

Abstract
Objectives
As a follow‐up to our previous reports showing that the G9a histone methyltransferase‐specific inhibitor BIX01294 enhances bone marrow cell cardiac potential, this drug was examined for its effects on cardiomyocytes and mouse cardiac progenitor cells (CPCs).
Materials and methods
Cardiomyocytes and cardiac explants were cultured ± BIX01294, and examined for changes in cardiac function, protein and gene expression. Additionally, enriched populations of CPCs, contained in the ‘phase bright cell’ component of explants, were harvested from non‐treated and BIX01294‐treated cardiac tissue, and assayed for differences in cell phenotype and differentiation potential. Mouse CPCs were cultured with rat cardiomyocytes to allow differentiation of the progenitors to be assayed using species‐specific PCR primers.
Results
While BIX01294 had no discernible effect on myocyte function and sarcomeric organization, treatment with this drug significantly increased CPC proliferation, as indicated by enhanced MTT metabolization and BrdUrd incorporation (4.1‐ and 2.0‐fold, respectively, P < 0.001) after 48 h labelling, and increased Ki67 expression (4.8‐fold, P < 0.001) after 7 days culture. Heart explants exposed to BIX01294 generated 3.6‐fold (P < 0.005) greater yields of CPCs by 2 weeks culture. Importantly, CPCs obtained from non‐treated and BIX01294‐treated cultures did not differ in phenotype or differentiation potential.
Conclusions
These data indicate that BIX01294 can expand CPCs without undermining their capacity as cardiac progenitors, and suggest that this drug may have utility for generating large numbers of CPCs for cardiac repair.
Introduction
The adult heart was long considered a static, terminally differentiated organ with a limited capacity for tissue regeneration. Yet in recent years, numerous investigations have identified small pools of progenitor cells within the adult heart that help replenish cardiac cells during normal homoeostasis and in response to injury 1, 2, 3, 4. A standard method for isolating cardiac progenitor cells (CPCs) is by long‐term culture of heart tissue, where an enriched population of CPCs appear as small round phase bright cells that reside on top of an adherent layer of cells 5, 6, 7, 8. A limitation in using CPCs for combating human disease is that their source are heart biopsies, which generate low amounts of these cells. Thus, there is the need to develop the means to rapidly expand the numbers of CPCs from tissue biopsies without altering their phenotype or differential potential.
In previous studies, we showed that exposing bone marrow stem cells to the G9a histone methyltransferase (G9a HMTase) inhibitor BIX01294 helped generate cells with a cardiopotent phenotype 9, 10. This outcome was indicated by the upregulation of the pre‐cardiac genes Mesp1 and brachyury in bone marrow cultures and the subsequent enhanced ability of the BIX01294‐treated cells to express early cardiomyocyte markers in response to the cardiac differentiation factor Wnt11. The present study was initially designed to examine whether BIX01294 would have any deleterious impact on cardiac function and/or differentiation that would compromise its utility in generating a plentiful and accessible source of stem cells that could be used for cardiac repair. Accordingly, we tested the effect of BIX01294 on cardiac myocytes and endogenous cardiac progenitors. Here, we report that BIX01294 elicited no negative effect on cardiomyocyte beating or sarcomeric organization. Moreover, CPCs displayed the same phenotype and differentiation potential when obtained from BIX01294‐treated or non‐treated cardiac explants. However, BIX01294 significantly increased the proliferation of CPCs and greatly enhanced their cell numbers in culture. These data indicate that BIX01294 can act as an expansion factor for endogenous CPCs and suggest this drug may have utility as an agent that can generate large numbers of native CPCs for treating heart disease.
Material and methods
Atrial tissue explants and generation of phase bright cells
Atrial cardiac tissue was isolated from 8‐ to 10‐week‐old C57BL/6 mice. The atria were excised from hearts and minced into 1–2 mm3 pieces, washed with Ca+‐ and Mg2+‐free phosphate‐buffered saline (PBS) and digested with 0.2% trypsin and 0.1% type IV collagenase for 10 min. Tissue fragments were then plated for 2 weeks on gelatin‐coated dishes in Iscove's Modified Dulbecco's Medium (IMDM) containing 20% FBS plus penicillin–streptomycin (100 U/ml) at 37 °C and 5% CO2. Cultures were fed twice weekly for 2 weeks prior to BIX01294 (Cayman Chemical, Ann Arbor, MI, USA) and control treatments.
Within 2 weeks in culture, explanted cardiac atria exhibited numerous small, loosely adherent ‘phase bright cells’ that appeared on top of the tissue. Phase bright cells were harvested using 0.5 nmol/l EDTA for no more than 2 min while visually monitoring cell detachment. Numbers of phase bright cells and the percentage of viable cells generated from the atrial cultures were assessed by trypan blue exclusion, as previously described 11. The significance of the differences in cell numbers generated from control and BIX01294‐treated cultures was determined by the paired t‐test. All statistical assessments of data within this study were calculated with the Macintosh Rosetta application InStat (GraphPad Software, La Jolla, CA, USA).
Isolation of neonatal rat cardiomyocytes
Cardiomyocytes were isolated from 1‐day‐old Wistar rats (Taconic Biosciences, Hudson, NY, USA), using previously described protocols 12, 13. In brief, hearts were minced into 1–3 mm3 pieces and digested for 10 min at 37 °C with 1% type IV collagenase (Worthington, Biochemical, Lakewood, NJ, USA), 2.5% trypsin, 10 μg/ml DNAse I and 0.1% chicken serum in pH 7.6 buffered Hank's balanced salt solution (Sigma, St. Louis, MO, USA). Dissociated cells were transferred to a separate tube containing 100% horse serum. This procedure was repeated up to 8 times until tissue was completely digested. Afterwards, the cell suspension was spun at 300 g for 10 min, with the cell pellets resuspended in Dulbecco's modified Eagle's medium/Nutrient Mixture F‐12 (DMEM/F12) media and incubated for 1 h in tissue culture plates. The supernatant was then collected, centrifuged at 300 g for 10 min, with the pelleted cardiomyocytes plated onto 35 mm dishes in 2% FBS/IMDM plus Pen/Strep and allowed to attach for at least 2 days prior to immunocytochemistry or co‐culture with mouse CPCs. For some experiments, rat cardiomyocytes were cultured in the presence or absence of 8 μm BIX01294 for 2 days and assayed for up to 9 days. In addition, 10 μm isoproterenol was added to a subset of these cultures as a positive inotropic factor.
Co‐cultures
Isolated primary neonatal rat cardiomyocytes were used for co‐culture with mouse atrial phase bright cells. Phase bright cells were harvested from BIX01294‐treated and non‐treated atrial explants, as described above. Freshly isolated phase bright cells were washed with PBS, counted and labelled with 20 μm carboxyfluorescein succinimidyl ester vital dye (CFSE; ThermoFisher Scientific, Waltham, MA, US) for 1 h at 37 °C. After extensive washing with PBS, these labelled mouse cells were plated onto beating neonate rat cardiomyocytes at a ratio of 1:10. Co‐cultures received fresh 2% FBS/IMDM medium on alternate days and after extended incubation periods were processed for immunofluorescent labelling or RNA extraction. The use of different species as source for these two distinct cell populations subsequently enabled CPC‐derived cells to be assayed for cardiac gene expression, using species‐specific PCR primers.
