

Abstract
With advances in technology and bioinformatics we are now positioned to view and manage cancer through an evolutionary lens. This perspective is critical as our appreciation for the role of tumor heterogeneity, tumor immune compartment, and tumor micro-environment on cancer pathogenesis and evolution grows. Here, we explore recent knowledge on the evolutionary basis of cancer pathogenesis and progression, viewing tumors as multi-lineage, multi-component organisms whose growth is regulated by sub-component fitness relationships. We propose reconsidering some current tenets of the cancer management paradigm in order to take better advantage of crucial fitness relationships to improve cancer patient outcomes.
Introduction
Genetic, epigenetic, and non-Darwinian adaptive evolutionary processes act in concert to shape biological landscapes and phenotypes across both physiologic and pathological disease states, including cancers. At their most basic level, cancer cells are normal cells that have acquired somatic genetic alterations and epigenetic changes and no longer follow their physiologic development plan. Acquisition of certain alterations initiates an evolutionary process that results in cellular adaptation and malignancy, the evolutionary trajectories of which are shaped by multiple forces that fuel cancer initiation and progression. These aberrant cells can diverge from the destined program by a multitude of mechanisms, including mutations in the DNA during replication, errors in the DNA replication, repair machinery, and exposure to mutagens. Cancer cells manifest and exploit canonical hallmarks of malignancy to persist (1,2). An additional dimension of complexity is overlaid on tumor cell biology when the contributions from an evolving tumor immune compartment and tumor microenvironment are considered. Micro-environmental changes in both immune and stromal compartments, epigenetic influences, and changes in the signaling patterns within and between cells impact tumor cell survival (Figure 1). The state(s) and role(s) of these compartments evolve during treatment and are frequently intertwined (Figure 2).
Figure 1. Tumor cell influences and evolution.
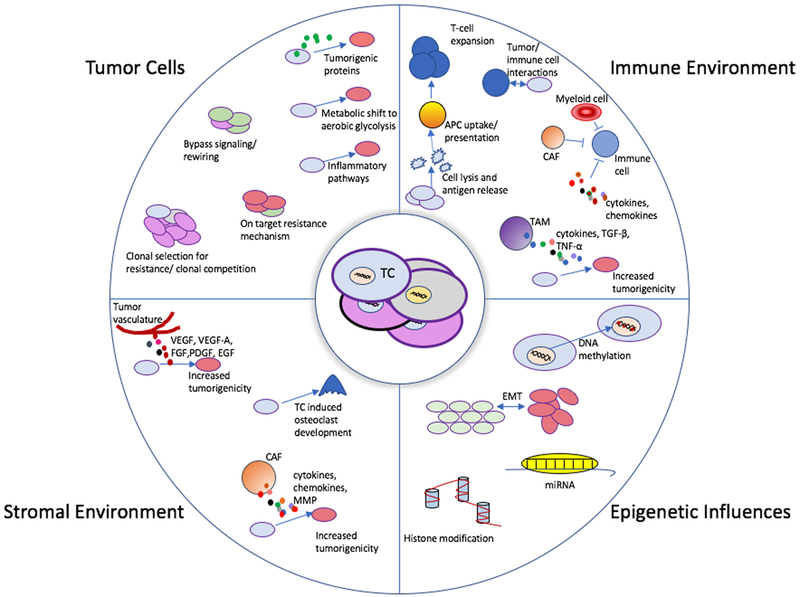
Depiction of the evolutionary paths of tumor cells. The center portion of this image depicts a group of heterogeneous tumor cells. There are four predominant, and often overlapping, categories of evolutionary influence that can impact tumor cells during cancer progression and treatment that impact response to therapies. Each category depicts several individual mechanisms of tumor promotion and evolution. These mechanisms are not mutually exclusive and can occur concurrently.
Abbreviations: APC- antigen present cell, CAF- Cancer associated Fibroblast, EGF – Endothelial growth factor, EMT – epithelial to mesenchymal transition, FGF – Fibroblast growth factor, miRNA- micro-RNA, MMP- Matrix metalloproteinases, PDGF – platelet derived growth factor, TAM – Tumor associated macrophage, TC- tumor cell, TGF- β – transforming growth factor beta, TNF- α – tumor necrosis factor alpha, VEGF/ VEGF-A - vascular endothelial growth factor (A),
Figure 2. Depiction of tumor evolution.
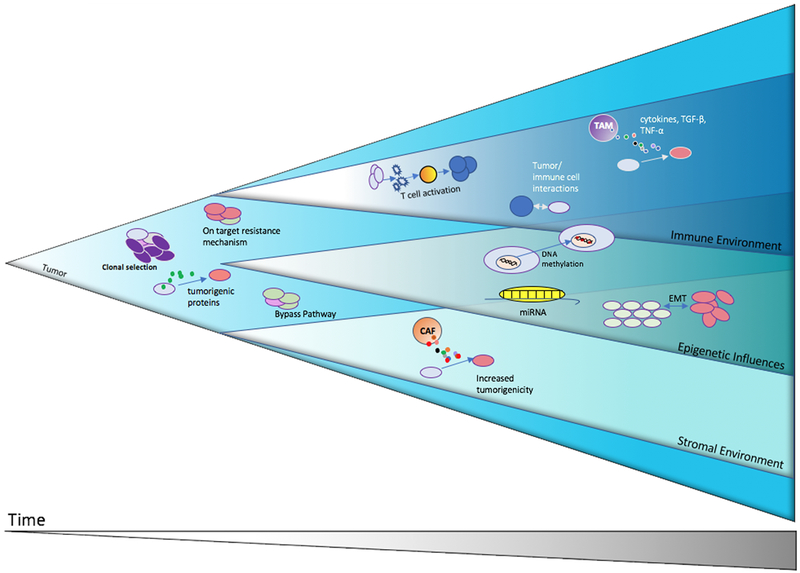
Shown is a theoretical illustration of tumor growth and evolution demonstrating how some of the different modes of tumor promotion and evolution in response to therapy may occur and evolve concurrently.
Abbreviations: CAF- Cancer associated Fibroblast, EMT – epithelial to mesenchymal transition, miRNA- micro-RNA, TAM – Tumor associated macrophage, TGF- β – transforming growth factor beta, TNF- α – tumor necrosis factor alpha
The evolution of cancer cells is critical for their survival in the complex and competitive niche environments in which they establish themselves either as primary tumors or metastases. It has become clear that there is significant heterogeneity within and between tumor deposits (3). This realization has resulted in a new understanding of the importance of the clonal and subclonal changes and the tumor micro-environment that can drive tumorigenesis and modulate responses to therapy. Additionally, non-genetic cellular phenotypes are influenced by signaling pathway activation and dynamic adaptation (via post-translational regulation) and impact cancer evolution and drug resistance (4).
Herein, we review the basic tenants of cancer evolution as well as our understanding of how the complex relationship between the tumor microenvironment and tumor and native cells evolves. Further, we explore different models and theories for engineering this multi-lineage evolutionary process in a controlled and pro-active manner to create opportunities for improved cancer management and treatment.
Characterizing the Genetic Basis and Heterogeneity in Cancer
Early efforts to characterize somatic mutations in human cancer genomes were limited by the available technology and analytics. Protein kinases were a natural focus given the efficacy of anticancer treatments aimed at protein kinases. Studies evaluating somatic mutation burden in the kinomes of colon cancer (5), breast cancer (6), gliomas (7), and testicular germ cell cancer (8), and multi- or pan-cancer analyses, demonstrated that the majority of somatic mutations in cancer are “passenger” mutations that are not active in driving the cancer (9).
As technology advanced, larger studies yielded additional candidate genes involved in cancer development, and more therapeutic targets, and pushed the evolution of bioinformatic approaches to counter the challenges of false discovery (10). These studies illustrate that oncogenic alterations are often differentially represented in distinct cancer types (11). A theme that emerged from a multitude of cancer genome sequencing efforts is that while somatic mutation burden and specific oncogenic driver genes are often inconsistently present across tumor types, common pathways are dysregulated. Thus, in cancer certain pathway traits are selected for from the genetically-rich substrate of the human genome.
In breast and colon cancer, Wood and colleagues comprehensively characterized the genomic landscape of 22 breast and colon cancers and illustrated the magnitude of the genomic landscape, highlighting the common features between cancers and that there are a finite number of pathways that become dysregulated during tumorigenesis (12). This work provided additional examples illustrating that cancer is a disease of pathway dysregulation with multiple genetic interactions converging on the dysregulation of a set of core pathways (13). In non-small cell lung cancer (NSCLC), Imiulinski et al. sequenced the whole genomes and/or exomes of 183 lung adenocarcinomas and matched normal adjacent tissue pairs (14). They reported a mean somatic mutation rate of 12 Mutations/Megabase (Mut/Mb). They re-verified 19 established oncogenic driver genes, with overlapping pathways, and identified an additional 6 potential oncogenes of new interest.
Larger, systematic mutational characterization followed in multiple cancer subtypes through the efforts of the Cancer Genome Atlas Research Network and other groups and have resulted in characterization of lung adenocarcinoma, small cell lung cancer, head and neck cancer, multiple myeloma, CLL, NHL, ovarian, colon cancer, and squamous cell lung cancer and glioblastoma multiforme (10,15–23). Through these efforts, putative cancer genes and their requisite pathways were identified within and between multiple malignancies and the challenge of tumor genomic heterogeneity was magnified. Multiple co-existing genetic alterations contributing to tumor evolution were often present in individual tumors and patients (10,15–26).
In landmark studies, two research teams characterized the mutational spectrum and heterogeneity within and between cancers. Published contemporaneously, these datasets demonstrate the complexity and depth of cancer somatic mutations. Lawrence MS, Stojanow P et al. evaluated the heterogeneity in mutation rates in a dataset of 3,083 tumor/normal pairs across 27 tumor types (27). This work identified two primary contributors: first, the importance of gene expression level; the mutation rate is correlated to gene expression level and replication time due to transcription-coupled DNA repair processes and second, DNA regions that are replicated later in the cell cycle have higher mutation rates. In parallel work, Alexandrov LB et al. clarified the challenging and sometimes divergent results of a multitude of prior studies by exploring mutational frequency across different cancer subtypes (28). They identified and classified nearly 5 million somatic mutations by comparing tumor versus normal tissue in 7,042 cancers. They determined that there was a high variability in the somatic mutation burden, ranging from 0.001 Mut/Mb to >400 Mut/Mb and demonstrated that cancers related to chronic mutagenic exposures exhibit the highest prevalence of somatic mutations. Further, they identified 21 distinct mutational signatures based on DNA base substitution classification and their application to the 30 cancer subtypes. Interestingly, they also identified localized regions with hypermutation, or kataegis. It is thought that a subfamily of the APOBEC (apolipo-protein B mRNA editing enzyme, catalytic polypeptide like) cytidine deaminases may be a causative factor in genomic regions with kataegis due to the similarity in the mutational signature (29). In breast cancer, kataegis signatures correlate with favorable prognosis and in lung cancer the APOBEC mutational signature, appears to underlie tumor subclonal diversification and be a predictive marker of response to immunotherapy (26,29,30).
Longitudinal and Evolutionary Views
To further investigate the interplay of pathway alterations and heterogeneity of tumors, several groups have developed large scale data monitoring projects to characterize and track intratumoral heterogeneity during treatment. Studies addressing this critical evolutionary question include breast (31,32), pancreatic (33,34), kidney (3), colorectal (35,36), prostate (37), lung cancers (38), urothelial cancers (39,40) and CLL (41). In metastatic renal cell cancer, Gerlinger et al. evaluated intra-tumoral heterogeneity in their pioneering study by analyzing tumor tissue from primary and metastatic disease sites within the same patient. This study provided a new appreciation for the complexity facing targeted cancer therapy, given that 63–69% of mutations identified were not found in all individually assayed samples from the same patient. The heterogeneity was present in all levels of analysis; genome, transcriptome and proteome (3).
