

Abstract
Influenza A annually infects 5–10% of the world’s human population resulting in one million deaths. Influenza causes annual epidemics and re-infects previously exposed individuals because of antigenic drift in the glycoprotein hemagglutinin. Due to antigenic drift, the immune system is simultaneously exposed to novel and conserved parts of the influenza virus via vaccination and/or infection throughout life. Preexisting immunity has long been known to augment subsequent hemagglutination inhibitory antibody (hAb) responses. However, the preexisting immunological contributors that influence hAb responses are not understood. Therefore, we adapted and developed sequential infection and immunization mouse models using drifted influenza strains to show that MHC Class II haplotype and T cell reactivity influence subsequent hAb responses. We found that CB6F1 mice infected with A/CA followed by immunization with A/PR8 have increased hAb responses to A/PR8 compared to C57BL/6 mice. Increased hAb responses in CB6F1 mice were CD4+ T cell and B cell dependent and corresponded to increased germinal center A/PR8-specific B and T follicular helper cells. These results suggest that conserved MHC Class II restricted epitopes within HA are essential for B cells to respond to drifting influenza and could be leveraged to boost hAb responses.
Keywords: Influenza, vaccinology, infectious diseases, preexisting immunity, immunological memory
Graphical Abstarct
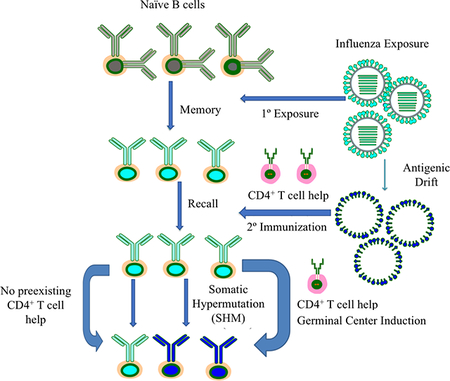
Reengagement of both memory B and CD4+ T cells from previous influenza exposure are necessary for updated and enhanced antibody responses to drifted HA immunization. Without HA-specific cognate CD4+ T cell help, established by initial influenza exposure, secondary B cell responses are greatly reduced.
INTRODUCTION
Influenza A virus is a zoonotic pathogen that continuously circulates between hosts, including humans, birds, horses, dogs and pigs (1, 2). The virus has a segmented, negative-sense, single-stranded RNA genome that encodes 12 proteins. Typically, influenza A viruses are subtyped based on their surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA), which are responsible for viral entry and release, respectively (3, 4). Currently, 18 HA and 10 NA subtypes circulate in birds, with two viral subtypes concurrently circulating in humans. Immune pressure from the infected host directs influenza HA and NA genes to undergo antigenic drift, characterized by non-synonymous point mutations. Importantly, the best correlate for protection in both pre-clinical models and clinical trials is the induction of hemagglutination inhibitory antibodies (hAbs) to non-conserved areas of HA (5–9). Accumulated mutations in these regions result in antigenically-distinct HAs, allowing drifted viruses to re-infect individuals that have previously been exposed. Despite the practice of annual immunization to elicit hAbs directed to the current circulating influenza, the vaccine is only 59% effective in healthy adults during well-matched years, presumably due to antigenic drift as well as diverse host factors including preexisting influenza exposure and age (10). Additionally, healthy adults who receive the trivalent inactivated vaccine (TIV) seroconvert an estimated 50–80% of the time based on immunogenicity studies (11–14). High seroconversion directly predicts protection, but the mechanism(s) of how preexisting immunity affects vaccine immunogenicity is not fully understood (15, 16). This limited understanding is compounded by diverse host histories and genetics, making it difficult to optimally elicit these classical hAbs across many populations (17–20).
Importantly, exposure to a drifted HA preferentially stimulates an individual’s memory B cell response to recurring epitopes at the expense of de novo responses to new epitopes. Recent technological advances in B cell sequencing demonstrates that reactivation of memory B cell responses to drifted influenza strains result in either: 1) memory B cells that undergo somatic hypermutation (SHM) and become more adapted to the drifted influenza HA antigen, or 2) the memory B cell repertoire does not adapt, and instead directly differentiates into a plasmablast to secrete antibodies that recognize the undrifted epitopes of previous strains (21). This latter phenomenon is termed original antigenic sin (OAS) and is phenotypically characterized by diminished antibody responses to novel parts of the drifted strain upon exposure to that strain and increased antibody responses to the initial pre-exposure strain (22–29). It is not clear what intrinsic or extrinsic factors influence whether memory B cells adapt to a drifted strain. One hypothesis is that the HA-specific memory B cells’ ability to present conserved HA-based cognate antigens via MHC Class II to preexisting HA-reactive memory CD4+ T cells initiates further adaptation by SHM of memory B cells to the drifted strain (12, 30–32). It has been shown that HA-specific B cells are solely helped by HA-reactive CD4+ T cells in the mouse model of influenza (33). Therefore, we postulate that if drifted HAs contain conserved MHC II epitopes, preexisting memory CD4+ T cells can help B cells adapt to the newer influenza strain.
RESULTS
hAb responses to drifted recombinant HA is dependent upon mouse MHC Class II haplotype
In order to assess how preexisting immunity augments subsequent hAb responses to drifted influenza immunization, we established mouse models by first exposing C57BL/6 and CB6F1 mice to 0.5 LD50 (Lethal Dose 50%) A/CA/7/09 (A/CA), the prototype H1N1 virus from the 2009 pandemic, allowing the mice to undergo infection, seroconversion, and recuperation for 28 days and then re-exposing mice to 1 μg of the drifted strain A/PR8 HA by intramuscular immunization (i.m.). Mice were weighed daily and bled at day 21 post-primary and secondary immunization to measure both hAb titer by HAI assay and HA titer by enzyme-linked immunosorbent assay (ELISA).
