

Abstract
Objectives
Generally, the secretory forms of FGF are known to regulate cell proliferation, differentiation and morphogenesis by binding to the extracellular domain of cell surface receptors. Intracellular FGFs (FGF11‐14) are expressed principally in the nervous system. FGF13 is a microtubule‐stabilizing protein that regulates neuronal polarization and migration. Previous studies have reported high expression of FGF13 in cultures of single muscle fibres. However, functions of FGF13 in muscle development have not been explored.
Materials and methods
Real‐time RT‐PCR was performed to detect expression of FGF13 during C2C12 muscle cell proliferation and differentiation. To further understand the role of FGF13, its effects on proliferation and differentiation were examined by western blot analyses of cells transfected with FGF13 siRNA or FGF13 expression plasmids, or treated with chemical MEK inhibitors. Effects of FGF13 on related signalling pathways in C2C12 cell proliferation and differentiation were determined.
Results
FGF13 inhibited C2C12 cell proliferation by up‐regulating p27 mRNA level and by down‐regulating Cyclin E protein expression, during cell proliferation. Additionally, FGF13 down‐regulated Spry1 protein expression, activating the ERK1/2 pathway by phosphorylation and leading to C2C12 cell differentiation inhibition. Consequently, FGF13 seemed to function as a repressor of myoblast differentiation via the ERK1/2 pathway. Although FGF13 inhibited Spry1 regardless of cell proliferation or differentiation, its pathway activation occurred only during the stage of myoblast differentiation.
Conclusions
FGF13 inhibited C2C12 cell proliferation and differentiation by down‐regulating Spry1. These findings indicate that FGF13 played a negative regulatory role in skeletal muscle development.
Introduction
Members of the fibroblast growth factor (FGF) family have been identified as signalling molecules in a variety of developmental processes, including cell proliferation, differentiation and morphogenesis 1, 2. FGFs can also be classified as intracrine, paracrine or endocrine signalling molecules based on their mechanisms of action. Intracrine FGFs (FGF11‐FGF14) are not secreted because they lack any signal peptide. Intracrine FGFs act as intracellular molecules in an FGFR‐independent manner 3. These four FGFs are expressed primarily in the nervous system; however, some of them are also expressed in other tissues.
FGF13 is widely distributed in the developing brain and has approximately 5‐fold higher expression level than FGF11, FGF12 or FGF14 4. FGF13 has high expression in brains and skeletal muscles of patients with Borjeson–Forssman–Lehmann syndrome (BFLS) 5. Accumulating evidence has demonstrated that FGF13 is important in regulating Nav channel activity. Intracellular FGFs often colocalize with Nav channels in initial segments of axons and nodes of Ranvier 6, 7, 8. Thus, these FGFs seem to modulate initiation and propagation of action potentials 9, 10, 11, 12. In addition, FGF13 is a microtubule‐stabilizing protein that regulates neuronal polarization and migration 13. One recent study reported that high expression of FGF13 was observed in culture of single muscle fibres 14. However, functions of intracellular FGF13 in skeletal muscle remain largely unknown.
ERK1/2 proteins are expressed ubiquitously in mammalian cells and have the ability to promote cell proliferation by transducing a variety of mitogenic signals. Activation of ERK1/2 involves Ras, Raf‐1 and MAPK/ERK kinases (MEK)‐1/2 15. Activated ERKs continue to activate downstream kinases and transcription factors 16. ERK1/2 signalling is required during G1 phase of the cell cycle for commitment of myoblasts to DNA synthesis, but is not required further in mitosis once cells enter S phase. Moreover, ERK1/2 proteins are required for myoblast proliferation but are dispensable for muscle gene expression and cell fusion 17. p38 MAPK inhibits the Raf/ERK pathway after inducing muscle differentiation, thereby facilitating withdrawal from the cell cycle 18.
Sprouty (Spry) proteins are a family of intracellular negative regulators of RTK signalling. The four Spry family members (Spry1, 2, 3 and 4) 19, 20 play critical roles in regulating cell proliferation, differentiation and survival in various cell types, including endothelial cells 21, 22, C2C12 myoblasts 23 and skeletal muscle satellite cells 24. Spry proteins have the capacity to repress p44/p42‐MAPK activation via certain growth factors. When Spry proteins are expressed ectopically, they cause attenuation of fibroblast proliferation and PC12 cells differentiation 25, 26. Spry1 expression has been found to be down‐regulated in cycling myogenic progenitors, raising the possibility that Spry1 normally inhibits RTK signals required for their proliferation 24. Spry2, a repressor of p44/p42‐MAPK activation by FGF, promotes myogenic differentiation in the presence of FGF223.
In this study, we found that FGF13 inhibited proliferation of C2C12 cells and induced G1 phase cell cycle arrest by up‐regulating p27 mRNA level and by down‐regulating Cyclin E protein expression during cell proliferation. Additionally, FGF13 inhibited Spry1 expression and promoted ERK1/2 activation by phosphorylation, which causes retardation of myogenic differentiation during C2C12 cell differentiation. This report is the first to document a clear biological function of FGF13 during proliferation and differentiation of skeletal muscle myoblasts.
Materials and methods
Cell culture and DM‐induced differentiation
Mouse C2C12 cell line was purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were cultured in growth medium [GM: Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA, USA)]. When the myoblasts reached 80% confluence, they were induced to differentiate using low serum DMEM medium (DM) supplemented with 2% horse serum (HS). A selective inhibitor of MEK1 and MEK2 (PD98059) signal transduction was obtained from Beyotime (Jiangsu, China) and used at concentration of 25 μm, on the first day of differentiation.
siRNA and DNA transfection
For myogenic differentiation studies, C2C12 cells were seeded in 60 mm plates (Nunc) or six‐well plates with growth medium, and transfected with siRNA on the first day of myogenic differentiation. Briefly, a predesigned FGF13 siRNA (Invitrogen; sequences of primers are provided in Table 1) was synthesized and pre‐diluted in Opti‐MEM medium (Invitrogen) with Lipofectamine 2000 (Invitrogen). Lipofectamine–siRNA mixture was added to each well immediately. After cells had been incubated for 6–8 h, medium was replaced with fresh regular DMEM supplemented with 2% HS.
