

Abstract
Abstract. Objectives: This study is to evaluate the effect of separase depletion on cell cycle progression of irradiated and non‐irradiated cells through the G2/M phases and consecutive cell survival. Materials and methods: Separase was depleted with siRNA in two human non‐small cell lung carcinoma (NSCLC) cell lines. Cell cycle progression, mitotic fraction, DNA repair, apoptotic and clonogenic cell death were determined. Results: By depletion of endogenous separase with siRNA in NSCLCs, we showed that separase affects progression through the G2 phase. In non‐irradiated exponentially growing cells, separase depletion led to an increased G2 accumulation from 17.2% to 29.1% in H460 and from 15.7% to 30.9% in A549 cells and a decrease in mitotic cells. Depletion of separase significantly (P < 0.01) increased the fraction of radiation‐induced G2 arrested cells 30–56 h after irradiation and led to decrease in the mitotic fraction. This was associated with increased double‐strand break repair as measured by γ‐H2AX foci kinetics in H460 cells and to a lesser extent in A549 cells. In addition, a decrease in the expression of mitotic linked cell death after irradiation was found. Conclusions: These results indicate that separase has additional targets involved in regulation of G2 to M progression after DNA damage. Prolonged G2 phase arrest in the absence of separase has consequences on repair of damaged DNA and cell death.
INTRODUCTION
Eukaryotic cells have evolved sophisticated mechanisms to ensure genomic integrity and survival after treatment with DNA damaging agents. They are able to do this by activating cell cycle checkpoints and repair pathways in order to arrest cells with damaged DNA, and allow for DNA repair. The G2 checkpoint permits cells to avoid segregation of defective chromosomes and is regulated by distinct checkpoint pathways, triggered either by DNA damage (ATM/ATR‐mediated pathway; Xu et al. 2002; Wang et al. 2003), by various stresses like osmotic shock and modification of chromatin structure (p38‐mediated pathway; Goldstone et al. 2001; Jha et al. 2002; , Xu et al. 2002) or by microtubule dynamics (Chfr‐mediated pathway; Rieder & Cole 2000; Scolnick & Halazonetis 2000).
Besides cell cycle checkpoints, which regulate entry into S and mitotic phases, respectively, the spindle assembly checkpoint guards progression through mitosis by regulating metaphase to anaphase transition. Separation of sister chromatids during anaphase ensures that a complete set of chromosomes is transmitted from one generation to the other; it is accompanied by proteolytic cleavage of the cohesion protein by separase. Separase itself is inhibited by phosphorylation of the cyclin B/Cdk1 complex (Stemmann et al. 2001) and by binding to an inhibitor protein called securin. Activation of separase is therefore mediated by proteolytic cleavage of securin and the cyclin B/Cdk1 complex, by a large multiunit protein complex consisting of ubiquitin ligases, known as anaphase promoting complexes. Although proteolysis of securin is necessary for separase activation, binding of securin to separase also has a positive role in activation of separase by promoting its nuclear localization (Yanagida 2000; Hornig et al. 2002). In addition to its role in regulating separase activity, securin also activates expression of p21WAF1/CIP1 (Mu et al. 2003) and interacts with p53 (Bernal et al. 2002) that implicates a role in the G2 checkpoint response.
The spindle assembly checkpoint is activated when chromosomes are misaligned or spindle attachment is perturbed by chemical or physical damage. In addition, any treatment altering chromatin structure may compromise the centromere region, which impacts kinetochores and also activates the spindle assembly checkpoint, leading to a delay in progression of mitosis (Mikhailov et al. 2002). The fate of cells with defects in sister chromatid separation is cell death via mitotic catastrophe and/or apoptosis, or survival with accumulation of genomic instabilities, which are thought to be the source of aneuploidy characteristic of most tumour cells (Cahill et al. 1998; Burum‐Auensen et al. 2007). To further characterize the mechanism that involves human separase in G2‐ and M‐phase progression, in apoptosis and in clonogenic survival after ionizing radiation‐induced DNA damage, separase was depleted with siRNA. We hypothesized that this in irradiated cells might decrease cell cycle progression by increasing the fraction of M‐phase‐arrested cells and therefore decreasing M‐phase progression with multiple mitotic failures. Here, we have tested this hypothesis by transient depletion of separase with siRNA techniques in non‐small cell lung carcinoma (NSCLC) cell lines by monitoring cell cycle progression, M‐phase fraction, late mitotic‐linked cell death and clonogenic survival of irradiated cells.
