

SUMMARY
Presynaptic dopamine (DA) transmission has been measured in schizophrenia using different paradigms aimed at providing estimates of the integrity or the activity of the presynaptic dopaminergic neuron. Researchers have measured: (1) DA synthesis capacity with [18F]DOPA, a measure of the activity of dopa decarboxylase, (2) DA release with studies measuring the impact of a DA releasing stimulant challenge on the binding of a D2 receptor radiotracer, (3) D2 baseline occupancy by DA, a measure of baseline intrasynaptic DA, assessed by the changes in binding of D2 radiotracer induced by DA depletion, and (4) the DA and the vesicular monoamine transporters, to assess the integrity of presynaptic terminals. The relationship between DA release and D2 receptor occupancy at baseline by DA has also been assessed in the same patients. Overall, these different imaging modalities have converged to show a dysregulation of presynaptic dopaminergic activity in schizophrenia, leading to excessive DA release in the striatum, particularly in the projection to the associative striatum, an area of integration between cognitive and limbic cortical inputs. Excessive striatal presynaptic DA is linked to the emergence of acute psychotic symptoms and to their response to treatment in schizophrenia. Understanding the etiology of this dysregulation and its consequences on the rest of the circuitry is important for future drug development.
Keywords: PET, Presynaptic dopamine, Schizophrenia, SPECT
Introduction
The dopamine (DA) hypothesis of schizophrenia was first formulated by Rossum et al. [1] based on the observation that antipsychotics may block DA receptors. It received support through the years from multiple lines of evidence. Initially, support derived from the findings that DA receptors are activated by psychostimulants, that non‐reserpine neuroleptics are DA receptor antagonists and that DA plays an important role in the extrapyramidal motor system [2]. The correlation between clinical doses of antipsychotic drugs and their potency to block DA D2 receptors [3, 4], as well as the observations of psychotogenic effects of DA enhancing drugs [for review, see Refs. 5 and 6], provided additional support for the link between excessive dopamine activity and the psychotic symptoms of schizophrenia. Subsequently, a deficiency in mesocortical DA function was suggested as a possible integrative explanation for many clinical observations regarding negative symptoms and cognitive deficits, leading to a reformulation of this hypothesis [7] to incorporate an imbalance between subcortical excess and cortical deficit. Finally, with the advent of molecular imaging, various aspects of presynaptic DA transmission have been measured to test this hypothesis.
This review will focus on indices of presynaptic transmission, including studies of enzymatic activity within the dopaminergic terminals, transmitter release, and DA transporter (DAT) availability, after a brief review of the methodologies for the imaging paradigms used.
One index of presynaptic DA transmission is the activity of enzymes within the dopaminergic synthesis pathway. DA synthesis occurs within the DA neurons. Tyrosine is transported into the cell via amino acid carriers in the blood–brain barrier and cell membranes. Once in the intracellular space it is hydroxylated to L‐3,4‐dihydroxyphenylalanine (L‐DOPA) by tyrosine hydroxylase (TH). L‐DOPA is then decarboxylated by aromatic acid decarboxylase (AADC) to DA (Figure 1). The first positron emission tomography (PET) tracer developed for studies of DA synthesis and/or storage in the human brain was [18F]DOPA [8, 9, 10]. [18F]DOPA, a radioactive analogue of L‐DOPA, the precursor of DA, is taken up by presynaptic monoaminergic neurons and is metabolized by AADC to 18F‐Dopamine, which is trapped and stored within vesicles in the nerve terminals. [18F]DOPA uptake, quantified as the influx constant Ki, can be used as a measure of AADC activity and vesicular storage capacity [11]. High values for [18F]DOPA Ki are observed in areas of dense DA nerve terminal innervation (e.g., the striatum), [18F]DOPA has been extensively used to probe the functional integrity of striatal dopaminergic neurons, particularly in Parkinson's disease and other movement disorders [12, 13, 14, 15] where [18F]DOPA uptake correlates with the number of surviving nigrostriatal cell numbers in both human studies and animal models [16, 17]. Although AADC is not the rate‐limiting step in the synthetic pathway for DA, it has been suggested that AADC activity may influence the rate of DA synthesis [18].
Figure 1.