In our models, C57BL/6 mice preexposed to A/CA/7/09 (A/CA), display little reactivity by HAI and ELISA to A/PR/8/34 (A/PR8) 21 days after A/PR8 immunization when compared to A/CA preexposed CB6F1 mice which display 32-fold increase in HAI and ELISA responses to the drifted A/PR8 HA immunization (Figure 1A-D). Importantly, both mouse models display HA binding cross-reactivity by ELISA to A/PR8 post primary infection with A/CA but do not display significant cross-reactive HAI titers, suggesting that they do not share neutralizing epitopes. To further test if the increased hAb titer to A/PR8 in CB6F1 mice resulted in enhanced protection, we then challenged all groups with 1000 LD50 of A/PR8. Unsurprisingly, protection from weight loss and lethality directly corresponded to hAb A/PR8 titer. Only the CB6F1 mice with high A/PR8 HAI titers demonstrated 100% protection from weight loss and lethality (Figure 1E-F). This finding that A/CA preexposed F1 but not B6 mice display enhanced antibody responses to immunization with a drifted HA, A/PR8, led us to hypothesize this discrepancy was possibly due to mouse genetic background.
Figure 1: Pre-exposed CB6F1 but not C57BL/6 mice have increased antibody responses to drifted influenza immunization.

C57BL/6 (n = 4 per group) and CB6F1 mice were intranasally exposed to A/CA (500 pfu/mouse) or PBS then 28 days later immunized with A/PR8 rHA (1 μg/mouse). Mice were bled at 21 days post primary and secondary immunization to determine A/CA and A/PR8 HAI and ELISA titers. Mice were then challenged with 1000 LD50 A/PR8 to determine protection capability of the secondary response. A/PR8 HAI titer in C57BL/6 mice is shown in panel (A), A/PR8 endpoint titer± SD (Log10) in C57BL/6 mice in panel (B), A/PR8 HAI titer in CB6F1 mice is shown in panel (C), A/PR8 endpoint titer± SD (Log10) in CB6F1 mice in panel (D), at both day 21 post primary and secondary influenza immunization. (*p<0.05, ****p<0.0001, One-way ANOVA). Weight loss of living mice in panel (E) and percent survival in panel (F) up to 10 days post 1000 LD50 A/PR8 challenge. Data are representative of three independent experiments (n ≥ 4 animals).
The increased hAb response to drifted influenza virus immunization has been recently reported in Balb/c mice, however the immunological basis for how prior exposure enhances secondary responses is unknown (34). We reasoned that because CB6F1 mice have an increased diversity of MHC class II genes (I-Ab, I-A d, I-Ed) compared to C57BL/6 mice (I- Ab), the differential secondary response to immunization with intra-subtype HA may be linked to MHC Class II reactive epitopes conserved across the drifted HAs. To that end, we established CD4+ T cell reactivity to A/CA peptide pools in both CB6F1 and C57BL/6 mice. Interestingly, A/CA exposed C57BL/6 mice display no significant CD4+ T cell reactivity with A/CA peptide pools as measured by IFN-γ production using flow cytometry (Supplementary Figure 1A), suggesting the absence of strong MHC Class II reactive epitopes in C57BL/6 mice (Figure 2A). This absence of robust HA-specific CD4+ T cell reactivity to A/CA HA in C57BL/6 mice was further confirmed by IFN-γ ELISPOT and HA reactivity to A/CA rHA in CB6F1 mice has been previously established (Supplementary Figure 1B) (35). Conversely, CB6F1 mice infected with either A/CA or A/PR8 demonstrated CD4+ T cell reactivity with A/CA peptides (Figure 2A). To further confirm that other genetic differences between the two mouse strains were not playing a significant role we repeated the challenge-immunization experiment with B10.D2 mice, which have C57BL/6 background but an I-Ad I-Ed MHC Class II haplotype, sharing an MHC Class II allele with CB6F1 mice but not C57BL/6 mice. Similar to the CB6F1 mice, B10.D2 mice displayed an enhanced hAb response to A/PR8 as compared to previously uninfected mice (Figure 2B). These results strongly suggest that the HAs of A/PR8 and A/CA share a common I-E/Ad, restricted epitope, but no shared I-Ab epitopes. Taken together, these two contrasting mouse models, CB6F1 and C57BL/6, suggest that MHC Class II conservation between HAs is critical for optimal hAb responses to immunization with a drifted intra-subtype strain.
Figure 2: Enhanced hAb responses correlated with CD4+ T cell cross-reactivity.

C57BL/6 (n = 5 per group) and CB6F1 mice were intranasally infected with either A/PR8 or A/CA. Day 14 post infection spleens were harvested and individually stimulated with pooled peptides (1–10) of A/CA HA. Cells were then stained for analysis by flow cytometry and CD4+CD44hiIFNγ+ cells frequency of parent percentage (FoP %) were quantified with in panel (A). Error bars indicate ± SD and data show results from one of two experiments with similar results (n = 5 animals per experiment). (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, Two-way ANOVA). Flow cytometry gating strategy and representative flow plots are shown in Supplementary Figure 1. B10.D2 (n = 10 per group) mice were infected with A/CA or mock infected with PBS then 28 days later immunized with A/PR8 HA (1 μg). HAI titer to A/PR8 in panel (B) were evaluated in serum samples 21 days post drifted A/PR8 rHA i.m. immunization. (**p<0.01, One-way ANOVA). Data show results from one of two independent experiments with similar results (n = 10 animals per experiment).