Table 1.
Primer sequences
| Genes name | Primers(5′–3′) |
|---|---|
| siRNA‐FGF13 |
Forward: GGGUAUAGUUACCAAACUATT Reverse: UAGUUUGGUAACUAUACCCTT |
| FGF13 |
Forward:GGGGTACCGAAGAAGTGGTGGTGGTGG Reverse:CCGCTCGAGCTCGTTTTGCCCTCACTGGC |
For transient DNA transfection, C2C12 cells were seeded in 60 mm plates or six‐well plates with culture medium and transfected on the first day of myogenic differentiation. Plasmids were pre‐diluted in Opti‐MEM medium (Invitrogen) containing Sofast transfection reagent (Sunma Biotechnology Co., Ltd., Xiamen, China). Before transfection, the cells had been grown in DMEM (without HS and antibiotics) for 12 h. At 48 h or 72 h post‐transfection, they were harvested for further myogenic differentiation analysis.
For C2C12 cell cycle assays, C2C12 cells were seeded in 60 mm plates (Nunc) at initial plating density of 5 × 104 cells per plate. After they had been cultured for 6–8 h, plasmid transfection was performed via the above‐described method. At 72 h post‐transfection, cells were harvested for flow cytometric analysis.
FGF13 expression plasmid construction
RNA samples from mouse muscle were reverse‐transcribed to cDNA, and full‐length FGF13 cDNA was amplified using primers FGF13F and FGF13R (sequences provided in Table 1). FGF13 PCR products were purified from agarose gels and cloned into the pcDNA3.1 plus vector between EcoRI and BamHI sites.
Cell cycle assay
Cell cycle assays were performed at 72 h post‐transfection. Briefly, C2C12 cells were spun at 700 × g for 5 min, and cell pellets were washed once in PBS at 4 °C. One millilitre of 70% ethanol at 4 °C was added dropwise to pellets with tubes placed on a vortex mixer. Cell suspensions were incubated at −20 °C overnight. Subsequently, the cells were maintained at 4 °C and spun at 800 × g for 5 min. Supernatants were removed, and 1 ml propidium iodide (PI) working solution was added to each tube to re‐suspend pellets. Re‐suspended cells were maintained in the dark in a 37 °C water bath for 30 min. Then, treated samples were analysed using a flow cytometer.
Immunocytochemistry
C2C12 myoblasts cultured in 12‐well plates were washed in PBS and fixed in 4% paraformaldehyde for 10 min, followed by 0.1% Triton X‐100 for 5 min. Non‐specific reactivity was blocked by incubating cells in 5% goat serum for 30 min. Then, cells were incubated in monoclonal antibody (MF‐20; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) to the myosin heavy chain (MyHC) at 37 °C for 1–2 h. Next, secondary goat anti‐mouse IgG (Alexa Fluor® 546; Invitrogen) was added, and cells were incubated at 37 °C for 40 min. Nuclei were stained with DAPI for 10 min and images were captured using a Nikon TE2000 microscope (Nikon, Tokyo, Japan).
RNA isolation and real‐time RT‐PCR
TRIzol reagent (TaKaRa, Dalian, China) was used to extract total RNA from cell cultures. First‐strand cDNA synthesis was performed using a reverse transcription kit (TaKaRa) and random primers. Real‐time quantitative PCR was performed in triplicate samples, using SYBR Green kit (TaKaRa) on a Bio‐Rad iQ5 system. Mouse GAPDH was used as reference gene. The 2−ΔΔCT algorithm was employed to estimate relative expression level of each gene. Sequences of primers used are listed in Table 2.
Table 2.
Primer sequences for real‐time PCR
| Genes | Primers(5′–3′) | Size,bp |
|---|---|---|
| FGF13 | Forward: CAATGAACAGCGAGGGATAC | 20 |
| Reverse: GGTTTGGGCAGAAAATGTG | 19 | |
| Spry1 | Forward: CCATCCCAGGTCATAGGTCAG | 21 |
| Reverse: CAGATGAACTTGTGCTGGGTG | 21 | |
| Spry2 | Forward: CTCAGAGTGGCAACGGGTCG | 20 |
| Reverse: CATTGGTGTTTCGGATGGCTC | 21 | |
| Spry3 | Forward: ATCTGATTGGTCCCTGGCTAC | 21 |
| Reverse: CAGTGGTGCTTTGGGACATTG | 21 | |
| Spry4 | Forward: AGAGCAGCGTCCCTGTGAATC | 21 |
| Reverse: CTGGTCAATGGGTAAGATGGTG | 22 |
Western blot detection
C2C12 cells were collected after culture and adequate treatment, and total protein extracts were isolated using lysis buffer (RIPA, Beyotime) containing protease inhibitor (Pierce, Waltham, MA, USA). Briefly, 25–50 μg aliquots of each protein sample were separated on SDS polyacrylamide gel and transferred to polyvinylidene difluoride (PVDF) membranes (CST, USA). Membranes were blocked with 5% bovine serum albumin (BSA) in TBS/0.5% Tween‐20 (TBST) and then incubated with appropriate primary antibodies. Antibodies to myogenin (MyOG) and myosin heavy chain (MyHC) were purchased from the Developmental Studies Hybridoma Bank (University of Iowa, IA, USA), and antibodies to p38 MAPK, p‐p38 MAPK, ERK1/2, p‐ERK1/2, Cyclin E, Cyclin B1 and GAPDH were purchased from Santa Cruz Biotechnology (Dallas, Texas, USA). The next day, PVDF membranes were washed in TBST, incubated with secondary antibody (goat anti‐mouse IgG, Invitrogen) at room temperature for 1 h and then washed three times in TBST. Image acquisition and quantitative analysis were performed using ImageJ.