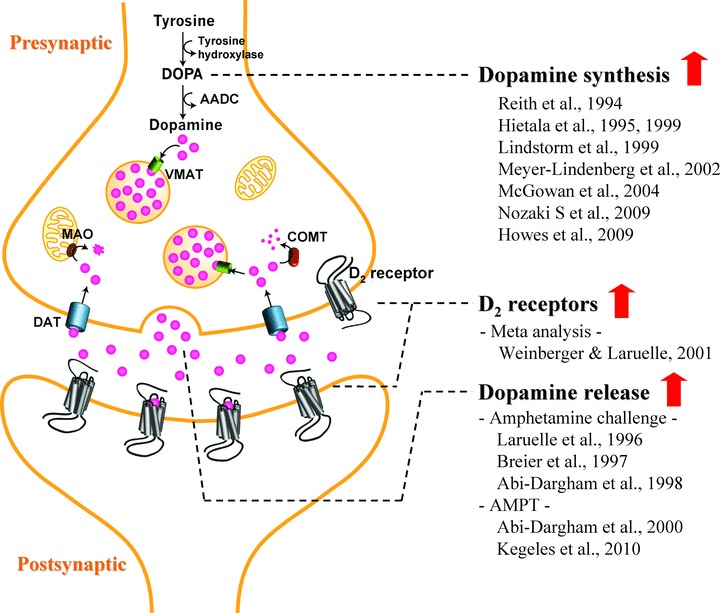
Summary of dopamine alterations in schizophrenia. Abbreviations: AADC, aromatic acid decarboxylase; AMPT, α‐methyl‐para‐tyrosine; COMT, catechol‐O‐methyltransferase; DAT, dopamine transporter; DOPA, 3,4‐dihydroxyphenylalanine; MAO, monoamine oxidase; VMAT, vesicular monoamine transporter.
Acute treatment with D2 receptor antagonists increases AADC activity in animal studies [19, 20, 21]. Conversely, acute treatment with the D2 agonist apomorphine decreases 11C‐DOPA influx in monkeys [22]. This is consistent with the electrophysiological data showing that acute administration of antipsychotics increases DA neuron firing, whereas chronic administration decreases the number of spontaneously active DA neurons in the rat substantia nigra due to depolarization block [23]. These together suggest that [18F]DOPA imaging provides a measure of the activity of AADC in presynaptic DA terminals because the activity of the enzyme changes in a similar direction to the DA firing in response to pharmacological interventions.
Another index of presynaptic activity is transmitter release, which can be indirectly measured by assessing changes in binding of D2 radiotracers after pharmacological manipulation with DA releasing or depleting agents. Administration of the DA releasing agent amphetamine produces a reduction in in vivo binding of D2 radiotracers such as [11C]raclopride and [123I]IBZM; this reduction in binding is a well‐validated measure of pharmacologically stimulated DA release. Conversely, administration of the dopamine depleting agent α‐methyl‐para‐tyrosine (AMPT) unmasks D2 receptors occupied by endogenous DA and results in a subsequent increase in in vivo binding of [11C]raclopride or [123I]IBZM (for review, see Ref. 24). The increased binding can be interpreted as measurement of baseline occupancy of D2 receptors by DA.
Some studies have attempted to validate these imaging paradigms used to probe neurotransmitter levels and have suggested that the changes in in vivo binding associated with these paradigms reflect changes in synaptic DA levels rather than general extrasynaptic levels. This interpretation is based on the lack of correlation between the magnitude of DA increase measured with microdialysis and the magnitude of displacement of D2 radiotracers across different challenge drugs [24]. Drugs that block the DAT, such as amphetamine and GBR12909, increase microdialysis‐measured DA more than DA‐releasing drugs that do not block the DAT, such as ketanserin and nicotine, yet the range of D2 radiotracer displacement is similar for all, suggesting that this measure is affected only by changes in intrasynaptic DA. Thus, the imaging measures of DA in the stimulated (obtained with the amphetamine challenge) or baseline condition (obtained with the AMPT depletion paradigm) are assumed to reflect measures of synaptic release of DA. Furthermore, synaptic levels are also assumed to be mostly influenced by phasic release, related to burst activity. However, contribution by basal tonic release, driven by single spike activity, cannot be excluded. More definitive validation will derive from combining in vivo imaging with ex vivo measurements in rodent models of altered DA activity to clarify the exact molecular equivalence of the imaging measures.
Studies in Patients with Schizophrenia
Most of the information regarding presynaptic DA transmission in schizophrenia derives from imaging studies, although some post mortem work has also shed light on this topic. One postmortem study of TH immunolabeling in patients with schizophrenia showed decreased TH labeled axons in layers 3 and 6 of the entorhinal cortex and in layer 6 of the prefrontal cortex [25, 26], supporting the view that cortical DA is deficient in schizophrenia. This finding was unrelated to premortem neuroleptic exposure. On the other hand, Benes et al. (1997) observed no significant changes in TH positive varicosities in the dorsolateral prefrontal cortex (DLPFC). In the anterior cingulate region (layer 2), these authors observed a significant shift in the distribution of TH varicosities from large neurons to small neurons. Another presynaptic index, DAT density, has been examined in the striatum of patients with schizophrenia, and does not appear to differ from that of controls [27, 28, 29, 30, 31, 32]. Postmortem studies of DA levels or DA metabolites have not yielded a conclusive set of findings.
