

Our study defines the allelic distribution of pfcrt, an important mediator of multidrug resistance in Plasmodium falciparum, in Africa and Asia. We leveraged whole-genome sequence analysis and gene editing to demonstrate how current drug combinations can select different allelic variants of this gene and shape region-specific parasite population structures. We document the ability of PfCRT mutations to modulate parasite susceptibility to current antimalarials in dissimilar, pfcrt allele-specific ways. This study underscores the importance of actively monitoring pfcrt genotypes to identify emerging patterns of multidrug resistance and help guide region-specific treatment options.
KEYWORDS: Plasmodium falciparum, drug resistance evolution, fitness, malaria, pfcrt
ABSTRACT
The global spread of Plasmodium falciparum chloroquine resistance transporter (PfCRT) variant haplotypes earlier caused the widespread loss of chloroquine (CQ) efficacy. In Asia, novel PfCRT mutations that emerged on the Dd2 allelic background have recently been implicated in high-level resistance to piperaquine, and N326S and I356T have been associated with genetic backgrounds in which resistance emerged to artemisinin derivatives. By analyzing large-scale genome sequencing data, we report that the predominant Asian CQ-resistant Dd2 haplotype is undetectable in Africa. Instead, the GB4 and previously unexplored Cam783 haplotypes predominate, along with wild-type, drug-sensitive PfCRT that has reemerged as the major haplotype. To interrogate how these alleles impact drug susceptibility, we generated pfcrt-modified isogenic parasite lines spanning the mutational interval between GB4 and Dd2, which includes Cam783 and involves amino acid substitutions at residues 326 and 356. Relative to Dd2, the GB4 and Cam783 alleles were observed to mediate lower degrees of resistance to CQ and the first-line drug amodiaquine, while resulting in higher growth rates. These findings suggest that differences in growth rates, a surrogate of parasite fitness, influence selection in the context of African infections that are frequently characterized by high transmission rates, mixed infections, increased immunity, and less recourse to treatment. We also observe that the Asian Dd2 allele affords partial protection against piperaquine yet does not directly impact artemisinin efficacy. Our results can help inform the regional recommendations of antimalarials, whose activity is influenced by and, in certain cases, enhanced against select PfCRT variant haplotypes.
INTRODUCTION
The pathogenesis of human malaria is initiated by Plasmodium asexual blood-stage parasites, with Plasmodium falciparum causing the most severe and lethal forms of disease. Recent reductions in the worldwide incidence of malaria can be largely attributed to the global deployment of artemisinin (ART)-based combination therapies (ACTs) to treat asexual blood-stage infections, combined with vector control strategies (1). ACTs consist of a fast-acting, rapidly eliminated ART compound, paired with a long-lived partner drug necessary to clear residual parasite biomass. P. falciparum resistance to certain ACT drugs has emerged in Southeast (SE) Asia, raising concerns that it might soon extend to Africa where malaria exerts its greatest impact (2, 3). This pattern of emerging resistance would mimic the prior spread from Asia to Africa of P. falciparum resistance to the former first-line drugs chloroquine (CQ) and pyrimethamine (4, 5). The global threat posed by multidrug resistance underscores the pressing need to characterize its genetic determinants and leverage that knowledge into improved methods of surveillance, containment, and treatment.
Artemether plus lumefantrine (ATM+LMF), artesunate plus amodiaquine (AS+ADQ), and dihydroartemisinin plus piperaquine (DHA+PPQ) are the three ACTs used most widely worldwide (2). The structural hallmark of these ART derivatives is an endoperoxide bridge that, upon cleavage by parasite-processed host heme-bound Fe2+, is potently parasiticidal. Once activated, ARTs can reduce the parasite biomass by up to 104-fold per ∼48-h cycle of intraerythrocytic development (6). Clinically, emerging resistance to ARTs is defined as extended parasite clearance times in AS- or ACT-treated patients, first reported in western Cambodia and now increasingly observed in neighboring countries (7–12). Slower clearance is associated with single nucleotide polymorphisms (SNPs) in the propeller domain of the P. falciparum k13 (kelch13) gene (13, 14). These mutations, including the most widespread C580Y mutation, as well as R539T, were shown in gene-edited parasites to confer ART resistance in vitro (15, 16). Resistance in vitro is defined as ≥1% survival of early ring-stage parasites (0 to 3 h postinvasion [hpi]) exposed for 6 h to the ART metabolite DHA, tested at the pharmacologically relevant concentration of 700 nM (ring-stage survival assay from 0 to 3 h [RSA0-3h]) (17). Molecular epidemiological and clinical data show that mutant K13 parasites are now widespread throughout SE Asia, but to date, they remain infrequent in Africa where ACTs currently retain excellent efficacy (9, 10, 18, 19).
The evolution of K13-mediated ART resistance in SE Asia has been attributed, in part, to the presence of a favorable genetic background that permits sustained parasite transmission in the field. Through a multicenter genome-wide association study, founder populations were identified that included mutations in parasite genes encoding the P. falciparum chloroquine resistance transporter (PfCRT), the apicoplast ribosomal protein S10, ferredoxin, and the P. falciparum multidrug resistance 2 transporter (20). Prior transfection-based studies have demonstrated that some PfCRT mutations can modestly modulate parasite ART susceptibility in vitro, as determined in 72-h dose-response assays that measure half-maximal growth inhibition concentrations (IC50 values) (21, 22). PfCRT mutations also impact the intracellular disposition of heme, which serves as the ART activator (23–25). PfCRT mutations are also known to modulate the potency of several ACT partner drugs, including LMF, mefloquine (MFQ), ADQ, and PPQ (2, 21, 26–31). Their selective pressures can therefore influence the regional prevalence of PfCRT haplotypes.
The role of mutant PfCRT in dictating P. falciparum resistance to the former first-line antimalarial CQ, along with the pleotropic impact of this transporter on multiple other drugs, has driven a detailed assessment of PfCRT evolution and its impact on drug resistance and fitness (22, 32). First discovered through the analysis of a P. falciparum genetic cross between the CQ-resistant Dd2 line (from SE Asia) and the CQ-sensitive HB3 line (from Honduras), mutant isoforms have been discovered to harbor four to nine SNPs, depending on their geographic origin (26, 33). Earlier studies of African isolates observed the FCB isoform, harboring seven mutations compared to the CQ-sensitive canonical 3D7 wild-type allele, along with a 6-amino-acid variant termed GB4 (Table 1) (26, 34, 35). These studies relied on samples that predate the use of ACTs in the region. Epidemiological reports have often examined only positions 72 to 76 in exon 2 of the pfcrt gene, without assigning complete haplotypes (36). Thus, the current distribution of pfcrt alleles in Africa and Asia, post-ACT treatment selection, has remained largely unknown.
TABLE 1.
PfCRT and K13 haplotypes of recombinant isogenic parasite linesa
| Parasite line | Amino acid in PfCRT at the following position: |
Amino acid in K13 at position 539 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 74 | 75 | 76 | 220 | 271 | 326 | 356 | 371 | ||
| Dd23D7 (wild type) | M | N | K | A | Q | N | I | R | R |
| Dd2Dd2 | I | E | T | S | E | S | T | I | R |
| Dd2Cam783 (Dd2Dd2 S326N) | I | E | T | S | E | N | T | I | R |
| Dd2FCB (Dd2Dd2 T356I) | I | E | T | S | E | S | I | I | R |
| Dd2GB4 (Dd2Dd2 S326N T356I) | I | E | T | S | E | N | I | I | R |
| Dd2R539T3D7 | M | N | K | A | Q | N | I | R | T |
| Dd2R539TDd2 | I | E | T | S | E | S | T | I | T |
| Dd2R539TCam783 (Dd2R539TDd2 S326N) | I | E | T | S | E | N | T | I | T |
| Dd2R539TFCB (Dd2R539TDd2 T356I) | I | E | T | S | E | S | I | I | T |
| Dd2R539TGB4 (Dd2R539TDd2 S326N T356I) | I | E | T | S | E | N | I | I | T |
The mutational status of PfCRT (PlasmoDB PF3D7_0709000) and K13 (PF3D7_1343700) is summarized for all recombinant parasite lines generated in this study. Mutations are shown in boldface type. The PfCRT haplotype encoded by Dd2 (wild-type K13) and Dd2R539T (K13 R539T mutant) parasites is indicated in superscript, with 3D7 designating the wild-type PfCRT haplotype.
Genetic and mathematical modeling studies that deconstructed the evolution of pfcrt alleles provided evidence that mutations within this gene likely occurred in bursts, with the preferred paths representing a balance between gaining stepwise resistance to CQ and minimizing the negative impact of reduced fitness, as determined from differences in parasite asexual blood-stage growth rates (22). An important role for fitness was highlighted by the finding in Malawi that complete removal of CQ pressure led to the virtual disappearance of mutant PfCRT as a result of it causing a decreased proliferation rate and the dominance of faster-growing wild-type parasites in these settings of frequent mixed infections (37, 38).
In this study, we leveraged recent advances in whole-genome sequence analysis of P. falciparum isolates across multiple sites in Asia and Africa (9) to explore the geographic distribution of pfcrt alleles and determine their impacts on asexual blood-stage parasite drug resistance and fitness, including the reported association of the PfCRT mutations N326S and I356T with emerging ART resistance (20). Our findings present novel insights into PfCRT haplotype distributions across Asia and Africa and how they might be impacted by regional differences in drug selection and parasite fitness.
RESULTS
Distribution of pfcrt alleles in Africa and Asia.
Although sub-Saharan Africa accounts for the vast majority of the global burden of malaria, SE Asia is critical in driving much of the emergence of multidrug resistance. Using the MalariaGEN Pf3k database of recently acquired P. falciparum whole-genome sequences that encompass these regions (9), we assessed the geographic distribution of pfcrt haplotypes by region and sampled country (Fig. 1 and Table 1; see also Tables S1, S2, and S3 in the supplemental material). In Africa, wild-type (3D7) PfCRT comprised 65.5% of the total haplotypes (513/783 genomes), consistent with reduced CQ use and the increased fitness of wild-type PfCRT in the absence of drug selection. Countries showed widely different percentages of wild-type PfCRT ranging from 100% in Malawi (all 147 genomes) to 11.5% in the Democratic Republic of Congo (6 of 52 genomes). These findings could be explained in part by differences in the local availability and use of CQ.
FIG 1.