In colorectal cancer, multiple studies have characterized heterogeneity within and between primary and metastatic disease sites as well as the evolution of advanced-stage cancers (42–47). These studies were critically important in demonstrating that there is not necessarily concordance between the primary tumor and metastatic site, even when sampled synchronously, and that in colorectal cancer clonal evolution can be tracked, as in other disease types (44,45,48,49). Also in colorectal cancer, cell-free DNA (cfDNA) was utilized to monitor the molecular evolution of acquired resistance and to overcome the challenge of tumor metastasis heterogeneity given that it is a sample of tumor DNA that is representative of the larger disease burden. Diaz et al. utilized cfDNA to demonstrate that KRAS wildtype patients treated with panitumumab can selectively evolve a resistant subclonal population and further that these resistance mutations are present before treatment. These findings highlight the importance of understanding baseline tumor heterogeneity and its dynamic evolution (36). Landmark work in colorectal cancer showed the utility of monitoring the dynamic changes and responses of intermittent drug schedules, as well as the value of assessing cfDNA to monitor treatment response and the emergence and regression of resistant subclones (50,51). Russo et al. demonstrated that utilization of molecular response data can inform treatment selection as new resistance-driving clones arise (51).
In high-grade serous ovarian cancer, Bashashat et. al profiled 31 spatially and temporally parsed ovarian cancer specimens obtained from 6 patients. Only a portion (51.5%, range 10.2–91.4%) of the mutations were present in each sample from a given patient (52), reinforcing the presence of extensive inter-tumor genomic heterogeneity within individual patients.
In lung cancer, Jamal-Hanjani et al reported on several aspects of the first 100 of an anticipated 842 early stage patients in the Tracking Cancer Evolution through Therapy (TRACERx) program (53,54). They found that a median of 30% of somatic mutations are subclonal and 48% of copy number alterations are subclonal, indicating that mutational processes are ongoing during tumor development. They further evaluated the timeline of genetic alterations and demonstrated the impact of chromosomal instability, which was the primary driver of parallel evolution (55). In a followup analysis, they explored allele specific human leukocyte antigen (HLA) loss and immune escape in the same cohort of patients and showed its presence in 40% of early-stage NSCLCs. The work also demonstrated that this is an evolutionarily-selected process associated with increased non-synonymous mutations and higher neo-antigen burden. Analysis of patients with stage IV disease demonstrated that this process occurs preferentially at metastatic sites (56). These findings link tumor genomic alterations and heterogeneity to immune escape and are parallel to findings in melanoma in which loss of heterozygosity in major histocompatibility complex class I antigen presentation components may contribute to resistance to certain immunotherapies (57).
Highlighting the genetic complexity present within individual tumors and patients, Blakely et. al evaluated 1,122 EGFR positive cfDNA samples from patients at different time-points during systemic therapy They demonstrated in a subset of patients with repeated assays during treatment that the tumor genomic complexity increased during treatment with an increasing number of somatic mutations (38). This observation suggests that in this disease treatment with a targeted therapy does not result in a purified genetic landscape, but instead may increase genetic diversity that contributes to ongoing tumor evolution and adaptation.
In renal cell carcinoma three topical analyses were published simultaneously (58–60). Turajlic et al evaluated 1,206 tumor regions from 101 patients and 106 tumors (all stages). Using a combination of driver panel sequencing, whole exome sequencing and whole genome sequencing, the group identified 740 somatic mutations. Correlation of non-synonymous mutations with known driver mutations identified a median of 3 driver mutations per tumor. They also found that von Hippel-Lindau related alterations were the only consistent clonal event, present in 95 of 106 tumors. Subclonal alterations (copy number alterations and mutations) were associated with known prognostic markers and patient outcome (59). The group analyzed 575 primary and 335 metastatic biopsies in 100 patients with metastatic renal cancer to characterize heterogeneity in metastatic versus primary (localized) disease. Their compelling findings demonstrated that the overall number of non-synonymous mutations and copy number alterations were more prevalent in the primary tumor than the metastasis. Further, they demonstrated that most genomic diversity was present in the primary tumor while only 5.4% of the genetic alterations arose de novo in the metastatic tumor site, reflecting an evolutionary bottleneck. Interestingly, as a parallel to the TRACERx lung findings, TRACERx renal also observed an increased imbalance at the HLA locus in selected clones (54,58).
The TRACERx prostate and melanoma groups have identified equally compelling data regarding intra-tumoral heterogeneity in prostate cancer and an evaluation of the tumor microenvironment in melanoma (61,62). A balanced focus on both the tumor and the microenvironment is critical given our increasing appreciation of the importance in the tumor microenvironment in modulating response to treatment and selective pressure from the microenvironment impacting tumor evolution.
A unifying theme across these studies is the presence of substantial diversity in populations that increases during the selective pressure of treatment and that this heterogeneity can be monitored over time. With targeted therapies, molecular heterogeneity dictates treatment response; however, tumor cells do not exist in isolation and the next frontier in understanding tumor biology and identifying treatment strategies requires a more comprehensive understanding of not only how tumor cells evolve with different treatments but also how the entire tumor ecosystem interacts and shifts in composition and cell state(s) during therapy.
Insights from Single Cell Organisms and Hematologic Malignancies
Many parallels to these findings have been identified in single cell organisms in which evolutionary studies are more feasible. For example, in yeast, steady state cultures of different yeast strains grown in chemostats can be monitored for hundreds of generations to characterize and manipulate evolution and apply selective pressure. Hope et. al demonstrated this by characterizing the frequency of different genetic pathways leading to an aggregation phenotype called flocculation (63). In these experiments, they identified the specific frequencies of the most common genetic changes leading to aggregation and determined that there was a favored evolutionary path to this trait, as opposed to several equally evolutionarily favored routes. Further, they characterized the most favorable adaptive route in different genetic backgrounds and the consequences of blocking it, which ultimately delayed, but did not prevent the development of the flocculation phenotype and revealed alternative pathways for phenotype development (63).
These results parallel the development of the EGFR T790M resistance mutation in EGFR mutated NSCLC when treated with a first or second generation TKI (64). There are several known mechanisms of resistance to first and second generation EGFR TKIs, but the most common mechanism is via the secondary mutation, T790M. When EGFR signaling is effectively blocked, drug resistance is attenuated, but the cancers evolve and find other routes to become resistant. This occurs regardless of oncogenic alteration or the targeted therapy. Separate from secondary mutations, bypass pathways can also become activated, such as various RTKs and MAPK and PI3K pathway components, that permit tumor progression despite effective blockade of the primary oncogenic target. On-target and off-target resistance mechanisms to targeted therapies in NSCLC have been reviewed in detail elsewhere (65). In colon cancer bypass pathways via biochemical requiring have also been identified. In BRAF V600E mutant melanoma, resistance to BRAF/MEK inhibitor treatment can occur through EGFR expression that is acquired through treatment. A mechanism of biochemical rewiring and resistance is via activation of the EGFR pathway by suppression of SOX10 and activation of TGF-β (66–68).
In bacteria, the evolutionary pressure of antimicrobials results in resistance via numerous different mechanisms, both intrinsic and extrinsic factors have been well characterized. Furthermore, the process of evolution in bacteria has been manipulated to evolve collateral sensitivity in which bacterial strains are designed to become more sensitive to subsequent treatments, based on prior treatments. Imamovic and Sommer demonstrated this in Escherichia coli with drug cycling protocols that avoid development of resistance using sensitivity profiles (69). Yen and Papin built on this work by investigating whether prior treatment constrains or potentiates response to subsequent treatment. They demonstrated that they could impact the evolution of Pseudomonas aeruginosa by demonstrating drug-order specific evolution and mutations. They not only observed different resistance levels of specific drugs based on prior treatment exposures but also identified genetic mutations and epigenetic processes underlying these responses (70). These findings have strong parallels to cancer biology and the idea of combinatorial and serial therapy designed to evolve tumor cells with collateral sensitivity. Furthermore, they demonstrate that there is ongoing inter-clone competition that evolves based on exposure to treatment and even environmental factors. This has been demonstrated in malignancy as well; in multiple myeloma, Keats et al. illustrated this point in a patient with two competing sub-clones that alternated dominance multiple times based on treatment until one clone underwent a dramatic linear evolution (71).
Another parallel between cancer biology and single cell organisms is with quorum sensing, in which bacteria use small secreted molecules (autoinducers) to facilitate interspecies communication and impact virulence factors, biofilm formation, and alter the microenvironment to interfere with other species by altering native flora and thus impact susceptibility to pathogens (72,73). Autoinducers and their receptors are important targets for disrupting pathogenic processes and an active area of research (74).The corollary in cancer is the secretion of factors by therapeutically stressed tumor cells or the micro-environment that can support tumor cell survival (75–77).
Though more complex than single cell organisms, hematologic malignancies contain fewer somatic mutations than most solid tumors (28). It is interesting to consider whether this translates to more robust responses to targeted therapies. In the solid tumors with the relevant genetic alteration, the complete response rate with targeted therapy (e.g. dabrafenib plus trametinib against mutant BRAF, osimertinib against mutant EGFR, and alectinib against ALK gene rearrangements) is low (<10%) (78–80). In hematologic tumors, the determination of response is measured differently; in CML, complete molecular response (CMR) and major molecular response (MMR) are similar counterparts to complete and partial response in solid tumors. The maximal rate of CMR is 20%, while the maximal MMR is 75% for nilotinib and the maximal CMR is 7% and 87% MMR for dasatinib (81,82). In patients with relapsed/refractory myeloid malignancies with mutant IDH2 treated with the IDH2 inhibitor enasidenib, the complete response rate was 20% while the rate of incomplete response was 18%(83). Further studies of AML patients treated with enasidenib demonstrated on a single cell level that resistance occurred through multiple mechanisms involving epigenetic or genetic mechanisms that promoted the differentiation arrest that was reverted on initial treatment (84).
There are several parallels between solid tumors and hematologic malignancies and a trend toward more complete responses in patients with hematologic malignancies treated with targeted therapies. The mechanisms of resistance are similar in that both off and on target mechanisms of resistance develop in clonal populations (85). Despite the appealing concept that clonal evolution in hematologic malignancies should follow a linear evolutionary trajectory given that the tumor cells are not compartmentalized, as in solid tumors, they actually have been found to frequently follow a branching evolutionary path. This implies that complex clonal interactions are ongoing and likely include tumor, immune, epigenetic and micro-environmental influences (85). The challenge is how to take advantage of this complexity and whether it is possible to stimulate an intra-tumoral fitness competition. For instance, different tumor cell subclones present with a tumor might act as cooperators or antagonists, with distinct therapeutic implications in each case (Figure 2). An interesting question for future study is whether the character or abundance of different subclonal tumor cell populations can be therapeutically controlled to engineer overall constraints on tumor progression or drug resistance.
Immunotherapy and treatment response evolution
Immunotherapy (immune checkpoint blockade) has revolutionized the treatment of many cancers. Unlike targeted therapies in which tumor cells develop resistance by acquiring mutations and/or activation of bypass signaling pathways, in immunotherapy, the tumor clonal landscape is changed due to complex interactions involving multiple factors that are impacted by the clonal composition and heterogeneity of the tumor (86,87).