Imaging DA Synthesis
Using PET and either [β‐11C]L‐DOPA or [18F]DOPA, most studies examining this index of DA synthesis capacity have found increased striatal DOPA uptake in patients with schizophrenia compared to controls [33, 34, 35, 36, 37, 38, 39, 40] (Figure 1). Of note, all investigations of patients experiencing an active psychotic episode at the time of scanning reported increased striatal DOPA uptake among patients [34, 35, 36]. Further, one study reported an association between positive symptoms and [18F]DOPA uptake in the putamen [35, for review and discussion, see Ref. 41]. Thus, overall this body of work provides evidence that as a group, individuals with schizophrenia are characterized by elevated presynaptic DA synthesis/storage, and that this abnormality may be most evident in patients with active psychosis [see Ref. 41]. In addition, Meyer‐Lindenberg et al. [39] found that reduced activation in the DLPFC during an executive functioning task, as measured by PET with [15O]H2O, was associated with increased striatal DOPA uptake in patients but not controls, providing additional support for the relation between prefrontal dysfunction and potentiated striatal DA activity in the illness.
Howes et al. [37] recently reported that striatal [18F]DOPA uptake was significantly increased among individuals experiencing prodromal symptoms of schizophrenia compared to controls, and that DOPA uptake correlated significantly with severity of prodromal symptoms. Of note, these differences were observed in the associative, but not in the limbic or sensorimotor, striatum. Importantly, this study provides evidence that increased DA synthesis activity may occur before the full syndrome of schizophrenia emerges among those displaying prodromal symptoms of the illness. Furthermore, a recent study reported increased striatal [18F]DOPA uptake among nonpsychotic first‐degree relatives of schizophrenia patients, suggesting that elevated presynaptic DA synthesis may be associated with the genetic vulnerability to the illness [42].
Imaging Stimulated DA Release
With imaging, synaptic DA release in the striatum, measured as the change in D2 radiotracer binding potential after administration of the DA‐releasing drug amphetamine, has been shown to be increased in schizophrenia [43, 44, 45] (Figure 1). Moreover, transient amphetamine‐induced exacerbation of positive symptoms observed in a subgroup of schizophrenia patients was associated with the degree of amphetamine‐induced reduction in striatal [123I]IBZM binding potential. In addition, patients experiencing an acute phase of illness, including those at first episode who were antipsychotic naïve, had a significantly greater degree of amphetamine‐induced striatal DA release compared to those in remission. The remitted group did not differ significantly from controls on this index of DA release, although they were numerically higher than controls [46]. The findings of these studies marked an important advance in schizophrenia research, as they more directly than any prior results supported the proposal that excessive DA transmission at striatal D2 receptors is involved in the experiencing of psychotic symptoms, and highlighted a role for a dysregulated, hyperresponsive subcortical DA system that fluctuates in its degree of dysregulation during different phases of the illness. Since this time, Abi‐Dargham and colleagues [47] have shown that as a group, patients with schizotypal personality disorder (SPD) are also characterized by a larger reduction in striatal [123I]IBZM binding potential following amphetamine compared to controls. The degree of amphetamine‐induced [123I]IBZM displacement observed in SPD was similar to that previously observed in remitted patients with schizophrenia but significantly lower than that seen in acutely ill schizophrenia patients [47]. Taken together, these results suggest that a more modest increase in DA release may characterize individuals at risk for psychosis, and that a larger degree of DA dysregulation characterizes those reaching the threshold of active psychosis. This is consistent with the data in first degree relatives showing elevation in DA synthesis capacity, [42], supporting the notion that dysregulation of striatal DA may have two components, a low‐grade increase present in patients between episodes and in at risk states, such as spectrum disorders or relatives, and a higher level of dysregulation present in acute psychotic episodes, including first episode of the illness.