MATERIALS AND METHODS
Cell culture and irradiation of cells
The non‐small cell lung carcinoma cell line H460 was obtained from American Type Culture Collection (Rockville, MD, USA) and was grown in RPMI 1640 containing 10% foetal calf serum, and 100 units/mL penicillin/streptomycin under an atmosphere of 5% CO2, 95% air at 37 °C. The A549 NSCLC cell line was obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany) and was grown in Eagle's minimal essential medium supplemented with 15% foetal calf serum, non‐essential amino acids and penicillin/streptomycin (100 units/mL, all from Gibco‐BRL, Paisley, UK). Cell cycles were synchronized by release from confluence. Accumulation of cells in the plateau phase was achieved through serum starvation and contact‐inhibited growth. Cultures were irradiated with a 60Co source at 2.1 Gy/min.
Cell cycle analysis
Cells were irradiated with 10 Gy 16 h after transfection and were harvested at various time points thereafter, fixed in 80% ethanol and stored for at least 24 at 4 °C. They were then washed in phosphate‐buffered saline (PBS) and stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) for 24 h at 4 °C. Cytograms were measured with a PAS II flow cytometer (Partec AG, Münster, Germany) and cell cycle analysis was performed using the software ‘Multicycle for Windows’ (Phoenixs Flow systems, San Diego, CA, USA).
Determination of mitotic and apoptotic cells
For mitotic enrichment, cells were re‐seeded on culture slides at 20 h after transfection at low cell density (~4 × 104 cells/cm2); they were then irradiated with 4 Gy at 6 h after re‐seeding and were treated with nocodazole (0.05–0.1 µg/mL, Sigma, Taufkirchen, Germany) 16 h thereafter. Cells were harvested at the times indicated after nocodazole, fixed in 4.5% formaldehyde followed by treatment in permeabilizing buffer (100 mm Tris‐HCl, pH 7.4; 50 mm EDTA, 0.5% Triton X‐100). After incubation in blocking buffer [3% bovine serum albumin, 0.1% Tween‐20, 4× standard saline citrate (SSC)] for at least 24 h at 4 °C, the primary mitosis specific antibodies, either rabbit antiphospho‐histone H3, or mouse antiphospho ser/thr‐prolin, mitotic protein monoclonal 2 (both antibodies from Upstate, Lake Placid, NY, USA) were added at dilutions of 1 : 2000 and 1 : 200, respectively, and were incubated overnight at 4 °C. Cells were further incubated with Alexa Fluor 488‐conjugated goat antimouse or goat antirabbit secondary antibody (Invitrogen, Karlsruhe, Germany) at a dilution of 1 : 400 for 90 min at room temperature. Cells were counterstained for DNA localization. Mitotic cells with uniformly intense staining patterns were evaluated by eye using a Zeiss fluorescence microscope (Wetzlar, Germany).
For flow cytometric analysis of mitotic fractions, cells were fixed in 80% ethanol, followed by treatment in permeabilizing buffer (0.25% Triton X‐100 in PBS). Cells were incubated with mouse antiphospho‐histone H3 and Alexa Fluor 488‐conjugated goat antimouse secondary antibody followed by DAPI staining. Dual labelled cytograms were analysed. The fraction of mitotic cells was determined by gating the G2/M region.
For measurement of late mitosis‐linked apoptosis, cells were irradiated with 20 Gy at 16 h after transfection and fractions of apoptotic cells 48 h after irradiation were determined as described elsewhere (Stapper et al. 1995).
Lipid‐mediated transfection of siRNA
Cells in the plateau phase were re‐seeded at a density of around 4 × 104 cells/cm2 for both H460, and A549 lines. Sequences for separase siRNA used were 5′‐GCUUGUGAUGCCAUCCUGATT‐3′ sense and 5′‐UCAGGAUGGCAUCACAAGCTT‐3′ antisense. Specificity of siRNA used was tested using scrambled siRNA (sense: 5′‐AGUCCUACCGUAGUGUUCGTT‐3′; antisense: 5′‐CGAACACUACGGUAGGACUTT‐3′), which showed no significant effect on separase expression. Each oligonucleotide contains a thymidine dimer at the 3′ end of the sequence and was purchased from Ambion (Austin, TX, USA). Annealed sense and antisense siRNA sequences were transfected with Oligofectamin (Invitrogen) 20–24 h after plating, when cells reached confluence of 60–80%, as described elsewhere for oligonucleotide transfection (Sak et al. 2003).