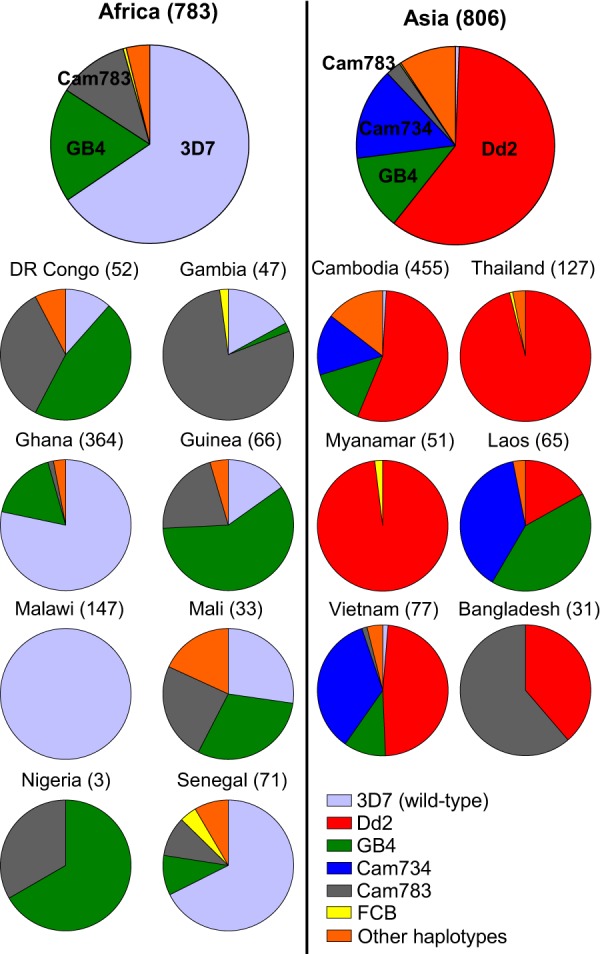
Predominant pfcrt alleles are distinct in Africa and Asia. Pie charts illustrate continent- and country-wide proportions of the indicated PfCRT haplotypes, as estimated based on whole-genome sequences present in the MalariaGEN Pf3k database (9). Analysis revealed the following haplotype proportions: in Africa, 3D7 (65.5%), GB4 (18.6%), Cam783 (11.5%), FCB (0.5%), other (3.8%); in SE Asia, 3D7 (0.6%), Dd2 (60%), GB4 (12.3%), Cam734 (15%), Cam783 (2.5%), FCB (0.2%), other (9.3%). Country-wide distribution of the pfcrt alleles is detailed in Tables S1 and S2 in the supplemental material. The mutational composition of the wild-type (3D7) and variant PfCRT haplotypes is shown in Table 1. Cam734 PfCRT consists of the following mutations: M74I, N75D, K76T, A144F, L148I, I194T, A220S, Q271E, and T333S.
Frequency distribution of PfCRT haplotypes from the Pf3K data set across Africa. Download Table S1, PDF file, 0.02 MB (19.2KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Frequency distribution of PfCRT haplotypes from the Pf3K data set across Asia. Download Table S2, PDF file, 0.02 MB (16.7KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PfCRT haplotype distribution across Asia and Africa, as derived from the Pf3K data set. Download Table S3, PDF file, 0.1 MB (151.4KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We found no evidence of the 8-amino-acid variant Dd2 haplotype in Africa (Fig. 1, Table 1, and Table S1). Instead, GB4 was the most common African mutant PfCRT haplotype (18.6% total prevalence, observed in 146 of 783 genomes), followed by the Cam783 haplotype (akin to Dd2 minus the N326S mutation; 11.5% of total, 90/783 genomes). Of note, not a single report has until now examined the Cam783 haplotype in parasites. The remaining portion of mutant haplotypes (4% of total) was comprised of various polymorphic haplotypes, with the FCB haplotype (akin to Dd2 minus the I356T mutation) being present at 0.5% prevalence (4 of 783 genomes). Among the remaining genomes, the genomes with reduced numbers of mutations (GB4-N75E and GB4-A220S) were the most common, suggesting selection against the more mutated isoforms (Table S3).
In Asia, the wild-type PfCRT haplotype had virtually disappeared, comprising only 0.6% of the total haplotypes (5/806 genomes) despite the removal of CQ pressure at least 20 years ago for the treatment of P. falciparum malaria in favor of more recent ACTs (39). Dd2 was the most common mutant haplotype (60.0% of total, 484/806 genomes), followed by the 9-amino-acid variant Cam734 (15.0% of total, 121/806 genomes), GB4 (12.3% of total, 99/806 genomes), Cam783 (2.5%; 20/806 genomes), and other mutant haplotypes (9.3% of total, 75/806 genomes; Tables S2 and S3).
Generation of isogenic parasites with variant pfcrt and k13 alleles.
In view of the highly significant association between the PfCRT mutations N326S and I356T and the emergence of ART resistance in SE Asia (with a P value of 7 × 10−10) (20), we tested whether these mutations could individually or collectively influence antimalarial susceptibility and/or parasite fitness in a way that would explain their local dominance across SE Asia. We note that these mutations are found together only in the Dd2 haplotype (Table 1 and Fig. 1). Using an established zinc-finger nuclease (ZFN)-based approach (Fig. S1), we generated isogenic parasites differing only by their pfcrt allele. The pfcrt sequences of these parasite lines were verified using primers listed in Table S4. Our PfCRT haplotype panel included full-length 3D7 (wild type), Dd2, GB4 (Dd2 minus the N326S and I356T mutations), and the two intermediates spanning the mutational interval between GB4 and Dd2, namely, the Cam783 allele (Dd2 minus the N326S mutation) and FCB (Dd2 minus the I356T mutation; see Table 1). Inclusion of the Cam783 haplotype allowed us to characterize the second most common mutant PfCRT haplotype in Africa (Fig. 1). We generated this panel of PfCRT haplotypes in two separate parasite backgrounds, namely, Dd2 and Dd2R539T, which encode the wild-type K13 (default) and R539T mutant isoforms (shown as a subscript), respectively (Table 1). The latter strain was previously reported as Dd2R539T, whose K13 mutation conferred one of the highest levels of ART resistance in Dd2 parasites, exceeding that of the more prevalent K13 C580Y mutation. We opted to use Dd2R539T in our studies as it therefore maximized our ability to measure small changes in the ring-stage survival assay (RSA) levels with different PfCRT haplotypes compared with wild-type K13 parasites (16). We included a K13 variant line in light of the rapid spread of mutant K13 across SE Asia (10) and the need to assess whether mutant K13 might alter the pleotropic impact of PfCRT on antimalarial drug susceptibility.
Allelic modification of pfcrt and validation of genetic editing. (A) Zinc-finger nuclease (ZFN)-based genetic engineering strategy. As detailed in Text S1, parasites were first enriched with a donor plasmid (pcrt-hdhfr) that encodes pfcrt exons 2 to 13 (e2-13) and includes mutations of interest (indicated in red). A comprehensive list of PfCRT haplotypes that were introduced into Dd2 or Dd2R539T parasites is found in Table 1. The pcrt-hdhfr plasmid also comprises the following elements: Plasmodiumberghei crt (pbcrt) 3′ UTR, human dhfr (hdhfr) selection cassette, as well as pfcrt-hdhfr-flanking homology regions (∼0.4 kb upstream of the intron 1-exon 2 junction and ∼1 kb native 3′ UTR downstream of hdhfr). Parasites were subsequently transfected with pZFNcrt-bsd. This plasmid facilitates calmodulin (cam) promoter-driven expression of a pair of pfcrt-targeting ZFNs (ZFNL and ZFNR) and includes the blasticidin S deaminase (bsd) selection cassette. Following ZFN-mediated introduction of a double-stranded break in pfcrt (indicated with a lightning bolt), parasites that employ the pcrt-hdhfr plasmid as a donor template incorporate mutations of interest through homologous recombination-based mechanisms. Genetic editing was evaluated by diagnostic PCR (see panel B), and successfully edited parasites were cloned by limiting dilution. (B) Blood PCR-based verification of pfcrt allelic exchange. Shown are representative results for Dd2 parasites into which the full-length GB4 pfcrt allele was introduced (Dd2GB4) as well as controls, namely, genetically unedited parental parasites (Dd2), unedited donor plasmid-enriched parasites (Dd2 + pcrt-hdhfr), and the sole donor plasmid (pcrt-hdhfr). All appropriately edited lines showed the expected DNA band migration patterns: 0.4 kb (p7+p8), 1.2 kb (p9+p8), 2.5 kb (p10+p11), and 2.7 kb (p9+p12). Primer (p) locations are illustrated in panel A. Download FIG S1, EPS file, 1.9 MB (1.9MB, eps) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental Materials and Methods. Download Text S1, PDF file, 0.2 MB (198.1KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download Table S4, PDF file, 0.02 MB (22.4KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CQ responses of pfcrt-modified lines and their chemosensitization by verapamil.
To assess the impact of these pfcrt alleles on parasite CQ responses, we subjected asexual blood-stage parasites to flow cytometry-based drug susceptibility assays and determined 50% and 90% growth-inhibitory antimalarial drug concentrations (IC50 and IC90, respectively). For CQ and its main in vivo metabolite, monodesethyl-CQ (md-CQ) (Fig. 2A and B; Table S5), mutant PfCRT isoforms Dd2, Cam783, FCB, and GB4 each conferred statistically significant increases in IC50 values compared with the isogenic comparator line expressing the wild-type 3D7 isoform (P < 0.002 in all instances by two-tailed Mann-Whitney U tests). Among the mutant isoforms, the highest and lowest levels of resistance to CQ and md-CQ were conferred by the Dd2 and GB4 PfCRT isoforms, respectively (Fig. 2A and B; Table S5). The reduced level of resistance associated with GB4 PfCRT (∼1.6-fold reduction versus Dd2 PfCRT for CQ and md-CQ) was significant in both the Dd2 and Dd2R539T parasite genetic backgrounds (P < 0.01 by two-tailed Mann-Whitney U tests; Fig. 2A and B; Table S5). These data suggest that GB4 might represent a mutational precursor of Dd2 PfCRT and that the latter proved more successful in the epidemiological context of SE Asia.
FIG 2.