Baseline DA Release
This paradigm is based on acutely depleting synaptic DA by orally administering the TH inhibitor AMPT over 48 h [48]. Using this technique with single photon emission computed tomography (SPECT) and the D2 radiotracer [123I]IBZM, Abi‐Dargham et al. [49] found that the increase in D2 receptor availability after acute DA depletion was significantly larger in patients with schizophrenia compared to controls, indicating increased occupancy of striatal D2 receptors by DA at baseline in schizophrenia. Of note, all of the patients of this study were experiencing an episode of illness exacerbation. It was further noted that AMPT exposure led to a significant reduction in severity of positive symptoms among patients, and that higher occupancy of striatal D2 receptors by DA was significantly associated with a greater reduction in positive symptoms after 6 weeks of antipsychotic treatment [49]. These results provided evidence that schizophrenia is characterized by excessive striatal D2 receptor stimulation by DA and that the degree of this dysregulation is associated with the degree of therapeutic response to antipsychotic medication observed in patients.
With the availability of high‐resolution PET scanners and the development of methods to examine DA receptor parameters in the various subregions of the striatum, it became possible to probe aspects of DA transmission separately for the functional subdivisions of the striatum [50, 51]. Using these methods with PET and [11C]raclopride, patients with schizophrenia displayed a significantly larger increase in D2 receptor availability after DA depletion compared to controls in the associative striatum (AST), but not in the limbic or sensorimotor striatal regions [52]. In particular, group differences were observed in the anterior (pre‐commissural) dorsal caudate. This finding has challenged the notion that in schizophrenia, alterations in subcortical DA transmission are most prominently localized to the ventral, rather than dorsal, striatum and stimulated thinking regarding the potential role of the AST in the illness. This striatal region receives prominent input from the DLPFC, and is thought to play an important role in regulating the circuitry from various cortical regions [52].
Correlation of Stimulated and Baseline DA Release
One study involving antipsychotic‐naïve patients with schizophrenia who participated in both the amphetamine‐challenge and DA depletion paradigms indicated that indices of stimulated and baseline DA release are strongly associated in patients but not controls [53]. These results suggest that these measures are indexing the same pathological process in patients. They also highlight the variability in this DA transmission alteration across patients, which is an observation that informs our understanding of schizophrenia pathophysiology and may be important in the design of targeted treatment interventions [53].
Imaging DA Transporters
DAT are located on the presynaptic membrane of DA terminals and regulate phasic DA transmission at the synapse by rapidly removing DA from the synaptic cleft through reuptake [54]. Because striatal DAT are located exclusively on DA terminals, its measurement has been used to index the density of dopaminergic terminals or innervation into the striatum. Numerous studies have now examined striatal DAT density in antipsychotic‐naïve or ‐free patients with schizophrenia using either PET or SPECT, and have failed to find significant differences between patients and controls [55, 56, 57, 58, 59]. These results suggest that the increased presynaptic striatal DA activity in schizophrenia indicated by other imaging parameters is not secondary to dysfunctional DAT or an elevation in the density of dopaminergic terminals. However, two studies using a dual‐isotope SPECT technique with first‐episode, antipsychotic‐naïve patients have reported a positive association between DAT and D2 receptor availability in patients but not controls [57, 60]. Further, using [99mTc]TRODAT‐1 and SPECT, researchers recently observed a significant increase in DAT availability in a subgroup of patients with predominantly positive symptoms compared to controls [60]. Pending further replication, most of the evidence at this point shows that the DAT is unaffected in schizophrenia. A similar picture has emerged for the vesicular monoamine transporter (VMAT) [61]. These together suggest integrity of striatal dopaminergic terminals.
Summary
Increased striatal DA function is one of the best established research findings in schizophrenia, observed with different imaging modalities and replicated by numerous independent labs. It seems most relevant to positive symptoms, as the severity of these correlates with magnitude of DA release [46, 62], and their response to treatment is also a function of the degree of dysregulation of striatal DA function [49]. Indeed, the most relevant site of therapeutic efficacy of antipsychotics is striatal, as measured by the strength of relationship between D2 occupancy in the striatum by antipsychotics and the response of positive symptoms [63, 64]. On the other hand, response of negative symptoms is unrelated to striatal occupancy, and response of positive symptoms does not seem to be coupled to degree of extrastriatal occupancy. These observations suggest that striatal dopaminergic function determines severity and treatment response of psychosis.
Nonetheless, striatal dopaminergic dysfunction can be reciprocally linked to a cortical dopaminergic dysfunction. Studies have documented that cortical DA affects subcortical DA dynamics. More recently, however, the reciprocal effect has been shown in a mouse model of striatal D2 overexpression that demonstrates cognitive impairment in tasks of working memory and behavioral flexibility, as well as altered prefrontal cortical DA levels, rates of DA turnover, and activation of prefrontal D1 receptors [65]. The mechanism by which this may occur is yet to be determined, but this finding suggests that flow of information in cortico‐striato‐thalamo‐cortical loops can be impaired at the striatal level, producing a bottom up impairment in cortical function.