Cell extracts and Western blot analysis
Cell lysis and electrophoresis were performed as previously described (Sak et al. 2003). For Western blot analysis, aliquots of total cell lysate (40 µg) were resolved in 4–12% pre‐cast polyacrylamide‐sodium dodecyl sulfate (SDS) gels, and subjected to Western blot analysis using separase (NB‐100–330, Zymed Laboratories Inc., San Francisco, CA, USA), β‐actin (sc‐1616‐R, Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies.
H2AX immunofluorescence staining
At specified times after irradiation cells were fixed and probed for γ‐H2AX; nuclear foci were evaluated as described previously (Sak et al. 2005).
Detection of mRNA by Northern blot analysis
At 20–24 h following transfection, cells were harvested mechanically and washed once with PBS. Total cellular RNA was extracted and Northern blot analysis was performed as previously described (Sak et al. 2005). A PCR‐amplified 1.25‐kbp fragment was the specific probe for separase. For synthesis of this probe, primers P1 (5′‐AGAGTCAACTTTGGGACTCTG‐3′) and P2 (5′‐GCCAAATCAACTATCTGACAG‐3′), positions 16–36 and 1242–1263, respectively (accession number AY455930) were used. As control for loading error and overall mRNA transcription activity, a plasmid probe for β‐actin or GAPDH gene was used (Ambion). Probes were labelled using a random prime labelling kit (Amersham) with α‐32P‐dCTP. Membranes were pre‐hybridized for 4 h at 58 °C in ExpressHyb hybridization buffer (Clontech Laboratories Inc., Heidelberg, Germany) and were hybridized overnight in fresh hybridization buffer containing 0.5–1 × 106 cpm/mL of hybridization probe at the same temperature. Membranes were washed once in 2× SSC, 0.1% SDS at 20 °C for 10 min, once at 42 °C for 10 min in the same buffer and once with 0.1× SSC, 0.1% SDS at 42 °C. The extinction signal was scanned and quantitatively analysed with Quantity One, version 4.2.1 software (Bio‐Rad, Munich, Germany).
Clonogenic survival assay
For measurement of clonogenic survival, transfected cells were harvested at 16–18 h following transfection and the assay was performed as previously described (Sak et al. 2002).
Data evaluation
All experiments were repeated at least three times, and data are provided as mean ± standard error for the independent experiments; graphs were constructed with the aid of Microcal Origin, version 4.1 (Microcal Software Inc., Northampton, MA, USA). Sign test for pair differences by Dixon & Mood (1946) was used to test the hypothesis that there would be no difference in measured endpoints between transfected and mock‐transfected cells.
RESULTS
Effect of separase depletion on non‐irradiated cells
Basal protein expression of separase was determined under different growth conditions, that is, enriched in G1 and G2/M phases and nocodazole was used to allow accumulation of cells in G2/M, cell cycle analysis and protein extraction having been performed at 0 and 20 h after treatment with nocodazole. Flow cytometry showed a clear accumulation of cells in G2/M after nocodazole treatment, with 14.9% and 97.6% of cells there after 0 and 20 h for H460 and 13.4% and 89.1% for A549, respectively. We found that full length separase protein (220 kDa) was the major form in G2/M but the processed forms (65 kDa and 150 kDa) mostly were present in G1/S and decreased in G2/M.