Statistical analysis
All data represent at least three independent experiments, and quantitative results are represented as mean ± SEM. Differences between groups were compared by variance analysis and significance tests using SPSS v11.5 software (SPSS, Chicago, IL, USA). Significance was accepted at P < 0.05.
Results
Presence of FGF13 slowed rate of C2C12 cell proliferation
Previous research has shown that levels of FGF13 mRNA expression are highest in the brain and that FGF13 acts as a microtubule‐stabilizing protein in neurons 13. However, a recent study reported that higher expression of FGF13 was observed during culture of single muscle fibres 14, but few studies have examined expression levels and functions of FGF13 in other tissues. Thus, we investigated expression pattern of FGF13 during C2C12 myogenic differentiation. FGF13 expression was clearly detectable during myogenic differentiation and reached its highest level on the fourth day of differentiation (Fig. 1a). Unexpectedly, FGF13 was not detectable during C2C12 cell proliferation (Fig. 1a). Thus, we focused on the roles of FGF13 in C2C12 cell proliferation. We transfected C2C12 cells with an FGF13 overexpression plasmid, harvested cells 72 h post‐transfection, and analysed these samples by flow cytometry. Some changes occurred during the cell cycle; proportion of G1 phase cells increased by 10% and proportion of S phase cells decreased by 10% compared to the control group treated with empty vector (Fig. 1b–d; P < 0.005). Thus, our preliminary speculation was that FGF13 induced G1/S cell cycle arrest and cause retardation of C2C12 cell proliferation. Meanwhile, protein levels of Cyclin E, Cyclin B1, ERK1/2 and p‐ERK1/2 were analysed by western blotting. We observed a more dramatic reduction in Cyclin E protein levels than Cyclin B1 levels at 72 h post‐transfection; however, ERK1/2 and p‐ERK1/2 protein levels did not show any remarkable changes (Fig. 1e). These data showed that FGF13 overexpression restrained G1 to S phase transition by down‐regulating Cyclin E protein expression. This result illustrated that FGF13 acted as a negative regulatory factor during C2C12 cell proliferation. Thus, we can explain the phenomenon of low FGF13 expression level in proliferating cells.
Figure 1.
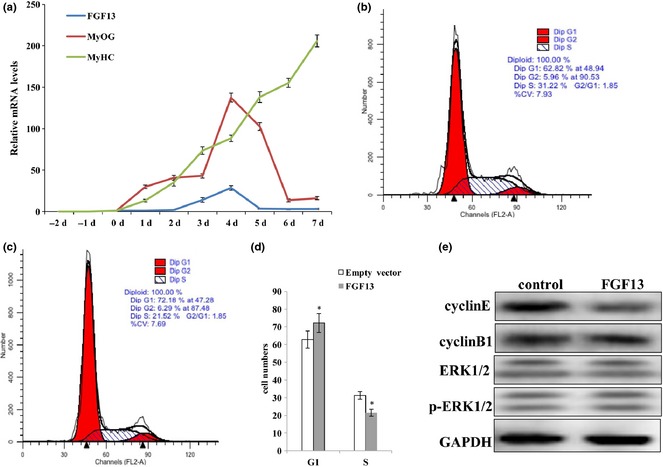
FGF13 affected C2C12 cell proliferation. (a) mRNA expression levels of FGF13, MyOG and MyHC during C2C12 myoblast differentiation. (b) C2C12 cells were transfected with empty vector during proliferation as a control group. (c) C2C12 cells were transfected with FGF13 overexpression plasmids during proliferation. The cells were harvested, and samples were analysed by flow cytometry at 72 h post‐transfection. (d) percentages of cells in G1 and S phases were analysed statistically in (b) and (c). (e) FGF13 was overexpressed in C2C12 cells during proliferation, and cyclin E and cyclin B1 protein levels were analysed by western blotting after 72 h. Results are presented as the mean ± SEM. n = 3. *P < 0.05.
FGF13 inhibited C2C12 myogenic differentiation and myotube hypertrophy
Next, we evaluated expression of MyOG and MyHC, markers of early and terminal differentiation of skeletal muscle cells respectively. Levels of MyHC mRNA expression in C2C12 cells increased steadily during myogenic differentiation, and MyOG expression reached its highest level on the fourth day of myogenic differentiation (Fig. 1a). These exciting findings indicated that the expression trend of FGF13 mRNA was consistent with levels of MyOG during myogenic differentiation (Fig. 1a). We inferred that FGF13 might have an effect on C2C12 cell differentiation over certain periods.
Then, we used an FGF13‐specific siRNA to knock‐down FGF13 on the first day of DM‐induced differentiation. Mouse C2C12 cells can be induced to differentiate under low serum (2% HS) conditions. We observed 65% reduction in level of FGF13 mRNA expression 24 h post‐transfection with FGF13‐specific siRNA (Fig. 2a; P < 0.001). Meanwhile, mRNA levels of MyOG and MyHC (markers of myogenic differentiation) increased by approximately 2‐ to 3‐fold compared to the negative control group (Fig. 2a; P < 0.001), and MyOG and MyHC protein levels increased by 1.75‐ and 2.2‐fold by 48 h post‐transfection respectively (Fig. 2b and 2c; P < 0.001). MyHC expression was analysed by immunofluorescence to examine myotube formation in cultured C2C12 cells at 72 h post‐transfection. We observed marked increase in MyHC+ cells and myotube sizes compared to the negative control group (Fig. 2d). These results clearly demonstrated that FGF13 knock‐down promoted myoblast differentiation in vitro. Hence, we hypothesized that FGF13 negatively affected myoblast differentiation.