The etiology of the striatal dopaminergic dysregulation is unknown, and most likely related to alterations in other systems that regulate DA transmission. Thus, DA dysregulation may not be a primary dysfunction in schizophrenia, but rather may represent a final common pathway to psychosis. In addition, DA dysregulation itself may play a role in other pathogenic effects, as the striatum represents an essential integrative node, by receiving input from the hippocampus and the cortex, two areas of pathology in schizophrenia, by modulating DA midbrain neurons, and by projecting indirectly back to the cortex. Pathological changes in hippocampal activity lead to dopaminergic dysregulation in animal models. Hippocampal pathology is also present in patients with schizophrenia [66, 67, 68], suggesting that the hippocampus may play an essential role in the genesis of the dopaminergic pathology in schizophrenia. This could be mediated by a disinhibition of dopaminergic cells in the ventral tegmental area via the pallidal projection, leading to an increased sensitivity to excitatory inputs and an increase in phasic firing [69]. Testing in patients for an association between hippocampal hyperactivity and dopaminergic phasic dysregulation is needed to confirm the relevance of these preclinical observations to the pathophysiology of the disorder. The striatal DA dysregulation in turn may lead to an impairment of cortical inputs, imbalancing the cortico–limbic integration that takes place within the striatum and explaining the different domains of pathology that exist within the schizophrenia spectrum.
Conclusion
Molecular imaging studies have provided robust support for the long‐held hypothesis positing subcortical DA hyperactivity in schizophrenia. Importantly, these findings converge in pointing to a hyperresponsive subcortical DA system that fluctuates in its degree of dysregulation across phases of the illness; further, DA dysregulation appears to be present to some degree during the prodromal period of the illness, before threshold psychotic symptoms emerge. Increased presynaptic DA functioning may be particularly evident in the dorsal caudate of the anterior striatum. Finally, a more direct characterization of cortical DA as well as extrastriatal DA transmission in schizophrenia is needed, as the evidence for cortical deficit is largely by inference. These studies are now feasible with the development of high affinity benzamide radiotracers that allow visualization of D2 receptors in low density areas of the brain and are sensitive to acute changes in DA tone.
Conflict of Interest
The authors have no conflict of interest.
References
- 1. van Rossum JM. The significance of dopamine receptor blockade for the mechanism of action of neuroleptic drugs. Arch Int Pharmacodyn Ther 1966;160:492–494. [PubMed] [Google Scholar]
- 2. Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3‐methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol 1963;20:140–144. [DOI] [PubMed] [Google Scholar]
- 3. Seeman P, Chau‐Wong M, Tedesco J, Wong K. Brain receptors for antipsychotic drugs and dopamine: Direct binding assays. Proc Natl Acad Sci USA 1975;72:4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 1976;19:481–483. [DOI] [PubMed] [Google Scholar]
- 5. Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology 1987;91:415–433. [DOI] [PubMed] [Google Scholar]
- 6. Angrist B, van Kammen DP. CNS stimulants as a tool in the study of schizophrenia. Trends Neurosci 1984;7:388–390. [Google Scholar]
- 7. Weinberger DR. Implications of the normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 1987;44:660–669. [DOI] [PubMed] [Google Scholar]
- 8. Volkow ND, Fowler JS, Gatley SJ, Logan J, Wang GJ, Ding YS, Dewey S. PET evaluation of the dopamine system of the human brain. J Nucl Med 1996;37:1242–1256. [PubMed] [Google Scholar]
- 9. Garnett ES, Firnau G, Chan PK, Sood S, Belbeck LW. [18F]fluoro‐dopa, an analogue of dopa, and its use in direct external measurements of storage, degradation, and turnover of intracerebral dopamine. Proc Natl Acad Sci USA 1978;75:464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garnett ES, Firnau G, Nahmias C. Dopamine visualized in the basal ganglia of living man. Nature 1983;305:137–138. [DOI] [PubMed] [Google Scholar]
