvious dopaminergic PC12 cell death. Our study shows that simvastatin prevented 6‐OHDA‐induced dopaminergic neuronal apoptosis and restored the downregulation of TH and DAT levels in 6‐OHDA‐lesioned PC12 cells (Figures 1, 2 and 4). This finding strongly implies that simvastatin provides a neuroprotective effect in PD. This result is consistent with previous studies, including our own, which demonstrated that statins decreased the rate of dopaminergic degeneration and may be of therapeutic benefit to PD patients 3, 17, 28.
Ye's group found that the enhanced formation of an mGluRI‐Homer‐PIKE‐L complex resulting from activating group I metabotropic glutamate receptors (mGluRIs) led to the activation of PI3 kinase activity and subsequently prevented neuronal apoptosis 32. Furthermore, Ye found that PIKE‐L directly regulated glycine‐induced NMDA receptor activation and was essential for glycine‐induced GluA2‐associated PI3K activation 33. There is a close interaction between glutamatergic NMDA receptors of the brain and the monoamine dopaminergic systems 34, 35. Our previous study showed that simvastatin upregulated the expression of NMDA receptors in a naive and 6‐OHDA‐lesioned PD rat brain and that simvastatin abolished the upregulation of NMDA NR1 receptors in 6‐OHDA‐incubated PC12 cells 4, 36. Based on our previous studies and Ye's studies, we reasonably hypothesized that the simvastatin‐mediated antiapoptotic actions may partially result from the activation of the PI3K/Akt signaling pathway. To test this hypothesis, we directly investigated the PI3K/Akt pathway in the current study.
Statins modulate the PI3K/Akt pathway in various cell lines, such as osteosarcoma, endothelia cells, and neurons 2, 37, 38, 39, 40, 41, indicating that statins have antiangiogenic, antiinflammatory, neurogenesis, and antiapoptotic effects and attenuate cerebral vasospasm. As an example of non‐neuronal cells, Dimmeler et al. 42 found that in endothelial progenitor cells, statins regulated the hematopoietic progenitor cell differentiation via activation of the PI3K/Akt pathway and inhibition of apoptosis. Similar results have been observed in Kureishi's study, which showed that simvastatin rapidly activated the protein kinase Akt/PKB in endothelial cells 43. Consistent with these studies, the current study showed that incubating PC12 with simvastatin caused an obvious increase in the amount of phosphorylated PI3K (Figure 3A). Because the downstream target of PI3K is Akt, we also observed increased phosphorylated Akt expression (Figure 3B), suggesting that simvastatin may regulate the dopaminergic neuronal survival via PI3K/Akt phosphorylation. Interestingly, in 6‐OHDA‐lesioned PC12, the amount of phosphorylated PI3K and Akt significantly decreased compared with the controls and was accompanied by obvious cell death and apoptosis; simvastatin restored this downregulation of phosphorylated PI3K and Akt, with a reduction in cell death and apoptosis (Figure 3A,B). Because the alterations in PI3K/Akt have been reported to be associated with neuronal dysfunction and neurodegeneration 44, 45, 46, our results strongly suggest that the enhanced phosphorylation of PI3K and Akt that occurs after the dopaminergic PC12 cells are incubated with simvastatin appears to contribute to simvastatin‐mediated cell survival 2, 41, 47.
The apoptosis executioner caspase 3 is downstream of PI3K/Akt, and its increase is correlated with neuronal death in PD 13, 48, 49, 50. Signore et al. indicated that in primary dopamine neurons, erythropoietin prevented neuronal apoptosis via activating the PI3K/Akt pathway and subsequently diminishing caspase 3 activation 51. Similarly, in dopaminergic SH‐SY5Y cell lines, the caspase 3 activation was found to be negatively correlated with Akt phosphorylation 52. Consistent with Signore and Fang's studies, our result showed that in dopaminergic PC12 cells, 6‐OHDA significantly increased the expression of caspase 3, which was accompanied by a decrease in PI3K/Akt phosphorylation. This finding strongly suggests that the PI3K/Akt/caspase 3 signaling pathway plays important roles in 6‐OHDA‐lesioned PC12 cell death. The upregulation of caspase 3 and the downregulation of PI3K/Akt phosphorylation resulting from the 6‐OHDA incubation were reversed after simvastatin treatment. Our results combined with other studies 51, 52 demonstrate that simvastatin mediates neuroprotection in the 6‐OHDA‐lesioned PC12 model by an antiapoptotic effect, at least partially, via the PI3K/Akt/caspase 3 pathway. To further verify the role of this signaling pathway in simvastatin‐mediated neuroprotection, LY294002, an inhibitor of PI3K, was incubated with simvastatin in 6‐OHDA‐lesioned PC12. We found that the administration of LY294002 with simvastatin led to a partial inhibition of the increased phosphorylation of Akt and of the increased expression of caspase 3 (Figure 3C). Our observation further verifies that simvastatin‐mediated neuroprotection in 6‐OHDA‐lesioned PC12 is, at least partially, dependent on the PI3K/Akt/caspase 3 pathway.