MTT assay
The quantification of cell metabolic activity, as an indirect indicator of cellular proliferation 14, 15, was performed using a colorimetric assay based on capability of enzyme mitochondrial reductase to reduce tetrazolium dye 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT; Sigma) to its insoluble formazan form. Phase bright cells were collected using 0.5 mm EDTA from 2‐, 5‐, 7‐ and 14‐day cultures that were incubated in the absence or presence of BIX01294. Pelleted cells were resuspended in 20% FBS/IMDM and samples seeded in 96‐well plates in duplicate. Cells were incubated for 2 days prior to media replacement with 100 μl of 9:1 mix of clear DMEM and MTT. After 4 h, MTT solution was replaced by 75 μl of 0.04N acidified isopropyl solution for 10 min. Quantification of the formazan dye produced was measured with an absorbance measurement of 560 nm, and reference wavelength of 690 nm using the Infinite M200 Microplate Reader (Tecan, Männedorf, Switzerland).
Bromodeoxyuridine (BrdUrd) incorporation assay
Atrial tissue was cultured for 2 weeks, followed by 7‐day treatments in the absence or presence of BIX01294, before exposing the cells to 4 × 10−5M 5‐bromo‐2′‐deoxyuridine (BrdUrd) at 37 °C for 48 h in the dark. Afterwards, phase bright cells were collected and cytospun onto histology slides for immunostaining. The cells were then washed with cold PBS, and after methanol fixation for 2 min, DNA was denatured by treatment with 2N hydrochloric acid (HCl) for 30 min. Cells were PBS washed and incubated for 1 h at 37 °C with rat anti‐BrdUrd antibody (Abcam, Cambridge, MA, USA). After three PBS washes, cells were incubated with fluorescein‐labelled goat anti‐rat secondary antibody (Jackson Immuno‐Research, West Grove, PA, USA) for 30 min at room temperature and counterstained with DAPI prior to analysis using a Zeiss LSM710 laser scanning confocal microscope. Percentage of cells that were BrdUrd‐positive was determined by manual counting of both immunolabelled and 4′, 6′‐diamidino‐2‐phenyindole (DAPI; Sigma)‐stained nuclei on digitized confocal microscope images using Image J software and the Cell Counter plug‐in (Kurt De Vos, University of Sheffield; kurt.devos@iop.kcl.ac.uk). Significance of differences in the prevalence of BrdUrd incorporation between cultures was assessed by ANOVA analysis using the Bonferroni procedure, as calculated with the InStat statistical application (GraphPad Software, La Jolla, CA, USA).
Immunofluorescent staining
Phase bright cells were collected and stained live on ice for 30 min with antibodies specific for CD90 (BD Biosciences, San Jose, CA, USA), Sca1 (eBioscience, San Diego, CA, USA) and connexin 43 (Cx43; ThermoFisher Scientific). Afterwards, cells were cytospun onto charged histology slides, prior to formalin fixation. For other molecules, cells were immunolabelled following their incubation on tissue culture plastic. Cells stained with mouse anti‐Islet‐1 (Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA) were subjected in sequence to formalin fixation for 10 min, Dent's fixation for 5 min, permeabilization with 0.3% triton/10%BSA/PBS, and overnight blocking with 1% BSA/0.3 m glycine/PBS. Staining with antibodies specific for connexin 40 (Cx40; ThermoFisher Scientific), muscle α‐actinin (Sigma) and Ki67 (Dako, Glostrup, Denmark) was performed on formalin‐fixed cultures that were incubated overnight with 5% BSA/PBS block. DyLight488‐conjugated secondary antibody was used to identify each of these molecules, with the cells counterstained with DAPI to identify nuclei, as previously described 10, 16.
Flow cytometry
Dissociated cells were incubated with monoclonal antibodies specific for Sca1 and Cx43, which were diluted in 1X PBS/1% BSA/2 mm EDTA at 4 °C for 30 min in the dark. Following washes with cold PBS, cells were resuspended for 30 min in DyLight488‐conjugated secondary antibody. As negative controls, cells were incubated with secondary antibody only. Labelled samples were analysed immediately using a BD Accuri C6 flow cytometer and the number of positive cells was determined over 50,000 events recorded for each sample.
RNA isolation and Polymerase Chain Reaction (PCR) amplification
Total RNA was extracted from cultured cells using Quick‐RNA MiniPrep Kits (Zymo Research, Irvine, CA, USA). Subsequently, cDNA was synthesized from RNA template using random primers and Moloney Murine Leukemia Virus Reverse Transcriptase (Promega, Madison, WI, USA). PCR amplification of expressed messages was carried out using Taq polymerase as previously described 9, 17, 18. Template concentrations were first normalized by PCR amplification of the housekeeping genes glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and hypoxanthine phosphoribosyltransferase (HPRT), using primers that recognize both mouse and rat, or mouse only sequences, respectively. Sequences and specificity of the oligonucleotide primers used in this study were: GAPDH (mouse and rat), AGGTCGGTGTGAACGGATTTG and TGTAGACCATGTAGTTGAGGTCA; Cardiac troponin I (mouse and rat), CTCTGCCAACTACCGAGCCTA and CTCTTCTGCCTCTCGTTCCAT; HPRT (mouse only), CAGCGTTTCTGAGCCATTGCT and ATCCTCGGCATAATGATTAGGT; Cardiac troponin T (mouse only), GTGTGCAGTCCCTGTTCAGA and ACCCTCAGGCTCAGGTTCA. To verify the size of the amplified DNA fragments, PCRs were run in parallel with molecular weight markers on gels made from DNA agar (Marine BioProducts, Delta, British Columbia, Canada) and containing GelRed nucleic acid stain (Biotium, Hayward, CA, USA).
Results
BIX01294 does not alter myocardial phenotype and function
As part of an effort to fully examine the utility of BIX01294 as a means to generate plentiful and accessible sources of stem cells that could be used for cardiomyocyte regeneration, we tested whether this drug may possess any negative effects on myocyte function and/or phenotype that would undermine its use for cardiac repair. For these experiments, cardiomyocytes were isolated from newborn rats, dissociated into a single cell suspension, and cultured in the absence or presence of 8 μm BIX01294, which was the optimal concentration of this drug used for enhancing the cardiocompetency of cells from the bone marrow 9, 10. Rat cardiomyocytes typically would not resume their contractile activity prior to 48 h of cultures, with peak beating rates being reached at 4 days of culture (Figs. 1a and S1). Thereafter, the beating rate would decrease, although consistent beating was still observed through day 9 of incubation. Neither the time course nor amplitude of myocyte beating rate displayed any variance between non‐treated and BIX01294‐treated rat cardiomyocyte cultures (Fig. 1a). The myofibrillar structure of individual myocytes within the non‐treated and BIX01294‐treated cultures was also examined. Immunofluorescent analysis of muscle α‐actinin demonstrated that BIX01294 did not disrupt sarcomeric structure, as myocytes cultured in the absence or presence of this drug displayed an identical myofibrillar architecture (Fig. 1b,c). Together, these experiments indicate that treatment with BIX01294 does not alter the muscle apparatus or functional activity of differentiated cardiomyocytes.
Figure 1.