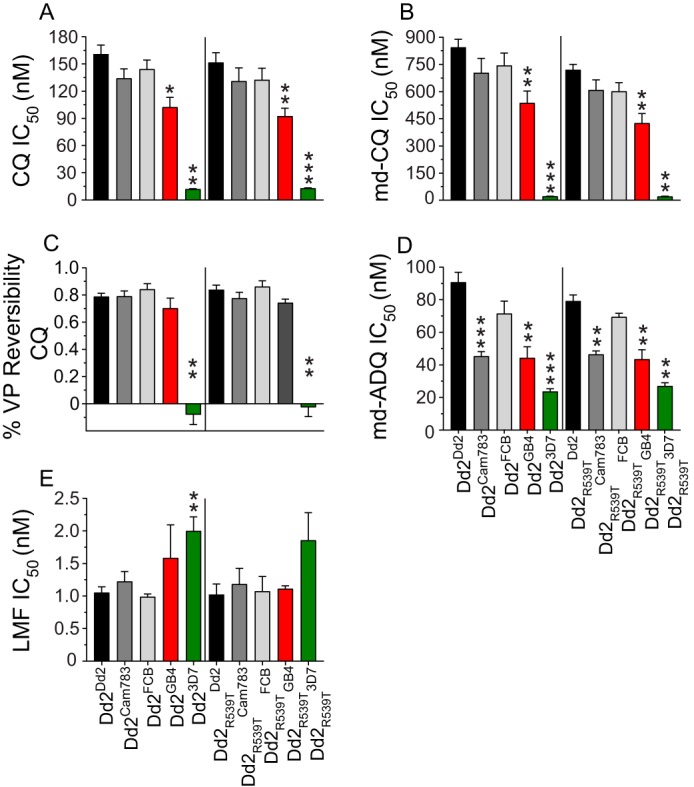
Responses of pfcrt-modified lines to various antimalarial drugs. Parasite susceptibilities to chloroquine (CQ), monodesethyl-CQ (md-CQ), md-amodiaquine (md-ADQ), and lumefantrine (LMF) were assessed by flow cytometry following exposure to the indicated drug for 72 h. (A, B, D, and E) Bar graphs show mean plus SEM IC50 values. (C) Percent CQ reversibility was determined as follows: 1 − CQ response modification index (RMI), with RMI being the IC50 value for CQ in the presence of 800 nM VP divided by the IC50 value for CQ alone. The bar graph shows mean plus SEM reversibility values. For all panels, results reflect 3 to 10 independent assays conducted in duplicate. Statistical significance was determined against Dd2Dd2 or Dd2R539TDd2 by two-tailed Mann-Whitney U tests using Graphpad Prism 7 software. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Antimalarial IC50 and IC90 values of pfcrt-modified parasite lines. Download Table S5, PDF file, 0.3 MB (283.6KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Chemosensitization of CQ-resistant parasites by the calcium channel blocker verapamil (VP) is a defining feature of parasite CQ resistance (40). VP reversibility can be quantified as the CQ response modification index (RMI), corresponding to the following equation: 1 − (IC50 value for CQ in the presence of 0.8 μM VP/IC50 value for CQ alone) (41). Comparison of the RMI values for our isogenic pfcrt-modified lines (Fig. 2C; Table S5) showed that the Dd2, Cam783, FCB, and GB4 isoforms yielded similar levels of VP reversibility, contrasting with isogenic parasites expressing the 3D7 isoform that showed no VP chemosensitization (21). Our observed CQ, md-CQ, and VP responses were equivalent between the k13 wild-type Dd2 and k13 mutant Dd2R539T lines.
Susceptibility of pfcrt-modified lines to clinically employed antimalarials.
We also examined our isogenic, pfcrt-modified lines for their susceptibility to a panel of clinically important drugs, namely, the ACT artemisinin compound AS, various ACT partner drugs (ADQ, PPQ, LMF, and pyronaridine [PND]), and the quinoline-type drug quinine (QN) (Fig. 2D and E; Fig. S2 and Table S5). All mutant pfcrt alleles conferred a degree of parasite resistance to the active ADQ metabolite md-ADQ compared with 3D7 pfcrt, with Dd2 pfcrt being the most resistant (Fig. 2D; Table S5). Interestingly, an important contributory role was identified for the N326S mutation. Mutant PfCRT haplotypes bearing S326N reversions (i.e., Cam783 and GB4; see Table 1) conferred a statistically significant reduction in md-ADQ resistance relative to the Dd2 isoform (P < 0.001 for Dd2 versus Cam783 and P < 0.01 for Dd2 versus GB4 by two-tailed Mann-Whitney U tests; Fig. 2D and Table S5). The notion that PfCRT residue 326 is an important contributor to ADQ resistance agrees with recent evidence that the N326D mutation, found among South American and western Pacific parasite isolates, contributes directly to md-ADQ resistance (22). We also observed a statistically significant ∼2-fold increase in the LMF IC50 value in the 3D7 allele compared to the Dd2 isoform in the K13 wild-type Dd2 background (Fig. 2E; Table S5). No statistically significant differences were observed between individual mutant PfCRT haplotypes for the compounds AS, PPQ, LMF, QN, and PND (Fig. 2E; Fig. S2 and Table S5).
Drug susceptibility of pfcrt-modified lines to antimalarial drugs. Parasite susceptibilities to artesunate (AS), piperaquine (PPQ), quinine (QN), and pyronaridine (PND) were assessed after 72-h exposures to the indicated drug. Parasitemia was measured using flow cytometry. Data are presented as mean ± SEM IC50 values for 3 to 10 independent assays performed in duplicate. Statistical significance was determined against Dd2Dd2 or Dd2R539TDd2 by two-tailed Mann-Whitney U tests using Graphpad Prism 7 software. Download FIG S2, EPS file, 0.8 MB (803.3KB, eps) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Survival of k13-mutant pfcrt-modified lines exposed to DHA.
Although our drug susceptibility assays did not reveal physiologic significant shifts in AS IC50 values among parasite lines, the utility of IC50-based studies for the detection of clinical ART resistance is restricted. A more clinically validated metric is parasite survival following drug pulses, which more closely mimics the in vivo pharmacology (17). For ART compounds, reduced susceptibility of early ring-stage parasites (0 to 3 hpi) to a 6-h pulse of 700 nM DHA in the in vitro RSAs correlates with ART resistance in the clinical setting, defined as lower rates of parasite clearance (17, 42). This phenotype has been associated with mutations in the parasite k13 gene, which is sufficient to mediate enhanced survival of DHA-pulsed parasites in RSAs (13, 16).
To examine whether PfCRT mutations could modulate the parasite’s ability to survive DHA pulses, we employed the k13-mutant Dd2R539T background (Table 1), which bears a K13 R539T mutation that confers in vitro RSA survival to DHA. We also included the ART-resistant mutant k13 parasite line Cam3.IIR539T, which carries the PfCRT Dd2 haplotype as a positive control (16). We also considered the possibility that PfCRT mutations may modulate ART resistance at subphysiologic levels of DHA in a stage-specific manner. Accordingly, we evaluated the survival of early ring (0 to 3 h) and mid-trophozoite (28 to 31 h) blood-stage parasites following exposure to a wide range of DHA concentrations (twofold dilutions spanning the range of 700 nM to 1.4 nM).
Results showed no impact of varying the PfCRT haplotype on parasite survival for any concentration examined (Fig. 3A). In contrast, in trophozoite-stage DHA survival assays (Fig. 3B), statistically significant differences were observed at the subphysiologic concentrations of 43.8 nM to 2.7 nM DHA. Parasites with 3D7 and GB4 PfCRT exhibited significantly increased protection from DHA toxicity compared to parasites with either Dd2 or FCB allele (P < 0.05 and P < 0.001 as determined by two-tailed Student’s t test). Compared to ring-stage parasites, trophozoites were considerably more susceptible to DHA exposure (e.g., compare parasite survival for 700 nM DHA in Fig. 3A and for 10.9 nM DHA in Fig. 3B, which show comparable survival rates). The highly DHA-resistant Cam3.IIR539T showed >40% survival in the ring stage, consistent with an earlier report (16), yet conferred no appreciable survival at the trophozoite stage.
FIG 3.
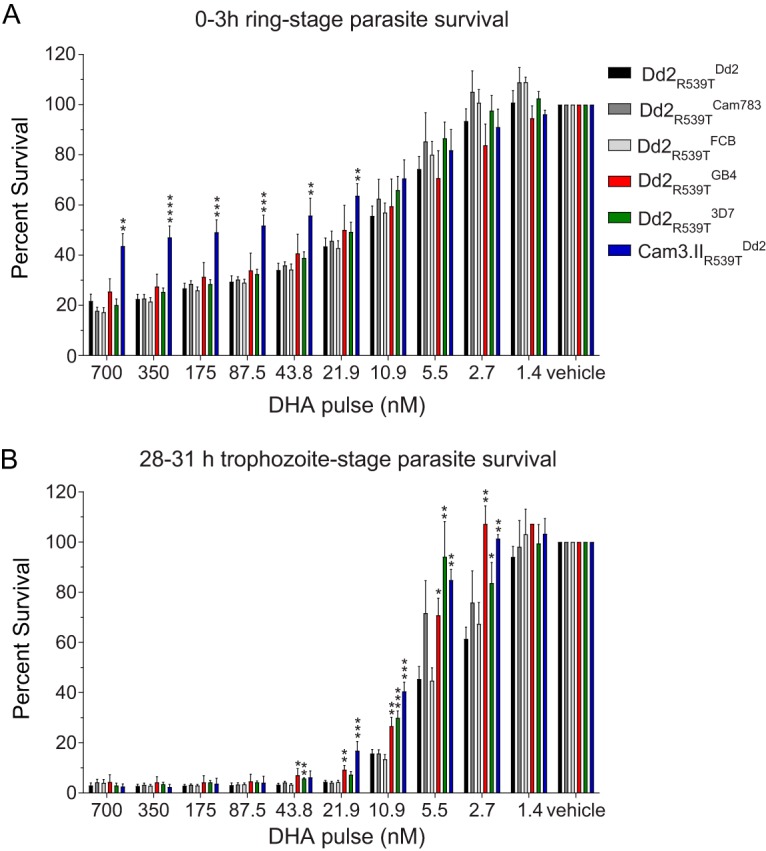
Dihydroartemisinin (DHA) survival of k13-mutant, pfcrt-modified lines. Tightly synchronized parasites at the early ring (0 to 3 hpi) (A) or mid-trophozoite (28 to 31 hpi) (B) developmental stage were exposed to a 3-h pulse of DHA at the indicated concentrations. Parasite viability was determined by flow cytometry at 72 hpi. Bar graphs correspond to the mean plus SEM percent survival, equivalent to the parasitemia of DHA-treated parasites divided by the parasitemia of vehicle control (DMSO)-treated parasites. Data are from three or four independent experiments, conducted in duplicate. Statistical significance was determined by two-tailed Student’s t tests (Graphpad Prism 7). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Survival of PPQ-exposed k13-mutant pfcrt-modified lines.
Similar to ART resistance, IC50-based drug susceptibility studies appear insufficient to reveal parasite resistance to the first-line ACT partner drug PPQ. This is apparent from ex vivo analyses of recrudescent parasites derived from DHA+PPQ treatment failures, which show a wide range of PPQ IC50 values that overlap with IC50 values of nonresistant isolates (43). Recent studies of PPQ-resistant parasites have highlighted the effectiveness of in vitro PPQ survival assays (PSAs) in uncovering resistance phenotypes that correlate with in vivo parasite recrudescence (43). In these assays, synchronized early ring-stage parasites are subjected to 200 nM PPQ for 48 h. Drug is then removed by washing, and parasite survival is assessed at the 72-h endpoint. The basis for a much longer duration of drug exposure compared to DHA-based assays is the relatively prolonged half-life of PPQ in blood following treatment (44).