Increased A/PR8-specific GC B cells and T follicular helper cells in previously exposed CB6F1 mice
Next, we investigated the impact of previous influenza exposure on the cellular composition of the draining lymph node following secondary exposure. We infected CB6F1 mice with A/CA or PBS, waited 28 days and subsequently immunized groups with drifted rHA A/PR8 (i.m.) or as a negative control a distinct shifted H3 rHA, X-31. At day 2 and 4 post immunization, we examined the immune populations comprising the draining inguinal lymph nodes by flow cytometry. At day 4, we saw an increase in germinal center (GC) B cells (B220+GL7+CD38lo) in previously exposed mice but not unexposed mice (Figure 3A and representative flow plots in Supplementary Figure 2A). To stain for A/PR8 specific B cells we attached a PE-avidin tetramer to a biotinylated dominant linear B cell epitope on A/PR8 HA that mapped to the receptor binding site (RBS) (36, 37). Using this staining tool, we were able to observe GC cells also became antigen-specific to A/PR8 (Figure 3B and representative flow plots in Supplementary Figure 2A). Further, as shown in representative flow plots (Supplementary Figure 2B) the GC cells in the previously exposed group upregulated the SMH initiation enzyme, activation-induced cytidine deaminase (AID) (Figure 3C). Last, there was a concurrent upregulation of T follicular helper (Tfh) cells (CD4+CD154hiCXCR5+PD1+) in previously exposed mice but not unexposed mice (Figure 3D and representative flow plots in Supplementary Figure 2C). While this suggests that memory B and CD4+ T cells from previous exposure enable adaptation to drifted HA, as previously unexposed mice do not exhibit enhanced Ab responses, it does not prove their necessity for this enhanced secondary response.
Figure 3: Increased Germinal Center A/PR8-specific B and T follicular helper cells in response to drifted immunization in previously exposed mice but not unexposed mice.

CB6F1 mice (n = 5 per group total, combined from two experiments with n ≥ 2 per group) were i.n. pre-exposed to A/CA or PBS then 28 days later immunized with rHA A/PR8 or rHA X-31, as a negative control. Day 4 post immunization inguinal lymph nodes were used for flow cytometry quantification of GC B cells (B220+GL7+CD38lo) in panel (A). PR8-specific GC cells in inguinal LNs (B220+CD95+GL7+CD38lo PR8-Tet+ CD138lo) in panel (B). Expression of activation-induced cytidine deaminase (AID) in GC cells determined by mean florescence intensity (MFI) shown in panel (C) and frequencies of Tfh cells (CD4+ CD154+ CD44+CXCR5hiPD1hi) in panel (D). (*p<0.05, **p<0.01, ****p<0.0001, One-way ANOVA). Representative flow cytometry plots and gating strategy are shown in Supplementary Figure 2.
CD4+ T cell are necessary for enhanced secondary responses to drifted immunization
We employed the CB6F1 mouse model to evaluate the requirement of memory CD4+ T cells for enhancement of antibody responses following boost with drifted HA. First, we depleted CD4+ T cells after contraction at day 27 and 28 post primary infection, confirmed depletion and then waited 36 days for reconstitution (day 65 post infection) of the naïve CD4+ T cell compartment prior to immunization with A/PR8 (Supplementary Figure 3). CD4+ T cell depletion was found to be accomplished effectively although incompletely, even in tissues, including lung, axial and brachial lymph nodes and spleen (Supplementary Figure 3C-E). Strikingly, CD4+ T cell depletion diminished the hAb response to drifted HA immunization as compared to the isotype control treated group, demonstrating the requirement for cross-reactive memory CD4+ T cells in mounting a hAb immune response to a drifted A/PR8 HA immunization (Figure 4A and B). These results strongly suggest that cross-reactive memory CD4+ T cells are crucial and necessary for enhancing responses to drifted influenza HA.
Figure 4: Increased hAb response to immunization in CB6F1 mice was dependent on memory CD4+ T cells.

CB6F1 mice (n = 5 per group per experiment) were intranasally exposed to A/CA (500 pfu/mouse) or PBS and then depleted of CD4+ T cells in between primary and secondary immunization using 500 μg Ab GK 1.5 given i.p., then allowed to reconstitute their naïve CD4+ T cell population prior to secondary i.m. exposure to rHA PR8 (1 μg/mouse) as shown in Supplementary Figure 3. Day 21 post rHA PR8 immunization A/PR8 HAI titer in panel (A), PR8 HA endpoint titer ± SD (Log10) in panel (B). (*p<0.05, One-way ANOVA). Data shown are representative of two independent experiments (n = 5 mice per experiment).
Recombinant HA immunization with adjuvant can stimulate memory CD4+ T cells for enhanced recall hAb responses
Adjuvants, including a fully synthetic lipid-A TLR4 agonist in a stable oil-in-water emulsion, SLA-SE, strongly enhance memory CD4+ T cell reactivity and induce Tfh cellsin the context of protein immunization (39). Therefore, we next determined if enhancement of hAb responses to immunization was dependent upon infection for inducing CD4+ T cells capable of aiding subsequent responses or if rHA protein in combination with adjuvant was capable of producing the immune memory necessary. Similar to previously published data, we found that primary exposure to rHA A/CA alone was unable to provide enhancement to secondary responses (Supplementary Figure 4) (34). Conversely, adjuvanted rHA A/CA immunized mice did display enhanced hAb responses to subsequent immunization (Supplementary Figure 4). To assess further the role of CD4+ T cells in this response, we again depleted CD4+ T cells as outlined above. The enhanced antibody response to secondary immunization was indeed dependent on memory CD4+ T cells as mice in the depleted group demonstrated decreased Ab reactivity to immunization with rHA PR8(Supplementary Figure 4). These results demonstrated that primary exposure with infection or adjuvanted protein immunization significantly affects subsequent responses due to CD4+ T cell reactivity conservation.
Memory B cells are partially necessary for increased magnitude of hAb response in CB6F1 mice
To determine if B cells are necessary for enhanced hAb responses to secondary drifted immunization in CB6F1 mice, we depleted B cells with an anti-CD20 antibody, confirmed depletion both in tissues (Lung, lymph nodes and spleen) and blood and then allowed reconstitution of the naïve B cell compartment before immunization with rHA immunization (Supplementary Figure 5). Anti-CD20 antibody eliminates all B cell subsets in circulation except for plasma cells. CB6F1 mice depleted of memory B cells demonstrated lower A/PR8 HAI and endpoint titers compared to mock depleted mice, albeit less impactful than CD4+ T cell depletion (Figure 5). Taken together with the depletion effects of memory CD4+ T cell depletion, this suggested that both memory B cell and CD4+ T cells are necessary for full enhancement of subsequent antibody immunization responses.
