

Abstract
This paper summarises how scaffold proteins affects and regulate the JNK signalling pathway. We believe that some of these scaffold proteins, by virtue of their anchoring and catalytic properties contribute to a high degree of specificity of intra cellular signalling pathways that regulate the progression through the cell cycle.
Introduction
The mitogen‐activated protein kinase (MAPK) pathway is one of the more intensely studied signalling cascades; it was one of the first links made between extracellular signals and transcriptional activation of early response genes. The MAPK pathway is known to be involved in the pleiotropic response that includes fundamentally different functions, such as proliferation, survival, locomotion and onset of differentiation (1). MAPKs (also known as Erks) are regulated by a cascade of protein phosphorylation where there are three distinct tiers of regulation (Fig. 1). At the top is MAPK kinase kinase (MAPKKK, also known as Raf), which is a serine/threonine kinase and operates by activating an enzyme at the next tier, which is a MAPK kinase (MAPKK or Mek). The MAPKKs can phosphorylate tyrosine as well as threonine residues and specifically activate MAPKs by phosphorylating a TxY motif in a vital part of their activation loop, thereby incapacitating the ATP binding site (2). MAPKK activates MAPK, which is the main effector of signal messages. Whereas MAPKK displays a high degree of specificity, it has been observed that MAPKs can phosphorylate a large number of substrate proteins. Moreover, MAPK not only phosphorylates cytoplasmic proteins, but has also been shown to enter the nucleus and phosphorylate transcription factors in situ (1). In this way, MAPK directly links extracellular activation to specific changes in gene transcription and has therefore attracted substantial attention (3). Figure 1 shows the key scaffold Ste5 in Saccharomyces cerevisiae. It assembles the three tiers of phosphorylating enzymes into a signalling unit. In yeast, Ste 5 links the heterotrimeric G protein, Gpa, to Ste1 (MAPKKK), Ste7 (MAPKK) and Fus3 (MAPK) in response to pheromone activation of the mating receptor GCPR, Ste2/3. More specifically, Ste5 can catalytically activate MAPK upon binding, by inducing autophosphorylation of the threonine residue in the TxY motif in Fus3 (4).
Figure 1.

Primary structures of scaffold proteins involved in JNK signalling. Schematic representation of domain structures of JIP1, IB2/JIP2, JSAP‐1/JIP3, JLP and POSH oriented from amino (N) to carboxy (C) termini. Predicted domains were obtained from the human version of these proteins in the ensembl database. One representative from each protein family is shown, not their isoform variants. The JNK binding domains were experimentally determined in vitro.
This archetypical model has since been used to explain the role of scaffold proteins in mammalian cell signalling (5, 6). Even though it has been difficult to identify mammalian homologues of Ste5, a plethora of scaffold proteins has since been identified (7). In theory, scaffold proteins play two partly related functional roles. First, scaffold proteins are tools by which cells can maintain a high degree of specificity in signalling pathways. This is mainly achieved through co‐localization of molecules that participate in the same signalling pathway in the same area of the cell. In other words, scaffold proteins increase efficacy of the pathway as well as ensure a high degree of specificity. Second, scaffold proteins can act as catalysts and thereby activate different components in the signalling pathway. Ste5, MP1 (MAPK partner 1), JIP‐1 (JNK interacting protein‐1), JSAP‐1 (JNK/SAPK activating protein 1) and KSR (kinase suppressor of Ras) were the first regulatory scaffold proteins that have had a catalytic role assigned to them (8). By optimizing both these roles, scaffold proteins are also instrumental in regulating positive as well as negative feedback loops.
Although the anchoring and catalytic roles of scaffold proteins are judged in the literature as combined entities, it is reasonable to believe that while they act in concert, they may well also act indirectly in their anchoring role. For example, this has been observed in non‐MAPK‐linked transduction of intracellular signals where the scaffold assembles proteins that are parts of the same signalling chain, to a unit, but where individual molecules do not directly act on each other. There are many examples of pure anchor scaffolds such as NDMA receptor‐associated protein (9), Ina‐D (10) and A kinase anchoring proteins (11). This category also comprises members of the tyrosine phosphorylated scaffold proteins, such as cytoplasmic domain of the PDGF‐receptor, insulin responsive substrate 1 (IRS‐1), T‐cell protein linkers for activation of T cells and Src homology 2 domain, containing leucocyte protein of 76 kDa (reviewed in (12)).
Nine distinct MAPKs have been described in mammalian cells, namely ERK1/2, ERK3, ERK4, ERK5, ERK6/p38MAPKg, ERK7, ERK8, JNK1/2/3 and p38MAPKa/b/d (13, 14, 15, 16). Of these, three groups have been studied in particular detail, namely ERK1/2, JNK1/2/3 and p38MAPKa/b/d (reviewed in (17)).
Here, we summarize recent findings on how scaffold proteins affect and, in some cases, regulate one of these crucial signalling pathways, the c‐Jun N‐terminal kinase (JNK) pathway. We believe that these scaffold proteins, by virtue of their anchoring and catalytic properties, are pivotal in maintaining a high degree of specificity in intracellular signalling pathways, that control aspects of the cell cycle, particularly transition from G1 to S‐phase.
Scaffold proteins in the JNK pathway
Five structurally distinct scaffold proteins have been shown to assemble the JNK signalling unit (Fig. 2). These proteins contain a variety of protein‐interacting motifs and constitute a family normally referred to as JIPs (JNK interacting proteins). In addition to anchoring a JNK signalling module, some members coordinate signalling of p38MAPK modules. The five archetype proteins are JIP‐1, JIP‐2, JIP‐3 (equivalent of JSAP‐1), JNK‐interacting leucine zipper protein (JLP) and plenty of SH3 (POSH). In addition to these core molecules, several variants, that are the result of alternative splicing, have been isolated and characterized (18, 19, 20, 21, 22, 23, 24, 25, 26).
Figure 2.
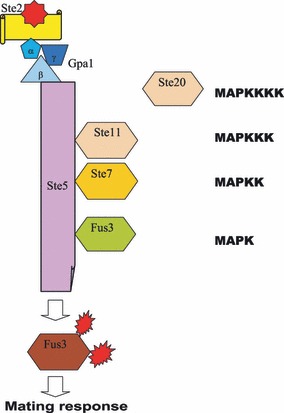
The archetypal MAP kinase model for mating response in budding yeast Saccharomyces cerevisiae.
JIP‐1
JIP‐1 was the first JIP to be isolated (27) and transcriptional analysis of adult mouse organs has revealed that it is expressed at high levels in the brain, at intermediate levels in testes and kidney and at low levels in muscle and lung (27). Shortly thereafter, IB1 was identified and shown to be a splice variant confined to the pancreas, where it potentiates the Glut2 promoter (28). To avoid confusion, JIP‐1 is sometimes referred to as JIP‐1a and IB1 as JIP‐1b. Both isoforms consist of an amino‐terminal JNK‐binding domain (JBD), a phosphotyrosine binding domain (PTB) and a Src homology 3 (SH3) domain. In addition, IB1 has an extra carboxy‐terminal PTB motif (27, 28).
