

Abstract
Background
This review is one of a series on drugs used to treat chronic neuropathic pain. Estimates of the population prevalence of chronic pain with neuropathic components range between 6% and 10%. Current pharmacological treatment options for neuropathic pain afford substantial benefit for only a few people, often with adverse effects that outweigh the benefits. There is a need to explore other treatment options, with different mechanisms of action for treatment of conditions with chronic neuropathic pain. Cannabis has been used for millennia to reduce pain. Herbal cannabis is currently strongly promoted by some patients and their advocates to treat any type of chronic pain.
Objectives
To assess the efficacy, tolerability, and safety of cannabis‐based medicines (herbal, plant‐derived, synthetic) compared to placebo or conventional drugs for conditions with chronic neuropathic pain in adults.
Search methods
In November 2017 we searched CENTRAL, MEDLINE, Embase, and two trials registries for published and ongoing trials, and examined the reference lists of reviewed articles.
Selection criteria
We selected randomised, double‐blind controlled trials of medical cannabis, plant‐derived and synthetic cannabis‐based medicines against placebo or any other active treatment of conditions with chronic neuropathic pain in adults, with a treatment duration of at least two weeks and at least 10 participants per treatment arm.
Data collection and analysis
Three review authors independently extracted data of study characteristics and outcomes of efficacy, tolerability and safety, examined issues of study quality, and assessed risk of bias. We resolved discrepancies by discussion. For efficacy, we calculated the number needed to treat for an additional beneficial outcome (NNTB) for pain relief of 30% and 50% or greater, patient's global impression to be much or very much improved, dropout rates due to lack of efficacy, and the standardised mean differences for pain intensity, sleep problems, health‐related quality of life (HRQoL), and psychological distress. For tolerability, we calculated number needed to treat for an additional harmful outcome (NNTH) for withdrawal due to adverse events and specific adverse events, nervous system disorders and psychiatric disorders. For safety, we calculated NNTH for serious adverse events. Meta‐analysis was undertaken using a random‐effects model. We assessed the quality of evidence using GRADE and created a 'Summary of findings' table.
Main results
We included 16 studies with 1750 participants. The studies were 2 to 26 weeks long and compared an oromucosal spray with a plant‐derived combination of tetrahydrocannabinol (THC) and cannabidiol (CBD) (10 studies), a synthetic cannabinoid mimicking THC (nabilone) (two studies), inhaled herbal cannabis (two studies) and plant‐derived THC (dronabinol) (two studies) against placebo (15 studies) and an analgesic (dihydrocodeine) (one study). We used the Cochrane 'Risk of bias' tool to assess study quality. We defined studies with zero to two unclear or high risks of bias judgements to be high‐quality studies, with three to five unclear or high risks of bias to be moderate‐quality studies, and with six to eight unclear or high risks of bias to be low‐quality studies. Study quality was low in two studies, moderate in 12 studies and high in two studies. Nine studies were at high risk of bias for study size. We rated the quality of the evidence according to GRADE as very low to moderate.
Primary outcomes
Cannabis‐based medicines may increase the number of people achieving 50% or greater pain relief compared with placebo (21% versus 17%; risk difference (RD) 0.05 (95% confidence interval (CI) 0.00 to 0.09); NNTB 20 (95% CI 11 to 100); 1001 participants, eight studies, low‐quality evidence). We rated the evidence for improvement in Patient Global Impression of Change (PGIC) with cannabis to be of very low quality (26% versus 21%;RD 0.09 (95% CI 0.01 to 0.17); NNTB 11 (95% CI 6 to 100); 1092 participants, six studies). More participants withdrew from the studies due to adverse events with cannabis‐based medicines (10% of participants) than with placebo (5% of participants) (RD 0.04 (95% CI 0.02 to 0.07); NNTH 25 (95% CI 16 to 50); 1848 participants, 13 studies, moderate‐quality evidence). We did not have enough evidence to determine if cannabis‐based medicines increase the frequency of serious adverse events compared with placebo (RD 0.01 (95% CI ‐0.01 to 0.03); 1876 participants, 13 studies, low‐quality evidence).
Secondary outcomes
Cannabis‐based medicines probably increase the number of people achieving pain relief of 30% or greater compared with placebo (39% versus 33%; RD 0.09 (95% CI 0.03 to 0.15); NNTB 11 (95% CI 7 to 33); 1586 participants, 10 studies, moderate quality evidence). Cannabis‐based medicines may increase nervous system adverse events compared with placebo (61% versus 29%; RD 0.38 (95% CI 0.18 to 0.58); NNTH 3 (95% CI 2 to 6); 1304 participants, nine studies, low‐quality evidence). Psychiatric disorders occurred in 17% of participants using cannabis‐based medicines and in 5% using placebo (RD 0.10 (95% CI 0.06 to 0.15); NNTH 10 (95% CI 7 to 16); 1314 participants, nine studies, low‐quality evidence).
We found no information about long‐term risks in the studies analysed.
Subgroup analyses
We are uncertain whether herbal cannabis reduces mean pain intensity (very low‐quality evidence). Herbal cannabis and placebo did not differ in tolerability (very low‐quality evidence).
Authors' conclusions
The potential benefits of cannabis‐based medicine (herbal cannabis, plant‐derived or synthetic THC, THC/CBD oromucosal spray) in chronic neuropathic pain might be outweighed by their potential harms. The quality of evidence for pain relief outcomes reflects the exclusion of participants with a history of substance abuse and other significant comorbidities from the studies, together with their small sample sizes.
Plain language summary
Cannabis products for adults with chronic neuropathic pain
Bottom line
There is a lack of good evidence that any cannabis‐derived product works for any chronic neuropathic pain.
Background
Neuropathic pain is pain coming from damaged nerves. It is different from pain messages that are carried along healthy nerves from damaged tissue (for example, a fall, or cut, or arthritic knee). Neuropathic pain is treated by different medicines to those used for pain from damaged tissue.
Several products based on the cannabis plant have been suggested as treatment for pain, including neuropathic pain. These products include inhaled herbal cannabis, and various sprays or tablets containing active cannabis ingredients obtained from the plant, or made synthetically.
Some people with neuropathic pain claim that cannabis‐based products are effective for them, and that is often highlighted in the media. Study characteristics
In November 2017 we searched for clinical trials that used cannabis products to treat conditions with chronic neuropathic pain in adults. We found 16 studies involving 1750 people. Studies lasted 2 to 26 weeks. Studies compared different cannabis‐based medicines. Ten studies compared an oromucosal (mouth) spray with a plant‐derived combination of tetrahydrocannabinol (THC), the principal psychoactive constituent of cannabis, and cannabidiol (CBD), an anti‐inflammatory ingredient of cannabis, against a fake medication (placebo). Two studies each compared inhaled herbal cannabis and cannabis plant‐derived THC with placebo, and one study compared a man‐made cannabinoid mimicking the effects of THC (nabilone) with placebo. One study compared nabilone with a pain killer (dihydrocodeine).
Key results and quality of the evidence
We rated the quality of the evidence from studies using four levels: very low, low, moderate, or high. Very low‐quality evidence means that we are very uncertain about the results. High‐quality evidence means that we are very confident in the results.
There was no high‐quality evidence.
All cannabis‐based medicines pooled together were better than placebo for the outcomes substantial and moderate pain relief and global improvement. All cannabis‐based medicines pooled together were better than placebo in reducing pain intensity, sleep problems and psychological distress (very low‐ to moderate‐quality evidence).
There was no difference between all cannabis‐based medicines pooled together and placebo in improving health‐related quality of life, stopping the medication because it was not effective, and in the frequency of serious side effects (low‐quality evidence).
More people reported sleepiness, dizziness and mental problems (e.g. confusion) with all cannabis‐based medicines pooled together than with placebo (low‐quality evidence). There was moderate‐quality evidence that more people dropped out due to side effects with cannabis‐based medicines than with placebo.
Herbal cannabis was not different from placebo in reducing pain and the number of people who dropped out due to side effects (very low‐quality evidence).
Summary of findings
Summary of findings 1. Cannabis‐based medicines compared with placebo for chronic neuropathic pain.
| Cannabis‐based medicines compared with placebo for chronic neuropathic pain | ||||||
|
Patient or population: adults with chronic neuropathic pain Settings: outpatient study centres and hospitals in Europe and North America Intervention: cannabis‐based medicines (smoked cannabis; oral plant‐based (dronabinol) or synthetic tetrahydrocannabinol (THC) (nabilone); oromucosal spray of THC and cannabidiol (CBD)) Comparison: placebo | ||||||
| Outcomes |
Probable outcome with intervention 95% CI |
Probable outcome with placebo |
Relative effect Risk difference (95% CI) |
No. of participants (studies) | Quality of the evidence (GRADE) | Comments |
| Participant‐reported pain relief of 50% or greater | 209 per 1000 (196 to 222) |
173 per 1000 | 0.05 (0.00 to 0.09) | 1001 (8 studies) | ⊕⊕⊝⊝ low1,2 |
NNTB 20 (11 to 100) |
| Patient Global Impression of Change much or very much improved | 261 per 1000 (246 to 276) |
211 per 1000 | 0.09 (0.01 to 0.17) | 1092 (6 studies) | ⊕⊝⊝⊝ very low1,3,4 |
NNTB 11 (6 to 100) |
| Withdrawals due to adverse events | 104 per 1000 (99 to 107) |
47 per 1000 | 0.04 (0.02 to 0.07) | 1848 (13 studies) | ⊕⊕⊕⊝ moderate1 |
NNTH 25 (16 to 50) |
| Serious adverse events | 66 per 1000 (63 to 69) |
52 per 1000 | 0.01 (‐0.01 to 0.03) | 1876 (13 studies) | ⊕⊕⊝⊝ low1,2 |
NNTH not calculated |
| Participant‐reported pain relief of 30% or greater | 377 per 1000 (358 to 396) |
304 per 1000 | 0.09 (0.03 to 0.15) | 1586 (10 studies) | ⊕⊕⊕⊝ moderate1 |
NNTB 11 (7 to 33) |
| Specific adverse events:nervous system disorder | 611 per 1000 (576 to 644) |
287 per 1000 | 0.38 (0.18 to 0.58) | 1304 (9 studies) | ⊕⊕⊝⊝ low1,3 |
NNTH 3 (2 to 6) |
| Specific adverse events:psychiatric disorders | 165 per 1000 (156 to 174) |
49 per 1000 | 0.10 (0.06 to 0.15) | 1314 (9 studies) | ⊕⊕⊝⊝ low1,3 |
NNTH 10 (7 to 16) |
| Abbreviations: CI: Confidence interval; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; RD: risk difference | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect; Moderate quality: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different; Low quality: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect; Very low quality: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded once: indirectness. People with current or historical substance abuse, or both, and major medical diseases excluded. 2 Downgraded once: imprecision. CI included zero. 3 Downgraded once: inconsistency. I²>50%.
4 Downgraded once: Publication bias. All studies funded by the manufacturer of the drug.
Background
The protocol for this review was based on a template for reviews of drugs used to relieve neuropathic pain. The aim is for all reviews to use the same methods, based on new criteria for what constitutes reliable evidence in chronic pain (Moore 2010a; Moore 2012; Appendix 1).
Description of the condition
The 2011 International Association for the Study of Pain definition of neuropathic pain is "pain caused by a lesion or disease of the somatosensory system" (Jensen 2011), and based on a definition agreed at an earlier consensus meeting (Treede 2008). Neuropathic pain is a consequence of a pathological maladaptive response of the nervous system to 'damage' from a wide variety of potential causes. It is characterised by pain in the absence of a noxious stimulus and may be spontaneous (continuous or paroxysmal) in its temporal characteristics or be evoked by sensory stimuli (dynamic mechanical allodynia where pain is evoked by light touch of the skin). Neuropathic pain is associated with a variety of sensory loss (numbness) and sensory gain (allodynia) clinical phenomena, the exact pattern of which vary between people and disease, perhaps reflecting different pain mechanisms operating in an individual person and, therefore, potentially predictive of response to treatment (Demant 2014; Helfert 2015; von Hehn 2012). Pre‐clinical research hypothesises a bewildering array of possible pain mechanisms that may operate in people with neuropathic pain, which largely reflect pathophysiological responses in both the central and peripheral nervous systems, including neuronal interactions with immune cells (Baron 2012; Calvo 2012; von Hehn 2012). Overall, the treatment gains in neuropathic pain, to even the most effective of available drugs, are modest (Finnerup 2015; Moore 2013a), and a robust classification of neuropathic pain is not yet available (Finnerup 2013).
Neuropathic pain is usually divided according to the cause of nerve injury. There may be many causes, but some common causes of neuropathic pain include diabetes (painful diabetic neuropathy (PDN)), shingles (postherpetic neuralgia), amputation (stump and phantom limb pain), neuropathic pain after surgery or trauma, stroke or spinal cord injury, trigeminal neuralgia, and HIV infection. Sometimes the cause is unknown.
Many people with neuropathic pain conditions are significantly disabled with moderate or severe pain for many years. Chronic pain conditions comprised five of the 11 top‐ranking conditions for years lived with disability in 2010 (Vos 2012), and are responsible for considerable loss of quality of life and employment, and increased healthcare costs (Moore 2014a). A study in the USA found that healthcare costs were three‐fold higher for people with neuropathic pain than matched control participants (Berger 2004). A UK study and a German study showed a two‐ to three‐fold higher level of use of healthcare services in people with neuropathic pain than those without (Berger 2009; Berger 2012). For postherpetic neuralgia, for example, studies demonstrate a large loss of quality of life and substantial costs (Scott 2006; Van Hoek 2009).
In systematic reviews, the overall prevalence of neuropathic pain in the general population is reported to be between 7% and 10% (Van Hecke 2014), and about 7% in a systematic review of studies published since 2000 (Moore 2014a). In individual countries, prevalence rates have been reported as 3.3% in Austria (Gustorff 2008), 6.9% in France (Bouhassira 2008), and up to 8% in the UK (Torrance 2006). Some forms of neuropathic pain, such as PDN and post‐surgical chronic pain (which is often neuropathic in origin), are increasing (Hall 2008).
Estimates of incidence vary between individual studies for particular origins of neuropathic pain, often because of small numbers of cases. In primary care in the UK, between 2002 and 2005, the incidences (per 100,000 person‐years' observation) were 28 (95% confidence interval (CI), 27 to 30) for PHN, 27 (95% CI, 26 to 29) for trigeminal neuralgia, 0.8 (95% CI, 0.6 to 1.1) for phantom limb pain, and 21 (95% CI, 20 to 22) for PDN (Hall 2008). Other studies have estimated an incidence of 4 in 100,000 per year for trigeminal neuralgia (Katusic 1991; Rappaport 1994), and 12.6 per 100,000 person‐years for trigeminal neuralgia and 3.9 per 100,000 person‐years for PHN in a study of facial pain in the Netherlands (Koopman 2009). One systematic review of chronic pain demonstrated that some neuropathic pain conditions, such as PDN, can be more common than other neuropathic pain conditions, with prevalence rates up to 400 per 100,000 person‐years (McQuay 2007).
Neuropathic pain is difficult to treat effectively, with only a minority of people experiencing a clinically relevant benefit from any one intervention (Kalso 2013; Moore 2013b). A multidisciplinary approach is now advocated, combining pharmacological interventions with physical or cognitive (or both) interventions. The evidence for interventional management is very weak, or non‐existent (Dworkin 2013). Conventional analgesics such as paracetamol and nonsteroidal anti‐inflammatory drugs (NSAIDs) are not thought to be effective, but without evidence to support or refute that view (Moore 2015a). Some people may derive some benefit from a topical lidocaine patch or low‐concentration topical capsaicin, although evidence about benefits is uncertain (Derry 2012; Derry 2014). High‐concentration topical capsaicin may benefit some people with PHN (Derry 2017). Treatment is often by so‐called pain modulators such as antidepressants (duloxetine and amitriptyline; Lunn 2014; Moore 2017; Moore 2015b; Sultan 2008), or antiepileptics (gabapentin or pregabalin; Moore 2009; Moore 2014b; Wiffen 2013). Evidence for efficacy of opioids is unconvincing (Gaskell 2016; Sommer 2015; Stannard 2016).
The proportion of people who achieve worthwhile pain relief (typically at least 50% pain intensity reduction; Moore 2013a) is small, generally only 10% to 25% more than with placebo, with numbers needed to treat for an additional beneficial outcome (NNTB) usually between 4 and 10 (Kalso 2013; Moore 2013b). Neuropathic pain is not particularly different from other chronic pain conditions in that only a small proportion of trial participants have a good response to treatment (Moore 2013b).
The current National Institute for Health and Care Excellence (NICE) guidance for the pharmacological management of neuropathic pain suggests offering a choice of amitriptyline, duloxetine, gabapentin, or pregabalin as initial treatment for neuropathic pain (with the exception of trigeminal neuralgia), with switching if the first, second, or third drugs tried are not effective or not tolerated (NICE 2013). This concurs with other recent guidelines (Finnerup 2015).
There is a need to explore other treatment options, with different mechanisms of action and from different drug categories, for treatment of neuropathic pain syndromes. Medical cannabis has been promoted by some patient organisations and advocates for the treatment of chronic pain refractory to conventional treatment and is available for pain management in some countries of the world, e.g. Canada and Israel (Ablin 2016). However, the use of cannabis for medical reasons is highly contested because of the adverse health effects of long‐term cannabis use for recreational purposes (Volkow 2014).
Description of the intervention
The cannabinoid system is ubiquitous in the animal kingdom, with multiple functions that move the organism back to equilibrium. A large body of evidence currently supports the presence of cannabinoid (CB) receptors and ligands in the peripheral and central nervous system, but also in other tissues such as bone and in the immune system (Owens 2015).
The endocannabinoid system has three broad and overlapping functions in mammals. The first is a stress recovery role, operating in a feedback loop in which endocannabinoid signalling is activated by stress and functions to return endocrine, nervous, and behavioural systems to homeostatic balance. The second is to control energy balance through regulation of the intake, storage, and utilisation of food. The third involves immune regulation; endocannabinoid signalling is activated by tissue injury and modulates immune and inflammatory responses (Hillard 2012). Thus, the endocannabinoid neuromodulatory system appears to be involved in multiple physiological functions, such as anti‐nociception, cognition and memory, endocrine function, nausea and vomiting, inflammation, and immune recognition (De Vries 2014; Hillard 2012). Cannabis is a genus of the flowering plant in the family Cannabaceae. The number of species within the genus is disputed. Three species may be recognized, Cannabis sativa, Cannabis indica and Cannabis ruderalis. These plants, commonly known as marijuana, have been used for pain relief for millennia, and have additional effects on appetite, sleep, and mood (Kalant 2001). Data from clinical trials with synthetic and plant‐based cannabis‐based medicines suggest a promising approach for the management of chronic neuropathic pain of different origins (De Vries 2014; Jensen 2015).
How the intervention might work
Cannabis contains over 450 compounds, with at least 70 classified as phytocannabinoids. Two are of particular medical interest. Delta 9‐tetrahydrocannabinol (delta 9‐THC) is the main active constituent, with psychoactive (e.g. reduction of anxiety and stress) and pain‐relieving properties. The second molecule of interest is cannabidiol (CBD), which has lower affinity for the cannabinoid (CB) receptors and the potential to counteract the negative effects of THC on memory, mood, and cognition, but also has an effect on pain modulation by anti‐inflammatory properties. The specific roles of currently identified endocannabis‐based medicines that act as ligands at CB receptors within the nervous system (primarily but not exclusively CB 1 receptors) and in the periphery (primarily but not exclusively CB 2 receptors) are only partially elucidated, but there are abundant pre‐clinical data to support their influence on nociception (Owens 2015).
It is also hypothesised that cannabis reduces alterations in cognitive and autonomic processing in chronic pain states (Guindon 2009). The frontal‐limbic distribution of CB receptors in the brain suggests that cannabis may preferentially target the affective qualities of pain (Lee 2013). In addition, cannabis may attenuate low‐grade inflammation, another postulate for the pathogenesis of neuropathic pain (Zhang 2015).
The content of THC and CBD in medical cannabis is highly variable and ranges from 1% to 22% THC and 0.05% to 9% CBD. In contrast the THC/CBD concentration in THC/CBD (nabiximols) oromucosal spray and the THC content in plant‐derived and synthetic THC are standardised (Häuser 2017).
Taking into consideration the poorly understood pathogenesis of chronic neuropathic pain syndromes, the complexity of symptom expression, and the absence of an ideal treatment, the potential for manipulation of the cannabinoid system as a therapeutic modality is attractive.
Why it is important to do this review
While recent guidance tends to be generally in agreement about the role of antidepressants and anticonvulsants in the management of chronic neuropathic pain (Finnerup 2015; NICE 2013), the role of opioids (Sommer 2015) and of cannabis‐based medicines (Häuser 2017, Häuser 2018) is under debate. Recent systematic reviews on the use of cannabis‐based medicines to treat chronic pain came to different conclusions on their importance in chronic neuropathic pain (Boychuk 2015; Finnerup 2015; Petzke 2016; Whiting 2015). This was probably due to the inclusion of different trials, different standards to evaluate the quality of evidence, and different weighting of the outcomes of efficacy, tolerability, and safety. Due to the conflicting conclusions of recent systematic reviews on the importance of cannabis‐based medicines in treating chronic neuropathic pain, as well as the public debate on the medical use of herbal cannabis for chronic pain (Ablin 2016; Fitzcharles 2014), we saw the need for a Cochrane Review applying the standards of Cochrane Pain, Palliative and Supportive Care (PaPaS).
Objectives
To assess the efficacy, tolerability, and safety of cannabis‐based medicines (herbal, plant‐based, synthetic) compared to placebo or conventional drugs for conditions with chronic neuropathic pain in adults.
Methods
Criteria for considering studies for this review
Types of studies
We included studies if they were randomised, double‐blind, controlled trials (RCTs) of at least two weeks' duration (drug titration and maintenance or withdrawal). We included studies with a parallel, cross‐over, and enriched enrolment randomised withdrawal (EERW) design with at least 10 participants per treatment arm. We required full journal publication, with the exception of online clinical trial results summaries of otherwise unpublished clinical trials, and abstracts with sufficient data for analysis. We did not include short abstracts. We excluded studies that were not randomised, studies of experimental pain, case reports, and clinical observations. We included studies that reported at least one outcome of efficacy and one of safety as defined below.
Types of participants
Studies included adults aged 18 years and above with one or more chronic (three months and more) neuropathic pain condition including (but not limited to):
cancer‐related neuropathy;
central neuropathic pain (e.g. multiple sclerosis);
complex regional pain syndrome (CRPS) Type II;
HIV neuropathy;
painful diabetic neuropathy;
peripheral polyneuropathy of other aetiologies, for example toxic (alcohol, drugs);
phantom limb pain;
postherpetic neuralgia;
postoperative or traumatic peripheral nerve lesions;
spinal cord injury;
nerve plexus injury;
trigeminal neuralgia.
Where included studies had participants with more than one type of neuropathic pain, we analysed results according to the primary condition. Studies had to state explicitly that they included people with neuropathic pain (by title). We excluded studies that assessed pain in people with neurological diseases without specifying that the pain assessed was of neuropathic nature. We excluded studies with fibromyalgia because the nature of fibromyalgia (neuropathic or not) is under debate (Clauw 2015); cannabis‐based medicines in fibromyalgia are the subject of another Cochrane Review (Häuser 2016). We excluded studies with 'mixed pain' (Baron 2004), because the concept is neither internationally accepted nor sufficiently validated and the focus of this review is only neuropathic pain.
Types of interventions
Cannabis‐based medicines, either herbal cannabis (hashish, marihuana), plant‐based cannabinoids (dronabinol: nabiximols), or pharmacological (synthetic) cannabinoids (e.g. levonantradol, nabilone), at any dose, by any route, administered for the relief of neuropathic pain and compared to placebo or any active comparator. We did not include studies with drugs under development that manipulate the endocannabinoid system by inhibiting enzymes that hydrolyse endocannabninoids and thereby boost the levels of the endogenous molecules (e.g. blockade of the catabolic enzyme fatty acid amide hydrolase (FAAH)) (Long 2009).
Types of outcome measures
The standards used to assess evidence in chronic pain trials have changed substantially in recent years, with particular attention being paid to trial duration, withdrawals, and statistical imputation following withdrawal, all of which can substantially alter estimates of efficacy. The most important change is the move from using mean pain scores, or mean change in pain scores, to the number of people who have a large decrease in pain (by at least 50%) and who continue in treatment, ideally in trials of eight to 12 weeks' duration or longer. These standards are set out in the PaPaS Author and Referee Guidance for pain studies of Cochrane Pain, Palliative and Supportive Care (Cochrane PaPaS 2012). This Cochrane Review assessed evidence using methods that make both statistical and clinical sense, and will use criteria for what constitutes reliable evidence in chronic pain (Moore 2010a).
We anticipated that studies would use a variety of outcome measures, with most studies using standard subjective scales (numerical rating scale (NRS) or visual analogue scale (VAS) for pain intensity or pain relief, or both). We were particularly interested in Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) definitions for moderate and substantial benefit in chronic pain studies (Dworkin 2008).
Primary outcomes
Participant‐reported pain relief of 50% or greater. We preferred composite neuropathic pain scores over single‐scale generic pain scores if both measures were used by studies;
PGIC (Patient Global Impression of Change) much or very much improved;
Withdrawals due to adverse events (tolerability);
Serious adverse events (safety). Serious adverse events typically include any untoward medical occurrence or effect that at any dose results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, is a congenital anomaly or birth defect, is an 'important medical event' that may jeopardise the person, or may require an intervention to prevent one of the above characteristics/consequences.
Secondary outcomes
Participant‐reported pain relief of 30% or greater. We preferred composite neuropathic pain scores over single‐scale generic pain scores if both measures were used by studies;
Mean pain intensity. We preferred composite neuropathic pain scores over single‐scale generic pain scores if both measures were used by studies;
Health‐related quality of life;
Sleep problems;
Fatigue;
Psychological distress;
Withdrawals due to lack of efficacy;
Any adverse event;
Specific adverse events, particularly nervous system (e.g. dizziness, somnolence, headache) and psychiatric disorders (e.g. confusion state; paranoia, psychosis, substance dependence) according to the Medical Dictionary for Regulatory Activities (MedDRA) (International Council for Harmonisation 2016).
