

Abstract
Abstract. Objectives: Previously, we have found that the ClC‐3 chloride channel is involved in endothelin‐1 (ET‐1)‐induced rat aortic smooth muscle cell proliferation. The present study was to investigate the role of ClC‐3 in cell cycle progression/distribution and the underlying mechanisms of proliferation. Materials and methods: Small interference RNA (siRNA) is used to silence ClC‐3 expression. Cell proliferation, cell cycle distribution and protein expression were measured or detected with cell counting, bromodeoxyuridine (BrdU) incorporation, Western blot and flow cytometric assays respectively. Results: ET‐1‐induced rat basilar vascular smooth muscle cell (BASMC) proliferation was parallel to a significant increase in endogenous expression of ClC‐3 protein. Silence of ClC‐3 by siRNA inhibited expression of ClC‐3 protein, prevented an increase in BrdU incorporation and cell number induced by ET‐1. Silence of ClC‐3 also caused cell cycle arrest in G0/G1 phase and prevented the cells’ progression from G1 to S phase. Knockdown of ClC‐3 potently inhibited cyclin D1 and cyclin E expression and increased cyclin‐dependent kinase inhibitors (CDKIs) p27KIP and p21CIP expression. Furthermore, ClC‐3 knockdown significantly attenuated phosphorylation of Akt and glycogen synthase kinase‐3β (GSK‐3β) induced by ET‐1. Conclusion: Silence of ClC‐3 protein effectively suppressed phosphorylation of the Akt/GSK‐3β signal pathway, resulting in down‐regulation of cyclin D1 and cyclin E, and up‐regulation of p27KIP and p21CIP. In these BASMCs, integrated effects lead to cell cycle G1/S arrest and inhibition of cell proliferation.
INTRODUCTION
Cell proliferation must at some time point lead to cell volume increase (Bortner & Cidlowski 2004; Lang et al. 2005). This evokes a regulatory volume decrease process through activation of various channels/transporters, including chloride (Cl−) and potassium (K+) channels, which induces effluxes of Cl−, K+ and H2O, and returns the cell volume to normal size (Wondergem et al. 2001; Wang et al. 2002; , Lang et al. 2005) and recent evidence suggests the emerging roles of the ClC‐3 Cl− channel in cell proliferation. Endothelin‐1(ET‐1) induced cell proliferation with an increase in endogenous ClC‐3 protein expression in rat aortic pulmonary artery smooth muscle cells and human prostate cancer epithelial cells (Wang et al. 2002; , Lemonnier et al. 2004; Dai et al. 2005). The antisense protein specific to ClC‐3 Cl− channels significantly inhibited cell proliferation (Wang et al. 2002); however, its roles in cell cycle progression and its underlying mechanisms still remain to be clarified.
Based on our previous findings on the roles of ClC‐3 in cell proliferation, this study aimed to explore how silence of the ClC‐3 channel by small interference RNA (siRNA) influenced basilar artery smooth muscle cells’ (BASMCs) cell cycle progression/distribution, expression of which key proteins are responsible for regulating cell cycle progression and the underlying mechanisms of these processes. We found that here deficiency in the ClC‐3 Cl− channel inhibits cell proliferation through interference in the cell cycle G1/S transition, which is related closely to the Akt/glycogen synthase kinase‐3β (GSK‐3β) signal transduction pathway.
MATERIALS AND METHODS
Cell culture
Male Sprague‐Dawley rats were supplied by the Experimental Animal Center of Sun Yat‐Sen University in Guangzhou, China. All procedures complied with the standards for care and use of animal subjects as stated in the Guide for the Care and Use of Laboratory Animals (issued by the Ministry of Science and Technology of China, Beijing). Basilar arteries were isolated aseptically and placed in Krebs buffer as described previously (Shi et al. 2007). After fat and connective tissue were cleaned away, the artery strip was cut into ~0.5 mm pieces and then was plated in Dulbecco's modified essential medium (DMEM)/F‐12 containing 20% foetal calf serum. BASMCs were cultured in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Cells were identified as smooth muscle cells by morphology and immunostaining with monoclonal antibody specific for smooth muscle α‐actin (Xiao et al. 2002). Passages 8–14 BASMCs at 70–90% confluence was in 35‐mm dishes or 96‐well plates and growth was arrested by incubation in 0.2% calf serum/DMEM/F‐12, 72 h before experiments.