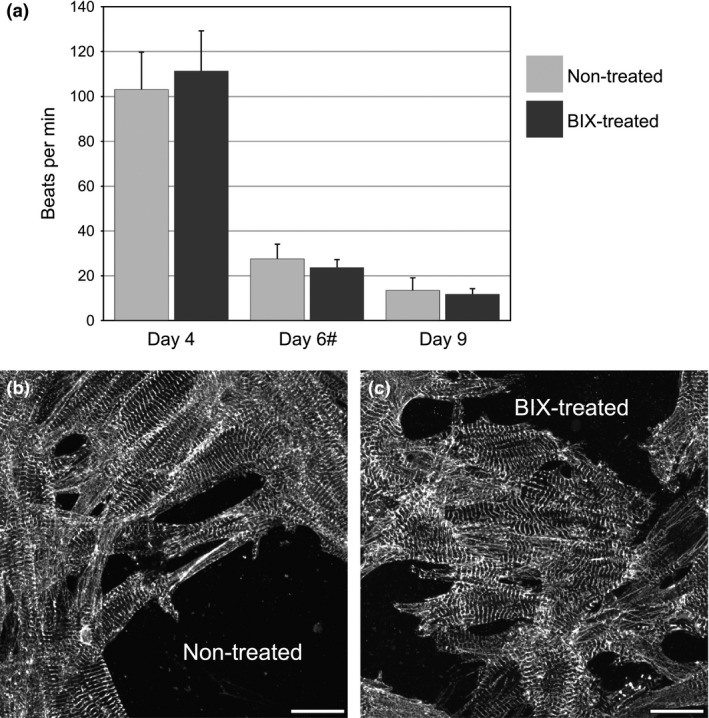
BIX 01294 does not alter myocardial function and phenotype. (a) Newborn rat cardiomyocytes were cultured in the absence or presence of BIX01294 and imaged on live video up to 9 days after initiation of culture in order to determine rates of myocyte beating. No significant difference was observed between the treated and non‐treated cultures at every time point. (#) Isoproterenol was added to both groups on day 6 for a positive inotropic effect. Beating data were compiled from four independent cultures for each time point. Parallel cultures of (b) non‐treated control and (c) BIX01294‐treated rat cardiomyocytes were also analysed for their sarcomeric architecture, as visualized by α‐actinin immunofluorescent staining. Note that BIX01294 did not noticeably affect the striated pattern of α‐actinin expression within individual myocytes. Scale bar = 20 μm.
BIX01294 increases the production of phase bright cells
In previous studies, we reported that BIX01294 can enhance the cardiac competency of bone marrow cells 9, 10. As a follow‐up to that earlier finding, we wanted to investigate whether BIX01294 may also have a positive effect on progenitor cells within the heart. CPCs can be routinely obtained from explanted mouse atrial tissue, as a cellular component of the phase bright cell population that appears within the first 2 weeks of culture 5, 6. Our results with non‐treated cultures were similar to what others have reported, as primary outgrowths of small stromal‐like cells grew from the cardiac atrial explants within the first 2 weeks, with phase bright cells first appearing above the stromal‐like cell layer at days 10–14 of incubation. When we exposed parallel cultures of atrial tissue to BIX01294, no discernible differences in the kinetics of this response were observed (data not shown). However, BIX01294‐treated tissue produced far greater amounts of phase bright cells than did the control cultures (Fig. 2). On average, a 7‐day treatment of atrial tissue explants with BIX01294 generated approximately 3.6‐fold more phase bright cells compared to non‐treated cultures (Fig. 2c), as assayed by trypan blue exclusion.
Figure 2.
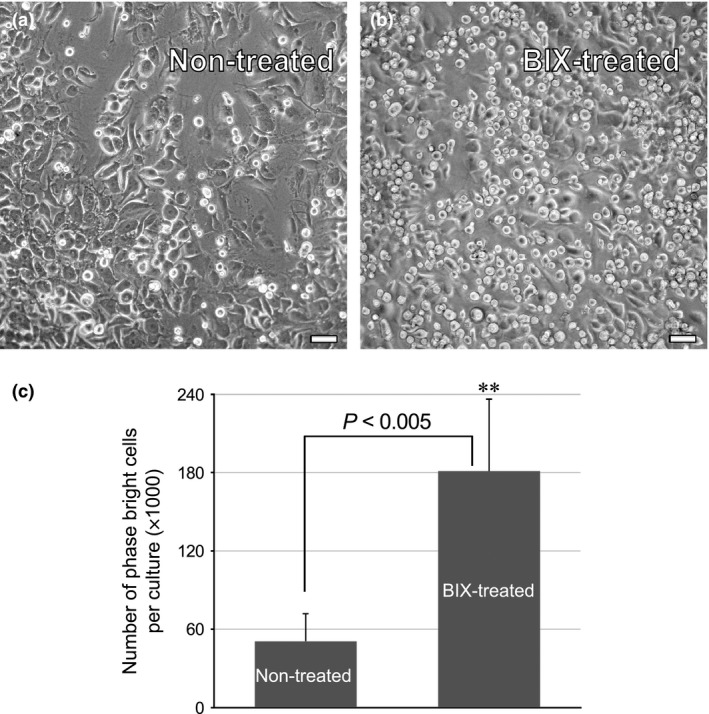
BIX 01294 promotes increased generation of phase bright cells. Adult mouse atrial tissue was cultured in the (a) absence or (b) presence of 8 μm BIX01294 for 7 days, and then imaged live at day 14 of incubation. It was evident by microscopic inspection that (a) the non‐treated control cultures contained far fewer phase bright cells than (b) were generated from BIX01294‐treated tissue. This observation was verified by (c) selectively harvesting phase bright cells from atrial cultures and counting viable cells using the trypan blue exclusion assay. Cardiac cultures treated with BIX01294 generated on average 3.6‐fold greater numbers of phase bright cells, as compared to control cultures. (**) The differences in cell numbers observed among the two distinct culture conditions had a statistical significance of P < 0.005, as determined by triplicate counts from 12 independent cultures. Scale bar = 50 μm.
BIX01294 stimulates proliferation of CPCs
Having demonstrated that BIX01294 promotes increased number of phase bright cells from atrial cultures, we next explored in greater detail the effect of this drug on this cell population. A standard method for assessing increases in cell proliferation is the MTT assay 15, 19, which measures cell metabolism as a function of mitochondrial reductase activity. For these experiments, atrial cultures were incubated over a time course of 2, 5 and 7 days in the absence or presence of BIX01294. In addition, the tissue was subjected to a longer 14‐day incubation period, consisting of an initial 7‐day treatment with or without BIX01294, and then maintained for an additional 7 days with fresh media without the drug. For all these cultures, phase bright cells were subsequently collected, replated in 96‐well plates at identical cell concentrations, and then assessed for relative metabolic activity after two further days of incubation. Thus, measurement of the reduction of the MTT substrate in each well was proportional to the number of viable cells that accumulated in these secondary cultures, and served as an indirect indicator for the rates of proliferation of the primary cultures. As shown in Fig. 3a, phase bright cells harvested from atrial explants that were exposed to BIX01294 for 5 and 7 days, displayed significantly higher levels of the reduced MTT substrate than non‐treated cells. Moreover, the results from the 14‐day treatment protocol indicated that the enhanced proliferative phenotype persisted for at least a week following withdrawal of BIX01294 (Fig. 3a). Corresponding dose–response analysis indicated that the optimal BIX01294 dosage for provoking cell proliferation was 8 μm (Fig. 3b). This concentration of BIX01294 generated levels of metabolic activity by the cultured phase bright cells that were approximately 4‐fold greater than that obtained from control cultures, with ANOVA testing confirming statistical significance (F 5,17 = 4.453, P < 0.005).
Figure 3.