The anti-tumor mechanism of action of immunotherapy is indirect compared to chemotherapy and targeted therapy. The complexity of the immune compartment and its interactions with different cell types is reviewed in detail elsewhere (87). Due to its integral role, it is important to characterize how immunotherapy impacts the tumor immune compartment, tumor cells and the tumor microenvironment. Preclinically, Li et al demonstrated in pancreatic adenocarcinoma mouse models that tumor cell intrinsic production of the chemokine CXCL1 modulated sensitivity to immunotherapy (88). Clinically, Riaz et al. characterized the genomic changes in the tumors of 68 patients with melanoma after progression of immunotherapy (89). In doing so, they identified a complex network of relationships between the host immune system, the tumor microenvironment and tumor cells. They described an association between mutational burden and response to therapy in treatment naïve patients that was not observed in patients who were previously treated with ipilimumab (an anti-CTLA4 antibody). They also demonstrated that after 4 weeks of nivolumab (an anti-PD-1 antibody) therapy there was a significant decrease in detectable mutation and neoantigen burden in patients with complete or partial tumor responses, but only a modest decrease in patients with stable or progressive disease. Further, they demonstrated that the tumor genetic clonal landscape became more contracted in patients whose tumors responded to treatment. They also evaluated the tumor microenvironment and demonstrated a correlation between T-cell expansion in proportion to neoantigen depletion in patients who were immunotherapy naïve (90). Interestingly, Jerby-Aron et al. recently demonstrated the ability to identify a gene expression signature predictive of immunotherapy treatment response in melanoma and how the immune response can be modulated by CDK4/6 (91). In NSCLC, Anagnostou et al demonstrated that after treatment with immune checkpoint inhibitors mutation-associated neoantigens are eliminated, thus altering the genomic landscape of tumors (92). In addition to neoantigen exposure to assist with immune targeting of tumor cells, immune-mediated tumor cell death requires activation of T-cells. This activation is also partially facilitated by activation of multiple epigenetic mechanisms; combining epigenetically targeted therapies and immunotherapies has the potential for significant clinical impact (86). Indeed, there are numerous ongoing efforts to determine the feasibility and efficacy of this emerging polytherapy strategy (93).
Epigenetic Landscape
Lineage heterogeneity as well as plasticity are important features of many cancers. Plasticity is enabled, in part, by the epigenetic landscape of tumor cells and its malleability during tumor progression or under drug therapy. Epigenetic influences refer to changes in gene expression and cellular function and phenotype without permanent changes in the genome. Many epigenetic mechanisms can impact cellular functions and can promote or inhibit tumor cell growth when dysregulated. Commonly recognized mechanisms include DNA methylation, and histone or chromatin modifications. While numerous studies have addressed somatic genetic heterogeneity of tumor cells, characterization of the heterogeneity of the epigenetic landscape is less well explored. The heterogeneity observed in tumors in response to treatment is a combined reflection of the heterogeneity of the tumor cell DNA, the micro-environment and the epigenetic forces shaping cell states and tumor evolution. Epigenetic influences can provide selective advantages to cancer cells given that epigenetic modifications can be applied and regulated on a more rapid time scale than genomic evolution (94).
These epigenetic mechanisms can result in the persistence of cancer cells during treatment and ultimately limit responses to treatment by facilitating resistance. Evidence of “persister” tumor cell populations has been previously discussed and their transient and reversible nature is a natural fit for the properties of epigenetic regulation. Sharma et al elucidated these drug tolerant persister cells elegantly in a NSCLC cell line and identified potential mechanisms by which to target them via IGF-1 and HDAC inhibitors (95). In BRAF V600E cancers, the ERK pathway is rapidly modulated in response to treatment (96). Unlike in melanoma, the clinical response to BRAF/MEK inhibitor treatment in BRAF V600E mutant colon cancers is relatively limited (97). One mechanism of this resistance is thought to be under epigenetic control via activation of the PI3K/AKT pathway (98).
Epithelial to mesenchymal transition (EMT) is another example of an adaptive, epigenetically influenced cellular process in which epithelial cells undergo a transition and lose polarity and cell-to-cell contacts and become resistant to apoptosis (99,100). In normal cells EMT is a critical component of wound healing and embryonic development. In tumors, EMT is thought to contribute to metastasis and treatment resistance (100). EMT is induced by transcription factors responding to extracellular cues such as TGF-β (100–102). Importantly, EMT is not a binomial state and instead is a spectrum that allows cells to adapt quickly to new environmental cues to maximize survival. In malignancy, EMT is often induced by receptor tyrosine kinase inhibition, resulting in adaptive selection for cells that have transition to EMT (100). In NSCLC, specifically this is a well explored mechanism of resistance to EGFR TKI treatment (103–105).
Beyond EMT, the utility of leveraging epigenetics in the management of cancer is apparent in the case of DNA methylation of the O-6-Methylguanine-DNA Methyltransferase (MGMT) gene in glioblastoma. The normal function of this gene is to repair DNA damage (106). When methylated in the promoter, MGMT gene expression is silenced and the cells have impaired DNA damage repair and are more susceptible to DNA damaging chemotherapy agents such as temozolomide. Patients with a methylated MGMT promoter demonstrated a survival advantage to those without (106). Conversely, DNA methylation of promoter regions in other genes can alter gene expression and drive resistance to chemotherapy, as observed in NSCLC and ovarian cancer (107–110).
Additional mechanisms of epigenetic alterations include histone modifications and micro-RNAs (miRNAs). Histone modifications have become a frequent area of focus and include histone acetylation, deacetylation, and methylation. Several histone deacetylase (HDAC) inhibitors are currently in clinical use in multiple different cancer subtypes with additional clinical trials ongoing (111,112). In NSCLC, EMT is impacted by histone acetylation (101). Several studies have demonstrated that HDAC inhibitors can also augment the antitumor effects of the EGFR TKIs and re-sensitize EGFR TKI resistant cells (113–118). Similar findings have been described in melanoma, in which BRAF inhibitor sensitivity was restored when an HDAC inhibitor was combined with a BRAF inhibitor (119). Furthermore, HDAC inhibitors can increase immune activation by upregulating the expression of MHC class I and II proteins as well as multiple costimulatory molecules (120). In the clinic, the combination of an EGFR TKI and HDAC inhibitor is tolerable; however, efficacy is unclear given the lack of phase II and III clinical trials and HDAC inhibitor (and other epigenetically active drugs)/immunotherapy drug combinations are being tested (93,121–123). Importantly, combinations such as these may need to be deployed in the first line treatment setting instead of after drug resistance fully develops since drug resistance is often multi-factorial in nature.
Micro-RNAs are another type of epigenetic mechanism that is more recently recognized. They are small endogenous non-coding RNA segments that can regulate gene expression, and inhibit or promote cancer by targeting messenger RNAs for activation or de-activation (111,112). Multiple preclinical studies have demonstrated their potential to both monitor therapeutic response and as a therapy target (124). As with other epigenetic influences specific miRNAs can also promote EMT (125,126)
Ultimately incorporating epigenetically-directed treatments could address a major component of tumor plasticity and thereby help to constrain tumor evolution under treatment. These treatments must be studied in different disease settings to understand the best strategy for clinical implementation to blunt this aspect of the plasticity shaping tumor evolution.
Tumor Microenvironment
The tumor microenvironment (TME) consists of the supporting structures and cells that surround and encase cancer cells (127). The TME includes numerous structures such as the vasculature, nervous tissue and extracellular matrix that is comprised of several cell types such as fibroblasts, macrophages, lymphocytes, endothelial cells and the biological materials they generate and secrete. As with epigenetic influences, the TME can inhibit or promote cancer growth. Its composition differs depending on the tissue type and a multitude of other factors. Prior to the identification of targetable oncogenes, a large focus area of cancer biology was the impact of the TME and how it can be manipulated to aid in the treatment of cancer (128). A classic example of this focus is development of angiogenesis inhibitors that alter the tumor microenvironment and modify (inhibit or correct) tumor blood and nutrient supply (129–131). Other examples include efforts at targeting hypoxia in the TME with agents that act upon hypoxia-inducible factor (HIF)-1alpha signaling in different types of cancers, and the approval of proteasome inhibitors which appear to act, in part, through re-programming certain pro-tumorigenic features of the TME in multiple myeloma and mantle cell lymphoma (132).
A critical and abundant component of the TME is fibroblast cells. These cells are compelling in their dual role, with both pro and anti-tumorigenic effects. In their anti-tumor state, native fibroblasts regulate the continuous remodeling of the extracellular matrix and can suppress cancer cell growth by contact inhibition and contact-independent suppression via secretion of soluble factors such as TGF alpha and beta, TNF, and IL6 (133–138). Conversely, cancer associated fibroblasts (CAFs) trans-differentiate from multiple different stromal cell types via several different epigenetic pathways (138). CAFs are often pro-tumorigenic and permit invasion and facilitate stromal dissemination and metastasis of cancer cells as well as produce numerous cytokines, chemokines, matrix metalloproteinases, other soluble factors that promote cancer growth and suppression of the host immune system (138). Many of these activities are known to directly induce resistance to cancer treatments through these and other paracrine mechanisms (139–142). Given their significant role in tumor surveillance and progression and metastasis, efforts are aimed at targeting this aspect of the TME; however, clinical activity to date is less than compelling for the tested drugs (143,144). In the preclinical setting, there is potential for targeting CAF nuclear receptors with small molecules; using this strategy increased sensitivity to cisplatin in tumor xenograft models (145). While other groups demonstrated promising targets such as the NRG1/HER3 pathway, NOX4 and SerpinB2, the clinical benefit is under investigation (145–147).
Tumor-associated macrophages (TAMs) are involved in tumor cell survival and may exhibit prognostic implications given pre-clinical evidence that macrophage-derived growth factors can activate multiple cancer promoting pathways in cancer cells in a paracrine manner within the TME (148). In multiple cancer subtypes, TAMs are involved in the EMT by secretion of multiple factors that promote this cell state transition(149).
With the identification of targetable oncogenes, research on the impact of manipulating the microenvironment in tumors with targetable oncogenes slowed. With the advent of clinically effective immunotherapy and the increased understanding of paracrine, pro-tumor cell signaling events emanating from different cell types within the TME, the importance and impact of the TME on survival, tumor progression, and therapy resistance is a renewed, important area of focus.
Tumor Energy Metabolism
A major current challenge is to identify strategies for integrating our knowledge of targetable oncogenes, tumor heterogeneity, tumor drug resistance and the influence of the TME in this setting. A further layer of complexity that we must incorporate is that each of these factors is dynamic during tumor growth and treatment. We must consider the biology of a tumor as an evolutionary process involving population dynamics and competing, or cooperative subpopulations of cells (including tumor, stromal, and immune cells). Work by Ibrahim-Hasim et al. demonstrated this principle in silico, in vitro, and in vivo and showed that small environmental perturbations of the TME extracellular pH can alter the balance of different cancer cell subtypes. This resulted in guided selection of either more invasive aerobic glycolysis phenotypes, or less invasive angiogenic tumor cells depending on the perturbation (150). This work demonstrated that different cancer models (prostate and breast) utilize identical cellular adaptive strategies. These findings highlight the need to renew investigative focus on the TME and the manipulation of the metabolic pathways of the tumor and TME. An ongoing question is whether these types of environmental controls exist in all types of malignancies and how to integrate them into preclinical experimental systems and clinical diagnostic and therapeutic modalities.
