

Abstract
Objectives
Huntington's disease (HD) is an inherited human neurodegenerative disorder characterized by uncontrollable movement, psychiatric disturbance and cognitive decline. Impaired proliferative/differentiational potentials of adult neural progenitor cells (ANPCs) have been thought to be a pathogenic mechanism involved in it. In this study, we aimed to elucidate intrinsic properties of ANPCs subjected to neurodegenerative condition in YAC128 HD mice.
Materials and methods
ANPCs were isolated from the SVZ regions of 4‐month‐old WT and YAC128 mice. Cell proliferation, migration and neuronal differentiation in vitro were compared between these two genotypes with/without Ca2+ inhibitors or ROS scavenger treatments. Differences in ANPC proliferation and differentiation capabilities in vivo between the two genotypes were evaluated using Ki‐67 and Doublecortin (DCX) immunofluorescence respectively.
Results
Compared to WT ANPCs, YAC128 ANPCs had significantly enhanced cell proliferation, migration and neuronal differentiation in vitro, accompanied by increased Ca2+ and ROS signals. Raised proliferation and migration in YAC128 ANPCs were abolished by Ca2+ signalling antagonists and ROS scavenging. However, in vivo, HD ANPCs failed to show any elevated proliferation or differentiation.
Conclusions
Increased Ca2+ signalling and higher level of ROS conferred HD ANPC enhancement of proliferation and migration potentials. However, the in vivo micro‐environment did not support endogenous ANPCs to respond appropriately to neuronal loss in these YAC128 mouse brains.
Introduction
Throughout adulthood, mammalian neurogenesis mainly occurs in two regions: the subventricular zone (SVZ) of brain lateral ventricles and subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) 1, 2. Newborn neurons form functional connections with existing circuitry to maintain and reorganize the olfactory bulb 3, 4, 5 and contribute to hippocampal‐dependent memory and behaviour 6, 7, 8. Importantly, neurogenesis can be up‐regulated by a variety of stimuli such as occurrence of seizures, either ischaemic or by stroke 9, 10, 11, 12. These findings spark considerable interest in endogenous cell repair strategies, and the adult neural progenitor cells (ANPCs) have been proposed to be an endogenous source of cells for treatment of neurodegenerative diseases 13, 14. Neurogenesis has been widely studied in a number of neurodegenerative disorders including Alzheimer's disease 15, 16, 17, Parkinson's disease 18, 19, 20 and Huntington's disease (HD) 21, 22, 23, 24.
HD is an inherited human neurodegenerative disorder caused by hugely expanded CAG repeats in the huntingtin (HTT) gene, characterized by neuronal cell death in discrete brain regions, resulting in movement disorders, psychiatric disturbance, and cognitive decline 25, 26. The striatum is the brain structure primarily affected, although other areas can also be affected in patients with advanced HD 27. Due to close proximity of the SVZ to the striatum, neurogenesis in the SVZ has consistently been studied in HD transgenic mouse models, as well as in HD patients. Several lines of evidence have demonstrated normal ANPC proliferation in the SVZ in multiple HD transgenic mouse brains in vivo 21, 22, 23, 24, while others have shown impaired proliferation capacity of neural stem cells (NSCs) isolated/cultured from embryonic HD mouse brains in vitro 28, 29. Interestingly, in human post‐mortem HD brains, increase has been found in ANPC proliferation in the SVZ, specially in patients with advanced HD 30. To date, there has not been any clear explanation for these conflicting observations.
Neurogenesis is a complex process which includes proliferation, migration and differentiation, and can be affected by intrinsic properties of endogenous neural progenitor cells (NPCs) and the micro‐environment in which they reside in vivo. YAC128 HD mouse model animals express the full‐length human mutant HTT gene with 128 CAG repeats, and faithfully recapitulates many features of the human condition 31, 32. In the present study, to determine whether intrinsic properties of ANPCs were changed in response to HD, we examined proliferation, migration and neuronal differentiation capacities of ANPCs isolated from SVZ regions of age‐matched WT and YAC128 HD mouse brains, in vitro. Somewhat surprisingly, we found that proliferation, migration and neuronal differentiation significantly increased in YAC128 ANPCs, and that enhanced Ca2+ and ROS signals were essential for enhancements of proliferation and migration in YAC128 ANPCs. Mutant HTT protein‐triggered elevated Ca2+ and ROS signalling have been previously reported to be harmful to mature neurons including striatal medium spiny neurons (MSNs) in HD 33, 34, 35, 36, 37, 38, 39. Thus, in contrast to their toxicities to mature neurons in HD, elevated Ca2+ and ROS signalling seem to deliver increased capabilities of proliferation and migration to HD ANPCs. Interestingly, unlike results in vitro, YAC128 HD mouse brains had similar levels of ANPC proliferation and differentiation to WT SVZ ones, in vivo, indicating that the micro‐environment in which the ANPCs were organized was the key limiting factor for adult neurogenesis in YAC128 HD mouse brains, rather than intrinsic proliferation and differentiation properties of the cells. Thus, to promote endogenous neurogenesis to counteract striatal neurodegeneration in HD brains, liberating ANPCs from their constrained micro‐environment is a key approach that should be considered.
Materials and methods
Isolation and culture of adult neural progenitor cells
YAC128 HD transgenic mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Generation and breeding of transgenic YAC128 HD mice (FVBN/NJ background strain) have previously been described 31. All animal experiments were reviewed and approved by our Institute of Zoology Institutional Animal Care and Use Committee and were conducted according to the committee's guidelines. 4‐month‐old wild type and YAC128 mice (FVB strain) were sacrificed by decapitation. Subventricular zones from individual animals was dissociated with accutase (Millipore, Temecula, CA, USA) for 10 min at 37 °C, and mechanically dissociated using a fire‐polished 1000 μl plastic tip. After centrifugation, cells were re‐suspended in growth medium consisting of neurobasal medium, 2% B27 supplement without vitamin A, 1% glutamax, 1 × penicillin/streptomycin (all from Invitrogen, Carlsbad, CA, USA) and 20 ng/ml of both epidermal growth factor (EGF) and basic fibroblast growth factor (FGF2) (Peprotech, Rocky Hill, NJ, USA). Cells were plated in growth medium either on to six‐well uncoated plastic plates for floating spheres, or on to poly‐L‐ornithine‐coated plastic plates for monolayer cultures. Half the volume of each culture medium was changed every 2 days, and cells were passaged every 4–6 days with accutase. After expanding and propagating them for at least 14 passages, monolayer cultured cells were fixed and probed with antibodies against Nestin (1:200; Millipore) and SOX2 (1:200; Abcam, London, UK).
Adult neural progenitor cell differentiation
Adult neural progenitor cells were induced to differentiate after plating on poly‐L‐ornithine‐coated glass coverslips. The detailed procedure was adapted from a protocol previously described, which requires withdrawal of EGF and slow reduction of FGF2 while introducing and increasing brain‐derived neurotrophic factor (BDNF) 40. Half the medium was exchanged after 72 h incubation in respective defined media. Cells were then fixed and probed with antibodies against microtubule‐associated protein (MAP2) (1:400; Chemicon, Temecula, CA, USA) and glial fibrillar acidic protein (GFAP) (1:400; Chemicon). Soma area neurite length of MAP2+ cells were measured using Image‐J software.
Cell proliferation assays
Self‐renewal was first assessed by dissociating neurospheres into single cells and re‐culturing them at low density of 1000 cells per well, in 24‐well plates. Size of secondary spheres after 8 days culture was measured using Image‐J software. Neurospheres with diameters >40 μm were assessed.
Growth curve experiments were also used to evaluate capability for cell proliferation. 5 × 104 cells per well were seeded in poly‐L‐ornithine‐coated six‐well plates in 2 ml growth medium, with or without testing agents (100 μm glutamate, 50 μm 2‐APB, 1 mm NAC, 1 μm/2 μm U73122), and each sample was plated in triplicate. Cell number was counted after 0, 2 and 4 days culture. At each time point, cells were dissociated using accutase then resuspended in growth medium. Aliquots of cells were incubated in trypan blue (Sigma, St. Louis, MO, USA) then counted with the aid of a haemocytometer.
