

Abstract
Dopaminergic neurons in the substantia nigra pars compacta and ventral tegmental area of the midbrain form the nigrostriatal and mesocorticolimbic dopaminergic pathways that, respectively, project to dorsal and ventral striatum (including prefrontal cortex). These midbrain dopaminergic nuclei and their respective forebrain and cortical target areas are well established as serving a critical role in mediating voluntary motor control, as evidenced in Parkinson's disease, and incentive‐motivated behaviors and cognitive functions, as exhibited in drug addiction and schizophrenia, respectively. Although it cannot be disputed that excitatory and inhibitory amino acid‐based neurotransmitters, such as glutamate and GABA, play a vital role in modulating activity of midbrain dopaminergic neurons, recent evidence suggests that acetylcholine may be as important in regulating dopaminergic transmission. Midbrain dopaminergic cell tonic and phasic activity is closely dependent upon projections from hindbrain pedunculopontine and the laterodorsal tegmental nuclei, which comprises the only known cholinergic inputs to these neurons. In close coordination with glutamatergic and GABAergic activity, these excitatory cholinergic projections activate nicotinic and muscarinic acetylcholine receptors within the substantia nigra and ventral tegmental area to modulate dopamine transmission in the dorsal/ventral striatum and prefrontal cortex. Additionally, acetylcholine‐containing interneurons in the striatum also constitute an important neural substrate to provide further cholinergic modulation of forebrain striatal dopaminergic transmission. In this review, we examine neurological and psychopathological conditions associated with dysfunctions in the interaction of acetylcholine and dopamine and conventional and new pharmacological approaches to treat these disorders.
Keywords: Addiction, Dopaminergic system, Mesopontine cholinergic system, Parkinson's disease, Schizophrenia
Introduction
Two major dopaminergic (DA) systems in the central nervous system are comprised of the nigrostriatal system with dopamine‐containing cell bodies in the substantia nigra pars compacta (SNc) of the midbrain that project predominantly to the caudate‐putamen in primates (referred to as the dorsal striatum in lower vertebrates, such as the rat), and the mesocorticolimbic system with dopamine‐containing cell bodies originating in the ventral tegmental area (VTA) of the midbrain and projecting to limbic structures, such as the nucleus accumbens (NAc), hippocampus, amygdala, and medial prefrontal cortex (mPFC) [1, 2, 3, 4]. Progressive degeneration of nigrostriatal DA neurons is known to be the major neuropathological characteristic of Parkinson's disease [5], while psychopathological conditions such as drug addiction [6] and schizophrenia [7] have been associated with aberrant alterations in the activity of DA neurons in the mesocorticolimbic system. Normal basal and phasic activity of these DA neuronal systems appear to be critically dependent upon the only known cholinergic projections to DA cells in the midbrain that arise from several acetylcholine (ACh)‐rich nuclei within the pons region of the hindbrain, particularly the laterodorsal (LDT) and pedunculopontine (PPT) tegmental nuclei. Furthermore, the cellular make‐up of the striatum includes ACh‐containing interneurons, which also constitute an additional neural substrate for dopamine–ACh interactions and serve as modulators of the striatal output necessary for production of fluid voluntary movements.
Cholinergic activation of neuronal elements in the CNS is mediated by two types of receptors, metabotropic muscarinic ACh receptors (mAChRs) and ionotropic nicotinic ACh receptors (nAChRs). Activation of mAChRs results in a slower, but potentially more sustained response [8]. Molecular cloning has identified five mAChR subtypes, including M1‐like (M1, M3, and M5) receptors, selectively linked to Gq proteins which decrease potassium conductance upon activation, and M2‐like (M2 and M4) receptors, coupled to Gi proteins that increase potassium conductance upon activation [9, 10, 11]. Thus, the activation of mAChRs has interestingly been shown to result in both excitation and inhibition of DA activity in the basal ganglia [12, 13], suggesting a complex modulatory role of mAChRs involving numerous subtypes at multiple levels of the nigrostriatal and mesocorticolimbic DA systems. Conversely, operating as ligand‐gated ion channels, nAChRs consist of different combinations of five α and β protein subunits, but also include a homomeric α7 subtype. The roles of nAChRs in the brain are diverse, but in general activation of nAChRs, which are commonly located presynaptically, results in a rapid increase in sodium and/or calcium channel conductance, which stimulates central neuronal activity and neurotransmitter release [14].
Preclinical and clinical evidence indicates that DA and cholinergic systems operate in a dynamic balance, with disruptions often leading to neurological, psychiatric, and drug‐addictive disorders. Restoration of the balance between these two and other neurotransmitter systems has been and remains the main approach for treating such disorders and ameliorating the associated symptoms. Thus, the present review discusses the cholinergic influence on DA systems via the modulatory roles of mAChRs and nAChRs, as well as, pharmaceutical approaches aimed at targeting cholinergic–DA interactions to alleviate the symptoms of neuropsychiatric disorders.
Nigrostriatal Dopaminergic System
Stimulation of dopamine‐containing cells in the SNc elicits fast excitatory responses in striatal neurons [15], while 6‐OHDA lesions of these SNc cells reduce basal levels of extracellular striatal dopamine concentrations [16]. In addition, dopamine content in the striatum is positively correlated with the degree of DA cell loss in the SNc, with a 95% loss in striatal dopamine content leading to the greatest reduction in basal dopamine release [17]. Dopamine transmission in the striatum is most commonly associated with voluntary movements [e.g.,18] and has been linked to the selection and initiation of contextually appropriate motor patterns [19, 20]. Reduced dopamine in the striatum is associated with motor symptoms of Parkinson's disease such as difficulty initiating and terminating movements, gait impairments, and muscular rigidity [21, 22, 23], whereas excess dopamine release in the striatum can lead to functional motor dysfunctions such as stereotypy, with the degree of intensity of stereotypical behaviors being positively correlated with striatal dopamine release [24].
The neurons of the striatum project to the internal segment of the globus pallidus (GPi) and the substantia nigra pars reticulata (SNr) via two pathways, a direct (monosynaptic) connection and an indirect pathway through the external segment of the globus pallidus (GPe) and subthalamic nucleus (STN) (Figure 1). Striatal neurons in the direct pathway utilize D1 receptors, whereas those in the indirect pathway utilize D2 receptors [25]. Activation of D1 receptors stimulates adenylate cyclase activity, thus activating the GABAergic substance P‐containing medium spiny output neurons, whereas activation of D2 receptors inhibits adenylate cyclase, thus inhibiting GABAergic enkephalin‐containing output neurons [26]. Thus, the direct (via D1) and indirect (via D2) pathways have opposing actions, but may reach the same net outcome of activating motor regions of the cortex. For example, activation of D1 receptors in the direct striatal GABAergic pathway leads to inhibition of GPi/SNr inhibitory GABAergic projections to the thalamus, subsequently increasing activity in the thalamus that, in turn, excites motor areas in the cortex [26, 27, 28]. Alternatively, activation of D2 receptors in the indirect pathway inhibits striatal inhibitory GABAergic neurons, resulting in disinhibition (excitation) of GPe inhibitory GABAergic neurons that project to the STN. As a consequence, decreased activity of the STN excitatory glutamatergic neurons that innervate the GPi/SNr, GPe, and SNc leads to less inhibitory drive of these nuclei to the thalamus, thereby indirectly increasing excitation of the motor areas in the cortex [26, 27, 28]. In sum, the net effect of striatal dopamine release from the nigrostriatal pathway increases thalamocortical activity via direct or indirect reduction of GPi/SNr activity consequently facilitating voluntary movements.
Figure 1.
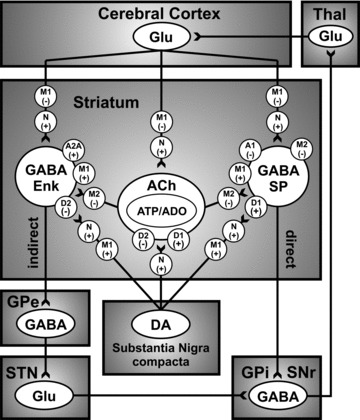
Simplified thalamocortical basal ganglia circuitry depicting the innervation of the striatum by the nigrostriatal DA system and mediation of excitatory and inhibitory influence, via dopamine D1‐like (D1/D5) and D2‐like (D2/D3/D4) receptors and acetylcholine (ACh) muscarinic M1‐like (M1/3/5) and M2‐like (M2/4) receptors, of direct and indirect GABAergic striatal output pathways to the globus pallidus internus/substantia nigra reticulata (GPi/SNr), respectively. Note that the direct GABAergic striatal output pathway contains both M1 and M2 receptors, whereas the indirect pathway has primarily M1 receptors [29]. Presynaptic nicotinic receptors (N), of subtypesincluding α4β2*, α6β2*, and α4α6β2β3*, may also modulate striatal dopamine release, as well as glutamate release via presynaptic α7 nicotinic and M1 (likely M3) muscarinic receptors [30]. Nigrostriatal dopamine may also interact with striatal cholinergic interneurons, via dopamine D1‐ and D2‐like receptors, to mediate the co‐release of adenosine triphosphate (ATP) and adenosine (ADO) to act on A1 and A2A receptors on direct and indirect GABAergic striatal output pathways, respectively. GABA: γ‐aminobutyric acid; Glu: glutamate; GPe: globus pallidus externus; Enk: enkephalin; SP: substance P; STN: subthalamic nucleus; Thal: thalamus. “+” and “–” depicts the excitatory and inhibitory influence of each receptor subtype on the activity of the dopaminergic.
Mesopontine Cholinergic Modulation of the Nigrostriatal Dopaminergic System
Modulation of Striatal Dopamine Release by Activation of Cholinergic Receptors in the PPT
The mesopontine cholinergic system arises from neurons located in the LDT and PPT of the hindbrain. The PPT consists of a diffuse group of ACh‐containing, as well as noncholinergic, neurons that surround ascending fibers of the superior cerebellar peduncle and stretch from the dorsal pons to the ventral midbrain. The LDT is a smaller cluster of 70% cholinergic neurons that lies in the floor of the fourth ventricle, just medial to the most caudal PPT neurons. Although the neuronal populations of the LDT and PPT may appear anatomically to form a continuous column, the two nuclei are considered distinct due to their differences in efferent and afferent innervations [31, 32, 33, 34]. Most significantly, these mesopontine nuclei represent the only known cholinergic projections to midbrain DA cells [35]. The cholinergic neurons of the PPT also project to basal ganglia nuclei, such as the STN and GPi (entopeduncular nucleus in rats) [31, 34, 36, 37, 38], suggesting a functional involvement of the PPT in sensorimotor‐related activities of the striatum.
In vivo electrochemistry studies have shown that electrical and chemical stimulation of the PPT enhances dopamine release in the rat striatum [39, 40]. As such, mAChRs located within the PPT have been implicated in the inhibition of striatal dopamine release. Intra‐PPT infusions of the nonselective mAChR antagonist scopolamine enhances striatal dopamine release and dopamine‐dependent behaviors such as locomotion and stereotypy; both of which can be blocked by the cholinergic agonist carbachol infused into the PPT [41, 42]. These mAChRs are most likely autoreceptors of the M2 family as M2 receptors have been localized presynaptically on PPT cholinergic neurons [43, 44], and intra‐PPT infusion of the M2/4 selective mAChR antagonist methoctramine has been shown to enhance striatal dopamine release [40]. Activation of M2‐like mAChRs in the PPT results in hyperpolarization of mesopontine cholinergic cells [45, 46] and a net decrease in excitation to SNc DA cells resulting in lowered extracellular levels of striatal dopamine [39]. Therefore, M2‐like mAChRs are thought to function as cholinergic autoreceptors involved in feedback inhibition at the level of PPT cholinergic cells, serving as regulators of information received by the PPT.
Modulation of Striatal Dopamine Release by Activation of Projections from the PPT
Activation of PPT cholinergic neurons could evoke striatal dopamine release through both direct and indirect neuronal circuits, as given in Figure 2. Excitatory cholinergic inputs, including glutamatergic inputs, from the PPT directly project to dopamine‐containing cell bodies in the SNc [31, 34, 39, 47, 48]. Electrical and chemical (GABA‐A antagonist bicuculline) activation of the PPT elicits burst‐firing in SNc DA cells [49, 50], a physiological effect that is critically dependant on an intact functional cholinergic input. Indeed, Kitai et al. [51] have shown that SNc DA cells in slice preparations only exhibit burst firing in the presence of bath applied cholinergic agonists. Previous work from our lab using in vivo chronoamperometry has shown that PPT stimulation elicits striatal dopamine release, in which an initial rapid increase in dopamine release was blocked by a combination of intra‐SNc infusions of nAChR and ionotropic glutamatergic receptor (iGluR) antagonists and a delayed, prolonged increase in striatal dopamine release was selectively blocked by mAChR antagonists infused into the SNc [39]. However, in addition to direct cholinergic activational inputs to SNc DA cells, the PPT may also activate DA cells to enhance striatal dopamine release via indirect PPT cholinergic/glutamatergic inputs to excitatory glutamatergic neurons in the STN that, in turn, innervate SNc DA cells (Figure 2) [36, 52].
Figure 2.
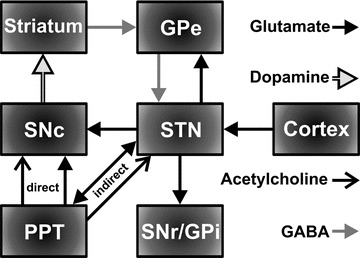
Simplified basal ganglia circuitry depicting direct innervation of dopaminergic cells in the substantia nigra compacta (SNc) by the cholinergic and glutamatergic neurons in the pedunculopontine tegmental nucleus (PPT) and indirect innervation of the SNc via glutamatergic neurons in the STN. SNr: substantia nigra reticulata; GPi: globus pallidus internus; GPe: globus pallidus externus. The direct GABAergic pathway from striatum to SNr/GPi has been omitted for clarity.
Although the neurochemical functional nature of the PPT‐STN‐SNc circuitry has received less attention compared to the PPT‐SNc projection, electrical stimulation of the PPT has been shown to activate STN neurons via cholinergic and glutamatergic projections [38, 53, 54]. Thus, in order to determine the role of the STN in the release of striatal dopamine following PPT stimulation, as well as the overall contribution of STN mAChR and nAChR activation, we used in vivo fixed potential amperometry to record dopamine release in the striatum of urethane anesthetized male mice while stimulating the PPT before and during a 1.0 μL intra‐STN infusion of the local anaesthetic lidocaine (4%) or a combination of the mAChR antagonist scopolamine (1.0 μg) and the nAChR antagonist mecamylamine (1.0 μg).
As shown in Figure 3, these studies involved stereotactic placement of a concentric bipolar stimulating electrode unilaterally into the PPT and a 31 g stainless‐steel guide infusion cannula implanted ipsilateral into the STN, with the tip of the guide cannula positioned 1 mm above site. To complete the electrometer circuit, an Ag/AgCl reference and stainless‐steel auxiliary electrode combination was placed in surface contact with contralateral cortical tissue approximately 2.0 mm posterior to bregma, and a carbon fiber recording microelectrode with an active recording surface of 250 μm (length) by 10 μm (o.d.) was then implanted ipsilateral into the dorsomedial striatum.
Figure 3.