Search methods for identification of studies
Electronic searches
We searched the following databases, without language restrictions:
The Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO) (searched 7 November 2017);
MEDLINE (via Ovid) (1946 to 7 November 2017);
Embase (via Ovid) (1974 to 7 November 2017).
Appendix 2 shows the search strategies.
Searching other resources
We reviewed the bibliographies of any RCTs identified and review articles, and searched the following clinical trials databases: US National Institutes of Health clinical trial register (www.ClinicalTrials.gov), European Union Clinical Trials Register (www.clinicaltrialsregister.eu), World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/), and International Association for Cannabinoid Medicines (IACM) databank (www.cannabis-med.org/studies/study.php) to identify additional published or unpublished data. We contacted trial investigators to request missing data.
Data collection and analysis
We performed separate analyses according to particular neuropathic pain conditions. We combined different neuropathic pain conditions in analyses for exploratory purposes only.
Selection of studies
Two review authors (WH, FP) determined eligibility by reading the abstract of each study identified by the search. We eliminated studies that clearly did not satisfy the inclusion criteria, and obtained full copies of the remaining studies. Two review authors (WH, FP) independently read these studies and reached agreement by discussion. We did not anonymise the studies before assessment. We created a PRISMA flow chart (Moher 2009).
Data extraction and management
Two review authors (WH, FP) extracted data independently using a standard form and checked for agreement before entering data into Review Manager 5 (RevMan 2014). Two review authors (WH, MM) extracted independently data calculated by imputation. We included information about the pain condition and number of participants treated, study setting, inclusion and exclusion criteria, demographic and clinical characteristics of the study samples (age, gender, race, pain baseline), prior recreational cannabis use, drug and dosing regimen, co‐therapies allowed, rescue medication, study design (placebo or active control), study duration and follow‐up, analgesic outcome measures and results, withdrawals, and adverse events (participants experiencing any adverse event or serious adverse event).
Assessment of risk of bias in included studies
Two review authors (WH, FP) independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a), and adapted from those used by Cochrane Musculoskeletal for recent reviews on drug therapy in fibromyalgia, with any disagreements resolved by discussion. We assessed the following for each study.
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (i.e. any truly random process, e.g. random number table; computer random number generator); unclear risk of bias (when the method used to generate the sequence was not clearly stated). We excluded studies at a high risk of bias that used a non‐random process (e.g. odd or even date of birth; hospital or clinic record number).
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as: low risk of bias (e.g. telephone or central randomisation; consecutively numbered, sealed, opaque envelopes); unclear risk of bias (when method was not clearly stated). We excluded studies that did not conceal allocation and were therefore at a high risk of bias (e.g. open list).
Blinding of participants and personnel/treatment providers (systematic performance bias). We assessed the methods used to blind participants and personnel/treatment providers from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that it was blinded and described the method used to achieve blinding, e.g. identical tablets; matched in appearance and smell); unclear risk of bias (study stated that it was blinded but did not provide an adequate description of how it was achieved); high risk of bias (blinding of participants was not ensured, e.g. tablets different in form or taste).
Blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study outcome assessors from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that outcome assessors were blinded to the intervention or exposure status of participants); unclear risk of bias (study stated that the outcome assessors were blinded but did not provide an adequate description of how it was achieved); high risk of bias (outcome assessors knew the intervention or exposure status of participants).
Incomplete outcome data (checking for possible attrition bias due to the amount, nature, and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk of bias (i.e. less than 10% of participants did not complete the study or used 'baseline observation carried forward' (BOCF) analysis, or both); unclear risk of bias (used 'last observation carried forward' analysis); or high risk of bias (used 'completer' analysis).
Reporting bias due to selective outcome reporting (reporting bias). We checked if an a priori study protocol was available and if all outcomes of the study protocol were reported in the publications of the study. There is low risk of reporting bias if the study protocol is available and all of the study's pre‐specified (primary and secondary) outcomes that are of interest in the review are reported in the pre‐specified way, or if the study protocol is not available but it is clear that the published reports include all expected outcomes, including those that are pre‐specified (convincing text of this nature may be uncommon). There is a high risk of reporting bias if not all of the study's pre‐specified primary outcomes are reported; one or more primary outcomes is reported using measurements, analysis methods or subsets of the data (e.g. subscales) that are not pre‐specified; one or more reported primary outcomes are not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect); one or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis; the study report did not include results for a key outcome that would be expected to have been reported for such a study. There is unclear risk of bias if insufficient information is available to permit judgement of ‘Low risk’ or ‘High risk’.
Group similarity at baseline (selection bias). We assessed similarity of the study groups at baseline for the most important prognostic clinical and demographic indicators. There is low risk of bias if groups are similar at baseline for demographic factors, value of main outcome measure(s), and important prognostic factors. There is an unclear risk of bias if important prognostic clinical and demographic indicators are not reported. There is high risk of bias if groups are not similar at baseline for demographic factors, value of main outcome measure(s), and important prognostic factors.
Size of study (checking for possible biases confounded by small size). We assessed studies as being at low risk of bias (200 participants or more per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); or high risk of bias (fewer than 50 participants per treatment arm).
Two review authors (WH, FP) assessed the included studies using the Cochrane 'Risk of bias' tool. We defined studies with zero to two unclear or high risks of bias to be high‐quality studies, with three to five unclear or high risks of bias to be moderate‐quality studies, and with six to eight unclear or high risks of bias to be low‐quality studies (Schaefert 2015).
Measures of treatment effect
We calculated numbers needed to treat for an additional beneficial outcome (NNTB) as the reciprocal of the absolute risk reduction (ARR; McQuay 1998). For unwanted effects, the NNTB becomes the number needed to treat for an additional harmful outcome (NNTH) and is calculated in the same manner. We used dichotomous data to calculate risk differences (RD) with 95% CIs using a fixed‐effect model unless we found significant statistical or clinical heterogeneity (see below). We set the threshold for a clinically relevant benefit or a clinically relevant harm for categorical variables by an NNTB or NNTH less than 10 (Moore 2008).
We calculated standardised mean differences (SMD) with 95% CIs for continuous variables using a fixed‐effect model unless we found significant statistical or clinical heterogeneity. We used Cohen's categories to evaluate the magnitude of the effect size, calculated by SMD, with Hedges' g value of 0.2 = small, 0.5 = medium, and 0.8 = large (Cohen 1988). We labelled a g value less than 0.2 to be a 'not substantial' effect size. We assumed a minimally important difference if the Hedges' g value was 0.2 or greater (Fayers 2014).
Unit of analysis issues
We split the control treatment arm between active treatment arms in a single study if the active treatment arms were not combined for analysis.
We included studies with a cross‐over design where separate data from the two periods were reported, data were presented that excluded a statistically significant carry‐over effect, or statistical adjustments were carried out in case of a significant carry‐over effect.
Dealing with missing data
We used intention‐to‐treat (ITT) analysis where the ITT population consisted of participants who were randomised, took at least one dose of the assigned study medication, and provided at least one post‐baseline assessment.
Where means or standard deviations (SDs) were missing, we attempted to obtain these data through contacting trial authors. Where SDs were not available from trial authors, we calculated them from t values, P values, CIs, or standard errors, where reported by the studies (Higgins 2011b). Where rates of pain relief of 30% and of 50% or greater were not reported or provided on request, we calculated them from means and SDs using a validated imputation method (Furukawa 2005).
Assessment of heterogeneity
We dealt with clinical heterogeneity by combining studies that examined similar conditions. We assessed statistical heterogeneity visually (L'Abbé 1987), and using the I2 statistic (Higgins 2003). When the I2 value was greater than 50%, we considered possible reasons for this.
Assessment of reporting biases
We assessed publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean an NNTB of 10 or higher; Moore 2008).
Data synthesis
We intended to use a fixed‐effect model for meta‐analysis. We used a random‐effects model using the inverse variance method in Review Manager 5 for meta‐analysis (RevMan 2014) because there was significant clinical heterogeneity due to the different types of neuropathic pain conditions included.
Quality of the evidence
Two review authors (WH, FP) independently rated the quality of the outcomes. We used the GRADE system to rank the quality of the evidence using the GRADEpro Guideline Development Tool software (GRADEpro GDT 2015), and the guidelines provided in Chapter 12.2 of the CochraneHandbook for Systematic Reviews of Interventions (Schünemann 2011).
The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence for each outcome. The GRADE system uses the following criteria for assigning grade of evidence:
high: we are very confident that the true effect lies close to that of the estimate of the effect;
moderate: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different;
low: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect;
very low: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
We decreased the grade rating by one (‐ 1) or two (‐ 2) if we identified:
serious (‐ 1) or very serious (‐ 2) limitation to study quality;
important inconsistency (‐ 1);
some (‐ 1) or major (‐ 2) uncertainty about directness;
imprecise or sparse data (‐ 1);
high probability of reporting bias (‐ 1).
In addition, there may be circumstances where the overall rating for a particular outcome needs to be adjusted as recommended by GRADE guidelines (Guyatt 2013a). For example, if there are so few data that the results are highly susceptible to the random play of chance, or if a study uses last observation carried forward (LOCF) imputation in circumstances where there are substantial differences in adverse event withdrawals, one would have no confidence in the result, and would need to downgrade the quality of the evidence by three levels, to very low quality. In circumstances where there were no data reported for an outcome, we planned to report the level of evidence as very low quality (Guyatt 2013b).
See also Appendix 3: GRADE: criteria for assigning grade of evidence.
'Summary of findings' table
We included one 'Summary of findings' table to present the main findings in a transparent and simple tabular format. In particular, we included key information concerning the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the outcomes. The 'Summary of findings' table includes the primary outcomes and the secondary outcomes of participant‐reported pain relief of 30% or greater, and nervous system disorders and psychiatric disorders as specific adverse events.
Subgroup analysis and investigation of heterogeneity
We performed subgroup analyses according to individual neuropathic pain syndromes because placebo response rates for the same outcome can vary between conditions, as can the drug‐specific effects (Moore 2013b). We performed subgroup analyses (different cannabis‐based medicines; very short‐term (less than four weeks), short‐term (four to 12 weeks), intermediate‐term (13 to 26 weeks), and long‐term (more than 26 weeks) study duration) where there were at least two studies available. We post‐hoc decided to perform subgroup analyses of studies with and without publication in peer‐reviewed journals. We performed subgroup analyses if at least two studies for a subgroup were available.
Sensitivity analysis
We planned no sensitivity analysis because the evidence base is known to be too small to allow reliable analysis.
Results
Description of studies
See Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification.
Results of the search
Appendix 2 shows the search strategies and hits retrieved for these databases. The searches (performed 7 November 2017) produced 1446 records after duplicates were removed. We identified 264 potentially relevant studies in CENTRAL, 949 in MEDLINE, 494 in Embase, three in the European Union Clinical Trials Register, 27 in the US National Institutes of Health clinical trials register, 116 in the WHO clinical trial register and 28 in the International Association for Cannabinoid Medicines (IACM) databank. After removing duplicates and reading the full reports, we included 16 studies involving 1750 participants into the qualitative and quantitative analysis (Bermann 2004; Ellis 2009; Frank 2008; Langford 2013; Lynch 2014; NCT00710424; NCT01606176; NCT01606202; Nurmikko 2007; Rog 2005; Schimrigk 2017; Selvarajah 2010; Serpell 2014; Schimrigk 2017Svendsen 2004; Toth 2012; Ware 2010) (see Figure 1). We excluded 15 studies. Of note, three studies from the database of the US National Institutes of Health have been not published in peer‐reviewed journals, and are awaiting classification. The results of three studies have not been published so far in the database of the US National Institutes of Health (NCT00710424; NCT01606176; NCT01606202).
1.
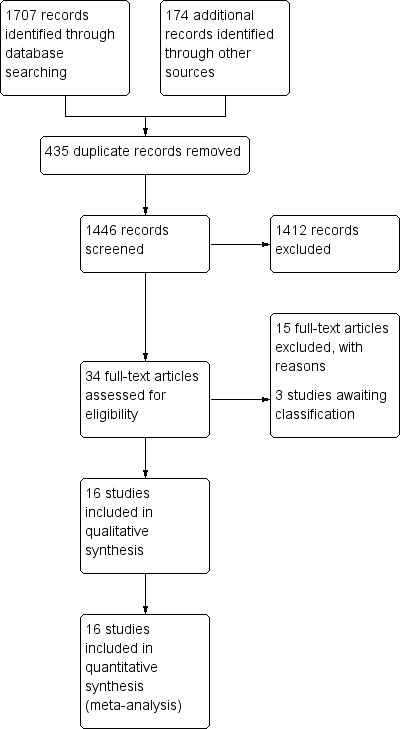
Study flow diagram
Included studies
Characteristics of the studies
Study design
Six studies used a cross‐over design (Bermann 2004; Ellis 2009; Frank 2008; Lynch 2014; Svendsen 2004; Ware 2010), nine a parallel design (Langford 2013; NCT00710424; NCT01606176; NCT01606202; Nurmikko 2007; Rog 2005; Schimrigk 2017; Selvarajah 2010; Serpell 2014) and two an enriched enrolment randomised withdrawal (EERW) design (Langford 2013;Toth 2012).
Study duration
Three studies were very short‐term studies (two to four weeks) (NCT01606176; NCT01606202; Ware 2010), eight were short‐term studies (four to 12 weeks) (Bermann 2004; Ellis 2009; Frank 2008; Lynch 2014; Nurmikko 2007; Rog 2005; Selvarajah 2010; Toth 2012), and five were intermediate‐term studies (12 to 26 weeks) (Langford 2013; NCT00710424; Schimrigk 2017; Serpell 2014; Svendsen 2004).
Study setting
Five studies were conducted in the UK (Bermann 2004; Frank 2008; NCT01606176; Rog 2005; Selvarajah 2010), three studies in Canada (Lynch 2014; Toth 2012; Ware 2010), three studies in multiple European countries (Langford 2013; NCT00710424; Nurmikko 2007), and one study in multiple countries of different continents (Serpell 2014), one study in USA (Ellis 2009), one study in Romania (NCT01606202), one study in Germany (Schimrigk 2017) and one study in Denmark (Svendsen 2004). Nine studies were single centre (Bermann 2004; Ellis 2009; Lynch 2014; Rog 2005;Schimrigk 2017; Selvarajah 2010; Svendsen 2004; Toth 2012; Ware 2010), and seven were multicentre (Frank 2008; Langford 2013; NCT00710424; NCT01606176; NCT01606202; Nurmikko 2007; Serpell 2014).
Sample sizes
The sample sizes ranged between 20 and 339.
Study periods
Study period was between 2000 and 2010 in seven studies (Bermann 2004; Frank 2008; Langford 2013; Schimrigk 2017; Serpell 2014; Svendsen 2004; Ware 2010). The remaining studies did not report the study period.
Study funding
Three studies were funded by public funds (Ellis 2009; Selvarajah 2010; Ware 2010), one study reported that there was no external funding (Lynch 2014), and the remaining studies were funded by the manufacturer of the drug. Four authors declared that they had no conflict of interest (Ellis 2009; Lynch 2014; Selvarajah 2010; Ware 2010). Six studies did not report on conflicts of interest (Bermann 2004; NCT00710424; NCT01606176; NCT01606202; Nurmikko 2007; Svendsen 2004). Six authors reported potential conflicts of interest by honoraria and/or funding received by the manufacturer of the drug studied (Frank 2008; Langford 2013; Rog 2005; Schimrigk 2017; Serpell 2014; Toth 2012).
Characteristics of the participants
Types of neuropathic pain
Five studies included participants with neuropathic pain associated with multiple sclerosis (Langford 2013; NCT01606176; Rog 2005; Schimrigk 2017; Svendsen 2004), three studies with mixed peripheral pain of various aetiologies (Nurmikko 2007; Serpell 2014; Ware 2010), three studies with diabetic polyneuropathy (NCT00710424; Selvarajah 2010; Toth 2012), and one study with plexus injury (Bermann 2004), one study with spinal cord injury (NCT01606202), one study with HIV‐neuropathy (Ellis 2009), one study with chemotherapy‐induced polyneuropathy (Lynch 2014), and one study with mixed central or peripheral pain of various aetiologies (Frank 2008).
Demographics
The mean age of the participants ranged between 34 and 61 years. The youngest mean age was in the studies with medical cannabis (Ellis 2009; Ware 2010). The percentage of men ranged between 17% and 100%.
Inclusion criteria
Nine studies required a pain score of 4 or above on a zero to 10 scale at baseline for inclusion (Bermann 2004; Ellis 2009; Frank 2008; Langford 2013; Lynch 2014; NCT00710424; Nurmikko 2007; Rog 2005; Schimrigk 2017). The remaining studies did not report on an inclusion criterion of a defined pain intensity. Five studies required for inclusion that the pain was refractory to previous analgesics without specifying the type and dosage of previous unsuccessful analgesic therapy (Ellis 2009; Langford 2013; NCT00710424; NCT01606176; Ware 2010).
Exclusion criteria
All studies excluded people with major medical diseases (heart, liver, kidney, seizures). Ten studies mentioned explicitly that they excluded people with a history of substance abuse (Bermann 2004; Ellis 2009; Frank 2008; Langford 2013; Lynch 2014; NCT00710424; NCT01606176; Nurmikko 2007; Rog 2005; Schimrigk 2017).
Previous experience of participants with herbal cannabis
Nine studies reported previous herbal cannabis experience of participants for medical and/or recreational use (Bermann 2004; Ellis 2009; Langford 2013; Lynch 2014; Nurmikko 2007; Rog 2005; Selvarajah 2010; Serpell 2014; Ware 2010). The percentage of participants with previous herbal cannabis experience ranged from 7% to 91%. Of note, the rates of previous herbal cannabis experience were the highest in the two studies with inhaled cannabis, with 91% in Ellis 2009 and 81% in Ware 2010. One study excluded people who had used marijuana in the month before study entry (Schimrigk 2017).
Characteristics of the treatment delivered
Types of cannabis‐based medicines
All studies used THC/CBD oromucosal spray except two studies that used oral synthetic THC (nabilone) (Frank 2008; Toth 2012), two studies that used plant‐based THC (dronabinol) (Schimrigk 2017; Svendsen 2004Schimrigk 2017 and two studies that used inhaled (by pipe or cigarette) herbal cannabis (Ellis 2009; Ware 2010). All studies compared to placebo except one study that compared to dihydrocodeine (DHC) (Frank 2008).
Rescue and Co‐medication
Two studies (Bermann 2004; Nurmikko 2007) did not allow rescue medication. Five studies used paracetamol (Frank 2008; Langford 2013; NCT01606202; Serpell 2014; Svendsen 2004) and one study tramadol (Schimrigk 2017). The remaining studies did not report details on rescue medication (Ellis 2009; Lynch 2014; NCT00710424; NCT01606176; Rog 2005; Selvarajah 2010; Toth 2012; Ware 2010). Four studies did not report if co‐medications were allowed (NCT00710424; NCT01606176; Selvarajah 2010; Toth 2012). The remaining studies allowed stable dosage of analgesic co‐medications.
Excluded studies
We excluded 15 studies for the following reasons: five studies because no definite statement was given that the pain was of neuropathic nature (Corey‐Bloom 2012; Novotna 2011; Wade 2004; Wissel 2006; Zajicek 2012); five studies because the study duration was less than two weeks (Abrams 2007; Karst 2003; Wallace 2015; Wilsey 2013; Wilsey 2008). one because the reports of the outcomes of efficacy did not meet the predefined inclusion criteria for efficacy (Zajicek 2003), two studies because there were fewer than 10 participants per treatment arm (Rintala 2010; Turcotte 2015), and one study each because participants with non‐neuropathic pain were included (Notcutt 2011) and participants without pain were included (Wade 2003).
Studies awaiting assessment
We found three studies with unpublished results or unknown status of which we received no information from the contacted authors. All three studies were conducted with nabilone by Canadian universities (NCT00699634; NCT01035281; NCT01222468). One of these studies was sponsored by the manufacturer of the drug (NCT00699634); the remaining two studies were funded by the university (NCT01035281; NCT01222468).
Risk of bias in included studies
The risk of bias of most domains was unclear in all studies: see Figure 2 and Figure 3 for a ’Risk of bias’ summary and graph and Characteristics of included studies for detailed information regarding 'Risk of bias' assessments of each study. The overall study quality according to the predefined criteria of the Cochrane 'Risk of bias' tool was low quality in two studies (Selvarajah 2010; Ware 2010), moderate quality in 12 studies (Bermann 2004; Ellis 2009; Frank 2008; Langford 2013; Lynch 2014; NCT00710424; NCT01606176; NCT01606202; Schimrigk 2017; Serpell 2014; Svendsen 2004; Toth 2012) and high quality in two studies (Nurmikko 2007; Rog 2005).
2.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
3.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study
Allocation
Random sequence generation
Random sequence generation was adequately described and therefore of low risk of bias in all studies except NCT00710424; NCT01606176; NCT01606202; Selvarajah 2010; Ware 2010, which did not adequately describe it (unclear risk of bias).
Allocation concealment
Allocation concealment was adequately described and therefore of low risk of bias in all studies except NCT00710424; NCT01606176; NCT01606202; Schimrigk 2017; Selvarajah 2010; Ware 2010, which did not adequately describe it (unclear risk of bias).
Blinding
Blinding of participants and personnel
Blinding of participants and personnel was adequately described and therefore of low risk of bias in all studies except Bermann 2004; Ellis 2009; Langford 2013; Lynch 2014; Schimrigk 2017; Selvarajah 2010; Ware 2010, which did not adequately describe it (unclear risk of bias).
Blinding of outcome assessor
Blinding of outcome assessment for adverse events was only adequately described by Nurmikko 2007 and Rog 2005. The remaining studies did not adequately describe it (unclear risk of bias).
Incomplete outcome data
Only one study performed intention‐to‐treat (ITT) analysis by baseline observation carried forward (BOCF) method (Svendsen 2004). Three studies performed completer analysis (Frank 2008; Selvarajah 2010; Ware 2010) (high risk of bias). The remaining studies performed ITT by last observation carried forward (LOCF) method and were therefore of unclear risk of bias.
Selective reporting
Two studies were of high risk of bias because they did not report all predefined outcomes (Ellis 2009; Selvarajah 2010). Four studies did not report on a study protocol and were therefore of unclear risk of bias (Bermann 2004; Lynch 2014; Svendsen 2004; Toth 2012). The remaining studies reported the outcomes as defined in a study protocol.
Other potential sources of bias
Group similarity at baseline
All studies had a low risk of bias because there were no significant differences in demographic and clinical variables at baseline except one study with a high risk of bias (Toth 2012).
Sample size
Sample size was of unclear risk of bias in seven studies (Frank 2008; Langford 2013; NCT00710424; NCT01606202; Nurmikko 2007; Schimrigk 2017; Serpell 2014), and of high risk of bias in nine studies (Bermann 2004; Ellis 2009; Lynch 2014; NCT01606176; Rog 2005; Selvarajah 2010; Svendsen 2004; Toth 2012; Ware 2010).
Effects of interventions
See: Table 1
All cannabis‐based medicines versus placebo ‐ studies with a cross‐over and parallel design
See Table 1.
Primary outcomes
The quailty of evidence was downgraded by one level due to indirectness (people with current or historical substance abuse, or both, and major medical diseases excluded) for all outcomes.
Participant‐reported pain relief of 50% or greater
We analysed eight studies with 1001 participants. One hundred and 10 of 526 (20.9%) participants in the cannabis‐based medicines and 82 of 475 (17.3%) participants in the placebo group reported pain relief of 50% or greater (risk difference (RD) 0.05, 95% CI 0.00 to 0.09); P value 0.04; I² = 29%). NNTB was 20 (11 to 100). According to the predefined categories, there was no clinically relevant benefit of cannabis‐based medicines (see Analysis 1.1). The quality of evidence was low, downgraded due to indirectness and imprecision (CI included zero).
1.1. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 1: Pain relief of 50% or greater
Patient Global Impression of Change much or very much improved
We analysed six studies with 1092 participants. One hundred and fifty‐six of 548 (28.4%) participants in the cannabis‐based medicines and 112 of 544 (22.1%) participants in the placebo group reported to be much or very much improved (RD 0.09 (95% CI 0.01 to 0.17; P value 0.02; I² = 58%). The NNTB was 11 (6 to 100). According to the predefined categories, there was no clinically relevant benefit of cannabis‐based medicines (see Analysis 1.2). The quality of evidence was very low, downgraded due to indirectness, inconsistency (I²>50%) and publication bias (all studies funded by the manufacturer of the drug).
1.2. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 2: Patient Global Impression much or very much improved
Withdrawals due to adverse events
We analysed 13 studies with 1848 participants. One hundred and three of 989 (10.4%) participants in the cannabis‐based medicines and 40 of 859 (4.7%) participants in the placebo group withdrew due to adverse events (RD 0.04, 95% CI 0.02 to 0.07; P value 0.0009; I² = 25%). The NNTH was 25 (16 to 50). According to the predefined categories there was no clinically relevant harm by cannabis‐based medicines (see Analysis 1.3). The quality of evidence was moderate, downgraded due to indirectness.
1.3. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 3: Withdrawals due to adverse events
Serious adverse events
We analysed 13 studies with 1876 participants. Sity‐six of 989 (6.7%) participants in the cannabis‐based medicines and 46 of 887 (5.2%) participants in the placebo group reported serious adverse events (RD 0.01, 95% CI ‐0.01 to 0.03; P value 0.29; I² = 0%) (see Analysis 1.4). The quality of evidence was low, downgraded due to indirectness and imprecision (CI included zero; low number of events).
1.4. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 4: Serious adverse events
Secondary outcomes
Participant‐reported pain relief of 30% or greater
We analysed 10 studies with 1586 participants. Three hundred and twenty‐three of 819 (39.4%) participants in the cannabis‐based medicines and 251 of 767 (32.7%) participants in the placebo group reported pain relief of 30% or greater (RD 0.09, 95% CI 0.03 to 0.15; P value 0.004; I² = 34%). NNTB was 11 (7 to 33). According to the predefined categories, there was no clinically relevant benefit by cannabis‐based medicines (see Analysis 1.5). The quality of evidence was moderate, downgraded due to indirectness.