Proliferation was further determined using 5‐bromodeoxyuridine (BrdU) incorporation. Dissociated cells were plated on glass coverslips coated with poly‐L‐ornithine (0.015 mg/ml) and cultured for 48 h. Cells were incubated for 5 h with 10 μm BrdU, and then fixed and washed. DNA was denatured by incubating the cells in 1 N HCl for 45 min at room temperature. Cells were washed and blocked with 10% bovine serum albumin for 60 min at room temperature, and primary mouse anti‐BrdU antibody (1:200, BD, San Jose, CA, USA) was applied and incubated for 60 min at 37 °C followed by goat anti‐mouse Alexa 546 antibody (1:500, Invitrogen). Nuclei were stained with 10 μg/ml Hoechst 33342 (Sigma). Images were acquired using a Leica inverted fluorescence microscope.
Migration assay
Neurospheres with similar diameters were selected and plated on poly‐L‐ornithine‐coated six‐well culture plates containing neurobasal medium supplemented with 2% B27 with or without NAC (5 mm) or 2‐APB (50 μm) or U73122 (2 μm/5 μm) for 12 or 24 h. Cell migration was evaluated by measuring distances ANPCs had migrated from edges of neurospheres towards exernal borders.
Ca2+ imaging
Intracellular Ca2+ measurements were performed on cells bathed in imaging buffer comprised of 140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm glucose and 20 mm HEPES (pH 7.3). Cells were loaded with 4 μm of Fura‐2 AM (Invitrogen) for 40 min at room temperature (RT), then allowed further 10 min after washing to de‐ester the dye before imaging. Images were captured using a Nikon inverted microscope (Eclipse Ti) and ×40 magnification oil‐immersion objective lens (N.A. = 1.40). Cells were alternatively excited at 340 nm and 380 nm, using a single‐band multi‐exciter filter set (FURA2‐C‐000, BrightLine®; Semrock, Rochester, New York, USA), emissions were collected through a 409 nm single‐edge dichroic beam splitter (BrightLine®, Semrock) and 510/84 nm single‐band emitter (BrightLine®, Semrock). Images were collected every 2 seconds for the duration of the experiment. Intracellular Ca2+ concentration ([Ca2+]i) within single cells was reported as ratio of the 510 nm emission excited at 340 and 380 nm (340/380). Glutamate (100 nm) was added at 100 s to induce calcium signaling and changes in Ca2+ levels were monitored for 400 s. To verify efficiency of 2‐APB (50 μm) and U73122 (2 μm), these drugs were added at 60 s before 100 μm glutamate was added at 200 s.
ROS measurement
Intracellular ROS level was assessed using 2′,7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA)(Beyotime, Shanghai, China), which can be hydrolysed to 2′,7′‐dichlorodihydrofluorescein (DCFH) by esterases; then DCFH is oxidized by ROS in the cells, yielding 2′,7′‐dichlorofluorescein (DCF). After treatment with NAC (1 mm, 18 h; 5 mm, 4 h), cells were washed with neurobasal medium, incubated in 10 μm of DCFH‐DA at 37 °C for 30 min, and washed twice in neurobasal medium. Then they were resuspended into a single cell suspension using accutase, and analysed by flow cytometry. To detect mitochondrial superoxide production, monolayer cultured cells were treated for 15 min at 37 °C with 5 μm of MitoSOX™ Red (Molecular Probes; Invitrogen), and washed three times, digested with accutase, collected by centrifugation, and analysed by flow cytometry. Flow cytometric analysis was performes immediately after dye treatment. All measurements were repeated at least three times.
Immunofluorescence
To examine cell proliferation and differentiation in vivo, a separate set of 4‐month‐ (4 WT, 5 YAC128) and 18‐month‐ (7 WT, 6 YAC128) old mice were sacrificed. Mice were deeply anaesthetized with chloral hydrate, perfused transcardially with PBS and fixed with 4% paraformaldehyde. Brains were excised, post‐fixed overnight at 4 °C, and transferred to 30% sucrose. 25 μm coronal brain sections were processed for detection of Ki‐67‐positive cells and DCX‐positive cells. After rinsing briefly in PBS, sections were treated for 2 h in 10% donkey serum and 0.3% Triton X‐100 in PBS then incubated in rabbit polyclonal antibody against Ki‐67 (1:200, Abcam) or a rabbit polyclonal antibody against DCX (1:200, Abcam) in PBS containing 5% donkey serum, for 12 hours at 4 °C. Sections were then rinsed three times in PBST (PBS plus 0.3% tween‐20) and incubated for 2 hours in Alexa Fluor 488‐labelled donkey anti‐rabbit IgG antibody (1:1000; Invitrogen). Hoechst33342 (10 μg/ml;, Invitrogen) labelled the positions of cell nuclei. Fluorescence‐stained sections were analysed using a confocal laser‐scanning microscope (Carl Zeiss LSM510).
Data collection and analyses
In experiments when cell counts or measurements were involved, a naive person collected all images, to minimize potential observer bias. All such experiments were repeated at least three times and a representative experiment is presented here. Data are shown as mean ± SE, statistical comparisons were made using the independent samples t‐test, and significance level was set at 5%.
Results
Characteristics of NPCs isolated from adult mouse SVZ
In our study, NPCs were isolated from SVZs of 4‐month‐old WT and YAC128 mice. NPCs were grown as either neurospheres in non‐coated plates (Fig. 1a) or monolayer cultures on poly‐L‐ornithine‐coated dishes (Fig. 1b) in the presence of EGF and FGF2. After expanding and propagating cells for at least 14 passages, they uniformly expressed Nestin and SOX2 (Fig. 1c), markers typically found in uncommitted neural stem cells 41, 42, indicating that our ANPCs maintained stem cell characteristics over the culture period and that these cell populations were highly homogeneous.
Figure 1.
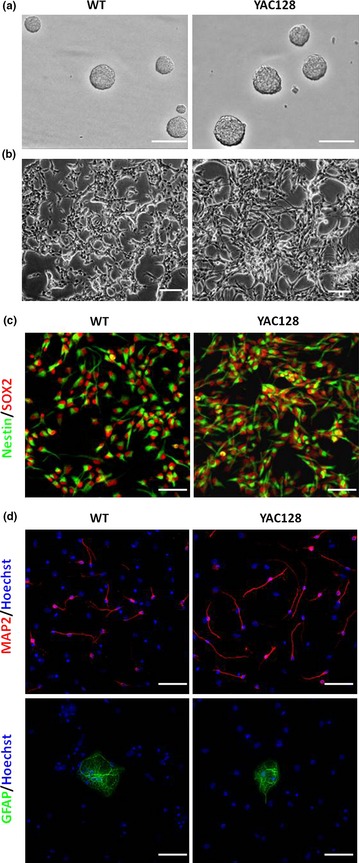
ANPC cultures and monolayer ANPC differentiation in vitro . (a) ANPCs were grown as neurospheres in a non‐coated six‐well plastic plate. Scale bar: 250 μm. (b) ANPCs were grown as monolayers in poly‐ornithine‐coated six‐well plastic plates. Scale bar: 250 μm. (c) Monolayer ANPCs were stained with Nestin (green) and SOX2 (red). Scale bar: 50 μm. (d) Monolayer ANPCs were able to differentiate into neurons (MAP2, red) and astrocytes (GFAP, green). Scale bar: 50 μm.
ANPCs were plated as monolayers and differentiated using a previously described protocol 40, in which EGF was withdrawn and FGF2 concentrations reduced while BDNF was increased. Differentiated cells were stained for lineage‐specific differentiation markers MAP2 (for neurons) and GFAP (for astrocytes) (Fig. 1d), demonstrating multipotencies of ANPCs isolated from SVZ regions of adult WT and YAC128 mouse brains.