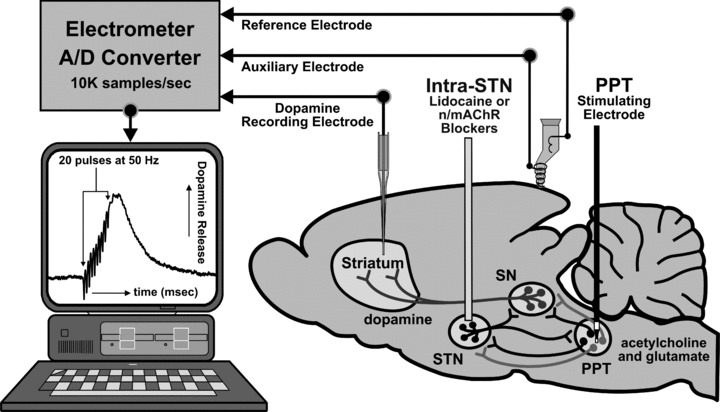
Schematic diagram of the mouse brain illustrating a typical setup for in vivo fixed potential amperometric recording of striatal dopamine release in mice (C57BL/6J mice, Jackson Labs.) evoked by electrical stimulation (20 pulses of 0.5 ms duration cathodal monophasic constant current pulses at 50 Hz applied every 30 sec at 800 μA). A carbon fiber microelectrode (Thornel Type P, Union Carbide) is positioned in the dorsomedial striatum, an Ag/AgCl reference and stainless‐steel auxiliary electrode combination is placed in contact with contralateral cortical tissue, a concentric bipolar stimulating electrode (SNE‐100; Rhodes Medical Co.) is implanted into the pedunculopontine tegmental nucleus (PPT), and a stainless‐steel guide cannula is placed in the STN for microinfusions of the axonal blocker lidocaine, nAChR, or mAChR antagonists. Coordinates (in mm) for the PPT, STN guide cannula, and striatum were (AP −4.7, −2.0, +1.4 from bregma, ML +1.25, +1.6, +1.4 from midline, and DV −2.7, −3.0, −2.5 from dura), respectively. In other studies examining STN stimulation and intra‐substantia nigra (SN) microinfusions described later, coordinates corresponded to (in mm) (AP −2.0, −3.1 from bregma, ML +1.6, +1.35 from midline, and DV −4.0, −2.8 from dura), respectively [55]. Black, light gray, and dark gray neuronal pathways correspond to glutamatergic, cholinergic, and dopaminergic neurons.
Fixed potential amperometry coupled with carbon fiber microelectrodes has been confirmed as a valid technique for real‐time monitoring of striatal dopamine oxidation current evoked by brief electrical stimulation of ascending nigrostriatal DA projections to the striatum and afferent inputs to the SNc [39, 56, 57]. Mean changes in dopamine oxidation current, corresponding to stimulation‐evoked dopamine release, were converted to mean dopamine concentrations (μM) by post‐experiment in vitro calibration of the carbon fiber electrode in solutions of dopamine (2–10 μM) using a flow injection system [58]. For each animal, changes in stimulation‐evoked dopamine concentration after infusion were expressed as mean percent changes with respect to pre‐infusion baseline responses (100%).
Using the same procedures for microinfusions as in our previously published studies [40, 57], intra‐STN infusions of 1.0 μL of sterile phosphate‐buffered saline (PBS, pH ∼ 7.4) in a separate group of mice did not significantly alter striatal dopamine release evoked by PPT stimulation (n = 4; −4.34 ± 3.3%, P > 0.05; Figure 4). Thus, the observed drug effects were not attributed to nonspecific effects of the infusion procedure. In contrast, compared to pre‐infusion baseline levels, intra‐STN lidocaine infusions significantly attenuated PPT stimulation‐evoked striatal dopamine release (n = 4; −49.95 ± 7.7%, P < 0.05; Figure 4), as well as intra‐STN infusion of a combination of the mAChR antagonist scopolamine and the nAChR antagonist mecamylamine (n = 4; −25.8 ± 3.4%, P < 0.05; Figure 4) with the peak decreases occurring 2 min postinfusion. Thus, PPT stimulation‐evoked striatal dopamine release is significantly dependent upon activation of glutamatergic STN cells that project to the SNc as seen by a 50% decrease in PPT stimulation‐evoked striatal dopamine release following intra‐STN lidocaine. These data have not been previously published. Additional studies examining the effects of GluR antagonists infused into the STN during PPT stimulation accounted for the remaining ∼24% of PPT excitatory input to the STN (data not shown). These findings highlight the importance of excitatory cholinergic, including glutamatergic, projections from the PPT to the STN in PPT stimulation‐evoked striatal dopamine release as they accounted for at least a quarter each of dopamine release in the striatum following PPT stimulation. Altogether, these studies suggest that STN AChRs and GluRs are equally involved in mediating excitatory cholinergic and glutamatergic inputs arising from the PPT.
Figure 4.
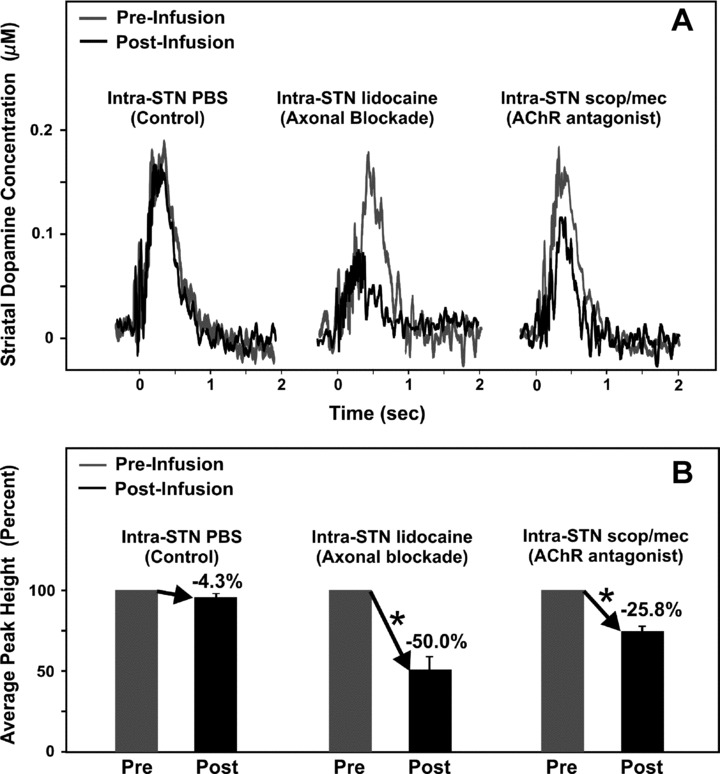
Mean amperometric recordings of dopamine release in the striatum evoked by electrical stimulation of the pedunculopontine tegmental nucleus (A) and corresponding mean peak percentages (B). Profiles illustrate mean peak effects in response to STN microinfusions of PBS, the local anaesthetic lidocaine, or a combination of the muscarinic acetylcholine receptor (mAChR) antagonist scopolamine (scop) and the nicotinic acetylcholine receptor (nAChR) antagonist mecamylamine (mec) (A). Time zero indicates the start of the train of 20 pulses at 50 Hz. *Significant change in striatal dopamine concentration after the infusion compared to pre‐infusion baseline responses (100%).
The use of a combination of broad‐spectrum mAChR and nAChR antagonists in this study prevented identification of the specific mAChR and nAChR subtypes utilized by PPT afferents to the STN. Cholinergic agonists such as carbachol have been shown to excite STN neurons [59], however, nAChR agonists alone had no apparent effect on neuronal cell activity in the STN [60]. Furthermore, the mAChR antagonist scopolamine, but not the nAChR antagonist mecamylamine, have been shown to block ACh‐evoked STN cell excitations [60]; thus, it may be postulated that STN AChRs are primarily muscarinic. M3 mAChR subtype, in particular, are prominently expressed in the STN [61], and Shen and Johnson [62] found that the mAChR agonist muscarine reduced the amplitude of GABA inhibitory postsynaptic currents, while the effect was blocked by the nonsubtype specific mAChR antagonist scopolamine as well as the M3 specific mAChR antagonist 4‐diphenylacetoxy‐N‐methylpiperidine methiodide (4‐DAMP). These investigators concluded that muscarinic agonists in the STN act at presynaptic M3 mAChRs on GABA afferents, causing disinhibition (excitation) of STN neurons, thereby permitting afferents from the PPT to have a greater excitatory influence on STN output. Thus, STN mAChRs, particularly of the M3 subtype, may be involved in the indirect activation of SNc DA cells via PPT–STN–SNc pathways [62].
Modulation of Striatal Dopamine Release by Activation of Afferent Projections to the PPT
Clearly, direct stimulation of the PPT elicits dopamine release in the striatum. However, less is known about afferent projections to the PPT and the mechanisms driving them. Similarly, stimulation of the STN has been shown to generate striatal dopamine release [63], but the extent to which the PPT is mediating STN‐evoked dopamine release or the contribution of mAChRs and nAChRs located on SNc DA neurons remains unknown. To address these issues, in a similar fashion as described above we used in vivo fixed potential amperometry to record dopamine release in the striatum of urethane anesthetized male mice while stimulating the STN before and during intra‐SNc 1.0 μL infusions of the mAChR antagonist scopolamine (1.0 μg) or the nAChR antagonist mecamylamine (1.0 μg).
Intra‐SNc infusions of 1.0 μL of PBS (pH ∼7.4) in a separate group of mice did not significantly alter striatal dopamine release evoked by stimulation of the STN (n = 4; −5.0 ± 3.4%, P > 0.05; Figure 5) again confirming that the observed drug effects were not attributed to nonspecific effects of the infusion procedure. In contrast, compared to pre‐infusion baseline levels, intra‐SNc infusions of the mAChR antagonist scopolamine or the nAChR antagonist mecamylamine significantly attenuated STN stimulation‐evoked striatal dopamine release (n = 4/drug; −27.9 ± 4.8% and −28.2 ± 5.4%, respectively; P < 0.05; Figure 5) with peak decreases occurring 2 min postinfusion. Together with evidence that pharmacological activation of mAChRs and/or nAChRs in the SNc stimulates DA neuronal activity and dopamine release in the striatum [39, 64], the present results suggest that excitatory cholinergic inputs arising from the PPT and activating both mAChRs and nAChRs in the SNc account for at least 60% of the excitatory drive of the STN on SNc DA activity, with the balance likely mediated by both direct (STN‐SNc) and indirect (STN‐PPT‐SNc) glutamatergic inputs (unpublished results).
Figure 5.
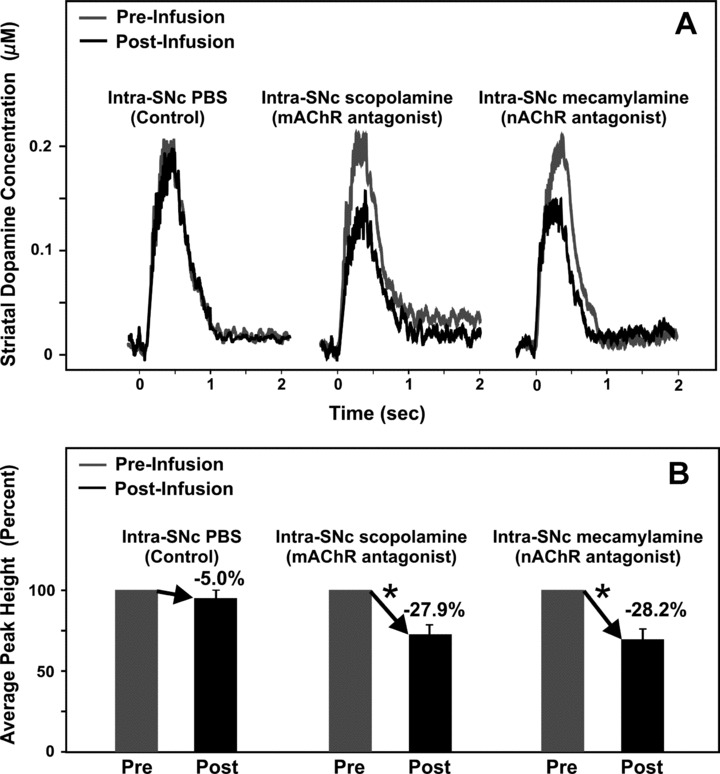
Mean amperometric recordings of dopamine release in the striatum evoked by electrical stimulation of the STN (A) and corresponding mean peak percentages (B). Profiles illustrate mean peak effects in response to substantia nigra pars compacta (SNc) microinfusions of PBS, the mAChR antagonist scopolamine, or the nAChR antagonist mecamylamine (A). Time zero indicates the start of the train of 20 pulses at 50 Hz. *Significant change in striatal dopamine concentration after the infusion compared to pre‐infusion baseline responses (100%).
The use of broad‐spectrum mAChR and nAChR antagonists in this additional study also did not permit identification of the specific mAChR and nAChR subtypes utilized by PPT afferents to the SNc. However, muscarine excitation of midbrain DA neurons has been shown to be mediated by M1‐like receptors [64], and given that relatively high expression levels of mRNA for the M5 mAChR subtype in the SNc and the finding that the M5 subtype is the only mAChR to be definitively localized on SNc DA cells [65, 66], SNc mAChRs of the M5 subtype are thought to be involved in the release of striatal dopamine following PPT stimulation [39]. Nicotine administered locally into the SNc increases the firing of SNc DA neurons and enhances concentrations of dopamine metabolites in the striatum [67, 68]. Several nAChR subunits, such as α3 to α7 and β2 to β3, have been shown to be present in the SNc [69, 70, 71, 72, 73]. In particular, cholinergic inputs from the PPT may enhance nigrostriatal DA transmission via activation of α4β2 and α7 nAChRs localized on DA cells in the SNc [30].
Striatal Cholinergic Modulation of the Nigrostriatal Dopaminergic System
Cholinergic projections from the PPT do not provide the only means by which ACh interacts with the nigrostriatal DA system. The striatum, with its intrinsic cholinergic and extrinsic DA innervation, is heavily dependent upon functional ACh–dopamine interactions. The majority of neurons within the striatum are spiny GABAergic projection neurons, with 2% of striatal neurons being large cholinergic interneurons and the rest being GABAergic interneurons [19, 74]. Although few in number, cholinergic interneurons have large, dense axonal arbours [14] and are noted as autonomous pacemakers, given their ability to spike in the absence of synaptic input activity. For this reason they are often referred to as “tonically active neurons”[75]. Together with their broad axonal terminal fields, the ongoing tonic activity of cholinergic interneurons translates to tonic ACh release, continuously activating mAChRs and nAChRs throughout the striatum [14, 76]. In the striatum, ACh regulates its own release via muscarinic autoreceptors on cholinergic interneurons. Endogenous ACh and mAChR agonists reduce ACh release in the striatum, most likely through activation of M2 and/or M4 mAChRs [77, 78], which are present on the somatodendritic areas as well as the axon terminals of striatal cholinergic interneurons [79, 80]. In addition, the relatively selective M2‐like mAChR antagonist AF‐DX 116 has been found to enhance ACh release in the striatum, most likely through blockade of autoinhibition [81]. Release of ACh from cholinergic interneuronal activity is also modulated by dopamine. Striatal cholinergic interneurons contain D2 receptors, in both the short (typically presynaptic autoreceptor) and long (most often found postsynaptically) isoforms, as well as D5 receptors, which are mainly in the somatodendritic areas [14, 82, 83]. D1‐like, most likely D5, receptor activation in the striatum leads to a depolarization of cholinergic interneurons and ACh release [84, 85]. However, activation of D2 receptors on these interneurons inhibits the autonomous spiking and subsequently decreases release of ACh in the striatum [86].