1.5. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 5: Pain relief of 30% or greater
Mean pain intensity
We analysed 14 studies with 1837 participants. Cannabis‐based medicines were superior to placebo in the reduction of mean pain intensity (standardised mean difference (SMD) ‐0.35, 95% CI ‐0.60 to ‐0.09; P value 0.008; I² = 84%). According to Cohen’s categories, there was a small effect size indicating a minimal clinically important improvement (see Analysis 1.6). The quality of evidence was low, downgraded due to indirectness and inconsistency (I²>50%).
1.6. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 6: Mean pain intensity
Health‐related quality of life
We analysed nine studies with 1284 participants. Cannabis‐based medicines were not superior to placebo in the improvement of health‐related quality of life (HRQoL) (SMD 0.02, 95% CI ‐0.10 to 0.13; P value 0.79; I² = 0%) (see Analysis 1.7). The quality of evidence was low, downgraded due to indirectness and inconsistency (CI included zero).
1.7. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 7: Health‐related quality of life
Sleep problems
We analysed eight studies with 1386 participants. Cannabis‐based medicines were superior to placebo in the reduction of sleep problems (SMD ‐0.47, 95% CI ‐0.90 to ‐0.04; P value 0.03; I² = 92%). According to Cohen’s categories, there was a small effect size indicating a minimal clinically important improvement (see Analysis 1.8). The quality of evidence was low, downgraded due to indirectness and inconsistency (I²>50%).
1.8. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 8: Sleep problems
Fatigue
The analysis was not possible because fatigue was assessed only by one study (Langford 2013).
Psychological distress
We analysed seven studies with 779 participants. Cannabis‐based medicines were statistically significantly superior to placebo in the reduction of psychological distress (SMD ‐0.32, 95% CI ‐0.61 to ‐0.02; P value 0.04; I² = 66%). According to Cohen’s categories, there was a small effect size indicating a minimal clinically important improvement (see Analysis 1.9). The quality of evidence was low, downgraded due to indirectness and inconsistency (I²>50%).
1.9. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 9: Psychological distress
Withdrawals due to lack of efficacy
We analysed nine studies with 1576 participants. There was no difference in the frequency of withdrawals due to lack of efficacy between cannabis‐based medicines and placebo. Twenty‐two of 818 (2.7%) participants in the cannabis‐based medicines and 31 of 758 (4.1%) participants in the placebo group withdrew due to lack of efficacy (RD ‐0.00, 95% CI ‐0.02 to 0.01; P value 0.79; I² = 0%) (see Analysis 1.10). The quality of evidence was low, downgraded due to indirectness and imprecision (CI included zero).
1.10. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 10: Withdrawals due to lack of efficacy
Any adverse event
We analysed seven studies with 1356 participants. Five hundred and sixty‐two of 684 (80.2%) participants in the cannabis‐based medicines and 441 of 672 (65.6%) participants in the placebo group reported adverse events (RD 0.19, 95% CI 0.12 to 0.27; P value < 0.0001; I² = 64%). NNTH was 5 (4 to 8). According to the predefined categories, there was a clinically relevant harm by cannabis‐based medicines (see Analysis 1.11). The quality of evidence was low, downgraded due to indirectness and inconsistency (I²>50%).
1.11. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 11: Any adverse event
Specific adverse events
Nervous system disorders
We analysed nine studies with 1304 participants. Four hundred and fourteen of 677 (61.1%) participants in the cannabis‐based medicines and 180 of 627 (28.7%) participants in the placebo group reported adverse events of the nervous system (RD 0.38, 95% CI 0.18 to 0.58; P value 0.0003; I² = 94%). NNTH was 3 (2 to 6). According to the predefined categories, there was a clinically relevant harm by cannabis‐based medicines (see Analysis 1.12). The quality of evidence was low, downgraded due to indirectness and inconsistency (I²>50%).
1.12. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 12: Specific adverse event: nervous system disorders
Psychiatric disorders
We analysed nine studies with 1314 participants. One hundred and twelve of 677 (16.5%) participants in the cannabis‐based medicines and 31 of 637 (4.9%) participants in the placebo group reported psychiatric adverse events (RD 0.10, 95% CI 0.06 to 0.15; P value < 0.0001; I² = 54%). NNTH was 10 (7 to 16). According to the predefined categories, there was no clinically relevant harm by cannabis‐based medicines (see Analysis 1.13). The quality of evidence was low, downgraded due to indirectness and inconsistency (I²>50%).
1.13. Analysis.

Comparison 1: Cannabis‐based medicines versus placebo at final treatment, Outcome 13: Specific adverse event: psychiatric disorders
Cannabis‐based medicines versus placebo ‐ studies with an enriched enrolment randomised withdrawal design (results of double‐blind phase)
We present a qualitative analysis of the study results (Langford 2013; Toth 2012) because the data were not suited for quantitative analysis. The quality of evidence for each outcome was very low, downgraded because of indirectness (people with current or historical substance abuse, or both, and major medical diseases excluded), imprecision (low number of events) and publication bias (all studies funded by manufacturer of the drug).
Primary outcomes
Participant‐reported pain relief of 50% or greater
We analysed one study with 26 participants. There was no difference between nabilone and placebo in the number of participants with a 50% pain relief or greater (31% versus 8%; P value 0.12).
We analysed one study with 42 participants. There was a difference between THC/CBD and placebo in the number of participants with a treatment failure (24% versus 57%; P value 0.04).
Patient Global Impression of Change much or very much improved
We analysed one study with 26 participants. Six of 13 participants in the nabilone and one of 13 participants in the placebo group reported to be much or very much improved (P value 0.04).
Withdrawals due to adverse events
We analysed two studies with 68 participants. There was no difference between cannabis‐based medicines and placebo. None of the 21 participants dropped out of the THC/CBD spray group and one of 21 dropped out of the placebo group. None dropped out in the nabilone (13 participants) or placebo (13 participants) groups.
Serious adverse events
We analysed two studies with 68 participants. There was no difference between cannabis‐based medicines and placebo. Three of 21 participants experienced a serious adverse event in the THC/CBD spray and one of 21 in the placebo group. None experienced a serious adverse event in the nabilone (13 participants) or placebo (13 participants) group.
Secondary outcomes
Participant‐reported pain relief of 30% or greater
We analysed one study with 26 participants. There was a difference between nabilone and placebo in the number of participants with pain relief of 30% or greater (85% versus 38%; P value 0.006).
Mean pain intensity
We analysed two studies with 68 participants. The estimated treatment difference between THC/CBD spray and placebo was ‐0.79 (P value 0.03). The average pain intensity was 3.5 ± 1.3 in the nabilone and 5.4 ± 1.7 in the placebo group (P value 0.005) (higher scores indicate more pain).
Health‐related quality of life
We analysed two studies with 68 participants. The estimated treatment difference between THC/CBD spray and placebo was 1.94 (P value 0.18) in one study. The HRQoL score was 0.74 ± 0.03 in the nabilone and 0.60 ± 0.8 in the placebo group (P value < 0.05) in one study (higher scores indicating a better HRQoL).
Sleep problems
We analysed two studies with 68 participants. The estimated treatment difference between THC/CBD spray and placebo was ‐0.99 (P value 0.02). The sleep problems score was 27.1 ± 2.1 in the nabilone and 33.0 ± 2.6 in the placebo group (P value < 0.05) (higher scores indicate more sleep problems).
Fatigue
Neither of these studies assessed this outcome.
Psychological distress
We analysed one study with 42 participants. The estimated treatment difference between THC/CBD spray and placebo was ‐0.56 (P value 0.73).
Withdrawals due to lack of efficacy
We analysed one study with 42 participants. None of the participants in the THC/CBD study dropped out due to lack of efficacy.
Any adverse event
We analysed two studies with 68 participants. Ten per cent of participants with THC/CBD spray and 24% of participants with placebo reported an adverse event. Fifty‐four per cent of the participants receiving nabilone and 46% of the participants receiving placebo reported at least one adverse event (P value 1.0).
Specific adverse events
Nervous system disorders
We analysed one study with 42 participants. None of the participants in the THC/CBD group reported adverse events of the nervous system.
Psychiatric disorders
We analysed one study with 42 participants. Five per cent of participants in both groups reported a psychiatric adverse event.
Cannabis‐based medicines versus any active other drug
Only one study compared nabilone with dihydrocodeine (DHC) in 73 participants (Frank 2008). We therefore present a qualitative analysis of the study results. The quality of evidence for each outcome was very low, downgraded because of indirectness (people with current or historical substance abuse, and major medical diseases excluded), imprecision (low number of events) and publication bias (all studies funded by manufacturer of the drug).
Primary outcomes
Participant‐reported pain relief of 50% or greater
Frank 2008 assessed this outcome, however the study authors reported only the mean pain intensity.
Patient Global Impression of Change much or very much improved
Frank 2008 did not assess this outcome.
Withdrawals due to adverse events
There was no difference between nabilone and DHC. Four of 96 participants dropped out in the nabilone group and 8/96 in the DHC group (P value 0.23).
Serious adverse events
No major adverse events occurred when participants took either drug.
Secondary outcomes
Participant‐reported pain relief of 30% or greater
Frank 2008 assessed this outcome, however the study authors reported only the mean pain intensity.
Mean pain intensity
There was no difference between nabilone (59.93 ± 24.42) and DHC (58.58 ± 24.08) (P value not reported).
Health‐related quality of life
There was no difference between nabilone and DHC with a treatment difference of 8.9 (P value 0.48).
Sleep problems
There was no difference between nabilone and DHC with a treatment difference of 0.2 (P value 0.28).
Fatigue
Frank 2008 did not assess this outcome.
Psychological distress
There was no difference between nabilone and DHC with a treatment difference of 2.5 (P value 0.35).
Withdrawals due to lack of efficacy
Frank 2008 did not assess this outcome.
Any adverse event
There were 334 adverse events reported in the nabilone and 305 in the DHC group (no difference).
Specific adverse events
Nervous system disorders
This outcome was not assessed.
Psychiatric disorders
This outcome was not assessed.
Assessment of publication bias
The planned assessment of publication bias was not possible because the NNTB of all cannabis‐based medicines pooled together versus placebo for all dichotomous primary and secondary outcomes surpassed the pre‐set level of an NNTB of 10 or less.
Subgroup analysis and investigation of heterogeneity
We post‐hoc decided to restrict subgroup analyses to the outcomes pain relief of 50% or greater, PGIC (Patient Global Impression of Change) much or very much improved, withdrawals due to adverse events, serious adverse events and mean pain intensity. A subgroup analysis was only performed with at least two studies available.
Different types of neuropathic pain syndromes
We excluded studies with mixed samples of central and/or peripheral neuropathic pain from subgroup analysis because we wanted to assess the effects of cannabis‐based medicines on distinctive neuropathic pain syndromes. We found no subgroup difference between different types of neuropathic pain syndromes in the outcomes pain relief of 50% or greater (P value 0.20), withdrawals due to adverse events (P value 0.13), serious adverse events (P value 0.97), and mean pain intensity (P value 0.46). There was a subgroup difference between different types of neuropathic pain syndromes in the outcome PGIC (P value 0.02).
Different types of cannabis‐based medicines
Participant‐reported pain relief of 50% or greater
THC/CBD oromucosal spray was not different to placebo. RD was 0.05 (95% CI ‐0.00 to 0.11) (P value 0.07) (seven studies with 737 participants. Dronabinol (two studies with 264 participants) was not different to placebo. RD was 0.05 (95% CI ‐0.05 to 0.15) (P value 0.31) This outcome could not be analysed for herbal cannabis.
Patient Global Impression of Change much or very much improved
THC/CBD oromucosal spray (six studies with 1092 participants) was superior to placebo. RD was 0.09 (95% CI 0.01 to 0.17) (P value 0.02). The trials with dronabinol and herbal cannabis did not report this outcome.
Withdrawals due to adverse events
THC/CBD oromucosal spray (nine studies with 1408 participants) was superior to placebo. RD was 0.05 (95% CI 0.01 to 0.08) (P value 0.007). Dronabinol (two studies with 264 participants) was not different to placebo. RD was 0.05 (95% CI ‐0.04 to 0.13) (P value 0.27). Herbal cannabis (two studies with 152 participants) was not different to placebo. RD was 0.00 (95% CI ‐0.08 to 0.08) (P value 0.71).
Serious adverse events
THC/CBD oromucosal spray (eight studies with 1436 participants) was not different to placebo. RD was 0.01 (95% CI ‐0.01 to 0.02) (P value 0.52). Dronabinol (two studies with 264 participants) was not different to placebo. RD was 0.04 (95% CI ‐0.02 to 0.11) (P value 0.16). Herbal cannabis (two studies with 152 participants) was not different to placebo. RD was 0.01 (95% CI ‐0.05 to 0.06) (P value 0.74).
Mean pain intensity
THC/CBD oromucosal spray (nine studies with 1433 participants) was superior to placebo. SMD was ‐0.40 (95% CI ‐0.75 to ‐0.05) (P value 0.03). Dronabinol (two studies with 264 participants) was not superior to placebo. SMD was ‐0.09 (95% CI ‐0.33 to 0.15) (P value 0.45). Herbal cannabis (two studies with 152 participants) was not superior to placebo. SMD was ‐0.28 (95% CI ‐0.64 to 0.08) (P value 0.13).
Very short‐term, short‐term and intermediate‐term duration studies
Participant‐reported pain relief of 50% or greater
Cannabis‐based medicines in short‐term studies were not superior to placebo (three studies with 840 participants). RD was 0.06 (95% CI ‐0.01 to 0.13) (P value 0.05). Cannabis‐based medicines in intermediate‐term studies were not superior to placebo (three studies with 603 participants). RD was 0.04 (95% CI ‐0.03 to 0.11) (P value 0.24).
Patient Global Impression of Change much or very much improved
Cannabis‐based medicines in very short‐term studies were not superior to placebo (two studies with 186 participants). RD was 0.17 (95% CI ‐0.18 to 0.51) (P value 0.34). Cannabis‐based medicines in intermediate‐term studies were not superior to placebo (three studies with 840 participants). RD was 0.05 (95% CI ‐0.00 to 0.11) (P value 0.05).
Withdrawals due to adverse events
Cannabis‐based medicines in very short‐term studies were not superior to placebo (three studies with 270 participants). RD was 0.03 (95% CI ‐0.03 to 0.09) (P value 0.34). Cannabis‐based medicines in short‐term studies were not superior to placebo (four studies with 478 participants). RD was 0.01 (95% CI ‐0.02 to 0.04) (P value 0.80). Cannabis‐based medicines in intermediate‐term studies were superior to placebo (five studies with 1120 participants). RD was 0.07 (95% CI 0.03 to 0.12) (P value 0.002).
Serious adverse events
Cannabis‐based medicines in very short‐term studies were not superior to placebo (three studies with 270 participants). RD was ‐0.01 (95% CI ‐0.05 to 0.34) (P value 0.59). Cannabis‐based medicines in short‐term studies were not superior to placebo (five studies with 435 participants). RD was 0.00 (95% CI ‐0.02 to 0.02) (P value 1.0). Cannabis‐based medicines in intermediate‐term studies were superior to placebo (five studies with 1120 participants). RD was 0.03 (95% CI 0.00 to 0.06) (P value 0.05).
Mean pain intensity
Cannabis‐based medicines in very short‐term studies were not superior to placebo (three studies with 268 participants). SMD was ‐0.13 (95% CI ‐0.38 to 0.12) (P value 0.31). Cannabis‐based medicines in short‐term studies were not superior to placebo (six studies with 453 participants). SMD was ‐0.63 (95% CI ‐1.31 to 0.05) (P value 0.07). Cannabis‐based medicines in intermediate‐term studies were not superior to placebo (five studies with 1109 participants). SMD was ‐0.09 (95% CI ‐0.20 to 0.03) (P value 0.31).
Published and unpublished trials with THC/CBD oromucosal spray
Participant‐reported pain relief of 50% or greater
An analysis was not possible because the outcome was not reported by the unpublished trials.
Patient Global Impression of Change much or very much improved
THC/CBD spray was superior to placebo in published trials (three studies with 655 participants). RD was 0.07 (95% CI 0.01 to 0.13) (P value 0.03). THC/CBD spray was not superior to placebo in unpublished trials (three studies with 437 participants). RD was 0.12 (95% CI ‐0.10 to 0.33) (P value 0.29).
Withdrawals due to adverse events
There was a difference between THC/CBD spray and placebo in published trials (six studies with 935 participants). RD was 0.03 (95% CI 0.00 to 0.07) (P value 0.03). There was no difference between THC/CBD spray and placebo in unpublished trials (three studies with 437 participants). RD was 0.06 (95% CI ‐0.03 to 0.15) (P value 0.17).
Serious adverse events
There was no difference between THC/CBD spray and placebo in published trials (six studies with 935 participants). RD was 0.01 (95% CI ‐0.01 to 0.03) (P value 0.48). There was no difference between THC/CBD spray and placebo in unpublished trials (three studies with 437 participants). RD was ‐0.00 (95% CI ‐0.04 to 0.04) (P value 1.0).
Mean pain intensity
THC/CBD spray was superior to placebo in published trials (eight studies with 1069 participants). SMD was ‐0.46 (95% CI ‐0.42 to ‐0.01) (P value 0.05). THC/CBD spray was not superior to placebo in unpublished trials (three studies with 437 participants). SMD was ‐0.08 (95% CI ‐0.26 to 0.10) (P value 0.39).
Studies with high and unclear risk of bias due to sample size
Five of the 10 studies that reported the outcome 30% or more pain relief had treatment group sizes below 50 participants and we considered them at high risk of bias. Analysis of these five studies with 328 participants (24% of the total) showed an RD for pain relief of 30% or greater of 0.17 (95% CI 0.06 to 0.27); 40% of participants reported this outcome with cannabis‐based medicines and 26% with placebo.
Five of the 10 studies that reported the outcome 30% or more pain relief had treatment group sizes above 50 but below 200 participants and we considered them at unclear risk of bias. Analysis of these four studies with 1018 participants (76% of the total) showed an RD for pain relief of 30% or greater of 0.05 (95% CI ‐0.00 to 0.11); 41% of participants reported this outcome with cannabis‐based medicines and 37% with placebo.
Heterogeneity
I² was less than 50% except for Patient Global Impression of Change (I² = 58%), mean pain intensity (I² = 55%), sleep problems (I² = 92%), psychological distress (I² = 66%), any adverse event (I² = 64%), nervous system disorders as adverse event (I² = 94%) and psychiatric disorders as adverse event (I² = 54%). We did not find clinical explanations for heterogeneity.
Discussion
Summary of main results
We included 16 studies, 2 to 26 weeks long, with 1750 participants. All studies compared cannabis‐based medicines with placebo except one study that compared synthetic THC with dihydrocodeine (DHC). Studies compared an oromucosal spray with a plant‐derived combination of THC and CBD (10 studies), inhaled herbal cannabis (two studies), synthetic THC (nabilone) (two studies) and plant‐derived THC (dronabinol) (two studies).
All cannabis‐based medicines (at any dose) pooled together were superior to placebo for substantial (50% and more) (low‐ quality evidence) and moderate (30% and more) pain relief (moderate‐quality evidence), for global improvement (very low‐quality evidence), and in reduction of mean pain intensity (low‐quality evidence), sleep problems (low‐quality evidence), and psychological distress (low‐quality evidence). The effect sizes of mean pain intensity, sleep problems and psychological distress were clinically relevant. There was moderate‐quality evidence that more people dropped out due to adverse events with cannabis‐based medicines compared to placebo. There was low‐quality evidence that more people reported any adverse event and adverse events of the central nervous system and psychiatric disorders with all cannabis‐based medicines pooled together than with placebo. The effect size of adverse events of the nervous system disorders was clinically relevant. There was no difference between all cannabis‐based medicines pooled together and placebo in the frequency of serious adverse events (low‐quality evidence), for improvement of health‐related quality of life (low‐quality evidence) and dropouts due to lack of efficacy (moderate‐quality evidence).
There was no high‐quality evidence suggesting that any cannabis‐based medicine (herbal cannabis, THC/CBD oromucosal spray, synthetic or plant‐based THC) was of value in treating people with chronic neuropathic pain.
Overall completeness and applicability of evidence
The overall completeness and applicability of the evidence were poor. The usefulness of the available evidence was limited because reporting quality was poor by current standards (Moore 2010a). The reliability of the pooled results in general and of findings on nabilone in particular was limited because the results of three studies with nabilone have not been published and the results were not provided by the study authors on request (NCT00699634; NCT01035281; NCT01222468). The applicability of the evidence to routine clinical care was limited because all the included studies excluded people with current or historical substance abuse, or both, and major medical diseases.
Quality of the evidence
We found the evidence for most outcomes to be low quality because of indirectness (people with major medical disorders excluded) and inconsistent results. Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. In addition, we found signs of publication bias. We found three industry‐sponsored studies of THC/CBD spray with negative results, which have not been fully published yet. We also found three studies of nabilone but the results were unknown; the study authors did not respond to our requests. Despite growing requirements for trial registration, full access to clinical trial data remains elusive (Mintzes 2015).
Six studies reviewed used a cross‐over design with a study duration between one to two weeks for each period, and cross‐over designs have methodological issues that could lead to bias (Elbourne 2002). The short study duration limits their applicability. In addition, there are issues about the time needed (if any) for washout between treatment periods. Poor reporting limits their use in meta‐analysis, possibly with some biases (Moore 2013b).
A large number of participants (7% to 91%) in the studies were former cannabis users. No subgroup comparisons (former cannabis users versus cannabis‐naive participants) were conducted by any study. A prospective observational study found that the rate of non‐serious adverse events among current cannabis users was lower than that among ex‐cannabis users or naive users (Ware 2015).Therefore we do not know if the study results on efficacy and safety of the RCTs reviewed are valid for cannabis‐naive participants.
People with chronic neuropathic pain exhibit a variety of pain‐related sensory symptoms and findings (Baron 2017). They use different descriptors for their pain (e.g. burning, tugging, pricking, cramping). None of the neuropathic pain scales available cover all potential descriptors of neuropathic pain (Thyson 2014). Eight of the studies reviewed used a neuropathic pain scale. However, none of the studies reported the effects of cannabis‐based medicines on the single dimensions of the neuropathic pain scales used. A recent study with botulinum toxin in peripheral neuropathic pain demonstrated a statistically significant effect on paroxysmal pain, but not on burning and deep pain (Attal 2016). Therefore we do not know the efficacy of cannabis‐based medicines for specific qualities of neuropathic pain.
Perhaps the biggest issue is that of the relatively small size of the studies. Nine of the 16 studies were at high risk of bias because of small size. There are issues over both random chance effects with small amounts of data, and potential bias in small studies, especially in pain (Dechartes 2013; Dechartres 2014; Moore 1998; Nüesch 2010; Thorlund 2011). Cochrane Reviews have been criticised for perhaps over‐emphasising results of underpowered studies or analyses (AlBalawi 2013; Turner 2013). On the other hand, it may be unethical to ignore potentially important information from small studies or to randomise more participants if a meta‐analysis including small, existing studies provided conclusive evidence. In this review, we chose to limit analyses to studies with a minimum of 10 participants per treatment group. Small studies may have influenced positive results in this review. For example, for moderate pain relief (at least 30% pain relief), the overall result was positive with an RD of 0.09 (0.03 to 0.15) in an analysis of 10 studies with 1566 participants, but the difference between cannabis‐based medicines and placebo was much larger in small studies. We had not initially planned this analysis, but examination of the forest plots demonstrated that for this and other outcomes, the elimination of small studies eliminated statistical significance. In view of the accumulating evidence regarding potential bias in small studies, the quality of the evidence for cannabis‐based medicines for treating neuropathic pain cannot be relied upon.
Potential biases in the review process
The absence of publication bias (unpublished trials showing no benefit of cannabis‐based medicines over placebo) can never be proved. We carried out a broad search for studies and feel it is unlikely that significant amounts of relevant data remain unknown to us.
We might have overestimated the risk of bias of some studies that did not report some details of methodology (e.g. randomisation and blinding procedures).
Most studies selected statistical methods (last observation carried forward, completer analysis) that bias results towards exaggerating the efficacy of drugs (Moore 2013b).
The influence of allowed co‐interventions (e.g. rescue medication) on positive effects and adverse events was unclear because type and dosage of co‐interventions were not clearly reported or controlled for.
This systematic review included 1750 participants. To capture rare and potentially severe adverse events a larger data set would have been necessary. For example, to capture an adverse event with a frequency of 1:100,000, 300,000 participants' observations would have been necessary (Andersohn 2008).
Agreements and disagreements with other studies or reviews
We cannot share the optimistic conclusions of some reviews that cannabis‐based medicines are effective, well‐tolerated and safe in the treatment of chronic neuropathic pain (Andreae 2015; Boychuk 2015; Lynch 2011). Lynch 2011 performed a qualitative systematic review on cannabis‐based medicines in chronic non‐cancer pain with 11 studies in chronic neuropathic pain and concluded that cannabis‐based medicines are "modestly" effective in neuropathic pain and did not lead to withdrawal from the study. Boychuk 2015 performed a qualitative analysis of 13 studies of cannabis‐based medicines in 771 participants with chronic neuropathic pain and concluded that cannabis‐based medicines should be considered as an alternative treatment for neuropathic pain. The authors made no definitive statement on tolerability and safety. Andreae 2015 performed an individual participant data analysis of 178 participants from five studies of inhaled cannabis. They calculated an NNTB of 6 (95% CI 3 to 14) for a more than 30% reduction in pain scores compared to placebo. Withdrawals due to adverse events were found to be rare. The differences to our rather cautious conclusions on the efficacy, tolerability and safety of cannabis‐based medicines in chronic neuropathic pain can be explained as follows.
We performed a quantitative analysis, which included unpublished studies with negative results. The authors of the above‐mentioned reviews did not include the data of studies that are only available in databases.
We excluded studies of very short‐term duration. Andreae 2015; Boychuk 2015 and Lynch 2011 included two, one‐day studies (Wilsey 2013; Wilsey 2008), which we excluded because of short study duration. The European Medicines Agency requires that study duration for chronic neuropathic pain trials should be at least 12 weeks after a stable dose is achieved in order to exclude a transient effect (European Medicines Agency 2007).
We excluded studies that did not explicitly state that the pain was of neuropathic nature. This exclusion criterion was applied to some large studies in people with multiple sclerosis with spasticity as a major outcome. There is moderate‐quality evidence for the efficacy of cannabis‐based medicines to reduce spasticity symptoms (Whiting 2015; Zettl 2016). However, spasticity‐associated pain should not be mixed with central neuropathic pain (Koppel 2014).