YAC128 ANPCs had higher cell proliferation, migration and neuronal differentiation capabilities
One major property of stem cell is self‐renewal 43, thus we first compared proliferation capabilities between WT and YAC128 ANPCs; neurosphere formation was evaluated as an indicator of this. To minimize effects of cell aggregation during growth of neurospheres, ANPCs were plated at low density. After 8 days of culture, diameters of neurospheres were measured. As shown in Fig. 2a, there was significant increase in size of the spheres, detected in YAC128 ANPCs compared to WT cells, suggesting that YAC128 ANPCs had much higher proliferative potential than WT ANPCs. This result was further confirmed by growth curve experimentation. YAC128 ANPCs had a much higher growth rates than WT ANPCs (Fig. 2b), confirming their higher potential for self‐renewal in HD ANPCs. Furthermore, percentages of BrdU‐positive cells in YAC128 ANPCs was significantly higher than that in WT cells as shown by BrdU incorporation assay (Fig. 2c,d).
Figure 2.
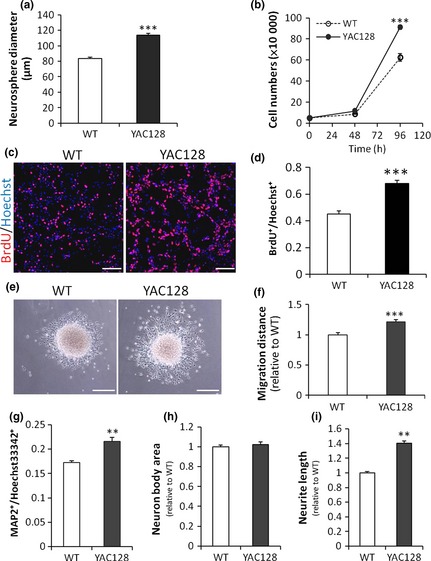
ANPC s derived from SVZ of HD mouse brain exhibit increased proliferation, migration and neurogenic differentiation compared with that of WT control. (a) Neurosphere assay. The size of neurospheres were determined after 8‐day culture in vitro (n = 70 neurospheres per group, ***P < 0.001). (b) YAC128 HD ANPCs had a much higher rate of growth compared with the WT control (n = 3, ***P < 0.001). (c,d) BrdU incorporation assay. The percentage of BrdU‐positive cells in YAC128 ANPCs was significantly higher than that in WT cells (n = 6, ***P < 0.001). Scale bar: 100 μm. (e) Neurospheres cultured on poly‐L‐ornithine dishes migrated outwards radially. Scale bar: 500 μm. (f) The radial migration distance of YAC128 HD neurospheres were significantly increased compared with the WT control after 12‐hour cultivation (n = 40 neurospheres per group, ***P < 0.001). (g) The percentage of neuronal cells (MAP2+) was quantified by normalizing the total MAP2+ cells to the total Hoechst33342+ cells after 21‐day differentiation (n = 3, **P < 0.01). (h) Quantification of the soma area of neurons derived from WT and YAC128 ANPCs (n = 50 neurons per group). (i) Neurite lengths of MAP2+ cells derived from WT and YAC128 ANPCs were measured. Independent t‐test showed a significant increase in neurite length in YAC128 neurons compared with WT neurons (n = 50 neurons per group, **P < 0.01). Data were shown as the mean ± SE.
To determine whether there was any difference in mobility between WT and YAC128 ANPCs, an in vitro migration assay was employed. After culturing on poly‐ornithine‐coated plates for 12 h, ANPCs from neurospheres migrated radially outwards (Fig. 2e). Distances from edges of neurospheres to borders to which the ANPCs migrated were measured to evaluate cell migration. Statistics analysis indicated that YAC128 ANPCs migrated significantly faster than WT ANPCs (Fig. 2f).
We next compared potentials for neuronal differentiation between WT and YAC128 ANPCs in vitro. ANPCs were differentiated using a previously described protocol 40. As shown in Figs. 1d and 2g, both genotypes of ANPCs were able to differentiate into neurons (MAP2+). Percentages of MAP2+ cells were then quantified by normalizing total MAP2+ cells to total Hoechst33342+ cells. YAC128 ANPCs differentiated for 21 days had significantly higher percentages of MAP2+ cells compared to WT ANPCs of identical passage and differentiation times (Figs. 1d,2g). Moreover, neurite length of differentiated YAC128 MAP2+ cells was significantly longer than that of WT MAP2+ cells (Fig. 2i). No significant difference was detected in soma area between YAC128 and WT neuronal like cells (Fig. 2h). Thus, YAC128 HD ANPCs were able to differentiate into greater numbers of neurons with longer neurites compared to WT ANPCs, indicating enhanced neuronal differentiation capacities in HD ANPCs.
ANPCs exhibited enhanced Ca2+ signals
It has already been demonstrated that polyQ expansion in HTT induces enhanced Ca2+ signalling in HD MSNs 33, 34, 35, 36, 37. To investigate whether this also exists in ANPCs, we compared glutamate‐induced Ca2+ signalling between WT and YAC128 ANPCs. Intracellular Ca2+ concentration was monitored using Fura‐2 imaging, and data are presented as 340/380 ratios (Fig. 3a–c). On average, basal Ca2+ levels before glutamate application were not significantly different from each other between genotypes of ANPC (Fig. 3a). Treatment with 100 nm glutamate caused significantly higher amplitude of transient cellular Ca2+ in YAC128 ANPCs over WT ANPCs (Fig. 3a–b). To quantify amounts of Ca2+ loaded into cytoplasm during glutamate stimulation, we calculated areas (ratio*s) below transient Ca2+ traces. Significantly higher amounts of Ca2+ were loaded into the cytoplasm in YAC128 ANPCs than WT ANPCs (Fig. 3c). Similarly, treatment with 100 μm glutamate also caused significantly higher transient Ca2+ and more Ca2+ load in YAC128 ANPCs than in WT ANPCs (Fig. 3d,3g–h). Additionally, both 2‐APB and U73122 significantly reduced Ca2+ levels in both ANPC genotypes (Fig. 3d–h).
Figure 3.
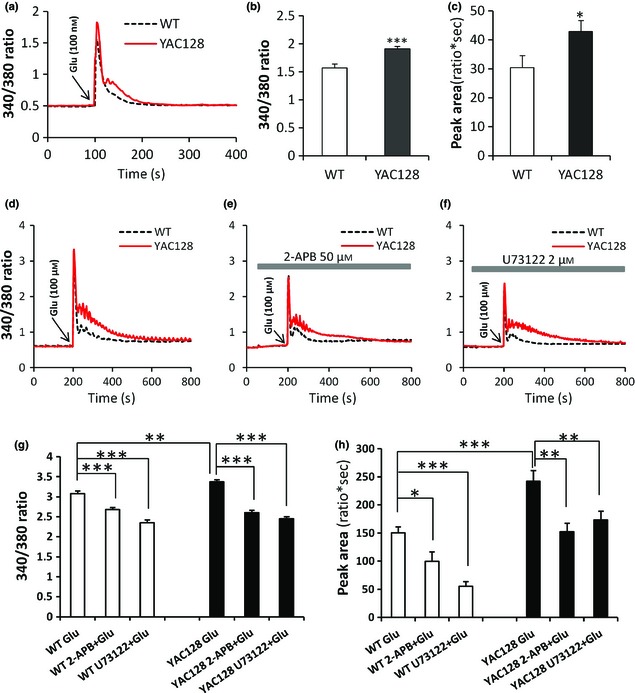
YAC 128 ANPC s exhibit increased Ca 2+ signalling compared with the WT controls. (a) 100 nm glutamate‐induced Ca2+ signals in ANPCs from the WT and YAC128 mouse brains. Intracellular Ca2+ concentration ([Ca2+]i) was presented as a 340/380 Fura‐2 ratio. The average 340/380 values (mean ± SE) were shown for WT (n = 23) and YAC128 (n = 30) ANPCs. Both peak values (b) and peak areas (c) were significantly higher in YAC128 ANPCs than in the WT controls (*P < 0.05, ***P < 0.001). (d) The 100 μM glutamate‐induced Ca2+ signals in ANPCs from the WT and YAC128 mouse brains. Both peak values (g) and peak areas (h) were significantly higher in YAC128 (n = 21) ANPCs than in the WT (n = 23) controls (**P < 0.01, ***P < 0.001). The 100 μm glutamate‐induced [Ca2+]i transients in WT and YAC128 ANPCs were significantly reduced in the presence of the InsP3R blocker 2‐APB (50 μm) (e) and PLC inhibitor U73122 (2 μm) (f). (g) Effect of 2‐APB and U73122 on the peak value of [Ca2+]i in WT and YAC128 ANPCs in response to 100 μm glutamate. Pre‐treatment with 2‐APB and U73122 significantly decreased the peak value of [Ca2+]i in both WT (n = 20 for 2‐APB and n = 23 for U73122) and YAC128 ANPCs (n = 26 for 2‐APB and n = 22 for U73122) when compared with the control group (***P < 0.001). (h) Effect of 2‐APB and U73122 on the peak area of [Ca2+]i in WT and YAC128 ANPCs in response to 100 μm glutamate. Pre‐treatment with 2‐APB and U73122 significantly decreased the peak area of [Ca2+]i in both WT and YAC128 ANPCs when compared with the control group (*P < 0.05, **P < 0.01,***P < 0.001).