Acetylcholine release in the striatum via cholinergic interneurons is known to modulate striatal dopamine release via striatal mAChRs and nAChRs. Acetylcholine activation of mAChRs facilitates dopamine release in the striatum [12, 87, 88]. Although all subtypes of mAChRs (M1–M5) are located within the striatum, the limited receptor subtype selectivity of mAChR agonists and antagonists has led to some conflicting results regarding the specific mAChR subtypes involved in modulating striatal dopamine release [80, 89]. Using genetically altered mice lacking functional M1–M5 mAChRs, Zhang and colleagues [80] found that the lack of M1 or M2 receptors had no effect on striatal dopamine release. However, the lack of M4 and M5 receptors significantly reduced striatal dopamine release, whereas the lack of M3 receptors significantly increased dopamine release in the striatum. These findings suggest indicate that M4 and M5 mAChR activation facilitates striatal dopamine release, but stimulation of M3 receptors inhibits such release [80]. The neuronal location of these mAChR subtypes determines their influence on striatal dopamine activity. M4 receptors are abundantly expressed on cell bodies of medium spiny GABAergic projection neurons comprising the direct pathway (Figure 1) [90, 91]. Activation of these M4 mAChRs inhibits GABA release in the striatum, perhaps mediated by collaterals of striatal GABAergic neurons [80, 92, 93], subsequently resulting in reduced GABAA receptor‐mediated inhibition of dopamine release from striatal DA nerve terminals [94]. mRNA for the M5 mAChR is found exclusively in SNc DA cells, allowing for expression of M5 receptors in both dopamine‐containing cell bodies in the SNc and nerve terminals in the striatum [65]. Thus, the findings that M5 mAChR‐deficient mice show reduced dopamine release in striatal brain slices suggest these presynaptic M5 mAChRs facilitate the release of dopamine [80]. In contrast, activation of presynaptic M3 mAChRs located on cortical glutamatergic inputs to GABAergic neurons in the striatum has been shown to inhibit glutamate release [95], which may in turn reduce striatal dopamine release, since glutamate in the striatum facilitates dopamine release via activation of presynaptic ionotropic glutamate receptors on DA nerve terminals [96].
In the striatum, nAChRs are expressed presynaptically on DA nerve terminals, and activation of these nAChRs initiates a depolarization and/or a calcium signal, enhancing dopamine release [14]. Nicotine infused directly into the striatum elevates dopamine release from nerve terminals [97]. Specifically, nigrostriatal DA terminals abundantly express β2 and α6 subunits which are incorporated in α4β2*, α6β2*, and α4α6β2β3* nAChR subtypes [30, 98, 99], both of which have been shown to be involved in ACh‐induced striatal dopamine release [100, 101]. Normal motor control depends on interactions between the nigrostriatal DA neurons and cholinergic neurons in the PPT and cholinergic interneurons in the striatum. Recognition of the cholinergic receptor subtypes on neurons within the nigrostriatal DA system is important for understanding basal ganglia function, particularly under neuropathological conditions such as Parkinson's disease in which AChRs may be effective targets in treating symptoms and restoring a functional balance in dopamine–ACh interactions.
Parkinson's Disease
Parkinson's disease is a neurological disorder characterized by a progressive degeneration of the nigrostriatal DA system [102]. The onset of clinical symptoms such as bradykinesia, tremor, and rigidity occur with at least 80% decrease in striatal dopamine content and at least 50% or greater loss of DA neurons in the SNc [103, 104]. A reduction in striatal DA transmission, as in the parkinsonian condition, is thought to increase the inhibitory output from the basal ganglia to the thalamus, resulting in impaired motor functioning. In order to compensate for the reduction in DA tone in the striatum, the most commonly used pharmaceuticals for Parkinson's disease include the dopamine precursor levodopa alone or in combination with peripheral aromatic l‐amino acid decarboxylase inhibitors carbidopa or benserazide (Sinemet and Prolopa, respectively) or COMT inhibitor entacapone (Comtan) and/or dopamine agonists, such as bromocriptine (Parlodel), pergolide (Permax), pramipexole (Mirapex), and ropinirole (Requip) [103, 105]. Dopamine synthesized from levodopa activates both D1 and D2 receptors in the striatum, which is important therapeutically as antiparkinsonian drugs with high D2 and low D1 affinity have been shown to be less effective in reversing motor symptoms compared to levodopa; however, as the disease progresses with time, there is a loss of efficacy of these DA drug treatments and oftentimes psychiatric complications occur with increased doses [28, 106]. Thus, novel pharmaceutical treatments are continuously being explored for better management of the motor symptoms associated with Parkinson's disease. Given the well known functional interactions of the striatal and mesopontine cholinergic systems with the nigrostriatal DA system, more selective agents acting on the various ACh receptor subtypes existing heterogeneously at key anatomical sites in the brain represent promising pharmaceutical targets in the treatment of this neurological disorder.
One direct link of the mesopontine cholinergic system in Parkinson's disease stems from the observation that cholinergic neurons in the PPT are reduced by nearly 40% in these patients [107]. Indeed, excitotoxic lesions of the PPT have been found to produce parkinsonian‐like postural deficits, hypokinesia, and locomotor deficits in primates [108], while a significant loss of PPT neurons has been found to correlate with the extent of neuronal loss of DA cells in the SNc and the severity of Parkinson's disease symptoms [109]. These findings suggest that reduced excitatory cholinergic output from the PPT may lead to less excitation of SNc DA cells, and thus in part contribute to the degenerative reduction in striatal dopamine transmission [48].
With regards to the striatum, acting on D2 receptors on striatal cholinergic interneurons dopamine or DA agents have been shown to inhibit the autonomous spiking of these cells leading to a decrease in striatal ACh release [86]. In the neuropathological condition of Parkinson's, reduced striatal dopamine would lead to overactivity of cholinergic interneurons and excess ACh release in the striatum. Nomoto et al. [110] have suggested that, as a result of decreased DA activity in the parkinsonian state, increased striatal cholinergic interneuronal activity may also lead to increased extracellular adenosine levels in the striatum, as adenosine is coreleased or synthesized from ATP coreleased with ACh in the striatum [111]. Adenosine A2A receptors coexist with dopamine D2 receptors on GABAergic neurons that comprise the indirect striatal output pathway, whereas adenosine A1 and dopamine D1 receptors are found on GABAergic neurons that form the direct striatal output pathway (Figure 1) [112]. Dopaminergic input from the SNc into the striatum inhibits the release of ACh through D2 receptors, and also stimulates its release through D1 receptors [113, 114]. These findings suggest alterations in cholinergic interneuronal release of adenosine in the parkinsonian striatum. Recent in vitro biochemical studies have suggested that A1 receptors may be important in modulating GABAergic transmission in limbic regions, rather than GABAergic output neurons in the striatum [115], as these receptors appear to be predominantly located presynaptically on intrinsic striatal interneurons [116]. In contrast, A2A receptor‐mediated modulation of the striatopallidal GABAergic output neurons appears to make a major contribution to A2A receptor‐mediated control of GABA transmission in both normal and parkinsonian animal model [117]. Local infusions of the adenosine A2A agonist CGS 21680 induce akinesia in rats [118], suggesting that increased adenosine in the striatum acting at A2A receptors may contribute to the induction of akinesia. All together, these findings suggest that adenosine antagonists, particularly those targeting the A2A adenosine receptor subtype, may be of therapeutic value in treating specific symptoms, such as akinesia, in Parkinson's disease [119]. In recent clinical trials, the A2A antagonist istradefylline has been shown to reduce “off” time in patients with Parkinson's receiving DA pharmacotherapy. However, these therapeutic effects have proven inconsistent and will require further clinical trials to establish the clinical utility of this drug class [120].
Historically, anticholinergics were the first available drugs for the alleviation of Parkinson's symptoms, and are still used as secondary treatments for Parkinson's disease in conjunction with other antiparkinsonian drugs [121]. Centrally‐acting anticholinergics, all specific for mAChRs, include benztropine (Cogentin), which is widely prescribed, and biperiden (Akineton), orphenadrine (Norflex), and procyclidine (no longer prescribed in the United States) [122]. Anticholinergic drugs have been used mainly in tremor‐predominant cases of Parkinson's disease and are thought to act by counterbalancing the reduced DA influence on the medium spiny GABAergic output neurons to the globus pallidus [123, 124]. Evidence suggests that a lack of striatal dopamine triggers a reduction in the efficacy of M4 autoreceptors on cholinergic interneurons, which would result in a net increase in ACh release (Figure 1) [125]. An elevation in ACh as a result of a loss of dopamine in Parkinson's disease then may act at mAChRs on medium spiny neurons in the striatum, thus justifying the use of antimuscarinic drugs in treating motor symptoms of Parkinson's disease [75]. In addition, parkinsonian symptoms in mice induced by mAChR agonists can be reduced by systemic administration of a broad‐spectrum mAChR antagonist, as well as a mAChR antagonist with moderate selectivity for the M4 mAChR [126]. These latter findings of blockade of striatal M4 receptors have been considered as functionally significant in reducing parkinsonian symptoms [126]. However, it is unlikely that mAChR antagonists are acting on postsynaptic M4 receptors in the striatum, as striatal M4 receptor blockade leads to a reduction in local dopamine release [88]. Nevertheless, M4 mAChRs may potentially be a pharmaceutical target for treating motor symptoms of Parkinson's disease, via a different cholinergic neuronal location. For example, M4 receptor deficient mice show exacerbated striatal DA activity, perhaps due to a loss of mAChR autoreceptor feedback on cholinergic neurons of the PPT, which would lead to an enhancement in excitatory cholinergic drive on surviving SNc DA neurons and to subsequent elevations in striatal dopamine release [40, 41, 127].
Although mildly effective in treating motor symptoms in Parkinson's disease, but rather prescribed mainly in these patients for bladder dysfunction, the use of antimuscarinic drugs has been shown to be associated with impaired neuropsychiatric and cognitive function [123, 128]. Clinical evidence suggests that M1 mAChR antagonism may account for these unwanted side effects, as M1 mAChR antagonists such as benztropine, orphenadrine, and trihexyphenidyl (Artane) have been shown to increase amyloid plaque and neurofibrillary tangle densities similar to that found with Alzheimer's disease pathology [128]. Thus, development of more selective mAChR antagonists may provide better symptomatic treatment of the motor symptoms in Parkinson's disease without the detrimental cognitive side effects.
Findings from animal studies also suggest that nicotine or nAChR agonists may be an effective treatment for the motor symptoms of Parkinson's disease. Stimulation of nAChRs has been shown to modulate locomotor activity in intact nonlesioned animals as well as ameliorate motor dysfunctions in animals with nigrostriatal damage [129, 130]. Additionally, studies have shown that people who smoke, or have smoked regularly, are 50% less likely to develop Parkinson's disease than those who have never smoked, and nicotine has been found to alleviate parkinsonian cognitive and motor symptoms once Parkinson's disease has developed [131]. The mechanisms underlying these therapeutically beneficial qualities of nicotine are not known. Smoking and nicotine treatment have been shown to protect the nigrostriatal DA neurons from degeneration following MPTP or 6‐OHDA treatment [17, 132]. However, acute or short‐term treatment with nicotine has shown little to no effects on motor activity in Parkinson's patients or parkinsonian animal models, suggesting that nicotine treatment may only provide a neuroprotective and/or restorative effect with chronic use [133].
With the progressive loss of presynaptic DA nerve terminals in Parkinson's disease, there is an accompanying decline in nAChRs [134, 135]. The exact nAChR subtypes lost in the brains of Parkinson's patients are not fully known [131]. Studies indicate a role for striatal α4 and α6 nAChRs in striatal dopamine release, with α6 having a more restricted localization than α4, which is widespread throughout the brain [101, 134, 136]. Despite a greater than 50% reduction in these receptor subtypes following nigrostriatal damage, striatal α4 and α6 nAChR function appears to remain at relatively normal levels [137]. These compensatory functional changes in the presence of significant nAChR loss suggest that subtype‐selective agonists may be beneficial despite nigrostriatal damage. Furthermore, administration of nAChR agonists in monkeys strongly potentiate the effects of levodopa, allowing for a reduction in the doses necessary for therapeutic benefits and perhaps, consequently, decreasing the occurrence of debilitating side effects [130]. Another finding supporting the potential of nAChR agonists as pharmaceutical treatments for Parkinson's disease is that the doses of epibatidine, a nonsubtype selective nAChR agonist, utilized to enhance nigrostriatal dopamine release does so without eliciting reinforcing or rewarding effects [131, 138]. These observations suggest the possibility of developing novel nAChR agonists that lack addictive properties for pharmacological treatment of Parkinson's disease.
Mesocorticolimbic Dopaminergic System
The mesocorticolimbic DA system projects from the VTA in the midbrain to forebrain limbic and cortical areas [3]. This system can be further divided into two subsystems, determined by the localization of dopamine‐containing cell bodies within the VTA and their projection targets. Dopaminergic cells of the paranigral VTA subdivision that project to the NAc, amygdala, and hippocampus constitute the mesolimbic DA pathway, while DA neurons of the parabrachial VTA subdivision that innervate cortical structures make up the mesocortical DA pathway [4]. The mesolimbic DA system has been classically associated with the acquisition of behaviors reinforced by natural and drug‐related rewarding stimuli [139]. A majority of DA neurons within the VTA project to the NAc but are not anatomically restricted to this nucleus; VTA DA neurons also project to the hippocampus, amygdala, and mPFC [6]; however, the involvement of the NAc in reward‐related behavior (e.g., brain‐stimulation reward as well as natural rewards such as food, water, and sexual interaction) is more established than the other VTA projection target nuclei [140, 141]. Dopaminergic activity in the NAc is considered to be critical for the establishment and maintenance of goal‐directed behaviors related to obtaining natural and artificial rewards [142, 143]. Dopamine release in the NAc is enhanced not only by natural rewards and drugs of abuse, but also upon presentation of conditioned stimuli [144, 145, 146]. Experimental studies have linked the NAc and the mesolimbic DA system to learning about stimulus‐reward relationships and maintenance of these learned behaviors. Animals have shown conditioned place preferences when opioids or psychostimulants, such as morphine or amphetamine, is directly infused into the NAc [147], but not when they are infused into the striatum [148]. Also, cytotoxic lesions of VTA DA neurons prevent the formation of conditioned place preferences to opiates [149] providing support for the critical role of DA projections from the VTA to the NAc in establishing stimulus‐reward associations.
The NAc GABAergic medium spiny neurons provide output to the ventral pallidum (VP) and subpallidum (SP) which relay the information to mediodorsal nuclei of the thalamus and the mesencephalic locomotor region of the hindbrain (Figure 6) [150, 151, 152]. The NAc is often subdivided into shell (medial NAc) and core (lateral NAc) subregions. The shell connects reciprocally to the extended amygdala, an interconnected set of limbic structures that mediate emotional responses, whereas the core is reciprocally innervated with the dorsal striatum involved in mediating sensory‐motor function [152]. The specific roles of the shell and core regions of the NAc in reward‐related behaviors are still not completely certain. The shell of the NAc has been shown to mediate hedonic states associated with sweet tasting food and intra‐shell, but not core, self‐administration of cocaine [153, 154]. Thus, the shell may be a critical neural substrate for the initiation of drug‐seeking behavior. On the other hand, the core seems to hold a more prevalent role in the conditioning processes associated with drug‐seeking behaviors [155], suggesting that the core may influence maintenance of drug seeking [156].
Figure 6.
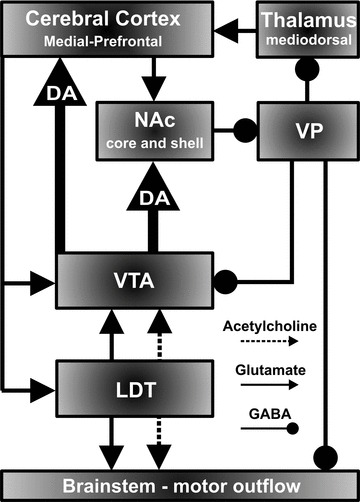
Simplified mesocorticolimbic circuitry depicting dopaminergic (DA) cells in the VTA projecting to the core and shell region of the nucleus accumbens (NAc) and medial prefrontal cortex and excitatory (acetylcholine/glutamate) inputs from the laterodorsal tegmental nucleus (LDT) and associated excitatory (glutamate) and inhibitory (γ‐aminobutyric acid: GABA) feedback from cortex and VP.