We performed a detailed analysis of adverse events and withdrawals due to adverse events.
On the other hand, our analyses do not support the conclusions of the Special Interest Group on Neuropathic Pain (NeuPSIG) of the International Association for the Study of Pain that cannabis‐based medicines are not effective in chronic neuropathic pain (Finnerup 2015). Our result that the use of cannabis‐based medicines is associated with an increased risk of short‐term adverse events, especially of the central nervous system, is in accordance with a systematic review of Whiting 2015 who analysed eight trials of cannabis‐based medicines in chronic neuropathic pain.
We did not find a long‐term RCT with cannabis‐based medicines answering the question of long‐term efficacy and safety. One study with dronabinol included in the review added a 32‐week, open‐label extension period to the randomised controlled period. The study authors reported that, during long‐term follow‐up, pain intensities remained at a low level (range 2.5 to 3.8 of a 0 to 10 scale). The number of adverse events and dropouts due to adverse events was lower in the long‐term than in the randomised‐controlled period. "Mild signs" of drug dependency were documented for one participant (Schimrigk 2017). THC/CBD oromucosal spray was investigated in a 38‐week, open‐label extension study. Three hundred and eighty participants with polyneuropathy associated with diabetes or allodynia entered this study from two previous RCTs. Participants received THC/CBD spray for a further 38 weeks in addition to their current analgesic therapy. The proportion of participants who reported at least a clinically relevant 30% improvement in pain continued to increase with time (up to nine months); at least half of all participants reported a 30% improvement at all time points. Improvements were observed for all secondary efficacy outcomes, including sleep quality, Patient Global Impression of Change and HRQoL. THC/CBD spray was well tolerated for the study duration and participants did not seek to increase their dose with time, with no new safety concerns arising from long‐term use (Hoggart 2015).
Authors' conclusions
Implications for practice.
For people with chronic neuropathic pain
There is no high‐quality evidence for the efficacy of any cannabis‐based product including herbal cannabis (marijuana) in any condition with chronic neuropathic pain. Some adverse events (particularly somnolence or sedation, confusion, psychosis) may limit the clinical usefulness of cannabis‐based medicines. It might be expected that, at best, a few people with neuropathic pain will benefit from long‐term use of cannabis‐based medicines.
Some current clinical guidelines and systematic reviews consider cannabis‐based medicines as third‐ or fourth‐line therapy for chronic neuropathic pain syndromes if established therapies (e.g. anticonvulsants, antidepressants) have failed (Moulin 2014; Petzke 2016).
For physicians
There is no high‐quality evidence for the efficacy of any cannabis‐based medicine (herbal cannabis, plant‐derived THC (dronabinol), synthetic THC (nabilone), plant‐derived THC/CBD combination) in any condition with chronic neuropathic pain. Some adverse events (particularly somnolence or sedation, confusion, psychosis) may limit the clinical usefulness of cannabis‐based medicines. It might be expected that, at best, a few people with neuropathic pain will benefit from long‐term use of cannabis‐based medicines. Since relatively few participants achieve a worthwhile response with cannabis‐based medicines, decisions to use these medicines may require stopping rules to avoid the unnecessary exposure to harms in the absence of benefit. .
The Canadian Pain Society recommended cannabis‐based medicines as third‐line therapy for chronic neuropathic pain syndromes if established therapies (e.g. anticonvulsants, antidepressants) had failed (Moulin 2014). The Special Interest Group on Neuropathic Pain (NeuPSIG) for the pharmacotherapy of neuropathic pain gave a weak recommendation against the use of cannabis‐based medicines (Finnerup 2015).
The status of approval of cannabis‐based medicines and reimbursement by health insurance companies for chronic pain differs from country to country (Ablin 2016; Krcevski‐Skvarc 2018).
For policy‐makers
There is no high‐quality evidence suggesting that cannabis‐based medicines (herbal cannabis plant‐derived THC (dronabinol), synthetic THC (nabilone), plant‐derived THC/CBD combination) are of value in treating people with chronic neuropathic pain. This needs to be explained to people requesting this treatment in jurisdictions where it is allowed, e.g. Canada, Germany and Israel.
The license of cannabis‐based medicines including herbal cannabis for people with chronic (neuropathic) pain is scheduled for some countries. A patient register to document the efficacy and risks of cannabis‐based medicines financed by public funds is preferable.
In the absence of high‐quality evidence of benefit, the use of cannabis‐based medicines at the discretion of a pain specialist with particular expertise in use of cannabis‐based medicines is desirable. Cannabis‐based medicines are no first‐line treatment of any condition with chronic neuropathic pain.
For funders
Since no single treatment is effective in a majority of individuals with chronic neuropathic pain, this relatively small number of people with neuropathic pain who benefit from cannabis‐based medicines may be considered worthwhile, particularly if switching rules are in place. The treatment should be supervised by a pain specialist.
Implications for research.
General
There may be differences in effect of different cannabis‐based medicines in different types of neuropathic pain. The optimal ratio of THC/CBD still needs to be determined. In addition, pure CBD products or the development of peripherally acting cannabinoid agonists may reduce central nervous system and psychiatric adverse events. To be certain of a result in terms of both direction and magnitude of effect would require very large clinical trials. These trials would need to have important design features.
Chronic neuropathic pain conditions that have not been included in previous trials, such as post‐stroke pain, need to be studied.
Study duration with a minimum of three months is recommended.
In those clinical conditions for which there is an established treatment option, a three‐arm study (study drug – standard drug treatment‐ placebo) is desirable, in order to allow the assessment of comparative efficacy and safety.
Outcomes of clinical utility, such as moderate and substantial benefit using neuropathic pain scales and Patient Global Impression of Change scale (PGIC), are recommended.
Imputation method are to be abandoned, as the outcome desired is that of adequate pain relief in the longer term, and for that people have to continue on therapy. Withdrawal for any reason has to be classified as treatment failure.
It is preferable that the study protocol defines that treating people with cannabis‐based medicines who do not have pain relief is unacceptable, so that there would be built‐in stopping rules linked to pain relief after an adequate trial of therapy.
It is valuable to design and analyse studies whether there are any predisposing features linked with treatment success or failure.
Study data have to be made available for review authors for individual participant data analyses.
Reporting the details of the assessment of adverse events (spontaneous reports, open questions, symptom questionnaires) is mandatory because the type and frequency of adverse events is influenced by the modes of assessment (Häuser 2012). Adverse events have to be reported using the International Conference on Harmonization guidelines, and coded within organ classes using the Medical Dictionary for Regulatory Activities (International Council for Harmonisation 2016). It is desirable that regulatory agencies standardise the assessment strategies of adverse events in randomised controlled trials.
Design
The key question is whether there are any people with neuropathic pain who do well on cannabis‐based medicines in the long term; that is, with a substantial reduction in pain and/or improvement of daily functioning maintained and tolerable adverse events. An alternative to clinical trials might be the use of registry studies.
Measurement (endpoints)
Reporting of average pain changes is inadequate, and the use of responder analyses (pain relief of 50% or greater or participants experiencing mild or no pain) is preferred.
The contextual details (e.g. type of pain (average, worst, least, current), time period to be rated, location of pain) of their administration are typically not standardised, nor well‐reported in the literature, resulting in trial results that are challenging to interpret. In an effort to standardise pain intensity assessment. The Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks (ACTTION) public‐private partnership has developed a training system for participants in clinical trials using a zero to 10 numerical rating scale (NRS) to rate pain intensity (Smith 2016).
The use of validated neuropathic pain scales and the reports of the effects of cannabis‐based medicines on all items of the neuropathic pain scale are recommended. In addition, a subgrouping of participants with neuropathic syndromes based on sensory profiles is possible and may be useful in clinical trial design to enrich the study population for treatment responders (Baron 2017).
Long‐term studies aiming to capture data on misuse and abuse of cannabis‐based medicines and cannabis‐induced mental disorders are valuable.
What's new
| Date | Event | Description |
|---|---|---|
| 20 July 2020 | Amended | Minor error corrected in search strategy. |
| 20 July 2020 | Review declared as stable | See Published notes. |
History
Protocol first published: Issue 5, 2016 Review first published: Issue 3, 2018
Notes
Assessed for updating in 2020
A restricted search in June 2020 did not identify any potentially relevant studies likely to change the conclusions. Therefore, this review has now been stabilised following discussion with the authors and editors. The review will be assessed for updating in two years. If appropriate we will update the review before this date if new evidence likely to change the conclusions is published, or if standards change substantially which necessitates major revisions.
Acknowledgements
Cochrane Review Group funding acknowledgement: this project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to Cochrane Pain, Palliative and Supportive Care (PaPaS). The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
The protocol followed the agreed template for neuropathic pain, which was developed in collaboration with Cochrane Musculoskeletal and Cochrane Neuromuscular Diseases. The editorial process was managed by Cochrane Pain, Palliative and Supportive Care.
We thank Joanne Abbott for conducting the searches. We thank Colin Cameron, MD and Alison Moore (consumer) for their valuable reviews.
Appendices
Appendix 1. Methodological considerations for chronic pain
There have been several recent changes in how the efficacy of conventional and unconventional treatments is assessed in chronic painful conditions. The outcomes are now better defined, particularly with new criteria for what constitutes moderate or substantial benefit (Dworkin 2008); older trials may only report participants with 'any improvement'. Newer trials tend to be larger, avoiding problems from the random play of chance. Newer trials also tend to be of longer duration, up to 12 weeks, and longer trials provide a more rigorous and valid assessment of efficacy in chronic conditions. New standards have evolved for assessing efficacy in neuropathic pain, and we are now applying stricter criteria for the inclusion of trials and assessment of outcomes, and are more aware of problems that may affect our overall assessment. Below we have summarised some of the recent insights that must be considered in this new review.
Pain results tend to have a U‐shaped distribution rather than a bell‐shaped distribution. This is true in acute pain (Moore 2011a; Moore 2011b), back pain (Moore 2010c), and arthritis (Moore 2010d), as well as in fibromyalgia (Straube 2010); in all cases average results usually describe the experience of almost no‐one in the trial. Data expressed as averages are potentially misleading, unless they can be proven to be suitable.
As a consequence, we have to depend on dichotomous results (the individual either has or does not have the outcome) usually from pain changes or patient global assessments. The Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) group has helped with their definitions of minimal, moderate, and substantial improvement (Dworkin 2008). In arthritis, trials of less than 12 weeks' duration, and especially those shorter than eight weeks, overestimate the effect of treatment (Moore 2010c); the effect is particularly strong for less effective analgesics, and this may also be relevant in neuropathic‐type pain.
The proportion of patients with at least moderate benefit can be small, even with an effective medicine, falling from 60% with an effective medicine in arthritis to 30% in fibromyalgia (Moore 2009; Moore 2010c; Moore 2010d; Moore 2013b; Moore 2017; Straube 2008; Sultan 2008). A Cochrane Review of pregabalin in neuropathic pain and fibromyalgia demonstrated different response rates for different types of chronic pain (higher in diabetic neuropathy and postherpetic neuralgia and lower in central pain and fibromyalgia) (Moore 2009). This indicates that different neuropathic pain conditions should be treated separately from one another, and that pooling should not be done unless there are good grounds for doing so.
Individual patient analyses indicate that patients who get good pain relief (moderate or better) have major benefits in many other outcomes, affecting quality of life in a significant way (Moore 2010b; Moore 2014a).
Imputation methods such as last observation carried forward (LOCF), used when participants withdraw from clinical trials, can overstate drug efficacy especially when adverse event withdrawals with drug are greater than those with placebo (Moore 2012).
Appendix 2. Databases, search strategies and hits retrieved
CENTRAL (CRSO)
#1 MESH DESCRIPTOR Cannabis
#2 ((cannabi* or hash* or hemp or marijuana or marihuana or ganja or bhang)):TI,AB,KY
#3 MESH DESCRIPTOR Dronabinol
#4 ((dronabinol or marinol or nabilone or cesamet or dexanabinol or tetrahydrocannabinol or sativex or "HU 211")):TI,AB,KY
#5 #1 OR #2 OR #3 OR #4
#6 MESH DESCRIPTOR Neuralgia EXPLODE ALL TREES
#7 ((pain* or neuralgia or neuropathic)):TI,AB,KY
#9 #6 OR#7
#10 #5 AND #9
May 2016: 202
November 2017: 62
MEDLINE (OVID)
| 1. Cannabis/ |
| 2. (cannabi* or hash* or hemp or marijuana or marihuana or ganja or bhang).tw. |
| 3. Dronabinol/ |
| 4. (dronabinol or marinol or nabilone or cesamet or dexanabinol or tetrahydrocannabinol or sativex or "HU 211").tw. |
| 5. or/1‐4 |
| 6. exp Neuralgia/ |
| 7. (pain* or neuralgia or neuropathic).tw. |
| 8. 6 or 7 |
| 9. 5 and 8 |
| 10. randomized controlled trial.pt. |
| 11. controlled clinical trial.pt. |
| 12. randomized.ab. |
| 13. placebo.ab. |
| 14. drug therapy.fs. |
| 15. randomly.ab. |
| 16. trial.ab. |
| 17. groups.ab. |
| 18. 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 |
| 19. exp animals/ not humans.sh. |
| 20. 18 not 19 |
| 21. 9 and 20 |
May 2016: 772
November 2017: 177
Embase (OVID)
| 1. Cannabis/ | |
| 2. (cannabi* or hash* or hemp or marijuana or marihuana or ganja or bhang).tw. | |
| 3. Dronabinol/ | |
| 4. (dronabinol or marinol or nabilone or cesamet or dexanabinol or tetrahydrocannabinol or sativex or "HU 211").tw. | |
| 5. or/1‐4 | |
| 6. exp Neuralgia/ | |
| 7. (pain* or neuralgia or neuropathic).tw. | |
| 8. 6 or 7 | |
| 9. 5 and 8 | |
| 10. random$.tw. | |
| 11. factorial$.tw. | |
| 12. crossover$.tw. | |
| 13. cross over$.tw. | |
| 14. cross‐over$.tw. | |
| 15. placebo$.tw. | |
| 16. (doubl$ adj blind$).tw. | |
| 17. (singl$ adj blind$).tw. | |
| 18. assign$.tw. | |
| 19. allocat$.tw. | |
| 20. volunteer$.tw. | |
| 21. Crossover Procedure/ | |
| 22. double‐blind procedure.tw. | |
| 23. Randomized Controlled Trial/ | |
| 24. Single Blind Procedure/ | |
| 25. or/10‐24 | |
| 26. (animal/ or nonhuman/) not human/ | |
| 27. 25 not 26 | |
| 28. 9 and 27 |
May 2016: 417
November 2017: 77
European Union clinical trial register
November 2017: Neuropathic pain AND (cannabis OR cannabinoids): 3
U.S. National Institutes of Health clinical trial register
November 2017: Neuropathic pain AND (cannabis OR cannabinoids): 27
World Health Organization (WHO) International Clinical Trials Registry Platform
November 2017: Neuropathic pain AND (cannabis OR cannabinoids); 116
International Association for Cannabinoid Medicines (IACM) databank
November 2017: Neuropathic pain and controlled study: 28
Appendix 3. GRADE: criteria for assigning grade of evidence
The GRADE system uses the following criteria for assigning a quality level to a body of evidence (Chapter 12, Schünemann 2011).
High: randomised trials; or double‐upgraded observational studies
Moderate: downgraded randomised trials; or upgraded observational studies
Low: double‐downgraded randomised trials; or observational studies
Very low: triple‐downgraded randomised trials; or downgraded observational studies; or case series/case reports
Factors that may decrease the quality level of a body of evidence are:
limitations in the design and implementation of available studies suggesting high likelihood of bias;
indirectness of evidence (indirect population, intervention, control, outcomes);
unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses);
imprecision of results (wide confidence intervals; confidence interval including zero; low number of events);
high probability of publication bias.
Factors that may increase the quality level of a body of evidence are:
large magnitude of effect;
all plausible confounding would reduce a demonstrated effect or suggest a spurious effect when results show no effect;
dose‐response gradient.
Data and analyses
Comparison 1. Cannabis‐based medicines versus placebo at final treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Pain relief of 50% or greater | 8 | 1001 | Risk Difference (IV, Random, 95% CI) | 0.05 [0.00, 0.09] |
| 1.1.1 Central pain ‐ multiple sclerosis | 4 | 669 | Risk Difference (IV, Random, 95% CI) | 0.08 [‐0.00, 0.15] |
| 1.1.2 Peripheral pain ‐ chemotherapy‐induced polyneuropathy | 1 | 36 | Risk Difference (IV, Random, 95% CI) | 0.11 [‐0.06, 0.28] |
| 1.1.3 Peripheral pain ‐ diabetic polyneuropathy | 1 | 30 | Risk Difference (IV, Random, 95% CI) | ‐0.20 [‐0.54, 0.14] |
| 1.1.4 Peripheral pain ‐ plexus injury | 1 | 141 | Risk Difference (IV, Random, 95% CI) | 0.01 [‐0.04, 0.06] |
| 1.1.5 Peripheral pain ‐ polyneuropathy of various aetiologies | 1 | 125 | Risk Difference (IV, Random, 95% CI) | 0.13 [0.00, 0.25] |
| 1.2 Patient Global Impression much or very much improved | 6 | 1092 | Risk Difference (IV, Random, 95% CI) | 0.09 [0.01, 0.17] |
| 1.2.1 Central pain ‐ multiple sclerosis | 2 | 397 | Risk Difference (IV, Random, 95% CI) | 0.06 [‐0.01, 0.14] |
| 1.2.2 Central pain ‐ spinal cord injury | 1 | 116 | Risk Difference (IV, Random, 95% CI) | 0.34 [0.17, 0.50] |
| 1.2.3 Peripheral pain ‐ diabetic polyneuropathy | 1 | 281 | Risk Difference (IV, Random, 95% CI) | 0.02 [‐0.09, 0.14] |
| 1.2.4 Peripheral pain ‐ polyneuropathy of various aetiologies | 1 | 228 | Risk Difference (IV, Random, 95% CI) | 0.08 [‐0.02, 0.17] |
| 1.2.5 Central or peripheral pain ‐ various aetiologies | 1 | 70 | Risk Difference (IV, Random, 95% CI) | ‐0.01 [‐0.22, 0.19] |
| 1.3 Withdrawals due to adverse events | 13 | 1848 | Risk Difference (IV, Random, 95% CI) | 0.04 [0.02, 0.07] |
| 1.3.1 Central pain ‐ multiple sclerosis | 4 | 693 | Risk Difference (IV, Random, 95% CI) | 0.04 [0.01, 0.08] |
| 1.3.2 Central pain ‐ spinal cord injury | 1 | 116 | Risk Difference (IV, Random, 95% CI) | 0.09 [0.01, 0.17] |
| 1.3.3 Peripheral pain ‐ chemotherapy‐induced polyneuropathy | 1 | 36 | Risk Difference (IV, Random, 95% CI) | 0.00 [‐0.10, 0.10] |
| 1.3.4 Peripheral pain ‐ diabetic polyneuropathy | 1 | 297 | Risk Difference (IV, Random, 95% CI) | 0.12 [0.04, 0.20] |
| 1.3.5 Peripheral pain ‐ HIV polyneuropathy | 1 | 68 | Risk Difference (IV, Random, 95% CI) | 0.00 [‐0.13, 0.13] |
| 1.3.6 Peripheral pain ‐ plexus injury | 1 | 141 | Risk Difference (IV, Random, 95% CI) | 0.01 [‐0.04, 0.06] |
| 1.3.7 Peripheral pain ‐ polyneuropathy of various aetiologies | 3 | 427 | Risk Difference (IV, Random, 95% CI) | 0.08 [0.02, 0.13] |
| 1.3.8 Central and peripheral pain ‐ various aetiologies | 1 | 70 | Risk Difference (IV, Random, 95% CI) | ‐0.06 [‐0.19, 0.07] |
| 1.4 Serious adverse events | 13 | 1876 | Risk Difference (IV, Random, 95% CI) | 0.01 [‐0.01, 0.03] |
| 1.4.1 Central pain ‐ multiple sclerosis | 4 | 693 | Risk Difference (IV, Random, 95% CI) | 0.03 [‐0.01, 0.06] |
| 1.4.2 Central pain ‐ spinal cord injury | 1 | 116 | Risk Difference (IV, Random, 95% CI) | 0.02 [‐0.05, 0.09] |
| 1.4.3 Peripheral pain ‐ chemotherapy‐induced neuropathy | 1 | 36 | Risk Difference (IV, Random, 95% CI) | 0.00 [‐0.10, 0.10] |
| 1.4.4 Peripheral pain ‐ diabetic polyneuropathy | 1 | 297 | Risk Difference (IV, Random, 95% CI) | 0.01 [‐0.05, 0.08] |
| 1.4.5 Peripheral pain ‐ HIV polyneuropathy | 1 | 68 | Risk Difference (IV, Random, 95% CI) | 0.03 [‐0.07, 0.13] |
| 1.4.6 Peripheral pain ‐ plexus injury | 1 | 141 | Risk Difference (IV, Random, 95% CI) | 0.00 [‐0.03, 0.03] |
| 1.4.7 Peripheral pain ‐ polyneuropathies of various aetiologies | 3 | 455 | Risk Difference (IV, Random, 95% CI) | 0.01 [‐0.02, 0.04] |
| 1.4.8 Central and peripheral pain ‐ various aetiologies | 1 | 70 | Risk Difference (IV, Random, 95% CI) | ‐0.06 [‐0.15, 0.03] |
| 1.5 Pain relief of 30% or greater | 10 | 1586 | Risk Difference (IV, Random, 95% CI) | 0.09 [0.03, 0.15] |
| 1.5.1 Central pain ‐ multiple sclerosis | 3 | 645 | Risk Difference (IV, Random, 95% CI) | 0.11 [‐0.03, 0.25] |
| 1.5.2 Peripheral pain ‐ chemotherapy‐induced polyneuropathy | 1 | 36 | Risk Difference (IV, Random, 95% CI) | 0.11 [‐0.16, 0.38] |
| 1.5.3 Peripheral pain ‐ diabetic polyneuropathy | 2 | 327 | Risk Difference (IV, Random, 95% CI) | ‐0.04 [‐0.14, 0.07] |
| 1.5.4 Peripheral pain ‐ HIV polyneuropathy | 1 | 56 | Risk Difference (IV, Random, 95% CI) | 0.29 [0.05, 0.52] |
| 1.5.5 Peripheral pain ‐ plexus injury | 1 | 141 | Risk Difference (IV, Random, 95% CI) | 0.10 [‐0.06, 0.25] |
| 1.5.6 Peripheral pain ‐ polyneuropathy of various aetiologies | 2 | 381 | Risk Difference (IV, Random, 95% CI) | 0.11 [0.03, 0.19] |
| 1.6 Mean pain intensity | 14 | 1837 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐0.60, ‐0.09] |
| 1.6.1 Central pain ‐ multiple sclerosis | 4 | 668 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.25, 0.05] |
| 1.6.2 Central pain ‐ spinal cord injury | 1 | 114 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.41, 0.33] |
| 1.6.3 Peripheral pain ‐ chemotherapy‐induced polyneuropathy | 1 | 36 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.86, 0.45] |
| 1.6.4 Peripheral pain ‐ diabetic polyneuropathy | 2 | 324 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐0.27, 0.17] |
| 1.6.5 Peripheral pain ‐ HIV polyneuropathy | 1 | 56 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.41 [‐0.94, 0.12] |
| 1.6.6 Peripheral pain ‐ plexus injury | 1 | 141 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐0.79, ‐0.08] |
| 1.6.7 Peripheral pain ‐ polyneuropathy of various aetiologies | 3 | 428 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐1.75, 0.44] |
| 1.6.8 Central and peripheral pain ‐ various aetiologies | 1 | 70 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.71, 0.23] |
| 1.7 Health‐related quality of life | 9 | 1284 | Std. Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.10, 0.13] |
| 1.7.1 Central pain ‐ multiple sclerosis | 2 | 363 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.27, 0.14] |
| 1.7.2 Central pain ‐ spinal cord injury | 1 | 113 | Std. Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.37, 0.37] |
| 1.7.3 Peripheral pain ‐ diabetic polyneuropathy | 2 | 303 | Std. Mean Difference (IV, Random, 95% CI) | 0.17 [‐0.06, 0.39] |
| 1.7.4 Peripheral pain ‐ plexus injury | 1 | 141 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.42, 0.28] |
| 1.7.5 Peripheral pain of various aetiologies | 2 | 300 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.26, 0.21] |
| 1.7.6 Central and peripheral pain ‐ various aetiologies | 1 | 64 | Std. Mean Difference (IV, Random, 95% CI) | 0.15 [‐0.35, 0.64] |
| 1.8 Sleep problems | 8 | 1386 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.47 [‐0.90, ‐0.04] |
| 1.8.1 Central pain ‐ multiple sclerosis | 1 | 339 | Std. Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.21, 0.22] |
| 1.8.2 Central pain ‐ spinal cord injury | 1 | 114 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.41, 0.32] |
| 1.8.3 Peripheral pain ‐ diabetic polyneuropathy | 1 | 274 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.38, 0.10] |
| 1.8.4 Peripheral pain ‐ plexus injury | 1 | 141 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐0.78, ‐0.07] |
| 1.8.5 Peripheral pain ‐ polyneuropathy of various aetiologies | 3 | 448 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.78 [‐2.17, 0.61] |
| 1.8.6 Central and peripheral pain ‐ various aetiologies | 1 | 70 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.78, 0.16] |
| 1.9 Psychological distress | 7 | 779 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.61, ‐0.02] |
| 1.9.1 Central pain ‐ multiple sclerosis | 2 | 363 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.65, 0.59] |
| 1.9.2 Peripheral pain ‐ chemotherapy‐induced polyneuropathy | 1 | 36 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.07 [‐1.78, ‐0.37] |
| 1.9.3 Peripheral pain ‐ diabetic polyneuropathy | 1 | 30 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.97, 0.47] |
| 1.9.4 Peripheral pain ‐ plexus injury | 1 | 141 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.62, 0.08] |
| 1.9.5 Peripheral pain ‐ polyneuropathy of various aetiologies | 2 | 209 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.48 [‐0.80, ‐0.16] |
| 1.10 Withdrawals due to lack of efficacy | 9 | 1576 | Risk Difference (IV, Random, 95% CI) | ‐0.00 [‐0.02, 0.01] |
| 1.10.1 Central pain ‐ multiple sclerosis | 4 | 697 | Risk Difference (IV, Random, 95% CI) | 0.00 [‐0.02, 0.02] |
| 1.10.2 Peripheral pain ‐ diabetic polyneuropathy | 1 | 297 | Risk Difference (IV, Random, 95% CI) | ‐0.01 [‐0.05, 0.03] |
| 1.10.3 Peripheral pain ‐ plexus injury | 1 | 141 | Risk Difference (IV, Random, 95% CI) | 0.00 [‐0.04, 0.04] |
| 1.10.4 Peripheral pain ‐ polyneuropathy of various aetiologies | 2 | 371 | Risk Difference (IV, Random, 95% CI) | ‐0.04 [‐0.09, 0.01] |
| 1.10.5 Central and peripheral pain ‐ various aetiologies | 1 | 70 | Risk Difference (IV, Random, 95% CI) | 0.00 [‐0.05, 0.05] |
| 1.11 Any adverse event | 7 | 1356 | Risk Difference (IV, Random, 95% CI) | 0.19 [0.12, 0.27] |
| 1.11.1 Central pain ‐ multiple sclerosis | 3 | 627 | Risk Difference (IV, Random, 95% CI) | 0.22 [0.05, 0.39] |
| 1.11.2 Central pain ‐ spinal cord injury | 1 | 116 | Risk Difference (IV, Random, 95% CI) | 0.34 [0.18, 0.50] |
| 1.11.3 Peripheral pain ‐ diabetic polyneuropathy | 1 | 297 | Risk Difference (IV, Random, 95% CI) | 0.12 [0.02, 0.22] |
| 1.11.4 Peripheral pain ‐ polyneuropathy of various aetiologies | 1 | 246 | Risk Difference (IV, Random, 95% CI) | 0.15 [0.05, 0.25] |
| 1.11.5 Central and peripheral pain ‐ various aetiologies | 1 | 70 | Risk Difference (IV, Random, 95% CI) | 0.21 [0.06, 0.36] |
| 1.12 Specific adverse event: nervous system disorders | 9 | 1304 | Risk Difference (IV, Random, 95% CI) | 0.38 [0.18, 0.58] |
| 1.12.1 Central pain ‐ multiple sclerosis | 3 | 453 | Risk Difference (IV, Random, 95% CI) | 0.33 [0.09, 0.58] |
| 1.12.2 Central pain ‐ spinal cord injury | 1 | 116 | Risk Difference (IV, Random, 95% CI) | 0.53 [0.38, 0.68] |
| 1.12.3 Peripheral pain ‐chemotherapy‐induced polyneuropathy | 1 | 36 | Risk Difference (IV, Random, 95% CI) | 1.00 [0.90, 1.10] |
| 1.12.4 Peripheral pain ‐ diabetic polyneuropathy | 1 | 297 | Risk Difference (IV, Random, 95% CI) | 0.26 [0.15, 0.37] |
| 1.12.5 Peripheral pain ‐ polyneuropathy of various aetiologies | 2 | 332 | Risk Difference (IV, Random, 95% CI) | 0.29 [0.19, 0.39] |
| 1.12.6 Central and peripheral pain ‐ various aetiologies | 1 | 70 | Risk Difference (IV, Random, 95% CI) | 0.37 [0.15, 0.58] |
| 1.13 Specific adverse event: psychiatric disorders | 9 | 1314 | Risk Difference (IV, Random, 95% CI) | 0.10 [0.06, 0.15] |
| 1.13.1 Central pain ‐ multiple sclerosis | 3 | 453 | Risk Difference (IV, Random, 95% CI) | 0.10 [0.05, 0.16] |
| 1.13.2 Central pain ‐ spinal cord injury | 1 | 116 | Risk Difference (IV, Random, 95% CI) | 0.00 [‐0.06, 0.07] |
| 1.13.3 Peripheral pain ‐ chemotherapy‐induced polyneuropathy | 1 | 36 | Risk Difference (IV, Random, 95% CI) | 0.11 [‐0.06, 0.28] |
| 1.13.4 Peripheral pain ‐ diabetic polyneuropathy | 1 | 297 | Risk Difference (IV, Random, 95% CI) | 0.05 [0.01, 0.09] |
| 1.13.5 Peripheral pain ‐ polyneuropathy of various aetiologies | 2 | 342 | Risk Difference (IV, Random, 95% CI) | 0.21 [0.14, 0.29] |
| 1.13.6 Central and peripheral pain ‐ various aetiologies | 1 | 70 | Risk Difference (IV, Random, 95% CI) | 0.11 [‐0.05, 0.27] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bermann 2004.