Enhanced Ca2+ signals were required for increased proliferation and migration of HD ANPCs
Ca2+ signalling has previously been discussed in regulating neural stem/progenitor cell proliferation 44, 45, 46, 47 and migration 48, 49, 50, 51. In our study, we first evaluated effects of glutamate‐induced Ca2+ signalling on ANPC proliferation in each genotype. After incubation with 100 μm glutamate for 4 days, both genotypes underwent significant increases in proliferative rate compared to their control groups, and YAC128 HD ANPCs proliferated more rapidly than WT ANPCs (Fig. 4a), indicating that elevation of cellular Ca2+ lead to higher ANPC growth rate. Thus, increased proliferation of YAC128 ANPCs could be associated with their enhanced Ca2+ signalling. To confirm this, Ca2+ signalling was inhibited either by 2‐APB [a cell‐permeable IP3R inhibitor 52] or U73122 [a specific PLC inhibitor 53], and cell proliferation was assessed under these conditions respectively. Treatment with 50 μm 2‐APB significantly reduced cell proliferation in both WT and YAC128 HD ANPCs, and HD ANPCs no longer exhibited any proliferative advantage (Fig. 4b). Similar results were obtained when Ca2+ signalling in ANPCs was inhibited by U73122 (Fig. 4c). When treating with 2 μm of U73122, YAC128 ANPCs exhibited similar cell proliferation rates as WT ANPCs (Fig. 4c). These results indicate that increased proliferation of YAC128 ANPCs was calcium‐dependent. In addition, treatments with 2‐APB and U73122 did not cause significant cell death, excluding the possibility that inhibition of proliferation was due to increased cell death. (Fig. S1).
Figure 4.
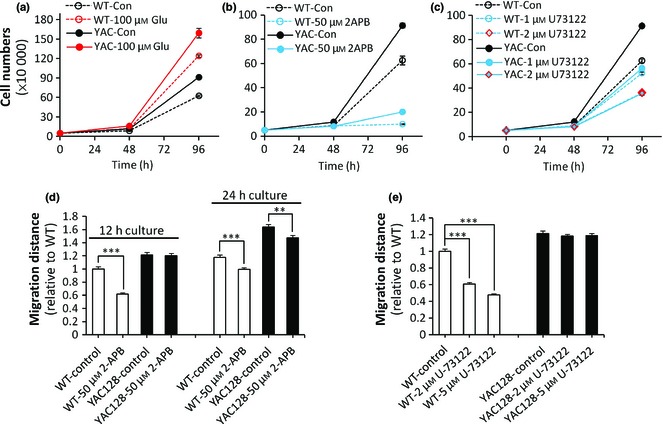
Enhanced Ca 2+ signals are required for the enhanced proliferation and migration in YAC 128 HDANPC s compared with WTANPC s. (a) Glutamate promoted the proliferation of both WT and YAC128 ANPCs. 2‐APB, an inhibitor of intracellular Ca2+ signal, strongly inhibited the proliferation (b) and migration (c) U‐73122, a PLC blocker, dose‐dependently inhibited the proliferation of ANPCs from WT and YAC128 mice. (d) of both WT ANPCs and YAC128 ANPCs (**P < 0.01; ***P < 0.001). WT ANPCs were more sensitive to 2‐APB than YAC128 ANPCs in cell migration. (e) U‐73122 inhibited the migration of WT ANPCs significantly (***P < 0.001), while the migration of YAC128 ANPCs was not affected by U‐73122. Data were presented as mean ± SE.
Ca2+ signalling has also been shown to play a role in neuronal motility, for example in migration of neural precursors, as well as elongation and branching of neurites 54. To test whether increased migration in YAC128 ANPCs could be attributed to enhanced Ca2+ signalling, we compared migration capabilities between WT and YAC128 ANPCs with/without Ca2+ signalling inhibitors. Neurospheres with similar diameters were plated into poly‐L‐ornithine‐coated dishes, cultured for 12 or 24 h with or without Ca2+ signalling inhibitors. As shown in Fig. 4d, 12 h after initiation of culture, 50 μm of 2‐APB significantly inhibited radial migration of both WT and YAC128 ANPCs, in a time‐dependent manner. Similar results were obtained using U73122 treatment (Fig. 4e). Both Ca2+ signalling inhibitors (2‐APB and U73122) were very effective in reducing migration of WT ANPCs but less of YAC128 ANPCs. These results indicated that reduction in Ca2+ signalling slowed ANPC migration, and relatively higher levels of Ca2+ signalling were essential for rapid migration. YAC128 ANPCs, which had much higher levels of Ca2+ signalling compared to WT ANPCs, may not be so sensitive to reduction in Ca2+ signalling during migration.
Taken together, these data indicate that enhanced Ca2+ signaling is essential for increased proliferation and migration capacities of YAC128 ANPCs.
YAC128 ANPCs had increased levels of reactive oxygen species
Marked changes in levels of reactive oxygen species have previously been identified in HD brain and in HD cell models 29, 38, 55, 56, 57. Primary cultured medium spiny neurons (MSNs) from YAC128 neonatal mouse brains have higher levels of reactive oxygen species (ROS) than WT cortical neurons 39. To determine if changes in ROS levels occurred in YAC128 ANPCs, ROS levels in live cells were measured by using DCFH‐DA. This is hydrolysed by esterases to DCFH, and DCFH is oxidized by ROS in the cells, yielding DCF. We found significant increase in DCF fluorescence in YAC128 compared to WT cells (Fig. 5a). Given the pivotal role of mitochondria in generation of ROS 58, mitochondrial ROS (mt‐ROS) were monitored by MitoSOX™ Red (Molecular Probes). Levels of mt‐ROS in YAC128 ANPCs were also significantly higher than in WT controls (Fig. 5b).
Figure 5.
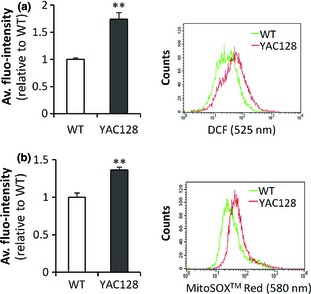
YAC 128 ANPC s show enhanced ROS signals. (a) DCF fluorescence was used as a marker of ROS and was measured in live cells with flow cytometry. Left, measurements of ROS in YAC128 ANPCs and WT ANPCs. YAC128 ANPCs had higher levels of ROS compared with WT controls. Right, scatter plot histogram of a representative experiment. (b) MitoSOX ™ Red fluorescence was used as an indicator of mitochondrial superoxide and was measured in live cells with flow cytometry. Left, measurement of mitochondrial superoxide in YAC128 ANPCs compared to WT ANPCs. YAC128 ANPCs had higher levels of mitochrondrial ROS compared with WT control. Right: scatter plot histogram of a representative experiment. Data were presented as mean ± SE (n = 3, **P < 0.01).