Four MAPKKKs can associate with JIP‐1, namely MEKK3, mixed lineage protein kinase 3 (MLK3), dual leucine zipper‐bearing kinase (DLK) and histidine protein kinase 1 (HPK1) (Fig. 3). In contrast, only one JNK‐specific MAPKK, MKK7, is capable of interacting with JIP‐1 (24, 26). In addition, two phosphatases, both M3/6 and MKP7, can bind to JIP‐1, albeit at different sites (29). This finding suggested that JIP‐1 controls JNK activation by balancing a MAPKKK‐driven activating phosphorylation loop against an inhibitory, attenuating phosphatase feedback mechanism (29). Earlier data have also suggested that JIP‐1 is involved in the Rho signalling pathway as JIP1 binds to the p190‐RhoGEF molecule in differentiated neurons (30). Considering recent data concerning the small GTP‐binding protein RhoA that regulates c‐Jun by a ROCK‐JNK signalling axis, it is tempting to propose a more direct role for JIP‐1. (31). The related JIP‐1b molecule has been suggested to be involved in pathogenesis of Alzheimer’s disease (32). Unlike JIP‐1a, JIP‐1b is required for Alzheimer’s beta‐amyloid precursor protein (APP) to interact with JNK, thereby providing the necessary scaffold for development of beta amyloid plaques that are hallmarks of Alzheimer’s (33).
Figure 3.
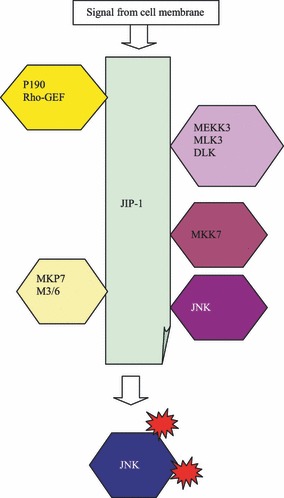
Scaffolding role of JIP‐1 in JNK activation.
JIP‐2
JIP‐2, which in many ways resembles JIP‐1, consists of a defined JNK‐binding motif, a PTB domain and an SH3 motif (26, 34). In the human adult, its coding gene is expressed in a variety of organs including brain, ovary, pancreas and prostate (26). Interestingly, its interacting role is much wider than that of JIP‐1. It is now clear that the JIP‐2 molecule can interact, not only with JNK1 and JNK2 as expected, but also with p38MAPK (35, 36). However, like JIP‐1, JIP‐2 specifically uses MKK7 as its MAPKK and cannot bind MKK4. Like JIP‐1, the MAPKKKs that interact with JIP‐2 include DLK and MLK3. However, JIP‐2 can also utilize MLK2 as its MAPKKK (26, 34, 36) (Fig. 4).
Figure 4.
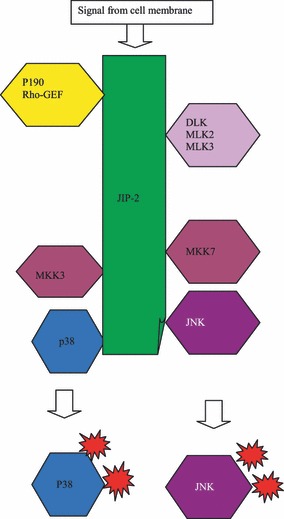
Scaffolding role of JIP‐2 in p38 and/or JNK activation.
JIP‐3 (JSAP‐1)
JIP‐3 was originally identified as a JNK/stress‐activated protein kinase‐associated protein (JSAP‐1) that bound to JNK1 as well as to JNK3 (18, 37). It differed in structure from JIP‐1 and JIP‐2, notably in that it was devoid of an SH3 domain and instead included a leucine zipper domain. This motif greatly enhances the capacity to form heterodimers, which makes it useful in combinatorial control of cellular processes. JIP‐3 can bind all three JNK molecules, JNK3 with the highest affinity (37). It can associate with either of two MAPKKs, namely MKK4 and MKK7. JIP3 is also capable of binding three different MAPKKKs: MEKK1 (18), MLK3 (37) and activator of S‐phase kinase (38) (Fig. 5). JIP‐3 exists in four different splice variants (39). Interestingly, all forms are expressed in brain tissue, but each of the four variants shows a different expression pattern in other organs and it has been proposed that different isoforms play a crucial role for recruitment of JNK1, 2 or 3 with a high degree of tissue specificity.
Figure 5.
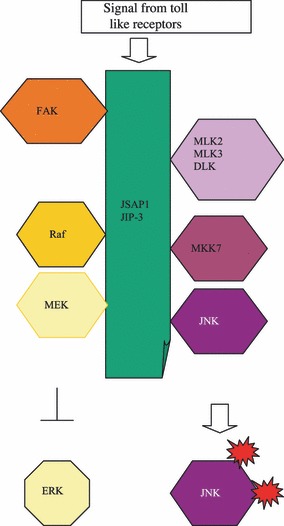
Scaffolding role of JSAP/JIP3 in ERK and/or JNK activation.
It was discovered at an early stage that JIP‐3 is involved in the ERK signalling pathway by binding Raf‐1 and MEK1 (18). Interestingly, these different kinase modules are inversely controlled, in that JIP‐3 expression increases JNK activity, but decreases ERK signalling, possibly by cleaving Raf‐1 and MEK from the ERK module (40. Apart from acting as a scaffold for the classical MAP kinase modules, JIP‐3 is capable of forming complexes with focal adhesion kinase (FAK) (41) and serves as a cooperative scaffold for activation of JNK in regulation of cell migration in response to fibronectin stimulation. JIP‐3 has been shown to mediate association between FAK and JNK, induced either by co‐expression of Src or by attachment of cells to fibronectin (42). Complex formation of FAK with JIP‐3 and p130 Crk‐associated substrate (p130Cas) results in increased FAK activity and phosphorylation of JIP‐3 and p130Cas, which require p130Cas hyperphosphorylation and is abrogated by inhibition of Src (42). JNK activation by fibronectin is enhanced by JIP‐3, which is suppressed by disrupting the FAK/p130Cas or by Src inhibition (42). Cell migration studies have revealed that JIP‐3 co‐localizes with JNK and phosphorylated FAK at the leading edge. Migration per se is stimulated by JIP‐3 expression, which depends on its JNK‐binding domain and is suppressed by inhibition of JNK (42).
A further interesting aspect of the multifaceted JIP‐3 scaffold protein is its association with Toll‐like receptors (TLRs) (43). TLRs recognize pathogen‐specific molecular motifs and induce a response involving MAP‐kinases. JIP‐3 associates with the N‐terminal region of TLR4, which is specific to JIP‐3; JIP‐1 and JIP‐2 are unable to bind to TLR4. JIP‐3 increases formation of TLR4‐JNK complexes, but it was clearly demonstrated that intact JIP‐3 molecule is required to elicit this response. Moreover, JIP3 was found to be associated with TLR2 and TLR9, but not with TLR1 and TLR6, indicating a pivotal role for this scaffold protein in TLR signalling (43).