Figure 5: Increased hAb response to immunization in CB6F1 mice was dependent on memory B cells.

CB6F1 mice (n = 4 per experiment) were depleted of B cells in between primary infection and secondary immunization using 500 μg anti-CD20 Ab given i.p., then allowed to reconstitute their naïve B cell population as outlined in Supplementary Figure 5. Day 21 post A/PR8 HA immunization A/PR8 HAI titer in panel (A) and endpoint titer ± SD (Log10) in panel (B). (*p<0.05, **p<0.01, One-way ANOVA). Data show results from one of two experiments with similar results (n = 4 animals per experiment).
Enhanced hAb responses can be elicited in C57BL/6 mice by conserving MHC Class II epitopes in HA
To investigate if conserved CD4+ T cell epitopes could enhance hAb responses in C57BL/6 mice we leveraged conserved MHC Class II epitopes within HA for both exposures, only changing the immunodominant B cell epitope. To do this, we replaced the A/PR8 HA RBS, a mapped immunodominant neutralizing B cell epitope, with the corresponding A/CA HA RBS, and subsequently rescued virus and confirmed the amino acid changes via sequencing (CA/PR8Δ). Using our C57BL/6 mouse model, we then exposed mice to PBS, A/CA or our engineered CA/PR8Δ virus, 28 days later we then immunized with A/PR8 i.m. and evaluated the antibody responses induced. Mice exposed to the CA/PR8Δ virus displayed increased germinal centers, compared to controls or A/CA infected mice at 4 days post-secondary immunization (Figure 6A). Likewise, these mice had increased titers, both by HAI and ELISA to A/PR8 upon secondary immunization (Figure 6B). Further, C57BL/6 mice pre-exposed to CA/PR8Δ and subsequently immunized with A/PR8 HA showed complete lethal and weight loss protection from challenge with 1000 LD50 (Figure 6D-E).
Figure 6: CA/PR8Δ exposed C57BL/6 mice have greater GC cell induction upon A/PR8 HA immunization in draining LN (inguinal) and increased hAb responses to drifted influenza immunization.

C57BL/6 mice (data are representative of two experiments with n = 3 per experiment) were pre-exposed to A/CA, CA/PR8Δ, or PBS then 28 days later i.m. immunized with A/PR8 rHA (1 μg). Day 4 post immunization inguinal LNs were harvested for analysis of percentage of GC cells (B220+GL7+CD38lo) shown in panel (A). A/PR8 HAI in panel (B) and HA endpoint titer ± SD (Log10) in panel (C) of C57BL/6 mice ≥ 5 (data are representative of two experiments with n ≥ 5 per experiment) day 21 post primary infection and drifted immunization with A/PR8 rHA (1μg). Challenge weight loss in panel (D) and survival from 1000 LD50 A/PR8 challenge in panel (E). (*p<0.05, ****p<0.0001, One-way ANOVA).
DISCUSSION
To better understand immunological responses to influenza, we have established that memory CD4+ T cell reactivity to HA is a prerequisite for immune adaptation to drifted HAs. Interestingly, we found that mice with CD4+ T cell reactivity to A/CA HA that cross-reacted with A/PR8 HA showed increased hAb responses to a drifted rHA upon immunization, whereas mice without this conserved reactivity did not show enhanced Ab responses. This CD4+ T cell reactivity was confirmed by mapping CD4+ T cell epitopes in HA; CB6F1 mice displayed CD4+ T cell reactivity to conserved HA epitopes in CA and PR8 HAs whereas C57BL/6 mice displayed no reactivity to epitopes of A/CA HA. These findings intimate that if cognate memory T cells are not available, B cell reactivity to drifted influenza will not robustly respond. These two divergent mouse models would be useful in explicitly defining and confirming this competition between memory and naïve B cells in both the absence and presence of cognate memory CD4+ T cell help.
The CD4+ T cell subset that has the potential to influence antibody responses by interacting with B cells has been identified as Tfh cells. Programming and activation of Tfh cells is dependent on antigen presentation by B cells (6, 42, 43). Further, memory B cells rapidly reactivate cognate memory Tfh cells providing accelerated antibody responses by the presenting memory B cell and directing reentry into GCs upon secondary immunization (44). However, it was unknown if memory B cells are necessary for the reactivation of memory Tfh cells upon drifted immunization with HA or if naïve B cells could also be directed to provide an enhanced hAb response. We found using our CB6F1 mouse model that memory B cells and CD4+ T cells are necessary for the magnitude of antibody responses observed, however memory CD4+ T cells are by far the larger contributor. Additionally, in the context of protein immunization priming, adjuvants have been shown to elicit Tfh induction and robust memory CD4+ T cells compared to protein immunization alone (39, 45, 46). Using our CB6F1 mouse model we found that adjuvant inclusion during initial protein immunization could induce memory CD4+ T cells that are capable of subsequently aiding heterologous hAb responses, as we have found with infection-induced priming, demonstrating the ability to establish memory CD4+ T cell help for subsequent responses. Further, we have shown here that the phenotype observed in C57BL/6 mice is reversible with preservation of conserved MHC Class II epitopes, demonstrating that B and T cell synergism is crucial for adaptation of B cells in the face of drifting neutralizing epitopes.