Similar to our DHA-based survival assays, we subjected pfcrt-modified Dd2R539T parasites to serial dilutions of PPQ starting from a maximum concentration of 200 nM. To probe for stage-specific effects, we exposed parasites to PPQ at either the early ring stage (0 to 3 hpi) or mid-trophozoite stage (28 to 31 hpi). Results for PPQ survival assays initiated during the early ring stage showed increased susceptibility of parasites expressing the pfcrt GB4 allele at 50 nM and 25 nM PPQ compared to isogenic parasites expressing the Dd2 allele (Fig. 4A; P < 0.05 and P < 0.01, respectively, as determined by two-tailed Student’s t test). At 25 nM PPQ concentration, our PSA data showed no significant differences between the Dd2 and FCB alleles and an increased PPQ susceptibility with the Cam783 allele (P < 0.05). These results implicate residue 326 as a contributor to increased survival to PPQ at lower concentrations.
FIG 4.
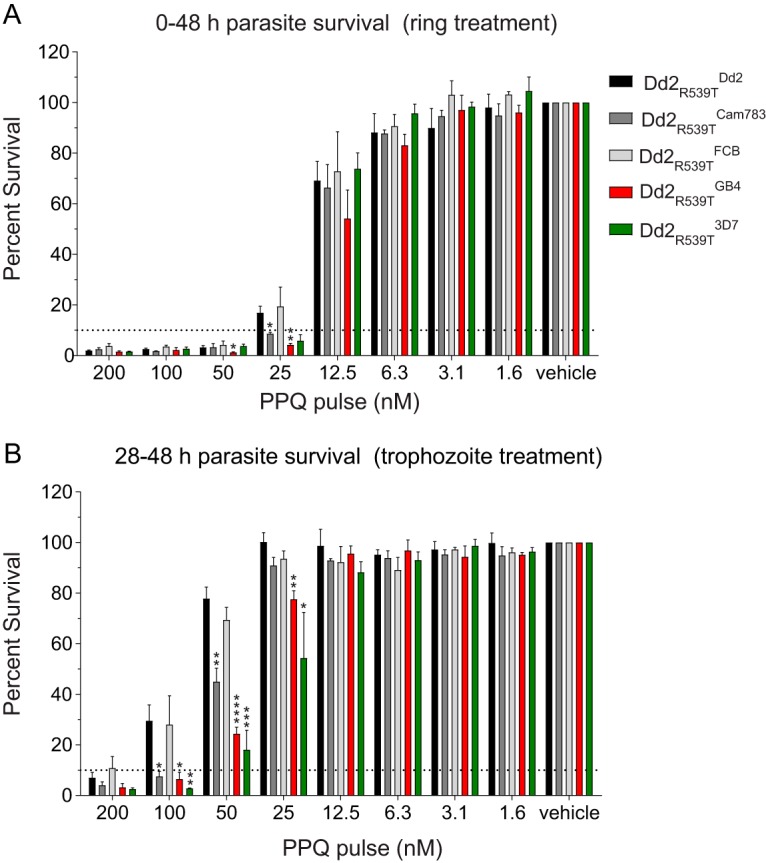
Piperaquine (PPQ) survival of k13-mutant, pfcrt-modified lines. Tightly synchronized parasites in the early ring (0 to 3 hpi) (A) or mid-trophozoite (28 to 31 hpi) (B) developmental stage were incubated at the indicated doses of PPQ until 48 hpi, after which drug was washed out. Parasite viability was determined by flow cytometry at 72 hpi. Bar graphs correspond to the mean plus SEM percent survival, equivalent to the parasitemia of PPQ-treated parasites divided by the parasitemia of vehicle control (0.5% lactic acid)-treated parasites. Data are from two to four independent experiments, conducted in duplicate. Statistical significance was determined by two-tailed Student’s t tests (Graphpad Prism 7). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Intriguingly, for PSAs initiated during the mid-trophozoite stage, when expression of PfCRT is maximal, we observed increased PPQ susceptibilities with the Cam783, GB4, and 3D7 alleles compared to the Dd2 allele at 100 nM, 50 nM, and 25 nM PPQ (Fig. 4B). The increased PPQ susceptibility at both the ring and trophozoite stages of the Cam783 allele, compared with the isogenic Dd2 pfcrt-expressing allele, suggests a role for the N326S mutation (present in Dd2) in reducing susceptibility to PPQ (Fig. 4B; P < 0.05 and P < 0.01 at 100 nM and 50 nM, respectively, as determined by two-tailed Student’s t test). We did not observe any differences in the survival of parasites with the FCB and Dd2 alleles, arguing against a role for residue 356 in modulating parasite susceptibility to PPQ (Fig. 4B).
In vitro growth of pfcrt-modified lines.
Plasmodium fitness is a multifaceted property that reflects the reproductive success of parasites over multiple rounds of infection. Previous analyses of isogenic parasites expressing variant pfcrt alleles have shed light on their ability to influence growth rates and have uncovered important inferences regarding their global spread (22, 45). Here, we used an established flow cytometry-based coculture assay that compares growth of green fluorescent protein (GFP)-negative (GFP–) pfcrt-modified test lines with that of a GFP-positive (GFP+) reporter line (22) (see Materials and Methods). The GFP– proportion of coculture was regularly determined for 10 parasite generations and then used to derive the per-generation selection coefficient (s) associated with a given pfcrt allele. This parameter was used as a reflection of asexual blood-stage fitness, with s = 0, s > 0, and s < 0 indicating fitness equal to, greater than, and less than that of Dd23D7 parasites (i.e., parasites with wild-type alleles at both the pfcrt and k13 loci).
Our results revealed several key differences in the fitness costs associated with mutant pfcrt alleles (Fig. 5; Fig. S3 and Table S6). Among PfCRT-matched lines expressing the 3D7, Cam783, or Dd2 haplotype, those harboring mutant R539T K13 showed a more pronounced, statistically significant growth defect compared to parasites with wild-type K13 (compare panels A and B in Fig. S3). In both the Dd2 and Dd2R539T genetic backgrounds, the Dd2 and pfcrt FCB alleles both conferred a notable fitness cost (range of mean s values, −0.16 to −0.24) compared with wild-type 3D7. Interestingly, the pfcrt GB4 and Cam783 alleles were associated with a less severe fitness cost (range of mean s values, −0.09 to −0.04) compared with the Dd2 and FCB alleles. Given that GB4 and Cam783 are the two predominant mutant PfCRT isoforms found in Africa (Fig. 1), this finding implicates fitness as a driver of haplotype selection in this region (46).
FIG 5.
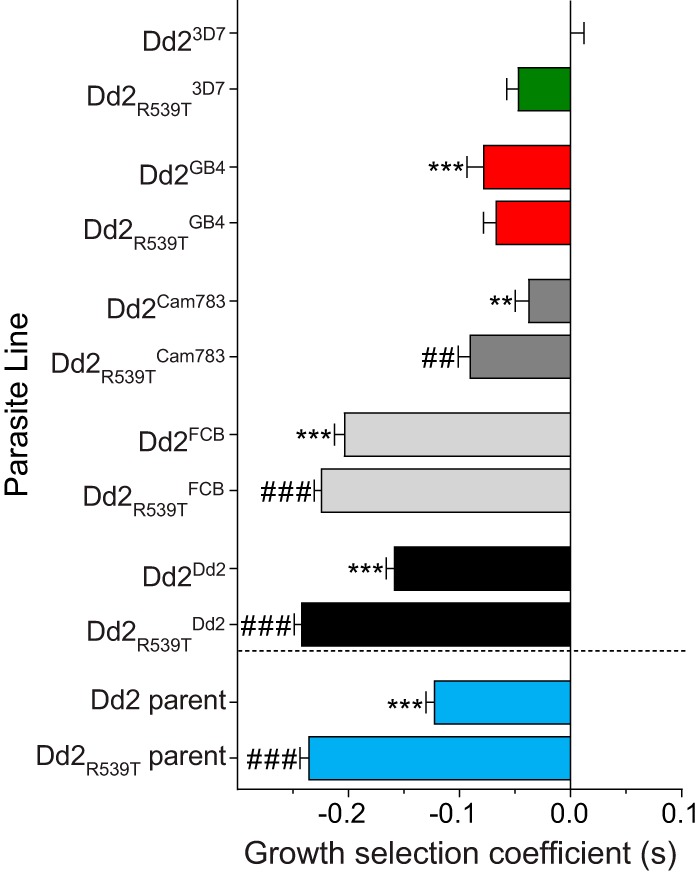
In vitro growth characteristics of pfcrt-modified and reference parasite lines. Cocultures consisting of a 1:1 ratio of the GFP– test strain and a GFP+ reporter strain (RGFP) were initiated at day 0 and were monitored by flow cytometry for 10 generations. The per-generation selection coefficient (s) for each test strain was determined as described in Text S1 and reflects parasite growth compared to Dd23D7 parasites, which carry the wild-type (3D7) pfcrt allele (i.e., s = 0 for Dd23D7). The bars depict mean plus SEM s values (detailed in Table S6), as determined in three independent assays, conducted in duplicate. Statistical significance between different PfCRT haplotypes was determined against Dd23D7 (**, P < 0.01; ***, P < 0.001) or Dd2R539T3D7 (##, P < 0.01; ###, P < 0.001) by two-way ANOVA with Sidak’s post hoc test using GraphPad Prism 7 software.
Fitness assay of pfcrt-modified lines. In vitro growth, used as a proxy of fitness of pfcrt-modified lines in wild-type Dd2 K13 (A) or a K13 R539T (B) genetic background. Parasite lines were cocultured with a GFP+ reporter Dd2 line in equal proportions for a period of 20 days (10 parasite generations). Parasitemia was measured using flow cytometry. Data are presented as means ± SEM from three independent experiments performed in duplicate. Download FIG S3, EPS file, 0.9 MB (931.6KB, eps) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In vitro growth selection coefficients of pfcrt-modified and reference parasite lines. Download Table S6, PDF file, 0.1 MB (111.6KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
By leveraging a large collection of whole-genome sequence data from African and Asian P. falciparum isolates, our study presents new insights into the global distribution of pfcrt alleles. In Africa, we observe that two-thirds of the sampled isolates now carry wild-type, CQ-sensitive pfcrt, with the CQ-resistant alleles being predominantly GB4 and Cam783. Notably, the Cam783 allele has previously not been reported in Africa, nor has it been dissected for its drug susceptibility profile. The presence of the Cam783 allele in Africa (ranging between 10% in Senegal to 79% in The Gambia; Fig. 1; see also Table S1 in the supplemental material) also indicates a possible migratory event from Asia to Africa that had remained undetected due to the low numbers of isolates sampled in prior studies (34). Alternatively, Cam783 might have evolved from the Dd2 allele via loss of the single N326S mutation, resulting in an allele that maintained a high level of CQ resistance and relatively improved parasite fitness (Fig. 2 and 5; Fig. S3 and Table S1).