JLP
JNK‐interacting leucine zipper protein is the latest member of the family of JNK‐interacting proteins to be described (22). JLP exists in two splice variants, JIP‐4 and sperm‐associated antigen 9 (SPAG9) (20, 21) that share common 3′ exons, but differ in the 5′ region. JIP‐4 and SPAG9 show fundamental differences in organ expression. JIP‐4 is mainly expressed in the brain, heart, liver, kidney and testis, whereas SPAG9 is only expressed in haploid germ cells during spermatogenesis. JLP has leucine zipper domains as well as a C‐terminal highly homologous with JIP‐3 (22). JLP contains three sulphydryl binding sites and also interacts with two transcription factors, Myc and Max; it was also shown that these two proteins have distinct binding domains on the JLP molecule (22). JLP is involved in JNK signalling as well as in p38 signalling, although one of the splice variants, SPAG9, only interacts with JNK 1, 2 or 3 but not with p38. The interacting sites of JNK1 and p38MAPK are quite distinct and it has been proposed that JLP in fact can tether both kinases. The observation that MKK4, which is an MAPKK that binds to JLP, can stimulate both pathways is intriguing and sheds further light on this interesting scaffold protein (44, 45) (Fig. 6).
Figure 6.
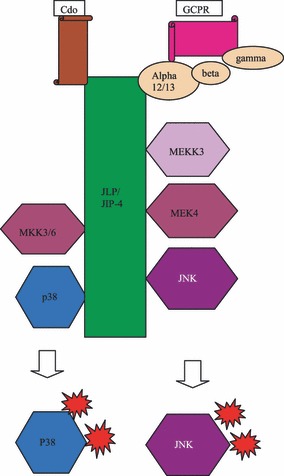
Scaffolding role of JLP/JIP‐4 in p38 and/or JNK activation.
Another unique feature is that JLP is the first scaffold protein shown to link a heterotrimeric G protein to a MAP kinase module. JLP therefore has been considered to be the mammalian equivalent of yeast Ste5. Recent evidence suggests that JLP binds alpha subunits of the G12 family of heterotrimeric G‐proteins (45, 46). The motifs that bind Ga12 or Ga13 have been mapped to the C‐terminal region of JLP molecule. Recently, the link between Ga13 binding and endodermal differentiation by JNK, in embryonal carcinoma cells has been established, which adds another interesting regulatory role to the repertoire of this scaffold protein (45). Finally, it has been suggested that JLP is also involved in spatiotemporal regulation of JNK signalling by associating with the kinesin light chain 1 (KLC1) (44).
Others
Plenty of SH3, POSH, was first identified as a Rac‐1 interacting protein (23) and has been recently found to form complexes with JIP‐1, and thus, is an important prerequisite for the JNK apoptotic pathway (47) (see below). Moreover, it was shown at an early stage that MEKK1 in itself can act as a JNK signalling pathway scaffold (48). MEKK1 binds MKK4 and JNK without any other scaffold protein involved (48). Finally, a marginal scaffolding role can be attributed to Beta‐arrestin 2 during transduction of G protein‐coupled receptor signalling (49).
JNK signalling and scaffold proteins in apoptosis
Although there is no immediate reason to believe that there is co‐regulation of growth factor and scaffold protein gene expression, some data regarding this matter have been published recently. In the study reporting the discovery of the first JIP‐1, it was shown that in vivo expression in adult mice differed grossly between organs (27). It was subsequently shown that these differences were less evident in the mouse embryo (50), thus differences in expression pattern pointed at some role for JIP‐1 in control of growth and development. It has been known for some time that insulin‐like growth factor 2 (IGF 2) is among the most prevalent factors promoting growth of the mammalian embryo (see 51, 52, 53, 54). By examining JIP‐1 gene expression in transgenic mice that were heterozygous for a functional Igf2 gene, it was possible to show that abrogation of IGF 2 expression was followed by decreased expression of JIP‐1 (50). Moreover, when expression patterns of genes coding for JIP‐1 and IGF 2 were examined in primary tumours of embryonic origin (germ cell tumours and Wilms’ tumour), it was found that irrespective of histological type, the two genes showed a high degree of co‐variation, in the sense that high IGF 2 expression was followed by high expression of JIP‐1 (55).
A slightly different pattern emerged when a Wilms’ tumour cell line came under scrutiny. It had been previously reported that insulin‐like growth factor can induce apoptosis in the Wilms’ tumour cell line WCCS‐1 (56). While this cell line had very low levels of intrinsic IGF 2 expression, it was found to display a level of JIP‐1 expression that was comparable with that of normal kidney (57). When exposed to physiological concentrations of IGF 2, WCCS‐1 cells underwent apoptosis and also significantly increased their JIP‐1 expression (57).
These data suggest that decrease in JIP‐1 expression in Wilms’ tumour cells may in some way, be linked to onset of apoptosis. It may also help explain why growth factor addition in this case acts to induce programmed cell death. All MAPK pathways have been implicated in regulation of apoptosis. More than a decade ago, it was shown that withdrawal of NGF from PC12 cells resulted in apoptosis and that this effect was mediated via activation of JNK (58). However, a number of studies has since shown that MAPK pathways can either stimulate or inhibit apoptosis (reviewed in (59)). In MKK‐4 knockout mice, it seems clear that JNK plays a protective role as these mice display increased rate of liver cell apoptosis (60). Survival signals ascribable to JNK are probably mediated by JunD, which in turn enhances transcriptional activity of numerous survival genes (61). In other systems, JNK has been implicated as a proapoptotic factor. JNK has been shown to phosphorylate the proapoptotic BH3 domain proteins, Bim and Bmf, that are normally sequestered, by binding to myosin and dynein (62). JNK‐induced phosphorylation releases these two proteins thereby initiating in mitochondria, facilitation of Bax‐dependent apoptosis (62). Moreover, JNK can phosphorylate and activate BAD, thereby coupling a stress‐activated signalling pathway to apoptosis (63). Another proposed role for JNK in promoting apoptosis is by inactivating protective proteins. Such targets include Bcl‐X and Bcl‐2 (64, 65) and it appears that this process depends upon Bax (66).
Interleukin 1‐beta acts as a proapoptotic stimulus in pancreatic islet beta cells (67). However, this activation process is paralleled by decrease in JIP‐1b expression (67). Conversely, overexpression of JIP‐1b significantly inhibits IL‐1 beta‐induced apoptosis (68). Presumably, pancreatic beta cells may constitute a special case where variations in JIP‐1b expression regulate susceptibility to cytokine‐induced apoptosis, irrespective of JNK signalling (69).
More recently, another JNK signalling scaffold protein, POSH, has been found to link active, GTP‐bound Rac1 to downstream JNK in an apoptotic pathway (70). Moreover, POSH interacts directly with JIP‐1 and Kukesov et al. (47) were able to demonstrate that POSH‐JIP‐1 complex is specifically involved in the apoptosis inducing process. Both POSH and JIP‐1 can independently interact with members of all three MAPK tiers. However, the direct link between POSH, JIP‐1 and JNK provides us with an interesting possibility to explain how scaffold proteins contribute to the apoptotic pathway.