A frequent therapeutic focus of tumor biology is aerobic glycolysis. It has long been appreciated that cancer cells are more reliant on aerobic glycolysis than oxidative phosphorylation, commonly known as the “Warburg effect” (151–154). The challenge is how to leverage this information to treat cancer. There is precedent for the feasibility of targeting metabolism across cancer subtypes and many oncogenic pathways stimulates glycolysis and promotes a more tumorigenic and invasive cellular environment. Mutations in KRAS, PIK3CA, and AKT can activate the AKT-mTOR pathway, which among other activities, promotes glycolysis (155). The tricarboxylic acid (TCA) cycle has also been targeted due to a significant association with diverse malignancies. The enzymes succinate dehydrogenase and fumarate hydratase can dysregulate the epigenomic landscape via alteration of metabolic processes in several cancers, including renal cell carcinoma (156), gastrointestinal stromal tumors (157,158), pheochromocytoma, and paraganglionomas (159,160). Another example, also from the TCA cycle, is the presence of mutations in isocitrate dehydrogenase (IDH) isoforms 1 and 2 which result in accumulation of high levels of the onco-metabolite 2-hydroxylglutarate (161,162). The first IDH2 inhibitor was approved by the US Food and Drug Administration in 2017 for use in IDH-mutated AML based on positive clinical trial findings (83).
Epigenetic mechanisms that modulate the Warburg effect were identified in multiple cancer subtypes. Interestingly, many of these mechanisms are via miRNA function and demonstrate the range of pro- and anti-tumor activities of these small nucleotides sequences. For example, the tumor suppressor miRNA miR-15b-5p is downregulated in osteosarcoma, which results in increased aerobic glycolysis (163). In lung cancer models, miR-31–5p can enhance glycolysis and cell proliferation(164). Conversely, in hepatocellular carcinoma miRNA-129–5p blocks glycolysis by targeting pyruvate dehydrogenase kinase 4 (PDK4), thus slowing carcinogenesis(165).
After the disappointing realization that due to tumor heterogeneity and evolution on treatment, targeting individual signaling pathways such as the PI3K/AKT/mTOR module generally proved suboptimal clinically, many groups are investigating rational combination therapies to improve the depth and duration of response in patients. The integration of metabolic, epigenetic and cellular programs and their heterogeneity and plasticity is a challenge to address, particularly in the context of tumor evolution during therapy (166). The potential to alter current treatment paradigms to a more “organismal-centric” strategy to control these different sources of heterogeneity and adaptability in cancers offers hope for blunting tumor evolution.
Clinical Trial Limitations and Opportunities for Innovation
One of the challenges in identifying rational combination therapies has been the traditional strategy of drug approval. As drugs are developed, the standard practice is to first determine the maximum tolerated dose (MTD), and then to proceed with efficacy assessments utilizing the MTD. The goal is to deliver the maximum amount of drug to kill cancer cells while avoiding toxic side effects. However, as noted by Gallaher et al., this strategy was developed before the full appreciation of molecular tumor heterogeneity and relies on the hypothesis that cells that are resistant to a given treatment are not present in the treatment naive population (167). We now recognize that tumor heterogeneity is a critical component of the development of resistance, demonstrating that one of the basic assumptions used for clinical trial design may be flawed. Multiple studies demonstrated equivalent outcomes in patients who received modified regimens of chemotherapy and targeted therapy with decreased dose intensity (168–170). This is particularly important to re-evaluate when considering combination therapies or switch therapies in which toxicity from combined agents may limit combination therapy options. A more adaptive approach may improve outcomes in certain cancer types. Several groups are modeling adaptive therapies, and in prostate cancer adaptive therapy approaches have demonstrated promising results (171). Clinical trials in multiple cancer subtypes are ongoing (NCT03630120, NCT03543969, NCT03511196, NCT02415621). It is critical to explore different drug treatment strategies that take advantage of our knowledge of multiple sub-populations of cancer cells to pro-actively engineer tumor evolution to a more indolent or drug sensitive state, or to drive the cancer into an evolutionary pause or “dead-end”.
In breast cancer, the benefit of modulating therapy during treatment to take advantage of evolutionary pressure and population dynamic to increase duration of response was demonstrated by Silva et al who described that the challenge of resistance and disease progression in metastatic breast cancer is the development of acquired resistance to chemotherapy by increased expression of the p-glycoprotein membrane pump that essentially removes chemotherapeutic agent from the cells. They demonstrated in silico and in vitro a prolonged time to resistance by combining standard chemotherapy, with dosing based on MTD and adaptive chemotherapy treatment with low dose verapamil and 2-deoxyglucose (172). Also in breast cancer, Enriquez-Navas et al. demonstrated a prolonged progression free survival (PFS) by modulating the dose of paclitaxel based on response to therapy in vivo (173).
This approach is also gaining traction in cancers that are treated with targeted therapy. In colon cancer, several studies have been implemented which take advantage of real-time assays of clonal predominance. Specifically, the CHRONOS (NCT03227926), RASINTRO (NCT03259009) and CRICKET (NCT02296203) trials are ongoing with the goal of identifying patients whose tumors may be sensitive to anti-EGFR therapy based on clonality results from cfDNA assessment.
Other strategies for monitoring clonal evolution before resistance development include the ongoing clinical trials in renal cell carcinoma (NCT01158521) and NSCLC (NCT03088930, NCT03433469) in which a patient’s tumor can be evaluated before and after a limited period of targeted therapy. Though subsequent therapy is not modulated based on the results in these cases, these studies are critical to assist in building an encyclopedia of response to treatment by tumors and the dynamic composition of the tumor ecosystem.
Combination Therapies
The challenges of single modality therapies are apparent in that the richness of tumor heterogeneity (including the tumor cells, the immune and micro-environment and epigenetic factors) essentially primes the tumor to evolve into a more molecularly complex and treatment-refractory organism. In NSCLC, activating mutations in EGFR predict for response to EGFR TKI treatment but most responses are incomplete and virtually all patients develop resistance that is mediated by multiple mechanisms (38,174–183). Jonsson, Blakely et al. highlighted the challenges of using targeted therapy in a heterogeneous tumor genomic environment given that the while targeted therapy is effective on the targeted clone, subclones are typically present that are less sensitive to the therapy. These subclones thrive in response to the selective pressure of a single-agent targeted therapy. This work demonstrated in silico and in vitro that utilizing dynamic treatment modulation both in response to changing subclone populations and in anticipation of subclone emergence, rational combination therapies can prolong the time to disease progression and in some cases via a non-intuitive switch therapy approach (184).
Other groups have demonstrated the benefit of combining targeted therapy with chemotherapy, integrating targeted therapy with antiangiogenic therapy and combination targeted therapies with improvements in PFS and OS. In the first randomized study to evaluate the benefit of combining an EGFR TKI with chemotherapy in patients with EGFR activating mutations, Sugawara et al evaluated the benefit of concurrent or sequential alternating gefitinib and chemotherapy in untreated NSCLC patients (NEJ005) (185). In 80 patients, the authors reported a median PFS of 18.3 months for concurrent treatment and 15.3 months and OS of 41.9 and 30.7 months, for the concurrent versus sequential alternating arms, respectively (185). In a follow up study, (NEJ009) the authors combined carboplatin/pemetrexed with gefitinib and demonstrated a superior OS compared with gefitinib monotherapy (52.2 months versus 38.8 months) (186). Similarly, in FASTACT-2 patients received doublet chemotherapy with either placebo or erlotinib on days 15–28 of up to six 28 day cycles followed by maintenance TKI. The PFS was 16.8 months in the chemotherapy with erlotinib group, compared with 6.9 months in the chemotherapy plus placebo group (HR 0.25 95% CI 0.16–0.39; p<0.001). The median OS was 31.4 months in the combination group and 20.6 months in the chemotherapy with placebo group despite 85% of the placebo group patients going on to receive and EGFR TKI as subsequent treatment (HR 0.48 95%CI 0.27–0.84; p=0.0092) (187).
In BRAF V600E melanoma and NSCLC, the combination of the two different targeted agents resulted in significant improvement in PFS and OS (188,189). The success of this combination is a testament to the benefit of combination therapy. Notably the lack of efficacy of dabrafenib and trametinib in the second-line setting after a BRAF-directed monotherapy highlights that tumors can become more complex under the pressure of therapy (190). These examples demonstrate the value of rational combination therapies given the superior OS noted in the experimental arms compared with standard sequential therapy.
A significant challenge in identifying effective combination therapies is related to the traditional clinical trial structure in that drug combinations need to be proven effective in late stage disease before advancing to the frontline setting. As we have described above, cancers evolve continuously therefore, we risk overlooking effective combination treatments by requiring their validation in heavily pre-treated patients.
The idea of switching or intercalating two therapies with different mechanisms of action is not unique to cancer. This approach has been successfully employed in infectious disease for treatment of HIV, malaria and tuberculosis with success (191). As in infectious disease, it critical to explore the benefit of radical treatment strategies at treatment initiation given that the more selective pressure cells undergo, the more genetically heterogeneous they become and ultimately, the more challenging the cancer will be to treat (38).
Longitudinal Monitoring of Evolution
Technical advances have revealed new ways to monitor treatment response and evolution of treatment resistance longitudinally. Serial biopsies at times of progression are important to monitor the somatic genomic and cellular changes as tumors evolve during treatment. Multiple repeat biopsies are not technically and logistically feasible in all patients and risk procedural complications. Furthermore, tissue biopsies can be molecularly biased given that they only reveal the tumor, immune and microenvironmental features of the individual sample, which may not be representative of the whole disease (3). Autopsy surveys are also often used in patients and prove invaluable information regarding tumor evolution and clonal population across disease sites globally and allow for reconstruction of evolutionary histories using phylogenetic analysis (192). However, the obvious issue with autopsy analyses is that it is a terminal endpoint and active modulation of therapy based on the findings is not feasible. Plasma sampling to evaluate cfDNA provides a minimally invasive way to assay the heterogeneity of the entire tumor cell population. Development of longitudinal plasma-based evaluation and the use of safe and informative tumor biopsy based serial protocols permits a more immediate approach to effectively monitor tumor evolution in the clinic (193,194). Limitations of plasma-based studies include the requirement of a baseline level of cfDNA present for detection and the challenge of recognizing complex genomic features (195). Several clinical trials are ongoing across multiple cancer types to evaluate the use of cfDNA in patients (NCT02443948, NCT03691012, NCT02245100, NCT03637686, NCT02883517).
In the preclinical setting, after the recognition of the importance of monitoring mutations and heterogeneity longitudinally the focus has shifted to understanding the transcriptome and how it evolves in concert with the genome. Given that the TME, immune cell, and tumor cell changes can influence the transcriptome, it is challenging to decipher the contributions of each of these populations. Bioinformatics analyses on total RNA sequencing can help in separating tumor, stromal, and immune cells. It is still difficult to prove whether this truly represents the individual contributions of these cell types (196).
