

SUMMARY
Multiple neurotransmitter circuits are disturbed in schizophrenia, and the dopamine hypothesis of schizophrenia prevails as the hypothesis with most empirical support. On the other hand, schizophrenia is highly heritable with a pattern consistent with both common and rare allelic variants and gene × environment interaction. Advances in the field of neuroimaging have expanded our knowledge of intermediate phenotypes, the neurobiological processes that convey the risk from the genes to the complex phenotype. In this article, we review the recent and continuously accumulating evidence from in vivo imaging studies aiming at characterizing neurochemical intermediate phenotypes of schizophrenia. Dopaminergic alterations in schizophrenia are shared by individuals at genetic risk who do not express the illness, suggesting a “dopamine hypothesis of schizophrenia vulnerability.” This hypothesis has the potential to help us better understand the dopaminergic dysfunction in the context of the complex pathophysiological process leading to schizophrenia. In the future, neurotransmitter imaging studies should investigate the gene × environment interaction in schizophrenia, and try to identify neurobiological correlates of heightened sensitivity to environmental stressors (e.g., cannabis, childhood trauma, and other psychosocial stress) in genetically vulnerable individuals.
Keywords: Dopamine, Genetics, Positron emission tomography, Schizophrenia
Introduction
Schizophrenia is a severe and debilitating mental illness that affects approximately 1% of the population throughout the world. It is a multifactorial disease with strong genetic contributions along with prominent gene × environment interactions [1]. Schizophrenia manifests as a disease with subtle structural and functional abnormalities of various brain structures, such as the striatum, hippocampus, and prefrontal cortex [2, 3]. At the molecular level, multiple neurotransmitter systems appear to be abnormal. Dopamine is traditionally associated with schizophrenia, owing to the propensity of dopamine D2 receptor antagonists to alleviate and dopamine‐releasing drugs to exacerbate positive symptoms of schizophrenia [4, 5]. The dopamine hypothesis of schizophrenia suggests that subcortical dopamine overactivity is a common feature brought on by multiple contributing factors from genes and environment and by dysfunctions of many brain circuits [4, 6]. Also, the cortical dopamine neurotransmission is suggested to be dysfunctional [7]. Dopamine abnormalities are tightly interwoven with glutamate and GABA deficits that are hypothesized to play a role in schizophrenia [3, 8].
Schizophrenia is highly heritable, as indicated by epidemiological studies [9, 10]. However, schizophrenia is neither a purely genetic disorder, nor caused by a single gene. Rather, it likely results from a combination of both rare and common genetic variants [11], environmental risk factors [12], and their interaction [1]. Research into intermediate phenotypes has become a popular strategy in the pursuit of identifying which factors convey the increased risk from the genes to the observed phenotype because intermediate phenotypes are more proximal to the underlying genes than complex clinical phenotypes. This strategy can be used to study the effects of previously identified genetic risk variants in the general population [13], or to study individuals who are at risk for developing the illness [14]. These studies have shown that risk variants for schizophrenia modulate neocortical and hippocampal structure, function, and connectivity in ways that resemble the pathophysiology of schizophrenia [13]—and that similar alterations are often found in individuals with genetic risk for schizophrenia but who do not express the illness [15, 16, 17]. The latter strategy is attractive since it allows the examination of inherited pathophysiology that is not confounded by antipsychotic medication or chronicity of illness.
The purpose of this review article is to summarize molecular imaging evidence for neurotransmitter abnormalities in individuals at genetic risk for schizophrenia. First, we briefly describe the most commonly used imaging strategies and discuss their potential. Next, we review the current literature on molecular imaging studies in schizophrenia, and discuss their consistency and implications for genetic studies. We then turn to actual evidence from imaging studies in high risk populations that target neurotransmitter systems relevant for schizophrenia. Finally, we attempt to form a synthesis of the emergent imaging data and suggest future directions.
Overview of Molecular Imaging Techniques
Molecular imaging techniques such as single‐photon emission computed tomography (SPECT) and positron emission tomography (PET) are based on the use of short‐lived radioactive isotopes attached to a molecule of interest. These techniques have unsurpassed sensitivity and specificity to measure different proteins in the human body. To quantify receptor binding, the principles of in vitro receptor binding techniques are applied to the in vivo situation—with obvious limitations. For example, radioligands are typically given at very small doses, or “tracer” doses, which are assumed to occupy only a minimal proportion of the target molecules. This technique of using one small radiotracer concentration cannot measure receptor density and affinity separately—only their product, which is often referred to as the binding potential [18]. These imaging techniques are typically used to measure the availability of the target (e.g., receptor, transporter, or enzyme) in a baseline condition, and the result is inferred to reflect the density of the target in brain tissue. However, another technique is based on the principle that endogenous neurotransmitters compete with the radioligand in binding to the target; therefore, changes in synaptic neurotransmitter concentrations can be estimated by scanning the same subject before and after a pharmacological challenge. For example, binding of the dopamine D2 receptor antagonist radioligands [ 11 C]raclopride and [123I]IBZM has been shown to be sensitive to increases and decreases in endogenous dopamine [19], although factors other than competitive inhibition are likely to be involved [20]. These “challenge study” paradigms are able to provide a more comprehensive view of the neurotransmitter abnormalities.
Neurotransmitter Abnormalities in Schizophrenia Revealed by Molecular Imaging
This selective and brief overview will focus on two neurotransmitter systems with most evidence from imaging studies: dopamine and serotonin.
Striatal presynaptic dopamine system is dysregulated in schizophrenia. First, majority of studies show increased presynaptic dopamine synthesis capacity in schizophrenia, as measured by the uptake of either fluorine‐18 or carbon‐11 labeled fluorodopa (FDOPA), a precursor for dopamine [21, 22, 23, 24, 25, 26, 27, 28]. Second, patients with schizophrenia have elevated amphetamine‐induced striatal dopamine release [29, 30, 31, 32]. This abnormality is also seen in schizotypal personality disorder [33], suggesting genetic contributions, since this condition is genetically related to schizophrenia [34]. Finally, patients with schizophrenia have increased baseline occupancy of D2 receptors by dopamine in the associative striatum [35, 36]. Thus, robust evidence suggest that schizophrenia is associated with increased presynaptic dopamine function, as implicated by increased synthesis capacity and higher tonic (baseline) and phasic (stimulated) dopamine release. The two latter phenomena appear to be correlated with each other in drug‐naïve patients [37]. This increased dopamine function does not reflect increased number of dopamine terminals, since the density of dopamine transporters is unaltered in drug‐naïve schizophrenia [38], but rather dysregulated function per existing dopaminergic neuron.