Transfection of BASMCs with stealth siRNA
The sequence of the stealth siRNA duplex oligoribonucleotides against rat ClC‐3 gene (GenBank Accession No. NM_053363) is 5′‐UCACCAAGGAAACUGCAAGAAAGGC‐3′, and its corresponding complementary strand 5′‐GCCUUUCUUGCAGUUUCCUUGGUGA‐3′ (Invitrogen Life Technologies Inc., Carlsbad, CA, USA). The ClC‐3 siRNA was transfected transiently with LipofectamineTM RNAiMAX according to the manufacturer's instructions and a negative stealth siRNA sequence was used as control. Briefly, siRNA and LipofectamineTM RNAiMAX were diluted in Opti‐MEMI, respectively (Invitrogen Life Technologies Inc.). Then, the diluted siRNA duplex and the diluted LipofectamineTM RNAiMAX were combined for 20 min at room temperature to allow formation of transfection complexes. Complexes were then added to the cells while they were in the quiescent state and all were swirled gently to ensure uniform distribution. After incubation for 8 h at 37 °C, transfection mixture was removed and the cells were further incubated with normal growth conditions before use for experiments. The BLOCK‐iT Fluorescent Oligo, siRNA‐labelled fluorescein isothiocyanate (FITC; Invitrogen Life Technologies Inc.) was used for determination of siRNA transfection efficiency.
Cell counting and incorporation of bromodeoxyuridine
Cell proliferation was measured by counting and by bromodeoxyuridine (BrdU) incorporation as described previously (Xiao et al. 2002; Zhang et al. 2006), and according to the manufacturer's instructions (BrdU kits from Calbiochem, San Diego, CA, USA). After incubation, cells were mixed with 10 mmol/L BrdU for 18 h, and then the DNA was denatured by treatment with fixative/denaturing solution for 30 min, in order to enable anti‐BrdU antibody binding to the incorporated BrdU. Subsequently, anti‐BrdU monoclonal antibody was incubated for 1 h at room temperature. After washing, samples were treated with horseradish peroxidase‐conjugated goat antimouse immunoglobulin G for 30 min 100 mmol/L of colourless 3,3′,5,5′‐tetramethylbensidine was then added as the substrate for horseradish peroxidase. The coloured reaction product was quantified by optical density determined at 450–540 nm, using a microplate reader.
Flow cytometry for cell cycle analysis
Cell cycle status was evaluated by flow cytometry as described previously (Zhang et al. 2006). Measurement of propidium iodide uptake was performed using fluorescence activated cell sorting. After appropriate treatment, BASMCs were collected in medium by centrifugation at 200 g for 5 min at 4 °C. Pellets were rinsed once with ice‐cold phosphate‐buffered saline (PBS) and were fixed in 70% ethanol for 24 h. Samples were then stained with staining buffer (PBS containing 50 µg/mL of propidium iodide, 10 µg/mL RNase A, 0.1% sodium citrate and 0.1 Triton X‐100). DNA content was analysed by flow cytometry (EPICS XL, Beckman Coulter, Miami, FL, USA) to characterize population fraction in each phase of the cell cycle.
Western blot analysis
Western blot analysis was performed as described previously (Wang et al. 2002). Briefly, rat BASMCs were rinsed with ice‐cold PBS and lysis buffer: Tris‐HCl 50 mmol/L, NaCl 150 mmol/L, NaN3 0.02%, Nonidet P‐40 1%, sodium dodecyl sulfate 0.1%, sodium deoxycholate 0.5% and 1% protease inhibitor cocktail (Sigma Chemical, St. Louis, MO, USA). Protein was separated by using 8% SDS‐PAGE and was transferred to polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Bedford, MA, USA). Membranes were blocked at room temperature for 2 h in PBST (in mmol/L: NaCl 130, KCl 2.5, Na2HPO4 10, KH2PO4 1.5, 0.1% Tween‐20, and 5% bovine serum albumin, pH 7.4), incubated 1 h at room temperature initially with primary antibodies, and then with the appropriate secondary peroxidase‐conjugated antibodies (HRP‐linked antirabbit or antimouse secondary antibody and HRP‐linked antibiotin antibody, 1 h at room temperature). Blots were developed using a chemiluminescence system (Cell Signalling Technology Inc., Beverly, MA, USA) and then were visualized by exposure to Kodak X‐ray film. Density of target bands was accurately determined using a computer‐aided 1‐D gel analysis system. In this study, anti‐ClC‐3 antibody was purchased from Alomone Laboratories (Jerusalem, Israel) and Santa Cruz Biotechnology (Santa Cruz, CA, USA), antibodies against β‐actin, cyclin D1, p27KIP, total GSK3β, phospho‐GSK3β (Ser9), total Akt, phospho‐Akt (Ser473 and Thr308), secondary peroxidase‐conjugated antibodies(HRP‐linked) were from Cell Signalling Technology Inc. Cyclin‐dependent kinase‐2 (CDK2) was from BD Transduction Laboratories (San Diego, CA, USA), and cyclin E, p21CIP1 were from Santa Cruz Biotechnology.
Statistical analysis
All data are represented as means ± SEM. Statistical significance was tested by Student's t‐test or one‐way analysis of variance followed by Duncan's multiple range tests. P < 0.05 was considered significant.
