

Abstract
Ras genes potently drive human cancers, with mutated proto-oncogene GTPase KRAS4B (K-Ras4B) being the most abundant isoform. Targeted inhibition of oncogenic gene products is considered the “holy grail” of present-day cancer therapy, and recent discoveries of small-molecule KRas4B inhibitors were made thanks to a deeper understanding of the structure and dynamics of this GTPase. Because interactions with biological membranes are key for Ras function, Ras–lipid interactions have become a major focus, especially because such interactions evidently involve both the Ras C terminus for lipid anchoring and its G-protein domain. Here, using NMR spectroscopy and molecular dynamics simulations complemented by biophysical- and cell-biology assays, we investigated the interaction between K-Ras4B with the signaling lipid phosphatidylinositol (4,5)-phosphate (PIP2). We discovered that the β2 and β3 strands as well as helices 4 and 5 of the GTPase G-domain bind to PIP2 and identified the specific residues in these structural elements employed in these interactions, likely occurring in two K-Ras4B orientation states relative to the membrane. Importantly, we found that some of these residues known to be oncogenic when mutated (D47K, D92N, K104M, and D126N) are critical for K-Ras–mediated transformation of fibroblast cells, but do not substantially affect basal and assisted nucleotide hydrolysis and exchange. Moreover, the K104M substitution abolished localization of K-Ras to the plasma membrane. The findings suggest that specific G-domain residues can critically regulate Ras function by mediating interactions with membrane-associated PIP2 lipids; these insights that may inform the future design of therapeutic reagents targeting Ras activity.
Keywords: Ras protein, protein structure, phosphoinositide, cancer, nuclear magnetic resonance (NMR), molecular dynamics, cell signaling, cell transformation, PIP2, protein-membrane interactions, Ras GTPase
Introduction
Ras GTPases regulate diverse signal transduction pathways, controlling cell proliferation, differentiation and apoptosis, organization of the cytoskeleton, vesicular transport, metabolism, and nuclear import (1, 2). Mutated Ras proteins are associated with ∼30% of all human cancers, where they drive oncogenic processes (3). Among the three main isoforms expressed human cells, N-Ras, H-Ras, and K-Ras (4–5), mutated K-Ras4B is the most abundant of the oncogenic GTPases (6, 7). The protein is comprised of two major domains: the G-domain and the hyper-variable region (HVR)4 (Fig. 1A). The G-domain includes the N-terminal residues 1–166, which associate with GTPase exchange factors (GEFs), and GTPase activating proteins (GAPs) (8).
Figure 1.

Structure of K-Ras. A, structure of K-Ras4B (G12V, 1–169) is shown in ribbon representation with lobes, nucleotide, and switch regions highlighted as follows: lobes 1 and 2, as well as the switch regions shown in cyan, green, and dark orange, respectively. The bound GTP is shown in stick representation and the Mg2+ ion as a cyan sphere. B, sequence alignment of the HVR domains of K-Ras, H-Ras, and N-Ras (formally starting at 167/170), with posttranslational lipid modifications, but not carboxy-methyl truncations indicated. Pal and Far refer to palmitoylation and farnesylation sites, respectively.
The G-domain is comprised of two lobes (Fig. 1A). Lobe 1, the catalytic subdomain, encompassing residues 1–86, contains the functionally critical switch regions whose conformation and dynamics is nucleotide dependent (switch 1 including residues 25–40, and switch 2 including residues 57–75), as well as the phosphate-binding region (P-loop, residues 10–17). The second lobe, comprised of residues 87–166, is a regulatory domain and contains allosteric regions such as helix 3 and loop 7. Intramolecular communication has been proposed between the allosteric domain and the catalytic domain at the other side of the protein (reviewed in Refs. 9 and 10). As found in all Ras GTPases, the HVR is located at the end of the second lobe, in K-Ras4B comprising a 24-residue segment with the very C-terminal CAAX sequence (C, cysteine; A, aliphatic amino acid; X, any amino acid), which undergoes farnesylation, truncation, and methylation and serves as a lipid anchor of the protein to the plasma membrane. In contrast to the high sequence identity in the G-domain, Ras isoforms differ significantly in the HVR and can undergo isoform-specific post-translational lipid modification (Fig. 1B). The K-Ras4B HVR undergoes a single farnesylation, whereas some other GTPases can utilize additional palmitoylation for further anchoring to membranes. The unique C-terminal polybasic region further aids in localizing K-Ras4B to lipid bilayers (11–13).
It has long been established that interactions with cellular membranes are critical for signaling by Ras GTPases. The major outcome of these interactions is thought to be a temporal and spatial sequestration of a multicomponent signaling complex in the proximity of an activated growth factor receptor (14). Although the majority of the research in Ras GTPase interaction with membranes has focused on their phosphatidylcholine and phosphatidylserine components, the interaction of Ras GTPases with phosphoinositides have been rarely studied.