Early studies on striatal dopamine D2 receptors found increased binding in patients with schizophrenia [39, 40, 41], but later studies conducted with a different class of radioligands failed to confirm this finding [42, 43]. Nevertheless, a meta‐analysis of all published studies [29, 30, 31, 35, 36, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54] suggests a small but significant increase in striatal D2 receptors in schizophrenia (Figure 1) [55], which may be partially masked by increased endogenous dopamine at baseline [35]. Although early theories emphasized striatal D2 receptors in schizophrenia, cortical and thalamic dopamine D2 receptors have later been recognized as important [56]. Although results from imaging studies have not all been consistent, most studies have shown decreased extrastriatal D2 receptor binding in schizophrenia, especially in the thalamus [53, 54, 57, 58, 59, 60, 61, 62]. Low thalamic D2 receptor density may contribute to sensory gating abnormalities in schizophrenia.
Figure 1.
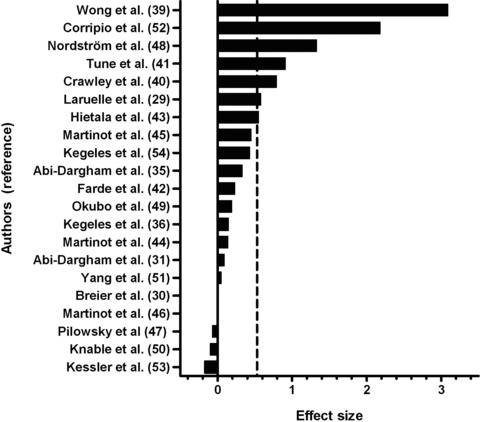
Meta‐analysis of all published studies (N = 21) reporting striatal dopamine D2 receptor binding in patients with schizophrenia (total of 294 patients) in comparisons with healthy subjects (total of 315 subjects). Effect size is calculated as the difference between the means of patients and controls, divided by the standard deviation in the control group. Dashed vertical line represents the average effect size 0.53 (standard deviation 0.80, 95% confidence interval 0.37–0.69), which corresponds to about 10% increase. The distribution of effect sizes is significantly different from that expected based on the null hypothesis of no overall differences between groups (one sample t‐test, P= 0.004).
Cortical dopamine D1 receptors are crucial for working memory performance, which is disturbed in schizophrenia [7]. Decreased cortical D1 receptor binding was initially reported [49], but was later not confirmed using the same radioligand, [11C]SCH 23390 [63]. In contrast, using another radioligand, [11C]NNC‐112, increased D1 receptor binding was reported [64], and high D1 receptor binding was correlated with poor cognitive performance. Potential reasons for this discrepancy include differential responses to chronic dopamine alterations of the two radioligands used [65], binding of these radioligands to cortical 5‐HT2A receptors [66], antipsychotic medication [67], and genetics [68]. Recently, decreased binding of both D1 receptor tracers was demonstrated in chronic and medicated schizophrenia [69], suggesting that choice of radioligand may not be relevant.
Studies on the serotonin system are far less abundant. Serotonin transporter density is unaltered in schizophrenia [70], whereas the stimulatory 5‐HT2A receptors are decreased in drug‐naïve patients [71]. Although the involvement of the inhibitory 5‐HT1A receptors was supported by postmortem studies showing increased densities [72], subsequent PET studies found both increased [73] and decreased [74] binding—and the latest study with the most rigorous quantification showed no change [75].
In summary, molecular imaging studies in humans have established that schizophrenia is associated with increased presynaptic dopamine synthesis capacity; increased baseline (tonic) synaptic concentrations of dopamine, which likely mask a small increase in postsynaptic dopamine D2 receptor binding; increased amphetamine‐induced (phasic) dopamine release; decreased thalamic D2 receptor binding; and possibly decreased cortical serotonin 5‐HT2A binding. However, brain imaging studies do not distinguish between state, trait or other phenomena, such as chronicity. Thus, it is possible that these abnormalities are associated with vulnerability to develop schizophrenia rather than the clinical phenotype of the illness; that is, are these abnormalities also present in the brains of individuals at risk of developing the illness.
Neurotransmitter Abnormalities and Genetic Risk for Schizophrenia
In general, in vivo molecular imaging studies looking at neurotransmitter abnormalities in individuals at genetic risk for schizophrenia are scarce, likely due to the challenging nature of both the imaging method and acquiring these individuals to participate in these studies. Nevertheless, the findings are beginning to show consistency in that some of the dopaminergic abnormalities are more likely due to vulnerability rather than expression of illness (Table 1). This section reviews the available literature from in vivo molecular imaging studies on two most commonly studied neurotransmitter systems: dopamine and serotonin.
Table 1.
Summary of evidence from molecular imaging studies of some of the neurotransmitter biomarkers in schizophrenia, high risk populations, and twins. See the text for details and references.
| Imaging biomarker | Drug‐naïve schizophrenia | Medicated schizophrenia | Genetic high risk | Clinical high risk | Heritability |
|---|---|---|---|---|---|
| Striatal D2 receptor | ↑ | ↑ | ↑ | ? | Yes |
| Thalamic D2 receptor | ↓ | ↓ | ? | ? | Yes |
| Cortical D1 receptor | ↑ | ↓ | ↑ | ? | Yes |
| Presynaptic dopamine synthesis | ↑ | ↑ | ↑ | ↑ | ? |
| Cortical 5‐HT2A receptor | ↓ | ↓ | ? | ↓ | Yes |
↑, higher than in healthy subjects; ↓, lower than in healthy subjects; ?, not enough data. Heritability refers to genetic contributions and is calculated as the intraclass correlation among identical twins.
To examine whether increased presynaptic dopamine function represents vulnerability or expression of schizophrenia, we recently examined [18F]FDOPA uptake in 17 first‐degree relatives of patients with schizophrenia (7 children and 10 siblings) in comparison with 17 healthy subjects [76]. These relatives were nonpsychotic as verified by structured psychiatric interviews and had mild (if any) and stable symptoms suggesting lack of any recent functional deterioration. The average age of these subjects was 34 years, meaning that they had predominantly passed the typical risk age for schizophrenia onset. We found about 20% higher striatal [18F]FDOPA influx values in the first‐degree relatives than in the healthy controls. This effect was more pronounced in the caudate than in the putamen. The effect size was roughly similar to that we previously found in patients with schizophrenia [21]. Curiously, one of the subjects who had two brothers and a father all diagnosed with schizophrenia had the highest [18F]FDOPA influx values of all; he also presented with moderate symptoms. This finding suggests that increased striatal presynaptic dopamine function is a phenomenon shared by unaffected siblings of patients with schizophrenia, and is therefore likely related to vulnerability than clinical expression of the illness. Further support for this hypothesis comes from studies showing increased [18F]FDOPA influx values in individuals at clinical risk for schizophrenia [4, 77, 78] and that [18F]FDOPA uptake is only marginally affected by the clinical state of the patient [79]. Preferential involvement in our study of the caudate nucleus, which is part of the associative striatum as opposed to limbic or sensorimotor striatum [80], is supported by recent findings of increased tonic dopamine levels in schizophrenia specifically in this region [36]; together, these findings are compatible with the hypotheses linking prefrontal dysfunction with striatal hyperdopaminergia in schizophrenia.