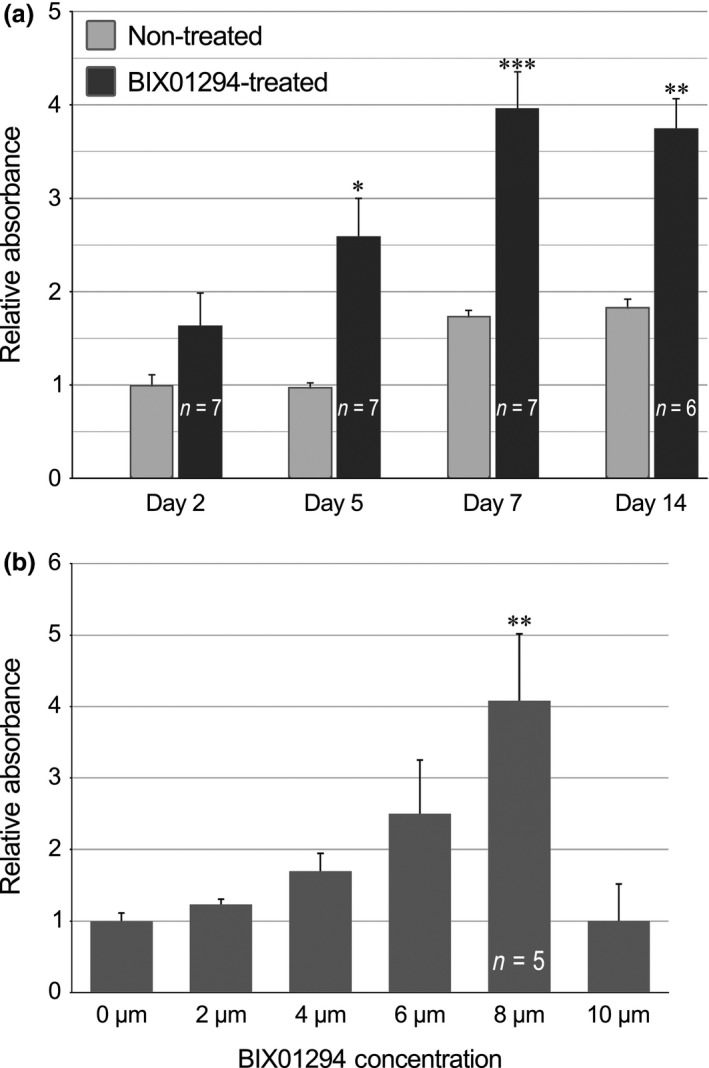
BIX 01294 stimulates proliferation of phase bright cells. The MTT assay was used as an indirect indicator of cell proliferation to assess (a) time course and (b) dose–response to BIX01294 treatments. (a) Time course studies were carried out by incubating atrial cultures for 2, 5 and 7 days in the absence or presence of 8 μm BIX01294. Additionally, there was a 14‐day time point where cultures were treated with or without BIX01294 for 7 days, and incubated for another 7 days in fresh medium without the drug. Statistical significance was determined by the t‐test by comparing BIX01294‐treated and the corresponding control cultures from the same day of incubation. (b) For dose–response analysis, atrial cultures were incubated in the absence or presence of various doses of BIX01294 for 7 days. For both time course and dose–response studies, phase bright cells were subsequently collected from cultures, replated at identical cell concentrations in 96‐well plates and then assessed for relative metabolic activity after two further days of incubation. For dosage experiments, ANOVA analysis was used to analyse statistical significance of each BIX01294 dosage versus the control cultures. F(5,17) = 4.453; *P < 0.05; **P < 0.005; ***P < 0.001.
Proliferation of cardiac cells was further assessed by uptake of the S‐phase marker BrdUrd. Atrial cultures were incubated with various doses of BIX01294 for 2 or 7 days, prior to a 48‐h exposure to BrdUrd. Phase bright cells were subsequently collected and immunofluorescently stained for BrdUrd incorporation. Comparative observation of immunostained phase bright cells obtained from non‐treated control (Fig. 4a,b) and BIX01294‐treated (Fig. 4c,d) cultures clearly indicate the far higher incorporation of the BrdUrd nucleoside within cells that had been exposed to the G9a HMTase inhibitor. Tabulation of BrdUrd‐positive cells obtained from the respective cultures verified that over a range of doses, BIX01294 enhanced the number of actively cycling phase bright cells, following both 2‐ and 7‐day treatments (Fig. 4e), with statistical significance confirmed by ANOVA analysis (F 3,32 = 41.573, P < 0.001).
Figure 4.
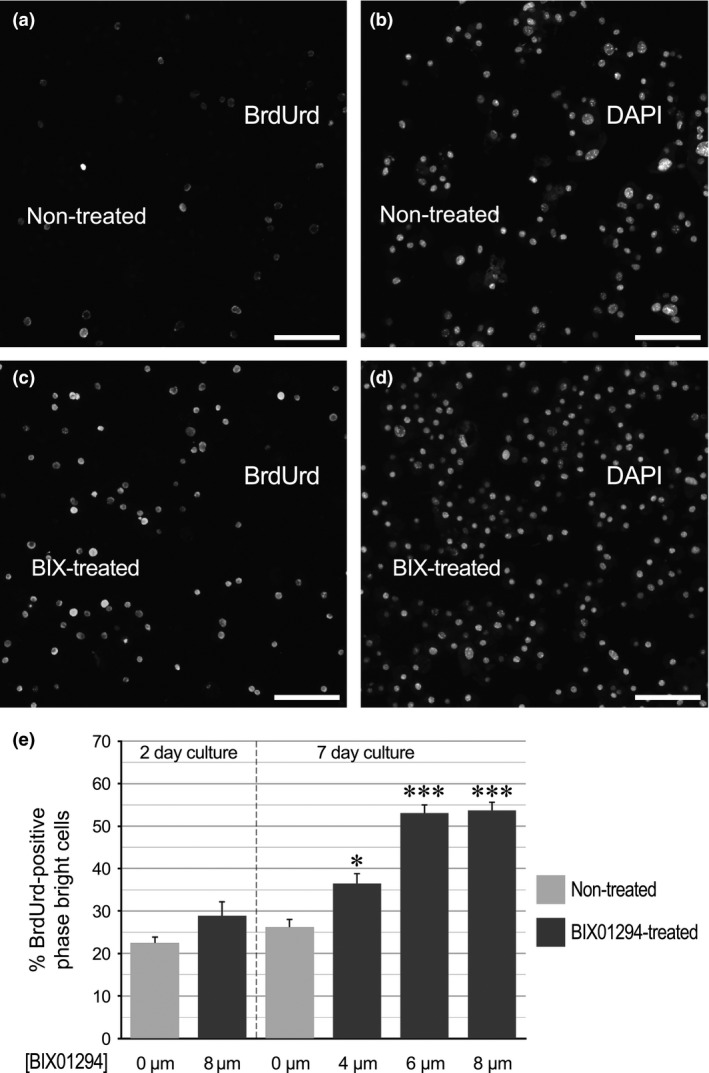
BIX 01294 treatment enhances BrdUrd incorporation by phase bright cells. Atrial tissue was exposed to BrdUrd for 48 h following a 7‐day incubation in the (a,b) absence or (c,d) presence of 8 μm BIX01294. Afterwards, phase bright cells were collected, cytospun onto glass slides, immunostained for BrdUrd incorporation and counterstained with DAPI to mark all cell nuclei. The corresponding BrdUrd and DAPI staining are showed side‐by‐side for individual fields of phase bright cells harvested from (a,b) non‐treated or (c,d) BIX01294‐treated tissue, respectively. Scale bar = 50 μm. (e) Calculation of per cent of BrdUrd‐positive phase bright cells in the absence or presence or various doses of BIX01294 for 2 (n = 8) and 7 days (n = 10). ANOVA analysis was used to analyse statistical significance of each BIX01294 dosage versus the control cultures. F(3,32) = 41.573; *P < 0.05; ***P < 0.001.