RESULTS
Effect of ClC‐3 siRNA on endogenous ClC‐3 protein expression
Here in BASMCs, transfection efficiency was detected by uptake of siRNA‐labelled FITC. As shown in Fig. 1a, under resting conditions fluorescence of the cells was negligible, while fluorescence was greatly increased in those treated with siRNA; this confirmed siRNA uptake by cells.
Figure 1.
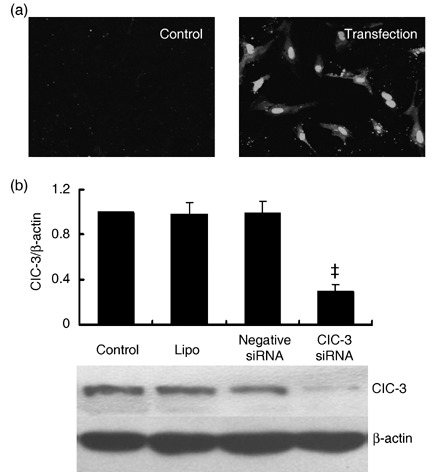
Effects of ClC‐3 siRNA transfection on endogenous ClC‐3 protein expression in BASMCs. (a) Representative image of fluorescence in BASMCs (200×). Stealth siRNA labelled with FITC was transfected to BASMCs, and fluorescence was detected in these cells after 6 h incubation (transfection), while there was no fluorescence detectable in untransfected (control) cells, indicating where siRNA had been successfully transfected. (b) Western blot results show that 40 nmol/L ClC‐3 siRNA decreased significantly endogenous ClC‐3 expression, but transfection of LipofectamineTM RNAiMAX (lipo) and negative siRNA control (negative siRNA) did not significantly change ClC‐3 expression (n = 5; ‡P < 0.01 versus control).
Endogenous expression of ClC‐3 protein was decreased by 72.5 ± 6.4% after treatment with 40 nm ClC‐3 siRNA for 48 h (P < 0.01, n = 5). Lipofectamine and negative siRNA transfection had no significant effects on ClC‐3 expression (Fig. 1b). Expression of ClC‐3 protein increased over 2‐fold in the rat BASMCs after 48‐h ET‐1 (10 nm) treatment. ClC‐3 siRNA at 40 nm suppressed ET‐1‐induced ClC‐3 protein expression by 66.4 ± 4.2% (Fig. 2a).
Figure 2.
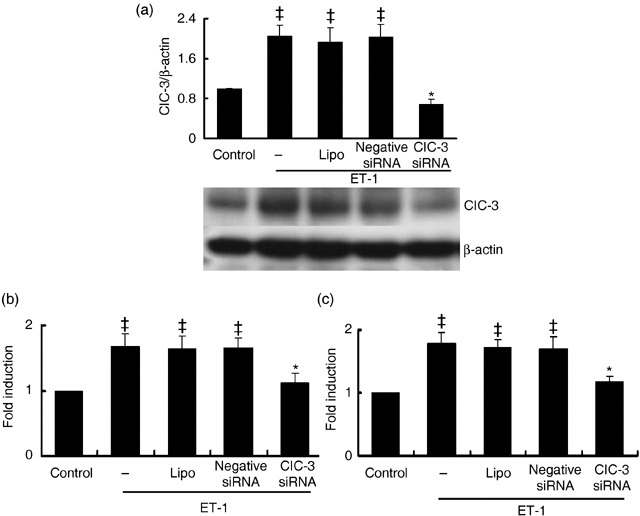
Effects of ClC‐3 knockdown on ET‐1‐dependent ClC‐3 protein expression and cell proliferation. (a) Densitometric analysis shows stealth siRNA inhibits ClC‐3 protein expression induced by 10 nmol/L ET‐1, while transfections of LipofectamineTM RNAiMAX and negative siRNA control had no significant effect on ClC‐3 protein expression (n = 5; *P < 0.01 versus control; **P < 0.01 versus ET‐1). Representative Western blots for ClC‐3 protein expression are shown in the bottom. (b) Increase in cell growth induced by 10 nmol/L ET‐1 was determined by counting the number of cells, and was significantly inhibited by incubation of ClC‐3 siRNA for 48 h, but not by transfection of LipofectamineTM RNAiMAX and negative siRNA. (c) The effects of ClC‐3 siRNA on cell proliferation induced by ET‐1 were further determinated by assaying BrdU incorporation. Incubation of ClC‐3 siRNA for 48 h also significantly decreased BrdU incorporation (n = 5; ‡P < 0.01 versus control; *P < 0.01 versus ET‐1). These results show that the inhibitory effect of ClC‐3 siRNA on ET‐1 induced ClC‐3 protein expression matches that of cell proliferation.