To look more closely downstream in the dopamine signaling pathway, and also to acquire better sensitivity in terms of genetic loading, we recently studied striatal dopamine D2 receptors as well as striatal and extrastriatal dopamine D1 receptors in twins discordant for schizophrenia [81, 82, 83]. The twin paradigm has the advantage of gradually increasing the genetic similarity from about 50% in fraternal (i.e., nonidentical) twins to 100% in identical twins. Fraternal twins have about 10% risk and identical twins have about 50% risk of developing schizophrenia, which greatly exceeds the about 1% risk in the normal population. These middle‐aged twins were recruited by cross‐referencing the National Twin Registry in Finland [84] including all same‐sex twin pairs born in Finland between 1940 and 1957 (N = 9692 pairs) with three national registers related to hospitalization, medications, and pensions [85]. This search identified 348 twin pairs with either or both cotwins having schizophrenia, and 9214 healthy twin pairs. We randomly sampled 11 discordant twin pairs (6 identical and 5 nonidentical) and 7 healthy twin (4 identical, 3 nonidentical) pairs for PET studies. D2 and D1 receptor binding was measured using PET and [11C]raclopride and [11C]SCH23390, respectively. We found that unaffected identical cotwins of patients with schizophrenia had higher D2 receptor densities that did healthy twins, specifically in the caudate nucleus (effect size 0.75) (Figure 2) [82]. Increased caudate D2 receptor density was negatively correlated with cognitive performance in the whole sample. Among identical healthy twins, we found high correlations in D2 receptor densities specifically in the caudate, consistent with major genetic influences. However, two recent studies in unaffected relatives of patients with schizophrenia did not replicate increased caudate D2 receptor binding at baseline [86, 87], probably owing to only modest genetic loading and small sample sizes in these studies, given the large interindividual variability of D2 measurements [88].
Figure 2.
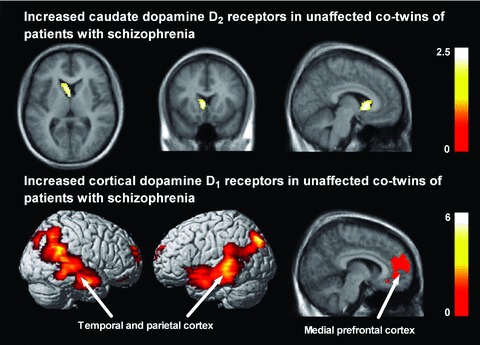
Increased caudate dopamine D2 receptors (top row) and increased cortical dopamine D1 receptors (bottom row) in unaffected cotwins of patients with schizophrenia. Results from a voxel‐based receptor mapping analysis are visualized on a magnetic resonance imaging template. Color scales represent T‐statistic values at voxel level. The right side of the brain appears on the right side of the images. See [82] and [81] for details.
The pattern of results from dopamine D1 receptor imaging in twins discordant for schizophrenia turned out to be more complex [81]. We found that unaffected identical cotwins had higher D1 receptor densities in the prefrontal, temporal, and parietal cortex than did healthy twins (Figure 2), and that nonidentical cotwins were intermediate between identical cotwins and healthy twins in D1 receptor binding. In these unaffected twins, high cortical D1 receptor binding predicted poor spatial working memory performance. Surprisingly, patients with schizophrenia had much lower D1 receptor binding in most regions in the brain than did their unaffected twins, and binding was negatively correlated with antipsychotic drug dose. Finally, cortical D1 receptors were under tight genetic control among healthy identical twins as indicated by high intraclass correlations. Taken together, it seems that there are distinct vulnerability and disease‐related contributions to cortical D1 receptors: receptors are increased in individuals at genetic risk, but decreased in patients who express the illness, possibly due to chronic antipsychotic medications. This finding is consistent with a recent results published in abstract form by Abi‐Dargham et al. [67] showing increased D1 binding only in drug‐naïve patients, but not in patients that had received prior antipsychotic drug treatment.
Unfortunately, there are no studies published to date on serotonergic markers in individuals at genetic risk for schizophrenia. However, there is preliminary evidence for abnormalities in these markers in people at clinical risk, and some of this evidence is compatible with theories on the role of these markers in the pathophysiology of schizophrenia. This evidence is for the involvement of the two major receptor subtypes: 5‐HT2A and 5‐HT1A.
As mentioned earlier, serotonin 5‐HT2A receptor is the major excitatory serotonin receptor in cortical pyramidal neurons [89] and is decreased in drug‐naïve schizophrenia [71]. Hurlemann et al. [90] recently studied serotonin 5‐HT2A receptors with PET and [18F]altanserin in 14 individuals at ultrahigh clinical risk for schizophrenia and in 21 healthy subjects. They found decreased 5‐HT2A receptor binding in individuals at risk for schizophrenia that was correlated with severity of the prodromal state. Binding was smallest in people who subsequently converted into first‐episode psychosis. These findings suggest that decreased 5‐HT2A may predate the onset of schizophrenia [71], but they also clearly indicate that 5‐HT2A receptor expression is at least partly state‐dependent in schizophrenia. Thus, the implications for the genetic mechanisms of schizophrenia remain unclear, but part of the decrease in 5‐HT2A receptors in the prodromal subjects may be attributable to genetic risk since some of the subjects had relatives with schizophrenia [90] and twin studies show that 5‐HT2A receptors are under strong genetic control [91].
The 5‐HT1A receptor, which occurs both pre‐ and postsynaptically and is the major inhibitory serotonin receptor, does not appear to be altered in patients with schizophrenia [75]. Despite widely studied in mood and anxiety disorders [92, 93], there are no published imaging studies on the 5‐HT1A receptor in individuals at genetic risk for schizophrenia. However, we have recently observed decreased 5‐HT1A receptor binding in unaffected first‐degree relatives of patients with schizophrenia compared with healthy subjects without such relatives (Huttunen, Hirvonen and Hietala, unpublished observations), using a similar study design as in our previous study on presynaptic dopamine function [76]. If confirmed, this preliminary finding suggests distinct vulnerability and disease‐related contributions to 5‐HT1A receptors in schizophrenia, and encourages further examination of this biomarker in twin studies.