These findings underscore the importance and complexity of immunological memory’s impact on subsequent responses when B and T cell reactivity is changing, as is the case with drifted influenza. Therefore, it is crucial to understand the mechanism by which preexisting immune responses shape future responses in order to optimize responses to vaccination using antigen design. Likewise, this memory B and T cell interaction has potential implications in altering Dengue and HIV responses, viruses that frequently have differing B cell and T cell reactivity and where antibody responses are critical for viral control (47–49). Additionally, this knowledge could be applied to enhance immunity to be more potent against differing strains or alternatively, employed to prevent adaptation of B cells, retaining broader responses, a goal of many universal vaccine candidates. Indeed, application of this knowledge is currently being pursued primarily by including adjuvants in avian influenza vaccine candidates to strongly elicit CD4+ T cell reactivity (50). Additionally, new influenza vaccine candidates are endeavoring to engineer universal T cell epitopes into Avian H7 HA (51–53). Undeniably, these techniques of T cell inclusion are challenging, as HA folding is sensitive to modification and structural integrity must be maintained to provide adequate B cell stimulation. Furthermore, in humans with many different MHC Class II haplotypes, finding a suitable T cell epitope poses further challenges (54). Theoretically, if inclusion of a universal T cell epitope did not interfere with the integrity of HA, this could be used with adjuvant prior to any influenza exposure to elicit foundational responses. These foundational responses could then be exploited to boost out any subsequent responses that contained the universal T cell epitope, eliciting a more robust and specific response toward the antigen of interest.
MATERIALS AND METHODS
Ethics
All animal experiments and protocols were approved by IDRI’s Institutional Animal Care and Use Committee (IACUC) protocol 2015–16. IDRI’s IACUC (A4337–01) animal welfare assurance is in accordance with the Public Health Service (PHS) Policy for Humane Care and Use of Laboratory Animals.
Mice
Female C57BL/6, CB6F1 and B10.D2 mice, 6–8 weeks of age were purchased from Jackson laboratory. All animals were housed in the IDRI Vivarium (Seattle, WA) under specific pathogen-free conditions.
Immunization and influenza virus infection of mice
Mice were immunized by intramuscular (i.m.) injection with 1 μg recombinant HA (rHA) from A/PR/8/34 or A/CA/07/09 or X-31(Protein Sciences Corporation), formulated with and without the adjuvant, SLA-SE (5 μg) or saline.
Infection Studies.
Mice were infected intranasally with 0.5 LD50 or 1000 LD50 dose of A/CA/07/09 and A/PR/8/34, in 25uL of phosphate-buffered saline (PBS). Mice were monitored for weight loss and other signs of virus induced morbidity daily and sacrificed if weight loss exceeded 20% of initial body weight.
CA/PR8Δ virus was made by replacing the RBS site (Sa/Sb region:WLTEKEGSYP) of the HA PR8 vaccine plasmid with corresponding RBS CA site (WLVKKGNSYP) via Q5 mutagenesis. CA/PR8Δ virus and was rescued using the vaccine plasmid system previously described (55–57). Virus had normal growth kinetics and mice were given 15 plaque forming units (pfu)/mouse to induce productive infection as observed by seroconversion and weight loss.
CD4+ and B cell depletion
InVivo Mab anti-mouse CD4+ (clone GK1.5) and isotype controls were purchased from BioXCell (West Lebanon, NH). Mice were intraperitoneally (i.p.) administered at a dose of 250 μg per mouse twice for depletion of CD4+ Cells. CD4+ T cell depletion was assessed by flow cytometry with anti-CD4 (clone RM4–5). Mouse Anti-CD20 antibody (clone 5D2, murine IgG2a) was graciously provided by Genentech. Mice were given two i.p. injections of 250 μg of CD20 antibody on consecutive days. Depletion was assessed by flow cytometry with an anti-B220 and anti-CD19 antibodies.
CD4+ T Cell ELISPOT
IFN-γ (R&D Systems, Minneapolis, MN, USA) ELISPOT analyses were conducted according to the manufacturer’s instructions. Spot images were collected using ImmunoCapture 6.4 and analyzed with ImmunoSpot 5.0 on an automated ELISPOT plate reader (C.T.L. Seri3A Analyzer; Cellular Technology, Shaker Heights, OH, USA).
Enzyme Linked Immunosorbent Assays (ELISA) Assays
Mouse sera were collected from individual mice and PR8 HA and CA HA reactive antibodies were determined by an enzyme-linked immunosorbent assay (ELISA) using rHA purchased from Protein Sciences Corporation as a coating antigen. Polysterene 96 well flat bottom immuno plates (NUNC) were coated overnight at 4°C with 0.1 μg rHA per well. Wells were washed three times with PBS- 0.5% Tween 20. Blocking buffer (1% bovine serum albumin (BSA) in PBS-Tween) was added to every well and incubated for 1h at RT. Wells were again washed and serial 2-fold sample dilutions were added to the plates in 0.5% BSA PBS and incubated at RT for 1 hr. Wells were again washed five times and incubated for 1 hr at RT with 100 μL/well horseradish peroxidase-conjugated goat anti-mouse secondary antibody specific for IgG (Southern Biotech) diluted in 1% BSA-PBS at a 1/4000 dilution. Subsequently, wells were washed five times and developed with 100 μL/well tetramethylbenzidine (TMB). Stop solution, H2S04 (1N), was then added at 100 μL/well to stop the reaction. The optical density was read at 450nm (OD450) using a Biotek Synergy 2.
Hemagglutination Inhibition Assays
Hemagglutinination Inhibition (HAI) activity specific to A/PR/8/34, A/CA/07/09 and A/X31 was performed as previously described in using 1% Turkey Red Blood Cells (TRBCs) (58). Briefly, each serum sample was treated with receptor-destroying enzyme (RDE) overnight at 37°C followed by heat inactivation to remove nonspecific inhibitors. HI titer was determined by the reciprocal of the highest dilution of sera that completely inhibited the agglutination of turkey RBCs following addition of 4 HAU (hemgglutination units) of virus, starting at a 1:10 dilution and serially diluting 2-fold down a 96 v-bottom plate.
B and T cell staining and stimulation
Antigen specific CD4+ T cell responses were measured by peptide and rHA stimulation and flow cytometry. Briefly, splenocytes or cells isolated from lymph nodes were isolated and cultured (2×106 cells per well) for 8hr with peptide or rHA (1 μg/well) in the presence of Brefeldin A (BD Biosciences). After stimulation, cells were permeabilized and stained with fluorochrome conjugated antibodies CD4 (clone RM4–5), CD8 (clone 53–6. 7), CD44 (clone IM7) and B220 (RA3–6B2) (BioLegend and eBioscience) in the presence of anti-CD16/32 (clone 93) for 15 minutes in the dark at room temperature. Cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences) for 30 minutes at room temperature in the dark. Cells were washed with Perm/Wash (BD Biosciences) and stained for 15 minutes with fluorochrome labeled antibodies to detect intracellular cytokines as follows: IFN-γ (clone XMG-1.2), IL-2 (JES6–5H4), TNF (MP6-XT22), IL-5 (clone: TRFK5) and IL-10 (clone: JES5–16E3) (BioLegend and eBioscience).