- 11. Brown WD, Taylor MD, Roberts AD, et al FluoroDOPA PET shows the nondopaminergic as well as dopaminergic destinations of levodopa. Neurology 1999;53:1212–8. [DOI] [PubMed] [Google Scholar]
- 12. Brooks DJ. Monitoring neuroprotection and restorative therapies in Parkinson's disease with PET. J Neural Transm Suppl 2000;60:125–137. [DOI] [PubMed] [Google Scholar]
- 13. Brooks DJ, Ibanez V, Sawle GV, et al Differing patterns of striatal 18F‐dopa uptake in Parkinson's disease, multiple system atrophy, and progressive supranuclear palsy. Ann Neurol 1990;28:547–555. [DOI] [PubMed] [Google Scholar]
- 14. Takikawa S, Dhawan V, Chaly T, et al Input functions for 6‐[fluorine‐18]fluorodopa quantitation in parkinsonism: Comparative studies and clinical correlations. J Nucl Med 1994;35:955–963. [PubMed] [Google Scholar]
- 15. Morrish PK, Sawle GV, Brooks DJ. Regional changes in [18F]dopa metabolism in the striatum in Parkinson's disease. Brain 1996;119(Pt 6):2097–2103. [DOI] [PubMed] [Google Scholar]
- 16. Pate BD, Kawamata T, Yamada T, et al Correlation of striatal fluorodopa uptake in the MPTP monkey with dopaminergic indices. Ann Neurol 1993;34:331–338. [DOI] [PubMed] [Google Scholar]
- 17. Snow BJ, Tooyama I, McGeer EG, et al Human positron emission tomographic [18F]fluorodopa studies correlate with dopamine cell counts and levels. Ann Neurol 1993;34:324–330. [DOI] [PubMed] [Google Scholar]
- 18. Cumming P, Gjedde A. Compartmental analysis of dopa decarboxylation in living brain from dynamic positron emission tomograms. Synapse 1998;29:37–61. [DOI] [PubMed] [Google Scholar]
- 19. Zhu MY, Juorio AV, Paterson IA, Boulton AA. Regulation of striatal aromatic L‐amino acid decarboxylase: Effects of blockade or activation of dopamine receptors. Eur J Pharmacol 1993;238:157–164. [DOI] [PubMed] [Google Scholar]
- 20. Cho S, Neff NH, Hadjiconstantinou M. Regulation of tyrosine hydroxylase and aromatic L‐amino acid decarboxylase by dopaminergic drugs. Eur J Pharmacol 1997;323:149–157. [DOI] [PubMed] [Google Scholar]
- 21. Danielsen EH, Smith D, Hermansen F, Gjedde A, Cumming P. Acute neuroleptic stimulates DOPA decarboxylase in porcine brain in vivo . Synapse 2001;41:172–175. [DOI] [PubMed] [Google Scholar]
- 22. Torstenson R, Hartvig P, Langstrom B, Bastami S, Antoni G, Tedroff J. Effect of apomorphine infusion on dopamine synthesis rate relates to dopaminergic tone. Neuropharmacology 1998;37:989–995. [DOI] [PubMed] [Google Scholar]
- 23. Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: A hypothesis for the etiology of schizophrenia. Neuroscience 1991;41:1–24. [DOI] [PubMed] [Google Scholar]
- 24. Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: A critical review. J Cereb Blood Flow Metab 2000;20:423–451. [DOI] [PubMed] [Google Scholar]
- 25. Akil M, Edgar CL, Pierri JN, Casali S, Lewis DA. Decreased density of tyrosine hydroxylase‐immunoreactive axons in the entorhinal cortex of schizophrenic subjects. Biol Psychiatry 2000;47:361–370. [DOI] [PubMed] [Google Scholar]
- 26. Akil M, Pierri JN, Whitehead RE, et al Lamina‐specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry 1999;156:1580–1589. [DOI] [PubMed] [Google Scholar]
- 27. Joyce JN, Lexow N, Bird E, Winokur A. Organization of dopamine D1 and D2 receptors in human striatum: Receptor autoradiographic studies in Huntington's disease and schizophrenia. Synapse 1988;2:546–557. [DOI] [PubMed] [Google Scholar]
- 28. Knable MB, Hyde TM, Herman MM, Carter JM, Bigelow L, Kleinman JE. Quantitative autoradiography of dopamine‐D1 receptors, D2 receptors, and dopamine uptake sites in postmortem striatal specimens from schizophrenic patients. Biol Psychiatry 1994;36:827–835. [DOI] [PubMed] [Google Scholar]
- 29. Hirai M, Kitamura N, Hashimoto T, et al [3H]GBR‐12935 binding sites in human striatal membranes: Binding characteristics and changes in parkinsonians and schizophrenics. Jpn J Pharmacol 1988;47:237–243. [DOI] [PubMed] [Google Scholar]