To determine whether separase was required for mitotic progression, we first depleted it by using siRNA. A549 and H460 cells were transfected with specific siRNA, and relative amounts of separase mRNA and protein expression were compared by Northern and Western blot analyses at 48 h after transfection, without synchronization with nocodazole. Cell cycle distribution at this time was exponential with 50–65% of cells in G1 phase, 20–30% in S phase and 10–15% in G2 phase. Transfection with specific siRNA decreased the steady state level of both separase mRNA and full length protein to 10–20% with respect to oligofectamin controls. Cell cycle progression was determined in A549 and H460 after depletion of separase. Figure 1 shows this at different times after transfection for non‐irradiated cells. Depletion of separase increased the fraction of cells in G2/M from around 17–29% at 30 h post‐transfection in A549 cells. The same effect was found in H460 cells with an increase in the G2/M fraction from 16% to 31%. Double labelling of cells for DNA (DAPI) and a mitosis related protein (phospho‐histone H3) showed that around 7–8% of the G2/M population of A549 cells at 56 h after sham irradiation consisted of mitotic and about 92% of G2 cells (inserts in Fig. 1). A similar propotion, around 5–10% mitotic and 90–95% G2 cells was found at that time in the G2/M fraction of H460 cells. Increased G2 arrest by separase depletion should lead to decrease in the fraction of mitotic cells. This was evaluated by quantifying nocodazole‐induced mitotic accumulation of transfected cells. The mitotic fraction was measured by means of H3‐positive cells in response to 24 h nocodazole treatment, in addition to separase depletion (Fig. 2); nocadazole had been started 48 h after transfection. Figure 2b shows depletion of separase in non‐irradiated cells without activation of the G2 checkpoint by DNA damage, reduced mitotic fraction from 13.6% to 6.0% in A549 cells; a decrease from 26.1% to 13.3% was found in H460 cells.
Figure 1.
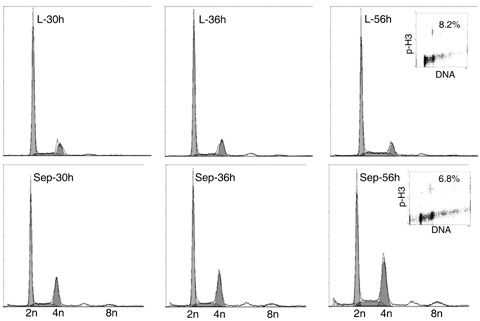
Influence of separase depletion on cell cycle progression. Flow cytometry data demonstrating cell cycle distribution in siRNA transfected A549 cells at 30, 36 and 56 h after sham irradiation. L, oligofectamin control; Sep, specific siRNA‐targeting separase.
Figure 2.
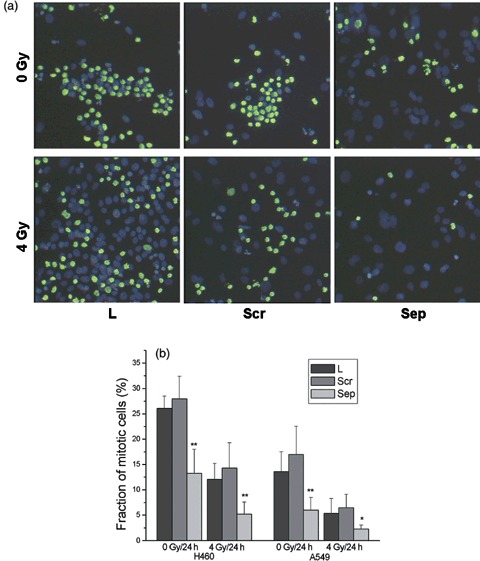
Influence of separase depletion on mitotic fraction (phosphorylated H3 at serine 10) in H460 and A549. (a) Respective immunofluorescence anlaysis of phospho‐histone H3‐positive A549 cells under the different treatment conditions (FITC for H3, DAPI for DNA; magnification ×200). (b) Mitotic fractions, with data points representing mean ± SEM of six independent experiments. Fractions of mitotic cells within the G2/M (4n) population as determined by double staining (DNA/phospho‐histone H3 staining) are shown for the 56 h time point for all treatments (inserted figures). L, oligofectamin control; Scr, control siRNA; Sep, specific siRNA‐targeting separase. Asterisks denote to the significance of the observed results between siRNA and mock‐transfected cells at *P < 0.05 and **P < 0.01.
Effect of separase depletion on irradiated cells
In addition, cell cycle progression after irradiation was determined after depletion of separase in order to evaluate its interaction with that of ionizing radiation, on G2 arrest. Irradiation with 10 Gy increased the G2/M fraction from 15% to 50% at 30 h after irradiation in both cell types. Depletion of separase after irradiation further increased the G2/M fraction to around 60% at 30 h (Fig. 3). Cell cycle analysis after double labelling for DNA (DAPI) and a mitosis‐related protein (phospho‐histone H3) showed that only a small fraction (around 3% of the G2/M population of A549 cells at 56 h after irradiation with 10 Gy) consisted of mitotic, and around 97% of G2 cells (inserts in Fig. 3a). A similar portion of in the region of 2% mitotic and 98% G2 cells was found at that time in the G2/M fraction of H460 cells.