B cells were stained with (1:200) to CD138 (clone281–2), GL7 (clone GL7), CD95 (clone Jo2), IgM (clone II/41), CD19 (clone 1D3 or 6D5), IgD (clone 11–26c.2a), CD38 (clone 90) and 1:100 CD16/32 (clone 93) for 15 minutes in the dark at 4°C. Non-B cell lineage cells were excluded by staining (1:200) and gating for Ly6G (clone 1A8), CD11b (clone M1/70), CD11c (clone N418), F4/80 (clone BM8), Ter119 (clone TER-119) and Thy1.2 (clone 53–2.1) hi populations, fixed and assessed by using a BD Fortessa flow cytometer, analyzed using FACSDiva (BD Bioscience) and FlowJo software (TreeStar).Flow Cytometry use and analysis adhered to the guidelines previously described (59).
A/PR8-specific B cells were first purified out of lymph nodes using a B cell isolation kit II (Miltenyi), then staining with PR8-tetramer-PE (WLTEKEGSYP-biotin), CD138 (clone281–2), GL7 (clone GL7), CD95 (clone Jo2), CD19 (clone 1D3 or 6D5), and CD38. PR8-tetramer was made by incubating 400 picomoles WLTEKEGSYP-biotin with 100 picomoles streptavidin-PE (prozyme) at room temperature for 30 minutes. The PR8-tetramer-PE was then diluted with DPBS to a working concentration of 1 μM PE and stored at 4°C (1 μL per sample for staining) as described previously (36, 37,60). PR8-tetramer stained splenic cells from MD4−/− Rag2−/− mice had minimal binding (data not shown).
Statistical Analysis
Statistical analysis was determined by one-way or two-way ANOVA with Bonferroni or Tukeys correction for multiple comparisons as appropriate. Graphs and statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA). P value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
The authors thank Drs. Richard Webby and Jesse Bloom for the influenza reverse genetic plasmids, Justin Taylor for technical assistance designing the A/PR/8/34 B cell tetramer and David Zeigler for critical reading of the manuscript. Genentech generously supplied the anti-CD20 antibody. The project described has been funded in whole or in part with Federal funds from the NIAID, NIH, DHHS, under Contract No. HHSN272201300029C.
Abbreviations:
- HA
hemagglutinin
- rHA
recombinant HA
- hAb
hemagglutination inhibitory antibody
- HAI
hemagglutination inhibition assay
- Abs
antibodies
- OAS
original antigenic sin
- A/CA
A/CA/07/09
- A/PR8
A/PR/8/34
- C57
C57BL/6
- F1
CB6F1
- LD50
Lethal Dose 50%
- i.m.
intramuscular injection
- Tfh
T follicular helper
- TIV
Trivalent Inactivated Vaccine
- RBS
Receptor Binding Site
Footnotes
Conflict of Interest Disclosure
RNC and NVH are in part employed by PAI Life Sciences. All other authors have declared that no competing interests exist.
References
- 1.Taubenberger JK, Kash JC. 2010. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 7:440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Air GM. 1981. Sequence relationships among the hemagglutinin genes of 12 subtypes of influenza A virus. Proc Natl Acad Sci U S A 78:7639–7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palese P 2004. Influenza: old and new threats. Nat Med 10:S82–87. [DOI] [PubMed] [Google Scholar]
- 4.Yewdell J, García-Sastre A. 2002. Influenza virus still surprises. Curr Opin Microbiol 5:414–418. [DOI] [PubMed] [Google Scholar]
- 5.Dormitzer PR, Galli G, Castellino F, Golding H, Khurana S, Del Giudice G, Rappuoli R. 2011. Influenza vaccine immunology. Immunol Rev 239:167–177. [DOI] [PubMed] [Google Scholar]
- 6.Victora GD, Wilson PC. 2015. Germinal center selection and the antibody response to influenza. Cell 163:545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu C, Wrammert J, Li GM, McCausland M, Wilson PC, Ahmed R. 2013. Cross-reactive humoral responses to influenza and their implications for a universal vaccine. Ann N Y Acad Sci 1283:13–21. [DOI] [PubMed] [Google Scholar]
- 8.Krammer F, Palese P, Steel J. 2015. Advances in universal influenza virus vaccine design and antibody mediated therapies based on conserved regions of the hemagglutinin. Curr Top Microbiol Immunol 386:301–321. [DOI] [PubMed] [Google Scholar]
- 9.Krammer F, Palese P. 2015. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov 14:167–182. [DOI] [PubMed] [Google Scholar]
- 10.Osterholm MT, Kelley NS, Sommer A, Belongia EA. 2012. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis 12:36–44. [DOI] [PubMed] [Google Scholar]
- 11.Jackson LA, Gaglani MJ, Keyserling HL, Balser J, Bouveret N, Fries L, Treanor JJ. 2010. Safety, efficacy, and immunogenicity of an inactivated influenza vaccine in healthy adults: a randomized, placebo-controlled trial over two influenza seasons. BMC Infect Dis 10:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson KJ, Liu Y, Roskin KM, Glanville J, Hoh RA, Seo K, Marshall EL, Gurley TC, Moody MA, Haynes BF, Walter EB, Liao HX, Albrecht RA, García-Sastre A, Chaparro-Riggers J, Rajpal A, Pons J, Simen BB, Hanczaruk B, Dekker CL, Laserson J, Koller D, Davis MM, Fire AZ, Boyd SD. 2014. Human responses to influenza vaccination show seroconversion signatures and convergent antibody rearrangements. Cell Host Microbe 16:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ovsyannikova IG, Oberg AL, Kennedy RB, Zimmermann MT, Haralambieva IH, Goergen KM, Grill DE, Poland GA. 2016. Gene signatures related to HAI response following influenza A/H1N1 vaccine in older individuals. Heliyon 2:e00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakaya HI, Wrammert J, Lee EK, Racioppi L, Marie-Kunze S, Haining WN, Means AR, Kasturi SP, Khan N, Li GM, McCausland M, Kanchan V, Kokko KE, Li S, Elbein R, Mehta AK, Aderem A, Subbarao K, Ahmed R, Pulendran B. 2011. Systems biology of vaccination for seasonal influenza in humans. Nat Immunol 12:786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wikramaratna PS, Rambaut A. 2015. Relationship between haemagglutination inhibition titre and immunity to influenza in ferrets. Vaccine 33:5380–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katz JM, Hancock K, Xu X. 2011. Serologic assays for influenza surveillance, diagnosis and vaccine evaluation. Expert Rev Anti Infect Ther 9:669–683. [DOI] [PubMed] [Google Scholar]