Functional evaluation of JNK‐interacting scaffold protein in vivo using mouse gene knockout models
In vitro and cell culture studies have elucidated specific interactions between scaffold proteins and signalling molecules. Cell processes dependent upon these interactions, such as apoptosis and differentiation, have been subsequently identified. In vivo studies have also proved valuable both for validation of these findings and more vitally, for revealing their physiological relevance in mammals by discovering the functional importance of these interactions and processes during development, and in maintenance and correct functioning of adult tissues.
Mouse gene knockout models have been made and analysed for several JNK‐interacting scaffold genes including JIP‐1 (Ib1), JIP‐2, JSAP1 (JIP‐3) and JLP. Because of the multiple protein–protein interaction domains present on each scaffold protein and overlap between sets of kinases and other molecules interacting with individual scaffold proteins, some degree of functional redundancy might be anticipated. Indeed, single JNK gene knockout mice have no apparently different phenotype, whereas combined knockout of JNK1 and JKN2 results in an embryonic lethal phenotype in which the neural tube fails to close due to defective regulation of apoptosis (71). Nevertheless, individual JNK‐interacting scaffold gene knockout mouse models have revealed distinct functions in specific tissues and cell types during development and in adults. All the reported mouse JNK‐interacting scaffold knockout models involve similar strategies based on deleting part of the relevant scaffold gene and its replacement with a pGK promoter‐driven neomycin resistance gene cassette.
JIP‐1 knockout mice
At least three independent Jip‐1 knockout mouse models have been made in which the genetic modification is similar, including deletion of sequences encoding the JNK binding domain, yet the severity of changes reported in the homozygous phenotypes are very different from each other. Despite this, all three models support the same conclusion, that levels of JIP‐1 govern JNK‐mediated apoptosis of neurons after brain insult. In one model (72), homozygous deletion of coding exons 3–8 of the mouse JIP‐1‐encoding gene resulted in embryonic lethality. This was observed very early, in pre‐implantation embryos, despite presence of Jip‐1 transcripts in both eggs and spermatozoa, taken to indicate a requirement for de novo Jip‐1 transcription in development of the zygote. However, in another Jip‐1 knockout model deletion of only exon 3 (73) and a later one, deletes sequences encoding the N‐terminal half of JIP‐1 (74). In these two models, homozygote knockout mice were viable and fertile and appeared normal at a gross level. The reason for this discrepancy with the Thompson et al. (72) model remains unknown. Thompson’s genetic modification was maintained on two different mouse genetic backgrounds, C57/BL6 and 129 SvJ, and embryonic lethality was associated with both strains. Whitmarsh et al. (73) and Im et al. (74) each maintained their genetic modifications on a C57Bl/6 strain background, and Im et al. also mention their use of 129 mice but do not report any result from these animals. Thus, the reason for different reported phenotypes cannot be readily attributed to the use of different strain backgrounds, although the homozygous animals studied by Im et al. (74) were generated after 6–7 generations of backcrossing to the C57/BL6 strain, whereas homozygous animals studied by Thompson et al. (72) were generated after only three generations of backcrossing to the same strain. It seems more likely that different targeting strategies used by each group could explain the different phenotypes, but further studies are needed to clarify this. For instance, it is not clear whether truncated JIP‐1 protein might be produced from any of the modified alleles. However, it is also worth noting that mutant mice used in Thompson et al.’s study (72) were derived from a single targeted ES cell clone and it cannot be excluded that a spontaneous mutation in these cells might contribute to the observed embryonic lethal phenotype.
Experiments on Jip‐1 −/− knockout mice used middle cerebral artery occlusion to induce brain ischaemia (74) or kainate injection to induce excitotoxic seizures in the hippocampus (73). In both cases, treatment of wild‐type mice caused an increase in JIP‐1 levels and induction of activated JNK, followed by cell death by apoptosis, in the brain region studied. In contrast, homozygous knockout mice showed either no induction of activated JNK (73) or only a short‐lived activation (74), followed by smaller infarcts or lesions containing fewer apoptotic cells than in the wild‐type mice. Mice heterozygous for the Whitmarsh and Im Jip‐1 knockout alleles are comparable with wild‐type mice in these experiments. In addition to neurites in the hippocampus, JIP‐1 is found in pancreatic islet β cells. Interestingly, pancreatic morphology and function are normal in Jip‐1 knockout homozygotes (73).
These results demonstrate that stress‐induced JNK‐mediated apoptosis is dependent upon JIP‐1, but only in the brain. Heterozygous Jip‐1 +/− mice made by Thompson et al. (72) were also used for a similar kainate‐based excitotoxic stress experiments (75). These mice had significantly lower levels of JIP‐1 protein in the hippocampus compared to wild‐type mice. In this case, treatment with kainate failed to upregulate JIP‐1 levels appreciably in heterozygotes, yet activation of JNK occurred. Contrary to homozygous knockout results, excitotoxic stress of these heterozygotes resulted in increased apoptosis in the hippocampus in comparison to wild‐type mice, suggesting that JIP‐1 can have a neuroprotective effect. Although these results seem contradictory, it is suggested that while low JIP‐1 levels in Thompson’s heterozygotes were insufficient to inhibit JNK signalling after brain insult, an absolute absence of JIP‐1 (as in Whitmarsh and Im’s homozygotes) was unable to activate JNK. Therefore, it is proposed that such exquisite modulation of JIP‐1 levels can result in either induction or inhibition JNK activation.
JSAP1 (JIP‐3) knockout mice
Several Jsap1 knockout mouse models have been reported. In one, exon 1 is deleted (76), whereas another has a larger deletion of DNA sequence encoding the N‐terminal part of the protein, including the leucine zipper protein interaction domain (77). In both cases, no JSAP1 protein was detectable, using Western blotting, in homozygous Jsap1 knockout embryos. Embryonic development in these animals appeared to be normal, but homozygous Jsap1 knockout pups died during the first day after birth, apparently from inability to breathe properly. Neonatal death was attributed to lack of JSAP1 neuronal function by studies of a later conditional Jsap1 knockout model. In this model, double transgenics homozygous for floxed exons 3 and 4 of the Jsap1 gene and carrying a nestin promoter‐driven cre transgene, gave rise to neuron‐specific Jsap1 deletion (78). The phenotypes of the conditional and conventional knockouts were essentially the same.
Jsap is expressed predominantly in neurons and neuroendocrine cells. A very detailed histological brain analysis was performed comparing homozygous Jsap1 knockout mice with wild type and heterozygous littermates, at embryonic day 18.5 (77). This showed that axon guidance had failed in the telencephalic commissures, fornix and optic nerve tract of homozygotes giving rise to subtle morphological abnormalities in the brain. Neuron axon tracts in these regions had high levels of activated JNK in wild‐type control mice, whereas only low levels were found in homozygous knockouts indicating that JNK activation here was dependent on JSAP1. Interestingly, transgenic PDGF promoter‐driven Jip‐1 (Tg‐Jip‐1) expression rescued these morphological phenotypes to some extent, although Tg‐Jip‐1/Jsap1 −/− mice still died on the day of birth. Jsap1 homozygous knockouts also had defective axon guidance in anterior regions of the thalamus and hippocampus, aberrant migration of cells in the cortical plate and disrupted axonal transport. These phenotypes were not rescued by expression of the Tg‐Jip‐1 transgene. Differences in rescue could reflect availability of protein binding partners common to JIP‐1 and JSAP1, in different brain regions.