The FCB allele, which was earlier suggested to have originally migrated from SE Asia to Africa (26, 34, 35), was observed at a prevalence of only 0.5% in our cohort of 783 genomes. Our analysis of African and Asian parasite genomes suggests that GB4 might have been an original migratory haplotype or might have evolved from FCB in Africa (via loss of the N326S mutation) and spread as a result of GB4’s improved fitness.
Using ZFN-based gene editing, we investigated the pleiotropic roles of PfCRT amino acid positions 326 and 356, which separate the majority of haplotypes circulating in Africa or Asia. Results show that the GB4 haplotype, which is wild type at these residues, is less resistant to CQ, md-CQ, and the ADQ metabolite md-ADQ compared to Dd2. The African Cam783 haplotype that is wild type at residue 326 also showed significant decrease in md-ADQ resistance compared to Dd2. Both the GB4 and Cam783 haplotypes nonetheless conferred higher md-ADQ IC50 values compared to the drug-sensitive wild-type 3D7 haplotype.
Most pfcrt alleles increased parasite susceptibility to LMF, as previously observed with the Dd2 haplotype and in agreement with clinical reports showing that LMF tends to select against pfcrt variant alleles in favor of the wild-type allele (27, 36, 47). Our drug susceptibility profiling data support the use of both ATM+LMF and AS+ADQ in regions of endemicity, as they appear to exert opposing selective pressure on pfcrt mutant and wild-type alleles, thus creating bottlenecks that could suppress multidrug resistance.
Earlier studies with isogenic, pfcrt-modified parasites support the notion that Dd2 PfCRT confers a substantial fitness cost to parasites, compared with the Cambodian Cam734 and South American/western Pacific 7G8 isoforms (45). Our current data suggest that parasites expressing Dd2 PfCRT persist in SE Asia, but not in Africa. Their persistence in Asia might be caused in part by the continued local use of CQ to treat Plasmodiumvivax infections, producing sufficient drug pressure to also select for mutant pfcrt in individuals exposed to both plasmodial species. Another explanation could be the reduced use of LMF in most countries in SE Asia, thus reducing selection pressure against Dd2 pfcrt. SE Asia also experiences a much lower level of overall transmission compared to Africa, translating in the former case to fewer mixed infections and greater population reliance on the use of drugs to resolve P. falciparum infections. We speculate that the epidemiological situation in Africa, where asymptomatic adults can frequently harbor multiple strains and use antimalarials relatively infrequently, provides for within-host selection that favors rapidly proliferating strains and selects against strains where resistance places them at a competitive growth disadvantage.
One impetus for our study was the report that the PfCRT N326S and I356T mutations present in Dd2 were associated with K13 mutations that have emerged in areas of emerging ART resistance (20). Therefore, we also constructed these mutant pfcrt alleles in a k13 mutant genetic background (K13 R539T) that affords elevated ART resistance, as defined using the in vitro RSA with very young rings. Our results showed no protective effect of mutant PfCRT in ring-stage survival, including at the 700 nM DHA concentration that has been validated as a surrogate for in vitro resistance and clinical efficacy (17). When tested with trophozoites, we observed that the pfcrt wild-type and GB4 alleles provided a small but significant survival advantage at lower concentrations of DHA compared to the Dd2 allele (Fig. 3B).
These findings suggest that the appearance in Asia of K13 mutations on the Dd2 background, beginning in Cambodia, might relate to other aspects of PfCRT function. For instance, ART toxicity to Plasmodium parasites is thought to possibly involve the induction of oxidative stress, as suggested by prior real-time studies of the parasite glutathione (GSH)-dependent redox state (48). Given the prospect that GSH is potentially one of the substrates of PfCRT (49, 50), it is plausible that the Dd2 form of PfCRT may help P. falciparum avert oxidative damage, perhaps through PfCRT-mediated transport of GSH into the parasite digestive vacuole (DV), where GSH has been proposed to degrade toxic heme (51, 52). Alternatively, these associations might simply reflect the initial emergence of mutant K13 on founder populations that harbored Dd2 PfCRT, whose N326S and I356T mutations are absent in the other common Asian haplotypes Cam734 and GB4 (Fig. 1 and Table 1).
Hemoglobin catabolism and heme detoxification, which occur primarily in the parasite’s DV and which furnish the parasite with its primary source of amino acids, are central to the modes of action and/or resistance to a wide array of ACT drugs (6, 23, 53, 54). PfCRT’s presence as a transporter on the DV membrane, where it helps regulate multiple physiological parameters, including hemoglobin-derived peptide and heme levels, DV pH, and ion homeostasis, in addition to its capacities to mediate drug efflux, position it to directly or indirectly alter interactions of multiple ACT drugs with heme moieties or hemozoin. Of these, the ability of certain PfCRT variant haplotypes to mediate parasite resistance to the CQ-like bisquinoline PPQ is of particular concern, as recent reports document a rapid emergence and spread of PPQ resistance. DHA+PPQ treatment failure rates exceeding 50% have now been reported in western Cambodia (55–58). Recent gene editing data, including Cambodian isolates, now provide compelling evidence that several novel pfcrt alleles that emerged on the Dd2 background have rapidly increased in prevalence in the past few years and can contribute to high-level PPQ resistance (30). These alleles were observed in parasites harboring multiple copies of the hemoglobinases plasmepsin II and III, with this gene amplification previously identified as a marker of PPQ-resistant parasites (59–62). Evidence suggests that plasmepsin II and III amplification might correct for fitness costs observed in pfcrt variants, exemplified by their abnormal DV morphologies and presumably a result of impaired peptide processing and efflux out of the DV (30). These novel pfcrt alleles are all derived from the Dd2 haplotype and include the additional mutations H97Y, F145I, and G353V. The use of Dd2 as the founder haplotype is consistent with our findings that this isoform mediates the highest level of parasite survival in PSAs conducted with trophozoites (Fig. 4B). Our study helps explain this finding by showing a role for the N326S mutation in particular in augmenting PSA survival beginning with trophozoites as well as ring stages. These data emphasize the value of delineating molecular markers that can help identify and contain the spread of PPQ resistance in SE Asia.
ADQ is a second ACT partner drug of particular interest with regard to mutant pfcrt alleles given the widespread use of AS+ADQ for the treatment and prevention of malaria in Africa. ADQ also comprises the formulation ADQ plus sulfadoxine-pyrimethamine, which is administered prophylactically to young children in some areas of sub-Saharan Africa during months of high transmission of malaria. Recent clinical studies have documented a lower incidence of parasite recrudescence following treatment with AS+ADQ compared to ATM+LMF, the two most widely deployed regimens that are known to select for mutant and wild-type pfcrt-expressing parasites, respectively (63). Based on the wide prevalence of the wild-type pfcrt allele in Africa and the paucity of pfcrt alleles that conferred a high degree of ADQ resistance in our study, the continued use of ADQ-based regimens in Africa seems justified. A meta-analysis of worldwide AS+ADQ efficacy showed a sevenfold-greater risk of parasite recrudescence after AS+ADQ treatment in Asia compared to Africa (64). Given these data, it is possible that only certain pfcrt alleles such as Dd2 are able to confer the minimum degree of ADQ resistance necessary to sustain resistant parasite populations, whereas other mutant pfcrt alleles (e.g., GB4 and Cam783) are more rapidly cleared and, in the context of appropriate dosing, pose less of a threat.
As malaria control efforts intensify in the coming years, their success will depend, in part, on the precise definition and monitoring of markers of parasite multidrug resistance. Our study supports the notion of PfCRT being a pleiotropic mediator of parasite multidrug resistance (21, 27, 33, 65, 66). The results suggest that opposing selective forces can be found in single ACT formulations; for example, in our survival assays, DHA and PPQ provided some evidence of selection for K76 and K76T, respectively (Fig. 3B and 4B). Likewise, these opposing selective forces may also be exerted by the long-lived partner drugs comprising distinct ACT formulations, such as PPQ or ADQ, in particular regions. Consistent with this, we document the ability of PfCRT mutations to modulate parasite LMF, ADQ, and PPQ susceptibility in dissimilar, pfcrt allele-specific ways. Our findings support the practice of antimalarial regimen cycling and underscore the importance of active monitoring of pfcrt genotypes to identify emerging patterns of multidrug resistance and help guide region-specific treatment options.
MATERIALS AND METHODS
Parasite genetic modification and cultivation.
Details regarding our ZFN-based pfcrt modification strategy, verification of appropriate genetic editing, and isolation of genetically modified parasite clones are found in Fig. S1 and Text S1 in the supplemental material. Unless stated otherwise, P. falciparum-infected human erythrocytes were cultured at 4% hematocrit in RPMI 1640 culture medium supplemented with 0.5% Albumax II (Invitrogen) (67). Cultures were incubated at 37°C in 5% O2/5% CO2/90% N2.
Analysis of MalariaGEN Pf3K field isolate genome data.
pfcrt sequence analyses were performed on genome data generated by the Plasmodium falciparum Community Genome Project (https://www.malariagen.net/projects/pf3k; pilot data release 3). This data set comprised 2,512 samples from 14 countries, acquired mostly in 2011-2012 (9). Assembly files were downloaded from the publicly accessible database ftp://ngs.sanger.ac.uk/production/pf3k/release_3/3.1/ , along with an additional 87 genomes obtained from Cambodian isolates in 2012 and 2013 deposited at the European Bioinformatics Institute (9, 20). SNPs were manually extracted using the Pf3D7 reference genome version 11.0 as previously described (68). Briefly, we identified all SNPs and rare variants present in the pfcrt genes of the field isolates based on the criteria of an alternate allele frequency of >0.4. To assign the correct PfCRT haplotypes, we filtered out samples where the alternate allele count at 12 major codon positions (74, 75, 76, 144, 148, 194, 220, 271, 326, 333, 356, and 371) was below 5. This yielded 783 African and 806 Asian isolates that were analyzed.
Drug susceptibility assays.
The concentrations of antimalarial drugs that were 50% and 90% growth inhibitory (i.e., IC50 and IC90) to asexual blood-stage parasites were determined using a flow cytometry-based assay that detects parasite nuclei and mitochondria using SYBR Green I and MitoTracker Deep Red, respectively (69). Parasites were subjected to treatment with drug (twofold serial dilutions of drug), and parasite growth was determined with an Accuri C6 flow cytometer after 72 h. Assays were performed with CQ with or without 0.8 μM VP, md-CQ, md-ADQ, AS, PPQ, LMF, QN, and PND. To determine VP-mediated reversibility of CQ resistance, the CQ+VP IC50 value was divided by the CQ IC50 value, yielding the CQ response modification index (RMI) (41). VP reversibility was defined as 1 − CQ RMI value. Statistical significance was determined via nonparametric, two-tailed Mann-Whitney U tests using GraphPad Prism 7 software.