Discussion
Molecular imaging studies provide evidence that neurotransmitter abnormalities schizophrenia are shared by individuals at risk for developing the illness, most available evidence pointing toward alterations in the dopamine system. Studies both in individuals at genetic risk and in individuals at clinical risk point toward increased striatal presynaptic synthesis, increased striatal D2 receptors, and increased cortical D1 receptors are potential intermediate phenotypes that convey risk for schizophrenia.
Consistent with the modern version of the dopamine hypothesis of schizophrenia [4, 5], it is likely that increased striatal D2 receptor binding in unaffected cotwins of patients with schizophrenia is secondary to abnormal regulation of subcortical dopamine by cortical and hippocampal afferents, rather than being a primary pathological change. Reduced markers of pyramidal cell integrity in the prefrontal cortex predict increased dopamine D2 receptors [94] and enhanced amphetamine‐induced dopamine release [95] in patients with schizophrenia, while abnormal prefrontal activity is associated with increased presynaptic striatal dopamine in both patients [28] and individuals at clinical risk [77]. Glutamate NMDA receptor antagonism is associated with increased amphetamine‐induced dopamine release in healthy humans [96], indicating the importance of glutamatergic control over subcortical dopamine. Regional specificity to the head of caudate is consistent with extensive connection of this striatal subregion with the prefrontal cortex [80]. Prefrontal pathology as a genetically mediated trait in schizophrenia is supported by findings of reduced gray matter volume [97] and deficits are frontally mediated cognitive functioning [98] in unaffected cotwins of patients with schizophrenia. Increased striatal D2 receptors may be more readily detected in individuals at genetic risk, because they probably lack the higher synaptic dopamine concentration that occurs in schizophrenia [35, 36] and that may occupy D2 receptors [99]. This hypothesis predicts that striatal dopamine D2 receptor may be a viable biomarker of vulnerability to schizophrenia. Whether this biomarker is useful for genetic association studies or clinical studies on early interventions remains to be seen.
If dopaminergic abnormalities in schizophrenia are genetically determined, which genes are involved? The gene for D2 receptors has many polymorphisms, some of which affect receptor binding in vivo. A recent meta‐analysis identified over‐representation of the C allele of the C957T polymorphism in schizophrenia [100] yet, this allele is associated with lower striatal D2 receptor binding in healthy subjects [101]. Another variant is the cysteine allele of the Ser311Cys polymorphism, which is associated with schizophrenia [102] but does not affect binding in vivo[103]. Effects of variation in the gene coding for the D1 receptor are less well understood [104]. On a larger scale, many schizophrenia risk genes, such as including dysbindin, neuregulin, and D‐amino‐acid oxidase and D‐amino‐acid oxidase activator, appear to converge onto dopaminergic or glutamatergic neurotransmission [3, 6] and could thus be implicated in the dopaminergic abnormalities in individuals at genetic risk. One candidate gene relevant in this context is that coding for catechol‐O‐monoamine transporter (COMT), which degrades cortical dopamine. This gene variant is interesting since it provides a plausible mechanism for cortical dopaminergic dysfunction as a genetically mediated trait for schizophrenia. Valine allele of the valine‐methionine polymorphism at amino acid 158 of this gene is putatively associated with lower cortical dopamine concentration and is largely implicated in prefrontal cortical pathology of schizophrenia [13, 105], although overall association to the clinical phenotype is modest at best [106]. Interestingly, the valine allele has also been shown to be associated with increased dopamine turnover rate postmortem [107] and in vivo[108] as well as with higher cortical dopamine D1 receptors in vivo[68]. Thus, the effects of the valine allele of the COMT gene would be consistent with higher striatal D2 receptors [82] and higher cortical D1 receptors [81] in unaffected cotwins of patients with schizophrenia. Although our results are consistent with the role of this genetic variant in the regulation of prefrontal function and subcortical dopamine, this variant is not associated with striatal or cortical D2 receptors in the healthy population [109], suggesting interactions with other genes or the environment in the risk population.
Conclusions and Future Directions
In conclusion, molecular imaging studies have provided robust evidence for dopaminergic abnormalities in schizophrenia. Similar alterations are now seen in individuals who are at genetic risk but who do not express the illness, suggesting that dopaminergic mechanisms may serve as intermediate phenotypes, part of the neural substrate that conveys the risk from the genotype to the complex phenotype. Thus, we propose a novel hypothesis to be tested further, the “dopamine hypothesis of schizophrenia vulnerability.” In addition to on‐going efforts of developing and applying novel molecular imaging probes for other neurotransmitter systems, we should aim at further characterizing the role of dopamine disturbance in the genetic risk for schizophrenia. For example, it is not known whether the alterations in tonic [35] and phasic [29] dopamine systems are genetically determined. On the other hand, by combining multiple imaging techniques and strategies, these neurochemical alterations should be integrated with the structural and functional deficits that characterize schizophrenia [77, 108]. Another avenue less frequently traveled is imaging the gene × environment interaction in schizophrenia, given that genetic and environmental factors (such as cannabis use, urbanicity, and childhood trauma) synergistically increase the risk of schizophrenia [1]. For example, individuals with preexisting genetically determined abnormalities may be at greater risk for triggering effects from the environment. Future neurotransmitter imaging studies should incorporate this design. The gene × environment interaction regarding cannabis is particularly interesting from the viewpoint of dopaminergic dysregulation in schizophrenia [110]. Cannabis is a risk factor for psychosis in the population [111], but those at genetic vulnerability to psychosis are at particular risk [112]. Interestingly, the COMT gene, which is closely associated with the sensitivity of the subcortical dopamine system (see above), modulates both short‐ and long‐term effects of cannabis [113, 114], and cannabis itself causes subcortical dopamine release [115]. Thus, neurotransmitter imaging might help unravel the intermediate phenotypes responsible for mediating the heightened sensitivity to this and other environmental stressors (e.g., childhood trauma and other psychosocial stress) in genetically vulnerable individuals. Ultimately, a comprehensive view of the intermediate phenotypes and how they interact with environmental factors could lead to discovery of novel pathophysiological mechanism and better treatment.
Conflicts of interest
The authors have no conflicts of interest relevant to this article.