Of the signaling lipid classes, phosphoinositides regulate key aspects of cell growth and proliferation. For example, PIP(4,5)P2 plays a critical role in endosomal vesicle trafficking to the apical as well as basolateral plasma membrane, assembly of the actin cytoskeleton, and communication with the extracellular milieu, whereas PI(3,5)P2 is responsible for late endosomal trafficking (15, 16). Under pathological conditions, phosphoinositides serve as key mediators of aberrant proliferation and survival signals, rendering them important targets for therapeutic interventions (17). In the context of cancer, phosphoinositides were shown to be major regulators of cell motility, invasion, and metastasis (18–20). Recent studies revealed several classes of transmembrane receptors and ion channels might be regulated by PIP2, or at least bind this signaling lipid with considerable affinity (21–23). Thus, PIP2 signaling may result in a cell-global concerted response to environmental changes.
It is, therefore, likely that localization if not activity of Ras GTPases is also affected by changes in nearby levels of PIP2 and PIP2 itself may be clustered locally by certain proteins (24, 25). Furthermore, PI3K is a major effector of Ras, which when activated by the GTPase generates PIP3 from PIP2(4,5) (26), whereas PLCβ and -γ are regulated by Ras to break down PIP2 (27–30). Potentially, PIP2 and Ras could be involved in positive or negative regulatory feedback loops. PIP2 is known to play a role in K-Ras localization in cells (31, 32) but the mechanism is debated and remains unclear (e.g. Ref. 33). At the protein level, we have the opportunity to understand the molecular (structural/dynamics) details of the interactions, and especially in the context of HVR isoform- and compartment-specific Ras signaling those rules are just beginning to be uncovered. For example, a number of studies have established that the dynamic interactions of the Ras HVR motifs with membranes modulate the targeting of Ras to the cell surface or to intracellular organelles (34–36). In addition, different HVR motifs in Ras isoforms also specify localization within plasma membrane subdomains (37, 38) and recently the Hancock laboratory (33) showed how mutations or phosphorylation in the K-Ras4B HVR can switch phosphatidylserine to PIP2-mediated membrane localization.
However, we and others have substantiated the view that the Ras G-domain–lipid interactions can also play a significant role in determining the protein's configuration (i.e. G-domain orientation) and dynamics at the membrane. For example, recent computational investigations suggested the participation of the K-Ras.G12V G-domain in binding with POPS membrane (39–42) and with PIP2 containing membranes (42, 43). Experimental work by Ikura and colleagues (44) also provided evidence that the K-Ras G-domain exists in several distinct orientations when contacting a bilayer of POPS lipid membrane. However, a detailed experimental study on K-Ras4B–PIP2 interactions has been missing.
Here, we utilized in vitro binding assays, NMR spectroscopy, computational simulations, and cell function experiments to study the K-Ras4B interaction with a PIP2 containing membrane. Our data clearly demonstrate a direct interaction between K-Ras4B and PIP2, also of the GTPases's G-domain and we provide structural details of the interactions and K-Ras4B orientations relative to the lipid bilayer. Last, the functional relevance of K-Ras4B–PIP2 interactions are corroborated when key residues are mutated and such PIP2 binding-compromised GTPases are examined in cell-transformation activity and intracellular localization experiments. Implications of these findings for Ras localization and function in cell signaling are discussed.
Results
Full-length as well as HVR-truncated K-Ras4B binds to PIP2 and other specific lipids
Lipid strip assays were first carried out to screen the lipids that can bind to unmodified K-Ras4B (the HVR is not lipidated or tuncated, as we are interested in the initial membrane interactions that the Ras G-domain may make). Fig. 2 shows the results for the binding of full-length (top) and HVR-truncated (bottom) K-Ras4B with different lipids. It can be seen that full-length K-Ras4B can bind to singly phosphorylated inositol phosphates, PIP2, PA (weakly), and phosphatidylserine (PS). Comparing the results of full-length and truncated K-Ras, overall, lipid binding with the full-length K-Ras is stronger. Additionally, when the HVR domain was truncated from K-Ras4B, the interactions with some lipids were abolished (e.g. PA and PI(3,4)P2 especially, in case of truncated K-Ras.S17N). These observations suggest that the HVR of K-Ras is necessary for binding these lipids. However, binding with singly phosphorylated inositol phosphates, PI(3,5)P2 and PI(4,5)P2, are maintained for HVR-truncated K-Ras proteins both in G12V.GTP and S17N.GDP forms, although the binding with PIP2s is clearly weaker, compared with the full-length protein. Nussinov, Gaponenko and co-workers (45) showed a lipid-strip assay of GDP-loaded K-Ras4B in a Current Opinions Review. This overall is consistent with our data showing that PA binding is associated with the full-length protein. In summary, our observations show that the K-Ras4B G-domain binds with selective lipids.
Figure 2.