- 17.Andrews SF, Huang Y, Kaur K, Popova LI, Ho IY, Pauli NT, Henry Dunand CJ, Taylor WM, Lim S, Huang M, Qu X, Lee JH, Salgado-Ferrer M, Krammer F, Palese P, Wrammert J, Ahmed R, Wilson PC. 2015. Immune history profoundly affects broadly protective B cell responses to influenza. Sci Transl Med 7:316ra192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, Lambkin-Williams R, Gilbert A, Oxford J, Nicholas B, Staples KJ, Dong T, Douek DC, McMichael AJ, Xu XN. 2012. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 18:274–280. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Myers JL, Bostick DL, Sullivan CB, Madara J, Linderman SL, Liu Q, Carter DM, Wrammert J, Esposito S, Principi N, Plotkin JB, Ross TM, Ahmed R, Wilson PC, Hensley SE. 2013. Immune history shapes specificity of pandemic H1N1 influenza antibody responses. J Exp Med 210:1493–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wheatley AK, Kent SJ. 2015. Prospects for antibody-based universal influenza vaccines in the context of widespread pre-existing immunity. Expert Rev Vaccines 14:1227–1239. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt AG, Do KT, McCarthy KR, Kepler TB, Liao HX, Moody MA, Haynes BF, Harrison SC. 2015. Immunogenic Stimulus for Germline Precursors of Antibodies that Engage the Influenza Hemagglutinin Receptor-Binding Site. Cell Rep 13:2842–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de St Groth Fazekas, Webster RG. 1966. Disquisitions on Original Antigenic Sin. II. Proof in lower creatures. J Exp Med 124:347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JH, Skountzou I, Compans R, Jacob J. 2009. Original antigenic sin responses to influenza viruses. J Immunol 183:3294–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linderman SL, Hensley SE. 2016. Antibodies with ‘Original Antigenic Sin’ Properties Are Valuable Components of Secondary Immune Responses to Influenza Viruses. PLoS Pathog 12:e1005806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kucharski AJ, Lessler J, Read JM, Zhu H, Jiang CQ, Guan Y, Cummings DA, Riley S. 2015. Estimating the life course of influenza A(H3N2) antibody responses from cross-sectional data. PLoS Biol 13:e1002082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lessler J, Riley S, Read JM, Wang S, Zhu H, Smith GJ, Guan Y, Jiang CQ, Cummings DA. 2012. Evidence for antigenic seniority in influenza A (H3N2) antibody responses in southern China. PLoS Pathog 8:e1002802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fonville JM, Wilks SH, James SL, Fox A, Ventresca M, Aban M, Xue L, Jones TC, Le NMH, Pham QT, Tran ND, Wong Y, Mosterin A, Katzelnick LC, Labonte D, Le TT, van der Net G, Skepner E, Russell CA, Kaplan TD, Rimmelzwaan GF, Masurel N, de Jong JC, Palache A, Beyer WEP, Le QM, Nguyen TH, Wertheim HFL, Hurt AC, Osterhaus ADME, Barr IG, Fouchier RAM, Horby PW, Smith DJ. 2014. Antibody landscapes after influenza virus infection or vaccination. Science 346:996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fonville JM, Fraaij PL, de Mutsert G, Wilks SH, van Beek R, Fouchier RA, Rimmelzwaan GF. 2016. Antigenic Maps of Influenza A(H3N2) Produced With Human Antisera Obtained After Primary Infection. J Infect Dis 213:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cobey S, Hensley SE. 2017. Immune history and influenza virus susceptibility. Curr Opin Virol 22:105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. 2011. Different B cell populations mediate early and late memory during an endogenous immune response. Science 331:1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor JJ, Jenkins MK, Pape KA. 2012. Heterogeneity in the differentiation and function of memory B cells. Trends Immunol 33:590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellebedy AH, Jackson KJ, Kissick HT, Nakaya HI, Davis CW, Roskin KM, McElroy AK, Oshansky CM, Elbein R, Thomas S, Lyon GM, Spiropoulou CF, Mehta AK, Thomas PG, Boyd SD, Ahmed R. 2016. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat Immunol 17:1226–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alam S, Knowlden ZA, Sangster MY, Sant AJ. 2014. CD4 T cell help is limiting and selective during the primary B cell response to influenza virus infection. J Virol 88:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JH, Liepkalns J, Reber AJ, Lu X, Music N, Jacob J, Sambhara S. 2016. Prior infection with influenza virus but not vaccination leaves a long-term immunological imprint that intensifies the protective efficacy of antigenically drifted vaccine strains. Vaccine 34:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldwin SL, Hsu FC, Van Hoeven N, Gage E, Granger B, Guderian JA, Larsen SE, Lorenzo EC, Haynes L, Reed SG, Coler RN. 2018. Improved Immune Responses in Young and Aged Mice with Adjuvanted Vaccines against H1N1 Influenza Infection. Front Immunol 9:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casares S, Brumeanu TD, Bot A, Bona CA. 1997. Protective immunity elicited by vaccination with DNA encoding for a B cell and a T cell epitope of the A/PR/8/34 influenza virus. Viral Immunol 10:129–136. [DOI] [PubMed] [Google Scholar]