As a third indicator of cell cycle progression, phase bright cells were immunostained for Ki67. This marker labels actively cycling cells, primarily in metaphase, but is absent in resting (G0 phase) cells. The pattern of Ki67 staining was consistent with the MTT and BrdUrd analysis, as atrial cultures incubated in BIX01294 for 7 days generated a 4.8‐fold increase in the percentage of phase bright cells that were Ki67‐positive, as compared to non‐treated controls (Fig. 5).
Figure 5.
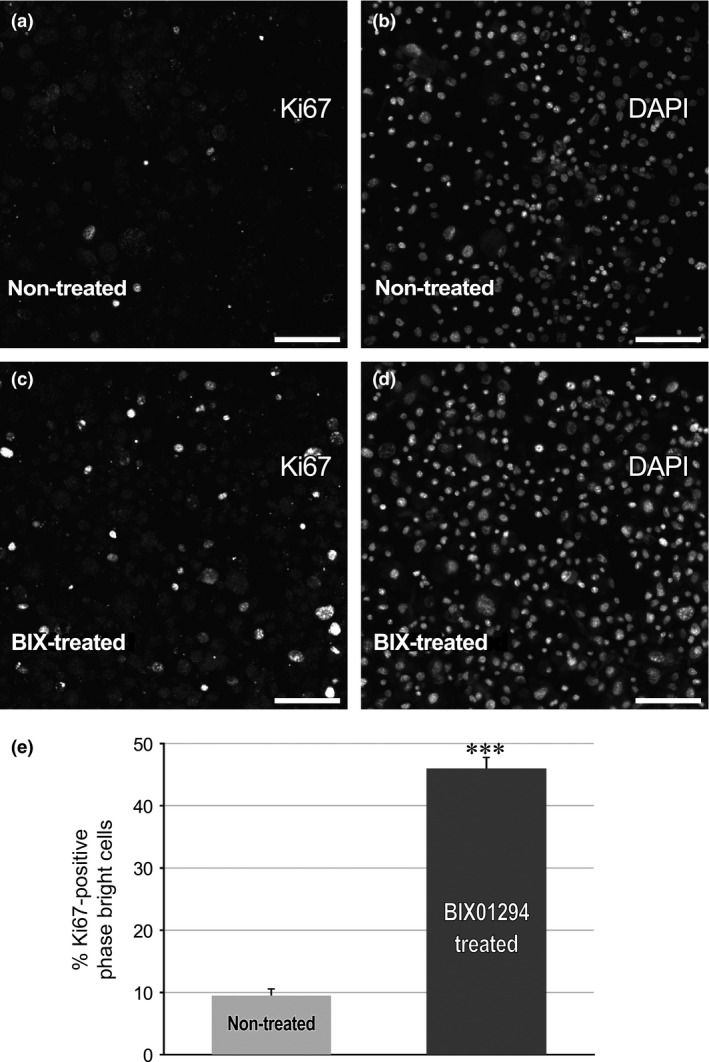
BIX 01294 treatment enhances Ki67 expression by phase bright cells. Following the incubation of atrial tissue for 7 days in the (a,b) absence or (c,d) presence of 8 μm BIX01294, phase bright cells were harvested and cytospun onto glass slides. Afterwards, the cells were immunostained for Ki67 expression and counterstained with DAPI. Individual fields of phase bright cells harvested from (a,b) non‐treated or (c,d) BIX01294‐treated tissue are displayed with the corresponding Ki67 and DAPI staining showed in adjacent panels. Scale bar = 50 μm. (e) Tabulation of per cent of Ki67‐positive phase bright cells in the absence or presence of BIX01294. Statistical significance of increased staining within BIX01294‐treated versus control cultures was determined by the t‐test. ***P < 0.001 (n = 9).
BIX01294 does not alter CPC phenotype
As BIX01294 exposure expanded numbers of phase bright cells, which normally contain enriched populations of CPCs, we next examined whether treatment with this drug provoked any changes in CPC phenotype. First, we compared phase bright cells collected from non‐treated control and BIX01294‐treated atrial tissue, for expression of CD90 and Sca1, which are stem cell markers associated with CPCs 5, 6, 20, 21. Immunofluorescent analysis demonstrated that neither the pattern nor prevalence of CD90 and Sca1 expression differed among CPCs derived from non‐treated and BIX01294‐treated cultures (Fig. 6a–d). Nor were there any differences apparent among CPCs obtained from control or treated cultures for the display of Islet1 (Fig. 6e–h), which is a transcription marker affiliated with cardiac progenitors 22, 23, 24. In addition, cells obtained from BIX01294‐treated and control cultures appeared to have identical distribution patterns of the CPC‐associated gap junction proteins, Cx40 and Cx43 (Fig. 7a–d). In accordance with this immunostaining data, flow cytometric analysis confirmed that BIX01294 did not affect the levels or proportion of cells that exhibited these CPC markers (Fig. 7e).
Figure 6.
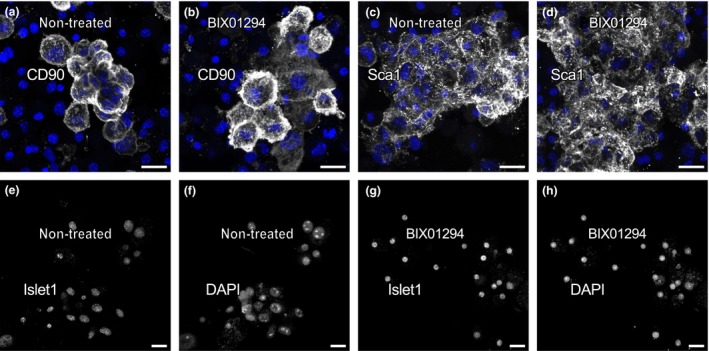
Expression of progenitor markers in atrial‐derived phase bright cells. Phase bright cells were collected from either (a,c,e,f) non‐treated control or (b,d,g,h) BIX01294‐treated atrial cultures, and immunofluorescently stained (grey) following (a–d) cytospinning onto glass slides or (e‐h) their attachment on culture plastic. Cultures were counterstained with DAPI to visualize nuclei (blue). Phase bright cells collected from control and treated cultures showed similar patterns and prevalence of expression for (a,b) CD90 and (c,d) Sca1, which are stem cell markers associated with CPCs. Differences were also not observed among phase bright cells obtained from (e,f) non‐treated or (g,h) BIX01294‐treated cultures, respectively, for the expression of cardiac progenitor marker Islet1. For this transcription marker, antibody and DAPI staining is shown independently for individual cellular fields, as indicated in the corresponding panels. Scale bar = 20 μm.
Figure 7.