References
- 1. van Os J, Rutten BP, Poulton R. Gene‐environment interactions in schizophrenia: Review of epidemiological findings and future directions. Schizophr Bull 2008;34:1066–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol Psychiatry 2005;10:40–68; image 45. [DOI] [PubMed] [Google Scholar]
- 3. Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit‐based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 2008;31:234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: Version III–the final common pathway. Schizophr Bull 2009;35:549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: A review and reconceptualization. Am J Psychiatry 1991;148:1474–1486. [DOI] [PubMed] [Google Scholar]
- 6. Tost H, Alam T, Meyer‐Lindenberg A. Dopamine and psychosis: Theory, pathomechanisms and intermediate phenotypes. Neurosci Biobehav Rev 2010;34:689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldman‐Rakic PS, Muly EC, 3rd , Williams GV. D(1) receptors in prefrontal cells and circuits. Brain Res Brain Res Rev 2000;31:295–301. [DOI] [PubMed] [Google Scholar]
- 8. Laruelle M, Kegeles LS, Abi‐Dargham A. Glutamate, dopamine, and schizophrenia: From pathophysiology to treatment. Ann N Y Acad Sci 2003;1003:138–158. [DOI] [PubMed] [Google Scholar]
- 9. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: Evidence from a meta‐analysis of twin studies. Arch Gen Psychiatry 2003;60:1187–1192. [DOI] [PubMed] [Google Scholar]
- 10. Tsuang M. Schizophrenia: Genes and environment. Biol Psychiatry 2000;47:210–220. [DOI] [PubMed] [Google Scholar]
- 11. Owen MJ, Craddock N, O’Donovan MC. Suggestion of roles for both common and rare risk variants in genome‐wide studies of schizophrenia. Arch Gen Psychiatry 2010;67:667–673. [DOI] [PubMed] [Google Scholar]
- 12. Cannon TD, van Erp TG, Bearden CE, et al Early and late neurodevelopmental influences in the prodrome to schizophrenia: Contributions of genes, environment, and their interactions. Schizophr Bull 2003;29:653–669. [DOI] [PubMed] [Google Scholar]
- 13. Meyer‐Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci 2006;7:818–827. [DOI] [PubMed] [Google Scholar]
- 14. Gottesman, II , Gould TD. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am J Psychiatry 2003;160:636–645. [DOI] [PubMed] [Google Scholar]
- 15. Cannon TD. Clinical and genetic high‐risk strategies in understanding vulnerability to psychosis. Schizophr Res 2005;79:35–44. [DOI] [PubMed] [Google Scholar]
- 16. Whalley HC, Whyte MC, Johnstone EC, Lawrie SM. Neural correlates of enhanced genetic risk for schizophrenia. Neuroscientist 2005;11:238–249. [DOI] [PubMed] [Google Scholar]
- 17. Kaymaz N, van Os J. Heritability of structural brain traits an endophenotype approach to deconstruct schizophrenia. Int Rev Neurobiol 2009;89:85–130. [DOI] [PubMed] [Google Scholar]
- 18. Innis RB, Cunningham VJ, Delforge J, et al Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 2007;27:1533–1539. [DOI] [PubMed] [Google Scholar]
- 19. Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: A critical review. J Cereb Blood Flow Metab 2000;20:423–451. [DOI] [PubMed] [Google Scholar]
- 20. Skinbjerg M, Liow JS, Seneca N, et al D2 dopamine receptor internalization prolongs the decrease of radioligand binding after amphetamine: A PET study in a receptor internalization‐deficient mouse model. Neuroimage 2010;50:1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hietala J, Syvalahti E, Vuorio K, et al Presynaptic dopamine function in striatum of neuroleptic‐naive schizophrenic patients. Lancet 1995;346:1130–1131. [DOI] [PubMed] [Google Scholar]
- 22. Hietala J, Syvalahti E, Vilkman H, et al Depressive symptoms and presynaptic dopamine function in neuroleptic‐naive schizophrenia. Schizophr Res 1999;35:41–50. [DOI] [PubMed] [Google Scholar]
- 23. Reith J, Benkelfat C, Sherwin A, et al Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci U S A 1994;91:11651–11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dao‐Castellana MH, Paillere‐Martinot ML, Hantraye P, et al Presynaptic dopaminergic function in the striatum of schizophrenic patients. Schizophr Res 1997;23:167–174. [DOI] [PubMed] [Google Scholar]
- 25. McGowan S, Lawrence AD, Sales T, Quested D, Grasby P. Presynaptic dopaminergic dysfunction in schizophrenia: A positron emission tomographic [18F]fluorodopa study. Arch Gen Psychiatry 2004;61:134–142. [DOI] [PubMed] [Google Scholar]
- 26. Elkashef AM, Doudet D, Bryant T, Cohen RM, Li SH, Wyatt RJ. 6‐(18)F‐DOPA PET study in patients with schizophrenia. Positron emission tomography. Psychiatry Res 2000;100:1–11. [DOI] [PubMed] [Google Scholar]
- 27. Lindstrom LH, Gefvert O, Hagberg G, Lundberg T, Bergstrom M, Hartvig P, Langstrom B. Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L‐(beta‐11C) DOPA and PET. Biol Psychiatry 1999;46:681–688. [DOI] [PubMed] [Google Scholar]
- 28. Meyer‐Lindenberg A, Miletich RS, Kohn PD, et al Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci 2002;5:267–271. [DOI] [PubMed] [Google Scholar]
- 29. Laruelle M, Abi‐Dargham A, van Dyck CH, et al Single photon emission computerized tomography imaging of amphetamine‐induced dopamine release in drug‐free schizophrenic subjects. Proc Natl Acad Sci U S A 1996;93:9235–9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Breier A, Su TP, Saunders R, et al Schizophrenia is associated with elevated amphetamine‐induced synaptic dopamine concentrations: Evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A 1997;94:2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abi‐Dargham A, Gil R, Krystal J, et al Increased striatal dopamine transmission in schizophrenia: Confirmation in a second cohort. Am J Psychiatry 1998;155:761–767. [DOI] [PubMed] [Google Scholar]
- 32. Laruelle M, Abi‐Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: Relationship to illness phases. Biol Psychiatry 1999;46:56–72. [DOI] [PubMed] [Google Scholar]
- 33. Abi‐Dargham A, Kegeles LS, Zea‐Ponce Y, et al Striatal amphetamine‐induced dopamine release in patients with schizotypal personality disorder studied with single photon emission computed tomography and [123I]iodobenzamide. Biol Psychiatry 2004;55:1001–1006. [DOI] [PubMed] [Google Scholar]