Figure 2.
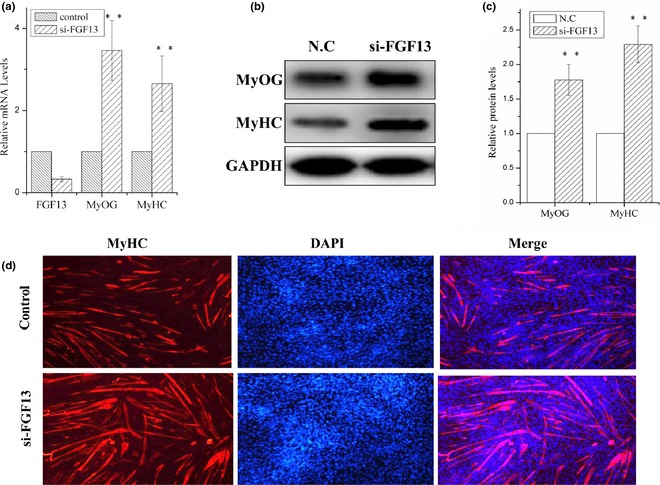
FGF13 knock‐down promoted myoblast differentiation. C2C12 cells were transfected with siRNA‐FGF13 (or NC siRNA) on the first day of differentiation for 6–8 h, and myogenic differentiation was continued. (a) interference efficiency and relative mRNA expression levels of MyoG and MyHC were measured by real‐time qPCR. (b) MyoG and MyHC protein levels were measured. (c) relative protein levels in (b) were analysed using ImageJ. (d) Immunofluorescence analysis of MyHC was performed in C2C12 cell myotubes. Results are presented as mean ± SEM. n = 3. **P < 0.01.
FGF13 affected MAPK–ERK1/2 signalling pathway during myoblast differentiation
To determine the effect of FGF13 overexpression on myogenic differentiation, C2C12 cells were transfected with pcDNA3.1‐FGF13 plasmids on the first day of DM‐induced differentiation. As expected, mRNA expression level of FGF13 was up‐regulated approximately 46 times, and MyOG and MyHC mRNA expression levels were down‐regulated approximately 50% and 40%, respectively, at 24 h post‐transfection (Fig. 3a; P < 0.001). Cells were harvested separately at 0 h, 24 h, 48 h and 72 h post‐transfection, and resulting samples were analysed by western blotting to measure protein levels of p38 MAPK, p‐p38 MAPK, ERK1/2 and p‐ERK1/2. These data demonstrated that FGF13 overexpression led to significant increase in p‐ERK1/2 protein levels by 48 and 72 h post‐transfection. However, we did not observe any changes in protein levels of p38 MAPK, p‐p38 MAPK or ERK1/2 (Fig. 3b). This result implies that FGF13 enhanced ERK1/2 activation by phosphorylation during myoblast differentiation.
Figure 3.
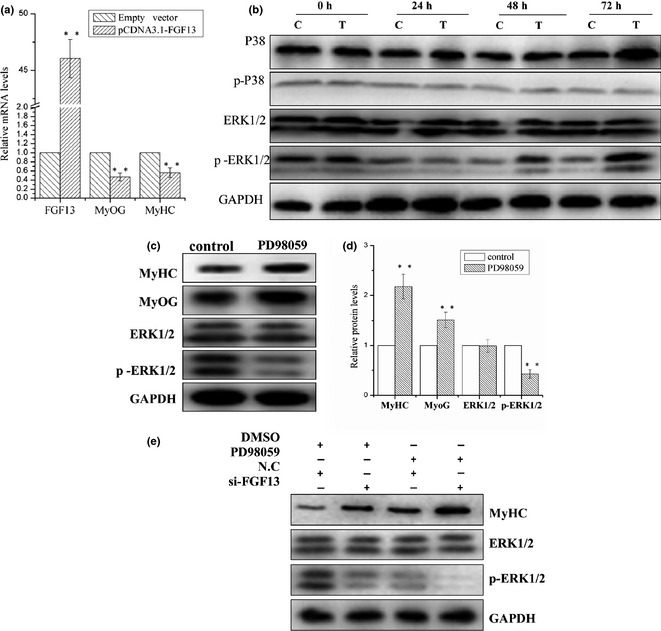
FGF13 induced ERK1/2 activation via phosphorylation to regulate myogenic differentiation. C2C12 cells were transfected with pcDNA3.1‐FGF13 (or empty vector) on the first day of differentiation, maintained for up to 8 h, and then induced to myogenic differentiation continuously in DMEM supplemented with 2% HS. (a) relative mRNA levels of MyoG, MyHC after FGF13 overexpression were measured by real‐time qPCR at 24 h post‐transfection. (b) MyHC, p38, p‐p38, ERK1/2 and p‐ERK1/2 protein expression levels from 0 to 72 h during myoblast differentiation were detected by western blotting. c: Empty vector group; T: pcDNA3.1‐FGF13 group. (c) C2C12 cells were treated with 25 μm PD98059 or DMSO (control) on the first differentiation day. MyOG, MyHC, ERK1/2 and p‐ERK1/2 protein levels were detected by western blotting after 48 h. (d) relative protein levels in (c) were analysed using ImageJ. (e) C2C12 cells were transfected simultaneously with si‐FGF13 and treated with 25 μm PD98059 on the first day of myogenic differentiation. MyHC, ERK1/2 and p‐ERK1/2 protein levels were detected by western blotting after 72 h. Results are presented as the mean ± SEM. n = 3. **P < 0.01.