Effect of ClC‐3 silence on cell proliferation
The effects of ClC‐3 knockdown on cell proliferation were determined by cell count and BrdU incorporation assay. Compared to control, 10 nm ET‐1 led to 1.6 ± 0.2‐fold and 1.8 ± 0.3‐fold (P < 0.01, n = 5) increments in cell number (Fig. 2b) and BrdU incorporation, respectively (Fig. 2c). ClC‐3 siRNA at 40 nm could produce 82.6 ± 6.4% and 79 ± 5.7% inhibition of ET‐1‐induced cell growth and BrdU incorporation, respectively, while treatments of lipofectamine and negative siRNA had no significant on ET‐1‐induced cell growth and BrdU incorporation, excluding the non‐specificity of the siRNA and/or transfection reagent.
Effect of ClC‐3 silence on cell cycle progression
As shown in Fig. 3, serum‐starved BASMCs in the control group were mostly in the G0/G1 (81.1%) and S phases (14.9%). This changed to a decrease from 81.1% to 62.6%, and an increase from 14.9% to 32.7% in S phase after treatment with ET‐1 (P < 0.01, n = 5), indicating that ET‐1 promotes cell cycle progression and accelerates cells’ entrance into S phase. ClC‐3 knockdown inhibited the effect of ET‐1 on cell cycle progression with a decrease in cell population in S phase from 32.7% to 17.0% and an increase in cell population in G0/G1 from 62.6% to 78.9% (Fig. 3). These results suggest that deficiency in ClC‐3 arrests the cell cycle in G0/G1 phase by preventing entrance into S phase in BASMCs.
Figure 3.
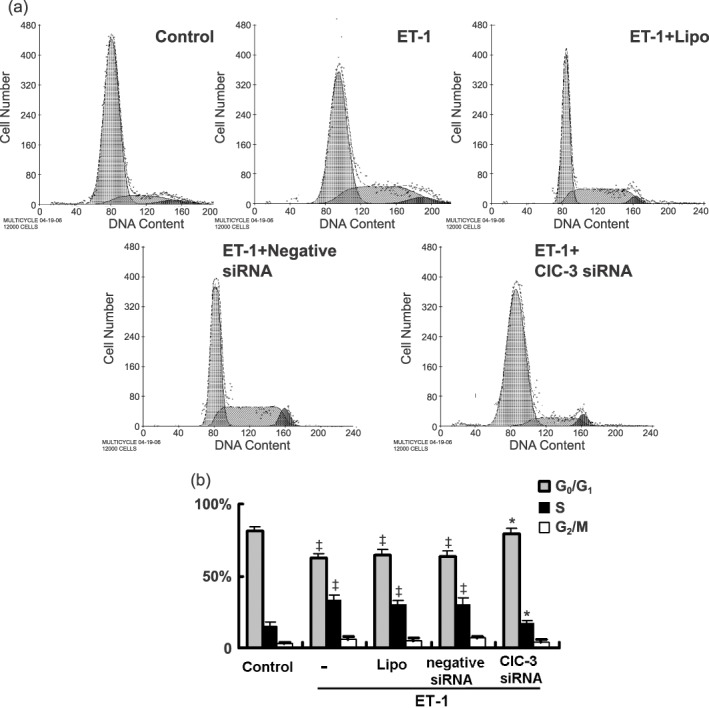
Flow cytometric analysis shows ET‐1 accelerated entrance into G1 and arrested entrance from G1 to S phase. (a) Representative images of cell cycle analysis. (b) Statistical analysis shows the percent distribution of cells in G0/G1, S, G2/M stages of the cell cycle (n = 5; ‡P < 0.01 versus control; *P < 0.01 versus ET‐1).
Effect of ClC‐3 silence on expression of cyclins and cyclin‐dependent kinase inhibitors
G1/S transition is governed by the balance of cyclins and cyclin‐dependent kinase inhibitors (CDKIs) (Braun‐Dullaeus et al. 1998). To explore the molecular mechanisms by which ClC‐3 knockdown arrests G1/S transition, we analysed the proteins regulating cell cycle progression in these cell phases, including cyclin D1, cyclin E, CDK2, p21CIP and p27KIP. As shown in Fig. 4, ET‐1 increased expressions of cyclin D1 and cyclin E to 2.2 ± 0.2‐fold and 1.6 ± 0.1‐fold and decreased expressions of p21CIP and p27KIP by 70.9 ± 6.4% and 61.6 ± 9.1%, respectively (P < 0.01, n = 5). Alterations induced by ET‐1 were completely reversed by ClC‐3 knockdown although no significant change of CDK2 was observed after ClC‐3 knockdown.
Figure 4.