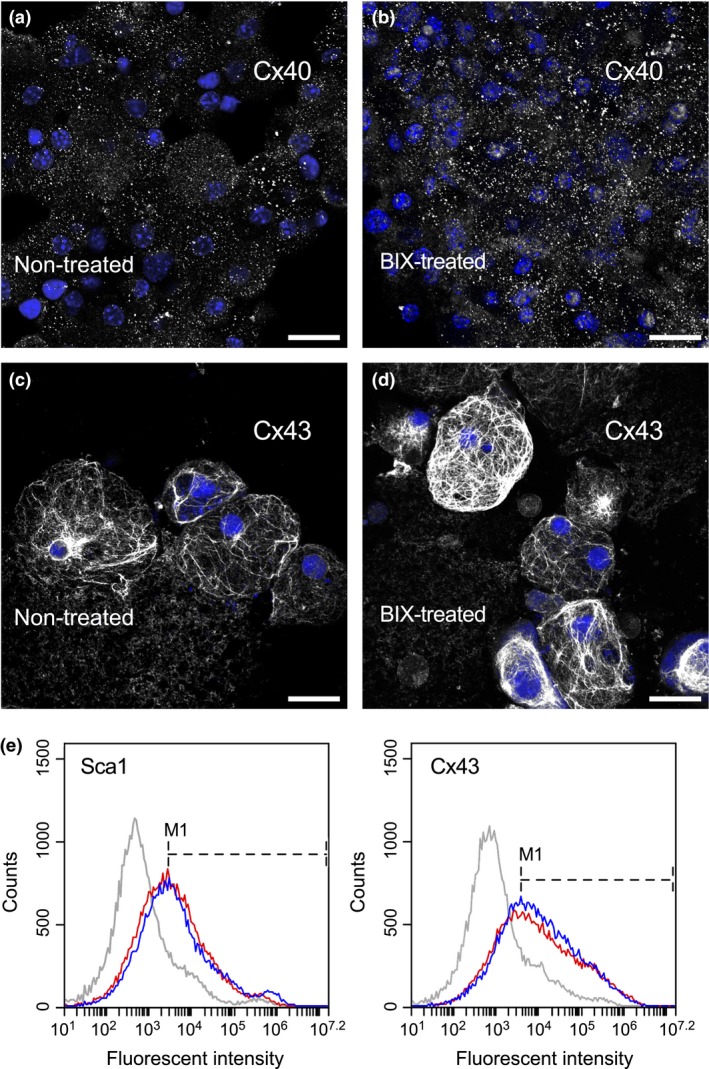
Cell membrane marker expression by phase bright cells. PBCs collected from either (a,c) non‐treated control or (b,d) BIX01294‐treated atrial cultures were immunostained (grey) for (a,b) Cx40 or (c,d) Cx43, and counterstained with DAPI (blue). Neither gap junction protein showed any comparative dissimilarities on their pattern of expression among the two culture conditions. Scale bar = 20 μm. (e) Flow cytometric analysis of Sca1 and Cx43 showed comparable histogram profiles. Plots for non‐treated control and BIX01294‐treated PBCs are shown in red and blue, respectively, with the grey lines representing cells stained with secondary antibody only. Placement of the M1 marker at peak fluorescence for each antigen indicate that Sca1‐positive cells represent 51.1% and 53.2%, and Cx43‐positive cells represent 61.0% and 65.0% of non‐treated and BIX01294‐treated populations of phase bright cells, respectively.
Native and BIX01294‐expanded CPCs display similar cardiac potential
A final issue to address is whether CPCs expanded by BIX01294 treatment retain the cardiac potential inherent in native CPCs. To compare the differentiation capacities of CPCs collected from non‐treated control and BIX01294‐treated tissue, these cells were co‐cultured with neonatal rat cardiomyocytes, which is an established culture model for testing the cardiac capacity of progenitor cell populations 25, 26. To distinguish the two sources of cells within the co‐culture, mouse CPC‐enriched cells were pre‐labelled with the fluorescent vital dye CFSE. After 2 weeks, co‐cultures were either immunostained to assess whether mouse CPCs gave rise to sarcomeric protein‐positive cells or harvested for RNA and analysed for mouse cell‐specific cardiac gene expression. As shown in Fig. 8a and 8b, CPCs obtained from non‐treated control and BIX01294‐treated mouse tissue appeared to display a similar capacity to form cardiomyocytes. This outcome is indicated by display of dual CFSE, muscle α‐actinin‐positive cells that integrated into clusters of beating myocytes. The generation of mouse‐derived cardiomyocytes was further supported by PCR analysis, where cardiac gene expression in mouse CPC/rat cardiomyocyte co‐cultures was compared to controls consisting of either mouse CPCs or rat cardiomyocytes cultured independently, or mouse heart and liver tissue (Fig. 8c,d). The cellular origin of cardiac gene transcription was discerned using primers that specifically detected mouse, but not rat cardiac genes. The PCR results (Fig. 8d) demonstrated that CPCs obtained from BIX01294 or non‐treated tissue were equally able to undergo myocardial differentiation as indicated by their similarly high levels of cardiac troponin T expression. Collectively, these data indicate that BIX01294 can expand numbers of adult heart CPCs without changing their phenotype or diminishing their cardiac capacity.
Figure 8.
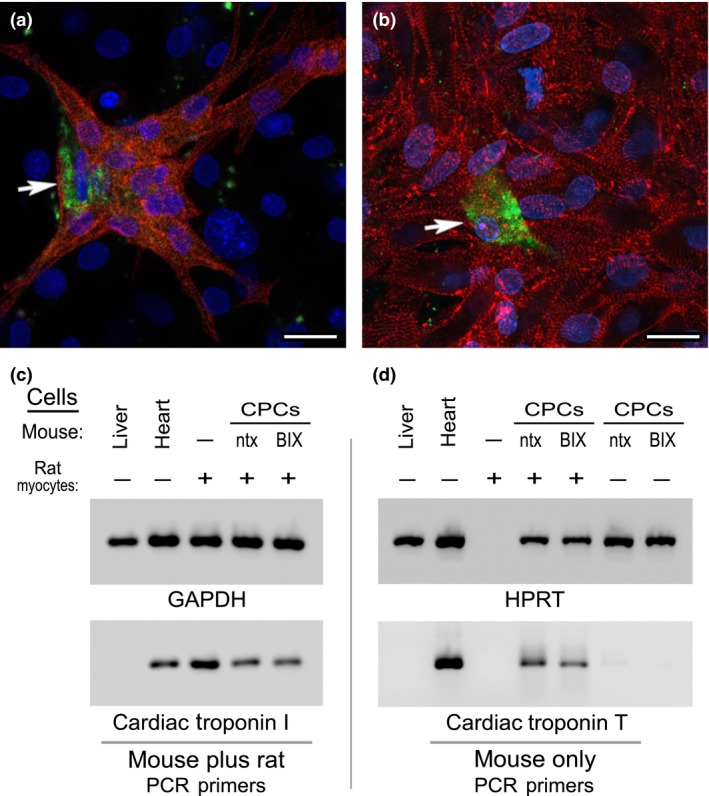
Co‐cultures of mouse CPC s and neonatal rat cardiomyocytes. Phase bright cells collected from (a) non‐treated or (b) BIX01294‐treated atrial tissue were labelled with the fluorescent vital dye CFSE (green), prior to 2 weeks of co‐culture with neonatal rat cardiomyocytes. Afterwards, co‐cultures were immunolabelled against muscle α‐actinin (red) and counterstained with DAPI (blue). Note that under both culture conditions, CPCs were able to undergo cardiac differentiation, as indicated by the presence of CFSE‐labelled, α‐actinin‐positive mouse‐derived cells (arrows) within large clusters of rat myocytes. These two representative images are indicative of the similar cardiac potential of mouse CPCs obtained from non‐treated or BIX01294‐treated atrial explants. Scale bar = 20 μm. This observation was substantiated by measurement of gene expression by PCR amplification of RNA harvested from the co‐cultures. (c) RNA concentrations of mouse liver, mouse heart or rat cardiomyocytes (CM) cultured independently or in the presence of control or BIX01294‐treated mouse CPCs were normalized to the housekeeping gene GAPDH (top panel), using primers that recognize the RNA from both species. Subsequent amplification with non‐species‐specific cardiac troponin I primers (bottom panel) verified that each of the samples (except the negative control liver) displayed strong myocardial gene expression. (d) RNA samples were amplified to the HPRT housekeeping gene using primers that strictly recognize this sequence only in mouse cells (top panel). The concentration of sample loaded per lane was normalized to the expression of HPRT expression, except the rat cardiomyocyte only control. For this rat RNA only sample, template volume was normalized to the co‐culture samples based on the expression displayed in panel C for non‐species‐specific GAPDH. Subsequently, normalized templates were amplified with primers that only recognize cardiac troponin T expressed solely in mouse cells (bottom panel). Among controls, mouse heart displayed expression of this myocardial gene as expected, while neither mouse liver or rat cardiomyocyte template was amplified with these mouse cardiac‐specific primers. However, when co‐cultured with the rat cardiomyocytes, control and BIX01294‐treated mouse CPCs showed near equivalent cardiac troponin T expression. Note that in the absence of the rat cardiomyocytes, neither population of mouse CPCs was positive for cardiac troponin T, which correlates with the progenitor phenotype of the phase bright cell‐derived cells.