In conclusion, our results clearly demonstrated that simvastatin provided robust neuroprotection against dopaminergic neurodegeneration, partially via antiinflammatory mechanisms such as inhibiting the inflammatory mediators COX2, IL‐6, and TNF‐α. This study indicates that simvastatin may have “therapeutic antiapoptosis” utility for PD via enhanced phosphorylation of the PI3K/Akt signaling pathway and impaired caspase 3 levels in 6‐OHDA‐lesioned PC12 cells, at least partially. The PI3K/Akt/caspase 3 pathway may play important roles in regulating dopaminergic neuronal survival during simvastatin‐mediated neuroprotection in PD; the modulation of this PI3K/Akt/caspase 3 pathway may provide a therapeutic benefit in other CNS diseases as well. Although it is not a complete phenocopy of the human disease, this 6‐OHDA‐lesioned PC12 model provides a useful means to study the pathomechanisms of clinical PD patients. A better understanding of the roles and relationships among statins, the PI3K/Akt/caspase 3 pathway, and the dopaminergic system may lead to new applications for the statin family in the modulation of psycho‐neurodegenerative disorders such as PD.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the 973 Project (2011CB510000), 985 Project grant (82000‐3281901), and National Natural Science Foundations of China (grant no: 81071031, 81271427) to Q.W.
The first two authors equally contributed to this work.
References
- 1. Dolga AM, Nijholt IM, Ostroveanu A, Ten Bosch Q, Luiten PG, Eisel UL. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J Alzheimers Dis 2008;13:111–122. [DOI] [PubMed] [Google Scholar]
- 2. Sugawara T, Ayer R, Jadhav V, Chen W, Tsubokawa T, Zhang JH. Simvastatin attenuation of cerebral vasospasm after subarachnoid hemorrhage in rats via increased phosphorylation of Akt and endothelial nitric oxide synthase. J Neurosci Res 2008;86:3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Q, Yan J, Chen X, et al. Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp Neurol 2011;230:27–34. [DOI] [PubMed] [Google Scholar]
- 4. Yan J, Xu Y, Zhu C, et al. Simvastatin prevents dopaminergic neurodegeneration in experimental parkinsonian models: The association with anti‐inflammatory responses. PLoS ONE 2011;6:e20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arboleda G, Morales LC, Benítez B, Arboleda H. Regulation of ceramide‐induced neuronal death: Cell metabolism meets neurodegeneration. Brain Res Rev 2009;59:333–346. [DOI] [PubMed] [Google Scholar]
- 6. Seaborn T, Masmoudi‐Kouli O, Fournier A, Vaudry H, Vaudry D. Protective effects of pituitary adenylate cyclase‐activating polypeptide (PACAP) against apoptosis. Curr Pharm Des 2011;17:204–214. [DOI] [PubMed] [Google Scholar]
- 7. Krafft PR, Altay O, Rolland WB, et al. α7 nicotinic acetylcholine receptor agonism confers neuroprotection through GSK‐3β inhibition in a mouse model of intracerebral hemorrhage. Stroke 2012;43:844–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mendelev N, Witherspoon S, Li PA. Overexpression of human selenoprotein H in neuronal cells ameliorates ultraviolet irradiation‐induced damage by modulating cell signaling pathways. Exp Neurol 2009;220:328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vazquez de la Torre A, Junyent F, Folch J, et al. Study of the pathways involved in apoptosis induced by PI3K inhibition in cerebellar granule neurons. Neurochem Int 2011;59:159–167. [DOI] [PubMed] [Google Scholar]
- 10. Dedoni S, Olianas MC, Onali P. Interferon‐β induces apoptosis in human SH‐SY5Y neuroblastoma cells through activation of JAK‐STAT signaling and down‐regulation of PI3K/Akt pathway. J Neurochem 2010;115:1421–1433. [DOI] [PubMed] [Google Scholar]
- 11. Chan CB, Liu X, Pradoldej S, et al. Phosphoinositide 3‐kinase enhancer regulates neuronal dendritogenesis and survival in neocortex. J Neurosci 2011;31:8083–8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahn JY, Rong R, Liu X, Ye Y. PIKE/nuclear PI 3‐kinase signaling mediates the antiapoptotic actions of NGF in the nucleus. EMBO J 2004;23:3995–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang G, Pan J, Chen SD. Kinases and kinase signaling pathways: Potential therapeutic targets in Parkinson's disease. Prog Neurobiol 2012;98:207–221. [DOI] [PubMed] [Google Scholar]