| Study characteristics | ||
| Methods |
Disease: plexus root avulsion with ≥ 1 root affected Study setting: single‐centre (orthopedic clinic), UK; 2001‐2002 Study design: cross‐over Study duration: 2‐week baseline, 3 cross‐over periods for 10‐14 days, no washout periods |
|
| Participants |
Inclusion criteria: pain ≥ 4 on 0‐10 scale, no cannabis use for 7 days prior to inclusion, Exclusion criteria: schizophrenia, other psychotic illness or significant psychiatric illness, other than depression associated with chronic illness; serious cardiovascular disease; significant renal or hepatic impairment; epilepsy or convulsions; significant history of substance abuse; known adverse reaction to cannabis or the product excipients; surgery within 2 months (6 months for nerve repair). Female patients who were pregnant, lactating or at risk of pregnancy were also excluded. Participants: N = 48, 46 male, 2 female, mean age 39 (23‐63 years). Pain baseline 7.5 (no SD reported) (scale 0‐10). 45.8% had used cannabis medicinally, 60.4 % recreationally. |
|
| Interventions |
Study medication: oromucosal spray THC only (27 mg/mL), THC/CBD mix (27/25 mg/mL), maximum 48 sprays/d; placebo spray Rescue medication: none Allowed co‐therapies: stable analgesic medication over 4 weeks (fentanyl not allowed, amitriptyline max. 75 mg/d, no further details provided) |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: reported (NRS 0‐10, average of the last 7 days) PGIC much or very much improved: not assessed Withdrawal due to AE: reported Serious AE attributed to medication: reported Participant‐reported pain relief ≥ 30%: not reported; calculated by imputation method (NRS 0‐10, average of the last 7 days) Mean pain intensity: NRS 0‐10, average of the last 7 days; SD calculated from P value HRQoL: Pain Disability Index 0‐70; SD calculated from P value Sleep problems: sleep quality 10‐0; SD calculated from P value Fatigue: not assessed Psychological distress: General Health Questionnaire‐12; SD calculated from P value Withdrawals due to lack of efficacy: reported Nervous system disorders‐related AE: incompletely reported (not suited for analysis) Psychiatric disorders‐related AE: incompeletely reported (not suited for analysis) Any adverse event: open question at each visit; VAS intoxication score for AE |
|
| Notes |
Funding: GW Pharmaceuticals and the Royal National Orthopaedic Hospital NHS Trust Conflicts of interest: not declared "No washout period was used between the three treatment periods. Any carry over effect was unlikely to be for greater than 2–3 days so the first week of titration for each period would be sufficient to counteract any carry over with efficacy comparisons being made by averaging the variables over the last 7 days of treatment". |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly allocated by a computer generated list to the six possible sequences of receiving the three study medications" |
| Allocation concealment (selection bias) | Low risk | "Although the treatment sequence was blinded, sealed code break envelopes, one for each patient, containing information on the treatment sequence were available if necessary. Blinding was maintained throughout the study". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Unclear risk | No study protocol available |
| Group similarity at baseline | Low risk | Identical demographic and clinical characteristics due to study design |
| Sample size bias | High risk | < 50 participants per treatment arm |
Ellis 2009.
| Study characteristics | ||
| Methods |
Disease: HIV neuropathy Study setting: single‐centre, university, USA; years of study not reported Study design: cross‐over Study duration: 2 weeks with 5 treatment days per each period, 2 weeks washout |
|
| Participants |
Inclusion criteria: adults with documented HIV infection, neuropathic pain refractory to ≥ 2 previous analgesics, and an average score of ≥ 5 on the pain intensity subscale of the Descriptor Differential Exclusion criteria: (1) current DSM‐IV substance use disorders; (2) lifetime history of dependence on cannabis; (3) previous psychosis with or intolerance to cannabis‐based medicines; (4) concurrent use of approved cannabinoid medications (i.e. Marinol); (5) positive urine toxicology screen for cannabis‐based medicines during the wash‐in week before initiating study treatment; and (6) serious medical conditions that might affect participant safety or the conduct of the trial. Individuals with a previous history of alcohol or other drug dependence were eligible provided that criteria for dependence had not been met within the last 12 months. Participants were excluded if urine toxicology demonstrated ongoing use of non prescribed, recreational drugs such as methamphetamine and cocaine Treatment group (delta‐9‐THC)/placebo group: N = 34 participants, mean age 49.1 years (SD 6.9); male 100%; pain baseline 11.1 (no SD reported) on a 0‐20 scale; 91% with previous cannabis experience |
|
| Interventions |
Study medication: smoked cannabis with THC ranging from 4% to 8% provided by the National Institute on Drug Abuse, depending on efficacy and tolerability. Cigarettes without THC. 4 smoking sessions in the 8‐h study day Rescue medication: not reported Allowed co‐therapies: stable regimen of opioids, anticonvulsants, antidepressants and analgesics |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported and not calculable by imputation method PGIC much or very much improved: not assessed Withdrawal due to AE: reported Serious AE: incompletely reported (not suited for meta‐analysis) Participant‐reported pain relief ≥ 30%: pain quality and impact descriptor differential scale 0‐20; NNTB reported; number of participants extracted from Andreae 2015 Mean pain intensity: pain quality and impact descriptor differential scale 0‐20; SD calculated from P values HRQoL: Sickness Impact profile; no details reported (not suited for meta‐analysis)** Sleep problems: not assessed Fatigue: not assessed Psychological distress: BSI** Withdrawals due to lack of efficacy: not reported Any adverse event: no details of assessment reported Nervous system disorders‐related AE: incompletely reported (not suited for meta‐analysis) Psychiatric disorders‐related AE: incompletely reported (not suited for meta‐analysis) |
|
| Notes |
Funding: Grant C00‐SD‐104 from the University of California, Center for Medicinal Cannabis Research Conflicts of interest: Heather Bentley and Ben Gouaux are employees of the Center for Medicinal Cannabis Research at the University of California, San Diego, the study sponsor. Ms Bentley is Project Manager for the CMCR and assisted the investigator with regulatory issues, oversight/monitoring, and preparation of the manuscript. Mr. Gouaux is a Research Associate with the CMCR and assisted the investigator with regulatory issues, oversight/monitoring, data preparation and analysis, and preparation and submission of the article. The study authors declare that over the past 3 years Dr. Atkinson has received compensation from Eli Lilly Pharmaceuticals. "There was no evident sequence effect" **No data shown; "As measured by the SIP and BSI, there were similar improvements in total mood disturbance, physical disability and quality of life for the cannabis and placebo treatment" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Random number generator" |
| Allocation concealment (selection bias) | Low risk | "Randomization was performed by a research pharmacist ... and the key to study assignment was withheld from investigators until completion statistical analyses". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Completer analysis of 30% pain reduction as reported by Andreae 2015 |
| Selective reporting (reporting bias) | High risk | Some outcomes were not reported |
| Group similarity at baseline | Low risk | Identical clinical and demographic characteristics due to study design |
| Sample size bias | High risk | < 50 participants per treatment arm |
Frank 2008.
| Study characteristics | ||
| Methods |
Disease: chronic central and PNP (radiculopathy, CRPS, diabetic neuropathy, posttraumatic or postsurgery, trigeminal neuralgia, PHN) Study setting: outpatient units of 3 hospitals in the UK, 2001‐2002 Study design: cross‐over Study duration: 1 week pre study, 6 weeks treatment, washout 2 weeks, 6 weeks treatment |
|
| Participants |
Inclusion criteria: pain, such as burning, stabbing, or paraesthesia within the distribution of a peripheral nerve and a clear clinical history of its cause (sensory abnormality, allodynia, burning pain, lancinating pain, sympathetic dysfunction), pain ≥ 40 on a 100 mm VAS, stable medication Exclusion criteria: DHC not stopped 2 weeks prior to inclusion, antipsychotics, benzodiazepines (except for night sedation), MAO inhibitors, legal action, ongoing cannabis‐based medicines, severe hepatic or renal disease, epilepsy, bipolar disorder, psychosis, or a history of substance misuse Participants: DHC then nabilone: N = 48 participants, mean age 50.6 (SD 15.2) years. 23 female. Mean pain baseline 69.6 (range 29‐95) on a 0‐100 scale. No reports on prior use of cannabis. Participants: nabilone then DHC: N = 48, mean age 49.7 (SD 12.0), 27 male, 21 female; Mean pain baseline 69.6 (range 29‐95) on a 0‐100 scale. No reports on prior use of cannabis. |
|
| Interventions |
Study medication: dose adjustment every week (twice first week) from 30‐240 mg DHC and 0.25‐2 mg nabilone Rescue medication: paracetamol 500 mg and codeine 30 mg throughout washout up to 8 times/d Allowed co‐therapies: "Stable analgesics" |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported, calculated by imputation method (daily pain score summarised as last bi‐weekly means VAS 0‐100) PGIC much or very much improved: not assessed Withdrawal due to AE: reported Serious AE attributed to medication: reported Participant‐reported pain relief ≥ 30%: not reported, calculated by imputation method (daily pain score summarised as last bi‐weekly means VAS 0‐100) HRQoL: SF‐36 physical functioning 50‐0 Sleep problems: NRS 0‐10; data reported not suited for meta‐analysis (P = 0.20) Fatigue: not assessed Psychological distress: SF‐36 Mental Health 50‐0; data reported not suited for meta‐analysis (P = 0.20) Withdrawals due to lack of efficacy: not reported Withdrawal due to AE: reported Any adverse event: "At each visit the patients filled in a side effects assessment form" Nervous system disorders‐related AE: incompletely reported, not suited for meta‐analysis Psychiatric disorders‐related AE:incompletely reported, not suited for meta‐analysis |
|
| Notes |
Funding: grant from Cambridge Laboratories Conflict of Interest: BF’s salary was provided as part of the above research grant although he was employed by the Newcastle upon Tyne University Hospitals Trust. "We excluded carry over by basing the analyses from the last two weeks of each treatment period". |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Treatment was allocated by random permuted blocks of 10, stratified by centre." |
| Allocation concealment (selection bias) | Low risk | "The pharmacies at the treatment centres, the patients, and all clinical personnel involved in the trial were unaware of treatment allocation at all times." Code breaking envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The pharmacy at St Mary’s Hospital supplied identical white capsules containing 250 μg nabilone or 30 mg dihydrocodeine." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Available cases analysis (all participants randomised, which provided data in each treatment period) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported as outlined in the study protocol ISRCTN15330757 |
| Group similarity at baseline | Low risk | Similar demographic and clinical characteristics at baseline |
| Sample size bias | Unclear risk | 50‐199 participants per treatment arm |
Langford 2013.
| Study characteristics | ||
| Methods |
Disease: central neuropathic pain in multiple sclerosis (MS) Study setting: multicentre, 33 sites in UK, Canada, Spain, France, Czech Republic; 2006‐2008 Study design: Patients who had failed to gain adequate analgesia from existing medication were treated with THC/CBD spray or placebo as an add‐on treatment in a double‐blind manner, for 14 weeks to investigate the efficacy of the medication in MS‐induced neuropathic pain. This parallel‐group phase of the study was then followed by an 18‐week randomised withdrawal study (14‐week, open‐label treatment period plus a double‐blind, 4‐week, randomised‐withdrawal phase) Study duration: Phase A: 1‐week baseline, 14‐week treatment. Phase B: 14‐week, open treatment phase with 2 weeks' titration and 12 weeks' stable dose, followed by a randomised withdrawal phase of four weeks (only in France and Czech Republic) |
|
| Participants |
Inclusion criteria: chronic neuropathic pain due to MS, of at least 3 months' duration. Participants were also to have a sum score of at least 24 on a pain 0–10 point NRS on the last 6 days during the baseline period. In addition, their analgesic regimen was to be stable for at least 2 weeks preceding the study entry day. For Phase B also: ≥ 3 sprays/d in last 7 days of phase A, and tolerability (that means no AE), stable medication Exclusion criteria: other somatic pain causes with severe pain, including PNP, significant psychiatric (except depression related to pain), renal, hepatic, cardiovascular, or convulsive disorders, sensitivity to cannabis‐based medicines Phase A, treatment group: N = 167, female/male (54/113), mean age 48.42 (SD 10.43), 11 (7%) with cannabis experience Placebo group: N = 172, male/female (55/117), mean age 49.51 (SD 10.50) 10 (6%) with cannabis experience Phase B, treatment group: N = 21; female/male (11/10), mean age 46.2 (10.39), 0 patients with cannabis experience Placebo group: N = 21, female/male 14/7, mean age 49.82 (9.75), 1 patient with cannabis experience |
|
| Interventions |
Study medication: THC/CBD oromucosal spray. Each actuation of active medication delivered 2.7 mg of THC and 2.5 mg of CBD to the oral mucosa. Placebo delivered the excipient plus colorants. Max. 12 sprays/24 h Rescue medication: paracetamol Allowed co‐therapies: pain medication: stable for at least 2 weeks |
|
| Outcomes |
Participant‐reported pain relief ≥ 50% (parallel): only OR reported, calculated by imputation method (NRS 0‐10 for mean daily chronic neuropathic pain, average over 7 days at baseline and final 7 days) PGIC much or very much improved (parallel): reported Withdrawal due to AE (parallel): reported Serious AE (parallel and EERW): reported Participant‐reported pain relief ≥ 30%:: reported Mean pain intensity (parallel): NRS 0‐10 for mean daily chronic neuropathic pain, average over 7 days at baseline and final 7 days; SD calculated from P value HRQoL (parallel): EQ‐5D VAS 0‐100 Sleep problems (parallel): NRS 0‐10; SD calculated from P value Fatigue: NRS 0‐10; SD calculated from P value Psychological distress (parallel): SF‐36 mental health: SD calculated from P value Withdrawals due to lack of efficacy (parallel): reported Any adverse event (parallel and EERW): reported. Details of assessment of AEs not reported. Nervous system disorders‐related AE (parallel and EERW): reported Psychiatric disorders‐related AE (parallel and EERW): reported |
|
| Notes |
Funding: GW Pharmaceuticals Conflicts of interest: R. Langford, J. Mares, A. Novotna, M. Vachora, I. Novakova, W. Notcutt, and S. Ratcliffe were all investigators in this study and their organizations received investigator fees from GW Pharma Ltd. accordingly for their participation in the study. R. Langford, W. Notcutt, and S. Ratcliffe have received consultancy and speaker fees from GW Pharma Ltd. to attend meetings. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization occurred using a pre‐determined computer generated randomisation code in which treatment allocation was stratified by centre, and used randomly permuted blocks of variable sizes. Separate randomisation schemes, using the same strategy, were produced for each part of the study." |
| Allocation concealment (selection bias) | Low risk | Separate randomisation schemes |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Low risk | Data reported as outlined in the study protocol NCT00391079 available |
| Group similarity at baseline | Low risk | Similar demographic and clinical characteristics at baseline |
| Sample size bias | Unclear risk | 50‐199 participants per treatment arm |
Lynch 2014.
| Study characteristics | ||
| Methods |
Disease: chemotherapy‐induced neuropathic pain Study setting: single centre, Canada; year of study not reported Study design: cross‐over design Study duration: 4 weeks each and 2 weeks washout |
|
| Participants |
Inclusion criteria: neuropathic pain persisting for 3 months after completing chemotherapy with paclitaxel, vincristine, or cisplatin. The average 7‐day intensity of pain had to be ≥ 4 on an 11‐point NRS. Participants also exhibited sensory abnormalities comprising allodynia, hyperalgesia, or hypethesia. Concurrent analgesics had to be stable for 14 days before entry into the trial. Exclusion criteria: ischaemic heart disease, ongoing epilepsy, a personal or family history of schizophrenia, or psychotic disorder or substance abuse or dependency within the previous 2 years. Exclusion criteria also included pregnancy or other medical condition that might compromise safety in the trial. Both groups: N = 18; mean age 58 (SD 11.34) years; 15/18 female; previous cannabis use 5/18 |
|
| Interventions |
Study medication: THC/CBD oromucosal spray. Each actuation of active medication delivered 2.7 mg of THC and 2.5 mg of CBD to the oral mucosa. Placebo delivered the excipient plus colorants. Max. 12 sprays/24 h Rescue medication: not reported Allowed co‐therapies: pain medication (anticonvulsants, antidepressants, NSAIDs, opioids): stable for at least 2 weeks |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported, calculated by imputation method. NRS (0‐10) for mean daily chronic neuropathic pain, average over 7 days at baseline and final 7 days PGIC much or very much improved: not assessed Withdrawal due to AE: reported Serious AE: reported Participant‐reported pain relief ≥ 30%: not reported, calculated by imputation method Mean pain intensity: reported HRQoL (parallel): SF‐36 physical component summary score 50‐0 Sleep problems: not assessed Fatigue: not assessed Psychological distress: SF‐36 mental health summary score 50‐0 Withdrawals due to lack of efficacy: not reported Any adverse event: not reported. No details of assessment of AEs reported. Nervous system disorders‐related AE: reported (summarised by the authors of the review) Psychiatric disorders‐related AE: reported (summarised by the authors of the review) |
|
| Notes |
Funding: none Conflicts of interest: the study authors declare no conflicts of interest "Thus, the two week washout was chosen to assure no carry over effect between study arms" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation schedule |
| Allocation concealment (selection bias) | Low risk | "Participants and study staff were blinded to the randomisation code, which was not broken until the completion of the study." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Unclear risk | No protocol reported |
| Group similarity at baseline | Low risk | Identical baseline characteristics due to study design |
| Sample size bias | High risk | < 50 participants per treatment arm |
NCT00710424.
| Study characteristics | ||
| Methods |
Disease: painful diabetic neuropathy Study setting: multicentre international trial, UK, Czech Republic, Romania; July 2005‐2006 Study design: parallel Study duration: 1 week baseline, 14 weeks treatment |
|
| Participants |
Inclusion criteria: ciagnosed with Type 1 or 2 diabetes mellitus as diagnosed according to the WHO criteria. Diagnosed with neuropathic pain due to distal symmetrical diabetic neuropathy of at least 6 months' duration, as defined by a NDS score of ≥ 4, and in whom pain was not wholly relieved with their current therapy. The NDS score must be attained from ≥ 2 different test parameters and not only the ankle jerk reflex. The last 6 daily diary 0‐10 NRS pain scores before randomisation summed to at least 24. Stable dose of regular pain medication and non‐pharmacological therapies (including TENS) for ≥ 14 days prior to the screening visit and willingness for these to be maintained throughout the study. Exclusion criteria: uncontrolled diabetes with HbA1c blood levels of > 11% at Visit 1, Day B1. Had used cannabinoid‐based medications within 60 days of study entry and were unwilling to abstain for the duration for the study. History of schizophrenia, other psychotic illness, severe personality disorder or other significant psychiatric disorder other than depression associated with their underlying condition, known or suspected history of alcohol or substance abuse. History of epilepsy or recurrent seizure, postural drop of 20 mmHg or more in systolic blood pressure at screening. Evidence of cardiomyopathy, MI, cardiac disease. QT interval; of > 450 ms (men) or > 470 ms (women) at Visit 1. Secondary or tertiary atrioventricular block or sinus bradycardia (HR < 50 bpm) or sinus tachycardia (HR > 110 bpm) at Visit 1. Diastolic blood pressure of < 50 mmHg or >105 mmHg in a sitting position at rest for 5 min prior to randomisation. Impaired renal hepatic function Treatment group: N = 149: mean age 60.8 (10.38 SD) years; female/male 56/93. No reports on baseline pain scores and on previous cannabis use. Placebo group: N = 148; mean age 58.2 (10.57 SD) years; female/male 58/90. No reports on baseline pain scores and on previous cannabis use. |
|
| Interventions |
Study medication: Sativex (DHC 27 mg/mL/CBD25 mg/mL), delivered in 100 µL actuations by mucosal spray, maximum max per 24 h: 65 mg TC/60 mg cannabidiol); placebo Rescue medication: no information provided Allowed co‐therapies: no information provided |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported and not calculable by imputation method. Mean Diabetic Neuropathy Pain 0‐10 NRS score at the end of treatment (average of last 7 days' treatment) (Your nerve pain over the last 24 h from 0‐10); PGIC much or very much improved: reported Withdrawal due to AE: reported Serious AE: reported Participant‐reported pain relief ≥ 30%:reported Mean pain intensity: Mean Diabetic Neuropathy Pain 0‐10 NRS score at the end of treatment (average of last 7 days' treatment) HRQoL:: EQ‐5D 0 ‐100 Sleep problems: NRS 0‐10 Fatigue: not assessed Psychological distress: not assessed Withdrawals due to lack of efficacy: reported Any adverse event: mean intoxication score. No details of assessment reported Nervous system disorders‐related AE: reported Psychiatric disorders‐related AE: reported |
|
| Notes |
Funding: GW Pharmaceuticals Conflicts of interest: not declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Low risk | All predefined outcomes reported |
| Group similarity at baseline | Low risk | Similar demographic characteristics at baseline |
| Sample size bias | Unclear risk | 50‐199 participants per treatment arm |
NCT01606176.