- 37.Frosch AE, Odumade OA, Taylor JJ, Ireland K, Ayodo G, Ondigo B, Narum DL, Vulule J, John CC. 2017. Decrease in Numbers of Naive and Resting B Cells in HIV-Infected Kenyan Adults Leads to a Proportional Increase in Total and. J Immunol 198:4629–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nayak JL, Alam S, Sant AJ. 2013. Cutting edge: Heterosubtypic influenza infection antagonizes elicitation of immunological reactivity to hemagglutinin. J Immunol 191:1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olafsdottir TA, Lindqvist M, Nookaew I, Andersen P, Maertzdorf J, Persson J, Christensen D, Zhang Y, Anderson J, Khoomrung S, Sen P, Agger EM, Coler R, Carter D, Meinke A, Rappuoli R, Kaufmann SH, Reed SG, Harandi AM. 2016. Comparative Systems Analyses Reveal Molecular Signatures of Clinically tested Vaccine Adjuvants. Sci Rep 6:39097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW, Swain SL. 2012. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest 122:2847–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown DM, Dilzer AM, Meents DL, Swain SL. 2006. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol 177:2888–2898. [DOI] [PubMed] [Google Scholar]
- 42.Crotty S 2015. A brief history of T cell help to B cells. Nat Rev Immunol 15:185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gitlin AD, Shulman Z, Nussenzweig MC. 2014. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, McHeyzer-Williams MG. 2015. Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat Immunol 16:296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asahi Y, Yoshikawa T, Watanabe I, Iwasaki T, Hasegawa H, Sato Y, Shimada S, Nanno M, Matsuoka Y, Ohwaki M, Iwakura Y, Suzuki Y, Aizawa C, Sata T, Kurata T, Tamura S. 2002. Protection against influenza virus infection in polymeric Ig receptor knockout mice immunized intranasally with adjuvant-combined vaccines. J Immunol 168:2930–2938. [DOI] [PubMed] [Google Scholar]
- 46.Faenzi E, Zedda L, Bardelli M, Spensieri F, Borgogni E, Volpini G, Buricchi F, Pasini FL, Capecchi PL, Montanaro F, Belli R, Lattanzi M, Piccirella S, Montomoli E, Ahmed SS, Rappuoli R, Del Giudice G, Finco O, Castellino F, Galli G. 2012. One dose of an MF59-adjuvanted pandemic A/H1N1 vaccine recruits pre-existing immune memory and induces the rapid rise of neutralizing antibodies. Vaccine 30:4086–4094. [DOI] [PubMed] [Google Scholar]
- 47.Priyamvada L, Cho A, Onlamoon N, Zheng NY, Huang M, Kovalenkov Y, Chokephaibulkit K, Angkasekwinai N, Pattanapanyasat K, Ahmed R, Wilson PC, Wrammert J. 2016. B Cell Responses during Secondary Dengue Virus Infection Are Dominated by Highly Cross-Reactive, Memory-Derived Plasmablasts. J Virol 90:5574–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiskopf D, Sette A. 2014. T-cell immunity to infection with dengue virus in humans. Front Immunol 5:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamper CT, Wilson PC. 2017. What Are the Primary Limitations in B-Cell Affinity Maturation, and How Much Affinity Maturation Can We Drive with Vaccination? Is Affinity Maturation a Self-Defeating Process for Eliciting Broad Protection? Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He F, Leyrer S, Kwang J. 2016. Strategies towards universal pandemic influenza vaccines. Expert Rev Vaccines 15:215–225. [DOI] [PubMed] [Google Scholar]
- 51.De Groot AS, McClaine E, Moise L, Martin W. 2010. Time for T?: Thoughts about the 2009 novel H1N1 influenza outbreak and the role of T cell epitopes in the next generation of influenza vaccines. Hum Vaccin 6:161–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Groot AS, Moise L, Liu R, Gutierrez AH, Terry F, Koita OA, Ross TM, Martin W. 2014. Cross-conservation of T-cell epitopes: now even more relevant to (H7N9) influenza vaccine design. Hum Vaccin Immunother 10:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu R, Moise L, Tassone R, Gutierrez AH, Terry FE, Sangare K, Ardito MT, Martin WD, De Groot AS. 2015. H7N9 T-cell epitopes that mimic human sequences are less immunogenic and may induce Treg-mediated tolerance. Hum Vaccin Immunother 11:2241–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bui HH, Peters B, Assarsson E, Mbawuike I, Sette A. 2007. Ab and T cell epitopes of influenza A virus, knowledge and opportunities. Proc Natl Acad Sci U S A 104:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffmann E, Krauss S, Perez D, Webby R, Webster RG. 2002. Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine 20:3165–3170. [DOI] [PubMed] [Google Scholar]
- 56.Lee CW. 2014. Reverse genetics of influenza virus. Methods Mol Biol 1161:37–50. [DOI] [PubMed] [Google Scholar]
- 57.Pleschka S, Jaskunas R, Engelhardt OG, Zürcher T, Palese P, García-Sastre A. 1996. A plasmid-based reverse genetics system for influenza A virus. J Virol 70:4188–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirst GK. 1942. THE QUANTITATIVE DETERMINATION OF INFLUENZA VIRUS AND ANTIBODIES BY MEANS OF RED CELL AGGLUTINATION. J Exp Med 75:49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cossarizza A, Chang HD, Radbruch A, Andrä I, Annunziato F, Bacher P, Barnaba V et al. 2017. Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur. J. Immunol. 47: 1584–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krishnamurty AT, Thouvenel CD, Portugal S, Keitany GJ, Kim KS, Holder A, Crompton PD, Rawlings DJ, Pepper M. 2016. Somatically Hypermutated Plasmodium-Specific IgM(+) Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 45:402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.