- 14. Zhang S, Wang XJ, Tian LP, et al. CD200‐CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson's disease. J Neuroinflammation 2011;8:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gui YX, Wang XY, Kang WY, et al. Extracellular signal‐regulated kinase is involved in alpha‐synuclein‐induced mitochondrial dynamic disorders by regulating dynamin‐like protein 1. Neurobiol Aging 2012;33:2841–2854. [DOI] [PubMed] [Google Scholar]
- 16. Selley ML. Simvastatin prevents 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res 2005;1037:1–6. [DOI] [PubMed] [Google Scholar]
- 17. Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE, Pahan K. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci 2009;29:13543–13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodriguez‐Blanco J, Martín V, Herrera F, García‐Santos G, Antolín I, Rodriguez C. Intracellular signaling pathways involved in post‐mitotic dopaminergic PC12 cell death induced by 6‐hydroxydopamine. J Neurochem 2008;107:127–140. [DOI] [PubMed] [Google Scholar]
- 19. Park HH, Lee KY, Kim SH, Lee YJ, Koh SH. L‐DOPA‐induced neurotoxicity is reduced by the activation of the PI3K signaling pathway. Toxicology 2009;265:80–86. [DOI] [PubMed] [Google Scholar]
- 20. Wasserman JK, Schlichter LC. Minocycline protects the blood‐brain barrier and reduces edema following intracerebral hemorrhage in the rat. Exp Neurol 2007;207:227–237. [DOI] [PubMed] [Google Scholar]
- 21. Naranjo‐Suárez S, Castellanos MC, Alvarez‐Tejado M, Vara A, Landázuri MO, del Peso L. Down‐regulation of hypoxia‐inducible factor‐2 in PC12 cells by nerve growth factor stimulation. J Biol Chem 2003;278:31895–31901. [DOI] [PubMed] [Google Scholar]
- 22. Iida J, Ishizaki H, Okamoto‐Tanaka M, et al. Synaptic scaffolding molecule alpha is a scaffold to mediate N‐methyl‐d‐aspartate receptor‐dependent RhoA activation in dendrites. Mol Cell Biol 2007;27:4388–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bannon MJ, Poosch MS, Xia Y, Goebel DJ, Cassin B, Kapatos G. Dopamine transporter mRNA content in human substantia nigra decreases precipitously with age. Proc Natl Acad Sci U S A 1992;89:7095–7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Afonso‐Oramas D, Cruz‐Muros I, Barroso‐Chinea P, et al. The dopamine transporter is differentially regulated after dopaminergic lesion. Neurobiol Dis 2010;40:518–530. [DOI] [PubMed] [Google Scholar]
- 25. Pan T, Zhu J, Hwu WJ, Jankovic J. The role of alpha‐synuclein in melanin synthesis in melanoma and dopaminergic neuronal cells. PLoS ONE 2012;7:e45183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Willey JZ, Elkind MS. 3‐Hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitors in the treatment of central nervous system diseases. Arch Neurol 2010;67:1062–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van der Most PJ, Dolga AM, Nijholt IM, Luiten PG, Eisel UL. Statins: Mechanisms of neuroprotection. Prog Neurobiol 2009;88:64–75. [DOI] [PubMed] [Google Scholar]
- 28. Hernández‐Romero MC, Argüelles S, Villarán RF, et al. Simvastatin prevents the inflammatory process and the dopaminergic degeneration induced by the intranigral injection of lipopolysaccharide. J Neurochem 2008;105:445–459. [DOI] [PubMed] [Google Scholar]
- 29. Jellinger KA. Interaction between α‐synuclein and tau in Parkinson's disease comment on Wills et al.: Elevated tauopathy and α‐synuclein pathology in postmortem Parkinson's disease brains with and without dementia. Exp Neurol 2011;227:13–18. [DOI] [PubMed] [Google Scholar]
- 30. Bezard E, Gross CE, Fournier MC, Dovero S, Bloch B, Jaber M. Absence of MPTP‐induced neuronal death in mice lacking the dopamine transporter. Exp Neurol 1999;155:268–273. [DOI] [PubMed] [Google Scholar]
- 31. Brooks DJ, Frey KA, Marek KL, et al. Assessment of neuroimaging techniques as biomarkers of the progression of Parkinson's disease. Exp Neurol 2003;184(Suppl 1):S68–S79. [DOI] [PubMed] [Google Scholar]
- 32. Rong R, Ahn JY, Huang H, et al. PI3 kinase enhancer‐Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci 2003;6:1153–1161. [DOI] [PubMed] [Google Scholar]