JLP knockout mice
Reported Jlp knockout involves deletion of the Jlp promoter and exon 1 (79). Despite in vitro studies suggesting roles for JLP in endodermal differentiation and cell migration, homozygous Jlp knockout mice have shown that JLP is indispensable only in later stages of sperm development. In comparison to wild‐type mice, male fertility, sperm number and sperm function in the “swim up” test were shown to be reduced in homozygous knockouts. Fertility of male jlp +/− heterozygotes did not differ from wild types, although it was not shown whether JLP protein levels were altered in the spermatids of this group. In wild‐type mice, JLP is expressed in a variety of organs, most strongly in the testis (79). Interestingly, no activated JNK could be detected in testes of homozygous knockouts, whereas activated JNK levels in the brain did not differ from heterozygotes or wild‐type mice suggesting that JNK activation is dependent upon JLP specifically in sperm development.
Genetically modified mouse models have proved that JNK scaffold proteins have distinct, essential functions in mammals. In addition to validating previous findings from cell culture and in vitro experiments, studies of JNK scaffold gene knockout mice have identified interesting questions concerning degeneracy and compensatory mechanisms between JNK scaffold proteins in vivo. In particular, it is notable that JNK scaffold gene knockout models have phenotypes corresponding to only a subset of the tissues in which the genes are normally expressed. Analysis of altered expression of downstream target genes in the knockout models will elucidate how deletion of each JNK scaffold gene causes its corresponding distinct phenotype. These models have shown an essential role for these genes both in mammalian development and in regulation of stress‐induced neuronal apoptosis.
Conclusions
The pivotal role of scaffold proteins in mediation of intracellular messages is now firmly established. By tethering individual pathway components together, different pathways can be insulated from one another. This becomes particularly useful when single components are used alternatively in different pathways, as was first shown for Ste11 in the MAPK pathway in S. cervisiae. Moreover, scaffold proteins can establish connections between pathways and help to distribute signals (17). This is observed, for example, when large signalling complexes are formed around tyrosine kinase receptors. Signal fluxes are optimized when scaffold proteins promote a more efficient use of local components and force them to interact. Some early mathematical modelling of the ERK pathway supports this hypothesis (80). In the ERK pathway, scaffolds can overcome double phosphorylation into an ordered sequence of events. This threshold mechanism has been used to explain why mammalian cells are not ultrasensitive to external signalling. The other side of the argument is that, in the presence of an excess of scaffold protein over its binding components, an incomplete complex will form and signalling will be obscured.
More recent modelling data suggest that depending on cellular conditions, spatial organization of kinases on scaffold proteins can either enhance or inhibit signal propagation through a kinase cascade. Specifically, scaffolds enhance signal propagation when prevailing conditions would lead to attenuation. Conversely, scaffolds inhibit propagation of signals that would otherwise be greatly amplified (81). In other words, scaffolds allow for controlled levels of signals that are delivered at appropriate times (82).
Another function of scaffold proteins is to organize spatial arrangement in signalling pathway architecture. Early data have demonstrated that the KSR scaffold complex is formed at the cell membrane (83), whereas MP‐1 scaffold complex that contains MEK‐ERK, is formed at endosomes through binding to an endosomal protein p14 (84). Using different scaffold proteins, the cell can arrange for signalling proteins to be distributed to different cellular compartments. Experimental data to support this notion come from the EGF receptor, where receptor internalization kinetics suggest that endosomal signalling is predominant at low growth factor levels (85, 86). The data also suggest that the MEK‐ERK complex acts as a sensor for EGF concentration. The difference in time required for receptor internalization and endosomal compartmentalization can thus account for two different activation phases. In addition, subcellular localization of JNK‐MAPK scaffolds has been implied in regulation of cell locomotion (87). Some recently published data have shed some further light on the compartmentalization issue. Blanco et al. (88) has shown that the activity of JIP‐1/JNK complexes in vivo can be efficiently down‐regulated by interleukin‐1 beta. In this situation, vaccinia‐related kinase (VRK2), an enzyme with different subcellular localization from IL‐1 beta, interacts stably with JIP‐1, TAK1 and MKK7 but not with JNK.
Thus, scaffold proteins can use both positive and negative signals to control and use different MAPK signalling components. Moreover, scattered data suggest that scaffold protein JIP‐1 can play a dual role in the apoptotic process including in vivo. Interestingly, POSH‐induced apoptosis can be efficiently counteracted by concomitant expression of serine/threonine kinase, Akt (89). Moreover, mutated POSH unable to bind Akt leads to increased activation of the JNK pathway (90). The relationship between Akt and JIP‐1 adds to the complexity of the picture as JIP‐1 increases Akt kinase activity in a dose‐dependent fashion (91). Conversely, a negative feedback from loop has been identified, in that Akt could be dissociated JIP‐1 during metabolic oxidative stress when JIP‐1 has been proposed to act as a negative regulator (92). Contradictory as it may seem, the general framework is now firmly established, however, many details concerning precise interaction between different related molecules remain to be elucidated.
Acknowledgements
The authors acknowledge the financial support of the Cancer Research Campaign (UK) and the Wellcome Trust. WE was the recipient of a UICC technical transfer fellowship.