To validate whether FGF13 is a regulator of the MAPK–ERK1/2 signalling pathway during myoblast differentiation, C2C12 cells were treated with an ERK1/2 inhibitor (PD98059, 25 μm). MyOG and MyHC protein levels were clearly up‐regulated by 48 h post‐treatment. ERK1/2 protein levels were not significantly affected, but protein level of p‐ERK1/2 decreased markedly compared to the control group (Fig. 3c and 3d; P < 0.001). These data suggest that the MAPK–ERK1/2 pathway played a negative regulatory role in myoblast differentiation, which is consistent with onr previous report 17. Then, C2C12 cells were transfected with si‐FGF13 and treated with 25 μm PD98059 on the first day of myoblast differentiation. We assessed MyHC, ERK1/2 and p‐ERK1/2 protein levels by western blot analysis after 72 h treatment. When FGF13 was knocked down, a significant increase in MyHC protein levels and marked reduction in p‐ERK1/2 levels were observed, similar to the PD98059 treatment group. When si‐FGF13 and PD98059 were treated simultaneously, this effect was dramatically enhanced (Fig. 3e). Therefore, FGF13 inhibition of ERK1/2 activity by phosphorylation was similar to that of the inhibitor PD98059. Thus, we can conclude that FGF13 inhibition of myogenic differentiation was indeed dependent on the ERK1/2 signalling pathway.
FGF13 activated the ERK1/2 signalling pathway by down‐regulating Spry1 during myogenic differentiation
All Spry family members have been shown to be negative regulators of RTK‐mediated ERK activation in various mammalian embryonic tissues, including brain, heart, gut and muscle 19, 20. We investigated mRNA expression levels of the four Spry family members when FGF13 was overexpressed during C2C12 cell proliferation. mRNA expression level of Spry1 decreased remarkably, but no other members of the family were affected (Fig. 4a; P < 0.001). Therefore, we knocked down Spry1 during C2C12 cell proliferation and differentiation. mRNA expression level of Spry1 was reduced by 65% during C2C12 cell proliferation. In addition, we measured mRNA expression levels of cell cycle regulatory factors p21 and p27 by real‐time qPCR; level of p27 mRNA expression increased significantly, but level of p21 mRNA expression showed no obvious change (Fig. 4b; P < 0.001). p21 and p27 are both cell cycle kinase inhibitors, and enhanced expression levels of the two proteins can retard cell proliferation 27. To confirm that Spry1 played a role in C2C12 cell differentiation, we knocked down Spry1 during myogenic differentiation and found that mRNA expression level of MyHC decreased by approximately 50% and that MEK1 and MEK2 mRNA expression levels clearly increased (Fig. 4c; P < 0.001). Moreover, during myogenic differentiation, protein level of p‐ERK1/2 increased, and protein level of MyHC decreased (Fig. 4g). Immunofluorescence analysis revealed significant reductions in MyHC+ cells and myotube sizes compared to the control group, after Spry1 knock‐down (Fig. 4d). Based on these results, we can confirm that Spry1 promoted myotube formation by inhibiting ERK1/2 pathways.
Figure 4.
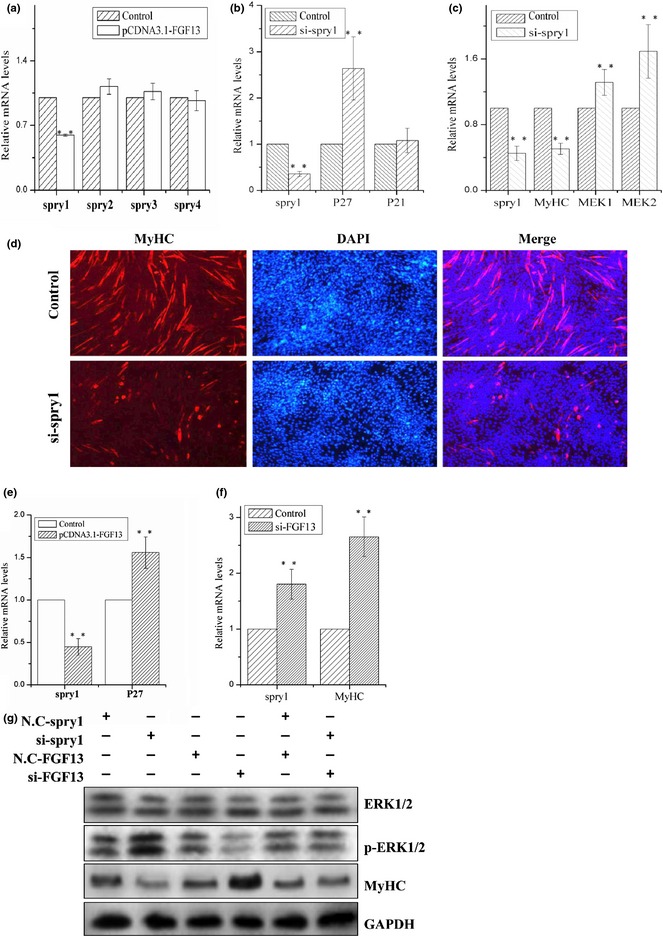
FGF13 regulated ERK1/2 signalling pathway by activating Spry1 during myogenic differentiation. (a) C2C12 cells were transfected with pcDNA3.1‐FGF13 or empty vector (control group) during cell proliferation, and relative mRNA expression levels of Spry1, Spry2, Spry3 and Spry4 were measured by real‐time qPCR at 24 h post‐transfection. C2C12 cells were transfected with empty vector during proliferation as a control group. (b) C2C12 cells were transfected with si‐Spry1 or NC‐siRNA (control group) during proliferation, and the relative mRNA expression levels of Spry1, p27 and p21 were measured by real‐time qPCR at 24 h post‐transfection. (c) C2C12 cells were transfected with si‐Spry1 or NC‐siRNA (control group) during differentiation, and relative mRNA expression levels of Spry1, MyHC, MEK1 and MEK2 were measured by real‐time qPCR at 24 h post‐transfection. (d) Immunofluorescence analysis of MyHC was performed in C2C12 cell myotubes 72 h after si‐Spry1 transfection. (e) C2C12 cells were transfected with siRNA‐FGF13 (or NC‐siRNA) on the first day of differentiation, and relative mRNA expression levels of Spry1 and MyHC were measured by real‐time qPCR at 24 h post‐transfection. (f) C2C12 cells were transfected with si‐FGF13 and si‐Spry1 separately or simultaneously, and the protein levels of MyHC, ERK1/2 and p‐ERK1/2 were measured by western blotting after 72 h. Results are presented as the mean ± SEM. n = 3. **P < 0.01.