Figure 3.
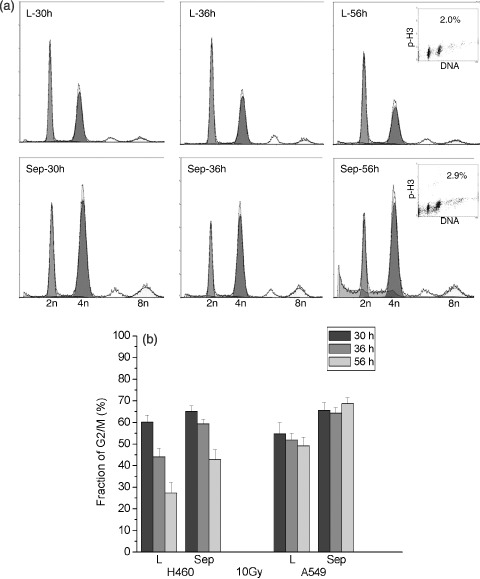
Influence of separase depletion on cell cycle progression after ionizing irradiation. (a) Flow cytometric data showing cell cycle distribution in siRNA‐transfected A549 cells at 30, 36 and 56 h after irradiation with 10 Gy. Cells in G1, G2/M and hyperploid phases are represented by 2n, 4n and 8n, respectively. (b) Summary of G2/M distribution from four to seven independent experiments after transfection and irradiation of A549 and H460 cells. L, oligofectamin control; Sep, specific siRNA‐targeting separase.
The study further showed that significantly less irradiated cells escaped from G2 arrest after separase depletion. Decrease in G2/M fraction from 30 h to 56 h after irradiation measured by flow cytometry was –22.3 ± 4.1% and +3.0 ± 0.4% for separase‐depleted H460 and A549 cells, respectively, as well as –32.9 ± 4.9% and –5.6 ± 1.4% for control‐transfected cells. These data show that depletion of separase delays progression of irradiated cells through the G2 phase.
Mitotic fraction was also measured after activation of the G2 checkpoint by irradiation. Cells were sham irradiated or irradiated at 4 Gy 24 h after transfection and mitotic fraction was measured by accumulation of cells in mitosis following a 24‐h nocodazole treatment that arrests them. Lower irradiation dose of 4 Gy was used in order to provide an opportunity for a considerable portion of the cells to escape from radiation‐induced G2 arrest into mitosis. Irradiation delayed mitotic entry, as measured by H3 expression, from 26.1% to 12.1% in H460 and from 13.6% to 5.4% in A549. Depletion of separase further reduced it by a factor of two (P < 0.01, sign test for 6 repeated pairs; Fig. 2). In summary, these data show that depletion of separase lead to decrease in mitotic fraction that resulted from accumulation of cells in the G2 phase of the cell cycle.
DNA double‐strand break repair, late apoptotic response and clonogenic survival
It would be expected that increased G2 arrest could also affect DNA repair and cell survival after irradiation. In order to evaluate the effect of separase depletion on repair of DNA double‐strand breaks, the number of γ‐H2Ax foci at 1 h and 4 h was determined in both cell lines. Transfection with siRNA had no significant effect on initial γ‐H2AX foci with 35.8 ± 6.7 and 36.5 ± 7.5 foci/cell at 1 h after irradiation with 4 Gy without/with siRNA in H460 and A549 cells, respectively. However, there was a significant decrease in number of γ‐H2AX foci remaining at 4 h after irradiation in H460 cells (three repeated measurements, P < 0.05, t‐test). Respective values were 27.9 ± 2.7 and 19.7 ± 2.1 foci/cell remaining without and with siRNA. A549 cells, which showed much lower escape from radiation‐induced G2 arrest, even without separase depletion, showed only a trend to decreased number of γ‐H2AX foci at 4 h after irradiation. In that cell line, a number of γ‐H2AX foci at 1 h were 39.1 ± 3.1 and 43.9 ± 3.6 without and with separase depletion after irradiation of 4 Gy. The respective number of foci at 4 h were 27.2 ± 4.0 and 23.7 ± 4.3.