The development of single cell analyses has been instrumental in allowing more accurate characterization of tumor and their microenvironment (197–204). In addition to characterization, these studies have identified interesting disease features that are important when considering treatment. For example, in head and neck cancer Puram et al analyzed the TME in primary and metastatic disease sites. They identified an EMT signature that was an independent predictor of more aggressive disease (202). In melanoma, Su et al, demonstrated the potential of incorporating single cell analysis into monitoring disease longitudinally by utilizing whole-transcriptome analysis and single cell proteomic profiling to investigate the responses of patient-derived BRAF mutation positive cell lines to BRAF inhibitor treatment (204). They utilized single cell protein signaling network analyses to demonstrate that adaptive resistance begins with activation of MEK and NFκB early in treatment, which allows cancer cells to persist despite BRAF inhibitor treatment. They demonstrated that with directed combinatorial therapy designed to inhibit specific protein signaling pathways, resistance was delayed. Similarly, Blakely et al demonstrated that NFκB is activated early in NSCLC treated with an EGFR TKI and that NFκB inhibition can attenuate resistance to treatment (205). These findings echo the results of Wei et al., in which the authors also employed single cell barcoded chip analysis to demonstrate the timeline and mode of adaptive signaling changes seen in response to treatment of a patient derived glioblastoma cell model (206). This type of analysis allows for determination of the protein levels of specifically queried proteins as well as protein-protein interactions within an individual cell. These studies highlight the value of auditing the proteome given that adaptive changes occur on a more rapid timeline and often involve post-transcriptional controls. Wei et al and Su et al demonstrated that protein networks evolve when treatment is applied resulting in increased functional heterogeneity of protein levels (more dispersion of functional protein levels) as well as increased activation of cell signaling networks that can confer resistance to targeted treatment (206). Adaptive responses to immunotherapy were identified in melanoma in which micro-environmental cues can lead to translational reprogramming and increased tumorigenesis and resistance to and anti-PD-1 treatment by repression of MITF and increased AXL (207). Adaptive signaling networks were characterized in CAFs in addition. Proteomic profiling and an in-depth bioinformatics analysis of CAF versus normal fibroblasts in patients with esophageal adenocarcinoma led to the characterization of the proteomic differences and possible therapeutic targets in the TME if there is a commonly active protein network (208).
Engineering Principles to Control the Multifactorial Basis of Tumor Evolution
A critical next step is to integrate and demonstrate these findings in patients, their convergence or privatization across cancer types, and to manipulate the causative protein signaling pathways before resistance develops to delay evolution and resistance to therapy. It is critical to determine how resistance changes signaling pathways and whether additional resistance mechanisms develop by manipulating evolution. Detailed work on modeling adaptive therapy approaches to treatment highlights the potential and challenges of this strategy to develop clinical trials (209,210). The primary challenge is applying the adaptive therapy models to patients and identifying clinically feasible ways for tracking tumor, epigenetic, and TME changes longitudinally during treatment at shorter intervals than imaging-based and tissue biopsies allow. One mechanism to implement clinically is “liquid biopsies” of tumor and micro-environmental features. In patients with targetable alterations, response to therapy in this way can be monitored in this way; utilizing this technology to guide therapeutic options has not been widely implemented. As described above clinical studies to explore this strategy are ongoing (211–215).
In addition to real-time monitoring, we should consider the potential benefits of alternative clinical trial designs including combinatorial trials in early stage disease and switch therapy trials. A critical component to switch therapy is the determination of the optimal time to switch treatment. In HIV, which is a corollary to our proposition, treatment changes are focused on maximizing time to response with combination therapies and therapy switching (216). This strategy is implemented to minimize the emergence and virulence of resistance. These methods rely on the determination of viral load and viral genotype to identify the optimal timing of switch therapy (216). Multiple groups have identified important axioms that need to be considered to implement this type of anticipatory, instead of reactive treatment strategy in cancer (184). Incorporation of “liquid biopsy”-based information shows promise in monitoring response and relapse to treatment in lung, gynecologic, colon and hematologic malignancies (211–215).
We propose an adaptive trial design in which patients are treated with therapy combinations with frequent molecular/heterogeneity re-assessment to determine if this approach can prolong treatment response by capitalizing on intra-tumoral fitness competition. Once therapy combinations are deemed safe they should be rapidly evaluated in the first-line treatment setting given that tumor heterogeneity increases during therapy and responses to treatment diminish after multiple lines of therapy (38).
An idealized concept of a clinical trial is depicted in Figure 3. Patient specimens undergo molecular profiling (DNA, RNA, epigenetic, proteomic) before treatment to determine their mutational liabilities. This assessment would also allow for comparison of individualized analyte measurements during treatment and would permit more timely and sensitive monitoring of tumor burden (217). Patients would first receive alternating targeted therapies in combination with an inhibitor of a known survival/bypass pathway, such as MAPK, AKT, or NFκB signaling (205). Based on the profile changes identified after the initial combination therapy, patients would then continue their current regimen, or switch to an alternate therapy as informed by the residual disease profile that is detected by serial molecular profiling. This treatment would then be subject to change to an alternative regimen dictated by the continued evolution of the molecular profile as assessed at defined intervals with ctDNA and tumor biopsy re-assessment.
Figure 3. Biological and learning based molecular treatment of cancer.
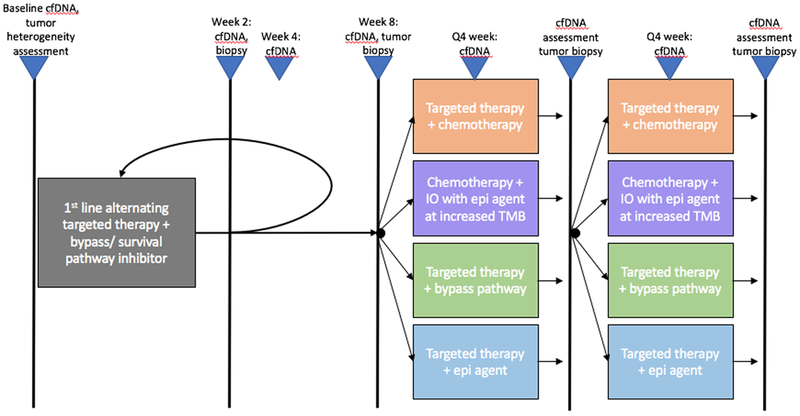
Depiction of a clinical trial design in which tumors and cfDNA are evaluated longitudinally during treatment to tailored therapy combinations based on the evolution of the persistent, or emergent clonal populations identified during active treatment and before treatment resistance is detected using more conventional clinical or radiographic assessments.
Abbreviations: cfDNA – cell-free DNA, IO – immunotherapy, TMB – tumor mutation burden, epi – epigenetically targeted agent.
By utilizing alternating therapies in the initial treatment setting, tumor growth could be better managed as a chronic disease. The potential of this approach is described above in multiple myeloma, in which two competing sub-clones alternated dominance based on treatment until a separate dominant mechanism of survival evolved (71). The potential for this same approach is seen in NSCLC with EGFR activating mutations. In this setting, EGFR T790M positive tumor cells (first generation EGFR TKI resistant but third generation EGFR TKI sensitive clones) grow more slowly than EGFR T790M negative tumor cells (218). A potential strategy for improved control of tumor cell subpopulation dynamics is to engineer a mixed population of EGFR T790M positive and negative tumor cells that would compete for dominance during alternating treatments with first and third generation EGFR TKIs. A visualization of this clonal interaction and competition is illustrated in Figure 4. To further complement the clonal competition, we have additionally proposed to include an inhibitor of a common bypass pathway likely to be active in both predominant subclonal populations.
Figure 4. Clonal predominance in alternating therapy.
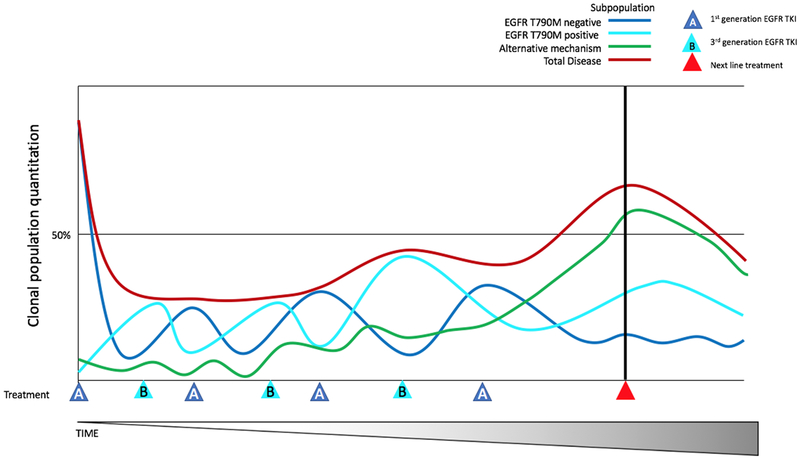
Depiction of clonal predominance switching during alternating treatment with 2 different EGFR TKI therapies to maintain a steady state population until outgrowth of a clonal population resistant to both therapies occurs, at which point a new therapy would be employed.
A challenge of this and other adaptive approaches is the continual re-assessment of tumor biology. This is more feasible in certain patients and cancers than in others. In addition to improving clinical outcomes, this clinical trial design would help to develop an encyclopedia of treatment response and resistance and determination of the minimum input needed to make treatment decisions effectively. The complexity of evolution in the setting of tumor heterogeneity makes this approach challenging. Yet, “realtime” monitoring in an evolutionary trial design is critical in order to leverage biological insights into tumor evolution to engineer principled therapeutic strategies for improved, chronic control of cancer.
Summary
Tumor biology is a much more complex and integrative process than originally recognized. Through the development and evolution of technology we now more fully appreciate the diversity between cancer types, within cancer types, subtypes, microenvironment and cancer cells themselves. We have learned that resistance to treatment is a synthetic phenotype, which must be considered when devising treatment and strategies to attenuate resistance to therapy. We must focus on principled approaches to engineer evolution to control the process and thereby advance cancer patient survival.
Statement of Significance.
Tumor and tumor immune compartment and micro-environment heterogeneity, and their evolution are critical disease features that impact treatment response. The impact and interplay of these components during treatment are viable targets to improve clinical response. In this article, we consider how tumor cells, the tumor immune compartment and microenvironment, and epigenetic factors interact and also evolve during treatment. We evaluate the convergence of these factors and suggest innovative treatment concepts that leverage evolutionary relationships to limit tumor growth and drug resistance.
Acknowledgements.
The authors acknowledge funding support from NIH / NCI U01CA217882, NIH / NCI U54CA224081, NIH / NCI R01CA204302, NIH / NCI R01CA211052, NIH / NCI R01CA169338, and the Pew-Stewart Foundations (to T.G.B.)