While subcortically projecting mesolimbic DA neurons have been shown to play a critical role in motivation and reward‐related behaviors, prefrontally projecting mesocortical DA cells are thought to facilitate neuronal activity in the mPFC thus modulating several aspects of cognitive function [157]. Dopaminergic neurons in the VTA constituting the mesocortical projections to the mPFC are thought to direct behaviors toward abstract goals [158]. Depletion of dopamine within the mPFC has been shown to impair performance on the delayed response task, a measurement of spatial working memory, to the same extent as surgical ablation of the mPFC [159]. This early finding suggests that mPFC dopamine activity mediates the maintenance of goal representations in working memory, which has been subsequently supported by neurophysiological data [160, 161]. The ability to regulate this aspect of cognitive function is critical for the active suppression and expression of irrelevant and relevant behaviors [158]. Excitatory glutamate‐containing pyramidal neurons in the mPFC also provide one of the principal corticofugal projections to VTA DA cells (Figure 6), more specifically VTA DA neurons that project exclusively to the mPFC (the mesocortical DA system), and not those that project to the NAc (the mesolimbic DA system) [162]. This reciprocal projection provides a feedback mechanism which may explain findings from in vivo electrophysiological studies that microiontophoresis of dopamine onto mPFC pyramidal cells generally suppresses their activity [163, 164]. A number of studies have suggested that the mPFC‐VTA projection may play a role in the response of the mesocortical DA pathway to stress [162, 165], in which both mPFC cells and mesocortical DA neurons are activated in conditions of stress [166, 167]. Furthermore, both rewarding and stressful situations stimulate dopamine release in the mPFC, which suggests that mesocortical DA neurons are involved in emotional arousal, regardless of its aversive or nonaversive nature [168].
Mesopontine Cholinergic Modulation of the Mesocorticolimbic Dopaminergic System
Modulation of NAc Dopamine Release by Activation of Cholinergic Receptors in the LDT
The LDT provides the principal source for cholinergic input to DA neurons of the VTA [32]. Input from the LDT is required for burst firing of DA cells in the VTA [169], which is thought to convey motivationally relevant information to forebrain areas, mostly the NAc, involved in the induction of reward and related processes [170]. Activity of LDT cholinergic neurons is modulated by mAChRs and nAChRs in the LDT. Stimulation of LDT mAChRs, most likely M2 autoreceptors, hyperpolarizes LDT cholinergic neurons [45], while mAChR antagonists infused into the LDT increases forebrain dopamine release [41, 171]. Nicotine infused into the LDT induces firing of LDT cholinergic neurons, increasing ACh outflow to DA neurons in the VTA. Direct nicotinic actions on LDT cholinergic neurons have been shown to be mediated by receptors containing α7, β2, and non‐α7 subunits [172].
Modulation of NAc Dopamine Release by Activation of Projections from the LDT
Electrical stimulation of the LDT as well as chemical stimulation of both mAChRs and nAChRs in the VTA has been shown to excite mesolimbic DA neurons [64, 173, 174], and to facilitate dopamine release in the NAc [175, 176, 177]. The excitatory effect of ACh acting on mAChRs and nAChRs in the VTA may function to influence incentive‐related behaviors driven by DA activity. The AChR agonist carbachol infused into the VTA induces conditioned place preference and is self‐administered by rats into this region [178], and blockade of mAChRs in the VTA disrupts responding for food reward [179]. A role of VTA nAChRs in dopamine‐related behaviors has also been demonstrated, such that locomotion is enhanced by intra‐VTA infusion of nicotine, the effects of which can be blocked by the nAChR antagonist mecamylamine [180].
As in our studies described earlier, with the same stimulation parameters and microinfusion procedures we used in vivo fixed potential amperometry to determine the relative contribution of mAChRs and nAChRs located on VTA DA neurons in eliciting stimulation‐evoked dopamine release in the NAc. In urethane anesthetized male mice the LDT was stimulated before and during a 1.0 μL intra‐STN infusion of the mAChR antagonist scopolamine (1.0 μg) or the nAChR antagonist mecamylamine (1.0 μg) [57]. Intra‐VTA infusions of 1.0 μL of sterile PBS (pH ∼7.4) in a separate group of mice did not significantly alter NAc dopamine release evoked by LDT stimulation (n = 4; −5.8 ± 3.6%, P > 0.05; Figure 7). However, intra‐VTA scopolamine or mecamylamine infusion significantly attenuated LDT stimulation‐evoked NAc dopamine release by blockade of VTA mAChRs and nAChRs (n = 4/drug; −69.4 ± 8.9% and −45.7 ± 2.8%, P < 0.05; Figure 7), respectively, with peak decreases occurring 5 min postinfusion [57]. The reported percentage decreases following blockade of mAChRs and nAChRs in the VTA contribute nearly 100% (taking standard error into account) of LDT‐evoked dopamine release in the NAc to excitatory cholinergic projections from the LDT, thus, suggesting that, at least in this paradigm, a glutamatergic projection from the LDT to the VTA plays a minor role in stimulating NAc dopamine release. Together with evidence that pharmacological activation of mAChRs and/or nAChRs in the VTA stimulates DA neuronal activity, as well as dopamine release in the striatum [64, 173, 175, 176], these results suggest that both mAChRs and nAChRs are involved in mediating excitatory cholinergic inputs arising from the LDT.
Figure 7.
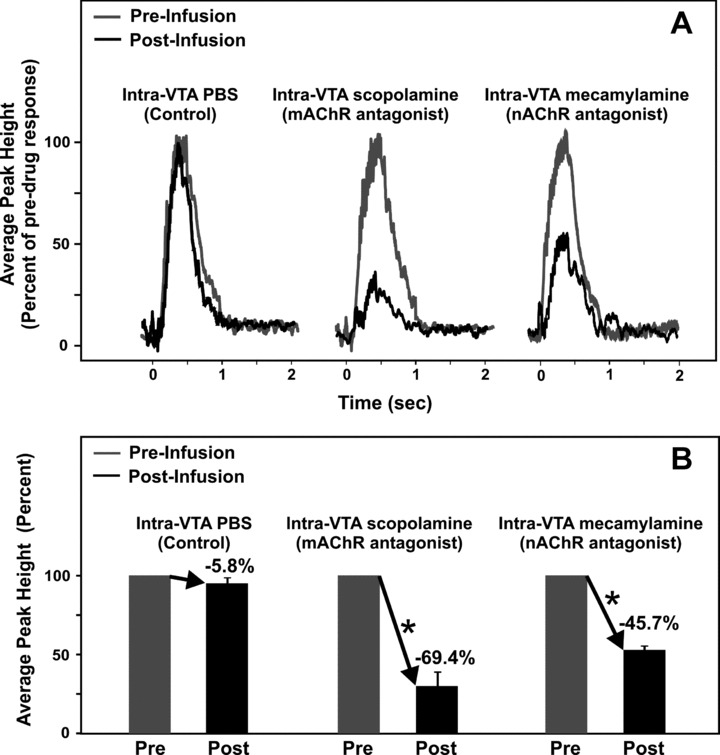
Mean amperometric recordings of dopamine release in the nucleus accumbens evoked by electrical stimulation of the laterodorsal tegmental nucleus (A) and corresponding mean peak percentages in bar graph form (B). Profiles illustrate mean peak effects in response to VTA microinfusions of PBS, the mAChR antagonist scopolamine, or the nAChR antagonist mecamylamine (A). Time zero indicates the start of the train of 20 pulses at 50 Hz. *Significant change in striatal dopamine concentration after the infusion compared to pre‐infusion baseline responses (100%). Coordinates in mm for the stimulating electrode in the LDT, guide infusion cannula in the VTA, and carbon fiber recording microelectrode in the NAc were: AP −1.0, +0.9, +1.5 from lambda, ML +0.4, +0.3, +1.0 from midline, and DV −2.4, −4.0, −4.0 from dura), respectively [55].
The mAChRs mediating LDT‐evoked striatal dopamine release may involve M3 and M5 subtypes that have been localized in the VTA. However M5 mAChRs are the only mAChRs to be definitively localized on DA cells in the VTA [65]. In addition, M3 mAChR activation has been shown to reduce, rather than enhance, excitatory transmission in DA midbrain cells by presynaptic mechanisms [181]. Indeed, studies using M5 receptor‐deficient mice or infusions of M5 antisense to reduce M5 receptor expression in the VTA in rats have shown that midbrain M5 mAChRs mediate prolonged facilitation of forebrain dopamine transmission and thereby maintenance of dopamine‐related reward [182, 183, 184]. Thus, it is likely that the observed attenuation in LDT stimulation‐evoked NAc dopamine release following intra‐VTA blockade of mAChRs is mediated by M5 mAChRs. In regards to nAChRs in the VTA, in situ hybridization and immunohistochemical findings suggest that α3‐α7 and β2‐β3 nAChR subtypes are present on dopamine‐containing cell bodies in the VTA, particularly α4 β2 and α7 subtypes [30, 69, 185, 186, 187]. It is important to note that the α7 nAChR subtype is also located presynaptically on glutamatergic afferents in the VTA. Activation of this receptor is thought to play an important role in mediating glutamate‐induced burst firing of VTA DA neurons [188, 189] and may therefore be involved in the reduction of phasic accumbal dopamine release observed following nAChR blockade in the VTA [57]. Overall, the present neurochemical recordings strongly suggest that excitatory cholinergic projections originating in the LDT may function to modulate VTA DA activity in the NAc thereby influencing incentive‐related behaviors, and in particular, mediate the addictive properties of psychomotor stimulants.
Psychomotor Stimulant Addiction
The mesolimbic DA pathway is well recognized as a central neural substrate involved in mediating the reinforcing properties of various drugs of abuse, such as the psychostimulants cocaine and amphetamines [190, 191]. Dopamine extracellular levels dramatically increase in the NAc following the self‐administration of cocaine, behavior that can be easily blocked or extinguished with dopamine receptor blockers [192]. Natural rewards, such as food, water and con‐specific mates, also stimulate mesolimbic dopamine release, but the magnitude of dopamine release generated by these appetitive stimuli do not rival the activational effects of psychostimulants [193]. Rapid “phasic” changes in extracellular levels of dopamine recorded in the NAc also coincide with cocaine‐seeking behaviors and cocaine‐related cues [194]. Brain imaging techniques have extended these observations to humans showing that psychostimulant‐induced elevations in NAc dopamine release correspond with self‐report measures of euphoria [195].
Because of the involvement of ACh in mesolimbic DA activity, the cholinergic system may play an important role in drug addiction. VTA ACh receptor activation is rewarding, as evidenced in that AChR agonists are self‐administered into the VTA [178]. Pharmacological stimulation of nAChRs and mAChRs in the VTA produces excitation of DA neurons [64, 173, 174] and facilitates dopamine release in the NAc [175, 176, 177]. As such, the excitatory effect of ACh acting on nAChRs and mAChRs in the VTA may function to influence incentive‐related behaviors driven by dopamine activity. For example, the rewarding effects of lateral hypothalamic brain stimulation in rats are enhanced by VTA microinfusion of ACh but attenuated by similar treatment with mAChR blockers and to a lesser extent by nAChR blockers [196, 197]. Also, the mixed nicotinic/muscarinic agonist carbachol microinfused into the VTA induces a conditioned place preference and is self‐administered by rats into this region [178], while blockade of mAChRs in the VTA disrupts responding for food reward [179].
Although nAChRs are expressed on DA terminals, the locomotor stimulant, reinforcing and dependence‐producing actions of nicotine appear to require activation of specific nAChR subtypes located on glutamatergic terminals in the VTA [198]. For example, locomotion is enhanced by intra‐VTA infusion and systemic injections of nicotine, the effects of which are blocked by the nAChR antagonist mecamylamine [180]. Also, blockade of nAChRs in the VTA decreases nicotine self‐administration [199, 200] and blocks the facilitatory effects of systemic nicotine on NAc dopamine release [201]. Evidence indicates that α7 and β2 type nAChRs in the VTA, known to be principally involved in nicotine's stimulatory effects on DA neuronal activity and dopamine release in the NAc, are integral in the reinforcing properties of this drug while the α7 subtype is also implicated in the nicotine withdrawal syndrome [198, 202, 203]. Together with evidence that α7 and β2 subunits modulate the reinforcing effects of cocaine [202, 204], these studies support the therapeutic value of nAChR antagonists in the treatment of psychostimulant addiction [205, 206]. However, evidence from a number of sources suggests that, rather than nAChRs, the rewarding effects of psychostimulants may be mediated mainly by mAChRs in the VTA, mostly likely of the M5 subtype [207].
As only M5 receptor mRNA appears exclusively localized to DA cells in the VTA [65, 90], the M5 mAChR subtype occupies a unique position to modulate behaviors driven by mesolimbic DA activity, including behaviors associated with the rewarding effects of psychostimulants. For example, cocaine is significantly less potent as a reinforcer in M5 receptor‐deficient mice, as evidenced by reduced cocaine self‐administration and significantly less time spent in a cocaine‐paired compartment compared to wild‐type mice [208, 209]. Moreover, the severity of withdrawal symptoms after chronic administration of cocaine is reduced in these animals [208]. Neurochemically, M5 receptor‐deficient mice exhibit a decrease in sustained NAc dopamine release following electrical stimulation of the LDT cholinergic neurons, which may account for the observed reduction in the rewarding effects of cocaine in animals lacking M5 mAChRs [184].
To further investigate the importance of mAChRs in the VTA in modulating dopamine transmission within the NAc, particularly during the facilitatory actions of cocaine, we examined the effect of intra‐VTA infusion of the mAChR antagonist scopolamine on the ability of cocaine to enhance LDT electrical stimulation‐evoked dopamine release in the NAc [210]. In a similar fashion to our fixed potential amperometric studies of PPT and STN stimulation‐evoked striatal dopamine release, we monitored temporal changes in LDT stimulation‐evoked dopamine release in the NAc of urethane anesthetized male mice in response to a 0.5 μL intra‐VTA scopolamine (10 μg) infusion either prior to or following systemic administration of cocaine (10 mg/kg, i.p.). With respect to pre‐infusion baseline levels (100%), LDT stimulation‐evoked dopamine release in the NAc was not significantly altered by intra‐VTA infusion of 1.0 μL of PBS administered either 5 min prior to (n = 3, 92.8 ± 2.2%, P= 0.83) or 15 min following (n = 3, 98.3 ± 2.9%, P= 0.62) systemic injection of saline (10 mL/kg, i.p.; Figure 8A). The temporal pattern of these drug vehicle responses provided control values against which to compare the effects of systemic cocaine injections and intra‐VTA scopolamine infusions (Figures 8B and C, gray lines). As expected, cocaine's ability to effectively block reuptake of synaptic dopamine [211] resulted in a significant increase in LDT stimulation‐evoked NAc dopamine release when administered either 5 min following intra‐VTA PBS (n = 3, 241.3 ± 13.6%, P < 0.05; Figure 8B) or 15 min before intra‐VTA PBS (n = 3, 253.1 ± 9.4%, P < 0.05; Figure 8C) with peak effects occurring 10–15 min postinjection. In contrast, scopolamine's ability to block mAChRs in the VTA [171, 176, 184] resulted in a significant decrease in LDT stimulation‐evoked dopamine release in the NAc, when infused in the VTA 5 min prior to (n = 4, −74.8 ± 2.8%, P < 0.05; Figure 8B) or 15 min following (n = 3, −69.4 s±4.8%, P < 0.05; Figure 8C) systemic injection of saline.
Figure 8.