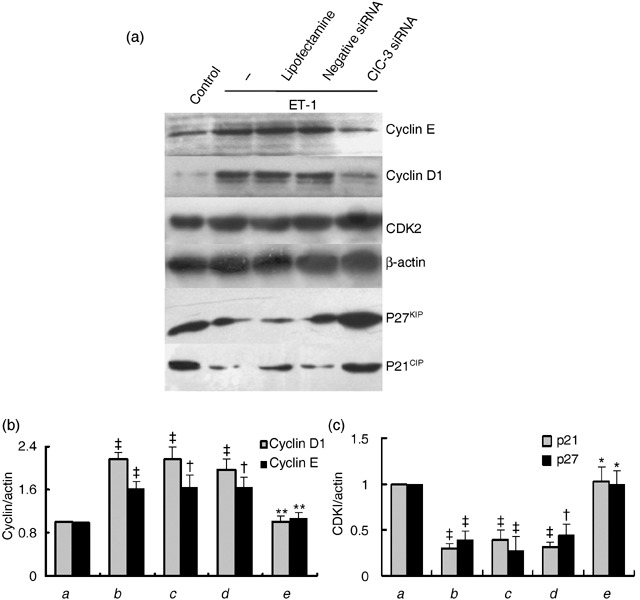
Effects of ClC‐3 knockdown by incubation of 40 nm ClC‐3 siRNA for 48 h, on protein expression of cyclin E, cyclin D1 and cyclin‐dependent kinase inhibitors (CDKIs) p21 and p27. (a) Representive Western blot images of cyclins and CDKIs. (b and c) Densitometric analysis of the effects of ClC‐3 knockdown on expression of cyclin D1 and cyclin E (b), and p21 and p27 (c), respectively. Expression of CDK2 was not changed by ClC‐3 knockdown. (a) control; (b) ET‐1; (c) ET‐1 + LipofectamineTM RNAiMAX; (d) ET‐1 + negative siRNA; (e) ET‐1 + ClC‐3 siRNA (n = 5; †P < 0.05, ‡P < 0.01 versus control; *P < 0.01 versus ET‐1).
Effect of ClC‐3 silence on Akt/GSK‐3β signal pathway
Phosphorylation of Akt and its downstream GSK‐3β has been documented as regulatory for cell cycle G1/S progression in many cells, through control of cyclin degradation (Hixon et al. 2000; Stabile et al. 2003; Sedding et al. 2005; Heron‐Milhavet et al. 2006). We investigated how deficiency in ClC‐3 affects the activation of the pathways. Our results have demonstrated that the time–course response of phosphorylation of Akt (Ser473) to ET‐1 was significantly impaired after 10‐min and 1‐h ET‐1 treatment, owing to loss of ClC‐3 (Fig. 5a). Moreover, deficiency in ClC‐3 failed to increase the phosphorylation of Akt at Thr308 and GSK‐3β at Ser9 evoked by ET‐1 (Fig. 5b). However, loss of ClC‐3 did not alter total Akt and GSK‐3β activity.
Figure 5.
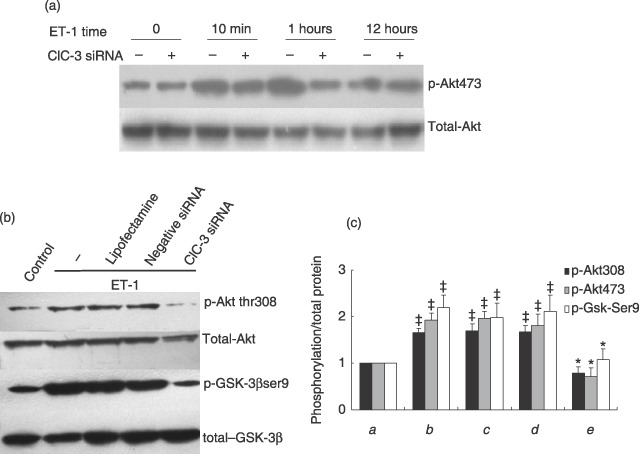
Effects of ClC‐3 knockdown by incubation of 40 nm ClC‐3 siRNA for 48 h, on phosphorylation of Akt and GSK‐3β. (a) Representative images of Western blots show that loss of ClC‐3 channel affects the time course response to ET‐1 after ClC‐3 was knocked own by siRNA, and ET‐1 was added to observe phosphorylation of Akt (Ser473) at 10‐min, 1‐h and 12‐h time point (n = 5). (b) Representative images of Western blots to show that ET‐1 increased phosphorylationsof Thr308 and its downstream target, GSK‐3β, at Ser9, but failed to increase this phosphorylation when ClC‐3 was knocked down. However, deficiency of ClC‐3 did not significantly alter protein levels of Akt and GSK‐3β. (c) Densitometric analysis of the effects of ClC‐3 knockdown on inactivation of Akt/GSK‐3β signal transduction pathway. (a) control; (b) ET‐1; (c) ET‐1 + LipofectamineTM RNAiMAX; (d) ET‐1 + negative siRNA; (e) ET‐1 + ClC‐3 siRNA; (n = 6; ‡P < 0.01 versus control; *P < 0.01 versus ET‐1).