To further verify the relationship between FGF13 and Spry1, we overexpressed FGF13 during C2C12 cell proliferation. Compared to the empty vector group, FGF13 overexpression group showed significant reduction in Spry1 mRNA expression and significant increase in p27 mRNA levels (Fig. 4e; P < 0.001). These data suggest that FGF13 promoted expression of cyclin kinase inhibitor p27 by suppressing Spry1, resulting in retardation of C2C12 cell proliferation. In contrast, inhibiting FGF13 expression increased mRNA expression levels of Spry1 and MyHC during myogenic differentiation (Fig. 4f; P < 0.001). After Spry1 knock‐down, western blot analysis showed that protein level of p‐ERK1/2 clearly increased and that protein level of MyHC significantly decreased (Fig. 4g), in contrast to the results of FGF13 knock‐down (Fig. 4g). We did not observe any changes in p‐ERK1/2 and MyHC protein levels when FGF13 and Spry1 were knocked down simultaneously (Fig. 4g). These results suggest that FGF13 and Spry1 play inverse roles during myogenic differentiation and that FGF13 inhibited myogenic differentiation by down‐regulating Spry1 expression. We conclude that FGF13 promoted ERK1/2 activation and phosphorylation by reducing Spry1 expression during C2C12 cell differentiation.
Discussion
To explore the functions of FGF13 in myoblasts, FGF13 was knocked down or overexpressed by transfected siRNA or DNA plasmid respectively. FGF13 expression reached its highest level on the fourth day of myogenic differentiation. Meanwhile, expression patterns of markers for early differentiation of skeletal muscle cells, such as MyOG, tended to be consistent with FGF13 expression (Fig. 1a). To achieve the best effect of interference or overexpression, transfection treatments were performed on the first day of myogenic differentiation. In fact, we attempted to transfect siRNA or DNA plasmid during different periods following the induction of C2C12 cell differentiation, and the effect of interference or overexpression on the first day of myogenic differentiation was better than at any other period (data not shown). This phenomenon may be due to short‐term effects of siRNAs or DNA plasmids post‐transfection.
Skeletal muscle development is a multistep process that includes myoblast generation from myogenic precursors, myoblast proliferation, subsequent cell cycle withdrawal, and multinucleated myotube differentiation 28. Previous studies have shown that intracellular FGF13 protein has higher expression levels in leg muscle, skeletal muscle and single muscle fibres of mouse 5, 14. However, the roles of FGF13 in skeletal muscle development remain unclear. Our data demonstrated that FGF13 plays a key role in early myogenic differentiation. The fourth differentiation day is a critical time point in myotube formation. Importantly, the expression level of FGF13 reached its highest point on the fourth day of myogenic differentiation and then reduced gradually (Fig. 1a). This finding suggested that FGF13 might be involved in muscle differentiation. Therefore, when FGF13 was knocked down on the first day of myogenic differentiation, the mRNA and protein levels of MyOG and MyHC significantly increased in C2C12 myoblasts. These data showed that FGF13 inhibits myogenic differentiation and myotube formation. In contrast, FGF13 overexpression reduced MyOG and MyHC expression. In addition, we also found that ERK1/2 activation by phosphorylation significantly increased on the third and fourth days of myogenic differentiation after FGF13 overexpression (Fig. 3b). Previous studies have shown that the Raf/ERK pathway must be inhibited during an early stage of myogenic differentiation; we also observed this inhibition (Fig. 3c–e). Thus, FGF13 promoted ERK1/2 activation by phosphorylation, thereby suppressing early stage of myogenic differentiation. Therefore, FGF13 seemed to play a negative regulatory role in the process of myogenic differentiation.
Spry family numbers Spry1, Spry2, Spry3 and Spry4 can form homodimers and heterodimers, and these dimers bind differentially to different components of the Ras–Raf–ERK pathway. Several earlier studies have demonstrated that Spry proteins inhibit Ras‐ERK‐MAPK signalling, specifically by RTKs, leaving the phosphoinositide 3‐kinase (PI3K) and other MAPK pathways unaffected 15, 16. Thus, we investigated expression levels of the four Spry family members when FGF13 was overexpressed during C2C12 cell proliferation and found that only Spry1 mRNA level decreased (Fig. 4a). One previous study showed that Spry1 and Spry4 heterodimers were the most potent at inhibiting ERK activation 29. However, the opposite phenomenon, where Spry1 had little effect on MAPK/ERK signalling but increased and maintained Akt activation and Spry4 expression, antagonized both MAPK/ERK and Akt signalling and suppressed VSMC differentiation 30. In this study, when Spry1 was knocked down, we found that MEK1 and MEK2 mRNA expression levels increased and that p‐ERK1/2 protein level increased. Meanwhile, MyHC expression and myotube formation decreased during myogenic differentiation after Spry1 knock‐down (Fig. 4c, 4d and 4g). These data further support Spry1 as a positive regulatory factor that promotes muscle formation by inhibiting ERK1/2 activation via phosphorylation during C2C12 cell differentiation.