Footnotes
Potential Conflicts of Interest. T.G.B is a consultant/advisor to Novartis, AstraZeneca, Revolution Medicines, Takeda, and Array Biopharma and has received research funding from Novartis, Revolution Medicines, and Ignyta. C.E.M. has received honoraria from Takeda, Guardant Health and research funding from Novartis and Revolution Medicines and personal fees from Novartis.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74 doi 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366(10):883–92 doi 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D, et al. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nature communications 2013;4:2467 doi 10.1038/ncomms3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science 2003;300(5621):949 doi 10.1126/science.1082596. [DOI] [PubMed] [Google Scholar]
- 6.Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet 2005;37(6):590–2 doi 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 7.Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 2006;66(8):3987–91 doi 10.1158/0008-5472.CAN-06-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bignell G, Smith R, Hunter C, Stephens P, Davies H, Greenman C, et al. Sequence analysis of the protein kinase gene family in human testicular germ-cell tumors of adolescents and adults. Genes Chromosomes Cancer 2006;45(1):42–6 doi 10.1002/gcc.20265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007;446(7132):153–8 doi 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008;455(7216):1069–75 doi 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010;466(7308):869–73 doi 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 12.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007;318(5853):1108–13 doi 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004;10(8):789–99 doi 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 14.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150(6):1107–20 doi 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463(7278):184–90 doi 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011;333(6046):1157–60 doi 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011;471(7339):467–72 doi 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 2011;365(26):2497–506 doi 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011;476(7360):298–303 doi 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474(7353):609–15 doi 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7 doi 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489(7417):519–25 doi 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455(7216):1061–8 doi 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014;346(6206):256–9 doi 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science 2016;352(6282):169–75 doi 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- 26.de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014;346(6206):251–6 doi 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499(7457):214–8 doi 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21 doi 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Antonio M, Tamayo P, Mesirov JP, Frazer KA. Kataegis Expression Signature in Breast Cancer Is Associated with Late Onset, Better Prognosis, and Higher HER2 Levels. Cell reports 2016;16(3):672–83 doi 10.1016/j.celrep.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang S, Jia M, He Z, Liu XS. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 2018;37(29):3924–36 doi 10.1038/s41388-018-0245-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell 2012;149(5):994–1007 doi 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012;486(7403):395–9 doi 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010;467(7319):1109–13 doi 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010;467(7319):1114–7 doi 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thirlwell C, Will OC, Domingo E, Graham TA, McDonald SA, Oukrif D, et al. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology 2010;138(4):1441–54, 54 e1–7 doi 10.1053/j.gastro.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 36.Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486(7404):537–40 doi 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, Walker DA, et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest 2013;123(11):4918–22 doi 10.1172/JCI70354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 2017;49(12):1693–704 doi 10.1038/ng.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Audenet F, Isharwal S, Cha EK, Donoghue MTA, Drill EN, Ostrovnaya I, et al. Clonal Relatedness and Mutational Differences between Upper Tract and Bladder Urothelial Carcinoma. Clin Cancer Res 2018. doi 10.1158/1078-0432.CCR-18-2039. [Epublication] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer G, Al-Ahmadie H, Schultz N, Hanrahan AJ, Ostrovnaya I, Balar AV, et al. Prevalence and co-occurrence of actionable genomic alterations in high-grade bladder cancer. J Clin Oncol 2013;31(25):3133–40 doi 10.1200/JCO.2012.46.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013;152(4):714–26 doi 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albanese I, Scibetta AG, Migliavacca M, Russo A, Bazan V, Tomasino RM, et al. Heterogeneity within and between primary colorectal carcinomas and matched metastases as revealed by analysis of Ki-ras and p53 mutations. Biochem Biophys Res Commun 2004;325(3):784–91 doi 10.1016/j.bbrc.2004.10.111. [DOI] [PubMed] [Google Scholar]
- 43.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A 2008;105(11):4283–8 doi 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vermaat JS, Nijman IJ, Koudijs MJ, Gerritse FL, Scherer SJ, Mokry M, et al. Primary colorectal cancers and their subsequent hepatic metastases are genetically different: implications for selection of patients for targeted treatment. Clin Cancer Res 2012;18(3):688–99 doi 10.1158/1078-0432.CCR-11-1965. [DOI] [PubMed] [Google Scholar]
- 45.Lee SY, Haq F, Kim D, Jun C, Jo HJ, Ahn SM, et al. Comparative genomic analysis of primary and synchronous metastatic colorectal cancers. PLoS One 2014;9(3):e90459 doi 10.1371/journal.pone.0090459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, et al. Comparative analysis of metazoan chromatin organization. Nature 2014;512(7515):449–52 doi 10.1038/nature13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie T, Cho YB, Wang K, Huang D, Hong HK, Choi YL, et al. Patterns of somatic alterations between matched primary and metastatic colorectal tumors characterized by whole-genome sequencing. Genomics 2014;104(4):234–41 doi 10.1016/j.ygeno.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 48.Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol 2014;15(8):454 doi 10.1186/s13059-014-0454-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vignot S, Lefebvre C, Frampton GM, Meurice G, Yelensky R, Palmer G, et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: evaluation of concordance between genomic and transcriptional profiles. Eur J Cancer 2015;51(7):791–9 doi 10.1016/j.ejca.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 50.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015;21(7):795–801 doi 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov 2016;6(2):147–53 doi 10.1158/2159-8290.CD-15-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bashashati A, Ha G, Tone A, Ding J, Prentice LM, Roth A, et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J Pathol 2013;231(1):21–34 doi 10.1002/path.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jamal-Hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, Quezada S, et al. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol 2014;12(7):e1001906 doi 10.1371/journal.pbio.1001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351(6280):1463–9 doi 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 2017;376(22):2109–21 doi 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 56.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017;171(6):1259–71 e11 doi 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 2017;8(1):1136 doi 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018;173(3):581–94 e12 doi 10.1016/j.cell.2018.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018;173(3):595–610 e11 doi 10.1016/j.cell.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018;173(3):611–23 e17 doi 10.1016/j.cell.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Linch M, Goh G, Hiley C, Shanmugabavan Y, McGranahan N, Rowan A, et al. Intratumoural evolutionary landscape of high-risk prostate cancer: the PROGENY study of genomic and immune parameters. Ann Oncol 2017;28(10):2472–80 doi 10.1093/annonc/mdx355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018;33(4):649–63 e4 doi 10.1016/j.ccell.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hope EA, Amorosi CJ, Miller AW, Dang K, Heil CS, Dunham MJ. Experimental Evolution Reveals Favored Adaptive Routes to Cell Aggregation in Yeast. Genetics 2017;206(2):1153–67 doi 10.1534/genetics.116.198895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19(8):2240–7 doi 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer 2017;17(11):637–58 doi 10.1038/nrc.2017.84. [DOI] [PubMed] [Google Scholar]
- 66.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483(7387):100–3 doi 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 67.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012;2(3):227–35 doi 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014;508(7494):118–22 doi 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 69.Imamovic L, Sommer MO. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci Transl Med 2013;5(204):204ra132 doi 10.1126/scitranslmed.3006609. [DOI] [PubMed] [Google Scholar]
- 70.Yen P, Papin JA. History of antibiotic adaptation influences microbial evolutionary dynamics during subsequent treatment. PLoS Biol 2017;15(8):e2001586 doi 10.1371/journal.pbio.2001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120(5):1067–76 doi 10.1182/blood-2012-01-405985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Federle MJ, Bassler BL. Interspecies communication in bacteria. J Clin Invest 2003;112(9):1291–9 doi 10.1172/JCI20195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xavier KB, Bassler BL. Interference with AI-2-mediated bacterial cell-cell communication. Nature 2005;437(7059):750–3 doi 10.1038/nature03960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mukherjee S, Moustafa DA, Stergioula V, Smith CD, Goldberg JB, Bassler BL. The PqsE and RhlR proteins are an autoinducer synthase-receptor pair that control virulence and biofilm development in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 2018;115(40):E9411–E8 doi 10.1073/pnas.1814023115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015;520(7547):368–72 doi 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487(7408):500–4 doi 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kessler BE, Mishall KM, Kellett MD, Clark EG, Pugazhenthi U, Pozdeyev N, et al. Resistance to Src inhibition alters the BRAF-mutant tumor secretome to promote an invasive phenotype and therapeutic escape through a FAK>p130Cas>c-Jun signaling axis. Oncogene 2018. doi 10.1038/s41388-018-0617-1. [Epublication] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018;378(2):113–25 doi 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 79.Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2017;377(9):829–38 doi 10.1056/NEJMoa1704795. [DOI] [PubMed] [Google Scholar]
- 80.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012;367(18):1694–703 doi 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cortes JE, Jones D, O’Brien S, Jabbour E, Konopleva M, Ferrajoli A, et al. Nilotinib as front-line treatment for patients with chronic myeloid leukemia in early chronic phase. J Clin Oncol 2010;28(3):392–7 doi 10.1200/JCO.2009.25.4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cortes JE, Jones D, O’Brien S, Jabbour E, Ravandi F, Koller C, et al. Results of dasatinib therapy in patients with early chronic-phase chronic myeloid leukemia. J Clin Oncol 2010;28(3):398–404 doi 10.1200/JCO.2009.25.4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130(6):722–31 doi 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quek L, David MD, Kennedy A, Metzner M, Amatangelo M, Shih A, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med 2018;24(8):1167–77 doi 10.1038/s41591-018-0115-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Landau DA, Carter SL, Getz G, Wu CJ. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia 2014;28(1):34–43 doi 10.1038/leu.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cruickshank B, Giacomantonio M, Marcato P, McFarland S, Pol J, Gujar S. Dying to Be Noticed: Epigenetic Regulation of Immunogenic Cell Death for Cancer Immunotherapy. Front Immunol 2018;9:654 doi 10.3389/fimmu.2018.00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24(5):541–50 doi 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018;49(1):178–93 e7 doi 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017;171(4):934–49 e16 doi 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017;171(4):934–49.e15 doi 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018;175(4):984–97 e24 doi 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov 2017;7(3):264–76 doi 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mazzone R, Zwergel C, Mai A, Valente S. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics 2017;9:59 doi 10.1186/s13148-017-0358-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 2014;54(5):716–27 doi 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010;141(1):69–80 doi 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012;22(5):668–82 doi 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J Clin Oncol 2015;33(34):4023–31 doi 10.1200/JCO.2015.63.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R Jr., Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res 2013;19(3):657–67 doi 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015;527(7579):472–6 doi 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15(3):178–96 doi 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 2006;66(2):944–50 doi 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- 102.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133(4):704–15 doi 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, Oyama T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res 2010;30(7):2513–7. [PubMed] [Google Scholar]