- 30. Czudek C, Reynolds GP. [3H] GBR 12935 binding to the dopamine uptake site in post‐mortem brain tissue in schizophrenia. J Neural Transm 1989;77:227–230. [DOI] [PubMed] [Google Scholar]
- 31. Pearce RK, Seeman P, Jellinger K, Tourtellotte WW. Dopamine uptake sites and dopamine receptors in Parkinson's disease and schizophrenia. Eur Neurol 1990;30(Suppl 1):9–14. [DOI] [PubMed] [Google Scholar]
- 32. Chinaglia G, Alvarez FJ, Probst A, Palacios JM. Mesostriatal and mesolimbic dopamine uptake binding sites are reduced in Parkinson's disease and progressive supranuclear palsy: A quantitative autoradiographic study using [3H]mazindol. Neuroscience 1992;49:317–327. [DOI] [PubMed] [Google Scholar]
- 33. Reith J, Benkelfat C, Sherwin A, et al Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci USA 1994;91:11651–11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hietala J, Syvalahti E, Vuorio K, et al Presynaptic dopamine function in striatum of neuroleptic‐naive schizophrenic patients. Lancet 1995;346:1130–1131. [DOI] [PubMed] [Google Scholar]
- 35. Hietala J, Syvalahti E, Vilkman H, et al Depressive symptoms and presynaptic dopamine function in neuroleptic‐naive schizophrenia. Schizophr Res 1999;35:41–50. [DOI] [PubMed] [Google Scholar]
- 36. Lindstrom LH, Gefvert O, Hagberg G, et al Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L‐(beta‐11C) DOPA and PET. Biol Psychiatry 1999;46:681–688. [DOI] [PubMed] [Google Scholar]
- 37. Howes OD, Montgomery AJ, Asselin MC, et al Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry 2009;66:13–20. [DOI] [PubMed] [Google Scholar]
- 38. McGowan S, Lawrence AD, Sales T, Quested D, Grasby P. Presynaptic dopaminergic dysfunction in schizophrenia: A positron emission tomographic [18F]fluorodopa study. Arch Gen Psychiatry 2004;61:134–142. [DOI] [PubMed] [Google Scholar]
- 39. Meyer‐Lindenberg A, Miletich RS, Kohn PD, et al Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci 2002;5:267–271. [DOI] [PubMed] [Google Scholar]
- 40. Nozaki S, Kato M, Takano H, et al Regional dopamine synthesis in patients with schizophrenia using L‐[beta‐11C]DOPA PET. Schizophr Res 2009;108:78–84. [DOI] [PubMed] [Google Scholar]
- 41. Howes OD, Montgomery AJ, Asselin MC, Murray RM, Grasby PM, McGuire PK. Molecular imaging studies of the striatal dopaminergic system in psychosis and predictions for the prodromal phase of psychosis. Br J Psychiatry Suppl 2007;51:s13–s18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huttunen J, Heinimaa M, Svirskis T, et al Striatal dopamine synthesis in first‐degree relatives of patients with schizophrenia. Biol Psychiatry 2008;63:114–117. [DOI] [PubMed] [Google Scholar]
- 43. Laruelle M, Abi‐Dargham A, van Dyck CH, et al Single photon emission computerized tomography imaging of amphetamine‐induced dopamine release in drug‐free schizophrenic subjects. Proc Natl Acad Sci USA 1996;93:9235–9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abi‐Dargham A, Gil R, Krystal J, et al Increased striatal dopamine transmission in schizophrenia: Confirmation in a second cohort. Am J Psychiatry 1998;155:761–767. [DOI] [PubMed] [Google Scholar]
- 45. Breier A, Su TP, Saunders R, et al Schizophrenia is associated with elevated amphetamine‐induced synaptic dopamine concentrations: Evidence from a novel positron emission tomography method. Proc Natl Acad Sci USA 1997;94:2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laruelle M, Abi‐Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: Relationship to illness phases. Biol Psychiatry 1999;46:56–72. [DOI] [PubMed] [Google Scholar]
- 47. Abi‐Dargham A, Kegeles L, Zea‐Ponce Y, et al Amphetamine‐induced dopamine release in patients with schizotypal personality disorders studied by SPECT and [123I]IBZM. Biol Psychiatry 2004;55:1001–1006. [DOI] [PubMed] [Google Scholar]
- 48. Laruelle M, DSouza CD, Baldwin RM, et al Imaging D‐2 receptor occupancy by endogenous dopamine in humans. Neuropsychopharmacology 1997;17:162–174. [DOI] [PubMed] [Google Scholar]
- 49. Abi‐Dargham A, Rodenhiser J, Printz D, et al Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA 2000;97:8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martinez D, Slifstein M, Broft A, et al Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: Amphetamine‐induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab 2003;23:285–300. [DOI] [PubMed] [Google Scholar]