DISCUSSION
Recent studies have found that the volume‐regulated Cl− channel (VRCC) current varies in different cell cycle phase, and Cl− channel blockers suppress cell proliferation and arrested cells in G0/G1 phase with inhibition of Cl− current (Duan et al. 1997; Shen et al. 2000; , Chen et al. 2002, 2007). These data indicate that the VRCC plays an essential role in cell cycle progression and cell proliferation through regulation of cell volume. Although the molecular nature of VRCC has yet to be conclusively identified, our recent study on A10 vascular smooth muscle cells (VSMCs) strongly supported that ClC‐3 is the molecular component involved in activation or regulation of VRCC (Zhou et al. 2005; , Guan et al. 2006). Given its potential importance for cell volume regulation in cell proliferation, its role attracts particular interest. In the present study, we found that ET‐1‐induced cell proliferation was accompanied by an increase in ClC‐3 expression; siRNA of ClC‐3 not only inhibited ET‐1‐induced expression of ClC‐3 protein, but also prevented BrdU incorporation and cell proliferation. Similar correlation between the increase in ClC‐3 expression and stimulation of mitogens has also been found in prostate cancer epithelial cells and pulmonary VSMCs (Lemonnier et al. 2004; , Dai et al. 2005). These results provide compelling evidence that ClC‐3 plays an important role in regulation of cell proliferation.
The BASMC proliferation is governed by passage through the cell cycle, and cell cycle progression is a tightly controlled event positively regulated by cyclins and CDKs, and negatively by CDKIs and tumour suppressor genes (Woo & Poon 2003). In this study, we found that knockdown of ClC‐3 inhibited entrance from G0/G1 to S phase and arrested the cell cycle at the G1/S checkpoint. To further clarify the mechanisms by which ClC‐3 knockdown induced G1/S arrest, we analysed expression of proteins responsible for the G1/S checkpoint. The results show that loss of ClC‐3 almost abolished ET‐1‐induced expression of cyclin D1 and cyclin E, but not the expression of CDK2. In addition, ClC‐3 knockdown also reversed the suppressive effects of ET‐1 on two CDKIs, p21CIP and p27KIP, which are widely reported to restrict G1/S transition in the cell cycle by inhibiting several cyclin‐CDK complexes (Sedding et al. 2003; , Barchiesi et al. 2006). Together, our results demonstrate that silencing the ClC‐3 Cl− channel prevents ET‐1‐induced cell proliferation through down‐regulation of cyclins and up‐regulation of CDKIs, and then results in restriction of G1/S transition.
It has been shown that Akt mediates cell cycle G1/S transition through increases in cyclin E and cyclin D1 expression (Suzuki et al. 1999; Haq et al. 2000; Hixon et al. 2000; Box & Demetrick 2004). Mutation of Akt at Thr308 and Ser473 abrogates vascular smooth muscle cell proliferation both in vitro and in vivo by delaying G1/S exit (Stabile et al. 2003). GSK‐3β is a downstream target of Akt, and phosphorylation of GSK‐3β at Ser9 saves cell cycle regulators, including cyclin D1, cyclin E, β‐catenin and c‐Myc, from their ubiquitin‐dependent destruction in many cancer cells (Haq et al. 2000; Park et al. 2003; , Box & Demetrick 2004). Activation of Akt or overexpression of its constitutively active forms decreases levels of p27 (Medema et al. 2000; Sedding et al. 2003; , Kotake et al. 2005; Barchiesi et al. 2006) and phosphorylation of GSK‐3β triggers p21 degradation (Rossig et al. 2002; , Mitchell et al. 2004), thereby promoting cell proliferation. Our results in this study have shown that ET‐1 failed to induce phosphorylation of Akt (Thr308, Ser473) and GSK‐3β (Ser9) when ClC‐3 was depleted by siRNA, suggesting that the ClC‐3 channel is one of the important regulators in this signal cascade. The data provide evidence to explain, at least partly, why ClC‐3 knockdown affects cyclins, such as cyclin D1 and cyclin E, and CDKIs, such as p27KIP, p21CIP and subsequent cell cycle G1/S progression. However, it is still not clear how ClC‐3 knockdown affects the Akt/GSK‐3β signal cascade.