Enhanced ROS signals were required for increased proliferation and migration of YAC128 ANPCs
ROS may play roles in regulating neural progenitor cell proliferation 59, 60, 61, 62. Thus, to determine potential relationships between enhanced ROS and increased proliferation in HD ANPCs, cells were cultured at the same density in the presence or absence of the ROS scavenger N‐acetylcysteine (NAC) and analysed for changes in proliferation. As expected, NAC significantly reduced cellular ROS levels of both WT and YAC128 ANPCs (Fig. 6a). In the absence of NAC, YAC128 ANPCs had more rapid proliferation than WT ANPCs (Fig. 6b), while in the presence of 1 mm NAC [which could not cause significant cell death during treatment (Fig. S1)], both WT and YAC128 ANPCs failed to proliferate (Fig. 6b), demonstrating ROS to be an essential signal for maintenance of ANPC self‐renewal.
Figure 6.
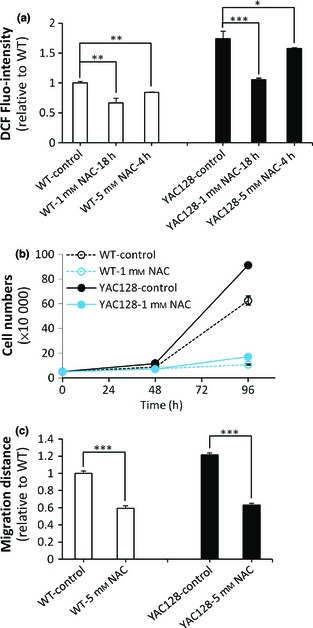
Increased ROS signals are required for enhanced proliferation and migration in YAC 128 ANPC s compared with WTANPC s. (a) Measurement of ROS levels in YAC128 and WT ANPCs pre‐treated with NAC, a ROS scavenger. NAC was able to decrease ROS signal significantly in both YAC128 and WT ANPCs (n = 3, *P < 0.05; **P < 0.01; ***P < 0.001). (b) NAC inhibited the proliferation of YAC128 and WT ANPCs. (c) NAC inhibited the migration of YAC128 and WT ANPCs significantly (n = 40 neuropheres per group, ***P < 0.001). Data were presented as mean ± SE.
ROS has also been linked to cell migration 63, 64, 65. Next, we investigated the relationship between redox state and ANPC migration. Chelating cellular ROS by NAC strongly inhibited migration of both WT and YAC128 HD ANPCs (Fig. 6c), indicating that ROS signalling is also required for ANPCs migration.
In summary, these observations indicated that increased proliferation and migration in YAC128 ANPCs were critically depended on enhanced ROS signalling.
YAC128 ANPCs exhibited normal proliferation and differentiation in the subventricular zone of mouse brains
Our experiments provide compelling evidence that YAC128 ANPCs manifest Ca2+/ROS signalling‐dependent enhancement in proliferation and migration in vitro. To discover any substantiation of our in vitro observations, experiments were undertaken to address whether ANPCs in HD mouse brains would have higher rates of cell proliferation than that in WT brains, in vivo. Antibody to Ki‐67, a nuclear antigen expressed during all stages of the cell cycle except G0, was used to label proliferating cells (Fig. 7a). There were no significant differences in cell proliferation between YAC128 and WT mouse brains at either 4 or 18 months of age, although there was significantly age‐dependent decline in numbers of proliferating cells in the SVZ of both genotypes (Fig. 7b). These results were consistent with previous reports 24, that proliferation of ANPCs appears to be no more than normal in SVZ of YAC128 HD mouse brains compared to WT brains. Moreover, numbers of immature neurons in the SVZ were also quantified by immunofluorescence for DCX (Fig. 7c). YAC128 mice displayed similar numbers of DCX‐positive cells in the SVZ as their WT controls (Fig 7d). We propose that micro‐environment in the SVZ constrained proliferation and differentiation capacities of HD ANPCs in vivo.
Figure 7.
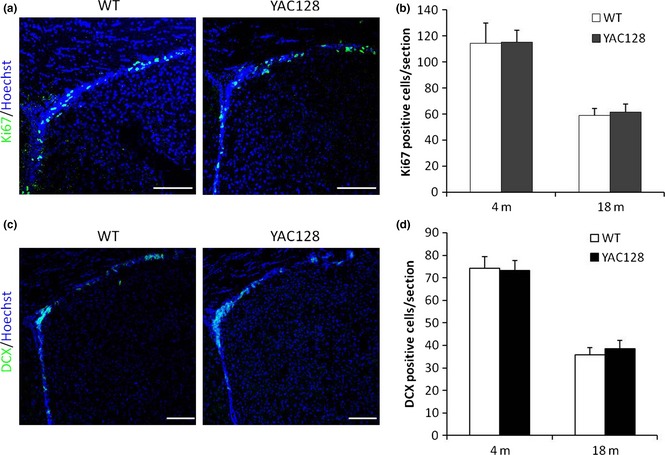
YAC 128 HD mice show similar levels of ANPC proliferation and differentiation as WT mice in SVZ. (a) Endogenous ANPC proliferation in the SVZ was assessed by immunofluorescence for cell cycle marker Ki67. (b) Statistic analysis of Ki‐67‐positive cells in SVZ. No significant difference between the genotypes in the number of Ki‐67‐positive cells was observed. (c) Endogenous ANPC neuronal differentiation in the SVZ was assessed by DCX immunofluorescence. (d) Statistic analysis of DCX‐positive cells in SVZ. No significant difference between the genotypes in the number of DCX‐positive cells was observed. Data were presented as mean ± SE. Scale bar: 100 μm.
Discussion
To evaluate intrinsic capacities of adult neural progenitor cells for proliferation and neuronal differentiation subjected to Huntington's disease conditions, we isolated neural progenitor cells from adult brains of mice expressing endogenous levels of full‐length human mutant HTT protein (YAC128). Both WT and YAC128 ANPCs were able to differentiate to both neurons and astrocytes. Compared to wild‐type ANPCs, YAC128 HDs manifested increased cell proliferation, migration, and neuronal differentiation capacities, accompanied by enhanced Ca2+ signalling and higher levels of intracellular reactive oxygen species (ROS). Here, we provide compelling evidence to demonstrate that enhanced Ca2+ and ROS signals (which are detrimental to mature medium spiny neurons), are essential for this intrinsic change in properties in HD ANPCs. This is the first report that mutant HTT protein triggers Ca2+ and ROS signalling‐dependent enhancement of proliferation and migration in HD ANPCs. Interestingly, increased proliferation and neuronal differentiation of HD ANPCs was largely constrained by the in vivo micro‐environment in mouse brains. Thus, our results also point to a new direction for development of cell‐based therapeutic strategies in HD: remodelling the in vivo micro‐environment is the key to liberate ANPC potential for promoting endogenous neurogenesis.
Previous studies have shown that Ca2+ elevations induced by growth factors and neurotransmitters tightly regulate proliferation of NPCs. For example, EGF, a well‐known regulator of NPC proliferation, mobilizes Ca2+ signalling by activation of receptor‐tyrosine‐kinase coupled to the PLC–InsP3 pathway 66; FGF2, another well‐known regulator of NPC proliferation, induces Ca2+ influx via TRPC1 channels 45. Likewise, glutamate, which triggers Ca2+ mobilization, has long been reported to promote neural progenitor cell proliferation 67, 68, 69, 70. In our study, increase in cellular Ca2+ induced by 100 μm glutamate led to significant increase in cell proliferation, while reduction in intracellular Ca2+ release with 50 μm 2‐APB resulted in significant inhibition of cell proliferation in both ANPC genotypes. Furthermore, inhibition of upstream of Ca2+ signalling by PLC inhibitor U73122 also significantly reduced cell proliferation. These findings indicated that appropriate higher intracellular Ca2+ concentration leads to higher proliferation of ANPCs. Importantly, inhibition of intracellular Ca2+ signalling abolished increased proliferation of HD ANPCs, strongly suggesting that enhanced Ca2+ signalling contributed to the higher rate of proliferation in HD ANPCs.