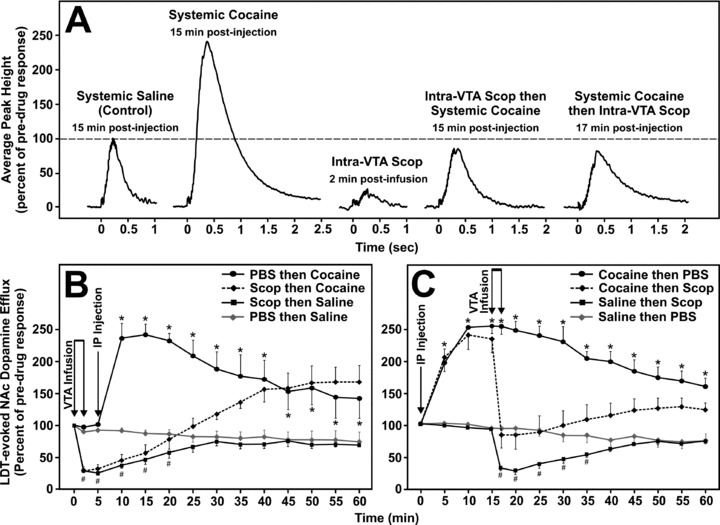
Mean amperometric recordings of dopamine release in the nucleus accumbens evoked by electrical stimulation of the laterodorsal tegmental nucleus (LDT) (A) and time courses of the effects of scopolamine prior to (B) or following (C) cocaine administration. Profiles illustrate mean peak effects in response to VTA microinfusions of PBS or scopolamine (scop, 10 Jg/0.5 Jl) prior to or following intraperitoneal injection (IP) of saline (10 mL/kg) or cocaine (10 mg/kg), with respect to pre‐drug baseline responses (100%) (A). Timezero indicates the start of the train of 20 pulses at 50 Hz. *Significantly higher dopamine levels following cocaine compared to saline injection. #Significantly lower dopamine levels following scopolamine compared to PBS infusion.
Intra‐VTA infusion of scopolamine 5 min before cocaine administration blocked cocaine‐induced elevations in LDT stimulation‐evoked dopamine release in the NAc (Figures 8A and B) and attenuated these increases when administered at the peak effect of cocaine (15 min postinjection) (Figures 8A and C). Using in vivo microdialysis, similar studies have reported inhibitory effects of atropine, a nonsubtype selective mAChR antagonist, on intravenous cocaine‐induced increases in extracellular NAc dopamine levels [201]. Thus, LDT cholinergic inputs to DA neurons in the VTA, via activation of mAChRs, are likely involved in modulating the enhancing effects of cocaine on dopamine synaptic concentrations in the NAc. These findings also offer a potential mechanistic explanation of early observations that rhesus monkeys trained to self‐administer cocaine decrease their intake when administered a mAChR antagonist [212]. Together with evidence that the M5 mAChR subtype is important in mediating cocaine‐associated reinforcement and withdrawal, this suggests that the development of antagonists aimed at selectively disrupting M5 mAChR function may be valuable in reducing abuse liability of psychostimulant drugs, and possibly may prove to be an important pharmacological target for the treatment of psychopathological diseases such as schizophrenia.
Schizophrenia
Schizophrenia is a complex and devastating neuropsychiatric disorder that affects approximately 0.5% of the world's population [213]. Schizophrenia is characterized by positive symptoms, such as delusions, hallucinations, and thought derailment and negative symptoms, such as lack of volition, agoraphobia, and flat affect, including cognitive impairments [214]. Coined “the dopamine hypothesis,” this initial theory for the neurochemical basis of schizophrenia centered on DA overactivity in the CNS which emerged from the observed correlation of therapeutic efficacy (particularly on positive symptoms of schizophrenia) of antipsychotic medication and DA receptor antagonism [215, 216, 217]. However, this simplified perspective of unilateral involvement of central DA systems in such a complex psychopathological disorder, has given way to inclusion of dysfunction in potentially more critical neurotransmitters such as glutamate and its interactions with DA systems [218, 219].
Regardless, although dopamine was originally regarded as a principle neurotransmitter involved in the pathogenesis of schizophrenia, compelling evidence remains implicating dysfunction in central cholinergic systems and their associated muscarinic and nicotinic receptors in schizophrenia [220]. Pharmacological investigations have demonstrated that AChsterase inhibitors, such as donepezil (Aricept), galantamine (Razadyne), rivastigmine (Exelon), and tacrine (Cognex), which reduce metabolism of ACh and thereby increase synaptic ACh levels, are effective in treating psychotic symptoms expressed in Alzheimer's patients and in Parkinson's‐induced dementia [221], while anticholinergic drugs, particularly mAChR antagonists administered at high doses, can induce psychosis [222]. Additionally, anticholinergic drugs have been shown to reduce certain negative symptoms in schizophrenia, while in contrast cholinergic agonists elicit negative type schizophrenic symptoms [223, 224, 225].
Together, these observations point to the involvement of alterations in the functioning of both central mAChRs and nAChRs in schizophrenia. For example, postmortem biochemical and histological analysis of schizophrenic brains has shown deficits in M2/M4 mAChRs, as well as a loss in striatal cholinergic interneurons [226]. We have previously shown that selective blockade of mesopontine M2 mACh autoreceptors results in excessive cholinergic activity at the level of DA cells in the midbrain and increases forebrain dopamine transmission [176], to a degree that would be expected to evoke a psychotic state that includes tactile, visual, auditory, and olfactory hallucinations [220, 227]. Also, deletion of M5 mAChRs has been shown to increase gene expression of dopamine D2 receptors, notably within the striatum, hypothalamus, hindbrain and tectum, although it is unclear whether presynaptic or postsynaptic D2 receptors are increased under these conditions [228]. This suggests a developmental compensatory mechanism whereby an upregulation of D2 receptor expression in the forebrain occurs in response to disruption of M5 mediated excitatory input from mesopontine cholinergic neurons to midbrain DA neurons and loss of M5 facilitatory effects in the striatum, which may be important since striatal D2 receptor densities have been shown to be upregulated in schizophrenic brains [229, 230]. Interestingly, M5 mutant mice show improved latent inhibition [228]. Since reduced dopamine transmission in the NAc, as a result of pharmacological D2 receptor blockade, is related to an improvement in latent inhibition [231, 232], the diminished LDT‐evoked NAc dopamine release observed in M5 mutant mice may account for these findings [184]. Thus, as poor latent inhibition has been observed in nonmedicated patients with acute schizophrenia [233], these latter findings suggest that increased M5 mAChR activity at the level of the SNc or striatum may be significant to this disorder. Indeed, several studies have shown that the human M5 gene is localized to chromosome 15q12–15 linked with schizophrenia [228, 234]. Thus, in light of evidence that mesopontine cholinergic hyperactivity may contribute to the pathophysiology of schizophrenia [220], several behavioral, eletrophysiological, and neurochemical studies have pointed towards the utility of muscarinic agents, in particular M5 selective blockers, in the treatment of schizophrenia [227].
Individuals with schizophrenia are also known to smoke cigarettes at much higher rates than normal, which may reflect an increased tendency to self‐medicate with nicotine, further implicating the cholinergic system [235, 236]. Patients with schizophrenia have been found to have fewer α4β2 and α7 nAChRs, and a gene locus harbouring the α7 nAChR has been linked with schizophrenia [237, 238, 239]. Furthermore, administration of α7 antagonists induces similar sensory gating deficits to those seen in schizophrenia [217, 240]. Thus, further development of specific nAChR partial agonists may prove beneficial in treating symptoms of schizophrenia, as recent trials of selective α7 agonists appear promising [241, 242].
Conclusion
Progressive degeneration of nigrostriatal DA neurons is well known to be the major pathological characteristic of Parkinson's disease, while disorders such as drug addiction and schizophrenia have been associated with complex changes in the activity of DA neurons in the mesocorticolimbic system. Neurotransmission in these DA neuronal systems appear to be critically dependent upon ACh‐rich nuclei within the pons region of the hindbrain, the PPT and LDT, which represent the only known cholinergic projections to midbrain DA cells in the SNc and VTA, respectively. Additionally, ACh‐containing interneurons in the striatum also constitute a neural substrate for dopamine‐ACh interactions and serve as modulators of striatal output necessary for production of fluid voluntary movements. Preclinical and clinical evidence suggests that DA and cholinergic systems operate in a dynamic balance, with disruptions often leading to neurological and psychiatric disorders. Treatments for ameliorating the symptoms associated with such disorders have been and remain focused on restoring the balance between these two and other neurotransmitter systems. However, the complex modulatory actions of ACh and the varying roles of mAChR and nAChR subtypes at multiple levels of the nigrostriatal and mesocorticolimbic DA systems obscure a definitive solution to restoring functional dopamine‐ACh interactions. Further research and development of subtype‐selective mAChR and nAChR pharmaceuticals that can differentially affect central DA systems at the level of their cellular sites of origin and terminal target structures will ultimately lead to improvements in the treatments of neuropsychiatric disorders associated with dysfunctional dopamine–ACh interactions.
Conflict of Interest
The authors have no conflict of interest.
References
- 1. Albanese A, Minciacchi D. Organization of the ascending projections from the ventral tegmental area: A multiple fluorescent retrograde tracer study in the rat. J Comp Neurol 1983;216:406–420. [DOI] [PubMed] [Google Scholar]
- 2. Bjorklund A, Lindvall O. Dopamine‐containing systems in the CNS. In: Bjorklund A, Hokfelt T, editors. Handbook of chemical neuroanatomy. Classical transmitters in the CNS (Volume 2). Amsterdam : Elsevier, 1984;55–122. [Google Scholar]
- 3. Fallon GH, Moore RY. Catecholamine innervation of the basal forebrain. IV. Topography of the dopamine projection to the basal forebrain and neostriatum. J Comp Neurol 1978;180:545–580. [DOI] [PubMed] [Google Scholar]
- 4. Le Moal M, Simon H. Mesocorticolimbic dopaminergic network: Functional and regulatory roles. Physiol Rev 1991;71:155–234. [DOI] [PubMed] [Google Scholar]
- 5. Jellinger KA. Recent developments in the pathology of Parkinson's disease. J Neural Transm Suppl 2002;62:347–376. [DOI] [PubMed] [Google Scholar]
- 6. Pierce RC, Kumaresan V. The mesolimbic dopamine system: The final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev 2006;30:215–238. [DOI] [PubMed] [Google Scholar]
- 7. Laruell M, Kegeles L, Abi‐Dargham A. Glutamate, dopamine, and schizophrenia: From pathophysiology to treatment. Ann NY Acad Sci 2003;103:138–158. [DOI] [PubMed] [Google Scholar]
- 8. Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry 2007;12:232–246. [DOI] [PubMed] [Google Scholar]
- 9. Bonner TI. Domains of muscarinic acetylcholine receptors that confer specificity of G protein coupling. Trends Pharmacol Sci 1992;13:48–50. [DOI] [PubMed] [Google Scholar]
- 10. Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 1998;50:279–290. [PubMed] [Google Scholar]
- 11. Hulme EC, Birdsall NJ, Buckley NJ. Muscarinic receptor subtypes. Annu Rev Pharmacol Toxicol 1990;30:633–673. [DOI] [PubMed] [Google Scholar]
- 12. Raiteri M, Leardi R, Marchi M. Heterogeneity of presynaptic muscarinic receptors regulating neurotransmitter release in the rat brain. J Pharmacol Exp Ther 1984;228:209–214. [PubMed] [Google Scholar]
- 13. Xu M, Mizobe F, Yamamoto T, Kato T. Differential effects of M1‐ and M2‐muscarinic drugs on striatal dopamine release and metabolism in freely moving rats. Brain Res 1989;495:232–242. [DOI] [PubMed] [Google Scholar]
- 14. Zhou F, Wilson C, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol 2002;53:590–605. [DOI] [PubMed] [Google Scholar]
- 15. Plenz D, Kitai ST. Organotypic cortex–striatum–mesencephalon cultures: The nigrostriatal pathway. Neurosci Lett 1996;209:177–180. [DOI] [PubMed] [Google Scholar]
- 16. Dentresangle C, Le Cavorsin M, Savasta M, Leviel V. Increased extracellular DA and normal evoked DA release in the rat striatum after a partial lesion of the substantia nigra. Brain Res 2001;893:178–185. [DOI] [PubMed] [Google Scholar]
- 17. Costa G, Abin‐Carriquiry J, Dajas F. Nicotine prevents striatal dopamine loss produced by 6‐hydroxydopamine lesion in the substantia nigra. Brain Res 2001;888:336–342. [DOI] [PubMed] [Google Scholar]
- 18. Wickens J. Striatal dopamine in motor activation and reward‐mediated learning: Steps towards a unifying model. J Neural Transm 1990;80:9–31. [DOI] [PubMed] [Google Scholar]
- 19. Hauber W. Involvement of basal ganglia transmitter systems in movement initiation. Prog Neurobiol 1998;56:507–540. [DOI] [PubMed] [Google Scholar]
- 20. Redgrave P, Prescott T, Gurney K. Is the short‐latency dopamine response too short to signal reward error? Trends Neurosci 1999;22:146–151. [DOI] [PubMed] [Google Scholar]
- 21. Knott C, Wilkin GP, Stern G. Astrocytes and microglia in the substantia nigra and caudate‐putamen in Parkinson's disease. Parkinsonism Relat Disord 1999;5:115–122. [DOI] [PubMed] [Google Scholar]
- 22. Lev N, Melamed E, Offen D. Apoptosis and Parkinson's disease. Prog Neuropsychopharmacol Biol Psychiatry 2003;27:245–250. [DOI] [PubMed] [Google Scholar]
- 23. Wolters EC, Francot C. Mental dysfunction in Parkinson's disease. Parkinsonism Relat Disord 1998;4:107–112. [DOI] [PubMed] [Google Scholar]
- 24. Sharp T, Zetterstrom T, Ljunberg T, Ungerstedt U. A direct comparison of amphetamine induced behaviours and regional brain dopamine release in the rat using intracerebral dialysis. Brain Res 1987;401:322–330. [DOI] [PubMed] [Google Scholar]
- 25. Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr., Sibley DR. D1 and D2 dopamine receptor‐regulated gene expression of striatonigral and striatopallidal neurons. Science 1990;250:1429–1432. [DOI] [PubMed] [Google Scholar]
- 26. Mink JW. The basal ganglia: Focused selection and inhibition of competing motor programs. Prog Neurobiol 1996;50:381–425. [DOI] [PubMed] [Google Scholar]
- 27. Galvan A, Wichmann T. Pathophysiology of Parkinsonism. Clin Neurophysiol 2008;119:1459–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wooten GF. Anatomy and function of dopamine receptors: Understanding the pathophysiology of fluctuations in Parkinson's disease. Parkinsonism Relat Disord 2001;8:79–83. [DOI] [PubMed] [Google Scholar]
- 29. Singer HS, Minzer K. Neurobiology of Tourette's syndrome: Concepts of neuroanatomic localization and neurochemical abnormalities. Brain Dev 2003;25:S70–S84. [DOI] [PubMed] [Google Scholar]
- 30. Livingstone PD, Wonnacott S. Nicotinic acetylcholine receptors and the ascending dopamine pathways. Biochem Pharmacol 2009;78:744–755. [DOI] [PubMed] [Google Scholar]
- 31. Oakman S, Faris P, Cozzari C, Hartman B. Characterisation of the extent of pontomesencephalic cholinergic neurons’ projections to the thalamus: Comparison with projections to midbrain dopaminergic groups. Neuroscience 1999;94:529–547. [DOI] [PubMed] [Google Scholar]