In addition, consequences of separase depletion on late apoptosis and clonogenic cell death were determined. Previously, we have shown that there is a distinct temporal relationship between the onset of late apoptosis and exit from radiation‐induced G2‐phase arrest. At 48 h after irradiation with 20 Gy, the apoptotic fraction was around 25% and 2.5% for H460 and A549 cells, respectively, as fewer A549 cells passed the G2 block. Depletion of separase in the H460 cell line resulted in a significant decrease in apoptosis, from 25.5 ± 1.8% or 22.5 ± 2.2% in oligofectamin or scrambled siRNA treated cells to 17.4 ± 2.3% for separase siRNA‐treated cells (P < 0.01, sign test for six repeated pairs). In addition, down‐regulation of separase slightly increased clonogenic survival after irradiation with 4 Gy from 14.0 ± 1.9% to 24.3 ± 3.2% in H460 cells and from 29.3 ± 3.6% to 38.5 ± 2.7% in A549 cells, respectively (P < 0.01, sign test for eight repeated pairs).
DISCUSSION
The spindle assembly checkpoint guards proper organization of the mitotic spindle and progression through mitosis. Separase is a key protein that regulates the spindle checkpoint and is responsible for separation of chromosomes, by proteolytic cleavage of the cohesion protein. Cleavage, as a regulatory mechanism, is not merely restricted to the target proteins of separase, but rather the activity of separase itself is regulated by a timely and co‐ordinated cleavage of separase. Analysis of human non‐cleavable separase isoforms has demonstrated no noticeable effect on its proteolytic activity (Waizenegger et al. 2002; Zou et al. 2002), but striking defects in mitotic entry, spindle assembly and metaphase chromosome alignment have been found in cell lines expressing non‐cleavable separase protein (Papi et al. 2005).
According to these effects of separase on mitotic entry and progression in eukaryotic cells and high mitotic activity of tumour cells, separase itself may be an attractive target for tumour therapy, especially in combination with radiotherapy. However, influence of separase depletion in combination with ionizing radiation has not yet been specified with respect to DNA damage response, cell cycle progression and clonogenic survival. Here, we show that down‐regulation of separase expression reduced mitotic entry by increasing G2 arrest of human NSCLC cell lines, especially after irradiation. This effect of separase depletion on G2 progression can be ascribed either to loss of mitosis‐promoting factor activity or to activation of distinct G2 checkpoint pathways, either by microtubule dynamics (Rieder & Cole 2000; Scolnick & Halazonetis 2000) or DNA damage (Xu et al. 2002; Wang et al. 2003).
It has been discussed that delay of mitotic entry after separase depletion depends on activity of the mitosis inhibiting factor Wee1 and/or on failure in chromosome condensation in late G2/early mitosis (Giménez‐Abián et al. 2005). Wee1 is directly destroyed by cleaved separase products, thereby promoting timely entry into mitosis (Papi et al. 2005). In addition, it has been shown that overriding the G2 checkpoint with caffeine, in cells with dysfunctional separase, did not restore normal mitotic response (Giménez‐Abián et al. 2005; Papi et al. 2005). A key step in regulating progression of cells through G2 into M is activation of cdc2–cyclin B1 complexes (Jin et al. 1998). It was shown that DNA damage prevents activation of cdc2–cyclin B1 complexes by a p53‐mediated pathway (Jin et al. 1998). Although p53‐dependent pathways are dispensable for initiation of G2 arrest, they play an essential role in sustaining G2 arrest after DNA damage (Bunz et al. 1998; Passalaris et al. 1999; Stuschke et al. 2002). In addition, the appearance of radiation‐induced late apoptosis was timely, related to the dissolution of the G2‐phase block (Bracey et al. 1997; Stuschke et al. 2002; Sak et al. 2003).
With respect to DNA damage response pathways, it has been proposed that securin, the inhibitor protein of separase, may connect DNA damage response to sister chromatid separation, delaying the onset of mitosis while DNA repair occurs (Romero et al. 2001, 2004). The protein coded for by the human securin gene (also called human pituitary tumour transforming gene), forms a complex with Ku70 and is subsequently phosphorylated by DNA‐PK in response to DNA double‐strand breaks. The phosphorylated form of securin then contributes to block sister chromatid separation by binding to separase (Romero et al. 2001). Securin protein also activates expression of p21WAF1/CIP1 (Mu et al. 2003) and interacts with p53 (Bernal et al. 2002; Zhou et al. 2003; Hamid & Kakar 2004). From these interaction modes of securin, reduction in expression of separase by siRNA protein would change the balance of securin/separase interactions in favour of free securin and facilitate its interaction with p53, p21WAF1/CIP1 and DNA‐PK.