| Study characteristics | ||
| Methods |
Disease: MS and other defects of neurological function Study setting: multicentre trial in the UK, no year of study reported Study design: parallel Study duration: baseline period, 3‐week treatment period |
|
| Participants |
Inclusion criteria: chronic refractory pain due to multiple sclerosis or other defects of neurological function. Neuropathic pain with a mean severity NRS score at ≥ 4 during last 7 days of the baseline period. Relatively stable neurology during the preceding 6 months. Stable medication regimen during the preceding 4 weeks. Had not used cannabis‐based medicines for at least the preceding 7 days and willing to abstain from any use of cannabis‐based medicines during the study Exclusion criteria: history of schizophrenia, other psychotic illness, severe personality disorder or other significant psychiatric disorder other than depression associated with their underlying condition. History of alcohol or substance abuse. Severe cardiovascular disorder, such as ischaemic heart disease, arrhythmias (other than well‐controlled atrial fibrillation), poorly controlled hypertension or severe heart failure. History of autonomic dysreflexia. History of epilepsy. Renal and liver problems Treatment group (delta‐9‐THC): N = 36, female/male 20/16, mean age 51.72 (SD 12.11), 24 in MS‐subset. No baseline pain scores reported. No reports on previous cannabis use Placebo group: N = 34, female/male 21/13, mean age 57.61 (SD 10.28), 19 in MS‐subset. No baseline pain scores reported. No reports on previous cannabis use |
|
| Interventions |
Study medication: each actuation of oromucosal spray delivers 2.5 mg THC and 2.5 mg CBD. The maximum permitted dose of was 8 actuations in any 3‐hour period, and 48 actuations in any 24‐h period (THC 120 mg:CBD 120 mg). Placebo same number of actuations possible Rescue medication: no details provided, but percentage of days with uses recorded as secondary outcome (less in active group) Allowed co‐therapies: no details provided |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported and not calculable by imputation method. NRS 0‐10, 3 measures/day, average of the last 7 days PGIC much or very much improved: reported Withdrawal due to AE: reported, systematic assessment Serious AE: reported, systematic assessment HRQoL: Spitzer Quality of life index 15‐0 Participant‐reported pain relief ≥ 30%: not reported and not calculable by imputation method Mean pain intensity: NRS 0‐10, 3 measures/day, average of the last 7 days Sleep problems: NRS 0‐10 Fatigue: not assessed Psychological distress: not assessed Withdrawals due to lack of efficacy: not reported Any adverse event: reported; systematic assessment, no details reported Nervous system disorders‐related AE: reported; systematic assessment Psychiatric disorders‐related AE: reported; systematic assessment |
|
| Notes |
Funding: GW Pharmaceuticals Conflicts of interest: not declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Identical placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Low risk | All predefined outcomes reported |
| Group similarity at baseline | Low risk | Similar demographic characteristics at baseline |
| Sample size bias | High risk | < 50 participants per treatment arm |
NCT01606202.
| Study characteristics | ||
| Methods |
Disease: intractable neuropathic pain associated with spinal cord Injury Study setting: multicentre study UK, Romania; no years of study reported Study design: parallel Study duration: 7‐21 days baseline period, 3 weeks treatment |
|
| Participants |
Inclusion criteria: diagnosis of non‐acute spinal cord injury, with central neuropathic pain not wholly relieved by current therapy. Central neuropathic pain with a mean severity NRS score ≥ 4 during last 7 days of the baseline period. Relatively stable neurology during the preceding 6 months. Stable medication regimen during the preceding 4 weeks. Had not used cannabis‐based medicines for at least the preceding 7 days and willing to abstain from any use of cannabis‐based medicines during the study Exclusion criteria: history of schizophrenia, other psychotic illness, severe personality disorder or other significant psychiatric disorder other than depression associated with their underlying condition. History of alcohol or substance abuse. Severe cardiovascular disorder, such as ischaemic heart disease, arrhythmias (other than well‐controlled atrial fibrillation), poorly controlled hypertension or severe heart failure. History of autonomic dysreflexia. History of epilepsy. Renal and liver problems Treatment group (delta‐9‐THC): N = 56, age 48.7 (12.97), female/male 13/43. No reports on pain baseline scores and on previous cannabis use Placebo group: N = 60, age 47.6 (12.69), female/male 12/48. No reports on pain baseline scores and on previous cannabis use |
|
| Interventions |
Study medication: THC (27 mg/mL): CBD (25 mg/mL) as extract of Cannabis sativa L., with peppermint oil, 0.05%, in ethanol:propylene glycol (50:50) excipient. Each actuation delivered 100 μL (THC 2.7 mg and CBD 2.5 mg). The maximum permitted dose of study medication was 8 actuations in any 3‐h period, and 48 actuations in any 24‐h period Rescue medication: paracetamol 500 mg Allowed co‐therapies: stable medication regimen |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported and not calculable by imputation method. NRS 0‐10 Neuropathic Pain Scale PGIC much or very much improved: reported Withdrawal due to AE: reported Serious AE: Participant‐reported pain relief ≥ 30%:: not reported and not calculable by imputation method Mean pain intensity: NRS 0‐10 Neuropathic Pain Scale HRQoL: Spitzer Quality of Life Index Score 15‐0 Sleep problems: sleep disturbance NRS 0‐10 Fatigue: not assessed Psychological distress: not assessed Withdrawals due to lack of efficacy: not reported Any adverse event: reported. No details of assessment reported Nervous system disorders‐related AE: reported Psychiatric disorders‐related AE: reported |
|
| Notes |
Funding: GW Pharmaceuticals Conflicts of interest: not declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided, but identical placebo |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Identical placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Low risk | All predefined outcomes reported |
| Group similarity at baseline | Low risk | Similar demographic characteristics of the study groups at baseline |
| Sample size bias | Unclear risk | 50‐199 participants per treatment arm |
Nurmikko 2007.
| Study characteristics | ||
| Methods |
Disease: pain and allodynia patients with unilateral neuropathic pain of peripheral origin of various aetiologies Study setting: multicentre (5 UK, 1 Belgium); study period not reported Study design: parallel Study duration: baseline 7‐10 days, therapy 5 weeks |
|
| Participants |
Inclusion criteria: unilateral PNP and allodynia, at least 6 months with identifiable nerve lesion, unilateral PNP and allodynia, CRPS type II, ≥ 4 on a NRS for spontaneous pain 4 out of 7 days during baseline. A stable medication regimen of analgesics for at least 2 weeks prior to study entry. Exclusion criteria: cannabinoid use < 7 days, failure to abstain, schizophrenia, psychosis, or other major psychiatric condition beyond depression with underlying condition. Concomitant severe non‐neuropathic pain or the presence of cancer‐related neuropathic pain or from diabetes mellitus, known history of alcohol or substance abuse, severe cardiovascular condition, poorly controlled hypertension, epilepsy, pregnancy, lactation, significant hepatic or renal impairment Treatment group (delta‐9‐THC): N = 63, female 35, mean age 52.4 (SD 15.8) years. Pain baseline 7.3 (SD 1.4) on 0‐10 scale. 13 (21%) prior cannabis use Placebo group: N = 62, female 39, age 54.3 (15.2) years; pain baseline 7.2 (SD 1.5) on 0‐10 scale. 2 (19%) prior cannabis use |
|
| Interventions |
Study medication: spray for sublingual and oro‐pharyngeal administration. Each 100 μL spray delivers 2.7 mg of THC and 2.5 mg of CBD, identically appearing placebo spray. Participants were allowed a maximum dose of 8 sprays per 3‐h interval and a maximum of 48 sprays per 24 h. Rescue medication: none Allowed co‐therapies: stable dose regimen |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: reported. NRS 0‐10 over 7 days PGIC much or very much improved: only average scores reported (not suited for meta‐analysis) Withdrawal due to AE: assessed Serious AE: assessed; only psychiatric serious AEs reported Participant‐reported pain relief ≥ 30%: reported. NRS 0‐10 over 7 days Mean pain intensity: neuropathic pain scale total score 0‐60 HRQoL: not assessed Sleep problems: NRS 0‐10; SD calculated from P value Fatigue: not assessed Psychological distress: General Health Questionnaire 0‐48: SD calculated from P value Any adverse event: not reported (details of assessment of AE not reported) Nervous system disorders‐related AE: incompeletely reported (not suited for analysis) Psychiatric disorders‐related AE: incompeletely reported (not suited for analysis) |
|
| Notes |
Funding: GW Pharmaceuticals Conflicts of interest: not declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "After eligibility was confirmed, patients were assigned to the next sequential randomisation number within each centre. The randomisation schedule had a 1:1 treatment allocation ratio with randomly permuted blocks stratified by centre and was generated using a computer based pseudo‐random number algorithm". |
| Allocation concealment (selection bias) | Low risk | "The randomisation schedule was held by the sponsor with a copy in patient‐specific sealed envelopes sent to the pharmacy in each centre." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "That the smell and taste of the cannabinoid preparation might lead to unblinding was averted by disguising them with addition of peppermint oil to both preparations." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The analyses were verified by an independent statistician. The principal investigator had full access to all the data and carried out further confirmatory analyses" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Low risk | All predefined outcomes reported |
| Group similarity at baseline | Low risk | Similar clinical and demographic characteristics at baseline |
| Sample size bias | Unclear risk | 50‐199 participants per treatment arm |
Rog 2005.
| Study characteristics | ||
| Methods |
Disease: central pain in MS Study setting: UK, single‐centre; study period not reported Study design: parallel, randomised, placebo‐controlled, parallel‐group study Study duration: 5 weeks, including 1 week baseline |
|
| Participants |
Inclusion criteria: at least 6 months after MS diagnosis, at least 3 months central pain with unlikely other cause, both with dysaesthetic characteristics or painful spasm, 2 weeks of stable analgesic regimen, no cannabinoid use the last 7 days Exclusion criteria: spasticity‐related pain, visceral pain, headache, acute MS‐related pain, major psychiatric disorder, other than pain‐related depression, severe concomitant illness, seizures, history or suspicion of substance abuse, diabetes mellitus, levodopa use, hypersensitivity to cannabis‐based medicines Treatment group (delta‐9‐THC/CBD): N = 34; 6 male/28 female, mean age 50.3 (SD 6.7) years; 15 with previous cannabis exposure Placebo group: N = 32; 8 male/24 female; mean age 48.1 (SD 9.7) years; 21 with previous cannabis exposure |
|
| Interventions |
Study medication: Oromucosal spray containing 2.7 mg THC and 2.5 mg CBD per 100 µL spray, max 48 sprays in 48 h, identically appearing placebo Rescue medication: not reported Allowed co‐therapies: amitriptylin maximally 75 mg/d |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported, calculated by imputation method. NRS 0‐10 for most troublesome neuropathic pain at daily maximum, mean of 7 days PGIC much or very much improved: reported Withdrawal due to AE: reported Serious AE attributed to medication: reported Participant‐reported pain relief ≥ 30%: not reported, calculated by imputation method Mean pain intensity: NRS 0‐10 for most troublesome neuropathic pain at daily maximum, mean of 7 days HRQoL: not assessed Sleep problems: sleep quality 10‐0; SD calculated from P value Fatigue: not assessed Psychological distress: General Health Questionnaire 0‐48: SD calculated from P value Withdrawals due to lack of efficacy: not reported Any adverse event: not reported. No details of assessment reported Nervous system disorders‐related AE: reported Psychiatric disorders‐related AE: reported |
|
| Notes |
Funding: GW Pharmaceuticals Conflicts of interest: Rog, Young and Nurmikko received funding and/or honoraria from GW pharmaceuticals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Pre‐determined randomisation code that remained unknown to study personnel throughout the trial. Randomised permuted blocks of 4 |
| Allocation concealment (selection bias) | Low risk | Pharmacist dispensed medication |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identically appearing placebo also for smell |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Secondary outcomes assessed by blinded nurses |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Low risk | Consistent reporting of all outcomes |
| Group similarity at baseline | Low risk | Similar demographic and clinical characteristics at baseline |
| Sample size bias | High risk | < 50 participants per study arm |
Schimrigk 2017.
| Study characteristics | ||
| Methods |
Disease: central neuropathic pain in MS Study setting: single‐centre (Neurology Department), Germany, study period 2007‐2010 Study design: parallel Study duration: dose titration of study medication over 2 weeks, 2 weeks' titration, followed by a 10‐week maintenance phase. 32 weeks open label |
|
| Participants |
Inclusion criteria: aged 18–70 years, met the McDonald criteria for definite MS and had stable disease symptoms and moderate‐severe central neuropathic pain (CNP) at maximal pain area for at least 3 months as reported by participants (Numerical Rating Scale (NRS) for pain ≥ 4). CNP was defined as initiated or caused by a primary lesion or dysfunction of the CNS. Exclusion criteria: any peripheral pain syndromes, pre‐existing psychotic disorders, severe cardiac diseases, or known substance abuse; dronabinol intake within the last 12 months prior to study entry or Marijuana use within 1 month prior to study entry Treatment group (dronabinol): N = 124, mean age 48.4 (SD 9.6) years, 88% female, time since CNP diagnosis 130 (96) months, pain score baseline (extracted from figure 6.6), previous cannabis use not reported Placebo group: N = 116, mean age 47.0 (SD 9.7) years, 87% female, time since CNP diagnosis 138 (98) months, pain score baseline (extracted from figure 6.8), previous cannabis use not reported |
|
| Interventions |
Study medication: dosing was increased every 5 days by 2.5 mg to reach a daily dose between 7.5 and 15.0 mg Rescue medication: oral intake of tramadol Allowed co‐therapies: amitriptyline and gabapentin, if started at least 3 months earlier with a stable dose |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%:: not reported. NRS 0‐10 mean weekly pain score. Calculated by imputation method. Baseline pain scores extracted from figure PGIC much or very much improved: not assessed Withdrawal due to AE: reported Serious AE attributed to medication: reported Participant‐reported pain relief ≥ 30%:: not reported. NRS 0‐10 mean weekly pain score. Calculated by imputation method. Baseline pain scores extracted from figure Mean pain intensity: NRS 0‐10 mean weakly pain score HRQoL: Short form health survey SF‐36. Mean changes without SD or P value reported* Sleep problems: not assessed Fatigue: not assessed Psychological distress: not assessed Withdrawals due to lack of efficacy: reported Any adverse event: for safety analysis, vital signs, laboratory parameters, (serious) AEs (SAEs) including (serious) adverse reactions (SARs) were regularly assessed during all 3 periods. Furthermore, participantss rated the global tolerability on a 4‐point rating scale (1 = very good to 4 = poor). If study medication intake was interrupted, the investigator documented withdrawal symptoms such as restlessness, irritability, sleep interference, decreased appetite, excessive sweating, or other drug‐dependence‐related symptoms Nervous system disorders‐related AE: not reported Psychiatric disorders‐related AE: not reported |
|
| Notes |
Funding: Bionorica research GmbH (Innsbruck, Austria) Conflicts of interest: CN, EMK, GW, and DA‐S are employees of Bionorica SE, Germany. SS has received grant support and speaker honoraria from Bayer Vital, Bionorica, Biogen, BMS, DIAMED, Genzyme, Novartis, Pfizer, Teva. MM has received lecture fees, travel grants and honoraria for consulting from Bayer Health Care AG, Biogen GmbH, Bionorica, Merck Serono, Novartis Pharma GmbH, Sanofi‐Aventis (Genzyme), and Teva *no significant difference |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomization code |
| Allocation concealment (selection bias) | Unclear risk | No details reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details reported ("Full analysis set") |
| Selective reporting (reporting bias) | Low risk | All outcomes as outlined in protocol NCT00959218 reported |
| Group similarity at baseline | Low risk | Similar demographic and clinical characteristics at baseline |
| Sample size bias | Unclear risk | 50‐199 participants per treatment arm |
Selvarajah 2010.
| Study characteristics | ||
| Methods |
Disease: Chronic painful diabetic peripheral polyneuropathy in diabetes mellitus type 1 and 2 Study setting: Single‐centre (Diabetes Research Department), UK; study period not reported Study design: Parallel Study duration: Dose titration of study medication over 2 weeks, followed by a 10‐week maintenance phase |
|
| Participants |
Inclusion criteria: Neuropathy Total Symptom Score 6 > 4 and < 16 for at least 6 months with stable glycaemic control (A1C 11%), persistent pain, despite an adequate trial of tricyclic antidepressants Exclusion criteria: Not reported Treatment group (delta‐9‐THC/CBD): N = 15, Mean age 58.2 (SD 8.8) years, 4 female, mean diabetes duration 11.2 ± 8.4 years, 2 with previous cannabis use Placebo group: N = 15, 7 female, mean age 54.4 (SD 11.6) years, mean diabetes duration 13.7 (SD 6) years; 2 with previous cannabis use |
|
| Interventions | Study medication: Sativex (tetrahydrocannabinol (27 mg/mL) and CBD (25 mg/mL)) as a pump‐action spray, sublingually, up to 4 doses per day Rescue medication: Not reported Allowed co‐therapies: Not reported |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: Reported. VAS 0‐10 PGIC much or very much improved: Not assessed Withdrawal due to AE: Reported, but not the proportion of patients in each group Serious AE attributed to medication: Not reported Participant‐reported pain relief ≥ 30%:: Not reported, calculated by imputation method (VAS 0‐10) Mean pain intensity: Neuropathic pain scale (VAS 0‐100) HRQoL: EQ‐5D health status index Sleep problems: Sleep quality 10‐0; SD calculated from P value Fatigue: Not assessed Psychological distress: General Health Questionnaire 0‐48: SD calculated from P value Withdrawals due to lack of efficacy: Not reported Any adverse event: Not reported. No details of assessment reported Nervous system disorders‐related AE: Not reported Psychiatric disorders‐related AE: Not reported |
|
| Notes |
Funding: Diabetes UK grant Conflicts of interest: The authors declared that they have no conflicts of interest relevant to the study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details reported |
| Allocation concealment (selection bias) | Unclear risk | No details reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | One patient excluded from ITT‐analysis |
| Selective reporting (reporting bias) | High risk | Tolerability and safety outcomes not reported |
| Group similarity at baseline | Low risk | Similar demographic and clinical characteristics at baseline |
| Sample size bias | High risk | < 50 participants per treatment arm |
Serpell 2014.
| Study characteristics | ||
| Methods |
Disease: post‐herpetic neuralgia, peripheral neuropathy, focal nerve lesion, radiculopathy or CRPS type 2 associated with allodynia Study setting: 21 centres in the UK, 7 centres in Czech Republic, 6 centres in Romania, 4 centres in Belgium 1 one centre in Canada; 2005‐2006 Study design: parallel Study duration: 15‐week (1‐week baseline and 14‐week treatment period) |
|
| Participants |
Inclusion criteria: age ≥18 years, mechanical allodynia within the territory of the affected nerve(s) (confirmed by either a positive response to stroking the allodynic area with a SENSELABTM Brush 05 (Somedic AB, Hörby, Sweden) or to force applied by a 5.07 g Semmes‐Weinstein monofilament), at least a 6‐month disease history (post‐herpetic neuralgia, peripheral neuropathy, focal nerve lesion, radiculopathy or CRPS CRPS type 2), receiving the appropriate treatment, sum score of at least 24 on a pain 0–10 NRS for more than 6 days (baseline days 2–7) during the baseline period (average 0–10 NRS score of 4/10), and pain that was not wholly relieved by their current therapy. Stable analgesic regimen for at least 2 weeks preceding study entry. Exclusion criteria: severe pain from other concomitant conditions; history of significant psychiatric, renal, hepatic, cardiovascular or convulsive disorders, or with a known hypersensitivity to the study medication; CRPS type 1, cancer‐related PNP or pain resulting from diabetes mellitus; receiving a prohibited medication (including cannabis or cannabinoid‐based medications (in the last year), any analgesics taken on a ‘PRN’ (when required) basis, the introduction of any new analgesic medication, or any alteration to the dosage of the patient’s concomitant analgesic medication (other than the rescue analgesia provided), or all paracetamol‐containing medications (stopped on the day the patient entered the baseline period)), patients unwilling to abstain for the study duration; patients with a known history of alcohol or substance abuse; women of child‐bearing potential or their partners unless willing to ensure effective contraception was used throughout the study, participants who had received an investigational medicinal product within 12 weeks of screening; pregnant or lactating women and those planning a pregnancy; people with any physical abnormality at screening (i.e. any abnormalities that, in the opinion of the investigator, would prevent the participant from safely participating in the study), or those intending to travel or donate blood during the study Treatment group (delta‐9‐THC): N = 128; 66% female; mean age 57.6 (mean age 14.4) years; 99% white; duration of neuropathic pain 6.3 (SD 6.7 years), 13 with cannabis exposure (10%) Placebo group: N = 118, 55% female; mean age 57 (SD 14.1) years; 98% white; duration of neuropathic pain 6.3 (SD 6.4) years, 12 with cannabis exposure (10%) |
|
| Interventions |
Study medication: pump action oromucosal spray, each 100 μL spray of THC/CBD delivered 2.7 mg of THC and 2.5 mg of CBD, each spray of placebo delivered the excipients plus colorants, both THC/CBD spray and placebo contained peppermint oil to blind the smell and taste, maximum of eight sprays in a 3‐h period up to a maximum of 24 sprays per 24‐h period Rescue medication: paracetamol 500 mg, max. Single dose 1 g, max. Daily dose 4 g Allowed co‐therapies: concomitant analgesic medication, with the exception of paracetamol (acetaminophen), provided that a stable dose was maintained throughout the study |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: NRS 0‐10. Only OR reported: not suited for meta‐analysis (P = 0.157) PGIC much or very much improved: reported Withdrawal due to AE: reported Serious AE: reported; systematic assessment Participant‐reported pain relief ≥ 30%: NRS 0‐10; only OR reported: not suited for meta‐analysis (P = 0.021) Mean pain intensity: Neuropathic pain scale: data not suited for meta‐analysis (P = 0.069) HRQoL: EQ‐5D Health Status 100 to 0 Sleep problems: sleep quality 10‐0; SD calculated from P value Fatigue: not assessed Psychological distress: General Health Questionnaire 0‐48: SD calculated from P value Withdrawals due to lack of efficacy: reported Any adverse event: reported; "systematic assessment" Nervous system disorders‐related AE: reported; systematic assessment Psychiatric disorders‐related AE: reported; systematic assessment |
|
| Notes |
Funding: GW Pharmaceuticals. GW Pharmaceuticals was involved in the study design, data collection and analysis, as well as in the preparation of this manuscript and publication decisions Conflicts of interest: all authors received investigator fees from GW Pharma Ltd (GW) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation code |
| Allocation concealment (selection bias) | Low risk | Treatment allocation by GW Biometrics department; sealed code break envelopes for each partcipant |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | THC/CBD and placebo spray contained peppermint oil to blind to taste and smell |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Low risk | Study protocol (NCT 00710554) available; all predefined outcomes reported |
| Group similarity at baseline | Low risk | Similar demographic and clinical characteristics at baseline |
| Sample size bias | Unclear risk | 50‐199 participants per treatment arm |
Svendsen 2004.
| Study characteristics | ||
| Methods |
Disease: MS (central pain) Study setting: outpatient clinic, University Hospital of Aarhus, Denmark; study period 2001 Study design: cross‐over Study duration: 15‐20 days with washout period of at least 21 days (actually 19‐57), 1 week baseline, 3 weeks intervention, 3 weeks washout, 3 weeks intervention |
|
| Participants |
Inclusion criteria: diagnosed with MS, aged 18‐55 years, pain ≥ 3 on 0‐10 NRS, investigators assessed central pain examination, central pain being a pain in a body territory with abnormal sensation to pinprick, touch, warmth, cold, ability to differentiate central from spasticity‐related pain Exclusion criteria: musculoskeletal disorders, PNP, visceral pain at max. pain site, hypersensitivity to cannabis‐based medicines or sesame oil, heart disease, mania, depression or schizophrenia, alcohol or drug misuse, no antidepressants, anticholinergic, antihistaminic agents or CNS depressants, use of analgesic drugs, (medications had to be stopped 1 week before first visit) pregnancy or lactation, sexually active women without reliable contraception, other clinical trials, lack of co‐operation, use of marijuana within 3 months before the study, unwillingness to abstain from marijuana use Treatment group (dronabinol) and placebo group: N = 24; 41.7% male, mean age 50 (23‐55) years, no ethnic group, current cannabis use not reported |
|
| Interventions |
Study medication: dronabinol starting with 1 x 2.5 mg capsules up to 2 x 5 mg/d Rescue medication: paracetamol Allowed co‐therapies: spasmolytic drugs and paracetamol |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: reported. NRS 0‐10 (end of treatment period) PGIC much or very much improved: not assessed Withdrawal due to AE: reported Serious AE: reported Participant‐reported pain relief ≥ 30%: not reported.Not calculable by imputation method because baseline values not reported Mean pain intensity: median spontaneous pain intensity NRS 0‐10 during the last week of treatment HRQoL: SF‐36 physical functioning (50‐0); data of first treatment period used for analysis; SD calculated from P value Sleep problems: not assessed Fatigue: not assessed Psychological distress: SF‐36 mental health (50‐0). Data of first treatment period used for analysis; SD calculated from P value Withdrawals due to lack of efficacy: reported Any adverse event: reported. "Patient used their own words to record AEs in diaries" Nervous system disorders‐related AE: reported Psychiatric disorders‐related AE: reported |
|
| Notes |
Funding: the study was supported by grants from the Danish Multiple Sclerosis Society (grant no 2002/71045), grant 900035 from manager Ejnar Jonasseon and his wife’s memorial grant, and the Warwara Larsen Foundation (grant no 664.28), Denmark. Solvay Pharmaceuticals provided study medication (dronabinol (Marinol) and placebo capsules), labelling, and packaging. In addition, the company provided financial support for study monitoring and data analysis. IPC‐Nordic, Denmark, packaged and labelled the study medication and monitored the study. These companies were not involved in the design or execution of the study or writing the manuscript. Conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "We assigned patients to treatment sequence by using a computer generated randomisation code with a block size of six prepared by IPC‐Nordic" |
| Allocation concealment (selection bias) | Low risk | "Investigators allocated patients consecutively by time of inclusion at the study site. One investigator (KBS) enrolled all participants and allocated them to treatment". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "We administered both active treatment and placebo as white capsules (soft gelatin capsules) in identical containers. The taste and smell of the capsules did not differ." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants terminated the study |
| Selective reporting (reporting bias) | Unclear risk | No study protocol reported |
| Group similarity at baseline | Low risk | No significant differences in demographic and clinical characteristics between the study groups because of study design |
| Sample size bias | High risk | < 25 participants per treatment arm |
Toth 2012.