- 32. Oakman S, Faris P, Kerr P, Cozzari C, Hartman B. Distribution of pontomesencephalic cholinergic neurons projecting to substantia nigra differs significantly from those projecting to ventral tegmental area. J Neurosci 1995;15:5859–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Semba K, Fibiger H. Afferent connections of the laterodorsal and the pedunculopontine tegmental nucleus in the rat: A retro‐ and antero‐grade transport and immunohistochemical study. J Comp Neurol 1992;323:387–410. [DOI] [PubMed] [Google Scholar]
- 34. Wang H, Morales M. Pedunculopontine and laterodorsal tegmental nuclei contain distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur J Neurosci 2009;29:340–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol 1991;37:475–524. [DOI] [PubMed] [Google Scholar]
- 36. Bevan MD, Bolam JP. Cholinergic, GABAergic, and glutamate‐enriched inputs from the mesopontine tegmentum to the subthalamic nucleus in the rat. J Neurosci 1995;15:7105–7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clarke NP, Bevan MD, Cozzari C, Hartman, BK , Bolam JP. Glutamate‐enriched cholinergic synaptic terminals in the entopeduncular nucleus and subthalamic nucleus of the rat. Neuroscience 1997;81:371–385. [DOI] [PubMed] [Google Scholar]
- 38. Woolf NJ, Butcher LL. Cholinergic systems in the rat brain: III. Projections from the pontomesencephalic tegmentum to the thalamus, tectum, basal ganglia, and basal forebrain. Brain Res Bull 1986;16:603–637. [DOI] [PubMed] [Google Scholar]
- 39. Forster GL, Blaha CD. Pedunculopontine tegmental stimulation evokes striatal dopamine efflux by activation of acetylcholine and glutamate receptors in the midbrain and pons of the rat. Eur J Neurosci 2003;17:751–762. [DOI] [PubMed] [Google Scholar]
- 40. Miller AD, Blaha CD. Nigrostriatal dopamine release modulated by mesopontine muscarinic receptors. Neuroreport 2004;15:1805–1808. [DOI] [PubMed] [Google Scholar]
- 41. Chapman CA, Yeomans JS, Blaha CD, Blackburn JR. Increased striatal dopamine efflux follows scopolamine administered systemically or to the tegmental pedunculopontine nucleus. Neuroscience 1997;76:177–186. [DOI] [PubMed] [Google Scholar]
- 42. Mathur A, Shandarin A, LaViolette SR, Parker J, Yeomans JS. Locomotion and stereotypy induced by scopolamine: Contributions of muscarinic receptors near the pedunculopontine tegmental nucleus. Brain Res 1997;775:144–155. [DOI] [PubMed] [Google Scholar]
- 43. Vilaró MT, Wiederhold KH, Palacios JM, Mengod G. Muscarinic M2 receptor mRNA expression and receptor binding in cholinergic and non‐cholinergic cells in the rat brain: A correlative study using in situ hybridization histochemistry and receptor autoradiography. Neuroscience 1992;47:367–393. [DOI] [PubMed] [Google Scholar]
- 44. Vilaró MT, Palacios JM, Mengod G. Multiplicity of muscarinic autoreceptor subtypes? Comparison of the distribution of cholinergic cells and cells containing mRNA for five subtypes of muscarinic receptors in the rat brain. Brain Res Mol Brain Res 1994;21:30–46. [DOI] [PubMed] [Google Scholar]
- 45. Luebke JI, McCarley RW, Greene RW. Inhibitory action of muscarinic agonists on neurons in the rat laterodorsal tegmental nucleus in vitro. J Neurophysiol 1993;70:2128–2135. [DOI] [PubMed] [Google Scholar]
- 46. Leonard CS, Llinás R. Serotonergic and cholinergic inhibition of mesopontine cholinergic neurons controlling REM sleep: An in vitro electrophysiological study. Neuroscience 1994;59:309–330. [DOI] [PubMed] [Google Scholar]
- 47. Moon‐Edley SM, Graybiel AM. The afferent and efferent connections of the feline nucleus tegmenti pedunculopontinus, pars compacta. J Comp Neurol 1983;217:187–215. [DOI] [PubMed] [Google Scholar]
- 48. Blaha CD, Winn P. Modulation of dopamine efflux in the striatum following cholinergic stimulation of the substantia nigra in intact and pedunculopontine tegmental nucleus‐lesioned rats. J Neurosci 1993;13:1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lokwan SJ, Overton PG, Berry MS, Clark D. Stimulation of the pedunculopontine tegmental nucleus in the rat produces burst firing in A9 dopaminergic neurons. Neuroscience 1999;92:245–254. [DOI] [PubMed] [Google Scholar]
- 50. Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci 2003;6:968–973. [DOI] [PubMed] [Google Scholar]
- 51. Kitai ST, Shepard PD, Callaway JC, Scroggs R. Afferent modulation of dopamine neuron firing patterns. Curr Opin Neurobiol 1999;9:690–697. [DOI] [PubMed] [Google Scholar]
- 52. Lee MS, Rinne JO, Marsden CD. The pedunculopontine nucleus: Its role in the genesis of movement disorders. Yonsei Med J 2000;41:167–184. [DOI] [PubMed] [Google Scholar]
- 53. Hammond C, Rouzaire‐Dubois B, Féger J, Jackson A, Crossman AR. Anatomical and electrophysiological studies on the reciprocal projections between the subthalamic nucleus and nucleus tegmenti pedunculopontinus in the rat. Neuroscience 1983;9:41–52. [DOI] [PubMed] [Google Scholar]
- 54. Lee HJ, Rye DB, Hallanger AE, Levey AI, Wainer BH. Cholinergic vs. noncholinergic efferents from the mesopontine tegmentum to the extrapyramidal motor system nuclei. J Comp Neurol 1988;275:469–492. [DOI] [PubMed] [Google Scholar]
- 55. Franklin KB, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego : Academic Press, 1997. [Google Scholar]
- 56. Dugast C, Suaud‐Chagny MF, Gonon F. Continuous in vivo monitoring of evoked dopamine release in the rat nucleus accumbens by amperometry. Neuroscience 1994;62:647–654. [DOI] [PubMed] [Google Scholar]
- 57. Lester DB, Miller AD, Pate TD, Blaha CD. Midbrain acetylcholine and glutamate receptors modulate accumbal dopamine release. Neuroreport 2008;19:991–995. [DOI] [PubMed] [Google Scholar]
- 58. Michael DJ, Wightman RM. Electrochemical monitoring of biogenic amine neurotransmission in real time. J Pharm Biomed Anal 1999;19:33–46. [DOI] [PubMed] [Google Scholar]
- 59. Flores G, Hernandez S, Rosales MG, Sierra A, Martines‐Fong D, Flores‐Hernandez J, Aceves J. M3 muscarinic receptors mediate cholinergic excitation of the spontaneous activity of subthalamic neurons in the rat. Neurosci Lett 1996;203:203–206. [DOI] [PubMed] [Google Scholar]
- 60. Feger J, Hammond C, Rouzaire‐Dubois B. Pharmacological properties of acetylcholine‐induced excitation of subthalamic nucleus neurones. Br J Pharmacol 1979;65:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Levey AI, Edmunds SM, Heilman CJ, Desmond TJ, Frey KA. Localization of muscarinic M3 receptor protein and M3 receptor binding in rat brain. Neuroscience 1994;63:207–221. [DOI] [PubMed] [Google Scholar]
- 62. Shen KZ, Johnson SW. Presynaptic dopamine D2 and muscarine M3 receptors inhibit excitatory and inhibitory transmission to rat subthalamic neurones in vitro. J Physiol 2000;525:331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee KH, Blaha CD, Harris BT, et al Dopamine efflux in the rat striatum evoked by electrical stimulation of the subthalamic nucleus: Potential mechanism of action in Parkinson's disease. Eur J Neurosci 2006;23:1005–1014. [DOI] [PubMed] [Google Scholar]
- 64. Lacey MG, Calabresi P, North RA. Muscarine depolarizes rat substantia nigra zona compacta and ventral tegmental neurons in vitro through M1‐like receptors. J Pharmacol Exp Ther 1990;253:395–400. [PubMed] [Google Scholar]
- 65. Vilaró MT, Palacios JM, Mengod G. Localization of m5 muscarinic receptor mRNA in rat brain examined by in situ hybridization histochemistry. Neurosci Lett 1990;114:154–159. [DOI] [PubMed] [Google Scholar]
- 66. Reever CM, Ferrari‐DiLeo G, Flynn DD. The M5 (m5) receptor subtype: Fact or fiction? Life Sci 1997;60:1105–1112. [DOI] [PubMed] [Google Scholar]
- 67. Lichtensteiger W, Felix D, Lienhart R, Hefti F. A quantitative correlation between single unit activity and fluorescence intensity of dopamine neurones in zona compacta of substantia nigra, as demonstrated under the influence of nicotine and physostigmine. Brain Res 1976;117:85–103. [DOI] [PubMed] [Google Scholar]
- 68. Lichtensteiger W, Hefti F, Felix D, Huwyler T, Melamed E, Schlumpf M. Stimulation of nigrostriatal dopamine neurones by nicotine. Neuropharmacology 1982;21:963–968. [DOI] [PubMed] [Google Scholar]
- 69. Göldner FM, Dineley KT, Patrick JW. Immunohistochemical localization of the nicotinic acetylcholine receptor subunit alpha6 to dopaminergic neurons in the substantia nigra and ventral tegmental area. Neuroreport 1997;8:2739–2742. [DOI] [PubMed] [Google Scholar]
- 70. Wonnacott S, Barik J, Dickinson J, Jones IW. Nicotinic receptors modulate transmitter cross talk in the CNS: Nicotinic modulation of transmitters. J Mol Neurosci 2006;30:137–140. [DOI] [PubMed] [Google Scholar]
- 71. Charpantier E, Barnéoud P, Moser P, Besnard F, Sgard F. Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. Neuroreport 1998;9:3097–3101. [DOI] [PubMed] [Google Scholar]
- 72. Champtiaux N, Han ZY, Bessis A, et al Distribution and pharmacology of alpha 6‐containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci 2002;22:1208–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Klink R, De Kerchove d’Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 2001;21:1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Parent A, Hazrati L. Functional anatomy of the basal ganglia. I. The cortico‐basal ganglia–thalamo–cortical loop. Brain Res Rev 1995;20:91–127. [DOI] [PubMed] [Google Scholar]
- 75. Pisani A, Bernardi G, Ding J, Surmeier DJ. Re‐emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci 2007;30:545–553. [DOI] [PubMed] [Google Scholar]
- 76. Apicella P. Leading tonically active neurons of the striatum from reward detection to context recognition. Trends Neurosci 2007;30:299–306. [DOI] [PubMed] [Google Scholar]
- 77. James MK, Cubeddu LX. Pharmacologic characterization and functional role of muscarinic autoreceptors in the rabbit striatum. J Pharmacol Exp Ther 1987;240:203–215. [PubMed] [Google Scholar]
- 78. Calabresi P, Centonze D, Pisani A, Sancesario G, North RA, Bernardi G. Muscarinic IPSPs in rat striatal cholinergic interneurones. J Physiol 1998;510:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bernard V, Laribi O, Levey AI, Bloch B. Subcellular redistribution of m2 muscarinic acetylcholine receptors in striatal interneurons in vivo after acute cholinergic stimulation. J Neurosci 1998;18:10207–10218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhang W, Yamanda M, Gomeza J, Basile AS, Wess J. Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1–M5 muscarinic receptor knock‐out mice. J Neurosci 2002;22:6347–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Galarraga E, Hernanez‐Lopez S, Reyes A, Miranda I, Bermudez‐Rattoni F, Vilchis C, Bargas J. Cholinergic modulation of neostriatal output: A functional antagonism between different types of muscarinic receptors. J Neurosci 1999;19:3629–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman‐Rakic PS. Regional, cellular, and subcellular variations in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci 1995;15:7821–7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N‐type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane‐delimited, protein‐kinase‐C‐insensitive pathway. J Neurophysiol 1997;77:1003–1015. [DOI] [PubMed] [Google Scholar]
- 84. Imperato A, Obinu MC, Casu MA, Mascia MS, Dazzi L, Gessa GL. Evidence that neuroleptics increase striatal acetylcholine release through stimulation of dopamine D1 receptors. J Pharmacol Exp Ther 1993;266:557–562. [PubMed] [Google Scholar]
- 85. DeBoer P, Abercrombie ED. Physiological release of striatal acetylcholine in vivo: Modulation by D1 and D2 dopamine receptor subtypes. J Pharmacol Exp Ther 1996;277:775–783. [PubMed] [Google Scholar]
- 86. DeBoer P, Heeringa MJ, Abercrombie ED. Spontaneous release of acetylcholine in striatum is preferentially regulated by inhibitory dopamine D2 receptors. Eur J Pharmacol 1996;317:257–262. [DOI] [PubMed] [Google Scholar]
- 87. Lehmann J, Langer SZ. Muscarinic receptors on dopamine terminals in the cat caudate nucleus: Neuromodulation of [3H]dopamine release in vitro by endogenous acetylcholine. Brain Res 1982;248:61–69. [DOI] [PubMed] [Google Scholar]
- 88. Smolders I, Bogaert L, Ebinger G, Michotte Y. Muscarinic modulation of striatal dopamine, glutamate, and GABA release, as measured with in vivo microdialysis. J Neurochem 1997;68:1942–1948. [DOI] [PubMed] [Google Scholar]
- 89. Zhou FM, Wilson C, Dani JA. Muscarinic and nicotinic cholinergic mechanisms in the mesostriatal dopamine systems. Neuroscientist 2003;9:23–36. [DOI] [PubMed] [Google Scholar]
- 90. Weiner DM, Levey AI, Brann MR. Expression of muscarinic acetylcholine and dopamine receptor mRNAs in rat basal ganglia. Proc Natl Acad Sci 1990;87:7050–7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bernard V, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing muscarinic receptor genes. J Neurosci 1992;12:3591–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kemel M‐L, Desban M, Glowinski J, Gauchyh C. Distinct presynaptic control of dopamine release in striosomal and matrix areas of the cat caudate nucleus. Proc Natl Acad Sci USA 1989; 86:9006–9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Marchi M, Sanguineti P, Raiteri M. Muscarinic receptors mediate direct inhibition of GABA release from rat striatal nerve terminals. Neurosci Lett 1990;116:347–351. [DOI] [PubMed] [Google Scholar]
- 94. Ronken E, Mulder AH, Schoffelmeer AN. Interacting presynaptic‐opioid and GABAA receptors modulate dopamine release from rat striatal synaptosomes. J Neurochem 1993;61:1634–1639. [DOI] [PubMed] [Google Scholar]
- 95. Sugita S, Uchimura N, Jiang ZG, North RA. Distinct muscarinic receptors inhibit release of gamma‐aminobutyric acid and excitatory amino acids in mammalian brain. Proc Natl Acad Sci USA 1991;88:2608–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kaiser S, Wonnacott S. α‐Bungarotoxin‐sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol 2000;58:312–318. [DOI] [PubMed] [Google Scholar]
- 97. Marshall DL, Redfern PH, Wonnacott S. Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: Comparison of naive and chronic nicotine‐treated rats. J Neurochem 1997;68:1511–1519. [DOI] [PubMed] [Google Scholar]
- 98. Jones IW, Bolam JP, Wonnacott S. Presynaptic localisation of the nicotinic acetylcholine receptor beta2 subunit immunoreactivity in rat nigrostriatal dopaminergic neurones. J Comp Neurol 2001;439:235–247. [DOI] [PubMed] [Google Scholar]
- 99. Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci 2001;4:1224–1229. [DOI] [PubMed] [Google Scholar]
- 100. Picciotto MR, Zoli M, Rimondini R, et al Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 1998;391:173–177. [DOI] [PubMed] [Google Scholar]