Significant reduction in numbers of migrating cells from neurospheres has been found in the presence of T‐type channel blockers 51, suggesting that entry of Ca2+ from the extracellular medium plays a role in neural progenitor cell migration. Intracellular Ca2+ stores have also been reported to affect migration of neural progenitor cells (NPC). One study from Pregno and colleagues showed that migratory activity induced by Neuregulin1 was through long‐lasting increase in intracellular calcium concentration, dependent on release of Ca2+ from internal stores 50. In our present study, inhibition of Ca2+ release from internal stores with 2‐APB or U73122, resulted in significantly reduced cell migration in both WT and YAC128 HD ANPCs, indicating that Ca2+ signalling was essential for active ANPC migration. Due to higher levels of Ca2+ signalling in YAC128 HD ANPCs, YAC128 ANPCs moved significantly faster than WT controls.
In mouse embryonic stem cells, Neuronatin promoted neural induction through increasing intracellular Ca2+ concentration, which in turn increased phosphorylation of Erk1/2, inhibited the BMP4 pathway and co‐operated with the FGF/Erk pathway to induce neural generation 71. In cultured ANPCs isolated from rodent hippocampus, evoked intracellular Ca2+ concentration was relayed to activation of transcription factor NeuroD, leading to neurogenesis 72. Similarly, isoxazole (a small molecule capable of triggering Ca2+ influx via Ca2+ channels and NMDA receptors), has been shown to induce robust neuronal differentiation in adult neural stem/progenitor cells 73. These findings indicate that increase in intracellular Ca2+ is critical for neural fate determination. In our present study, due to higher levels of Ca2+ signals in YAC128 HD ANPCs, YAC128 HD ANPCs differentiated into higher amounts of neurons with longer neurites than did WT controls, indicating that mutant HTT‐induced elevated Ca2+ signals actually enhanced potentials of HD ANPCs for neuronal differentiation and neuritogenesis.
Reactive oxygen species (ROS) overproduction has been implicated in pathogenesis of various neurodegenerative disorders such as Parkinson's 74, Alzheimer's 75 and Huntington's diseases 76. Besides their well‐known toxicity, ROS can also play roles as second messengers, regulating various cellular functions including proliferation of NPCs/NSCs. Yoneyama et al. have observed that antioxidant treatments significantly inhibit hippocampal progenitor proliferation 60. Le Belle and colleagues reported that increases in endogenous ROS levels consistently enhanced neurosphere generation of neural stem cells (NSCs) derived from CNS, and addition of exogenous agents that elevate ROS levels increased production of neurospheres 61. In addition, endogenous production of ROS has been demonstrated to be necessary to maintain neural stem populations in the brain 77. In agreement with these findings, we found that YAC128 ANPCs manifested higher levels of endogenous ROS compared to WT cells. More importantly, HD ANPCs exhibited ROS signal‐dependent enhancement of proliferation, as ROS scavenging abolished proliferation of both YAC128 and WT ANPCs. ROS might also be linked to cell migration. In haematopoietic stem cells, it has been shown that low level of ROS can retain stem cell quiescence, while high levels of ROS promote their migration 78. In our study, YAC128 ANPCs had higher ROS levels than WT cells; accordingly, they also showed ROS signal‐dependent increases in cell migration capability.
Our experiments demonstrate that YAC128 HD ANPCs not only maintained their “stemness”, but also had increased capabilities for proliferation, migration and neuronal differentiation in vitro. But in vivo, YAC128 mice brains failed to show any enhancement in ANPC proliferation/neuronal differentiation in the SVZ compared to WT mice, even by 18‐months of age, a time point at which the YAC128 mice not only exhibited behavioural and neuropathological changes (including neuronal loss) but also presented huntingtin inclusions in striatal cells 31. These observations strongly suggest that the striatal micro‐environment severely constrained neurogenesis in the SVZ, and failed to provide appropriate signals for generating new neurons to replace dead ones in YAC128 mouse brains. Given that the intrinsic potentials of HD ANPCs for proliferation and differentiation had not been impaired (were actually enhanced), endogenous cell repair strategies are potentially possible in HD, and may require exogenous factors to up‐regulate ANPC proliferation and neurogenesis 72, 79, 80, 81, 82, 83. However, our present study suggests that the SVZ micro‐environment in which ANPCs are organized limits adult neurogenesis in HD mouse brains. Thus, remoulding the micro‐environment into a suitable condition for endogenous neurogenesis, is critical for cell repair strategies.
Under neurodegenerative conditions in the SVZ of YAC128 mouse brains, intrinsic properties of HD ANPCs were not impaired. In contrast, mutant HTT‐induced elevated Ca2+ and ROS signalling conferred HD ANPCs with enhanced capabilities of proliferation, migration and neuronal differentiation. Additional pathways could also contribute to these phenotypes. A recent unbiased RNAi screen for modifiers of HD pathogenesis identified RRAS signalling as an affected pathway, which plays a role in both cell migration and neurite outgrowth 84. Interestingly, the in vivo micro‐environment did not support endogenous ANPCs to respond appropriately to neuronal loss in YAC128 mouse brains. Thus, how to liberate ANPCs from their constrained micro‐environment, by remoulding the niche, is a crucial step for cell replacement therapy in HD.
Competing interests
The authors of the manuscript declare no conflict of interests.
Supporting information
Fig. S1 Effect of 2‐APB (50 μm), U73122 (2 μm) and NAC (1 mm) on the apoptosis and necrosis of WT and YAC128 ANPCs. (a) Flow cytometry results after staining cells with Annexin V and PI. Cells were classified as healthy (Annexin V−,PI−), apoptotic (Annexin V+, PI−) and necrotic (Annexin V+, PI+). (b) Statistic analysis of the percentage of Annexin V‐positive cells (n = 3). All of these drugs did not cause much cell death, and there were no significant differences between the genotypes in the percentage of Annexin V+ cells.
Acknowledgements
We apologize to those investigators whose work we could not cite due to the space limit, and gratefully acknowledge their contributions to Huntington's disease field. We thank Zhongyang Qv and Yihan Wang for help with maintaining the YAC128 mouse colony, Ilya Bezprozvanny for facilitating transportation of mice strains, Zhifeng Xiao and Baoyang Hu for help with neural progenitor cell culture, and Caixia Guo for helpful discussion. This work was supported by grants from the National Basic Research Program of China (2011CB965003, 2012CB944702), NSFC (81371415, 31401151, 81300982), and the CAS/SAFEA International Partnership Program for Creative Research Teams.