- 51. Mawlawi O, Martinez D, Slifstein M, et al Imaging human mesolimbic dopamine transmission with positron emission tomography: I. Accuracy and precision of D2 receptor parameter measurements in ventral striatum. J Cereb Blood Flow Metab 2001;21:1034–1057. [DOI] [PubMed] [Google Scholar]
- 52. Kegeles L, Abi‐Dargham A, Frankle W, et al Increased synaptic dopamine in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry 2010;67:231–239. [DOI] [PubMed] [Google Scholar]
- 53. Abi‐Dargham A, Giessen EV, Slifstein M, Kegeles LS, Laruelle M. Baseline and amphetamine‐stimulated dopamine activity are related in drug‐naive schizophrenic subjects. Biol Psychiatry 2009;65:1091–1093. [DOI] [PubMed] [Google Scholar]
- 54. Grace AA. Dopamine In: Davis KL, Charney DS, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology‐The Fifth Generation of Progress. Philadelphia : Lippincott, Williams, & Wilkins, 2002;119–132. [Google Scholar]
- 55. Lavalaye J, Linszen DH, Booij J, et al Dopamine transporter density in young patients with schizophrenia assessed with [123]FP‐CIT SPECT. Schizophr Res 2001;47:59–67. [DOI] [PubMed] [Google Scholar]
- 56. Hsiao MC, Lin KJ, Liu CY, Tzen KY, Yen TC. Dopamine transporter change in drug‐naive schizophrenia: An imaging study with 99mTc‐TRODAT‐1. Schizoph Res 2003;65:39–46. [DOI] [PubMed] [Google Scholar]
- 57. Yang YK, Yu L, Yeh TL, Chiu NT, Chen PS, Lee IH. Associated alterations of striatal dopamine D2/D3 receptor and transporter binding in drug‐naive patients with schizophrenia: A dual‐isotope SPECT study. Am J Psychiatry 2004;161:1496–1498. [DOI] [PubMed] [Google Scholar]
- 58. Laakso A, Vilkman H, Alakare B, et al Striatal dopamine transporter binding in neuroleptic‐naive patients with schizophrenia studied with positron emission tomography. Am J Psychiatry 2000;157:269–271. [DOI] [PubMed] [Google Scholar]
- 59. Schmitt GJ, Frodl T, Dresel S, et al Striatal dopamine transporter availability is associated with the productive psychotic state in first episode, drug‐naive schizophrenic patients. Eur Arch Psychiatry Clin Neurosci 2006;256:115–121. [DOI] [PubMed] [Google Scholar]
- 60. Schmitt GJ, la Fougere C, Dresel S, et al Dual‐isotope SPECT imaging of striatal dopamine: First episode, drug naive schizophrenic patients. Schizophr Res 2008;101:133–141. [DOI] [PubMed] [Google Scholar]
- 61. Taylor SF, Koeppe RA, Tandon R, Zubieta JK, Frey KA. In vivo measurement of the vesicular monoamine transporter in schizophrenia. Neuropsychopharmacology 2000;23:667–675. [DOI] [PubMed] [Google Scholar]
- 62. Laruelle M, Abi‐Dargham A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J Psychopharmacol 1999;13:358–371. [DOI] [PubMed] [Google Scholar]
- 63. Kegeles LS, Slifstein M, Frankle WG, et al Dose‐occupancy study of striatal and extrastriatal dopamine D(2) receptors by aripiprazole in schizophrenia with PET and [(18)F]Fallypride. Neuropsychopharmacology 2008;33:3111–3125. [DOI] [PubMed] [Google Scholar]
- 64. Agid O, Mamo D, Ginovart N, et al Striatal vs extrastriatal dopamine D(2) receptors in antipsychotic response – a double‐blind PET study in schizophrenia. Neuropsychopharmacology 2006;32:1209–1215. [DOI] [PubMed] [Google Scholar]
- 65. Kellendonk C, Simpson EH, Polan HJ, et al Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron 2006;49:603–615. [DOI] [PubMed] [Google Scholar]
- 66. Heckers S, Rauch SL, Goff D, et al Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat Neurosci 1998;1:318–323. [DOI] [PubMed] [Google Scholar]
- 67. Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: Contrasts in schizophrenia. Hippocampus 2001;11:543–550. [DOI] [PubMed] [Google Scholar]
- 68. Meyer‐Lindenberg AS, Olsen RK, Kohn PD, et al Regionally specific disturbance of dorsolateral prefrontal‐hippocampal functional connectivity in schizophrenia. Arch Gen Psychiatry 2005;62:379–386. [DOI] [PubMed] [Google Scholar]
- 69. Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci 2007;27:11424–11430. [DOI] [PMC free article] [PubMed] [Google Scholar]