- 34. Siever LJ, Davis KL. The pathophysiology of schizophrenia disorders: Perspectives from the spectrum. Am J Psychiatry 2004;161:398–413. [DOI] [PubMed] [Google Scholar]
- 35. Abi‐Dargham A, Rodenhiser J, Printz D, et al Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A 2000;97:8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kegeles LS, Abi‐Dargham A, Frankle WG, et al Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry 2010;67:231–239. [DOI] [PubMed] [Google Scholar]
- 37. Abi‐Dargham A, van de Giessen E, Slifstein M, Kegeles LS, Laruelle M. Baseline and amphetamine‐stimulated dopamine activity are related in drug‐naive schizophrenic subjects. Biol Psychiatry 2009;65:1091–1093. [DOI] [PubMed] [Google Scholar]
- 38. Laakso A, Vilkman H, Alakare B, et al Striatal dopamine transporter binding in neuroleptic‐naive patients with schizophrenia studied with positron emission tomography. Am J Psychiatry 2000;157:269–271. [DOI] [PubMed] [Google Scholar]
- 39. Wong DF, Wagner HN, Jr ., Tune LE, et al. Positron emission tomography reveals elevated D2 dopamine receptors in drug‐naive schizophrenics. Science 1986;234:1558–1563. [DOI] [PubMed] [Google Scholar]
- 40. Crawley JC, Owens DG, Crow TJ, et al Dopamine D2 receptors in schizophrenia studied in vivo . Lancet 1986;2:224–225. [DOI] [PubMed] [Google Scholar]
- 41. Tune LE, Wong DF, Pearlson G, et al Dopamine D2 receptor density estimates in schizophrenia: A positron emission tomography study with 11C‐N‐methylspiperone. Psychiatry Res 1993;49:219–237. [DOI] [PubMed] [Google Scholar]
- 42. Farde L, Wiesel FA, Stone‐Elander S, Halldin C, Nordstrom AL, Hall H, Sedvall G. D2 dopamine receptors in neuroleptic‐naive schizophrenic patients. A positron emission tomography study with [11C]raclopride. Arch Gen Psychiatry 1990;47:213–219. [DOI] [PubMed] [Google Scholar]
- 43. Hietala J, Syvalahti E, Vuorio K, et al Striatal D2 dopamine receptor characteristics in neuroleptic‐naive schizophrenic patients studied with positron emission tomography. Arch Gen Psychiatry 1994;51:116–123. [DOI] [PubMed] [Google Scholar]
- 44. Martinot JL, Peron‐Magnan P, Huret JD, et al Striatal D2 dopaminergic receptors assessed with positron emission tomography and [76Br]bromospiperone in untreated schizophrenic patients. Am J Psychiatry 1990;147:44–50. [DOI] [PubMed] [Google Scholar]
- 45. Martinot JL, Paillere‐Martinot ML, Loc’h C, et al The estimated density of D2 striatal receptors in schizophrenia. A study with positron emission tomography and 76Br‐bromolisuride. Br J Psychiatry 1991;158:346–350. [DOI] [PubMed] [Google Scholar]
- 46. Martinot JL, Paillere‐Martinot ML, Loc’h C, et al Central D2 receptors and negative symptoms of schizophrenia. Br J Psychiatry 1994;164:27–34. [DOI] [PubMed] [Google Scholar]
- 47. Pilowsky LS, Costa DC, Ell PJ, Verhoeff NP, Murray RM, Kerwin RW. D2 dopamine receptor binding in the basal ganglia of antipsychotic‐free schizophrenic patients. An 123I‐IBZM single photon emission computerised tomography study. Br J Psychiatry 1994;164:16–26. [DOI] [PubMed] [Google Scholar]
- 48. Nordstrom AL, Farde L, Eriksson L, Halldin C. No elevated D2 dopamine receptors in neuroleptic‐naive schizophrenic patients revealed by positron emission tomography and [11C]N‐methylspiperone. Psychiatry Res 1995;61:67–83. [DOI] [PubMed] [Google Scholar]
- 49. Okubo Y, Suhara T, Suzuki K, et al Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 1997;385:634–636. [DOI] [PubMed] [Google Scholar]
- 50. Knable MB, Egan MF, Heinz A, Gorey J, Lee KS, Coppola R, Weinberger DR. Altered dopaminergic function and negative symptoms in drug‐free patients with schizophrenia. [123I]‐iodobenzamide SPECT study. Br J Psychiatry 1997;171:574–577. [DOI] [PubMed] [Google Scholar]
- 51. Yang YK, Yu L, Yeh TL, Chiu NT, Chen PS, Lee IH. Associated alterations of striatal dopamine D2/D3 receptor and transporter binding in drug‐naive patients with schizophrenia: A dual‐isotope SPECT study. Am J Psychiatry 2004;161:1496–1498. [DOI] [PubMed] [Google Scholar]
- 52. Corripio I, Perez V, Catafau AM, Mena E, Carrio I, Alvarez E. Striatal D2 receptor binding as a marker of prognosis and outcome in untreated first‐episode psychosis. Neuroimage 2006;29:662–666. [DOI] [PubMed] [Google Scholar]
- 53. Kessler RM, Woodward ND, Riccardi P, et al Dopamine D2 receptor levels in striatum, thalamus, substantia nigra, limbic regions, and cortex in schizophrenic subjects. Biol Psychiatry 2009;65:1024–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kegeles LS, Slifstein M, Xu X, et al Striatal and extrastriatal dopamine D(2)/D(3) receptors in schizophrenia evaluated with [(18)F]fallypride positron emission tomography. Biol Psychiatry 2010;68:634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Laruelle M. Imaging dopamine transmission in schizophrenia. A review and meta‐analysis. Q J Nucl Med 1998;42:211–221. [PubMed] [Google Scholar]
- 56. Lidow MS, Williams GV, Goldman‐Rakic PS. The cerebral cortex: A case for a common site of action of antipsychotics. Trends Pharmacol Sci 1998;19:136–140. [DOI] [PubMed] [Google Scholar]
- 57. Suhara T, Okubo Y, Yasuno F, et al Decreased dopamine D2 receptor binding in the anterior cingulate cortex in schizophrenia. Arch Gen Psychiatry 2002;59:25–30. [DOI] [PubMed] [Google Scholar]
- 58. Yasuno F, Suhara T, Okubo Y, et al Low dopamine d(2) receptor binding in subregions of the thalamus in schizophrenia. Am J Psychiatry 2004;161:1016–1022. [DOI] [PubMed] [Google Scholar]
- 59. Talvik M, Nordstrom AL, Olsson H, Halldin C, Farde L. Decreased thalamic D2/D3 receptor binding in drug‐naive patients with schizophrenia: A PET study with [11C]FLB 457. Int J Neuropsychopharmacol 2003;6:361–370. [DOI] [PubMed] [Google Scholar]
- 60. Tuppurainen H, Kuikka J, Viinamaki H, Husso‐Saastamoinen M, Bergstrom K, Tiihonen J. Extrastriatal dopamine D 2/3 receptor density and distribution in drug‐naive schizophrenic patients. Mol Psychiatry 2003;8:453–455. [DOI] [PubMed] [Google Scholar]