Reference
- 1. Alvarez‐Buylla A, Lim DA (2004) For the long run: maintaining germinal niches in the adult brain. Neuron 41, 683–686. [DOI] [PubMed] [Google Scholar]
- 2. Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132, 645–660. [DOI] [PubMed] [Google Scholar]
- 3. Gheusi G, Cremer H, McLean H, Chazal G, Vincent JD, Lledo PM (2000) Importance of newly generated neurons in the adult olfactory bulb for odor discrimination. Proc. Natl Acad. Sci. USA 97, 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carleton A, Petreanu LT, Lansford R, Alvarez‐Buylla A, Lledo PM (2003) Becoming a new neuron in the adult olfactory bulb. Nat. Neurosci. 6, 507–518. [DOI] [PubMed] [Google Scholar]
- 5. Imayoshi I, Sakamoto M, Ohtsuka T, Takao K, Miyakawa T, Yamaguchi M et al (2008) Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 11, 1153–1161. [DOI] [PubMed] [Google Scholar]
- 6. Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E (2001) Neurogenesis in the adult is involved in the formation of trace memories. Nature 410, 372–376. [DOI] [PubMed] [Google Scholar]
- 7. van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH (2002) Functional neurogenesis in the adult hippocampus. Nature 415, 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kitamura T, Saitoh Y, Takashima N, Murayama A, Niibori Y, Ageta H et al (2009) Adult neurogenesis modulates the hippocampus‐dependent period of associative fear memory. Cell 139, 814–827. [DOI] [PubMed] [Google Scholar]
- 9. Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 8, 963–970. [DOI] [PubMed] [Google Scholar]
- 10. Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N et al (2002) Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110, 429–441. [DOI] [PubMed] [Google Scholar]
- 11. Liu F, You Y, Li X, Ma T, Nie Y, Wei B et al (2009) Brain injury does not alter the intrinsic differentiation potential of adult neuroblasts. J. Neurosci. 29, 5075–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martino G, Pluchino S, Bonfanti L, Schwartz M (2011) Brain regeneration in physiology and pathology: the immune signature driving therapeutic plasticity of neural stem cells. Physiol. Rev. 91, 1281–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM et al (2010) Discovery of a proneurogenic, neuroprotective chemical. Cell 142, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsieh J, Eisch AJ (2010) Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: unraveling the genome to understand the mind. Neurobiol. Dis. 39, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC et al (2004) Increased hippocampal neurogenesis in Alzheimer's disease. Proc. Natl Acad. Sci. USA 101, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verret L, Jankowsky JL, Xu GM, Borchelt DR, Rampon C (2007) Alzheimer's‐type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J. Neurosci. 27, 6771–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li B, Yamamori H, Tatebayashi Y, Shafit‐Zagardo B, Tanimukai H, Chen S et al (2008) Failure of neuronal maturation in Alzheimer disease dentate gyrus. J. Neuropathol. Exp. Neurol. 67, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoglinger GU, Rizk P, Muriel MP, Duyckaerts C, Oertel WH, Caille I et al (2004) Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 7, 726–735. [DOI] [PubMed] [Google Scholar]
- 19. Crews L, Mizuno H, Desplats P, Rockenstein E, Adame A, Patrick C et al (2008) Alpha‐synuclein alters Notch‐1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J. Neurosci. 28, 4250–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winner B, Rockenstein E, Lie DC, Aigner R, Mante M, Bogdahn U et al (2008) Mutant alpha‐synuclein exacerbates age‐related decrease of neurogenesis. Neurobiol. Aging 29, 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gil JM, Mohapel P, Araujo IM, Popovic N, Li JY, Brundin P et al (2005) Reduced hippocampal neurogenesis in R6/2 transgenic Huntington's disease mice. Neurobiol. Dis. 20, 744–751. [DOI] [PubMed] [Google Scholar]
- 22. Lazic SE, Grote H, Armstrong RJ, Blakemore C, Hannan AJ, van Dellen A et al (2004) Decreased hippocampal cell proliferation in R6/1 Huntington's mice. NeuroReport 15, 811–813. [DOI] [PubMed] [Google Scholar]
- 23. McCollum MH, Leon RT, Rush DB, Guthrie KM, Wei J (2013) Striatal oligodendrogliogenesis and neuroblast recruitment are increased in the R6/2 mouse model of Huntington's disease. Brain Res. 1518, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simpson JM, Gil‐Mohapel J, Pouladi MA, Ghilan M, Xie Y, Hayden MR et al (2011) Altered adult hippocampal neurogenesis in the YAC128 transgenic mouse model of Huntington disease. Neurobiol. Dis. 41, 249–260. [DOI] [PubMed] [Google Scholar]
- 25. Li S, Li XJ (2006) Multiple pathways contribute to the pathogenesis of Huntington disease. Mol. Neurodegener. 1, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gil JM, Rego AC (2008) Mechanisms of neurodegeneration in Huntington's disease. Eur. J. Neurosci. 27, 2803–2820. [DOI] [PubMed] [Google Scholar]
- 27. Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME et al (2003) Evidence for more widespread cerebral pathology in early HD: an MRI‐based morphometric analysis. Neurology 60, 1615–1620. [DOI] [PubMed] [Google Scholar]
- 28. Molero AE, Gokhan S, Gonzalez S, Feig JL, Alexandre LC, Mehler MF (2009) Impairment of developmental stem cell‐mediated striatal neurogenesis and pluripotency genes in a knock‐in model of Huntington's disease. Proc. Natl Acad. Sci. USA 106, 21900–21905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ritch JJ, Valencia A, Alexander J, Sapp E, Gatune L, Sangrey GR et al (2012) Multiple phenotypes in Huntington disease mouse neural stem cells. Mol. Cell Neurosci. 50, 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Curtis MA, Penney EB, Pearson AG, van Roon‐Mom WM, Butterworth NJ, Dragunow M et al (2003) Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc. Natl Acad. Sci. USA 100, 9023–9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y et al (2003) Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12, 1555–1567. [DOI] [PubMed] [Google Scholar]
- 32. Ehrnhoefer DE, Butland SL, Pouladi MA, Hayden MR (2009) Mouse models of Huntington disease: variations on a theme. Dis. Model Mech. 2, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P et al (2002) Increased sensitivity to N‐methyl‐D‐aspartate receptor‐mediated excitotoxicity in a mouse model of Huntington's disease. Neuron 33, 849–860. [DOI] [PubMed] [Google Scholar]
- 34. Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL et al (2003) Huntingtin and huntingtin‐associated protein 1 influence neuronal calcium signaling mediated by inositol‐(1,4,5)\ triphosphate receptor type 1. Neuron 39, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinas R et al (2005) Disturbed Ca2 + signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc. Natl Acad. Sci. USA 102, 2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fan MM, Fernandes HB, Zhang LY, Hayden MR, Raymond LA (2007) Altered NMDA receptor trafficking in a yeast artificial chromosome transgenic mouse model of Huntington's disease. J. Neurosci. 27, 3768–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bezprozvanny I (2009) Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 15, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perez‐Severiano F, Rios C, Segovia J (2000) Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington's disease. Brain Res. 862, 234–237. [DOI] [PubMed] [Google Scholar]
- 39. Wang JQ, Chen Q, Wang X, Wang QC, Wang Y, Cheng HP et al (2013) Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 288, 3070–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Spiliotopoulos D, Goffredo D, Conti L, Di Febo F, Biella G, Toselli M et al (2009) An optimized experimental strategy for efficient conversion of embryonic stem (ES)‐derived mouse neural stem (NS) cells into a nearly homogeneous mature neuronal population. Neurobiol. Dis. 34, 320–331. [DOI] [PubMed] [Google Scholar]
- 41. Suh H, Consiglio A, Ray J, Sawai T, D'Amour KA, Gage FH (2007) In vivo fate analysis reveals the multipotent and self‐renewal capacities of Sox2 + neural stem cells in the adult hippocampus. Cell Stem Cell 1, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thier M, Worsdorfer P, Lakes YB, Gorris R, Herms S, Opitz T et al (2012) Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell 10, 473–479. [DOI] [PubMed] [Google Scholar]
- 43. Gage FH (2000) Mammalian neural stem cells. Science 287, 1433–1438. [DOI] [PubMed] [Google Scholar]
- 44. Ryu JK, Choi HB, Hatori K, Heisel RL, Pelech SL, McLarnon JG et al (2003) Adenosine triphosphate induces proliferation of human neural stem cells: role of calcium and p70 ribosomal protein S6 kinase. J. Neurosci. Res. 72, 352–362. [DOI] [PubMed] [Google Scholar]