- 104.Chung JH, Rho JK, Xu X, Lee JS, Yoon HI, Lee CT, et al. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 2011;73(2):176–82 doi 10.1016/j.lungcan.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 105.Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol 2011;6(7):1152–61 doi 10.1097/JTO.0b013e318216ee52. [DOI] [PubMed] [Google Scholar]
- 106.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352(10):997–1003 doi 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 107.Zhang YW, Zheng Y, Wang JZ, Lu XX, Wang Z, Chen LB, et al. Integrated analysis of DNA methylation and mRNA expression profiling reveals candidate genes associated with cisplatin resistance in non-small cell lung cancer. Epigenetics 2014;9(6):896–909 doi 10.4161/epi.28601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li A, Song J, Lai Q, Liu B, Wang H, Xu Y, et al. Hypermethylation of ATP-binding cassette B1 (ABCB1) multidrug resistance 1 (MDR1) is associated with cisplatin resistance in the A549 lung adenocarcinoma cell line. Int J Exp Pathol 2016;97(6):412–21 doi 10.1111/iep.12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zeller C, Dai W, Steele NL, Siddiq A, Walley AJ, Wilhelm-Benartzi CS, et al. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 2012;31(42):4567–76 doi 10.1038/onc.2011.611. [DOI] [PubMed] [Google Scholar]
- 110.Chou JL, Su HY, Chen LY, Liao YP, Hartman-Frey C, Lai YH, et al. Promoter hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest 2010;90(3):414–25 doi 10.1038/labinvest.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Faleiro I, Leao R, Binnie A, de Mello RA, Maia AT, Castelo-Branco P. Epigenetic therapy in urologic cancers: an update on clinical trials. Oncotarget 2017;8(7):12484–500 doi 10.18632/oncotarget.14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.van Kampen JG, Marijnissen-van Zanten MA, Simmer F, van der Graaf WT, Ligtenberg MJ, Nagtegaal ID. Epigenetic targeting in pancreatic cancer. Cancer Treat Rev 2014;40(5):656–64 doi 10.1016/j.ctrv.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 113.Zhang W, Peyton M, Xie Y, Soh J, Minna JD, Gazdar AF, et al. Histone deacetylase inhibitor romidepsin enhances anti-tumor effect of erlotinib in non-small cell lung cancer (NSCLC) cell lines. J Thorac Oncol 2009;4(2):161–6 doi 10.1097/JTO.0b013e318194fae7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen MC, Chen CH, Wang JC, Tsai AC, Liou JP, Pan SL, et al. The HDAC inhibitor, MPT0E028, enhances erlotinib-induced cell death in EGFR-TKI-resistant NSCLC cells. Cell Death Dis 2013;4:e810 doi 10.1038/cddis.2013.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Greve G, Schiffmann I, Pfeifer D, Pantic M, Schuler J, Lubbert M. The pan-HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR-mutated and -wildtype non-small cell lung cancer cells. BMC Cancer 2015;15:947 doi 10.1186/s12885-015-1967-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Smith DL, Acquaviva J, Sequeira M, Jimenez JP, Zhang C, Sang J, et al. The HSP90 inhibitor ganetespib potentiates the antitumor activity of EGFR tyrosine kinase inhibition in mutant and wild-type non-small cell lung cancer. Target Oncol 2015;10(2):235–45 doi 10.1007/s11523-014-0329-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu CY, Wu CY, Petrossian K, Huang TT, Tseng LM, Chen S. Treatment for the endocrine resistant breast cancer: Current options and future perspectives. J Steroid Biochem Mol Biol 2017;172:166–75 doi 10.1016/j.jsbmb.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 118.Tanimoto A, Takeuchi S, Arai S, Fukuda K, Yamada T, Roca X, et al. Histone Deacetylase 3 Inhibition Overcomes BIM Deletion Polymorphism-Mediated Osimertinib Resistance in EGFR-Mutant Lung Cancer. Clin Cancer Res 2017;23(12):3139–49 doi 10.1158/1078-0432.CCR-16-2271. [DOI] [PubMed] [Google Scholar]
- 119.Gallagher SJ, Gunatilake D, Beaumont KA, Sharp DM, Tiffen JC, Heinemann A, et al. HDAC inhibitors restore BRAF-inhibitor sensitivity by altering PI3K and survival signalling in a subset of melanoma. Int J Cancer 2018;142(9):1926–37 doi 10.1002/ijc.31199. [DOI] [PubMed] [Google Scholar]
- 120.Ansari J, Shackelford RE, El-Osta H. Epigenetics in non-small cell lung cancer: from basics to therapeutics. Transl Lung Cancer Res 2016;5(2):155–71 doi 10.21037/tlcr.2016.02.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gerber DE, Boothman DA, Fattah FJ, Dong Y, Zhu H, Skelton RA, et al. Phase 1 study of romidepsin plus erlotinib in advanced non-small cell lung cancer. Lung Cancer 2015;90(3):534–41 doi 10.1016/j.lungcan.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gray JE, Haura E, Chiappori A, Tanvetyanon T, Williams CC, Pinder-Schenck M, et al. A phase I, pharmacokinetic, and pharmacodynamic study of panobinostat, an HDAC inhibitor, combined with erlotinib in patients with advanced aerodigestive tract tumors. Clin Cancer Res 2014;20(6):1644–55 doi 10.1158/1078-0432.CCR-13-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Reguart N, Rosell R, Cardenal F, Cardona AF, Isla D, Palmero R, et al. Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer 2014;84(2):161–7 doi 10.1016/j.lungcan.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 124.Ji W, Sun B, Su C. Targeting MicroRNAs in Cancer Gene Therapy. Genes (Basel) 2017;8(1) doi 10.3390/genes8010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen X, Gao J, Yu Y, Zhao Z, Pan Y. Long non-coding RNA UCA1 targets miR-185–5p and regulates cell mobility by affecting epithelial-mesenchymal transition in melanoma via Wnt/beta-catenin signaling pathway. Gene 2018;676:298–305 doi 10.1016/j.gene.2018.08.065. [DOI] [PubMed] [Google Scholar]
- 126.Watt K, Newsted D, Voorand E, Gooding RJ, Majewski A, Truesdell P, et al. MicroRNA-206 suppresses TGF-beta signalling to limit tumor growth and metastasis in lung adenocarcinoma. Cell Signal 2018;50:25–36 doi 10.1016/j.cellsig.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 127.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19(11):1423–37 doi 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kallinowski F, Schlenger KH, Runkel S, Kloes M, Stohrer M, Okunieff P, et al. Blood flow, metabolism, cellular microenvironment, and growth rate of human tumor xenografts. Cancer Res 1989;49(14):3759–64. [PubMed] [Google Scholar]
- 129.Blumberg N Tumor angiogenesis factor. Speculations on an approach to cancer chemotherapy. Yale J Biol Med 1974;47(2):71–81. [PMC free article] [PubMed] [Google Scholar]
- 130.Carter SK. Clinical strategy for the development of angiogenesis inhibitors. Oncologist 2000;5 Suppl 1:51–4. [DOI] [PubMed] [Google Scholar]
- 131.Rosen LS. Angiogenesis inhibition in solid tumors. Cancer J 2001;7 Suppl 3:S120–8. [PubMed] [Google Scholar]
- 132.Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res 2013;73(16):4965–77 doi 10.1158/0008-5472.CAN-13-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stoker MG, Shearer M, O’Neill C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J Cell Sci 1966;1(3):297–310. [DOI] [PubMed] [Google Scholar]
- 134.Mehta PP, Bertram JS, Loewenstein WR. Growth inhibition of transformed cells correlates with their junctional communication with normal cells. Cell 1986;44(1):187–96. [DOI] [PubMed] [Google Scholar]
- 135.Kirk D, Szalay MF, Kaighn ME. Modulation of growth of a human prostatic cancer cell line (PC-3) in agar culture by normal human lung fibroblasts. Cancer Res 1981;41(3):1100–3. [PubMed] [Google Scholar]
- 136.Paland N, Kamer I, Kogan-Sakin I, Madar S, Goldfinger N, Rotter V. Differential influence of normal and cancer-associated fibroblasts on the growth of human epithelial cells in an in vitro cocultivation model of prostate cancer. Mol Cancer Res 2009;7(8):1212–23 doi 10.1158/1541-7786.MCR-09-0073. [DOI] [PubMed] [Google Scholar]
- 137.Alkasalias T, Flaberg E, Kashuba V, Alexeyenko A, Pavlova T, Savchenko A, et al. Inhibition of tumor cell proliferation and motility by fibroblasts is both contact and soluble factor dependent. Proc Natl Acad Sci U S A 2014;111(48):17188–93 doi 10.1073/pnas.1419554111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alkasalias T, Moyano-Galceran L, Arsenian-Henriksson M, Lehti K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int J Mol Sci 2018;19(5) doi 10.3390/ijms19051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cui Q, Wang B, Li K, Sun H, Hai T, Zhang Y, et al. Upregulating MMP-1 in carcinoma-associated fibroblasts reduces the efficacy of Taxotere on breast cancer synergized by Collagen IV. Oncol Lett 2018;16(3):3537–44 doi 10.3892/ol.2018.9092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med 2009;15(1):68–74 doi 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- 141.Vaquero J, Lobe C, Tahraoui S, Claperon A, Mergey M, Merabtene F, et al. The IGF2/IR/IGF1R Pathway in Tumor Cells and Myofibroblasts Mediates Resistance to EGFR Inhibition in Cholangiocarcinoma. Clin Cancer Res 2018;24(17):4282–96 doi 10.1158/1078-0432.CCR-17-3725. [DOI] [PubMed] [Google Scholar]
- 142.Yoshida T, Ishii G, Goto K, Neri S, Hashimoto H, Yoh K, et al. Podoplanin-positive cancer-associated fibroblasts in the tumor microenvironment induce primary resistance to EGFR-TKIs in lung adenocarcinoma with EGFR mutation. Clin Cancer Res 2015;21(3):642–51 doi 10.1158/1078-0432.CCR-14-0846. [DOI] [PubMed] [Google Scholar]
- 143.Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003;26(1):44–8 doi 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 144.Narra K, Mullins SR, Lee HO, Strzemkowski-Brun B, Magalong K, Christiansen VJ, et al. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol Ther 2007;6(11):1691–9. [DOI] [PubMed] [Google Scholar]
- 145.Chan JSK, Sng MK, Teo ZQ, Chong HC, Twang JS, Tan NS. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene 2018;37(2):160–73 doi 10.1038/onc.2017.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ogier C, Colombo PE, Bousquet C, Canterel-Thouennon L, Sicard P, Garambois V, et al. Targeting the NRG1/HER3 pathway in tumor cells and cancer-associated fibroblasts with an anti-neuregulin 1 antibody inhibits tumor growth in pre-clinical models of pancreatic cancer. Cancer Lett 2018;432:227–36 doi 10.1016/j.canlet.2018.06.023. [DOI] [PubMed] [Google Scholar]
- 147.Hanley CJ, Mellone M, Ford K, Thirdborough SM, Mellows T, Frampton SJ, et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. J Natl Cancer Inst 2018;110(1) doi 10.1093/jnci/djx121. [Epublication] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Choi H, Sheng J, Gao D, Li F, Durrans A, Ryu S, et al. Transcriptome analysis of individual stromal cell populations identifies stroma-tumor crosstalk in mouse lung cancer model. Cell Rep 2015;10(7):1187–201 doi 10.1016/j.celrep.2015.01.040. [DOI] [PubMed] [Google Scholar]
- 149.Chen Y, Tan W, Wang C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. Onco Targets Ther 2018;11:3817–26 doi 10.2147/OTT.S168317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ibrahim-Hashim A, Robertson-Tessi M, Enriquez-Navas PM, Damaghi M, Balagurunathan Y, Wojtkowiak JW, et al. Defining Cancer Subpopulations by Adaptive Strategies Rather Than Molecular Properties Provides Novel Insights into Intratumoral Evolution. Cancer Res 2017;77(9):2242–54 doi 10.1158/0008-5472.CAN-16-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Warburg O On the origin of cancer cells. Science 1956;123(3191):309–14. [DOI] [PubMed] [Google Scholar]