Although Spry proteins are believed to have similar structures, the mechanisms by which they inhibit downstream RTK signalling differ. As reported in this study, only Spry1 is involved in this process. Therefore, what is the relationship between FGF13 and Spry1 during myogenic differentiation? Knocking down FGF13 during C2C12 cell differentiation increased Spry1 and MyHC mRNA levels, decreased p‐ERK 1/2 protein levels, and increased MyHC protein levels (Fig. 4f and 4g). We confirmed that Spry1 promoted muscle formation by inhibiting ERK1/2 activation via phosphorylation. Therefore, knocking down FGF13 up‐regulated Spry1 expression, which may be beneficial for myogenic differentiation. Thus, functions of FGF13 and Spry1 were cancelled out when they were knocked down simultaneously (Fig. 4g). One further report confirmed that FGF induces Spry1 and Spry2 expression through Ca2+‐dependent pathways that require PLCγ 31. Here, we demonstrated that FGF13 promotes ERK1/2 activation via phosphorylation by down‐regulating Spry1 expression during C2C12 cell differentiation.
Additionally, we found some surprising results when examining C2C12 cell proliferation. First, FGF13 expression was not detectable during C2C12 cell proliferation (Fig. 1a). Thus, we overexpressed FGF13 and observed G1/S cell cycle arrest and C2C12 cell proliferation obstruction, together with down‐regulation of Cyclin E and up‐regulation of p27 mRNA expression (Figs. 1e and 4e). These data indicate that FGF13 inhibits G1 to S phase transition by down‐regulating Cyclin E and up‐regulating p27. Early studies demonstrated that accumulation of Cyclin E is a key effector of G1/S transition in normal cells 27. Second, we also found that Spry1 knock‐down up‐regulated p27 expression (Fig. 4b). The most crucial point is that FGF13 overexpression suppressed Spry1 and p27 mRNA expression (Fig. 4e). Thus, FGF13 down‐regulation of cyclin E and up‐regulation of p27 expression lead to slower cell proliferation by inhibiting Spry1. Accordingly, FGF13 is a negative regulator of C2C12 cell proliferation. ERK1/2 pathway activation appears to be both necessary and sufficient for transcriptional induction of Cyclin D and Cyclin E genes during G1 phase 27. However, in our study, inexplicably, the ERK1/2 pathway did not seem to affect cell proliferation, which differs from cell differentiation. Other factors may be related to C2C12 cell proliferation in the presence of FGF13.
However, FGF13 differs from other FGF family members in that it is not secreted from living cells via a classical pathway and cannot interact with FGF receptor tyrosine kinases 32. Earlier studies have demonstrated that FGF12 is structurally and functionally similar to FGF13; FGF12 is associated with MAP kinase (MAPK) scaffold protein Islet Brain‐2 (IB2) in the brain, and in some specific cell lines 33. Therefore, we hypothesized that FGF13 may interact with one or more intracellular proteins to regulate Spry protein activity. In summary, our work identified that FGF13 down‐regulated Spry1 expression, thereby activating the ERK1/2 pathway via phosphorylation and inhibiting C2C12 cell differentiation. Consequently, FGF13 may participate as a repressor during myoblast differentiation via the ERK1/2 pathway. In addition, FGF13 is a negative regulator of C2C12 cell proliferation. Although FGF13 inhibited Spry1 regardless of cell proliferation or differentiation, ERK1/2 pathway activation occurred only during C2C12 cell differentiation. Generally, FGF13 can inhibit C2C12 cell proliferation and differentiation by down‐regulating Spry1. However, future studies are needed to reveal this signalling pathway.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. U1201213) and Major Projects for Genetically Modified Organism Breeding (No. 2014ZX08009‐047B).