- 45. Fiorio Pla A, Maric D, Brazer SC, Giacobini P, Liu X, Chang YH et al (2005) Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor‐1‐induced Ca2 + entry and embryonic rat neural stem cell proliferation. J. Neurosci. 25, 2687–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gilley JA, Kernie SG (2011) Excitatory amino acid transporter 2 and excitatory amino acid transporter 1 negatively regulate calcium‐dependent proliferation of hippocampal neural progenitor cells and are persistently upregulated after injury. Eur. J. Neurosci. 34, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Somasundaram A, Shum AK, McBride HJ, Kessler JA, Feske S, Miller RJ et al (2014) Store‐operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J. Neurosci. 34, 9107–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scemes E, Duval N, Meda P (2003) Reduced expression of P2Y1 receptors in connexin43‐null mice alters calcium signaling and migration of neural progenitor cells. J. Neurosci. 23, 11444–11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McKinney MC, Kulesa PM (2011) In vivo calcium dynamics during neural crest cell migration and patterning using GCaMP3. Dev. Biol. 358, 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pregno G, Zamburlin P, Gambarotta G, Farcito S, Licheri V, Fregnan F et al (2011) Neuregulin1/ErbB4‐induced migration in ST14A striatal progenitors: calcium‐dependent mechanisms and modulation by NMDA receptor activation. BMC Neurosci. 12, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Louhivuori LM, Louhivuori V, Wigren HK, Hakala E, Jansson LC, Nordstrom T et al (2013) Role of low voltage activated calcium channels in neuritogenesis and active migration of embryonic neural progenitor cells. Stem Cells Dev. 22, 1206–1219. [DOI] [PubMed] [Google Scholar]
- 52. Estrada M, Cardenas C, Liberona JL, Carrasco MA, Mignery GA, Allen PD et al (2001) Calcium transients in 1B5 myotubes lacking ryanodine receptors are related to inositol trisphosphate receptors. J. Biol. Chem. 276, 22868–22874. [DOI] [PubMed] [Google Scholar]
- 53. Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA et al (2012) Activation of neuronal P2X7 receptor‐pannexin‐1 mediates death of enteric neurons during colitis. Nat. Med. 18, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zheng JQ, Poo MM (2007) Calcium signaling in neuronal motility. Annu. Rev. Cell Dev. Biol. 23, 375–404. [DOI] [PubMed] [Google Scholar]
- 55. Charvin D, Vanhoutte P, Pages C, Borrelli E, Caboche J (2005) Unraveling a role for dopamine in Huntington's disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc. Natl Acad. Sci. USA 102, 12218–12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li X, Valencia A, Sapp E, Masso N, Alexander J, Reeves P et al (2010) Aberrant Rab11‐dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington's disease. J. Neurosci. 30, 4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dong G, Ferguson JM, Duling AJ, Nicholas RG, Zhang D, Rezvani K et al (2011) Modeling pathogenesis of Huntington's disease with inducible neuroprogenitor cells. Cell. Mol. Neurobiol. 31, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. [DOI] [PubMed] [Google Scholar]
- 59. Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B et al (2000) Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J. Neurosci. 20, 7377–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yoneyama M, Kawada K, Gotoh Y, Shiba T, Ogita K (2010) Endogenous reactive oxygen species are essential for proliferation of neural stem/progenitor cells. Neurochem. Int. 56, 740–746. [DOI] [PubMed] [Google Scholar]
- 61. Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD et al (2011) Proliferative neural stem cells have high endogenous ROS levels that regulate self‐renewal and neurogenesis in a PI3K/Akt‐dependant manner. Cell Stem Cell 8, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Noble M, Proschel C, Mayer‐Proschel M (2011) Oxidative‐reductionist approaches to stem and progenitor cell function. Cell Stem Cell 8, 1–2. [DOI] [PubMed] [Google Scholar]
- 63. Hurd TR, DeGennaro M, Lehmann R (2012) Redox regulation of cell migration and adhesion. Trends Cell Biol. 22, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shi D, Li X, Chen H, Che N, Zhou S, Lu Z et al (2014) High level of reactive oxygen species impaired mesenchymal stem cell migration via overpolymerization of F‐actin cytoskeleton in systemic lupus erythematosus. Pathol. Biol. (Paris) 62, 382–390. [DOI] [PubMed] [Google Scholar]
- 65. Yang CM, Hsieh HL, Yu PH, Lin CC, Liu SW (2015) IL‐1beta Induces MMP‐9‐Dependent Brain Astrocytic Migration via Transactivation of PDGF Receptor/NADPH Oxidase 2‐Derived Reactive Oxygen Species Signals. Mol. Neurobiol. 52, 303–317. [DOI] [PubMed] [Google Scholar]
- 66. Maric D, Maric I, Chang YH, Barker JL (2003) Prospective cell sorting of embryonic rat neural stem cells and neuronal and glial progenitors reveals selective effects of basic fibroblast growth factor and epidermal growth factor on self‐renewal and differentiation. J. Neurosci. 23, 240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brazel CY, Nunez JL, Yang Z, Levison SW (2005) Glutamate enhances survival and proliferation of neural progenitors derived from the subventricular zone. Neuroscience 131, 55–65. [DOI] [PubMed] [Google Scholar]
- 68. Di Giorgi‐Gerevini V, Melchiorri D, Battaglia G, Ricci‐Vitiani L, Ciceroni C, Busceti CL et al (2005) Endogenous activation of metabotropic glutamate receptors supports the proliferation and survival of neural progenitor cells. Cell Death Differ. 12, 1124–1133. [DOI] [PubMed] [Google Scholar]
- 69. Suzuki M, Nelson AD, Eickstaedt JB, Wallace K, Wright LS, Svendsen CN (2006) Glutamate enhances proliferation and neurogenesis in human neural progenitor cell cultures derived from the fetal cortex. Eur. J. Neurosci. 24, 645–653. [DOI] [PubMed] [Google Scholar]
- 70. Nochi R, Kato T, Kaneko J, Itou Y, Kuribayashi H, Fukuda S et al (2012) Involvement of metabotropic glutamate receptor 5 signaling in activity‐related proliferation of adult hippocampal neural stem cells. Eur. J. Neurosci. 36, 2273–2283. [DOI] [PubMed] [Google Scholar]
- 71. Lin HH, Bell E, Uwanogho D, Perfect LW, Noristani H, Bates TJ et al (2010) Neuronatin promotes neural lineage in ESCs via Ca(2 + ) signaling. Stem Cells 28, 1950–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Deisseroth K, Singla S, Toda H, Monje M, Palmer TD, Malenka RC (2004) Excitation‐neurogenesis coupling in adult neural stem/progenitor cells. Neuron 42, 535–552. [DOI] [PubMed] [Google Scholar]
- 73. Schneider JW, Gao Z, Li S, Farooqi M, Tang TS, Bezprozvanny I et al (2008) Small‐molecule activation of neuronal cell fate. Nat. Chem. Biol. 4, 408–410. [DOI] [PubMed] [Google Scholar]
- 74. Przedborski S, Jackson‐Lewis V, Vila M, Wu DC, Teismann P, Tieu K et al (2003) Free radical and nitric oxide toxicity in Parkinson's disease. Adv. Neurol. 91, 83–94. [PubMed] [Google Scholar]
- 75. Zhu X, Raina AK, Lee HG, Casadesus G, Smith MA, Perry G (2004) Oxidative stress signalling in Alzheimer's disease. Brain Res. 1000, 32–39. [DOI] [PubMed] [Google Scholar]
- 76. Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- 77. Dickinson BC, Peltier J, Stone D, Schaffer DV, Chang CJ (2011) Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 7, 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ludin A, Gur‐Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C et al (2014) Reactive oxygen species regulate hematopoietic stem cell self‐renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 21, 1605–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jin K, LaFevre‐Bernt M, Sun Y, Chen S, Gafni J, Crippen D et al (2005) FGF‐2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington's disease. Proc. Natl Acad. Sci. USA 102, 18189–18194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cho SR, Benraiss A, Chmielnicki E, Samdani A, Economides A, Goldman SA (2007) Induction of neostriatal neurogenesis slows disease progression in a transgenic murine model of Huntington disease. J. Clin. Invest. 117, 2889–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Duan W, Peng Q, Masuda N, Ford E, Tryggestad E, Ladenheim B et al (2008) Sertraline slows disease progression and increases neurogenesis in N171‐82Q mouse model of Huntington's disease. Neurobiol. Dis. 30, 312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Peng Q, Masuda N, Jiang M, Li Q, Zhao M, Ross CA et al (2008) The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington's disease mouse model. Exp. Neurol. 210, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Decressac M, Wright B, Tyers P, Gaillard A, Barker RA (2010) Neuropeptide Y modifies the disease course in the R6/2 transgenic model of Huntington's disease. Exp. Neurol. 226, 24–32. [DOI] [PubMed] [Google Scholar]
- 84. Miller JP, Yates BE, Al‐Ramahi I, Berman AE, Sanhueza M, Kim E et al (2012) A genome‐scale RNA‐interference screen identifies RRAS signaling as a pathologic feature of Huntington's disease. PLoS Genet. 8, e1003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Effect of 2‐APB (50 μm), U73122 (2 μm) and NAC (1 mm) on the apoptosis and necrosis of WT and YAC128 ANPCs. (a) Flow cytometry results after staining cells with Annexin V and PI. Cells were classified as healthy (Annexin V−,PI−), apoptotic (Annexin V+, PI−) and necrotic (Annexin V+, PI+). (b) Statistic analysis of the percentage of Annexin V‐positive cells (n = 3). All of these drugs did not cause much cell death, and there were no significant differences between the genotypes in the percentage of Annexin V+ cells.