- 152.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006;66(18):8927–30 doi 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 153.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324(5930):1029–33 doi 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wong CC, Qian Y, Yu J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene 2017;36(24):3359–74 doi 10.1038/onc.2016.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Altomare DA, Khaled AR. Homeostasis and the importance for a balance between AKT/mTOR activity and intracellular signaling. Curr Med Chem 2012;19(22):3748–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ricketts CJ, Shuch B, Vocke CD, Metwalli AR, Bratslavsky G, Middelton L, et al. Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. J Urol 2012;188(6):2063–71 doi 10.1016/j.juro.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A 2011;108(1):314–8 doi 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov 2013;3(6):648–57 doi 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hensen EF, Bayley JP. Recent advances in the genetics of SDH-related paraganglioma and pheochromocytoma. Fam Cancer 2011;10(2):355–63 doi 10.1007/s10689-010-9402-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009;325(5944):1139–42 doi 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yen K, Travins J, Wang F, David MD, Artin E, Straley K, et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov 2017;7(5):478–93 doi 10.1158/2159-8290.CD-16-1034. [DOI] [PubMed] [Google Scholar]
- 162.Shih AH, Meydan C, Shank K, Garrett-Bakelman FE, Ward PS, Intlekofer AM, et al. Combination Targeted Therapy to Disrupt Aberrant Oncogenic Signaling and Reverse Epigenetic Dysfunction in IDH2- and TET2-Mutant Acute Myeloid Leukemia. Cancer Discov 2017;7(5):494–505 doi 10.1158/2159-8290.CD-16-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Weng Y, Shen Y, He Y, Pan X, Xu J, Jiang Y, et al. The miR-15b-5p/PDK4 axis regulates osteosarcoma proliferation through modulation of the Warburg effect. Biochem Biophys Res Commun 2018;503(4):2749–57 doi 10.1016/j.bbrc.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 164.Zhu B, Cao X, Zhang W, Pan G, Yi Q, Zhong W, et al. MicroRNA-31–5p enhances the Warburg effect via targeting FIH. FASEB J 2019;33(1):545–56 doi 10.1096/fj.201800803R. [DOI] [PubMed] [Google Scholar]
- 165.Han H, Li W, Shen H, Zhang J, Zhu Y, Li Y. microRNA-129–5p, a c-Myc negative target, affects hepatocellular carcinoma progression by blocking the Warburg effect. J Mol Cell Biol 2016. doi 10.1093/jmcb/mjw010. [Epublication] [DOI] [PubMed] [Google Scholar]
- 166.Bivona TG, Doebele RC. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat Med 2016;22(5):472–8 doi 10.1038/nm.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA. Spatial Heterogeneity and Evolutionary Dynamics Modulate Time to Recurrence in Continuous and Adaptive Cancer Therapies. Cancer Res 2018;78(8):2127–39 doi 10.1158/0008-5472.CAN-17-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Inal A, Kaplan MA, Kucukoner M, Isikdogan A. Docetaxel and Cisplatin Plus Fluorouracil compared with Modified Docetaxel, Cisplatin, and 5-Fluorouracil as first-line therapy for advanced gastric cancer: a retrospective analysis of single institution. Neoplasma 2012;59(2):233–6. [DOI] [PubMed] [Google Scholar]
- 169.Shah MA, Janjigian YY, Stoller R, Shibata S, Kemeny M, Krishnamurthi S, et al. Randomized Multicenter Phase II Study of Modified Docetaxel, Cisplatin, and Fluorouracil (DCF) Versus DCF Plus Growth Factor Support in Patients With Metastatic Gastric Adenocarcinoma: A Study of the US Gastric Cancer Consortium. J Clin Oncol 2015;33(33):3874–9 doi 10.1200/JCO.2015.60.7465. [DOI] [PubMed] [Google Scholar]
- 170.Xie X, Wu Y, Luo S, Yang H, Li L, Zhou S, et al. Efficacy and Toxicity of Low-Dose versus Conventional-Dose Chemotherapy for Malignant Tumors: a Meta-Analysis of 6 Randomized Controlled Trials. Asian Pac J Cancer Prev 2017;18(2):479–84 doi 10.22034/APJCP.2017.18.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang J, Cunningham JJ, Brown JS, Gatenby RA. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat Commun 2017;8(1):1816 doi 10.1038/s41467-017-01968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Silva AS, Kam Y, Khin ZP, Minton SE, Gillies RJ, Gatenby RA. Evolutionary approaches to prolong progression-free survival in breast cancer. Cancer Res 2012;72(24):6362–70 doi 10.1158/0008-5472.CAN-12-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Enriquez-Navas PM, Kam Y, Das T, Hassan S, Silva A, Foroutan P, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med 2016;8(327):327ra24 doi 10.1126/scitranslmed.aad7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England journal of medicine 2005;352(8):786–92 doi 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 175.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine 2005;2(3):e73 doi 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proceedings of the National Academy of Sciences of the United States of America 2007;104(52):20932–7 doi 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine 2011;3(75):75ra26 doi 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Doebele RC, Aisner DL, Le AT, Berge EA, Pilling AB, Kutateladze TG, et al. Analysis of resistance mechanisms to ALK kinase inhibitors in ALK+ NSCLC patients. J Clin Oncol 2012;30, 2012 (suppl; abstr 7504). [Google Scholar]
- 179.Sorensen BS, Wu L, Wei W, Tsai J, Weber B, Nexo E, et al. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer 2014;120(24):3896–901 doi 10.1002/cncr.28964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Soucheray M, Capelletti M, Pulido I, Kuang Y, Paweletz CP, Becker JH, et al. Intratumoral Heterogeneity in EGFR-Mutant NSCLC Results in Divergent Resistance Mechanisms in Response to EGFR Tyrosine Kinase Inhibition. Cancer Res 2015;75(20):4372–83 doi 10.1158/0008-5472.CAN-15-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov 2016;6(10):1118–33 doi 10.1158/2159-8290.CD-16-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, et al. Resistance Mechanisms to Targeted Therapies in ROS1(+) and ALK(+) Non-small Cell Lung Cancer. Clin Cancer Res 2018;24(14):3334–47 doi 10.1158/1078-0432.CCR-17-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.McCoach CE, Blakely CM, Banks KC, Levy B, Chue BM, Raymond VM, et al. Clinical Utility of Cell-Free DNA for the Detection of ALK Fusions and Genomic Mechanisms of ALK Inhibitor Resistance in Non-Small Cell Lung Cancer. Clin Cancer Res 2018;24(12):2758–70 doi 10.1158/1078-0432.CCR-17-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jonsson VD, Blakely CM, Lin L, Asthana S, Matni N, Olivas V, et al. Novel computational method for predicting polytherapy switching strategies to overcome tumor heterogeneity and evolution. Sci Rep 2017;7:44206 doi 10.1038/srep44206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sugawara S, Oizumi S, Minato K, Harada T, Inoue A, Fujita Y, et al. Randomized phase II study of concurrent versus sequential alternating gefitinib and chemotherapy in previously untreated non-small cell lung cancer with sensitive EGFR mutations: NEJ005/TCOG0902. Ann Oncol 2015;26(5):888–94 doi 10.1093/annonc/mdv063. [DOI] [PubMed] [Google Scholar]
- 186.Nakamura A, Inoue A, Morita S, Hosomi Y, Kato H, Fukuhara T, et al. Phase III study comparing gefitinib monotherapy (G) to combination therapy with gefitinib, carboplatin, and pemetrexed (GCP) for untreated patients (pts) with advanced non-small cell lung cancer (NSCLC) with EGFR mutations (NEJ009). J Clin Oncol 36, 2018 (suppl; abstr 9005) 2018. [Google Scholar]
- 187.Wu YL, Lee JS, Thongprasert S, Yu CJ, Zhang L, Ladrera G, et al. Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): a randomised, double-blind trial. Lancet Oncol 2013;14(8):777–86 doi 10.1016/S1470-2045(13)70254-7. [DOI] [PubMed] [Google Scholar]
- 188.Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland A, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 2017;18(10):1307–16 doi 10.1016/S1470-2045(17)30679-4. [DOI] [PubMed] [Google Scholar]
- 189.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372(1):30–9 doi 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 190.Johnson DB, Flaherty KT, Weber JS, Infante JR, Kim KB, Kefford RF, et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol 2014;32(33):3697–704 doi 10.1200/JCO.2014.57.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Goldberg DE, Siliciano RF, Jacobs WR Jr. Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell 2012;148(6):1271–83 doi 10.1016/j.cell.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pisapia DJ, Salvatore S, Pauli C, Hissong E, Eng K, Prandi D, et al. Next-Generation Rapid Autopsies Enable Tumor Evolution Tracking and Generation of Preclinical Models. JCO Precis Oncol 2017;2017 doi 10.1200/PO.16.00038. [Epublication] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dagogo-Jack I, Brannon AR, Ferris LA, Campbell CD, Lin JJ, Schultz KR, et al. Tracking the Evolution of Resistance to ALK Tyrosine Kinase Inhibitors through Longitudinal Analysis of Circulating Tumor DNA. JCO Precis Oncol 2018;2018 doi 10.1200/PO.17.00160. [Epublication] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ramesh N, Sei E, Gao R, Tsai PC, Logothetis C, Zurita AJ, et al. Abstract 3665: Longitudinal monitoring of prostate cancer evolution by plasma genome sequencing. Cancer Research 2018;78(13 Supplement):3665. [Google Scholar]
- 195.Hench IB, Hench J, Tolnay M. Liquid Biopsy in Clinical Management of Breast, Lung, and Colorectal Cancer. Front Med (Lausanne) 2018;5:9 doi 10.3389/fmed.2018.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nature communications 2013;4:2612 doi 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401 doi 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kim KT, Lee HW, Lee HO, Kim SC, Seo YJ, Chung W, et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol 2015;16:127 doi 10.1186/s13059-015-0692-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352(6282):189–96 doi 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nature communications 2017;8:15081 doi 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017;169(4):750–65 e17 doi 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017;171(7):1611–24 e24 doi 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Horning AM, Wang Y, Lin CK, Louie AD, Jadhav RR, Hung CN, et al. Single-Cell RNA-seq Reveals a Subpopulation of Prostate Cancer Cells with Enhanced Cell-Cycle-Related Transcription and Attenuated Androgen Response. Cancer research 2018;78(4):853–64 doi 10.1158/0008-5472.CAN-17-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Su Y, Wei W, Robert L, Xue M, Tsoi J, Garcia-Diaz A, et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proceedings of the National Academy of Sciences of the United States of America 2017;114(52):13679–84 doi 10.1073/pnas.1712064115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Blakely CM, Pazarentzos E, Olivas V, Asthana S, Yan JJ, Tan I, et al. NF-kappaB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep 2015;11(1):98–110 doi 10.1016/j.celrep.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wei W, Shin YS, Xue M, Matsutani T, Masui K, Yang H, et al. Single-Cell Phosphoproteomics Resolves Adaptive Signaling Dynamics and Informs Targeted Combination Therapy in Glioblastoma. Cancer Cell 2016;29(4):563–73 doi 10.1016/j.ccell.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Falletta P, Sanchez-Del-Campo L, Chauhan J, Effern M, Kenyon A, Kershaw CJ, et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev 2017;31(1):18–33 doi 10.1101/gad.290940.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Manousopoulou A, Hayden A, Mellone M, Garay-Baquero DJ, White CH, Noble F, et al. Quantitative proteomic profiling of primary cancer-associated fibroblasts in oesophageal adenocarcinoma. Br J Cancer 2018;118(9):1200–7 doi 10.1038/s41416-018-0042-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gluzman M, Scott JG, Vladimirsky A. Optimizing adaptive cancer therapy: dynamic programming and evolutionary game theory. bioRxiv 2018:491829 doi 10.1101/491829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.West J, You L, Brown J, Newton PK, Anderson ARA. Towards multi-drug adaptive therapy. bioRxiv 2018:476507 doi 10.1101/476507. [DOI] [Google Scholar]
- 211.Vandeputte C, Kehagias P, El Housni H, Ameye L, Laes JF, Desmedt C, et al. Circulating tumor DNA in early response assessment and monitoring of advanced colorectal cancer treated with a multi-kinase inhibitor. Oncotarget 2018;9(25):17756–69 doi 10.18632/oncotarget.24879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Pereira E, Camacho-Vanegas O, Anand S, Sebra R, Catalina Camacho S, Garnar-Wortzel L, et al. Personalized Circulating Tumor DNA Biomarkers Dynamically Predict Treatment Response and Survival In Gynecologic Cancers. PLoS One 2015;10(12):e0145754 doi 10.1371/journal.pone.0145754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tabernero J, Lenz HJ, Siena S, Sobrero A, Falcone A, Ychou M, et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol 2015;16(8):937–48 doi 10.1016/S1470-2045(15)00138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018;131(22):2413–25 doi 10.1182/blood-2017-11-812073. [DOI] [PubMed] [Google Scholar]
- 215.Shepherd F, Papadimitrakoupoulou V, Mok T, Wu Y, Han J, Ahn M, et al. Early clearance of plasma EGFR mutations as a predictor of response to osimertinib in the AURA3 trial. J Clin Oncol 36, 2018 (suppl; abstr 9027) 2018. [Google Scholar]
- 216.Luo R, Piovoso MJ, Martinez-Picado J, Zurakowski R. Optimal antiviral switching to minimize resistance risk in HIV therapy. PLoS One 2011;6(11):e27047 doi 10.1371/journal.pone.0027047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20(5):548–54 doi 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med 2011;3(90):90ra59 doi 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]