References
- 1. Morrison DK, Davis RJ (2003) Regulation of MAP kinase signalling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 19, 91–118. [DOI] [PubMed] [Google Scholar]
- 2. Chen Z, Gibson TB, Robinson F, Silvestro I, Pearson G, Xu B (2001) MAP kinases. Chem. Rev. 101, 2449–2476. [DOI] [PubMed] [Google Scholar]
- 3. Boldt S, Kolch W (2004) Targeting MAPK signalling: prometheus fire or pandoras box. Curr. Pharm. Des. 10, 1885–1905. [DOI] [PubMed] [Google Scholar]
- 4. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ (2007) Macrophage glucocorticoid receptors regulate Toll like receptor 4‐mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 109, 4313–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi KY, Satterberg B, Lyons DM, Elion EA (1994) Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S cerevisiae. Cell 78, 499–512. [DOI] [PubMed] [Google Scholar]
- 6. Marcus S, Polverino A, Barr M, Wigler M (1994) Complexes between STE5 and components of the pheromone responsive mitogen activated Protein kinase module. Proc. Natl. Acad. Sci. USA 91, 7762–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elton EA (2001) The Ste5p scaffold. J. Cell Sci. 114, 3967–3978. [DOI] [PubMed] [Google Scholar]
- 8. Burack WR, Shaw AS (2000) Signal transduction; hanging on a scaffold. Curr. Opin. Cell Biol. 12, 211–216. [DOI] [PubMed] [Google Scholar]
- 9. Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser IDC, Langeberg LK et al. (1999) Regulation of NMDA receptors by an associated phosphatase kinase signalling complex. Science 285, 93–96. [DOI] [PubMed] [Google Scholar]
- 10. Tsunoda S, Sierralta J, Sun YM, Bodner R, Suzuki E, Becker A et al. (1997) A multivalent PDZ domain protein assembles signalling complexes in a G‐protein coupled cascade. Nature 388, 243–249. [DOI] [PubMed] [Google Scholar]
- 11. Schillace RV, Scott JD (1999) Organisation of kinases, phosphatases and receptor signalling complexes. J. Clin. Invest. 103, 761–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rudd CE (1999) Adaptors and molecular scaffolds in immune cell signalling. Cell 96, 5–8. [DOI] [PubMed] [Google Scholar]
- 13. Bogoyevitch MA, Court NW (2004) Counting on mitogen activated protein kinases ERKs 3,4,5,6,7 and 8. Cell. Signal. 16, 1345–1354. [DOI] [PubMed] [Google Scholar]
- 14. Bogoyevitch MA (2006) The isoform specific functions of the c‐Jun N‐terminal Kinases (JNKs) differences revealed by gene targeting. Bioessays 28, 923–934. [DOI] [PubMed] [Google Scholar]
- 15. Coulombe P, Meloche S (2006) Atypical mitogen activated protein kinases. Structure, regulation and function. Biochim. Biophys. Acta 1773, 1376–1387. [DOI] [PubMed] [Google Scholar]
- 16. Zarubin T, Han J (2005) Activation and signalling of the p38 MAP kinase pathway. Cell Res. 15, 11–18. [DOI] [PubMed] [Google Scholar]
- 17. Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP (2007) Scaffold proteins of MAP kinase modules. Oncogene 26, 3185–3202. [DOI] [PubMed] [Google Scholar]
- 18. Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K et al. (1999) JSAP1, a novel jun N‐terminal protein kinase (JNK binding protein that functions as a Scaffold factor in the JNK signalling pathway). Mol. Cell. Biol. 19, 7539–7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jagadish N, Rana R, Mishra D, Garg M, Chaurasiya D, Hasegawa A et al. (2005) Immunogenicity and contraceptive potential of recombinant human sperm associated antigen (SPAG9). J. Reprod. Immunol. 67, 69–76. [DOI] [PubMed] [Google Scholar]
- 20. Jagadish N, Rana R, Selvi R, Mishra D, Garg M, Yadav S et al. (2005) Characterisation of a novel human sperm associated antigen 9 (SPAG9) having structural homology with c‐Jun N‐terminal kinase interacting protein. Biochem. J. 389, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelkar N, Standen CL, Davis RJ (2005) Role of JIP4 scaffold protein in the regulation of mitogen activated protein kinase signalling pathways. Mol. Cell. Biol. 25, 2733–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee CL, Onesime D, Reddy CD, Dhanasekaran N, Reddy EP (2002) JLP, a scaffolding protein that tethers JNK/p38MAPK signalling modules and transcription factors. Proc. Natl. Acad. Sci. USA 99, 14189–14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tapon N, Nagata K, Lamarche N, Hall A (1998) A new rac target POSH is an SH3 containing scaffold protein involved in the JNK and NF kappaB signalling pathways. EMBO J. 17, 1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Whitmarsh AJ, Davis RJ (1998) Structural organisation of MAP kinase signalling modules by scafold proteins in yeast and mammals. Trends Biochem. Sci. 23, 481–485. [DOI] [PubMed] [Google Scholar]
- 25. Whitmarsh AJ, Davis RJ (1999) Signal transduction by MAP kinases; regulation by phosphorylation dependent switches. Sci. STKE 1999, PE1. [DOI] [PubMed] [Google Scholar]
- 26. Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ (1999) The JIP group of mitogen activated protein kinase scaffold proteins. Mol. Cell. Biol. 19, 7245–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dickens M, Rogers JS, Cavanagh J, Rairano A, Xia Z, Halpern JR et al. (1997) A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277, 693–696. [DOI] [PubMed] [Google Scholar]
- 28. Bonny C, Oberson A, Steinmann M, Schorderet DF, Nicod P, Weber G (2000) IB1 reduces cytokine induced apoptosis of insulin secreting cells. J. Biol. Chem. 275, 16466–16472. [DOI] [PubMed] [Google Scholar]
- 29. Willoughby EA, Perkins GR, Collins MK, Whitmarsh AJ (2003) The JNK interacting protein 1 scaffold protein targets MAPK phosphatase 7 to dephosphorylate JNK. J. Biol. Chem. 278, 10731–10736. [DOI] [PubMed] [Google Scholar]
- 30. Meyer D, Liu A, Margolis B (1999) Interactuion of c‐Jun amino‐terminal kinase interacting protein 1 with p190 rhoGEF and its localisation in differentiated neurons. J. Biol. Chem. 174, 35113–35118. [DOI] [PubMed] [Google Scholar]
- 31. Marinissen MJ, Chiarello M, Tanos T, Bernard O, Narumiya S, Gutkind JS (2004) The small GTP binding protein RhoA regulates c‐jun by a ROCK‐JNK signalling axis. Mol. Cell 14, 29–41. [DOI] [PubMed] [Google Scholar]
- 32. Ferrer I (2004) Stress kinases involved in tau phosphorylation in Alzheimers disease, tauopathies and APP transgenic mice. Neurotox. Res. 6, 469–475. [DOI] [PubMed] [Google Scholar]
- 33. Scheinfeld MH, Roncarati R, Vito P, Lopez PA, Abdallah M, D′Adamio L (2002) Jun NH2 terminal kinase interacting protein 1 (JIP1) binds to the cytoplasmic domain of the Alzheimer beta amyloid precursorprotein (APP). J. Biol. Chem. 277, 3767–3775. [DOI] [PubMed] [Google Scholar]