Discussion
The adult heart is believed to contain endogenous stem cells, often referred to broadly as CPCs, which contribute to myocyte replacement during normal homoeostasis. However, the utility of CPCs for treating diseased hearts is limited by small quantities of these cells that can be obtained from patient biopsies. Thus, a major objective in optimizing the therapeutic utility of CPCs is to expand their numbers without changing their phenotype and sacrificing their competency to form differentiated myocytes.
In previous studies 9, 10, our laboratory has shown that suppression of G9a HMTase activity using the small molecule inhibitor BIX01294 can enhance the cardiac competency of stem cells obtained from the bone marrow. The present investigation was initially undertaken as a follow‐up to the bone marrow reports, as we aimed to explore whether BIX01294 would have any adverse consequences on differentiated cardiac tissue that would negate any beneficial effect that this drug would have on enhancing the ability of adult stem cells to generate functional cardiac muscle. When differentiated cardiac myocytes were incubated with BIX01294, we did not observe any obvious detrimental effects on their beating or sarcomeric organization. However, when BIX01294 was applied to explanted cardiac tissue fragments, there was a striking change that was readily evident by a simple visual inspection under the microscope, as these cultures showed a substantial increase in the amount of phase bright cells that were generated in response to this drug. This observation was confirmed mathematically and statistically by the trypan blue exclusion assay, which indicated that BIX01294 treatments produced on average a 3.6‐fold increase in phase bright cell numbers after 2 weeks of culture. Expansion and increased proliferative activity of this cell population in response to BIX01294 was further confirmed by measuring cell metabolic activity using the MTT assay, incorporation of BrdUrd and expression of the cell cycle marker Ki67. The significance of these results is that the phase bright cell population that arises from cardiac cultures is known to be highly enriched for CPCs. Importantly, the BIX01294‐mediated expansion of this cell population did not produce cells that differed in phenotype or differentiation potential from those CPCs that arose from non‐treated control cultures. Both the distribution and prevalence of stem cell markers (e.g. CD90, Sca1 and Islet‐1) and other CPC‐associated proteins (e.g. Cx40 and Cx43) among the non‐treated control and BIX01294‐treated cell populations were very similar. Moreover, co‐culture with neonatal rat cardiomyocytes indicated that CPCs obtained from non‐treated and BIX01294‐treated cardiac explants showed comparable capacities to undergo cardiac differentiation. Thus, the pronounced expansion of CPCs provoked by BIX01294 does not appear to undermine their capacity as cardiac progenitors.
As CPCs were first characterized in the adult heart, methods have been developed to grow these cells in vitro and expand their numbers in culture 6, 8, 27. These expansion protocols involve multi‐week incubation under high serum and/or cytokine environment. Certainly, our data show that in the absence of BIX01294, adult CPCs are proliferative and will expand in culture. However, administering BIX01294 to the original source tissue further enhanced their proliferative phenotype, without compromising their identity or capabilities as CPCs. The increased expansion in response to this drug is notable due to the limited number of cells that can be obtained from biopsies of human patients. The results presented in this study are promising, as treatments with BIX01294 did not appear to cause any deleterious effect on the function of CPCs or differentiated cardiac myocytes. Interesting follow‐up studies would be to determine if any of the cytokines that have been characterized for supporting CPC growth in culture 6, 8, 28 – such as stem cell factor, bFGF, EGF and cardiotrophin‐1 – would act in concert for further promoting the expansion of CPCs in cultures. Future studies should also test the capacity of CPCs derived from BIX01294‐treated cardiac tissue to treat the heart in situ, by repairing cardiac tissue that has been damaged in response to injury.
In previous studies, we noted that BIX01294 exposure enhanced the number of progenitor cells generated from bone marrow cultures 10. This raises the possibility that the positive effect of this drug on cell proliferation may be applicable for many cell types due to its inhibition of G9a HMTase. Yet reports in the literature portray the opposite role for this enzyme as a cell cycle regulator. In several types of cancers, the highly proliferative malignant phenotype has been associated with elevated levels of G9a HMTase 29, 30, 31, 32. Other laboratories have reported experimentation with cultured cells, where BIX01294 treatments stifled cell proliferation 33, 34. With the cancer diagnosis, this conundrum may be explained by the inhibition of tumour suppressor gene expression and activation of the serine–glycine oncogenic metabolic pathway that results from the abnormally high levels of G9a HMTase 30, 31. Thus, the overall level of G9a HMTase may dictate its effect on cell proliferation, with both abnormally high levels and reduction of its normal endogenous enzymatic activity causing an increase in cell cycle.
At first glance, our results may seem harder to reconcile with the previous cell culture investigations that G9a HMTase inhibition repressed cellular proliferation 33, 34. Yet, an effect we noticed is that the BIX01294 influence on cell behaviour is based on the overall culture environment. In the present study, BIX01294 was used to treat non‐dissociated tissue chunks, from which emerged the phase bright cells. While our previous studies with bone marrow used dissociated cells, those cultures were fully confluent when treated with BIX01294 9, 10. Under those conditions, the drug promoted a proliferative phenotype. In contrast, cells responded poorly when exposed to BIX01294 at subconfluent densities, as they were clearly stressed and therefore, non‐proliferative. Another contrast with studies describing BIX01294 as negatively influencing proliferation was that the cells in those reports were assayed within 24 h of exposure to the drug 33, 34. In the present study, that time point was too early to ascertain any increase in cell proliferation, as we assayed time courses of up to 2 weeks. Thus, BIX01294 does not act on cell proliferation as a simple on–off switch, but influences cell behaviour based on the overall environment of the cells and tissues.
In summary, our results indicate that BIX01294 can act as an expansion factor for endogenous CPCs from the adult heart. This drug is able to significantly enhance the number of CPCs obtained from cardiac tissue fragments, without compromising CPC phenotype or differentiation potential. Moreover, BIX01294 did not negatively impact the function or sarcomeric structure of fully differentiated cardiac myocytes, which indicates that this drug is not deleterious to cardiogenesis. Together, these data suggest that this drug may have utility as an agent for generating large numbers of native CPCs, which potentially could be used for treating heart disease.
Supporting information
Fig. S1. BIX01294 treatment does not alter cardiomyocyte beating. Newborn rat cardiomyocytes cultured in the (a) absence or (b) presence of 8 μm BIX01294 displayed similar beating rates, as demonstrated in videos of day 4 cultures.
Acknowledgements
The authors are grateful for the support of the New York Medical College/Westchester Medical Center Translational Stem Cell Center. Funding for this work was provided by the New York Medical College Castle‐Krob Research Endowment in support of the New York Medical College Intramural Research Support Program.