In people, development of hypertension and atherosclerosis are associated closely with vascular remodelling, which involves phenotypic changes to arterial smooth muscle cells from a contractile to a proliferative phenotype and enhancement of VSMC proliferation (Intengan & Schiffrin 2001; , Mandegar et al. 2004). Recently, we have found that volume‐regulated Cl− movement is augmented in rat BASMCs in proportion to the severity of hypertension and cerebral vascular remodelling (Shi et al. 2007). Our present results provide further evidence to support the suggestion that ClC‐3 Cl− channel is a potential target modulating BASMC proliferation in the process of cerebrovascular remodelling.
In summary, depletion of ClC‐3 Cl− channel protein effectively suppressed phosphorylation of Akt/GSK‐3β signal pathway, resulting in down‐regulation of cyclin D1 and cyclin E. In contrast, depletion of ClC‐3 Cl− channel protein produced up‐regulation of p27KIP and p21CIP. Integrated effects on these key cell cycle regulated proteins result in G1/S cell cycle arrest in BASMCs.
ACKNOWLEDGEMENTS
This work was sponsored by the National Natural Science Foundation of China (No. 30472021, No. 30500616 and Key Grant No. 30730105) and the Science Foundation of the Ministry of Education in China (No. 20050558072).
REFERENCES
- Barchiesi F, Jackson EK, Fingerle J, Gillespie DG, Odermatt B, Dubey RK (2006) 2‐Methoxyestradiol, an estradiol metabolite, inhibits neointima formation and smooth muscle cell growth via double blockade of the cell cycle. Circ. Res. 99, 266–274. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Cidlowski JA (2004) The role of apoptotic Volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pflugers Arch. 448, 313–318. [DOI] [PubMed] [Google Scholar]
- Box AH, Demetrick DJ (2004) Cell cycle kinase inhibitor expression and hypoxia‐induced cell cycle arrest in human cancer cell lines. Carcinogenesis 25, 2325–2335. [DOI] [PubMed] [Google Scholar]
- Braun‐Dullaeus RC, Mann MJ, Dzau VJ (1998) Cell cycle progression: new therapeutic target for vascular proliferative disease. Circulation 98, 82–89. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang L, Zhu L, Nie S, Zhang J, Zhong P, Cai B, Luo H, Jacob TJ (2002) Cell cycle‐dependent expression of volume‐activated chloride currents in nasopharyngeal carcinoma cells. Am. J. Physiol. Cell Physiol. 283, C1313–C1323. [DOI] [PubMed] [Google Scholar]
- Chen LX, Zhu LY, Jacob TJ, Wang LW (2007) Roles of volume‐activated Cl− currents and regulatory volume decrease in the cell cycle and proliferation in nasopharyngeal carcinoma cells. Cell Prolif. 40, 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai YP, Bongalon S, Hatton WJ, Hume JR, Yamboliev IA (2005) ClC‐3 chloride channel is up‐regulated by hypertrophy and inflammation in rat and canine pulmonary artery. Br. J. Pharmacol. 145, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B (1997) Molecular identification of a volume‐regulated chloride channel. Nature 390, 417–421. [DOI] [PubMed] [Google Scholar]
- Guan YY, Wang GL, Zhou JG (2006) The ClC‐3 Cl− channel in cell volume regulation, proliferation and apoptosis in vascular smooth muscle cells. Trends Pharmacol. Sci. 27, 290–296. [DOI] [PubMed] [Google Scholar]
- Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J, Hajjar R, Michael A, Force T (2000) Glycogen synthase kinase‐3beta is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol. 151, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron‐Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, Fernandez A, Lamb NJ (2006) Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol. Cell. Biol. 26, 8267–8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hixon ML, Muro‐Cacho C, Wagner MW, Obejero‐Paz C, Millie E, Fujio Y, Kureishi Y, Hassold T, Walsh K, Gualberto A (2000) Akt1/PKB upregulation leads to vascular smooth muscle cell hypertrophy and polyploidization. J. Clin. Invest. 106, 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intengan HD, Schiffrin EL (2001) Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension 38, 581–587. [DOI] [PubMed] [Google Scholar]
- Kotake Y, Nakayama K, Ishida N, Nakayama KI (2005) Role of 10 phosphorylation in p27 stabilization revealed by analysis of p27 knock‐in mice harboring a 10 mutation. J. Biol. Chem. 280, 1095–1102. [DOI] [PubMed] [Google Scholar]