In summary, we have demonstrated that depletion of separase with siRNA inhibits cell cycle progression of sham irradiated and irradiated human lung carcinoma cells (which have functional p53) by increasing G2‐phase arrest and consequently inhibiting mitotic entry. Increase in the irradiation‐induced G2 checkpoint response after separase depletion had consequences on the fraction of γ‐H2AX foci remaining at 4‐h, mitotic‐linked cell death and can also modify clonogenic survival.
ACKNOWLEDGEMENT
This study was supported by grants from the Deutsche Krebshilfe, project number 10‐2222.
REFERENCES
- Bernal JA, Luna R, Espina A, Lazaro I, Ramos‐Morales F, Romero F, Arias C, Silva A, Tortolero M, Pintor‐Toro JA (2002) Human securin interacts with p53 and modulates p53‐mediated transcriptional activity and apoptosis. Nat. Genet. 32, 222–224. [DOI] [PubMed] [Google Scholar]
- Bracey TS, Williams AC, Paraskeva C (1997) Inhibition of radiation induced G2 delay potentiates cell death by apoptosis and/or induction of giant cells in colorectal tumor cells with disrupted p53 function. Clin. Cancer Res. 3, 1371–1381. [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldmann T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Burum‐Auensen E, Deangelis PM, Schjølberg AR, Røislien J, Mjåland O, Clausen OPF (2007) Reduced levels of the spindle checkpoint protein BUB1B is associated with aneuploidy in colorectal cancers. Cell Prolif. 40, 595–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303. [DOI] [PubMed] [Google Scholar]
- Dixon WJ, Mood AM (1946) The statistical sign test. J. Am. Stat. Assoc. 41, 557–566. [DOI] [PubMed] [Google Scholar]
- Giménez‐Abián J, Diaz‐Martinez L, Waizenegger IC, Giménez‐Martin G, Clarke DJ (2005) Separase is required at multiple pre‐anaphase cell cycle stages in human cells. Cell Cycle 4, 1576–1584. [DOI] [PubMed] [Google Scholar]
- Goldstone S, Pavey S, Forrest A, Sinnamon J, Gabrielli B (2001) Cdc25‐dependent activation of cyclin A/cdk2 is blocked in G2 phase arrested cells independently of ATM/ATR. Oncogene 20, 921–932. [DOI] [PubMed] [Google Scholar]
- Hamid T, Kakar SS (2004) PTTG/securin activates expression of p53 and modulates its function. Mol. Cancer 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornig NC, Knowles PP, McDonald NQ, Uhlmann F (2002) The dual mechanism of separase regulation by securin. Curr. Biol. 12, 973–982. [DOI] [PubMed] [Google Scholar]
- Jha MN, Bamburg JR, Bernstein BW, Bedford JS (2002) Caffeine eliminates gamma‐ray‐induced G2‐phase delay in human tumor cells but not in normal cells. Radiat. Res. 157, 26–31. [DOI] [PubMed] [Google Scholar]
- Jin P, Hardy S, Morgan DO (1998) Nuclear localisation of cyclin B1 controls mitotic entry after DNA damage. J. Cell Biol. 141, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov A, Cole RW, Rieder CL (2002) DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle‐assembly checkpoint. Curr. Biol. 12, 1797–1806. [DOI] [PubMed] [Google Scholar]
- Mu JM, Oba K, Yanase T, Ito T, Ashida K, Goto K, Morinaga H, Ikuyama S, Takayanagi R, Nawata H (2003) Human pituitary tumor transforming gene (hPTTG) inhibits human lung cancer A549 cell growth through activation of p21(WAF1/CIP1). Endocr. J. 50, 771–781. [DOI] [PubMed] [Google Scholar]
- Papi M, Berdougo E, Randall CL, Ganguly S, Jallepalli PV (2005) Multiple roles for separase auto‐cleavage during the G2/M transition. Nat. Cell Biol. 7, 930–932. [DOI] [PubMed] [Google Scholar]