| Study characteristics | ||
| Methods |
Disease: diabetic peripheral polyneuropathy Study setting: single‐centre, Canada; study period not reported Study design: EERW Study duration: single‐blind for 4 weeks, double‐blind randomised withdrawal for 5 weeks |
|
| Participants |
Inclusion criteria: DPN pain questionnaire score ≥ 4, pain duration at least 3 months, pain severity with averaged scores of P40 mm on the 100‐mm VAS of the short‐Form McGill Pain Questionnaire Exclusion criteria: participants with other causes of pain, including PHN, lumbar radiculopathy, central neuropathic pain, CRPSs I or II, or significant osteoarthritis, were excluded. Any skin conditions over the area of DPN which could hinder examination, led to exclusion. Any current diagnoses of schizophrenia, psychotic disorder, bipolar affective disorder, obsessive compulsive disorder, or major depressive disorder were also exclusionary. Clinically significant unstable medical conditions that could compromise participation, such as with poor diabetic control (haemoglobin A1C ≥ 11%), history of substance abuse or dependence, malignancy other than squamous cell carcinoma in the last 2 years, elevation of liver enzymes above 3 times the upper limit of normal, or an anticipated need for surgery or hospitalisation within the next 16 weeks after screening led to exclusion at the discretion of the investigator. Those participants previously exposed to nabilone were excluded. Any use of self‐obtained cannabis‐based medicines or other illicit drugs during the study was prohibited, and participants with a positive urinary illicit drug screen (including detection of 11‐nor‐delta‐9‐ tetrahydrocannabinol‐9‐carboxylic acid) were excluded at screening. Treatment group (nabilone (delta‐9‐THC)): N = 13; mean age 61.6 (SD 14.6) years; 69% male; 92% white; duration of diabetes 10 (SD 12.6) years. No reports on previous cannabis use Placebo group: N = 13; mean age 60.8 (SD 15.2) years; 38% male; 92% white; duration of diabetes 9.7 (SD 13.1) years. No reports on previous cannabis use |
|
| Interventions |
Study medication: nabilone 1 mg‐5 mg/d orally Rescue medication: placebo drug Allowed co‐therapies: no details provided |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: reported (NRS 0‐10 over the preceding 24 h) PGIC much or very much improved: reported (in figure) Withdrawal due to AE: reported Serious AE: reported Participant‐reported pain relief ≥ 30%: reported Mean pain intensity: average pain intensity (VAS 0‐10) HRQoL: Euro‐QOL VAS 100‐0 Sleep problems: Medical Outcomes Study Sleep problems index: reported Fatigue: not assessed Withdrawals due to lack of efficacy: reported Any adverse event: reported; "All spontaneously reported and observed AEs were recorded at each clinic visit and during telephone follow‐up visits" Nervous system disorders‐related AE: incompletely reported. Not suited for meta‐analysis Psychiatric disorders‐related AE: reported |
|
| Notes |
Funding: Valeant Conflicts of interest: Dr. Toth received honoraria from Valeant Canada for educational lectures. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Electronic randomization system was used to randomise individual subjects without block randomisation as developed by an outside coordinator" |
| Allocation concealment (selection bias) | Low risk | "Randomization was concealed from subjects, clinical coordinator, and assessing physicians" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Medication was blinded for placebo using capsules of identical size, colour, taste, and smell." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT by LOCF |
| Selective reporting (reporting bias) | Unclear risk | Study protocol available (NCT01035281) but no outcomes reported |
| Group similarity at baseline | High risk | Significant difference in sex ratio at baseline |
| Sample size bias | High risk | < 25 participants per treatment arm |
Ware 2010.
| Study characteristics | ||
| Methods |
Disease: non HIV neuropathy > 3 months duration caused by trauma, surgery; with pain ≥ 40/100 VAS, stable analgesic regimen Study setting: single‐centre university, Canada; 2003‐2006 Study design: 4 periods cross‐over Study duration: 2 weeks with 5 treatment days per each period, 9 days' washout |
|
| Participants |
Inclusion criteria: men and women aged ≥ 18 years with neuropathic pain of at least 3 months in duration caused by trauma or surgery, with allodynia or hyperalgesia, and with an average weekly pain intensity score > 4 on a 10‐cm VAS. Participants had a stable analgesic regimen and reported not having used cannabis during the year before the study Potential participants had to have normal liver function (defined as aspartate aminotransferase < 3 times normal), normal renal function (defined as a serum creatinine level < 133 μmol/L), normal haematocrit (> 38%) and a negative result on β human chorionic gonadotropin pregnancy test (if applicable). Exclusion criteria: pain due to cancer or nociceptive causes, presence of significant cardiac or pulmonary disease, current substance abuse or dependence (including abuse of or dependence on cannabis), history of psychotic disorder, current suicidal ideation, pregnancy or breastfeeding, participation in another clinical trial within 30 days of enrolment, and ongoing insurance claims Treatment group (delta‐9‐THC)/placebo group): N = 23 participants, mean age: 45.4 years (SD 12.3); gender (male/female): 11/12; 18 (81%) with previous cannabis exposure, but not within the year prior to the study |
|
| Interventions |
Study medication: 3 different potencies of THC (2.5%, 6%, 9.4%) from whole herb in gelatine capsules inhaled through pipe. Placebo cigarettes underwent ethanolic extraction. Dose estimate: 0, 1.625, 3.9 and 5.85 mg/d (average) THC per period Rescue medication: not reported Allowed co‐therapies: "Stable regimen" |
|
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported and not calculable by imputation method. Average daily pain Intensity on 0‐10 NRS average over 5 treatment days PGIC much or very much improved: not assessed Withdrawal due to AE: reported Serious AE attributed to study medication: reported Participant‐reported pain relief ≥ 30%: not reported and not calculable by imputation method Mean pain intensity: average daily pain intensity on 0‐10 NRS HRQoL: EQ‐5D state of health VAS 100‐0 Sleep problems: sleep quality Leeds Sleep Evaluation Questionnaire 0‐10 Fatigue: not assessed Psychological distress: Profile of Mood States total mood disturbance 0‐200 Withdrawals due to lack of efficacy: not reported Any adverse event: reported; No details of assessment reported Nervous system disorders‐related AE: reported Psychiatric disorders‐related AE: reported |
|
| Notes |
Funding: Canadian Institutes of Health (JHM 50014) and Louise and Alan Wards Foundation Conflicts of interest: the study authors declare that they have not conflict of interest. "We found no evidence of significant carry‐over effect for any outcome" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details reported |
| Allocation concealment (selection bias) | Unclear risk | No details for investigators reported. Participants correctly guessed allocation at the end of the trial |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No ITT |
| Selective reporting (reporting bias) | Low risk | Consistent reporting according to study protocol (ISRCT68314063) |
| Group similarity at baseline | Low risk | Identical demographic and baseline characteristics due to study protocol |
| Sample size bias | High risk | < 25 participants per treatment arm |
AE: adverse events; bpm: beats per minute; BSI: Brief Symptom Inventory; CBD: cannabidiol; CNS: central nervous system; CRPS: complex regional pain syndrome; DHC: dihydrocodeine; DPN: diabetic peripheral neuropathic; DSM: Diagnostic and Statistical Manual of Mental Disorders; EERW: enriched enrolment randomised withdrawal; EQ‐5D: EuroQol quality of life instrument; HR: heart rate; HRQoL: Health‐related quality of life; ITT: intention‐to‐treat; LOCF: last observation carried forward; mg: milligrams; MAO: monoamine oxidase; MI: myocardial infarction; μL = microlitre; mL = millilitre; µmol/L: micromoles per litre; MS: multiple sclerosis; N: number; NDS: Neuropathy Disability Score; NNTB: number needed to treat for an additional beneficial outcome; NRS: numerical rating scale; NSAIDs: non‐steroidal anti‐inflammatory drugs; OR: odds ratio; PGIC: Patient Global Impression of Change; PHN: postherpetic neuralgia; PNP: peripheral neuropathic pain; SD; standard deviation; SIP: Sickness Impact Profile;SF‐36: short‐form 36 quality of life instrument; TENS: transcutaneous electrical nerve stimulation; THC: tetrahydrocannabinol; VAS: visual analogue scale; WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abrams 2007 | Cannabis cigarettes or placebo cigarettes in 55 participants. HIV‐associated neuropathy; study duration < 2 weeks |
| Corey‐Bloom 2012 | Smoked cannabis or placebo cigarettes in 30 participants with MS for 2 weeks; no definite statement that the pain was of neuropathic nature |
| Karst 2003 | Synthetic THC or oral placebo in 21 participants with chronic neuropathic central and peripheral pain of various aetiologies; study duration < 2 weeks |
| Notcutt 2011 | 34 ‘N of 1’ studies with THC, CBD and THC/CBD or placebo over 12 weeks; 2 participants with non‐neuropathic pain included |
| Novotna 2011 | 572 participants with MS were treated with THC/CBD spray for 12 weeks; participants were selected because of spasticity refractory to conventional treatment; no definite statement that the pain was of neuropathic nature |
| Rintala 2010 | Randomised, controlled, double‐blind, cross‐over pilot study with 7 participants with spinal cord injury and neuropathic pain comparing dronabinol with diphenhydramine; < 10 participants per treatment arm |
| Turcotte 2015 | 15 participants with MS‐induced neuropathic pain were treated with nabilone as an adjunctive to gabapentin for 9 weeks; < 10 participants per treatment arm |
| Wade 2003 | 20 participants with neurogenic symptoms due to lesions of the central or peripheral nervous system were treated with plant‐based THC/CBD for 2 weeks in a cross‐over design. 13 of 20 participants with pain. No statement or analysis that carry‐over effects were excluded |
| Wade 2004 | 160 participants with MS treated with THC/CBD spray or placebo spray of 6 weeks; no definite statement that the pain was of neuropathic nature |
| Wallace 2015 | Inhaled cannabis in 16 participants with painful diabetic polyneuropathy for 4 single dosing sessions. Study duration < 2 weeks |
| Wilsey 2008 | Vaporised cannabis (1.3% and 3.5%) or placebo in 39 participants with central and peripheral neuropathic pain for 1 day (experimental study) |
| Wilsey 2013 | 38 participants with central or peripheral neuropathic pain were treated with smoked cannabis or placebo. Study duration < 1 week |
| Wissel 2006 | Nabilone or placebo in 11 participants with MS und upper motor neuron disease‐associated spasticity‐related pain for 4 weeks; no definite statement that the pain was of neuropathic nature |
| Zajicek 2003 | 667 participants with MS were treated with oral cannabis extract (THC) or delta 9‐THC or placebo for 15 weeks. Spasticity was the primary outcome. Pain was a secondary outcome; only around 65% of participants had pain, with no pain intensity at baseline reported |
| Zajicek 2012 | 275 patients with MS were treated for 12 weeks with plant‐derived THC 2.5‐15 mg/d orally or placebo. No definite statement that the pain was of neuropathic nature |
CBD: cannabidiol; mg: milligrams; µmol/L: micromoles per litre;MS: multiple sclerosis; THC: tetrahydrocannabinol;
Characteristics of studies awaiting classification [ordered by study ID]
NCT00699634.
| Methods |
Disease: phantom limb pain Study setting: single‐centre university, Canada; 2009‐2011 Study design: parallel Study duration: 6 weeks |
| Participants |
Inclusion criteria
Exclusion criteria:
Treatment group nabilone/placebo group: N = not reported |
| Interventions |
Study medication: nabilone 0.5 mg at bedtime for 1 week, then 0.5 mg twice daily for 1 week. After a reassessment of the outcome measures, the dose is increased to 0.5 mg in the morning and 1 mg at hs for 1 week, followed by an increase to 1 mg twice daily in the last week of the study. Rescue medication: not reported Allowed co‐therapies: not reported |
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported PGIC much or very much improved: not assessed Withdrawal due to AE: not reported Serious AE attributed to study medication: not reported Participant‐reported pain relief ≥ 30%: not reported Mean pain intensity: VAS for pain; not reported HRQoL:: SF‐36 not reported Sleep problems: Groningen Sleep Quality Scale; not reported Fatigue: not assessed Psychological distress: Hospital Anxiety and Depression Scale not reported Withdrawals due to lack of efficacy: not reported Any adverse event: reported; no details of assessment reported Nervous system disorders‐related AE: not reported Psychiatric disorders‐related AE: not reported |
| Notes |
Funding: Valeant, University of Manitoba Conflicts of interest: not declared |
NCT01035281.
| Methods |
Disease: diabetic neuropathic pain Study setting: single‐centre university, Canada; start 2009; the recruitment status of this study is unknown because the information has not been verified recently. Study design: EERW Study duration: all participants who experienced at least a 30% reduction in their weekly mean pain score during the 4‐week, single‐blind flexible dosing phase considered a responder, and further continued in the study. During the double‐blind portion of the study, participants randomised to nabilone continued on the dose of nabilone achieved at the completion of the single‐blind phase, and this dose was maintained throughout the double‐blind phase. Participants randomised to placebo received 1 mg of nabilone daily for 1 week, followed by 4 consecutive weeks of placebo. This dose of nabilone permitted a tapering for those participants achieving a higher daily dose of nabilone during the single‐blind phase, or maintained those who were taking only 1 mg/d in the single‐blind phase, preventing an abrupt termination of treatment in participants who were randomised into the placebo portion. |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions |
Study medication:nabilone, flexible dosing nabilone at 0.5 mg‐4 mg/d Rescue medication: not reported Allowed co‐therapies: not reported |
| Outcomes |
Participant‐reported pain relief ≥ 50%: no information provided PGIC much or very much improved: no information provided Withdrawal due to AE: no information provided Serious AE attributed to study medication: no information provided Participant‐reported pain relief ≥ 30%: no information provided Mean pain intensity: no information provided HRQoL:: no information provided Sleep problems: no information provided Fatigue: no information provided Psychological distress: no information provided Withdrawals due to lack of efficacy: no information provided Any adverse event: no information provided Nervous system disorders‐related AE: no information provided Psychiatric disorders‐related AE: no information provided |
| Notes |
Funding: University of Calgary Conflicts of interest: not declared |
NCT01222468.
| Methods |
Disease: neuropathic pain in spinal cord injured persons Study setting: single‐centre university, Canada; 2012‐2015 Study design: cross‐over Study duration: 11 weeks each period |
| Participants |
Inclusion criteria
Exclusion criteria
Treatment group nabilone/placebo group: N = not reported |
| Interventions |
Study medication: nabilone 0.5 mg tablets od titrated to a maximum daily dose of 3 mg by mouth over an 11‐week phase; placebo 0.5 mg by mouth daily, dose titrated to a maximum daily dose of 3.0 mg by mouth over an 11‐week phase Rescue medication: not reported Allowed co‐therapies: not reported |
| Outcomes |
Participant‐reported pain relief ≥ 50%: not reported PGIC much or very much improved: not reported Withdrawal due to AE: not reported Serious AE attributed to study medication: not reported Participant‐reported pain relief ≥ 30%: not reported Mean pain intensity: VAS for pain and Neuropathic Pain Questionnaire; not reported HRQoL:: SF‐36 not reported Sleep problems: Pittsburgh Sleep Quality Index; not reported Fatigue: not assessed Psychological distress: not assessed Withdrawals due to lack of efficacy: not reported Any adverse event: reported; no details of assessment reported Nervous system disorders‐related AE: not reported Psychiatric disorders‐related AE: not reported |
| Notes |
Funding: University of Manitoba The Manitoba Spinal Cord Injury Research Fund Canadian Paraplegic Association Health Sciences Centre Foundation, Manitoba Conflicts of interest: not declared |
AE; adverse events; ALT: alanine aminotransferase; AST: aspartate aminotransferase; DSM: Diagnostic and Statistical Manual of Mental Disorders; EERW: enriched enrolment randomised withdrawal; GGT: gamma‐glutamyl transferase; HRQoL: health‐related quality of life; mg: milligrams; MI: myocardial infarction; N: number; PGIC: Patient Global Impression of Change; SF‐36: short‐form 36 quality of life instrument; THC: tetrahydrocannabinol; VAS: visual analogue scale
Differences between protocol and review
We changed the title of the review from "Cannabinoids" to "Cannabis‐based medicines" because medical cannabis contains compounds other than phytocannabinioids, for example, terpenoids. We updated the Background to reflect new template text. We specified in primary and secondary outcome measures that we preferred composite neuropathic pain scores over single‐scale generic pain scores if both measures were used by studies. We added mean pain intensity as secondary outcome measure. We included the European Union clinical trial register into our search. We added publication bias (all studies funded by the manufacturer of the drug) into the GRADE rating of the quality of evidence, and described our approach to assigning 'very low quality' in some circumstances. We post hoc decided to restrict subgroup analyses to the outcomes as reported in the 'Summary of findings' table. We post hoc decided to perform subgroup analyses of studies with and without publication in peer‐reviewed journals and of studies with high and unclear sample size bias. In the 'Summary of findings' table, we substituted the outcome health‐related quality of life with nervous system disorders and psychiatric disorders as specific adverse events. We removed the planned analysis by tiers of evidence as this is largely replaced by GRADE.
Contributions of authors
FP and WH drafted the protocol.
WH developed the search strategy together with Joanne Abbott (PaPaS Information Specialist).
MM, FP and WH selected studies for inclusion and extracted data from the studies.
WH, FP, and MM entered data into Review Manager 5 and carried out the analysis (RevMan 2014).
All review authors interpreted the analysis.
WH drafted the final review.
Sources of support
Internal sources
-
Technische Universität München, Germany
General institutional support
External sources
-
The National Institute for Health Research (NIHR), UK
NIHR Cochrane Programme Grant: 13/89/29 ‐ Addressing the unmet need of chronic pain: providing the evidence for treatments of pain
Declarations of interest
MM: none known; MM is a specialist in palliative care who treats patients with chronic neuropathic pain.
TP: none known; TP is a specialist pain physician and manages patients with neuropathic pain.
LR: none known; PR is a specialist in palliative care who treats patients with chronic neuropathic pain.
FP is a specialist in pain medicine who treats patients with chronic neuropathic pain. He has received speaking fees for one educational lecture for Janssen‐Cilaq (2015) on fibromyalgia and participated in an advisory board for the same company focusing on an unrelated product (2015).
WH is a specialist in general internal medicine, psychosomatic medicine and pain medicine, who treats patients with fibromyalgia and chronic neuropathic pain. He is a member of the medical board of the German Fibromyalgia Association. He is the head of the steering committee of the German guideline on fibromyalgia and a member of the steering committee of the European League Against Rheumatism (EULAR) update recommendations on the management of fibromyalgia. He received speaking fees for one educational lecture from Grünenthal (2015) on pain management.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Bermann 2004 {published data only}
- Berman JS, Symonds C, Birch R. Efficacy of two cannabis based medicinal extracts for relief of central neuropathic pain from brachial plexus avulsion: results of a randomised controlled trial. Pain 2004;112:299-306. [DOI] [PubMed] [Google Scholar]
Ellis 2009 {published data only}
- Ellis RJ, Toperoff W, Vaida F, Van den Brande G, Gonzales J, Gouaux B, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology 2009;34(3):672-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Frank 2008 {published data only}
- Frank B, Serpell MG, Hughes J, Matthews JN, Kapur D. Comparison of analgesic effects and patient tolerability of nabilone and dihydrocodeine for chronic neuropathic pain: randomised, crossover, double blind study. BMJ 2008;336:199-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Langford 2013 {published data only}
- Langford RM, Mares J, Novotna A, Vachova M, Novakova I, Notcutt W, et al. A double-blind, randomized, placebo-controlled, parallel-group study of THC/CBD oromucosal spray in combination with the existing treatment regimen, in the relief of central neuropathic pain in patients with multiple sclerosis. Journal of Neurology 2013;260:984-97. [DOI] [PubMed] [Google Scholar]
Lynch 2014 {published data only}
- Lynch ME, Cesar-Rittenberg P, Hohmann AG. A double-blind, placebo-controlled, crossover pilot trial with extension using an oral mucosal cannabinoid extract for treatment of chemotherapy-induced neuropathic pain. Journal of Pain and Symptom Management 2014;47:166-73. [DOI] [PubMed] [Google Scholar]
NCT00710424 {published data only}
- NCT 00710424. A study of Sativex® for pain relief due to diabetic neuropathy. clinicaltrials.gov/ct2/results?term=NCT00710424+&Search=Search (first Posted 4 July 2008).
NCT01606176 {published data only}
- NCT01606176. A Study to Evaluate the Effects of Cannabis Based Medicine in Patients With Pain of Neurological Origin. clinicaltrials.gov/ct2/results?cond=&term=NCT01606176&cntry1=&state1=&recrs= (first posted 25 May 2012).
NCT01606202 {published data only}
- NCT 01606202. A study of cannabis based medicine extracts and placebo in patients with pain due to spinal cord injury. clinicaltrials.gov/ct2/results?term= NCT 01606202&Search=Search (first posted 25 May 2012).
Nurmikko 2007 {published data only}
- Nurmikko TJ, Serpell MG, Hoggart B, Toomey PJ, Morlion BJ, Haines D. Sativex successfully treats neuropathic pain characterised by allodynia: a randomised, double-blind, placebo-controlled clinical trial. Pain 2007;133:210-20. [DOI] [PubMed] [Google Scholar]
Rog 2005 {published data only}
- Rog DJ, Nurmikko TJ, Friede T, Young CA. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology 2005;65:812-9. [DOI] [PubMed] [Google Scholar]
Schimrigk 2017 {published data only}
- Schimrigk S, Marziniak M, Neubauer C, Kugler EM, Werner G, Abramov-Sommariva D. Dronabinol is a safe long-term treatment option for neuropathic pain patients. European Neurology 2017;78:320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Selvarajah 2010 {published data only}
- Selvarajah D, Gandhi R, Emery CJ, Tesfaye S. Randomized placebo-controlled double-blind clinical trial of cannabis-based medicinal product (Sativex) in painful diabetic neuropathy: depression is a major confounding factor. Diabetes Care 2010;33:128-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Serpell 2014 {published data only}
- Serpell M, Ratcliffe S, Hovorka J, Schofield M, Taylor L, Lauder H, et al. A double-blind, randomized, placebo-controlled, parallel group study of THC/CBD spray in peripheral neuropathic pain treatment. European Journal of Pain 2014;18:999-1012. [DOI] [PubMed] [Google Scholar]
Svendsen 2004 {published data only}
- Svendsen KB, Jensen TS, Bach FW. Does the cannabinoid dronabinol reduce central pain in multiple sclerosis? Randomised double blind placebo controlled crossover trial. BMJ 2004;329:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Toth 2012 {published data only}
- Toth C, Mawani S, Brady S, Chan C, Liu C, Mehina E, et al. An enriched-enrolment, randomized withdrawal, flexible-dose, double-blind, placebo-controlled, parallel assignment efficacy study of nabilone as adjuvant in the treatment of diabetic peripheral neuropathic pain. Pain 2012;153:2073-82. [DOI] [PubMed] [Google Scholar]
Ware 2010 {published data only}
- Ware MA, Wang T, Shapiro S, Robinson A, Ducruet T, Huynh T, et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. Canadian Medical Association Journal 2010;184:E694-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Abrams 2007 {published data only}
- Abrams DI, Jay CA, Shade SB, Vizoso H, Reda H, Press S, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology 2007;68:515-21. [DOI] [PubMed] [Google Scholar]
Corey‐Bloom 2012 {published data only}
- Corey-Bloom J, Wolfson T, Gamst A, Jin S, Marcotte TD, Bentley H, et al. Smoked cannabis for spasticity in multiple sclerosis: a randomized, placebo-controlled trial. Canadian Medical Association Journal 2012;184:1143-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karst 2003 {published data only}
- Karst M, Salim K, Burstein S, Conrad I, Hoy L, Schneider U. Analgesic effect of the synthetic cannabinoid CT-3 on chronic neuropathic pain: a randomized controlled trial. JAMA 2003;290:1757-62. [DOI] [PubMed] [Google Scholar]
Notcutt 2011 {published data only}
- Notcutt W, Price M, Miller R, Newport S, Phillips C, Simmons S, et al. Initial experiences with medicinal extracts of cannabis for chronic pain: results from 34 'N of 1' studies. Anaesthesia 2004;59:440-52. [DOI] [PubMed] [Google Scholar]
Novotna 2011 {published data only}
- Novotna A, Mares J, Ratcliffe S, Novakova I, Vachova M, Zapletalova O, et al. Arandomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®)), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. European Journal of Neurology 2011;18:1122-31. [DOI] [PubMed] [Google Scholar]
Rintala 2010 {published data only}
- Rintala DH, Fiess RN, Tan G, Holmes SA, Bruel BM. Effect of dronabinol on central neuropathic pain after spinal cord injury: a pilot study. American Journal of Physical Medicine & Rehabilitation 2010;89:840-8. [DOI] [PubMed] [Google Scholar]
Turcotte 2015 {published data only}
- Turcotte D, Doupe M, Torabi M, Gomori A, Ethans K, Esfahani F, et al. Nabilone as an adjunctive to gabapentin for multiple sclerosis-induced neuropathic pain: a randomized controlled trial. Pain Medicine 2015;16:149-59. [DOI] [PubMed] [Google Scholar]
Wade 2003 {published data only}
- Wade DT, Robson P, House H, Makela P, Aram J. A preliminary controlled study to determine whether whole-plant cannabis extracts can improve intractable neurogenic symptoms. Clinical Rehabilitation 2003;17:21-9. [DOI] [PubMed] [Google Scholar]
Wade 2004 {published data only}
- Wade DT, Makela P, Robson P, House H, Bateman C. Do cannabis-based medicinal extracts have general or specific effects on symptoms in multiple sclerosis? A double-blind, randomized, placebo-controlled study on 160 patients. Multiple Sclerosis 2004;10:434-41. [DOI] [PubMed] [Google Scholar]
Wallace 2015 {published data only}
- Wallace MS, Marcotte TD, Umlauf A, Gouaux B, Atkinson JH. Efficacy of inhaled cannabis on painful diabetic neuropathy. Journal of Pain 2015;16:616-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilsey 2008 {published data only}
- Wilsey B, Marcotte T, Tsodikov A, Millman J, Bentley H, Donaghe H. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. Journal of Pain 2008;9:506-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilsey 2013 {published data only}
- Wilsey B, Marcotte T, Deutsch R, Gouaux B, Sakai S, Gouaux B, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. Journal of Pain 2013;14:136-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wissel 2006 {published data only}
- Wissel J, Haydn T, Müller J, Brenneis C, Berger T, Berger T. Low dose treatment with the synthetic cannabinoid Nabilone significantly reduces spasticity-related pain: a double-blind placebo-controlled cross-over trial. Journal of Neurology 2006;253:1337-41. [DOI] [PubMed] [Google Scholar]
Zajicek 2003 {published data only}
- Zajicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A, et al. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomized placebo-controlled trial. Lancet 2003;362:1517-26. [DOI] [PubMed] [Google Scholar]
Zajicek 2012 {published data only}
- Zajicek JP, Hobart JC, Slade A, Barnes D, Mattison PG, MUSEC Research Group. Multiple sclerosis and extract of cannabis: results of the MUSEC trial. Journal of Neurology, Neurosurgery and Psychiatry 2012;83:1125-32. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
NCT00699634 {published data only}
- NCT00699634. Nabilone for the treatment of phantom limb pain. clinicaltrials.gov/ct2/show/NCT00699634?term=NCT00699634&rank=1 (first posted 18 June 18).