- 61. Tuppurainen H, Kuikka JT, Laakso MP, Viinamaki H, Husso M, Tiihonen J. Midbrain dopamine D2/3 receptor binding in schizophrenia. Eur Arch Psychiatry Clin Neurosci 2006;256:382–387. [DOI] [PubMed] [Google Scholar]
- 62. Lehrer DS, Christian BT, Kirbas C, et al (18)F‐Fallypride binding potential in patients with schizophrenia compared to healthy controls. Schizophr Res 2010;122:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Karlsson P, Farde L, Halldin C, Sedvall G. PET study of D(1) dopamine receptor binding in neuroleptic‐naive patients with schizophrenia. Am J Psychiatry 2002;159:761–767. [DOI] [PubMed] [Google Scholar]
- 64. Abi‐Dargham A, Mawlawi O, Lombardo I, et al Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci 2002;22:3708–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Abi‐Dargham A, Moore H. Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist 2003;9:404–416. [DOI] [PubMed] [Google Scholar]
- 66. Catafau AM, Searle GE, Bullich S, et al Imaging cortical dopamine D1 receptors using [11C]NNC112 and ketanserin blockade of the 5‐HT 2A receptors. J Cereb Blood Flow Metab 2010;30:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Abi‐Dargham A, Xu XY, Thompson JL, et al Effect of antipsychotic treatment on cortical D1 receptors. Neuroimage 2010;52:S41–S41. [Google Scholar]
- 68. Slifstein M, Kolachana B, Simpson EH, et al COMT genotype predicts cortical‐limbic D1 receptor availability measured with [11C]NNC112 and PET. Mol Psychiatry 2008;13:821–827. [DOI] [PubMed] [Google Scholar]
- 69. Kosaka J, Takahashi H, Ito H, et al Decreased binding of [11C]NNC112 and [11C]SCH23390 in patients with chronic schizophrenia. Life Sci 2010;86:814–818. [DOI] [PubMed] [Google Scholar]
- 70. Frankle WG, Narendran R, Huang Y, et al Serotonin transporter availability in patients with schizophrenia: A positron emission tomography imaging study with [11C]DASB. Biol Psychiatry 2005;57:1510–1516. [DOI] [PubMed] [Google Scholar]
- 71. Rasmussen H, Erritzoe D, Andersen R, et al Decreased frontal serotonin2A receptor binding in antipsychotic‐naive patients with first‐episode schizophrenia. Arch Gen Psychiatry 2010;67:9–16. [DOI] [PubMed] [Google Scholar]
- 72. Hashimoto T, Nishino N, Nakai H, Tanaka C. Increase in serotonin 5‐HT1A receptors in prefrontal and temporal cortices of brains from patients with chronic schizophrenia. Life Sci 1991;48:355–363. [DOI] [PubMed] [Google Scholar]
- 73. Tauscher J, Kapur S, Verhoeff NP, et al Brain serotonin 5‐HT(1A) receptor binding in schizophrenia measured by positron emission tomography and [11C]WAY‐100635. Arch Gen Psychiatry 2002;59:514–520. [DOI] [PubMed] [Google Scholar]
- 74. Yasuno F, Suhara T, Ichimiya T, Takano A, Ando T, Okubo Y. Decreased 5‐HT1A receptor binding in amygdala of schizophrenia. Biol. Psychiatry 2004;55:439–444. [DOI] [PubMed] [Google Scholar]
- 75. Frankle WG, Lombardo I, Kegeles LS, et al Serotonin 1A receptor availability in patients with schizophrenia and schizo‐affective disorder: A positron emission tomography imaging study with [11C]WAY 100635. Psychopharmacology 2006;189:155–164. [DOI] [PubMed] [Google Scholar]
- 76. Huttunen J, Heinimaa M, Svirskis T, et al Striatal dopamine synthesis in first‐degree relatives of patients with schizophrenia. Biol Psychiatry 2008;63:114–117. [DOI] [PubMed] [Google Scholar]
- 77. Fusar‐Poli P, Howes OD, Allen P, et al Abnormal frontostriatal interactions in people with prodromal signs of psychosis: A multimodal imaging study. Arch Gen Psychiatry 2010;67:683–691. [DOI] [PubMed] [Google Scholar]
- 78. Stone JM, Howes OD, Egerton A, et al Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of Psychosis. Biol Psychiatry 2010;68:599–602. [DOI] [PubMed] [Google Scholar]
- 79. Valli I, Howes O, Tyrer P, McGuire P, Grasby PM. Longitudinal PET imaging in a patient with schizophrenia did not show marked changes in dopaminergic function with relapse of psychosis. Am J Psychiatry 2008;165:1613–1614. [DOI] [PubMed] [Google Scholar]
- 80. Joel D, Weiner I. The connections of the dopaminergic system with the striatum in rats and primates: An analysis with respect to the functional and compartmental organization of the striatum. Neuroscience 2000;96:451–474. [DOI] [PubMed] [Google Scholar]
- 81. Hirvonen J, van Erp TG, Huttunen J, et al Brain dopamine d1 receptors in twins discordant for schizophrenia. Am J Psychiatry 2006;163:1747–1753. [DOI] [PubMed] [Google Scholar]
- 82. Hirvonen J, van Erp TG, Huttunen J, et al Increased caudate dopamine D2 receptor availability as a genetic marker for schizophrenia. Arch Gen Psychiatry 2005;62:371–378. [DOI] [PubMed] [Google Scholar]
- 83. Hirvonen J, van Erp TG, Huttunen J, et al Striatal dopamine D1 and D2 receptor balance in twins at increased genetic risk for schizophrenia. Psychiatry Res 2006;146:13–20. [DOI] [PubMed] [Google Scholar]
- 84. Kaprio J, Koskenvuo M. Genetic and environmental factors in complex diseases: The older Finnish Twin Cohort. Twin Res 2002;5:358–365. [DOI] [PubMed] [Google Scholar]
- 85. Cannon TD, Kaprio J, Lonnqvist J, Huttunen M, Koskenvuo M. The genetic epidemiology of schizophrenia in a Finnish twin cohort. A population‐based modeling study. Arch Gen Psychiatry 1998;55:67–74. [DOI] [PubMed] [Google Scholar]
- 86. Lee KJ, Lee JS, Kim SJ, et al Loss of asymmetry in D2 receptors of putamen in unaffected family members at increased genetic risk for schizophrenia. Acta Psychiatr Scand 2008;118:200–208. [DOI] [PubMed] [Google Scholar]
- 87. Brunelin J, d’Amato T, Van Os J, Costes N, Suaud Chagny MF, Saoud M. Increased left striatal dopamine transmission in unaffected siblings of schizophrenia patients in response to acute metabolic stress. Psychiatry Res 2010;181:130–135. [DOI] [PubMed] [Google Scholar]
- 88. Farde L, Hall H, Pauli S, Halldin C. Variability in D2‐dopamine receptor density and affinity: A PET study with [11C]raclopride in man. Synapse 1995;20:200–208. [DOI] [PubMed] [Google Scholar]