- 34. Negri S, Oberson A, Steinmann M, Sauser C, Nicod P, Waeber G et al. (2000) cDNA cloning and mapping of a novel islet brain/JNK interacting protein. Genomics 64, 324–330. [DOI] [PubMed] [Google Scholar]
- 35. Buchsbaum RJ, Connolly BA, Feig LA (2002) Interaction of Rac exchange factors TIAM1 and RasGRF with a scaffold for the p38 mitogen‐activated protein kinase cascade. Mol. Cell. Biol. 22, 4073–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schoorlemmer J, Goldfarb M (2002) Fibroblast growth factor homologous factors and the islet brain scaffold protein regulate activation of a stress activated protein kinase. J. Biol. Chem. 277, 49111–49119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kelkar N, Gupta S, Dickens M, Davis RJ (2000) Interaction of a mitogen‐activated protein kinase signalling module with the neuronal protein JIP3. Mol. Cell. Biol. 20, 1030–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsuura H, Nishitoh H, Takeda K, Matsuzawa A, Amagasa T, Ito M et al. (2002) Phosphorylation dependent scaffolding role of JSAP1/JIP‐3 in the ASK1/JNK signalling pathway. A new mode of regulation of the MAP kinase cascade. J. Biol. Chem. 277, 40703–40709. [DOI] [PubMed] [Google Scholar]
- 39. Ito M, Akechi M, Hirose R, Ichimura M, Takamatsu N, Xu P et al. (2000) Isoforms of JSAP1 scaffold protein generated through alternative splicing. Gene 255, 229–234. [DOI] [PubMed] [Google Scholar]
- 40. Kuboki Y, Ito M, Takamatsu N, Yamamoto KI, Shiba T, Yoshioka K (2000) A scaffold protein in the c‐Jun NH2 terminal kinase signalling pathways suppresses the extracellular signal regulated kinase signalling pathways. J. Biol. Chem. 275, 39815–39818. [DOI] [PubMed] [Google Scholar]
- 41. Takino T, Yoshioka K, Miyamori H, Yamada KM, Sato H (2002) A scaffold protein in the jun N‐terminal kinase signalling pathway is associated with focal adhesion kinase and tyrosine phosphorylated. Oncogene 21, 6488–6497. [DOI] [PubMed] [Google Scholar]
- 42. Takino T, Nakada M, Miyamori H, Watanabe Y, Sato T, Gantulga D et al. (2005) JSAP1/JIP3 cooperates with focal adhesion kinase to regulate c‐jun N‐terminal kinase and cell migration. J. Biol. Chem. 280, 37772–37781. [DOI] [PubMed] [Google Scholar]
- 43. Matsuguchi T, Masuda A, Sugimoto K, Nagai Y, Yoshikai Y (2003) JNK interacting protein 3 associates with Toll like receptor 4 and is involved in LPS mediated JNK activation. EMBO J. 22, 4455–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nguyen Q, Lee CM, Reddy EP (2005) JLP associates with kinesin light chain 1 through a novel leucine zipper like domain. J. Biol. Chem. 280, 30185–30191. [DOI] [PubMed] [Google Scholar]
- 45. Kashef K, Xu H, Reddy EP, Dhanasekaran DN (2006) Endodermal differentiation of murine embryonic carcinoma cells by retinoic acid requires JLP a JNK scaffolding protein. J. Cell. Biochem. 98, 715–722. [DOI] [PubMed] [Google Scholar]
- 46. Kashef K, Lee CM, Ha JH, Reddy EP, Dhanasekaran DN (2005) JNK interacting leucine zipper protein is a novel scaffolding protein is a novel scaffolding protein in the Ga13 signalling pathway. Biochemistry 44, 14090–14096. [DOI] [PubMed] [Google Scholar]
- 47. Kukekov NV, Xu Z, Greene LA (2006) Direct interaction of the molecular scaffolds POSH and JIP is required for apoptotic activation of JNKs. J. Biol. Chem. 281, 15517–15524. [DOI] [PubMed] [Google Scholar]
- 48. Xu S, Cobb MH (1997) MEKK1 binds directly to the c‐jun N‐terminal kinases/stress activated protein kinases. J. Biol. Chem. 272, 32056–32060. [DOI] [PubMed] [Google Scholar]
- 49. Lefkovitz RJ, Shenoy SK (2005) Transduction of receptor signals by beta‐arrestins. Science 308, 512–517. [DOI] [PubMed] [Google Scholar]
- 50. Rohbe L, Larsson M, Rising A, Grip S, Burns J, Engström W (2004) Expression of JNK interacting protein JIP‐1 is down regulated in liver from mouse embryos with a disrupted insulin like growth factor II gene. In vivo 18, 643–648. [PubMed] [Google Scholar]
- 51. Ward A, Bierke P, Pettersson E, Engström W (1994) Insulin like growth factors; growth, transgenes and imprinting. Zool. Sci. 11, 167–174. [PubMed] [Google Scholar]
- 52. Engström W, Shokrai A, Otte K, Granerus M, Gessbo Å, Bierke P et al. (1998) Transcriptional regulation and biological significance of the insulin like growth factor 2 gene. Cell Prolif. 31, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dell G, Ward A, Shokrai A, Madej A, Engström W (1999) Regulation of the IGF system by glucocorticoids. Zool. Sci. 16, 377–385. [Google Scholar]
- 54. Smith FM, Garfield AS, Ward A (2006) Regulation of growth and metabolism by imprinted genes. Cytogenet. Genome Res. 113, 279–291. [DOI] [PubMed] [Google Scholar]
- 55. Engström W, Rising A, Grip S (2005) The JNK interacting protein JIP‐1 and insulin like growth factor II genes are co‐expressed in human embryonic tumours. Anticancer Res. 25, 1075–1078. [PubMed] [Google Scholar]
- 56. Granerus M, Johannisson A, Ekblom P, Engström W (1991) Insulin like growth factors I and II induce cell death in Wilms tumour cells. Mol. Pathol. 54, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Engström W, Granerus M (2009) Expression of JNK interacting protein JIP‐1 and insulin like growth factor II in Wilms tumour cell lines and primary Wilms Tumours. Anticancer Res. 29, 2467–2472. [PubMed] [Google Scholar]
- 58. Xia Z, Dickens M, Raingeaud J, Davis RJ (1995) Opposing effects of ERK and JNK‐p38 MAP‐kinases on apoptosis. Science 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- 59. Boldt S, Weidle UH, Kolch W (2003) The kinase domain of MEKK1 induces apoptosis by dysregulation of MAP kinase pathways. Exp. Cell Res. 283, 80–90. [DOI] [PubMed] [Google Scholar]
- 60. Nishina H, Vaz C, Billia P, Nghiem M, Sasaki T, De La Pompa JL (1999) Defective liver formation and liver cell apoptosis in mice lacking the stress signalling kinase SEK1/MKK4. Development 126, 505–516. [DOI] [PubMed] [Google Scholar]
- 61. Lamb JA, Ventura JJ, Hess P, Flavell RA, Davis RJ (2003) JunD mediates survival signalling by the JNK signal transduction pathway. Mol. Cell 11, 1479–1489. [DOI] [PubMed] [Google Scholar]
- 62. Lei K, Davis RJ (2003) JNK phosphorylation of Bim‐related members of the Bcl2 family induces Bax‐dependent apoptosis. Proc. Natl. Acad. Sci. USA 100, 2432–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Donovan N, Becker EB, Konishi Y, Bonni A (2002) JNK phosphorylation and activation of BAD couples the stress activated signalling pathway to the cell death machinery. J. Biol. Chem. 277, 40944–40949. [DOI] [PubMed] [Google Scholar]