References
- 1. Guillemot F, Zimmer C (2011) From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 71, 574–588. [DOI] [PubMed] [Google Scholar]
- 2. Itoh N, Ornitz DM (2008) Functional evolutionary history of the mouse Fgf gene family. Dev. Dyn. 237, 18–27. [DOI] [PubMed] [Google Scholar]
- 3. Kettunen P, Furmanek T, Chaulagain R, Kvinnsland IH, Luukko K (2011) Developmentally regulated expression of intracellular Fgf11‐13, hormone‐like Fgf15 and canonical Fgf16, ‐17 and ‐20 mRNAs in the developing mouse molar tooth. Acta Odontol. Scand. 69, 360–366. [DOI] [PubMed] [Google Scholar]
- 4. Smallwood PM, Munoz‐Sanjuan I, Tong P, Macke JP, Hendry SH, Gilbert DJ et al (1996) Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc. Natl Acad. Sci. USA 93, 9850–9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gecz J, Baker E, Donnelly A, Ming JE, McDonald‐McGinn DM, Spinner NB et al (1999) Fibroblast growth factor homologous factor 2 (FHF2): gene structure, expression and mapping to the Borjeson‐Forssman‐Lehmann syndrome region in Xq26 delineated by a duplication breakpoint in a BFLS‐like patient. Hum. Genet. 104, 56–63. [DOI] [PubMed] [Google Scholar]
- 6. Lou JY, Laezza F, Gerber BR, Xiao M, Yamada KA, Hartmann H et al (2005) Fibroblast growth factor 14 is an intracellular modulator of voltage‐gated sodium channels. J. Physiol. 569, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X et al (2007) Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage‐gated sodium channels. Neuron 55, 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang C, Hennessey JA, Kirkton RD, Wang C, Bryson V, Rosenberg PB et al (2011) FGF13 is a regulator of the cardiac voltage‐gated sodium channel Nav1.5. Biophys. J . 100, 420a–421a.21244838 [Google Scholar]
- 9. Kole MH, Stuart GJ (2012) Signal processing in the axon initial segment. Neuron 73, 235–247. [DOI] [PubMed] [Google Scholar]
- 10. Debanne D (2011) The nodal origin of intrinsic bursting. Neuron 71, 569–570. [DOI] [PubMed] [Google Scholar]
- 11. Kole MH (2011) First node of Ranvier facilitates high‐frequency burst encoding. Neuron 71, 671–682. [DOI] [PubMed] [Google Scholar]
- 12. Thaxton C, Pillai AM, Pribisko AL, Dupree JL, Bhat MA (2011) Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron 69, 244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu QF, Yang L, Li S, Wang Q, Yuan XB, Gao X et al (2012) Fibroblast growth factor 13 is a microtubule‐stabilizing protein regulating neuronal polarization and migration. Cell 149, 1549–1564. [DOI] [PubMed] [Google Scholar]
- 14. Chakkalakal JV, Jones KM, Basson MA, Brack AS (2012) The aged niche disrupts muscle stem cell quiescence. Nature 490, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kolch W (2000) Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J 351(Pt 2), 289–305. [PMC free article] [PubMed] [Google Scholar]
- 16. Robinson MJ, Cobb MH (1997) Mitogen‐activated protein kinase pathways. Curr. Opin. Cell Biol. 9, 180–186. [DOI] [PubMed] [Google Scholar]
- 17. Jones NC, Fedorov YV, Rosenthal RS, Olwin BB (2001) ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J. Cell. Physiol. 186, 104–115. [DOI] [PubMed] [Google Scholar]
- 18. Lee J, Hong F, Kwon S, Kim SS, Kim DO, Kang HS et al (2002) Activation of p38 MAPK induces cell cycle arrest via inhibition of Raf/ERK pathway during muscle differentiation. Biochem. Biophys. Res. Commun. 298, 765–771. [DOI] [PubMed] [Google Scholar]
- 19. Edwin F, Anderson K, Ying C, Patel TB (2009) Intermolecular interactions of Sprouty proteins and their implications in development and disease. Mol. Pharmacol. 76, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mason JM, Morrison DJ, Basson MA, Licht JD (2006) Sprouty proteins: multifaceted negative‐feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 16, 45–54. [DOI] [PubMed] [Google Scholar]
- 21. Impagnatiello MA, Weitzer S, Gannon G, Compagni A, Cotten M, Christofori G (2001) Mammalian sprouty‐1 and ‐2 are membrane‐anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. J. Cell Biol. 152, 1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee SH, Schloss DJ, Jarvis L, Krasnow MA, Swain JL (2001) Inhibition of angiogenesis by a mouse sprouty protein. J. Biol. Chem. 276, 4128–4133. [DOI] [PubMed] [Google Scholar]
- 23. de Alvaro C, Martinez N, Rojas JM, Lorenzo M (2005) Sprouty‐2 overexpression in C2C12 cells confers myogenic differentiation properties in the presence of FGF2. Mol. Biol. Cell 16, 4454–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shea KL, Xiang W, LaPorta VS, Licht JD, Keller C, Basson MA et al (2010) Sprouty1 regulates reversible quiescence of a self‐renewing adult muscle stem cell pool during regeneration. Cell Stem Cell 6, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maximy AA, Nakatake Y, Moncada S, Itoh N, Thiery JP, Bellusci S (1999) Cloning and expression pattern of a mouse homologue of drosophila sprouty in the mouse embryo. Mech. Dev. 81, 213–216. [DOI] [PubMed] [Google Scholar]
- 26. Hanafusa H, Torii S, Yasunaga T, Nishida E (2002) Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 4, 850–858. [DOI] [PubMed] [Google Scholar]
- 27. Meloche S, Pouyssegur J (2007) The ERK1/2 mitogen‐activated protein kinase pathway as a master regulator of the G1‐ to S‐phase transition. Oncogene 26, 3227–3239. [DOI] [PubMed] [Google Scholar]
- 28. Kuang S, Kuroda K, Le Grand F, Rudnicki MA (2007) Asymmetric self‐renewal and commitment of satellite stem cells in muscle. Cell 129, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ozaki K, Miyazaki S, Tanimura S, Kohno M (2005) Efficient suppression of FGF‐2‐induced ERK activation by the cooperative interaction among mammalian Sprouty isoforms. J. Cell Sci. 118, 5861–5871. [DOI] [PubMed] [Google Scholar]
- 30. Yang X, Gong Y, Tang Y, Li H, He Q, Gower L et al (2013) Spry1 and Spry4 differentially regulate human aortic smooth muscle cell phenotype via Akt/FoxO/myocardin signaling. PLoS ONE 8, e58746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abe M, Naski MC (2004) Regulation of sprouty expression by PLCgamma and calcium‐dependent signals. Biochem. Biophys. Res. Commun. 323, 1040–1047. [DOI] [PubMed] [Google Scholar]
- 32. Olsen SK, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M et al (2003) Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 278, 34226–34236. [DOI] [PubMed] [Google Scholar]
- 33. Okada T, Murata K, Hirose R, Matsuda C, Komatsu T, Ikekita M et al (2013) Upregulated expression of FGF13/FHF2 mediates resistance to platinum drugs in cervical cancer cells. Sci. Rep. 3, 2899. [DOI] [PMC free article] [PubMed] [Google Scholar]