DHA and PPQ survival assays.
Stage-specific survival assays were conducted following published protocols, with minor modifications (16, 17, 43, 70). Briefly, 15 ml of parasite culture was synchronized with 5% sorbitol and cultivated through the mature schizont stage (≥0.5% schizont parasitemia). Cultures were heparinized for 30 min at 37°C in RPMI 1640 medium containing 15 U/ml sodium heparin (Millipore) with occasional mild vortexing. Parasites were subsequently subjected to a gradient of 75% Percoll (Sigma-Aldrich) and centrifuged at 3,400 rpm for 15 min. The schizont fraction was washed with RPMI 1640 medium and incubated with human red blood cells (RBCs) for 3 h to initiate a new asexual blood-stage cycle, followed by synchronization with 5% sorbitol to eliminate non-ring parasite forms. Serial dilutions of DHA or PPQ were prealiquoted into 96-well plates using a BioTek precision pipetting system. Parasite cultures at early ring (0 to 3 hpi) or mid-trophozoite (18 to 21 hpi) stages were added to plates containing drug dilutions, at 0.5% parasitemia, 2% hematocrit, and 200-μl final volume per well. Duplicate wells were included for each parasite line and drug concentration. Vehicle controls for DHA and PPQ survival assays were DMSO and 0.5% lactic acid, respectively. For the RSA0-3h, parasites were exposed to 700 nM DHA for 3 h. For the PSA0-3h, parasites were exposed to 200 nM PPQ for 48 h. Following drug incubation, parasites were washed three times with RPMI 1640 medium and cultured in fresh medium until the assay endpoint of 72 h. Parasitemia was assessed by flow cytometry, which correlated with microscopic assessment of parasitemia. Parasite survival was calculated as the parasitemia in the presence of drug divided by the parasitemia in the vehicle control well, expressed as a percentage.
In vitro growth assays.
Growth of asexual blood-stage parasites was assessed in vitro using a previously described method that employs flow cytometric detection of GFP and the far-red fluorescent dye SYTO 61 (22). The latter was used to further separate infected from uninfected red blood cells. Briefly, 1:1 cocultures were established with a GFP-positive (GFP+) fluorescent reporter line (RGFP) and a single GFP-negative (GFP–) pfcrt-modified test line. RGFP in these assays corresponds to the previously generated CQ-resistant Dd2BiP-eGFP line (71). This line uses a BiP promoter to drive enhanced GFP (eGFP) expression and has a genetic background comparable to that of our pfcrt-modified test lines. Using flow cytometry, we calculated the proportion of GFP– parasites (corresponding to the pfcrt-modified test line) every 3 days for 20 days (i.e., 10 asexual blood-stage parasite generations). Cultures were regularly monitored and diluted such that parasitemias never exceeded 8%. Per-generation selection coefficients (s) associated with pfcrt alleles were calculated as described in Text S1. Statistical significance was assessed via two-way ANOVA with Sidak’s post hoc test using GraphPad Prism 7 software.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (R01 AI50234, AI109023, and AI124678 to D.A.F.). S.J.G. is a recipient of a F30 predoctoral fellowship from the NIH (F30 AI114070) and is grateful to the leadership of the Columbia University Medical Scientist Training Program for training support (T32 GM007367). S.M. was supported by a Human Frontier Science Program (HFSP) postdoctoral fellowship (LT000976/2016-L). J.L.S.-S. gratefully acknowledges the Columbia Integrated Training Program in Infectious Diseases Research (T32 AI100852; PI, S. Hammer) for training grant support.
S.K.D., S.J.G., and D.A.F. conceived and designed the study. S.K.D., S.J.G., T.Y., P.P.H., and S.M. acquired and analyzed the data. S.K.D., S.J.G., J.L.S.-S., and D.A.F. wrote the manuscript with input from all authors.
Footnotes
[In the originally published version, there was a typesetting error in the abstract. The article was updated online on 7 May 2019.]
Citation Dhingra SK, Gabryszewski SJ, Small-Saunders JL, Yeo T, Henrich PP, Mok S, Fidock DA. 2019. Global spread of mutant PfCRT and its pleiotropic impact on Plasmodium falciparum multidrug resistance and fitness. mBio 10:e02731-18. https://doi.org/10.1128/mBio.02731-18.
REFERENCES
- 1.World Health Organization. 2018. World Malaria Report 2018. World Health Organization, Geneva, Switzerland. https://www.who.int/malaria/publications/world-malaria-report-2018/en/.
- 2.Blasco B, Leroy D, Fidock DA. 2017. Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat Med 23:917–928. doi: 10.1038/nm.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sa JM, Chong JL, Wellems TE. 2011. Malaria drug resistance: new observations and developments. Essays Biochem 51:137–160. doi: 10.1042/bse0510137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. 2004. Intercontinental spread of pyrimethamine-resistant malaria. Science 305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- 5.Miller LH, Ackerman HC, Su XZ, Wellems TE. 2013. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med 19:156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klonis N, Creek DJ, Tilley L. 2013. Iron and heme metabolism in Plasmodium falciparum and the mechanism of action of artemisinins. Curr Opin Microbiol 16:722–727. doi: 10.1016/j.mib.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dondorp AM, Fairhurst RM, Slutsker L, Macarthur JR, Breman JG, Guerin PJ, Wellems TE, Ringwald P, Newman RD, Plowe CV. 2011. The threat of artemisinin-resistant malaria. N Engl J Med 365:1073–1075. doi: 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MalariaGEN Plasmodium falciparum Community Project. 2016. Genomic epidemiology of artemisinin resistant malaria. Elife 5:e08714. doi: 10.7554/eLife.08714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menard D, Khim N, Beghain J, Adegnika AA, Shafiul-Alam M, Amodu O, Rahim-Awab G, Barnadas C, Berry A, Boum Y, Bustos MD, Cao J, Chen JH, Collet L, Cui L, Thakur GD, Dieye A, Djalle D, Dorkenoo MA, Eboumbou-Moukoko CE, Espino FE, Fandeur T, Ferreira-da-Cruz MF, Fola AA, Fuehrer HP, Hassan AM, Herrera S, Hongvanthong B, Houze S, Ibrahim ML, Jahirul-Karim M, Jiang L, Kano S, Ali-Khan W, Khanthavong M, Kremsner PG, Lacerda M, Leang R, Leelawong M, Li M, Lin K, Mazarati JB, Menard S, Morlais I, Muhindo-Mavoko H, Musset L, Na-Bangchang K, Nambozi M, Niare K, Noedl H, et al. 2016. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N Engl J Med 374:2453–2464. doi: 10.1056/NEJMoa1513137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, Wells TNC. 2017. Malaria. Nat Rev Dis Primers 3:17050. doi: 10.1038/nrdp.2017.50. [DOI] [PubMed] [Google Scholar]
- 12.Woodrow CJ, White NJ. 2017. The clinical impact of artemisinin resistance in Southeast Asia and the potential for future spread. FEMS Microbiol Rev 41:34–48. doi: 10.1093/femsre/fuw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Ménard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale J-C, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Ménard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han K-T, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. 2014. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 32:819–821. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- 16.Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Menard D, Fidock DA. 2015. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, Kim S, Lim P, Mao S, Sopha C, Sam B, Anderson JM, Duong S, Chuor CM, Taylor WR, Suon S, Mercereau-Puijalon O, Fairhurst RM, Menard D. 2013. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 13:1043–1049. doi: 10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor SM, Parobek CM, DeConti DK, Kayentao K, Coulibaly SO, Greenwood BM, Tagbor H, Williams J, Bojang K, Njie F, Desai M, Kariuki S, Gutman J, Mathanga DP, Martensson A, Ngasala B, Conrad MD, Rosenthal PJ, Tshefu AK, Moormann AM, Vulule JM, Doumbo OK, Ter Kuile FO, Meshnick SR, Bailey JA, Juliano JJ. 2015. Absence of putative artemisinin resistance mutations among Plasmodium falciparum in Sub-Saharan Africa: a molecular epidemiologic study. J Infect Dis 211:680–688. doi: 10.1093/infdis/jiu467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamau E, Campino S, Amenga-Etego L, Drury E, Ishengoma D, Johnson K, Mumba D, Kekre M, Yavo W, Mead D, Bouyou-Akotet M, Apinjoh T, Golassa L, Randrianarivelojosia M, Andagalu B, Maiga-Ascofare O, Amambua-Ngwa A, Tindana P, Ghansah A, MacInnis B, Kwiatkowski D, Djimde AA. 2015. K13-propeller polymorphisms in Plasmodium falciparum parasites from sub-Saharan Africa. J Infect Dis 211:1352–1355. doi: 10.1093/infdis/jiu608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, Lim P, Mead D, Oyola SO, Dhorda M, Imwong M, Woodrow C, Manske M, Stalker J, Drury E, Campino S, Amenga-Etego L, Thanh TN, Tran HT, Ringwald P, Bethell D, Nosten F, Phyo AP, Pukrittayakamee S, Chotivanich K, Chuor CM, Nguon C, Suon S, Sreng S, Newton PN, Mayxay M, Khanthavong M, Hongvanthong B, Htut Y, Han KT, Kyaw MP, Faiz MA, Fanello CI, Onyamboko M, Mokuolu OA, Jacob CG, Takala-Harrison S, Plowe CV, Day NP, Dondorp AM, Spencer CC, McVean G, Fairhurst RM, White NJ, Kwiatkowski DP. 2015. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 47:226–234. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sidhu AB, Verdier-Pinard D, Fidock DA. 2002. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabryszewski SJ, Modchang C, Musset L, Chookajorn T, Fidock DA. 2016. Combinatorial genetic modeling of pfcrt-mediated drug resistance evolution in Plasmodium falciparum. Mol Biol Evol 33:1554–1570. doi: 10.1093/molbev/msw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Combrinck JM, Mabotha TE, Ncokazi KK, Ambele MA, Taylor D, Smith PJ, Hoppe HC, Egan TJ. 2013. Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem Biol 8:133–137. doi: 10.1021/cb300454t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis IA, Wacker M, Olszewski KL, Cobbold SA, Baska KS, Tan A, Ferdig MT, Llinas M. 2014. Metabolic QTL analysis links chloroquine resistance in Plasmodium falciparum to impaired hemoglobin catabolism. PLoS Genet 10:e1004085. doi: 10.1371/journal.pgen.1004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee AH, Dhingra SK, Lewis IA, Singh MK, Siriwardana A, Dalal S, Rubiano K, Klein MS, Baska KS, Krishna S, Klemba M, Roepe PD, Llinas M, Garcia CRS, Fidock DA. 2018. Evidence for regulation of hemoglobin metabolism and intracellular ionic flux by the Plasmodium falciparum chloroquine resistance transporter. Sci Rep 8:13578. doi: 10.1038/s41598-018-31715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE. 2000. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 6:861–871. doi: 10.1016/S1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sisowath C, Petersen I, Veiga MI, Martensson A, Premji Z, Bjorkman A, Fidock DA, Gil JP. 2009. In vivo selection of Plasmodium falciparum parasites carrying the chloroquine-susceptible pfcrt K76 allele after treatment with artemether-lumefantrine in Africa. J Infect Dis 199:750–757. doi: 10.1086/596738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sa JM, Twu O, Hayton K, Reyes S, Fay MP, Ringwald P, Wellems TE. 2009. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci U S A 106:18883–18889. doi: 10.1073/pnas.0911317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhingra SK, Redhi D, Combrinck JM, Yeo T, Okombo J, Henrich PP, Cowell AN, Gupta P, Stegman ML, Hoke JM, Cooper RA, Winzeler E, Mok S, Egan TJ, Fidock DA. 2017. A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. mBio 8:e00303-17. doi: 10.1128/mBio.00303-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross LS, Dhingra SK, Mok S, Yeo T, Wicht KJ, Kumpornsin K, Takala-Harrison S, Witkowski B, Fairhurst RM, Ariey F, Menard D, Fidock DA. 2018. Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat Commun 17:3314. doi: 10.1038/s41467-018-05652-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin RE, Shafik SH, Richards SN. 2018. Mechanisms of resistance to the partner drugs of artemisinin in the malaria parasite. Curr Opin Pharmacol 42:71–80. doi: 10.1016/j.coph.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Gabryszewski SJ, Dhingra SK, Combrinck JM, Lewis IA, Callaghan PS, Hassett MR, Siriwardana A, Henrich PP, Lee AH, Gnadig NF, Musset L, Llinas M, Egan TJ, Roepe PD, Fidock DA. 2016. Evolution of fitness cost-neutral mutant PfCRT conferring P. falciparum 4-aminoquinoline drug resistance is accompanied by altered parasite metabolism and digestive vacuole physiology. PLoS Pathog 12:e1005976. doi: 10.1371/journal.ppat.1005976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Callaghan PS, Hassett MR, Roepe PD. 2015. Functional comparison of 45 naturally occurring isoforms of the Plasmodium falciparum chloroquine resistance transporter (PfCRT). Biochemistry 54:5083–5094. doi: 10.1021/acs.biochem.5b00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorsey G, Kamya MR, Singh A, Rosenthal PJ. 2001. Polymorphisms in the Plasmodium falciparum pfcrt and pfmdr-1 genes and clinical response to chloroquine in Kampala, Uganda. J Infect Dis 183:1417–1420. doi: 10.1086/319865. [DOI] [PubMed] [Google Scholar]
- 35.Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ, Su XZ. 2002. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 418:320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- 36.Venkatesan M, Gadalla NB, Stepniewska K, Dahal P, Nsanzabana C, Moriera C, Price RN, Mårtensson A, Rosenthal PJ, Dorsey G, Sutherland CJ, Guérin P, Davis TME, Ménard D, Adam I, Ademowo G, Arze C, Baliraine FN, Berens-Riha N, Björkman A, Borrmann S, Checchi F, Desai M, Dhorda M, Djimdé AA, El-Sayed BB, Eshetu T, Eyase F, Falade C, Faucher J-F, Fröberg G, Grivoyannis A, Hamour S, Houzé S, Johnson J, Kamugisha E, Kariuki S, Kiechel J-R, Kironde F, Kofoed P-E, LeBras J, Malmberg M, Mwai L, Ngasala B, Nosten F, Nsobya SL, Nzila A, Oguike M, Otienoburu SD, Ogutu B, Ouédraogo J-B, et al. 2014. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am J Trop Med Hyg 91:833–843. doi: 10.4269/ajtmh.14-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, Taylor TE, Plowe CV. 2006. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med 355:1959–1966. doi: 10.1056/NEJMoa062032. [DOI] [PubMed] [Google Scholar]
- 38.Frosch AE, Laufer MK, Mathanga DP, Takala-Harrison S, Skarbinski J, Claassen CW, Dzinjalamala FK, Plowe CV. 2014. Return of widespread chloroquine-sensitive Plasmodium falciparum to Malawi. J Infect Dis 210:1110–1114. doi: 10.1093/infdis/jiu216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eastman RT, Fidock DA. 2009. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat Rev Microbiol 7:864–874. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin SK, Oduola AM, Milhous WK. 1987. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science 235:899–901. doi: 10.1126/science.3544220. [DOI] [PubMed] [Google Scholar]
- 41.Mehlotra RK, Fujioka H, Roepe PD, Janneh O, Ursos LM, Jacobs-Lorena V, McNamara DT, Bockarie MJ, Kazura JW, Kyle DE, Fidock DA, Zimmerman PA. 2001. Evolution of a unique Plasmodium falciparum chloroquine-resistance phenotype in association with pfcrt polymorphism in Papua New Guinea and South America. Proc Natl Acad Sci U S A 98:12689–12694. doi: 10.1073/pnas.221440898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amaratunga C, Witkowski B, Khim N, Menard D, Fairhurst RM. 2014. Artemisinin resistance in Plasmodium falciparum. Lancet Infect Dis 14:449–450. doi: 10.1016/S1473-3099(14)70777-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duru V, Khim N, Leang R, Kim S, Domergue A, Kloeung N, Ke S, Chy S, Eam R, Khean C, Loch K, Ken M, Lek D, Beghain J, Ariey F, Guerin PJ, Huy R, Mercereau-Puijalon O, Witkowski B, Menard D. 2015. Plasmodium falciparum dihydroartemisinin-piperaquine failures in Cambodia are associated with mutant K13 parasites presenting high survival rates in novel piperaquine in vitro assays: retrospective and prospective investigations. BMC Med 13:305. doi: 10.1186/s12916-015-0539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tarning J, Ashley EA, Lindegardh N, Stepniewska K, Phaiphun L, Day NP, McGready R, Ashton M, Nosten F, White NJ. 2008. Population pharmacokinetics of piperaquine after two different treatment regimens with dihydroartemisinin-piperaquine in patients with Plasmodium falciparum malaria in Thailand. Antimicrob Agents Chemother 52:1052–1061. doi: 10.1128/AAC.00955-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petersen I, Gabryszewski SJ, Johnston GL, Dhingra SK, Ecker A, Lewis RE, de Almeida MJ, Straimer J, Henrich PP, Palatulan E, Johnson DJ, Coburn-Flynn O, Sanchez C, Lehane AM, Lanzer M, Fidock DA. 2015. Balancing drug resistance and growth rates via compensatory mutations in the Plasmodium falciparum chloroquine resistance transporter. Mol Microbiol 97:381–395. doi: 10.1111/mmi.13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nair S, Li X, Arya GA, McDew-White M, Ferrari M, Nosten F, Anderson T. 2018. Fitness costs and the rapid spread of kelch13-C580Y substitutions conferring artemisinin resistance. Antimicrob Agents Chemother 62:e00605-18. doi: 10.1128/AAC.00605-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Some AF, Sere YY, Dokomajilar C, Zongo I, Rouamba N, Greenhouse B, Ouedraogo JB, Rosenthal PJ. 2010. Selection of known Plasmodium falciparum resistance-mediating polymorphisms by artemether-lumefantrine and amodiaquine-sulfadoxine-pyrimethamine but not dihydroartemisinin-piperaquine in Burkina Faso. Antimicrob Agents Chemother 54:1949–1954. doi: 10.1128/AAC.01413-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasozi D, Mohring F, Rahlfs S, Meyer AJ, Becker K. 2013. Real-time imaging of the intracellular glutathione redox potential in the malaria parasite Plasmodium falciparum. PLoS Pathog 9:e1003782. doi: 10.1371/journal.ppat.1003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juge N, Moriyama S, Miyaji T, Kawakami M, Iwai H, Fukui T, Nelson N, Omote H, Moriyama Y. 2015. Plasmodium falciparum chloroquine resistance transporter is a H+-coupled polyspecific nutrient and drug exporter. Proc Natl Acad Sci U S A 112:3356–3361. doi: 10.1073/pnas.1417102112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patzewitz EM, Wong EH, Muller S. 2012. Dissecting the role of glutathione biosynthesis in Plasmodium falciparum. Mol Microbiol 83:304–318. doi: 10.1111/j.1365-2958.2011.07933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Famin O, Krugliak M, Ginsburg H. 1999. Kinetics of inhibition of glutathione-mediated degradation of ferriprotoporphyrin IX by antimalarial drugs. Biochem Pharmacol 58:59–68. doi: 10.1016/S0006-2952(99)00059-3. [DOI] [PubMed] [Google Scholar]
- 52.Lehane AM, McDevitt CA, Kirk K, Fidock DA. 2012. Degrees of chloroquine resistance in Plasmodium - is the redox system involved? Int J Parasitol Drugs Drug Resist 2:47–57. doi: 10.1016/j.ijpddr.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawley SR, Bray PG, Mungthin M, Atkinson JD, O’Neill PM, Ward SA. 1998. Relationship between antimalarial drug activity, accumulation, and inhibition of heme polymerization in Plasmodium falciparum in vitro. Antimicrob Agents Chemother 42:682–686. doi: 10.1128/AAC.42.3.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorka AP, de Dios A, Roepe PD. 2013. Quinoline drug-heme interactions and implications for antimalarial cytostatic versus cytocidal activities. J Med Chem 56:5231–5246. doi: 10.1021/jm400282d. [DOI] [PubMed] [Google Scholar]
- 55.Saunders DL, Vanachayangkul P, Lon C, US Army Military Malaria Research Program, National Center for Parasitology, Entomology, and Malaria Control (CNM), Royal Cambodian Armed Forces. 2014. Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med 371:484–485. doi: 10.1056/NEJMc1403007. [DOI] [PubMed] [Google Scholar]