NCT01035281 {published data only}
- NCT01035281. Efficacy study of nabilone in the treatment of diabetic peripheral neuropathic pain. clinicaltrials.gov/ct2/show/NCT01035281?term=NCT01035281&rank=1 (first posted 18 December 2009).
NCT01222468 {published data only}
- NCT01222468. Effect of cannabinoids on spasticity and neuropathic pain in spinal cord injured persons. clinicaltrials.gov/ct2/show/NCT01222468?term=NCT01222468&rank=1 (first posted 18 October 2010).
Additional references
Ablin 2016
- Ablin J, Ste-Marie PA, Schäfer M, Häuser W, Fitzcharles MA. Medical use of cannabis products: lessons to be learned from Israel and Canada. Schmerz 2016;30:3-13. [DOI] [PubMed] [Google Scholar]
AlBalawi 2013
- AlBalawi Z, McAlister FA, Thorlund K, Wong M, Wetterslev J. Random error in cardiovascular meta-analyses: how common are false positive and false negative results? Interational Journal of Cardiology 2013;168(2):1102-7. [DOI: 10.1016/j.ijcard.2012.11.048] [DOI] [PubMed] [Google Scholar]
Andersohn 2008
- Andersohn F, Garbe E. Pharmacoepidemiological research with large health databases. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 2008;51:1134-55. [DOI] [PubMed] [Google Scholar]
Andreae 2015
- Andreae MH, Carter GM, Shaparin N, Suslov K, Ellis RJ, Ware MA, et al. Inhaled cannabis for chronic neuropathic pain: a meta-analysis of individual patient data. Journal of Pain 2015;16(12):1221-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Attal 2016
- Attal N, Andrade DC, Adam F, Ranoux D, Teixeira MJ, Galhardoni R, et al. Safety and efficacy of repeated injections of botulinum toxin A in peripheral neuropathic pain (BOTNEP): a randomised, double-blind, placebo-controlled trial. Lancet Neurology 2016;15:555-65. [DOI] [PubMed] [Google Scholar]
Baron 2004
- Baron R, Binder A. How neuropathic is sciatica? The mixed pain concept [Wie neuropathisch ist die lumboischialgie? Das gemischte schmerz konzept]. Orthopäde 2004;33:568-75. [DOI] [PubMed] [Google Scholar]
Baron 2012
- Baron R, Wasner G, Binder A. Chronic pain: genes, plasticity, and phenotypes. Lancet Neurology 2012;11(1):19-21. [DOI: 10.1016/S1474-4422(11)70281-] [DOI] [PubMed] [Google Scholar]
Baron 2017
- Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, et al. Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. Pain 2017;158:261-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Berger 2004
- Berger A, Dukes EM, Oster G. Clinical characteristics and economic costs of patients with painful neuropathic disorders. Journal of Pain 2004;5(3):143-9. [DOI: 10.1016/j.jpain.2003.12.004] [DOI] [PubMed] [Google Scholar]
Berger 2009
- Berger A, Toelle T, Sadosky A, Dukes E, Edelsberg J, Oster G. Clinical and economic characteristics of patients with painful neuropathic disorders in Germany. Pain Practice 2009;9(1):8-17. [DOI: 10.1111/j.1533-2500.2008.00244.x] [DOI] [PubMed] [Google Scholar]
Berger 2012
- Berger A, Sadosky A, Dukes E, Edelsberg J, Oster G. Clinical characteristics and patterns of healthcare utilization in patients with painful neuropathic disorders in UK general practice: a retrospective cohort study. BMC Neurology 2012;12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bouhassira 2008
- Bouhassira D, Lantéri-Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008;136:380-7. [DOI] [PubMed] [Google Scholar]
Boychuk 2015
- Boychuk DG, Goddard G, Mauro G, Orellana MF. The effectiveness of cannabinoids in the management of chronic nonmalignant neuropathic pain: a systematic review. Journal of Oral and Facial Pain and Headache 2015;29(1):7-14. [DOI] [PubMed] [Google Scholar]
Calvo 2012
- Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurology 2012;11(7):629-42. [DOI: 10.1016/S1474-4422(12)70134-5] [DOI] [PubMed] [Google Scholar]
Clauw 2015
- Clauw D. What is the meaning of "small fiber neuropathy" in fibromyalgia? Pain 2015;156(11):2115-6. [DOI] [PubMed] [Google Scholar]
Cochrane PaPaS 2012
- Cochrane Pain, Palliative and Supportive Care Group. PaPaS author and referee guidance. papas.cochrane.org/papas-documents (accessed 5 June 2015).
Cohen 1988
- Cohen J. Statistical Power Analysis for the Behavioral Sciences. Hillsdale: Lawrence Erlbaum Associates, 1988. [Google Scholar]
De Vries 2014
- De Vries M, Van Rijckevorsel DC, Wilder-Smith OH, Van Goor H. Dronabinol and chronic pain: importance of mechanistic considerations. Expert Opinion on Pharmacotherapy 2014;15:1525-34. [DOI] [PubMed] [Google Scholar]
Dechartes 2013
- Dechartres A, Trinquart L, Boutron I, Ravaud P. Influence of trial sample size on treatment effect estimates: meta-epidemiological study. BMJ 2013;346:f2304. [DOI: 10.1136/bmj.f2304] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dechartres 2014
- Dechartres A, Altman DG, Trinquart L, Boutron I, Ravaud P. Association between analytic strategy and estimates of treatment outcomes in meta-analyses. JAMA 2014;312:623-30. [DOI: 10.1001/jama.2014.8166] [DOI] [PubMed] [Google Scholar]
Demant 2014
- Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. Pain 2014;155(11):2263-73. [DOI: 10.1016/j.pain.2014.08.014] [DOI] [PubMed] [Google Scholar]
Derry 2012
- Derry S, Moore RA. Topical capsaicin (low concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 9. Art. No: CD010111. [DOI: 10.1002/14651858.CD010111] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2014
- Derry S, Wiffen PJ, Moore RA, Quinlan J. Topical lidocaine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2014, Issue 7. Art. No: CD010958. [DOI: 10.1002/14651858.CD010958.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2017
- Derry S, Rice ASC, Cole P, Tan T, Moore RA. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2017, Issue 1. Art. No: CD007393. [DOI: 10.1002/14651858.CD007393.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105-21. [DOI: 10.1016/j.jpain.2007.09.005] [DOI] [PubMed] [Google Scholar]
Dworkin 2013
- Dworkin RH, O'Connor AB, Kent J, Mackey SC, Raja SN, Stacey BR, et al. Interventional management of neuropathic pain: NeuPSIG recommendations. Pain 2013;154:2249-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140-9. [DOI] [PubMed] [Google Scholar]
European Medicines Agency 2007
- Guideline on clinical medicinal products intended for the treatment of neuropathic pain. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003478.pdf 2007 (Accessed May 2, 2015).
Fayers 2014
- Fayers PM, Hays RD. Don't middle your MIDs: regression to the mean shrinks estimates of minimally important differences. Quality of Life Research 2014;23:1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Finnerup 2013
- Finnerup NB, Scholz J, Attal N, Baron R, Haanpää M, Hansson P, et al. Neuropathic pain needs systematic classification. European Journal of Pain 2013;17(7):953-6. [DOI: 10.1002/j.1532-2149.2012.00282.x] [DOI] [PubMed] [Google Scholar]
Finnerup 2015
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurology 2015;14(2):162-73. [DOI: 10.1016/S1474-4422(14)70251-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fitzcharles 2014
- Fitzcharles MA, Clauw DJ, Ste-Marie PA, Shir Y. The dilemma of medical marijuana use by rheumatology patients. Arthritis Care and Research 2014;66:797-801. [DOI] [PubMed] [Google Scholar]
Furukawa 2005
- Furukawa TA, Cipriani A, Barbui C, Brambilla P, Watanabe N. Imputing response rates from means and standard deviations in meta-analyses. International Clinical Psychopharmacology 2005;20:49-52. [DOI] [PubMed] [Google Scholar]
Gaskell 2016
- Gaskell H, Derry S, Stannard C, Moore RA. Oxycodone for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 7. Art. No: CD010692. [DOI: 10.1002/14651858.CD010692.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015. Available at gradepro.org.
Guindon 2009
- Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS & Neurological Disorders Drug Targets 2009;8:403-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gustorff 2008
- Gustorff B, Dorner T, Likar R, Grisold W, Lawrence K, Schwarz F, et al. Prevalence of self-reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiologica Scandinavica 2008;52:132-6. [DOI] [PubMed] [Google Scholar]
Guyatt 2013a
- Guyatt G, Oxman AD, Sultan S, Brozek J, Glasziou P, Alonso-Coello P, et al. GRADE guidelines: 11. Making an overall rating of confidence in effect estimates for a single outcome and for all outcomes. Journal of Clinical Epidemiology 2013;66:151-7. [DOI] [PubMed] [Google Scholar]
Guyatt 2013b
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables-binary outcomes. Journal of Clinical Epidemiology 2013;66:158-72. [DOI] [PubMed] [Google Scholar]
Hall 2008
- Hall GC, Carroll D, McQuay HJ. Primary care incidence and treatment of four neuropathic pain conditions: a descriptive study, 2002-2005. BMC Family Practice 2008;9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Helfert 2015
- Helfert SM, Reimer M, Höper J, Baron R. Individualized pharmacological treatment of neuropathic pain. Clinical Pharmacology and Therapeutics 2015;97(2):135-42. [DOI: 10.1002/cpt.19] [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327:557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hillard 2012
- Hillard CJ, Weinlander KM, Stuhr KL. Contributions of endocannabinoid signaling to psychiatric disorders in humans: genetic and biochemical evidence. Neuroscience 2012;204:207-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hoggart 2015
- Hoggart B, Ratcliffe S, Ehler E, Simpson KH, Hovorka J, Lejčko J, et al. A multicentre, open-label, follow-on study to assess the long-term maintenance of effect, tolerance and safety of THC/CBD oromucosal spray in the management of neuropathic pain. Journal of Neurology 2015;262:27-40. [DOI] [PubMed] [Google Scholar]
Häuser 2012
- Häuser W, Bartram C, Bartram-Wunn E, Tölle T. Adverse events attributable to nocebo in randomized controlled drug trials in fibromyalgia syndrome and painful diabetic peripheral neuropathy: systematic review. Clinical Journal of Pain 2012;28:437-51. [DOI] [PubMed] [Google Scholar]
Häuser 2016
- Walitt B, Klose P, Fitzcharles MA, Häuser W. Cannabinoids for fibromyalgia. Cochrane Database of Systematic Reviews 2016, Issue 7. Art. No: CD011694. [DOI: 10.1002/14651858.CD011694] [DOI] [PMC free article] [PubMed] [Google Scholar]
Häuser 2017
- Häuser W, Fitzcharles MA, Radbruch L, Petzke F. Efficacy, tolerability and safety of cannabinoids in pain medicine and palliative care: an overview of systematic reviews and of prospective observational studies. Deutsches Ärzteblatt International 2017;114:627-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Häuser 2018
International Council for Harmonisation 2016
- International Council for Harmonisation. Medical Dictionary for Regulatory Activities Version 19.1. www.meddra.org/news-and-events/news/all-translations-meddra-version-191-are-now-available 2016.
Jensen 2011
Jensen 2015
- Jensen B, Chen J, Furnish T, Wallace M. Medical marijuana and chronic pain: a review of basic science and clinical evidence. Current Pain Headache Reports 2015;19:524. [DOI] [PubMed] [Google Scholar]
Kalant 2001
- Kalant H. Medicinal use of cannabis: history and current status. Pain Research & Management 2001;6:80-91. [DOI] [PubMed] [Google Scholar]
Kalso 2013
- Kalso E, Aldington DJ, Moore RA. Drugs for neuropathic pain. BMJ 2013;347:f7339. [DOI] [PubMed] [Google Scholar]
Katusic 1991
- Katusic S, Williams DB, Beard CM, Bergstralh EJ, Kurland LT. Epidemiology and clinical features of idiopathic trigeminal neuralgia and glossopharyngeal neuralgia: similarities and differences, Rochester, Minnesota,1945-1984. Neuroepidemiology 1991;10:276-81. [DOI] [PubMed] [Google Scholar]
Koopman 2009
- Koopman JS, Dieleman JP, Huygen FJ, De Mos M, Martin CG, Sturkenboom MC. Incidence of facial pain in the general population. Pain 2009;147:122-7. [DOI] [PubMed] [Google Scholar]
Koppel 2014
- Koppel BS, Brust JC, Fife T, Bronstein J, Youssof S, Gronseth G, et al. Systematic review: efficacy and safety of medical marijuana in selected neurologic disorders: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2014;82:1556-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Krcevski‐Skvarc 2018
L'Abbé 1987
- L'Abbé KA, Detsky AS, O'Rourke K. Meta-analysis in clinical research. Annals of Internal Medicine 1987;107:224-33. [DOI] [PubMed] [Google Scholar]
Lee 2013
- Lee MC, Ploner M, Wiech K, Bingel U, Wanigasekera V, Brooks J, et al. Amygdala activity contributes to the dissociative effect of cannabis on pain perception. Pain 2013;134:123-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Long 2009
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proceedings of the National Academy of Science of the United States of America 2009;106:20,270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lunn 2014
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database of Systematic Reviews 2014, Issue 1. Art. No: CD007115. [DOI: 10.1002/14651858.CD007115.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lynch 2011
- Lynch M, Campbell F. Cannabinoids for treatment of chronic non-cancer pain; a systematic review of randomized trials. British Journal of Clinical Pharmacology 2011;72:735-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
McQuay 1998
- McQuay H, Moore R. An Evidence-Based Resource for Pain Relief. Oxford: Oxford University Press, 1998. [ISBN: 0-19-263048-2] [Google Scholar]
McQuay 2007
- McQuay HJ, Smith LA, Moore RA. Chronic pain. In: In: Stevens A, Raftery J, Mant J, Simpson S, editor(s). Health Care Needs Assessment, 3rd Series. Radcliffe Publishing, 2007. [Google Scholar]
Mintzes 2015
- Mintzes B, Lexchin J, Quintano AS. Clinical trial transparency: many gains but access to evidence for new medicines remains imperfect. British Medical Bulletin 2015;116:43-53. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group (2009). Preferred reporting items for systematic reviews and meta-analyses: The PRISMA Statement. PLoS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramèr MR, Collins SL, McQuay HJ. Size is everything - large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209-16. [DOI: 10.1016/S0304-3959(98)00140-7] [DOI] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. In: McQuay HJ, Kalso E, Moore RA, editors(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle: IASP Press, 2008:15-24. [ISBN: 978-0-931092-69-5] [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. Art. No: CD007076. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010a
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, et al. "Evidence" in chronic pain - establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386-9. [DOI: 10.1016/j.pain.2010.05.011] [DOI] [PubMed] [Google Scholar]
Moore 2010b
- Moore RA, Straube S, Paine J, Phillips CJ, Derry S, McQuay HJ. Fibromyalgia: moderate and substantial pain intensity reduction predicts improvement in other outcomes and substantial quality of life gain. Pain 2010;149(2):360-4. [DOI: 10.1016/j.pain.2010.02.039] [DOI] [PubMed] [Google Scholar]
Moore 2010c
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta-analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374-9. [DOI: 10.1136/ard.2009.107805] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010d
- Moore RA, Smugar SS, Wang H, Peloso PM, Gammaitoni A. Numbers-needed-to-treat analyses - do timing, dropouts, and outcome matter? Pooled analysis of two randomized, placebo-controlled chronic low back pain trials. Pain 2010;151(3):592-7. [DOI: 10.1016/j.pain.2010.07.013] [DOI] [PubMed] [Google Scholar]
Moore 2011a
- Moore RA, Straube S, Paine J, Derry S, McQuay HJ. Minimum efficacy criteria for comparisons between treatments using individual patient meta-analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain 2011;152(5):982-9. [DOI] [PubMed] [Google Scholar]
Moore 2011b
- Moore RA, Mhuircheartaigh RJ, Derry S, McQuay HJ. Mean analgesic consumption is inappropriate for testing analgesic efficacy in post-operative pain: analysis and alternative suggestion. European Journal of Anaesthesiology 2011;28(6):427-32. [DOI: 10.1097/EJA.0b013e328343c569] [DOI] [PubMed] [Google Scholar]
Moore 2012
- Moore RA, Straube S, Eccleston C, Derry S, Aldington D, Wiffen P, et al. Estimate at your peril: imputation methods for patient withdrawal can bias efficacy outcomes in chronic pain trials using responder analyses. Pain 2012;153(2):265-8. [DOI: 10.1016/j.pain.2011.10.004] [DOI] [PubMed] [Google Scholar]
Moore 2013a
- Moore RA, Straube S, Aldington D. Pain measures and cut-offs - 'no worse than mild pain' as a simple, universal outcome. Anesthesia 2013;68:400-12. [DOI] [PubMed] [Google Scholar]
Moore 2013b
- Moore A, Derry S, Eccleston C, Kalso E. Expect analgesic failure; pursue analgesic success. BMJ 2013;346:f2690. [DOI: 10.1136/bmj.f2690] [DOI] [PubMed] [Google Scholar]
Moore 2014a
- Moore RA, Derry S, Taylor RS, Straube S, Phillips CJ. The costs and consequences of adequately managed chronic non-cancer pain and chronic neuropathic pain. Pain Practice 2014;14(1):79-94. [DOI: 10.1111/papr.12050] [DOI] [PubMed] [Google Scholar]
Moore 2014b
- Moore RA, Cai N, Skljarevski V, Tölle TR. Duloxetine use in chronic painful conditions - individual patient data responder analysis. European Journal of Pain 2014;18(1):67-75. [DOI: 10.1002/j.1532-2149.2013.00341.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2015a
- Moore RA, Chi CC, Wiffen PJ, Derry S, Rice ASC. Oral nonsteroidal anti-inflammatory drugs for neuropathic pain. Cochrane Database of Systematic Reviews 2015, Issue 10. Art. No: CD010902. [DOI: 10.1002/14651858.CD010902.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2015b
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2015, Issue 7. Art. No: CD008242. [DOI: 10.1002/14651858.CD008242.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2017
- Wiffen PJ, Derry S, Bell RF, Rice ASC, Tölle TR, Phillips T, et al. Gabapentin for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2017, Issue 6. Art. No: CD007938. [DOI: 10.1002/14651858.CD007938.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moulin 2014
- Moulin D, Boulanger A, Clark AJ, Clarke H, Dao T, Finley GA, et al. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Research & Management 2014;19:328-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2013
- National Institute for Health and Care Excellence (NICE). Neuropathic pain - pharmacological management: the pharmacological management of neuropathic pain in adults in non-specialist settings, 2013. www.nice.org.uk/guidance/cg173 (accessed 19 October 2014).
Nüesch 2010
- Nüesch E, Trelle S, Reichenbach S, Rutjes AW, Tschannen B, Altman DG, et al. Small study effects in meta-analyses of osteoarthritis trials: meta-epidemiological study. BMJ 2010;341:c3515. [DOI: 10.1136/bmj.c3515] [DOI] [PMC free article] [PubMed] [Google Scholar]
Owens 2015
- Owens B. Drug development: the treasure chest. Nature 2015;525:S6-S8. [DOI] [PubMed] [Google Scholar]
Petzke 2016
- Petzke F, Enax-Krumova EK, Häuser W. Efficacy, tolerability and safety of cannabinoids for chronic neuropathic pain: a systematic review of randomized controlled studies. Schmerz 2016;30:62-88. [DOI] [PubMed] [Google Scholar]
Rappaport 1994
- Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self-sustaining discharge in the trigeminal ganglion. Pain 1994;56:127-38. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Schaefert 2015
- Schaefert R, Welsch P, Klose P, Sommer C, Petzke F, Häuser W. Opioids in chronic osteoarthritis pain. A systematic review and meta-analysis of efficacy, tolerability and safety in randomized placebo-controlled studies of at least 4 weeks duration [Opioide bei chronischem arthroseschmerz. Systematische übersicht und metaanalyse der wirksamkeit, verträglichkeit und sicherheit in randomisierten, placebokontrollierten studien über mindestens 4 wochen]. Schmerz 2015;29:47-59. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Scott 2006
- Scott FT, Johnson RW, Leedham-Green M, Davies E, Edmunds WJ, Breuer J. The burden of herpes zoster: a prospective population based study. Vaccine 2006;24(9):1308-14. [DOI: 10.1016/j.vaccine.2005.09.026] [DOI] [PubMed] [Google Scholar]
Smith 2016
- Smith SM, Amtmann D, Askew RL, Gewandter JS, Hunsinger M, Jensen MP, et al. Pain intensity rating training: results from an exploratory study of the ACTTION PROTECCT system. Pain 2016;157(5):1056-64. [DOI] [PubMed] [Google Scholar]
Sommer 2015
- Sommer C, Welsch P, Klose P, Schaefert R, Petzke F, Häuser W. Opioids in chronic neuropathic pain. A systematic review and meta-analysis of efficacy, tolerability and safety in randomized placebo-controlled studies of at least 4 weeks duration [Opioide bei chronischem neuropathischem schmerz. Systematische übersicht und metaanalyse der wirksamkeit, verträglichkeit und sicherheit in randomisierten, placebokontrollierten studien über mindestens 4 wochen]. Schmerz 2015;29:35-46. [DOI] [PubMed] [Google Scholar]
Stannard 2016
- Stannard C, Gaskell H, Derry S, Aldington D, Cole P, Cooper TE, et al. Hydromorphone for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 5. Art. No: CD011604. [DOI: 10.1002/14651858.CD011604.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2008
- Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrolment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. British Journal of Clinical Pharmacology 2008;66(2):266-75. [DOI: 10.1111/j.1365-2125.2008.03200.] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2010
- Straube S, Derry S, Moore RA, Paine J, McQuay HJ. Pregabalin in fibromyalgia - responder analysis from individual patient data. BMC Musculoskeletal Disorders 2010;11:150. [DOI: 10.1186/1471-2474-11-150] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sultan 2008
- Sultan A, Gaskell H, Derry S, Moore RA. Duloxetine for painful diabetic neuropathy and fibromyalgia pain: systematic review of randomised trials. BMC Neurology 2008;8:29. [DOI: 10.1186/1471-2377-8-] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Imberger G, Walsh M, Chu R, Gluud C, Wetterslev J, et al. The number of patients and events required to limit the risk of overestimation of intervention effects in meta-analysis--a simulation study. PLOS One 2011;6(10):e25491. [DOI: 10.1371/journal.pone.0025491] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thyson 2014
- Tyson SF, Brown P. How to measure pain in neurological conditions? A systematic review of psychometric properties and clinical utility of measurement tools. Clinical Rehabilitation 2014;28:669-86. [DOI] [PubMed] [Google Scholar]
Torrance 2006
- Torrance N, Smith BH, Bennett MI, Lee AJ. The epidemiology of chronic pain of predominantly neuropathic origin. Results from a general population survey. Journal of Pain 2006;7:281-9. [DOI] [PubMed] [Google Scholar]
Treede 2008
- Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 2008;70(18):1630-5. [DOI: 10.1212/01.wnl.0000282763.29778.59] [DOI] [PubMed] [Google Scholar]
Turner 2013
- Turner RM, Bird SM, Higgins JP. The impact of study size on meta-analyses: examination of underpowered studies in Cochrane Reviews. PLOS One 2013;8(3):e59202. [DOI: 10.1371/journal.pone.0059202] [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Hecke 2014
- Van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 2014;155:654-62. [DOI] [PubMed] [Google Scholar]
Van Hoek 2009
- Van Hoek AJ, Gay N, Melegaro A, Opstelten W, Edmunds WJ. Estimating the cost-effectiveness of vaccination against herpes zoster in England and Wales. Vaccine 2009;27(9):1454-67. [DOI: 10.1016/j.vaccine.2008.12.024] [DOI] [PubMed] [Google Scholar]
Volkow 2014
- Volkow ND, Baler RD, Compton WM, Weiss SR. Adverse health effects of marijuana use. New England Journal of Medicine 2014;370:2219-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
von Hehn 2012
- Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012;73(4):638-52. [DOI: 10.1016/j.neuron.2012.02.008] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vos 2012
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2163-96. [DOI: 10.1016/S0140-6736(12)61729-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ware 2015
- Ware MA, Wang T, Shapiro S, Collet JP, COMPASS study team. Cannabis for the management of pain: assessment of safety study (COMPASS). Journal of Pain 2015;16:1233-42. [DOI] [PubMed] [Google Scholar]
Whiting 2015
- Whiting PF, Wolff RF, Deshpande S, Di Nisio M, Duffy S. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA 2015;313:2456-73. [DOI] [PubMed] [Google Scholar]
Wiffen 2013
- Wiffen PJ, Derry S, Moore RA, Aldington D, Cole P, Rice ASC, et al. Antiepileptic drugs for neuropathic pain and fibromyalgia - an overview of Cochrane reviews. Cochrane Database of Systematic Reviews 2013, Issue 11. Art. No: CD010567. [DOI: 10.1002/14651858.CD010567.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zettl 2016
- Zettl UK, Rommer P, Hipp P, Patejdl R. Evidence for the efficacy and effectiveness of THC-CBD oromucosal spray in symptom management of patients with spasticity due to multiple sclerosis. Therapeutic Advances in Neurological Disorders 2016;9:9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2015
- Zhang J, Echeverry S, Lim TK, Lee SH, Shi XQ, Huang H. Can modulating inflammatory response be a good strategy to treat neuropathic pain? Current Pharmaceutical Design 2015;21:831-9. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Mücke 2016
- Mücke M, Phillips T, Radbruch L, Petzke F, Häuser W. Cannabinoids for chronic neuropathic pain. Cochrane Database of Systematic Reviews 2016, Issue 5. Art. No: CD012182. [DOI: 10.1002/14651858.CD012182] [DOI] [PMC free article] [PubMed] [Google Scholar]