- 89. Puig MV, Watakabe A, Ushimaru M, Yamamori T, Kawaguchi Y. Serotonin modulates fast‐spiking interneuron and synchronous activity in the rat prefrontal cortex through 5‐HT1A and 5‐HT2A receptors. J. Neurosci. 2010;30:2211–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hurlemann R, Matusch A, Kuhn KU, et al 5‐HT2A receptor density is decreased in the at‐risk mental state. Psychopharmacology (Berl) 2008;195:579–590. [DOI] [PubMed] [Google Scholar]
- 91. Pinborg LH, Arfan H, Haugbol S, et al The 5‐HT2A receptor binding pattern in the human brain is strongly genetically determined. Neuroimage 2008;40:1175–1180. [DOI] [PubMed] [Google Scholar]
- 92. Savitz J, Lucki I, Drevets WC. 5‐HT1A receptor function in major depressive disorder. Prog. Neurobiol. 2009;88:17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Parsey RV, Ogden RT, Miller JM, et al Higher serotonin 1A binding in a second major depression cohort: Modeling and reference region considerations. Biol. Psychiatry 2010;68:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bertolino A, Knable MB, Saunders RC, et al The relationship between dorsolateral prefrontal N‐acetylaspartate measures and striatal dopamine activity in schizophrenia. Biol Psychiatry 1999;45:660–667. [DOI] [PubMed] [Google Scholar]
- 95. Bertolino A, Breier A, Callicott JH, et al The relationship between dorsolateral prefrontal neuronal N‐acetylaspartate and evoked release of striatal dopamine in schizophrenia. Neuropsychopharmacology 2000;22:125–132. [DOI] [PubMed] [Google Scholar]
- 96. Kegeles LS, Abi‐Dargham A, Zea‐Ponce Y, et al Modulation of amphetamine‐induced striatal dopamine release by ketamine in humans: Implications for schizophrenia. Biol Psychiatry 2000;48:627–640. [DOI] [PubMed] [Google Scholar]
- 97. Cannon TD, Thompson PM, van Erp TG, et al. Cortex mapping reveals regionally specific patterns of genetic and disease‐specific gray‐matter deficits in twins discordant for schizophrenia. Proc Natl Acad Sci U S A 2002;99:3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cannon TD, Huttunen MO, Lönnqvist J, et al. The inheritance of neuropsychological dysfunction in twins discordant for schizophrenia. Am J Hum Genet 2000;67:369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Seeman P, Guan HC, Niznik HB. Endogenous dopamine lowers the dopamine D2 receptor density as measured by [3H]raclopride: Implications for positron emission tomography of the human brain. Synapse 1989;3:96–97. [DOI] [PubMed] [Google Scholar]
- 100. Monakhov M, Golimbet V, Abramova L, Kaleda V, Karpov V. Association study of three polymorphisms in the dopamine D2 receptor gene and schizophrenia in the Russian population. Schizophr Res 2008;100:302–307. [DOI] [PubMed] [Google Scholar]
- 101. Hirvonen M, Laakso A, Nagren K, Rinne JO, Pohjalainen T, Hietala J. C957T polymorphism of the dopamine D2 receptor (DRD2) gene affects striatal DRD2 availability in vivo . Mol Psychiatry 2005;10:889. [DOI] [PubMed] [Google Scholar]
- 102. Glatt SJ, Faraone SV, Tsuang MT. Meta‐analysis identifies an association between the dopamine D2 receptor gene and schizophrenia. Mol Psychiatry 2003;8:911–915. [DOI] [PubMed] [Google Scholar]
- 103. Pohjalainen T, Cravchik A, Gejman PV, et al. Antagonist binding characteristics of the Ser311 – >Cys variant of human dopamine D2 receptor in vivo and in vitro. Biochem Biophys Res Commun 1997;232:143–146. [DOI] [PubMed] [Google Scholar]
- 104. Wong AH, Buckle CE, Van Tol HH. Polymorphisms in dopamine receptors: What do they tell us? Eur J Pharmacol 2000;410:183–203. [DOI] [PubMed] [Google Scholar]
- 105. Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Vall 08/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A 2001;98:6917–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Munafo MR, Bowes L, Clark TG, Flint J. Lack of association of the COMT (Val158/108 Met) gene and schizophrenia: A meta‐analysis of case‐control studies. Mol Psychiatry 2005;10:765–770. [DOI] [PubMed] [Google Scholar]
- 107. Akil M, Kolachana BS, Rothmond DA, Hyde TM, Weinberger DR, Kleinman JE. Catechol‐O‐methyltransferase genotype and dopamine regulation in the human brain. J Neurosci 2003;23:2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Meyer‐Lindenberg A, Kohn PD, Kolachana B, et al. Midbrain dopamine and prefrontal function in humans: Interaction and modulation by COMT genotype. Nat Neurosci 2005;8:594–596. [DOI] [PubMed] [Google Scholar]
- 109. Hirvonen MM, Nagren K, Rinne JO, Pesonen U, Vahlberg T, Hagelberg N, Hietala J. COMT Val158Met genotype does not alter cortical or striatal dopamine D2 receptor availability in vivo . Mol Imaging Biol 2010;12:192–197. [DOI] [PubMed] [Google Scholar]
- 110. Henquet C, Di Forti M, Morrison P, Kuepper R, Murray RM. Gene‐environment interplay between cannabis and psychosis. Schizophr. Bull. 2008;34:1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Moore THM, Zammit S, Lingford‐Hughes A, Barnes TRE, Jones PB, Burke M, Lewis G. Cannabis use and risk of psychotic or affective mental health outcomes: A systematic review. Lancet 2007;370:319–328. [DOI] [PubMed] [Google Scholar]
- 112. Henquet C, Krabbendam L, Spauwen J, Kaplan C, Lieb R, Wittchen HU, van Os J. Prospective cohort study of cannabis use, predisposition for psychosis, and psychotic symptoms in young people. BMJ 2005;330:11–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Caspi A, Moffitt T, Cannon M, et al Moderation of the effect of adolescent‐onset cannabis use on adult psychosis by a functional polymorphism in the catechol‐O‐methyltransferase gene: Longitudinal evidence of a gene X environment interaction. Biol. Psychiatry 2005;57:1117–1127. [DOI] [PubMed] [Google Scholar]
- 114. Henquet C, Rosa A, Krabbendam L, et al An experimental study of catechol‐O‐methyltransferase Val158Met moderation of Δ‐9‐tetrahydrocannabinol‐induced effects on psychosis and cognition. Neuropsychopharmacology 2006;31:2748–2757. [DOI] [PubMed] [Google Scholar]
- 115. Bossong MG, van Berckel BNM, Boellaard R, et al Δ9‐Tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology 2008;34:759–766. [DOI] [PubMed] [Google Scholar]