- 56.Chaorattanakawee S, Saunders DL, Sea D, Chanarat N, Yingyuen K, Sundrakes S, Saingam P, Buathong N, Sriwichai S, Chann S, Se Y, Yom Y, Heng TK, Kong N, Kuntawunginn W, Tangthongchaiwiriya K, Jacob C, Takala-Harrison S, Plowe C, Lin JT, Chuor CM, Prom S, Tyner SD, Gosi P, Teja-Isavadharm P, Lon C, Lanteri CA. 2015. Ex vivo drug susceptibility and molecular profiling of clinical Plasmodium falciparum isolates from Cambodia in 2008-2013 suggest emerging piperaquine resistance. Antimicrob Agents Chemother 59:4631–4643. doi: 10.1128/AAC.00366-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leang R, Taylor WR, Bouth DM, Song L, Tarning J, Char MC, Kim S, Witkowski B, Duru V, Domergue A, Khim N, Ringwald P, Menard D. 2015. Evidence of Plasmodium falciparum malaria multidrug resistance to artemisinin and piperaquine in western Cambodia: dihydroartemisinin-piperaquine open-label multicenter clinical assessment. Antimicrob Agents Chemother 59:4719–4726. doi: 10.1128/AAC.00835-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amaratunga C, Lim P, Suon S, Sreng S, Mao S, Sopha C, Sam B, Dek D, Try V, Amato R, Blessborn D, Song L, Tullo GS, Fay MP, Anderson JM, Tarning J, Fairhurst RM. 2016. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis 16:357–365. doi: 10.1016/S1473-3099(15)00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Witkowski B, Duru V, Khim N, Ross LS, Saintpierre B, Beghain J, Chy S, Kim S, Ke S, Kloeung N, Eam R, Khean C, Ken M, Loch K, Bouillon A, Domergue A, Ma L, Bouchier C, Leang R, Huy R, Nuel G, Barale J-C, Legrand E, Ringwald P, Fidock DA, Mercereau-Puijalon O, Ariey F, Ménard D. 2017. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype-genotype association study. Lancet Infect Dis 17:174–183. doi: 10.1016/S1473-3099(16)30415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amato R, Lim P, Miotto O, Amaratunga C, Dek D, Pearson RD, Almagro-Garcia J, Neal AT, Sreng S, Suon S, Drury E, Jyothi D, Stalker J, Kwiatkowski DP, Fairhurst RM. 2017. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype-phenotype association study. Lancet Infect Dis 17:164–173. doi: 10.1016/S1473-3099(16)30409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Agrawal S, Moser KA, Morton L, Cummings MP, Parihar A, Dwivedi A, Shetty AC, Drabek EF, Jacob CG, Henrich PP, Parobek CM, Jongsakul K, Huy R, Spring MD, Lanteri CA, Chaorattanakawee S, Lon C, Fukuda MM, Saunders DL, Fidock DA, Lin JT, Juliano JJ, Plowe CV, Silva JC, Takala-Harrison S. 2017. Association of a novel mutation in the Plasmodium falciparum chloroquine resistance transporter with decreased piperaquine sensitivity. J Infect Dis 216:468–476. doi: 10.1093/infdis/jix334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bopp S, Magistrado P, Wong W, Schaffner SF, Mukherjee A, Lim P, Dhorda M, Amaratunga C, Woodrow CJ, Ashley EA, White NJ, Dondorp AM, Fairhurst RM, Ariey F, Menard D, Wirth DF, Volkman SK. 2018. Plasmepsin II-III copy number accounts for bimodal piperaquine resistance among Cambodian Plasmodium falciparum. Nat Commun 9:1769. doi: 10.1038/s41467-018-04104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeka A, Kigozi R, Conrad MD, Lugemwa M, Okui P, Katureebe C, Belay K, Kapella BK, Chang MA, Kamya MR, Staedke SG, Dorsey G, Rosenthal PJ. 2016. Artesunate/amodiaquine versus artemether/lumefantrine for the treatment of uncomplicated malaria in Uganda: a randomized trial. J Infect Dis 213:1134–1142. doi: 10.1093/infdis/jiv551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.WorldWide Antimalarial Resistance Network (WWARN) AS-AQ Study Group, Adjuik MA, Allan R, Anvikar AR, Ashley EA, Ba MS, Barennes H, Barnes KI, Bassat Q, Baudin E, Bjorkman A, Bompart F, Bonnet M, Borrmann S, Brasseur P, Bukirwa H, Checchi F, Cot M, Dahal P, D'Alessandro U, Deloron P, Desai M, Diap G, Djimde AA, Dorsey G, Doumbo OK, Espie E, Etard JF, Fanello CI, Faucher JF, Faye B, Flegg JA, Gaye O, Gething PW, Gonzalez R, Grandesso F, Guerin PJ, Guthmann JP, Hamour S, Hasugian AR, Hay SI, Humphreys GS, Jullien V, Juma E, Kamya MR, Karema C, Kiechel JR, Kremsner PG, Krishna S, Lameyre V, et al. 2015. The effect of dosing strategies on the therapeutic efficacy of artesunate-amodiaquine for uncomplicated malaria: a meta-analysis of individual patient data. BMC Med 13:66. doi: 10.1186/s12916-015-0301-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pelleau S, Moss EL, Dhingra SK, Volney B, Casteras J, Gabryszewski SJ, Volkman SK, Wirth DF, Legrand E, Fidock DA, Neafsey DE, Musset L. 2015. Adaptive evolution of malaria parasites in French Guiana: reversal of chloroquine resistance by acquisition of a mutation in pfcrt. Proc Natl Acad Sci U S A 112:11672–11677. doi: 10.1073/pnas.1507142112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Schalkwyk DA, Nash MN, Shafik SH, Summers RL, Lehane AM, Smith PJ, Martin RE. 2016. Verapamil-sensitive transport of quinacrine and methylene blue via the Plasmodium falciparum chloroquine resistance transporter reduces the parasite's susceptibility to these tricyclic drugs. J Infect Dis 213:800–810. doi: 10.1093/infdis/jiv509. [DOI] [PubMed] [Google Scholar]
- 67.Fidock DA, Nomura T, Wellems TE. 1998. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol Pharmacol 54:1140–1147. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- 68.Veiga MI, Dhingra SK, Henrich PP, Straimer J, Gnadig N, Uhlemann AC, Martin RE, Lehane AM, Fidock DA. 2016. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat Commun 7:11553. doi: 10.1038/ncomms11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ekland EH, Schneider J, Fidock DA. 2011. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J 25:3583–3593. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amaratunga C, Neal AT, Fairhurst RM. 2014. Flow cytometry-based analysis of artemisinin-resistant Plasmodium falciparum in the ring-stage survival assay. Antimicrob Agents Chemother 58:4938–4940. doi: 10.1128/AAC.02902-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, Proto WR, Blagborough AM, Meister S, Wirjanata G, Ruecker A, Upton LM, Abraham TS, Almeida MJ, Pradhan A, Porzelle A, Martinez MS, Bolscher JM, Woodland A, Norval S, Zuccotto F, Thomas J, Simeons F, Stojanovski L, Osuna-Cabello M, Brock PM, Churcher TS, Sala KA, Zakutansky SE, Jimenez-Diaz MB, Sanz LM, Riley J, Basak R, Campbell M, Avery VM, Sauerwein RW, Dechering KJ, Noviyanti R, Campo B, Frearson JA, Angulo-Barturen I, Ferrer-Bazaga S, Gamo FJ, Wyatt PG, Leroy D, Siegl P, Delves MJ, Kyle DE, Wittlin S, Marfurt J, Price RN, et al. 2015. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522:315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Frequency distribution of PfCRT haplotypes from the Pf3K data set across Africa. Download Table S1, PDF file, 0.02 MB (19.2KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Frequency distribution of PfCRT haplotypes from the Pf3K data set across Asia. Download Table S2, PDF file, 0.02 MB (16.7KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PfCRT haplotype distribution across Asia and Africa, as derived from the Pf3K data set. Download Table S3, PDF file, 0.1 MB (151.4KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Allelic modification of pfcrt and validation of genetic editing. (A) Zinc-finger nuclease (ZFN)-based genetic engineering strategy. As detailed in Text S1, parasites were first enriched with a donor plasmid (pcrt-hdhfr) that encodes pfcrt exons 2 to 13 (e2-13) and includes mutations of interest (indicated in red). A comprehensive list of PfCRT haplotypes that were introduced into Dd2 or Dd2R539T parasites is found in Table 1. The pcrt-hdhfr plasmid also comprises the following elements: Plasmodiumberghei crt (pbcrt) 3′ UTR, human dhfr (hdhfr) selection cassette, as well as pfcrt-hdhfr-flanking homology regions (∼0.4 kb upstream of the intron 1-exon 2 junction and ∼1 kb native 3′ UTR downstream of hdhfr). Parasites were subsequently transfected with pZFNcrt-bsd. This plasmid facilitates calmodulin (cam) promoter-driven expression of a pair of pfcrt-targeting ZFNs (ZFNL and ZFNR) and includes the blasticidin S deaminase (bsd) selection cassette. Following ZFN-mediated introduction of a double-stranded break in pfcrt (indicated with a lightning bolt), parasites that employ the pcrt-hdhfr plasmid as a donor template incorporate mutations of interest through homologous recombination-based mechanisms. Genetic editing was evaluated by diagnostic PCR (see panel B), and successfully edited parasites were cloned by limiting dilution. (B) Blood PCR-based verification of pfcrt allelic exchange. Shown are representative results for Dd2 parasites into which the full-length GB4 pfcrt allele was introduced (Dd2GB4) as well as controls, namely, genetically unedited parental parasites (Dd2), unedited donor plasmid-enriched parasites (Dd2 + pcrt-hdhfr), and the sole donor plasmid (pcrt-hdhfr). All appropriately edited lines showed the expected DNA band migration patterns: 0.4 kb (p7+p8), 1.2 kb (p9+p8), 2.5 kb (p10+p11), and 2.7 kb (p9+p12). Primer (p) locations are illustrated in panel A. Download FIG S1, EPS file, 1.9 MB (1.9MB, eps) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental Materials and Methods. Download Text S1, PDF file, 0.2 MB (198.1KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download Table S4, PDF file, 0.02 MB (22.4KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Antimalarial IC50 and IC90 values of pfcrt-modified parasite lines. Download Table S5, PDF file, 0.3 MB (283.6KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Drug susceptibility of pfcrt-modified lines to antimalarial drugs. Parasite susceptibilities to artesunate (AS), piperaquine (PPQ), quinine (QN), and pyronaridine (PND) were assessed after 72-h exposures to the indicated drug. Parasitemia was measured using flow cytometry. Data are presented as mean ± SEM IC50 values for 3 to 10 independent assays performed in duplicate. Statistical significance was determined against Dd2Dd2 or Dd2R539TDd2 by two-tailed Mann-Whitney U tests using Graphpad Prism 7 software. Download FIG S2, EPS file, 0.8 MB (803.3KB, eps) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Fitness assay of pfcrt-modified lines. In vitro growth, used as a proxy of fitness of pfcrt-modified lines in wild-type Dd2 K13 (A) or a K13 R539T (B) genetic background. Parasite lines were cocultured with a GFP+ reporter Dd2 line in equal proportions for a period of 20 days (10 parasite generations). Parasitemia was measured using flow cytometry. Data are presented as means ± SEM from three independent experiments performed in duplicate. Download FIG S3, EPS file, 0.9 MB (931.6KB, eps) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In vitro growth selection coefficients of pfcrt-modified and reference parasite lines. Download Table S6, PDF file, 0.1 MB (111.6KB, pdf) .
Copyright © 2019 Dhingra et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.