- 64. Yamamoto K, Ichijo H, Korsmeyer SJ (1999) Bcl2 is phosphorylated and inactivated by an ASK1/JUN N‐terminal kinase pathway normally activated at G2/M. Mol. Cell. Biol. 19, 8469–8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Basu A, Haldar S (2002) Identification of a novel Bcl‐xL phosphorylation site regulating the sensitivity of taxol‐ or 2‐methoxyestradiol induced apoptosis. FEBS Lett. 538, 41–47. [DOI] [PubMed] [Google Scholar]
- 66. Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB et al. (2002) The Bax subfamily of Bcl2 related proteins is essential for apoptotic signal transduction by JNK. Mol. Cell. Biol. 22, 4929–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Haefliger JA, Tawadros T, Meylan L, Gurun SL, Roehrich ME, Martin D et al. (2003) The scaffold protein IB1/JIP‐1 is a critical mediator of cytokine‐induced apoptosis in pancreatic beta cells. J. Cell Sci. 116, 1463–1469. [DOI] [PubMed] [Google Scholar]
- 68. Nikulina MA, Sandhu N, Shamim Z, Andersen NA, Oberson A, Dupraz P et al. (2003) The JNK binding domain of islet brain 1 inhibits IL‐1 induced JNK activity and apoptosis but not the transcription of key proapoptotic or protective genes in insulin secreting cell lines. Cytokine 24, 13–24. [DOI] [PubMed] [Google Scholar]
- 69. Ling Z, Van De Casteele M, Dong J, Helmberg H, Haeflinger JA, Waeber G et al. (2003) Variations in IB1/JIP1 expression regulate susceptibility of beta cells to cytokine induced apoptosis irrespective of c‐jun Nh2 terminal kinase signalling. Diabetes 52, 2497–2502. [DOI] [PubMed] [Google Scholar]
- 70. Xu Z, Kukekov NV, Greene LA (2003) POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J. 22, 252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22, 667–676. [DOI] [PubMed] [Google Scholar]
- 72. Thompson NA, Haefliger J‐A, Senn A, Tawadros T, Magara F, Ledermann B et al. (2001) Islet‐brain1/JNK‐interacting protein‐1 is required for early embryogenesis in mice. J. Biol. Chem. 276, 27745–27748. [DOI] [PubMed] [Google Scholar]
- 73. Whitmarsh AJ, Kuan C‐Y, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP et al. (2001) Requirement of the JIP‐1 scaffold protein for stress induces JNK activation. Genes Dev. 15, 2421–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Im J‐Y, Lee K‐W, Kim MH, Lee SH, Ha H‐Y, Cho I‐H et al. (2003) Repression of Phospho‐JNK and infarct volume in ischemic brain of JIP1‐deficient mice. J. Neurosci. Res. 74, 326–332. [DOI] [PubMed] [Google Scholar]
- 75. Magara F, Haefliger J‐A, Thompson N, Riederer B, Welker E, Nicod P et al. (2003) Increased vulnerability of kainic acid‐induced epileptic seizures in mice underexpressing the scaffold protein Islet‐Brain 1/JIP‐1. Eur. J. Neurosci. 17, 2602–2610. [DOI] [PubMed] [Google Scholar]
- 76. Kelkar N, Delmotte MH, Weston CR, Barrett T, Sheppard BJ, Flavell RA et al. (2003) Morphogenesis of the telencephalic commissure requires scaffold protein JNK‐interacting protein 3 (JIP3). Proc. Natl. Acad. Sci. USA 100, 9843–9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ha H‐Y, Cho I‐H, Lee K‐W, Lee K‐W, Song J‐Y, Kim K‐S et al. (2005) The exon guidance defect of the telencephalic commissures of the JSAP1‐deficient brain was partially rescued by the transgenic expression of JIP1. Dev. Biol. 277, 184–199. [DOI] [PubMed] [Google Scholar]
- 78. Iwanaga A, Sato T, Sugihara K, Hirao A, Takakura N, Okamoto H et al. (2007) Neural‐specific ablation of the scaffold protein JSAP1 in mice causes neonatal death. Neurosci. Lett. 429, 43–48. [DOI] [PubMed] [Google Scholar]
- 79. Iwanaga A, Wang G, Gantulga D, Sato T, Baljinnyam T, Shimizu K et al. (2008) Ablation of the scaffold protein JLP causes reduced fertility in male mice. Transgenic Res. 17, 1045–1056. [DOI] [PubMed] [Google Scholar]
- 80. Levchenko A, Bruck J, Sternberg PW (2000) Scaffold proteins may biphasically affect the levels of mitogen‐activated protein kinase signalling and reduce its threshold properties. Proc. Natl. Acad. Sci. USA 97, 5818–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Locasale JW, Shaw AS, Chakraborty AK (2007) Scaffold proteins confer diverse regulatory properties to protein kinase cascades. Proc. Natl. Acad. Sci. USA 104, 13307–13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Locasale JW, Chakraborty AK (2008) Regulations of signal duration and the statistical dynamics of kinase activation by scaffold proteins. PLoS Comput. Biol. 4, e10000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Muller J, Ory S, Copeland T, Piwnica‐Worms H, Morrisson DK (2001) CTAK1 regulates Ras signalling by phosphorylating the MAPK scaffold KSR1. Mol. Cell 8, 983–993. [DOI] [PubMed] [Google Scholar]
- 84. Teis D, Wunderlich W, Huber LA (2002) Localisation of the MP‐1/MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev. Cell 3, 803–814. [DOI] [PubMed] [Google Scholar]
- 85. Resat H, Ewald JA, Dixon DA, Wiley HS (2003) An integrated model of epidermal growth factor receptor trafficking and signal transduction. Biophys. J. 85, 730–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schoeberl B, Eichler‐Jonsson C, Giles ED, Muller G (2003) Computational modelling of the dynamics of the MAP kinase cascade activated by surface and internalised EGF‐receptors. Nat. Biotechnol. 20, 370–375. [DOI] [PubMed] [Google Scholar]
- 87. Huang C, Jacobson K, Schaller MD (2004) MAP‐kinases and cell migration. J. Cell Sci. 117, 4619–4628. [DOI] [PubMed] [Google Scholar]
- 88. Blanco S, Sanz‐Garcia M, Santos CR, Lazo PA (2008) Modulation of interleukin 1 transcriptional response by the interaction between VRK2 and the JIP1 scaffold protein. PLoS One 3, e1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lyons TR, Thorburn J, Ryan PW, Thorburn A, Anderson SM, Kassenbrock CK (2007) Regulation of the Pro‐apoptotic Scaffolding Protein POSH by Akt. J. Biol. Chem. 282, 21987–21997. [DOI] [PubMed] [Google Scholar]
- 90. Figueroa C, Tarras S, Taylor J, Vojtek AB (2003) Akt2 negatively regulates assembly of the POSH‐MLK‐JNK signalling complex. J. Biol. Chem. 278, 47922–47927. [DOI] [PubMed] [Google Scholar]
- 91. Kim AH, Sasaki T, Chao MV (2003) JNK‐interacting protein 1 promotes Akt1 activation. J. Biol. Chem. 278, 29830–29836. [DOI] [PubMed] [Google Scholar]
- 92. Song JJ, Lee YJ (2005) Dissociation of Akt1 from its negative regulator JIP‐1 is mediated through the ASK1‐MEK‐JNK signal transduction pathway during metabolic oxidative stress. A negative feedback loop. J. Cell Biol. 170, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]