- 101. Quik M, McIntosh JM. Striatal alpha6* nicotinic acetylcholine receptors: Potential targets for Parkinson's disease therapy. J Pharmacol Exp Ther 2006;316:481–489. [DOI] [PubMed] [Google Scholar]
- 102. Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci 1999;22:123–144. [DOI] [PubMed] [Google Scholar]
- 103. Samii A, Nutt JG, Ransom BR. Parkinson's disease. Lancet 2004;363:1783–1793. [DOI] [PubMed] [Google Scholar]
- 104. Fearnley JM, Lees AJ. Ageing and Parkinson's disease: Substantia nigra regional selectivity. Brain 1991;114:2283–2301. [DOI] [PubMed] [Google Scholar]
- 105. Olanow CW, Agid Y, Mizuno Y, et al Levodopa in the treatment of Parkinson's disease: Current controversies. Mov Disord 2004;19:997–1005. [DOI] [PubMed] [Google Scholar]
- 106. Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clark CE, Lang AE. A five year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. N Engl J Med 2000;342:1484–1491. [DOI] [PubMed] [Google Scholar]
- 107. Rinne JO, Ma SY, Lee MS, Collan Y, Röyttä M. Loss of cholinergic neurons in the pedunculopontine nucleus in Parkinson's disease is related to disability of the patients. Parkinsonism Relat Disord 2008;14:553–557. [DOI] [PubMed] [Google Scholar]
- 108. Pahapill PA, Lozano AM. The pedunculopontine nucleus and Parkinson's disease. Brain 2000;123:1767–1783. [DOI] [PubMed] [Google Scholar]
- 109. Zweig RM, Jankel WR, Hedreen JC, Mayeux R, Price DL. The pedunculopontine nucleus in Parkinson's disease. Ann Neurol 1989;26:41–46. [DOI] [PubMed] [Google Scholar]
- 110. Nomoto M, Kaseda S, Iwata S, Shimizu T, Fukuda T, Nakagawa S. The metabolic rate and vulnerability of dopaminergic neurons, and adenosine dynamics in the cerebral cortex, nucleus accumbens, caudate nucleus, and putamen of the common marmoset. J Neurol 2000;247(Suppl 5):V16–V22. [DOI] [PubMed] [Google Scholar]
- 111. James S, Richardson PJ. Production of adenosine from extracellular ATP at the striatal cholinergic synapse. J Neurochem 1993;60:219–227. [DOI] [PubMed] [Google Scholar]
- 112. Schiffmann SN, Libert F, Vassart G, Vanderheghen JJ. Distribution of adenosine A2 receptor mRNA in the human brain. Neurosci Lett 1991;130:177–181. [DOI] [PubMed] [Google Scholar]
- 113. Damsma G, Tham CS, Robertson GS, Fibiger HC. Dopamine D1 receptor stimulation increases striatal acetylcholine release in the rat. Eur J Pharmacol 1990;186:335–338. [DOI] [PubMed] [Google Scholar]
- 114. Bertorelli R, Consolo S. D1 and D2 dopaminergic regulation of acetylcholine release from striata of freely moving rats. J Neurochem 1990;54:2145–2148. [DOI] [PubMed] [Google Scholar]
- 115. Mayfield RD, Jones BA, Miller HA, Simosky JK, Larson GA, Zahniser NR. Modulation of endogenous GABA release by an antagonistic adenosine A1/dopamineD1 receptor interaction in rat brain limbic regions but not basal ganglia. Synapse 1999;33:274–281. [DOI] [PubMed] [Google Scholar]
- 116. Geiger JD. Localization of [3H]cyclohexyladenosine and [3H]nitrobenzylthioinosine binding sites in rat striatum and superior colliculus. Brain Res 1986;363:404–407. [DOI] [PubMed] [Google Scholar]
- 117. Ochi M, Shiozaki S, Kase H. Adenosine A(2A) receptor‐mediated modulation of GABA and glutamate release in the output regions of the basal ganglia in a rodent model of Parkinson's disease. Neuroscience 2004;127:223–231. [DOI] [PubMed] [Google Scholar]
- 118. Ferré S, Rubio A, Fuxe K. Stimulation of adenosine A2 receptors induces catalepsy. Neurosci Lett 1991;130:162–164. [DOI] [PubMed] [Google Scholar]
- 119. Pinna A. Novel investigational adenosine A(A2) receptor antagonists for Parkinson's disease. Expert Opin Investig Drugs 2009;18:1619–1631. [DOI] [PubMed] [Google Scholar]
- 120. Morelli M, Carta AR, Jenner P. Adenosine A2A receptors and Parkinson's disease. Handb Exp Pharmacol 2009;193:589–615. [DOI] [PubMed] [Google Scholar]
- 121. Katzenschlager R, Sampaio C, Costa J, Lees A. Anticholinergics for symptomatic management of Parkinson's disease. Cochrane Database Syst Rev 2003;2:CD003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Deleu D, Northway MG, Hanssens Y. An evidence‐based review of dopamine receptor agonists in the treatment of Parkinson's disease. Saudi Med J 2002;23:1165–1175. [PubMed] [Google Scholar]
- 123. Lees A. Alternatives to levodopa in the initial treatment of early Parkinson's disease. Drugs Aging 2005;22:731–740. [DOI] [PubMed] [Google Scholar]
- 124. Clarke CE. Medical management of Parkinson's disease. J Neurol Neurosurg Psychiatry 2002;72(Suppl 1):I22–I27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ding J, Guzman JN, Tkatch T, et al RGS4‐dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat Neurosci 2006;9:832–842. [DOI] [PubMed] [Google Scholar]
- 126. Betz AJ, McLaughlin PJ, Burgos M, Weber SM, Salamone JD. The muscarinic receptor antagonist tropicamide suppresses tremulous jaw movements in a rodent model of parkinsonian tremor: Possible role of M4 receptors. Psychopharmacology 2007;194:347–359. [DOI] [PubMed] [Google Scholar]
- 127. Tzavara ET, Bymaster FP, Davis RJ, et al M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: Relevance to the pathophysiology and treatment of related CNS pathologies. FASEB J 2004;18:1410–1412. [DOI] [PubMed] [Google Scholar]
- 128. Perry EK, Kilford L, Lees AJ, Burn DJ, Perry RH. Increased Alzheimer pathology in Parkinson's disease related to antimuscarinic drugs. Ann Neurol 2003;54:235–238. [DOI] [PubMed] [Google Scholar]
- 129. Meshul CK, Kamel D, Moore C, Kay TS, Krentz L. Nicotine alters striatal glutamate function and decreases the apomorphine‐induced contralateral rotations in 6‐OHDA‐lesioned rats. Exp Neurol 2002;175:257–274. [DOI] [PubMed] [Google Scholar]
- 130. Schneider JS, Van Velson M, Menzaghi F, Lloyd GK. Effects of the nicotinic acetylcholine receptor agonist SIB‐1508Y on object retrieval performance in MPTP‐treated monkeys: Comparison with levodopa treatment. Ann Neurol 1998;43:311–317. [DOI] [PubMed] [Google Scholar]
- 131. Janhunen S, Ahtee L. Differential nicotinic regulation of the nigrostriatal and mesolimbic dopaminergic pathways: Implications for drug development. Neurosci Biobehav Rev 2007;31:287–314. [DOI] [PubMed] [Google Scholar]
- 132. Parain K, Hapdey C, Rousselet E, Marchand V, Dumery B, Hirsch EC. Cigarette smoke and nicotine protect dopaminergic neurons against the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine Parkinsonian toxin. Brain Res 2003;984:224–232. [DOI] [PubMed] [Google Scholar]
- 133. Quik M, Bordia T, O’Leary K. Nicotinic receptors as CNS targets for Parkinson's disease. Biochem Pharmacol 2007;74:1224–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Quik M, Bordia T, Forno L, McIntosh JM. Loss of alphaconotoxinMII‐ and A85380‐sensitive nicotinic receptors in Parkinson's disease striatum. J Neurochem 2004;88;668–679. [DOI] [PubMed] [Google Scholar]
- 135. Bohr IJ, Ray MA, McIntosh JM, et al Cholinergic nicotinic receptor involvement in movement disorders associated with Lewy body diseases. An autoradiography study using [(125)l]alpha‐conotoxinMII in the striatum and thalamus. Exp Neurol 2005;191:292–300. [DOI] [PubMed] [Google Scholar]
- 136. Gotti C, Clementi F. Neuronal nicotinic receptors: From structure to pathology. Prog Neurobiol 2004;74:363–396. [DOI] [PubMed] [Google Scholar]
- 137. McCallum SE, Parameswaran N, Bordia T, McIntosh JM, Grady SR, Quik M. Decrease in apha3*/alpha6* nicotinic receptors but not nicotine‐evoked dopamine release in monkey brain after nigrostriatal damage. Mol Pharmacol 2005;68:737–746. [DOI] [PubMed] [Google Scholar]
- 138. Janhunen S, Ahtee L. Comparison of the effects of nicotine and epibatidine on the striatal extracellular dopamine. Eur J Pharmacol 2004;494:167–177. [DOI] [PubMed] [Google Scholar]
- 139. Spanagel R, Weiss F. The dopamine hypothesis of reward: Past and current status. Trends Neurosci 1999;22:521–527. [DOI] [PubMed] [Google Scholar]
- 140. Blaha CD, Phillips AG. Application of in vivo electrochemistry to the measurement of changes in dopamine release during intracranial self‐stimulation. J Neurosci Meth 1990;34:125–133. [DOI] [PubMed] [Google Scholar]
- 141. Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol 1989;40:191–225. [DOI] [PubMed] [Google Scholar]
- 142. Bardo MT. Neuropharmacological mechanisms of drug reward: Beyond dopamine in the nucleus accumbens. Crit Rev Neurobiol 1998;12:37–67. [DOI] [PubMed] [Google Scholar]
- 143. Salamone JD. The behavioral neurochemistry of motivation: Methodological and conceptual issues in studies of the dynamic activity of nucleus accumbens dopamine. J Neurosci Meth 1996;64:137–149. [DOI] [PubMed] [Google Scholar]
- 144. Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 1988;85:5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Di Ciano P, Blaha CD, Phillips AG. Conditioned changes in dopamine oxidation currents in the nucleus accumbens of rats by stimuli paired with self‐administration or yoked‐administration of d‐amphetamine. Eur J Neurosci 1998;10:1121–1127. [DOI] [PubMed] [Google Scholar]
- 146. Pontieri FE, Tanda G, Di Chiara G. Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc Natl Acad Sci USA 1995;92:12304–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Schildein S, Agmo A, Huston JP, Schwarting RK. Intraaccumbens injections of substance P, morphine and amphetamine: Effects on conditioned place preference and behavioral activity. Brain Res 1998;790:185–194. [DOI] [PubMed] [Google Scholar]
- 148. Carr GD, White NM. Conditioned place preference from intra‐accumbens but not intra‐caudate amphetamine injections. Life Sci 1983;33:2551–2557. [DOI] [PubMed] [Google Scholar]
- 149. Phillips AG, LePaine FG, Fibiger HC. Dopaminergic mediation of reward produced by direct injection of enkephalin into the ventral tegmental area of the rat. Life Sci 1983;33:2505–2511. [DOI] [PubMed] [Google Scholar]
- 150. Groenenwegen HJ, Berendse HW, Meredith GE, Haber SN, Voorn P, Wolters JG, Lohman AHM. Functional anatomy of the ventral, limbic system‐innervated striatum. In: Willner P, Scheel‐Kruger J, editors. The mesolimbic dopamine system: From motivation to action. Chichester : Wiley, 1991;19–60. [Google Scholar]
- 151. Mogenson GJ, Brudzynski SM, Wu M, Yang CR, Yim CCY. From motivation to action: A review of limbic to nucleus accumbens to ventral pallidum to pedunculopontine tegmental nucleus circuitries involved in limbic‐motor integration. In: Kalivas PW, editors. Limbic motor circuits and neuropsychiatry. Boca Raton : CRC Press, 1993;193–236. [Google Scholar]
- 152. Usuda I, Tanaka K, Chiba T. Efferent projections of the nucleus accumbens in the rat with special reference to subdivision of the nucleus: Biotinylated dextran amine study. Brain Res 1998;797:73–93. [DOI] [PubMed] [Google Scholar]
- 153. Pecina S, Berridge KC. Opioid site in nucleus accumbens shell mediates eating and hedonic “liking” for food: Map based on microinjection Fos plumes. Brain Res 2000;863:71–86. [DOI] [PubMed] [Google Scholar]
- 154. Rodd‐Henricks ZA, McKinzie DL, Li TK, Murphy JM, McBride WJ. Cocaine is self‐administered into the shell but not the core of the nucleus accumbens of Wistar rats. J Pharmacol Exp Ther 2002;303:1216–1226. [DOI] [PubMed] [Google Scholar]
- 155. Ito R, Dalley JW, Howes SR, Robbins TW, Everitt BJ. Dissociation in conditioned dopamine release in the nucleus accumbens core and shell in response to cocaine cues and during cocaine‐seeking behavior in rats. J Neurosci 2000;20:7489–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Di Chiara G. Nucleus accumbens shell and core dopamine: Differential role in behavior and addiction. Behav Brain Res 2002;137:75–114. [DOI] [PubMed] [Google Scholar]
- 157. Williams SM, Goldman‐Rakic PS. Widespread origin of the primate mesofrontal dopamine system. Cereb Cortex 1998;8:321–345. [DOI] [PubMed] [Google Scholar]
- 158. Cools R. Role of dopamine in the motivational and cognitive control of behavior. Neuroscientist 2008;14:381–395. [DOI] [PubMed] [Google Scholar]
- 159. Brozoski TJ, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science 1979;205:929–932. [DOI] [PubMed] [Google Scholar]
- 160. Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted‐U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci 2007;10:376–384. [DOI] [PubMed] [Google Scholar]
- 161. Wang Y, Goldman‐Rakic PS. D2 receptor regulation of synaptic burst firing in prefrontal cortical pyramidal neurons. Proc Natl Acad Sci USA 2004;101:5093–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Carr D, Sesack S. Projections from the rat prefrontal cortex to the ventral tegmental area: Target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci 2000;20:3864–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Sesack SR, Bunney BS. Pharmacological characterization of the receptor mediating electrophysiological responses to dopamine in the rat medial prefrontal cortex: A microiontophoretic study. J Pharmacol Exp Ther 1989;248:1323–1333. [PubMed] [Google Scholar]
- 164. Thierry AM, Mantz J, Milla C, Glowinski J. Influence of the mesocortical/prefrontal dopamine neurons on their target cells. Ann NY Acad Sci 1988;537:101–111. [DOI] [PubMed] [Google Scholar]
- 165. Horger BA, Roth RH. The role of mesoprefrontal dopamine neurons in stress. Crit Rev Neurobiol 1996;10:395–418. [DOI] [PubMed] [Google Scholar]
- 166. Mantz J, Milla C, Glowinski J, Thierry A. Differential effects of ascending neurons containing dopamine and noradrenaline in the control of spontaneous activity and of evoked responses in the rat prefrontal cortex. Neuroscience 1988;27:517–526. [DOI] [PubMed] [Google Scholar]
- 167. Mantz J, Thierry AM, Glowinski J. Effect of noxious tail pinch on the discharge rate of mesocortical and mesolimbic dopamine neurons: Selective activation of the mesocortical system. Brain Res 1989;476:377–381. [DOI] [PubMed] [Google Scholar]
- 168. Imperato A, Puglisi‐Allegra S, Casolini P, Angelucci L. Changes in brain dopamine and acetylcholine release during and following stress are independent of the pituitary‐adrenocortical axis. Brain Res 1991;538:111–117. [DOI] [PubMed] [Google Scholar]
- 169. Lodge DJ, Grace AA. The laterodorsal tegmentum is essential for burst firing of ventral tegmental area dopamine neurons. Proc Natl Acad Sci 2006;103:5167–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cooper DC. The significance of action potential bursting in the brain reward circuit. Neurochem Int 2002;41:333–340. [DOI] [PubMed] [Google Scholar]
- 171. Forster GL, Blaha CD. Laterodorsal tegmental stimulation elicits dopamine efflux in the rat nucleus accumbens by activation of acetylcholine and glutamate receptors in the ventral tegmental area. Eur J Neurosci 2000;12:3596–3604. [DOI] [PubMed] [Google Scholar]