References
- 1. Chong JJ, Forte E, Harvey RP (2014) Developmental origins and lineage descendants of endogenous adult cardiac progenitor cells. Stem Cell Res. 13, 592–614. [DOI] [PubMed] [Google Scholar]
- 2. Finan A, Richard S (2015) Stimulating endogenous cardiac repair. Front. Cell Dev. Biol. 3, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leri A, Rota M, Pasqualini FS, Goichberg P, Anversa P (2015) Origin of cardiomyocytes in the adult heart. Circ. Res. 116, 150–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nadal‐Ginard B, Ellison GM, Torella D (2014) The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult. Stem Cell Res. 13, 615–630. [DOI] [PubMed] [Google Scholar]
- 5. Carr CA, Stuckey DJ, Tan JJ, Tan SC, Gomes RS, Camelliti P et al (2011) Cardiosphere‐derived cells improve function in the infarcted rat heart for at least 16 weeks–an MRI study. PLoS ONE 6, e25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davis DR, Zhang Y, Smith RR, Cheng K, Terrovitis J, Malliaras K et al (2009) Validation of the cardiosphere method to culture cardiac progenitor cells from myocardial tissue. PLoS ONE 4, e7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leinonen JV, Emanuelov AK, Platt Y, Helman Y, Feinberg Y, Lotan C et al (2013) Left atrial appendages from adult hearts contain a reservoir of diverse cardiac progenitor cells. PLoS ONE 8, e59228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F et al (2004) Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ. Res. 95, 911–921. [DOI] [PubMed] [Google Scholar]
- 9. Mezentseva NV, Yang J, Kaur K, Iaffaldano G, Rémond MC, Eisenberg CA et al (2013) The histone methyltransferase inhibitor BIX01294 enhances the cardiac potential of bone marrow cells. Stem Cells Dev. 22, 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang J, Kaur K, Ong LL, Eisenberg CA, Eisenberg LM (2015) Inhibition of G9a histone methyltransferase converts bone marrow mesenchymal stem cells to cardiac competent progenitors. Stem Cells Int. 2015, 270428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eisenberg CA, Bader DM (1996) Establishment of the mesodermal cell line QCE‐6. A model system for cardiac cell differentiation. Circ. Res. 78, 205–216. [DOI] [PubMed] [Google Scholar]
- 12. Edwards JG, Bahl JJ, Flink IL, Cheng SY, Morkin E (1994) Thyroid hormone influences beta myosin heavy chain (beta MHC) expression. Biochem. Biophys. Res. Commun. 199, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 13. Hicks S, Labinskyy N, Piteo B, Laurent D, Mathew JE, Gupte SA et al (2013) Type II diabetes increases mitochondrial DNA mutations in the left ventricle of the Goto‐Kakizaki diabetic rat. Am. J. Physiol. Heart Circ. Physiol. 304, H903–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vishnoi T, Kumar A (2013) Conducting cryogel scaffold as a potential biomaterial for cell stimulation and proliferation. J. Mater. Sci. Mater. Med. 24, 447–459. [DOI] [PubMed] [Google Scholar]
- 15. Zhang P, Guo Z, Wu Y, Hu R, Du J, He X et al (2015) Histone deacetylase inhibitors inhibit the proliferation of gallbladder carcinoma cells by suppressing AKT/mTOR signaling. PLoS ONE 10, e0136193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaur K, Yang J, Eisenberg CA, Eisenberg LM (2014) 5‐azacytidine promotes the transdifferentiation of cardiac cells to skeletal myocytes. Cell. Reprogram. 16, 324–330. [DOI] [PubMed] [Google Scholar]
- 17. Eisenberg CA, Burch JB, Eisenberg LM (2006) Bone marrow cells transdifferentiate to cardiomyocytes when introduced into the embryonic heart. Stem Cells 24, 1236–1245. [DOI] [PubMed] [Google Scholar]
- 18. Martin LK, Bratoeva M, Mezentseva NV et al (2012) Inhibition of heart formation by lithium is an indirect result of the disruption of tissue organization within the embryo. Dev. Growth Differ. 54, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen M, Lv Z, Huang L , Zhang W , Lin X, Shi J et al (2015) Triptolide inhibits TGF‐β1‐induced cell proliferation in rat airway smooth muscle cells by suppressing Smad signaling. Exp. Cell Res. 331, 362–368. [DOI] [PubMed] [Google Scholar]
- 20. Uchida S, De Gaspari P, Kostin S, Jenniches K, Kilic A, Izumiya Y et al (2013) Sca1‐derived cells are a source of myocardial renewal in the murine adult heart. Stem Cell Rep. 1, 397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Hu Q, Nakamura Y, Lee J, Zhang G, From AH et al (2006) The role of the sca‐1 + /CD31‐ cardiac progenitor cell population in postinfarction left ventricular remodeling. Stem Cells 24, 1779–1788. [DOI] [PubMed] [Google Scholar]
- 22. Di Felice V, Zummo G (2013) Stem cell populations in the heart and the role of Isl1 positive cells. Eur. J. Histochem. 57, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mishra R, Vijayan K, Colletti EJ, Harrington DA, Matthiesen TS, Simpson D et al (2011) Characterization and functionality of cardiac progenitor cells in congenital heart patients. Circulation 123, 364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ye J, Boyle A, Shih H, Sievers RE, Zhang Y, Prasad M et al (2012) Sca‐1 + cardiosphere‐derived cells are enriched for Isl1‐expressing cardiac precursors and improve cardiac function after myocardial injury. PLoS ONE 7, e30329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E et al (2007) Regenerative potential of cardiosphere‐derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115, 896–908. [DOI] [PubMed] [Google Scholar]
- 26. Tufan H, Zhang XH, Haghshenas N, Sussman MA, Cleemann L, Morad M (2012) Cardiac progenitor cells engineered with Pim‐1 (CPCeP) develop cardiac phenotypic electrophysiological properties as they are co‐cultured with neonatal myocytes. J. Mol. Cell. Cardiol. 53, 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen L, Pan Y, Zhang L, Wang Y, Weintraub N, Tang Y (2013) Two‐step protocol for isolation and culture of cardiospheres. Methods Mol. Biol. 1036, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vajravelu BN, Hong KU, Al‐Maqtari T, Cao P, Keith MC, Wysoczynski M et al (2015) C‐Kit promotes growth and migration of human cardiac progenitor cells via the PI3K‐AKT and MEK‐ERK pathways. PLoS ONE 10, e0140798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Casciello F, Windloch K, Gannon F, Lee JS (2015) Functional role of G9a histone methyltransferase in cancer. Front Immunol. 6, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ding J, Li T, Wang X, Zhao E, Choi JH, Yang L et al (2013) The histone H3 methyltransferase G9A epigenetically activates the serine‐glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 18, 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hua KT, Wang MY, Chen MW, Wei LH, Chen CK, Ko CH et al (2014) The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer. 13, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lehnertz B, Pabst C, Su L, Miller M, Liu F, Yi L et al (2014) The methyltransferase G9a regulates HoxA9‐dependent transcription in AML. Genes Dev. 28, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ke XX, Zhang D, Zhu S, Xia Q, Xiang Z, Cui H (2014) Inhibition of H3K9 methyltransferase G9a repressed cell proliferation and induced autophagy in neuroblastoma cells. PLoS ONE 9, e106962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang Q, Lu Z, Singh D, Raj JU (2012) BIX‐01294 treatment blocks cell proliferation, migration and contractility in ovine foetal pulmonary arterial smooth muscle cells. Cell Prolif. 45, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. BIX01294 treatment does not alter cardiomyocyte beating. Newborn rat cardiomyocytes cultured in the (a) absence or (b) presence of 8 μm BIX01294 displayed similar beating rates, as demonstrated in videos of day 4 cultures.