- Lang F, Foller M, Lang KS, Lang PA, Ritter M, Gulbins E, Vereninov A, Huber SM (2005) Ion channels in cell proliferation and apoptotic cell death. J. Membr. Biol. 205, 147–157. [DOI] [PubMed] [Google Scholar]
- Lemonnier L, Shuba Y, Crepin A, Roudbaraki M, Slomianny C, Mauroy B, Nilius B, Prevarskaya N, Skryma R (2004) Bcl‐2‐dependent modulation of swelling‐activated Cl− current and ClC‐3 expression in human prostate cancer epithelial cells. Cancer Res. 64, 4841–4848. [DOI] [PubMed] [Google Scholar]
- Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX (2004) Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc. Res. 68, 75–103. [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM (2000) AFX‐like Forkhead transcription factors mediate cell‐cycle regulation by Ras and PKB through p27kip1. Nature 404, 782–787. [DOI] [PubMed] [Google Scholar]
- Mitchell D, Rodgers K, Hanly J, McMahon B, Brady HR, Martin F, Godson C (2004) Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am. J. Pathol. 164, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KW, Yang HM, Youn SW, Yang HJ, Chae IH, Oh BH, Lee MM, Park YB, Choi YS, Kim HS, Walsh K (2003) Constitutively active glycogen synthase kinase‐3beta gene transfer sustains apoptosis, inhibits proliferation of vascular smooth muscle cells, and reduces neointima formation after balloon injury in rats. Arterioscler. Thromb. Vasc. Biol. 23, 1364–1369. [DOI] [PubMed] [Google Scholar]
- Rossig L, Badorff C, Holzmann Y, Zeiher AM, Dimmeler S (2002) Glycogen synthase kinase‐3 couples AKT‐dependent signaling to the regulation of p21Cip1 degradation. J. Biol. Chem. 277, 9684–9689. [DOI] [PubMed] [Google Scholar]
- Sedding DG, Hermsen J, Seay U, Eickelberg O, Kummer W, Schwencke C, Strasser RH, Tillmanns H, Braun‐Dullaeus RC (2005) Caveolin‐1 facilitates mechanosensitive protein kinase B (Akt) signaling in vitro and in vivo . Circ. Res. 96, 635–642. [DOI] [PubMed] [Google Scholar]
- Sedding DG, Seay U, Fink L, Heil M, Kummer W, Tillmanns H, Braun‐Dullaeus RC (2003) Mechanosensitive p27Kip1 regulation and cell cycle entry in vascular smooth muscle cells. Circulation 108, 616–622. [DOI] [PubMed] [Google Scholar]
- Shen MR, Droogmans G, Eggermont J, Voets T, Ellory JC, Nilius B (2000) Differential expression of volume‐regulated anion channels during cell cycle progression of human cervical cancer cells. J. Physiol. 529 (Pt. 2), 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi XL, Wang GL, Zhang Z, Liu YJ, Chen JH, Zhou JG, Qiu QY, Guan YY (2007) Alteration of volume‐regulated chloride movement in rat cerebrovascular smooth muscle cells during hypertension. Hypertension 49, 1371–1377. [DOI] [PubMed] [Google Scholar]
- Stabile E, Zhou YF, Saji M, Castagna M, Shou M, Kinnaird TD, Baffour R, Ringel MD, Epstein SE, Fuchs S (2003) Akt controls vascular smooth muscle cell proliferation in vitro and in vivo by delaying G1/S exit. Circ. Res. 93, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Suzuki E, Nagata D, Kakoki M, Hayakawa H, Goto A, Omata M, Hirata Y (1999) Molecular mechanisms of endothelin‐1‐induced cell‐cycle progression: involvement of extracellular signal‐regulated kinase, protein kinase C, and phosphatidylinositol 3‐kinase at distinct points. Circ. Res. 84, 611–619. [DOI] [PubMed] [Google Scholar]
- Wang GL, Wang XR, Lin MJ, He H, Lan XJ, Guan YY (2002) Deficiency in ClC‐3 chloride channels prevents rat aortic smooth muscle cell proliferation. Circ. Res. 91, E28–E32. [DOI] [PubMed] [Google Scholar]
- Wondergem R, Gong W, Monen SH, Dooley SN, Gonce JL, Conner TD, Houser M, Ecay TW, Ferslew KE (2001) Blocking swelling‐activated chloride current inhibits mouse liver cell proliferation. J. Physiol. 532, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo RA, Poon RY (2003) Cyclin‐dependent kinases and S phase control in mammalian cells. Cell Cycle 2, 316–324. [PubMed] [Google Scholar]
- Xiao GN, Guan YY, He H (2002) Effects of Cl− channel blockers on endothelin‐1‐smooth muscle cells. Life Sci. 70, 2233–2241. [DOI] [PubMed] [Google Scholar]
- Zhang HN, Zhou JG, Qiu QY, Ren JL, Guan YY (2006) ClC‐3 chloride channel prevents apoptosis induced by thapsigargin in PC12 cells. Apoptosis 11, 327–336. [DOI] [PubMed] [Google Scholar]
- Zhou JG, Ren JL, Qiu QY, He H, Guan YY (2005) Regulation of intracellular Cl− concentration through volume‐regulated ClC‐3 chloride channels in A10 vascular smooth muscle cells. J. Biol. Chem. 280, 7301–7308. [DOI] [PubMed] [Google Scholar]