- 172. Ishibashi M, Leonard CS, Kohlmeier KA. Nicotinic activation of laterodorsal tegmental neurons: Implications for addiction to nicotine. Neuropsychopharmacology 2009;34:2529–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Calabresi P, Lacey MG, North RA. Nicotinic excitation of rat ventral tegmental neurones in vitro studied by intracellular recording. Br J Pharmacol 1989;98:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Gronier B, Rasmussen K. Activation of midbrain presumed dopaminergic neurones by muscarinic cholinergic receptors: An in vivo electrophysiological study in the rat. Br J Pharmacol 1998;124:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Blaha CD, Allen LF, Das S, Inglis WL, Latimer MP, Vincent SR, Winn P. Modulation of dopamine efflux in the nucleus accumbens after cholinergic stimulation of the ventral tegmental area in intact, pedunculopontine tegmental nucleus‐lesioned, and laterodorsal tegmental nucleus‐lesioned rats. J Neurosci 1996;16:714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Miller AD, Blaha CD. Midbrain muscarinic receptor mechanisms underlying regulation of mesoaccumbens and nigrostriatal dopaminergic transmission in the rat. Eur J Neurosci 2005;21:1837–1846. [DOI] [PubMed] [Google Scholar]
- 177. Nisell M, Nomikos GG, Svensson TH. Systemic nicotine‐induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse 1994;16:36–44. [DOI] [PubMed] [Google Scholar]
- 178. Ikemoto S, Wise RA. Rewarding effects of the cholinergic agents carbachol and neostigmine in the posterior ventral tegmental area. J Neurosci 2002;22:9895–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Ikemoto S, Panksepp J. Dissociations between appetitive and consummatory responses by pharmacological manipulations of reward‐relevant brain regions. Behav Neurosci 1996;110:331–345. [DOI] [PubMed] [Google Scholar]
- 180. Reavill C, Stolerman IP. Locomotor activity in rats after administration of nicotinic agonists intracerebrally. Br J Pharmacol 1990;99:273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Grillner P, Bonci A, Svensson TH, Bernardi G, Mercuri NB. Presynaptic muscarinic (M3) receptors reduce excitatory transmission in dopamine neurons of the rat mesencephalon. Neuroscience 1999;91:557–565. [DOI] [PubMed] [Google Scholar]
- 182. Yeomans JS, Takeuchi J, Baptista M, et al Brain‐stimulation reward thresholds raised by an antisense oligonucleotide for the M5 muscarinic receptor infused near dopamine cells. J Neurosci 2000;20:8861–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Yeomans J, Forster G, Blaha C. M5 muscarinic receptors are needed for slow activation of dopamine neurons and for rewarding brain stimulation. Life Sci 2001;68:2449–2456. [DOI] [PubMed] [Google Scholar]
- 184. Forster GL, Yeomans JS, Takeuchi J, Blaha CD. M5 muscarinic receptors are required for prolonged accumbal dopamine release after electrical stimulation of the pons in mice. J Neurosci 2002;22:RC190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW. The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (alpha 5) in the rat central nervous system. Brain Res 1990;526:45–53. [DOI] [PubMed] [Google Scholar]
- 186. Le Novère N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci 1996;8:2428–2439. [DOI] [PubMed] [Google Scholar]
- 187. Arroyo‐Jimenez MM, Bourgeois JP, Marubio LM, et al Ultrastructural localization of the alpha4‐subunit of the neuronal acetylcholine nicotinic receptor in the rat substantia nigra. J Neurosci 1999;19:6475–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Schilström B, Fagerquist MV, Zhang X, Hertel P, Panagis G, Nomikos GG, Svensson TH. Putative role of presynaptic alpha7* nicotinic receptors in nicotine stimulated increases of extracellular levels of glutamate and aspartate in the ventral tegmental area. Synapse 2000;38:375–383. [DOI] [PubMed] [Google Scholar]
- 189. Schilström B, Rawal N, Mameli‐Engvall M, Nomikos GG, Svensson TH. Dual effects of nicotine on dopamine neurons mediated by different nicotinic receptor subtypes. Int J Neuropsychopharmacol 2003;6:1–11. [DOI] [PubMed] [Google Scholar]
- 190. Di Chiara G. The role of dopamine in drug abuse viewed from the perspective of its role in motivation. Drug Alcohol Depend 1995;38:95–137. [DOI] [PubMed] [Google Scholar]
- 191. Koob GF. Drugs of abuse: Anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci 1992;13:177–184. [DOI] [PubMed] [Google Scholar]
- 192. Chang JY, Sawyer SF, Lee RS, Woodward DJ. Electrophysiological and pharmacological evidence for the role of the nucleus accumbens in cocaine self‐administration in freely moving rats. J Neurosci 1994;14:1224–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Blaha CD, Phillips AG. A critical assessment of electrochemical procedures applied to the measurement of dopamine and its metabolites during drug‐induced and species‐typical behaviours. Behav Pharmacol 1996;7:675–708. [PubMed] [Google Scholar]
- 194. Phillips PE, Stuber GD, Heien ML, Wightman RM, Carelli RM. Subsecond dopamine release promotes cocaine seeking. Nature 2003;422:614–618. [DOI] [PubMed] [Google Scholar]
- 195. Drevets WC, Gautier C, Price JC, et al Amphetamine‐induced dopamine release in human ventral striatum correlates with euphoria. Biol Psychiatry 2001;49:81–96. [DOI] [PubMed] [Google Scholar]
- 196. Redgrave P, Horrell RI. Potentiation of central reward by localised perfusion of acetylcholine and 5‐hydroxytryptamine. Nature 1976;262:305–307. [DOI] [PubMed] [Google Scholar]
- 197. Yeomans J, Baptista M. Both nicotinic and muscarinic receptors in ventral tegmental area contribute to brain‐stimulation reward. Pharmacol Biochem Behav 1997;57:915–921. [DOI] [PubMed] [Google Scholar]
- 198. Nomikos GG, Schilström B, Hildebrand BE, Panagis G, Grenhoff J, Svensson TH. Role of alpha7 nicotinic receptors in nicotine dependence and implications for psychiatric illness. Behav Brain Res 2000;113:97–103. [DOI] [PubMed] [Google Scholar]
- 199. Corrigall WA, Coen KM, Adamson KL. Self‐administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res 1994;653:278–284. [DOI] [PubMed] [Google Scholar]
- 200. Ikemoto S, Qin M, Liu ZH. Primary reinforcing effects of nicotine are triggered from multiple regions both inside and outside the ventral tegmental area. J Neurosci 2006;26:723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Sziráki I, Sershen H, Hashim A, Lajtha A. Receptors in the ventral tegmental area mediating nicotine‐induced dopamine release in the nucleus accumbens. Neurochem Res 2002;27:253–261. [DOI] [PubMed] [Google Scholar]
- 202. Panagis G, Kastellakis A, Spyraki C, Nomikos G. Effects of methyllycaconitine (MLA), an alpha 7 nicotinic receptor antagonist, on nicotine‐ and cocaine‐induced potentiation of brain stimulation reward. Psychopharmacology (Berl) 2000;149:388–396. [DOI] [PubMed] [Google Scholar]
- 203. Picciotto MR, Corrigall WA. Neuronal systems underlying behaviors related to nicotine addiction: Neural circuits and molecular genetics. J Neurosci 2002;22:3338–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Zachariou V, Caldarone BJ, Weathers‐Lowin A, et al Nicotine receptor inactivation decreases sensitivity to cocaine. Neuropsychopharmacology 2001;24:576–589. [DOI] [PubMed] [Google Scholar]
- 205. Reid MS, Mickalian JD, Delucchi KL, Berger SP. A nicotine antagonist, mecamylamine, reduces cue‐induced cocaine craving in cocaine‐dependent subjects. Neuropsychopharmacology 1999;20:297–307. [DOI] [PubMed] [Google Scholar]
- 206. Rose JE, Behm FM, Westman EC, Levin ED, Stein RM, Ripka GV. Mecamylamine combined with nicotine skin patch facilitates smoking cessation beyond nicotine patch treatment alone. Clin Pharmacol Ther 1994;56:86–99. [DOI] [PubMed] [Google Scholar]
- 207. Williams MJ, Adinoff B. The role of acetylcholine in cocaine addiction. Neuropsychopharmacology 2008;33:1779–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Fink‐Jensen A, Fedorova I, Wörtwein G, et al Role for M5 muscarinic acetylcholine receptors in cocaine addiction. J Neurosci Res 2003;74:91–96. [DOI] [PubMed] [Google Scholar]
- 209. Thomsen M, Woldbye DP, Wörtwein G, Fink‐Jensen A, Wess J, Caine SB. Reduced cocaine self‐administration in muscarinic M5 acetylcholine receptor‐deficient mice. J Neurosci 2005;25:8141–8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Lester DB, Miller AD, Blaha CD. Muscarinic receptor blockade in the ventral tegmental area attenuates cocaine enhancement of laterodorsal tegmentum stimulation‐evoked accumbens dopamine efflux in the mouse. Synapse 2009;64:216–223. [DOI] [PubMed] [Google Scholar]
- 211. Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science 1988;242:715–723. [DOI] [PubMed] [Google Scholar]
- 212. Wilson MC, Schuster CR. Cholinergic influence on intravenous cocaine self‐administration by rhesus monkeys. Pharmacol Biochem Behav 1973;1:643–649. [DOI] [PubMed] [Google Scholar]
- 213. Bhugra D. The global prevalence of schizophrenia. PLoS Med 2005;2:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Andreasen NC. Schizophrenia: The fundamental questions. Brain Res Brain Res Rev 2000;31:106–112. [DOI] [PubMed] [Google Scholar]
- 215. Carlsson A, Hansson LO, Waters N, Carlsson ML. Neurotransmitter aberrations in schizophrenia: New perspectives and therapeutic implications. Life Sci 1997;61:75–94. [DOI] [PubMed] [Google Scholar]
- 216. Emilien G, Maloteaux JM, Geurts M, Hoogenberg K, Cragg S. Dopamine receptors–physiological understanding to therapeutic intervention potential. Pharmacol Ther 1999;84:133–156. [DOI] [PubMed] [Google Scholar]
- 217. Stone JM, Pilowsky LS. Novel targets for drugs in schizophrenia. CNS Neurol Disord Drug Targets 2007;6:265–272. [DOI] [PubMed] [Google Scholar]
- 218. Coyle JT. Glutamate and schizophrenia: Beyond the dopamine hypothesis. Cell Mol Neurobiol 2006;26:365–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Stone JM, Morrison PD, Pilowsky LS. Glutamate and dopamine dysregulation in schizophrenia – a synthesis and selective review. J Psychopharmacol 2007;21:440–452. [DOI] [PubMed] [Google Scholar]
- 220. Yeomans JS. Role of tegmental cholinergic neurons in dopaminergic activation, antimuscarinic psychosis and schizophrenia. Neuropsychopharmacology 1995;12:3–16. [DOI] [PubMed] [Google Scholar]
- 221. Wynn ZJ, Cummings JL. Cholinesterase inhibitor therapies and neuropsychiatric manifestations of Alzheimer's disease. Dement Geriatr Cogn Disord 2004;17:100–108. [DOI] [PubMed] [Google Scholar]
- 222. Terry AV Jr. Role of the central cholinergic system in the therapeutics of schizophrenia. Curr Neuropharmacol 2008;6:286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Janowsky DS, Davis JM, Huey L, Judd LL. Adrenergic and cholinergic drugs as episode and vulnerability markers of affective disorders and schizophrenia. Psychopharmacol Bull 1979;15:33–34. [PubMed] [Google Scholar]
- 224. Tandon R, Greden JF. Cholinergic hyperactivity and negative schizophrenic symptoms. A model of cholinergic/dopaminergic interactions in schizophrenia. Arch Gen Psychiatry 1989;46:745–753. [DOI] [PubMed] [Google Scholar]
- 225. Tandon R, Greden JF, Haskett RF. Cholinergic hyperactivity and negative symptoms: Behavioral effects of physostigmine in normal controls. Schizophr Res 1993;9:19–23. [DOI] [PubMed] [Google Scholar]
- 226. Crook JM, Dean B, Pavey G, Copolov D. The binding of [3H]AF‐DX 384 is reduced in the caudate‐putamen of subjects with schizophrenia. Life Sci 1999;64:1761–1771. [DOI] [PubMed] [Google Scholar]
- 227. Bymaster FP, Shannon HE, Rasmussen K, et al Potential role of muscarinic receptors in schizophrenia. Life Sci 1999;64:527–534. [DOI] [PubMed] [Google Scholar]
- 228. Wang H, Ng K, Hayes D, Gao X, Forster G, Blaha C, Yeomans J. Decreased amphetamine‐induced locomotion and improved latent inhibition in mice mutant for the M5 muscarinic receptor gene found in the human 15q schizophrenia region. Neuropsychopharmacology 2004;29:2126–2139. [DOI] [PubMed] [Google Scholar]
- 229. Laruelle M, Abi‐Dargham A, Van Dyck CH, et al Single photon emission computerized tomography imaging of amphetamine‐induced dopamine release in drug‐free schizophrenic subjects. Proc Natl Acad Sci USA 1996;93:9235–9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Abi‐Dargham A, Gil R, Krystal J, et al Increased striatal dopamine transmission in schizophrenia: Confirmation in a second cohort. Am J Psychiatry 1998;155:761–767. [DOI] [PubMed] [Google Scholar]
- 231. Joseph MH, Peters SL, Moran PM, Grigoryan GA, Young AM, Gray JA. Modulation of latent inhibition in the rat by altered dopamine transmission in the nucleus accumbens at the time of conditioning. Neuroscience 2000;101:921–930. [DOI] [PubMed] [Google Scholar]
- 232. Moser PC, Hitchcock JM, Lister S, Moran PM. The pharmacology of latent inhibition as an animal model of schizophrenia. Brain Res Brain Res Rev 2000;33:275–307. [DOI] [PubMed] [Google Scholar]
- 233. Gray NS, Pilowsky LS, Gray JA, Kerwin RW. Latent inhibition in drug naive schizophrenics: Relationship to duration of illness and dopamine D2 binding using SPET. Schizophr Res 1995;17:95–107. [DOI] [PubMed] [Google Scholar]
- 234. De Luca V, Wang H, Squassina A, Wong GW, Yeomans J, Kennedy JL. Linkage of M5 muscarinic and alpha7‐nicotinic receptor genes on 15q13 to schizophrenia. Neuropsychobiology 2004;50:124–127. [DOI] [PubMed] [Google Scholar]
- 235. Lohr JB, Flynn K. Smoking and schizophrenia. Schizophr Res 1992;8:93–102. [DOI] [PubMed] [Google Scholar]
- 236. Nisell M, Nomikos GG, Svensson TH. Nicotine dependence, midbrain dopamine systems and psychiatric disorders. Pharmacol Toxicol 1995;76:157–162. [DOI] [PubMed] [Google Scholar]
- 237. Freedman R, Hall M, Adler LE, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry 1995;38:22–33. [DOI] [PubMed] [Google Scholar]
- 238. Breese CR, Lee MJ, Adams CE, et al Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology 2000;23:351–364. [DOI] [PubMed] [Google Scholar]
- 239. Ripoll N, Bronnec M, Bourin M. Nicotinic receptors and schizophrenia. Curr Med Res Opin 2004;20:1057–1074. [DOI] [PubMed] [Google Scholar]
- 240. Crook JM, Tomaskovic‐Crook E, Copolov DL, Dean B. Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia: A study of Brodmann's areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. Am J Psychiatry 2001;158:918–925. [DOI] [PubMed] [Google Scholar]
- 241. Olincy A, Harris JG, Johnson LL, et al Proof‐of‐concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry 2006;63:630–638. [DOI] [PubMed] [Google Scholar]
- 242. Gray JA, Roth BL. Molecular targets for treating cognitive dysfunction in schizophrenia. Schizophr Bull 2007;33:1100–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]