- 33. Chan CB, Chen Y, Liu X, et al. PIKE‐mediated PI3‐kinase activity is required for AMPA receptor surface expression. EMBO J 2011;30:4274–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Bartolomeis A, Fiore G, Iasevoli F. Dopamine‐glutamate interaction and antipsychotics mechanism of action: Implication for new pharmacological strategies in psychosis. Curr Pharm Des 2005;11:3561–3594. [DOI] [PubMed] [Google Scholar]
- 35. Pickel VM, Colago EE, Mania I, Molosh AI, Rainnie DG. Dopamine D1 receptors co‐distribute with N‐methyl‐D‐ aspartic acid type‐1 subunits and modulate synaptically‐evoked N‐methyl‐D‐aspartic acid currents in rat basolateral amygdala. Neuroscience 2006;142:671–690. [DOI] [PubMed] [Google Scholar]
- 36. Wang Q, Zengin A, Deng C, et al. High dose of simvastatin induces hyperlocomotive and anxiolytic‐like activities: The association with the up‐regulation of NMDA receptor binding in the rat brain. Exp Neurol 2009;216:132–138. [DOI] [PubMed] [Google Scholar]
- 37. Cerezo‐Guisado MI, Garcia‐Marin LJ, Lorenzo MJ, Bragado MJ. Lovastatin inhibits the growth and survival pathway of phosphoinositide 3‐kinase/protein kinase B in immortalized rat brain neuroblasts. J Neurochem 2005;94:1277–1287. [DOI] [PubMed] [Google Scholar]
- 38. Zhao TT, Trinh D, Addison CL, Dimitroulakos J. Lovastatin inhibits VEGFR and AKT activation: Synergistic cytotoxicity in combination with VEGFR inhibitors. PLoS ONE 2010;5:e12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsubaki M, Yamazoe Y, Yanae M, et al. Blockade of the Ras/MEK/ERK and Ras/PI3K/Akt pathways by statins reduces the expression of bFGF, HGF, and TGF‐β as angiogenic factors in mouse osteosarcoma. Cytokine 2011;54:100–107. [DOI] [PubMed] [Google Scholar]
- 40. Wu H, Lu D, Jiang H, et al. Simvastatin‐mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma 2008;25:130–139. [DOI] [PubMed] [Google Scholar]
- 41. Sugawara T, Ayer R, Jadhav V, Chen W, Tsubokawa T, Zhang JH. Mechanisms of statin treatment in cerebral vasospasm. Acta Neurochir Suppl 2011;110(Pt 2):9–11. [DOI] [PubMed] [Google Scholar]
- 42. Dimmeler S, Aicher A, Vasa M, et al. HMG‐CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3‐kinase/Akt pathway. J Clin Invest 2001;108:391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kureishi Y, Luo Z, Shiojima I, et al. The HMG‐CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 2000;6:1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Colin E, Régulier E, Perrin V, et al. Akt is altered in an animal model of Huntington's disease and in patients. Eur J Neurosci 2005;21:1478–1488. [DOI] [PubMed] [Google Scholar]
- 45. van der Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: Learning to survive. Prog Neurobiol 2006;79:205–221. [DOI] [PubMed] [Google Scholar]
- 46. Sun ZK, Yang HQ, Pan J, et al. Protective effects of erythropoietin on tau phosphorylation induced by beta‐amyloid. J Neurosci Res 2008;86:3018–3027. [DOI] [PubMed] [Google Scholar]
- 47. Sugawara T, Jadhav V, Ayer R, Zhang J. Simvastatin attenuates cerebral vasospasm and improves outcomes by upregulation of PI3K/Akt pathway in a rat model of subarachnoid hemorrhage. Acta Neurochir Suppl 2008;102:391–394. [DOI] [PubMed] [Google Scholar]
- 48. Liot G, Gabriel C, Cacquevel M, et al. Neurotrophin‐3‐induced PI‐3 kinase/Akt signaling rescues cortical neurons from apoptosis. Exp Neurol 2004;187:38–46. [DOI] [PubMed] [Google Scholar]
- 49. Takuma K, Baba A, Matsuda T. Astrocyte apoptosis: Implications for neuroprotection. Prog Neurobiol 2004;72:111–127. [DOI] [PubMed] [Google Scholar]
- 50. Li Q, Ren J. Chronic alcohol consumption alters mammalian target of rapamycin (mTOR), reduces ribosomal p70s6 kinase and p4E‐BP1 levels in mouse cerebral cortex. Exp Neurol 2007;204:840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Signore AP, Weng Z, Hastings T, et al. Erythropoietin protects against 6‐hydroxydopamine‐induced dopaminergic cell death. J Neurochem 2006;96:428–443. [DOI] [PubMed] [Google Scholar]
- 52. Fang CX, Yang X, Sreejayan N, Ren J. Acetaldehyde promotes rapamycin‐dependent activation of p70(S6K) and glucose uptake despite inhibition of Akt and mTOR in dopaminergic SH‐SY5Y human neuroblastoma cells. Exp Neurol 2007;203:196–204. [DOI] [PubMed] [Google Scholar]