- Passalaris TM, Benanti JA, Gewin L, Kiyono T, Galloway DA (1999) The G(2) checkpoint is maintained by redundant pathways. Mol. Cell. Biol. 19, 5872–5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Cole R (2000) Microtubule disassembly delays the G2‐M transition in vertebrates. Curr. Biol. 10, 1067–1070. [DOI] [PubMed] [Google Scholar]
- Romero F, Gil‐Bernabe AM, Saez C, Japon MA, Pintor‐Toro JA, Tortolero M (2004) Securin is a target of the UV response pathway in mammalian cells. Mol. Cell. Biol. 24, 2720–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F, Multon MC, Ramos‐Morales F, Dominquez A, Bernal JA, Pintor‐Toro JA, Tortolero M (2001) Human securin, hPTTG, is associated with Ku heterodimer, the regulatory subunit of the DNA‐dependent protein kinase. Nucleic Acids Res. 29, 1300–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sak A, Stueben G, Groneberg M, Böcker W, Stuschke M (2005) Targeting Rad51‐dependent homologous recombination: implications for the radiosensitivity of human lung cancer cell lines. Br. J. Cancer 92, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sak A, Stuschke M, Wurm R, Schroeder G, Sinn B, Wolf G, Budach V (2002) Selective inactivation of DNA‐dependent protein kinase with antisense oligonucleotides: consequences for the rejoining of radiation‐induced DNA double‐strand breaks and radiosensitivity of human cancer cell lines. Cancer Res. 62, 6621–6624. [PubMed] [Google Scholar]
- Sak A, Wurm R, Elo B, Grehl S, Pottgen C, Stuben G, Sinn B, Wolf G, Budach V, Stuschke M (2003) Increased radiation‐induced apoptosis and altered cell cycle progression of human lung cancer cell lines by antisense oligodeoxynucleotides targeting p53 and p21 (WAF1/CIP1). Cancer Gene Ther. 10, 926–934. [DOI] [PubMed] [Google Scholar]
- Scolnick DM, Halazonetis TD (2000) Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature 406, 430–435. [DOI] [PubMed] [Google Scholar]
- Stapper NJ, Stuschke M, Sak A, Stuben G (1995) Radiation‐induced apoptosis in human sarcoma and glioma cell lines. Int. J. Cancer 62, 58–62. [DOI] [PubMed] [Google Scholar]
- Stemmann O, Zou H, Gerber SA, Gygi SP, Kirchner MW (2001) Dual inhibition of sister chromatid separation at metaphase. Cell 107, 715–726. [DOI] [PubMed] [Google Scholar]
- Stuschke M, Sak A, Wurm R, Sinn B, Wolf G, Stuben G, Budach V (2002) Radiation‐induced apoptosis in human non‐small‐cell lung cancer cell lines is secondary to cell‐cycle progression beyond the G2‐phase checkpoint. Int. J. Radiat. Biol. 78, 807–819. [DOI] [PubMed] [Google Scholar]
- Waizenegger I, Giménez‐Abián JF, Wernic D, Peters M (2002) Regulation of human separase by securin binding and autocleavage. Curr. Biol. 12, 1368–1378. [DOI] [PubMed] [Google Scholar]
- Wang X, Khadpe J, Hu B, Iliakis G, Wang Y (2003) An overactivated ATR/CHK1 pathway is responsible for the prolonged G2 accumulation in irradiated AT cells. J. Biol. Chem. 278, 30869–30874. [DOI] [PubMed] [Google Scholar]
- Xu B, Kim ST, Lim DS, Kastan MB (2002) Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol. Cell. Biol. 22, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagida M (2000) Cell cycle mechanisms of sister chromatid separation; roles of Cut1/separin and Cut2/securin. Genes Cells 5, 1–8. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Mehta KR, Choi AP, Scolavino S, Zhang X (2003) DNA damage‐induced inhibition of securin expression is mediated by p53. J. Biol. Chem. 278, 462–470. [DOI] [PubMed] [Google Scholar]
- Zou H, Stemman O, Anderson JS, Mann M, Kirschner MW (2002) Anaphase specific auto‐cleavage of separase. FEBS Lett. 528, 246–250. [DOI] [PubMed] [Google Scholar]