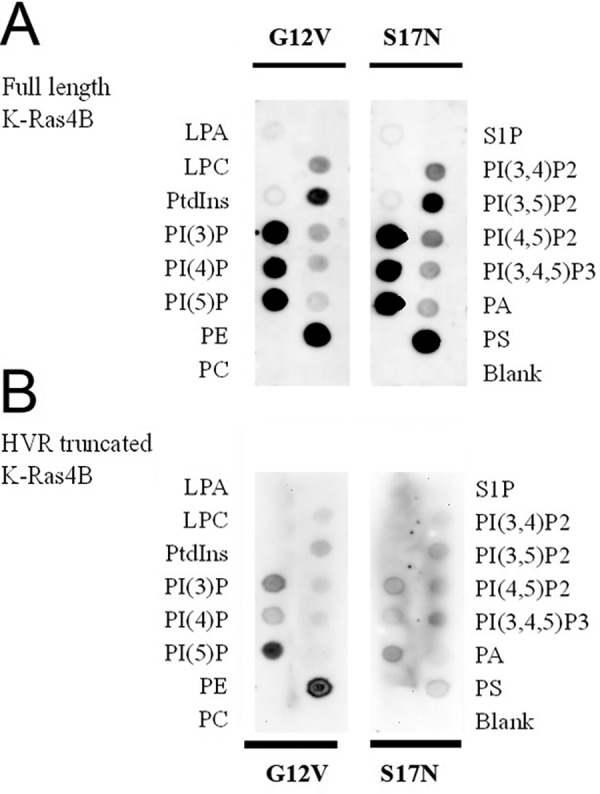
Lipid-strip assay shows binding and that the interactions are lipid, HVR but less so GTPase-state selective. A, full-length K-Ras4B (1–188); and B, HVR-truncated K-Ras4B (1–169) as (i) G12V constitutively active mutant initially loaded with GTP, (ii) dominant-negative mutant S17N, bound to GDP. Note that images in B were collected for 8 min, whereas in A, a 2-min data collection was used.
K-Ras4B binds to PI(4,5)P2
Our lipid-strip assays showed that K-Ras4B can interact with PIP2s including PI(3,4)P2, PI(3,5)P2, and PI(4,5)P2. This study specifically focuses on the binding between K-Ras4B with signaling lipid PI(4,5)P2. To further confirm the binding between K-Ras4B and PI(4,5)P2, microscale thermophoresis (MST) was employed, also for measuring the binding affinity. The purified K-Ras4B protein was fluorescently labeled with NT-647 dye, and then titrated against PIP2-doped liposomes (5% PIP2, 95% DOPC). A representative dataset for full-length unmodified and HVR-truncated K-Ras4B is shown in Fig. 3, and the fitted Kd is 28 ± 7 μm, indicating a moderate binding between K-Ras4B and PI(4,5)P2 in vitro. However, the binding of HVR-truncated K-Ras to these liposomes is significantly decreased (about 9-fold) pointing to the importance of the HVR to enhance affinity. We similarly studied a number of G-domain mutants, designed to disrupt PIP2 binding (see below).
Figure 3.

MST measurement of full-length K-Ras4B and HVR-truncated K-Ras4B interaction with 5% PI(4,5)P2-doped DOPC lipid vesicles. A DOPC liposome doped with fluorescently labeled and unlabeled PI(4,5)P2 was titrated with a serial dilution of full-length K-Ras4B (1–188) G12V (black curve) or HVR truncated K-Ras4B (1–169) G12V (red curve) in a 2-fold dilution series.
K-Ras4B on PI(4,5)P2 membrane-interface and orientations studied by NMR
The NMR spectrum of HVR-truncated K-Ras4B.GDP has been assigned (46) and we first sought to characterize PIP2 lipid binding to the K-Ras4B G-domain by use of the lipid headgroup, IP3. However, the perturbations to the NMR spectra were both small and distributed across the protein, making it unlikely that all changes reflect the true configurational states at a lipid bilayer surface (see Fig. S1 and Table S1). Recent developments in NMR have made it possible to study integral membrane proteins using lipid bilayer-like discs, bicelles or nanodiscs, up to 10 nm in diameter (47, 48). Here, we next used PIP2-doped nanodiscs. Slightly smaller perturbations were seen compared with the lipid headgroup, overall affecting a similar number of resonances (Fig. S2) and the data were not analyzed further. It is possible that nanodiscs are too small to allow good diffusion of the lipid spin label (approximately 2 per disc), which may on average also occupy a noninteracting side. However, increasing the spin label concentration did not improve the discrimination between interacting and noninteracting residues, suggesting, as an alternative that K-Ras may interact nonspecifically and transiently with the disc-bounding peptide in the absence of membrane anchoring. Thus, we decided to go to an even bigger membrane model system, large unilamellar vesicles, or liposomes.
NMR spectroscopy is a unique method for the characterization of weak and dynamic interactions (49, 50). As mentioned by contrast to the studies of Ikura and colleagues (44), we do not have an anchoring of K-Ras4B to the membrane via a lipidated C terminus. The latter would likely lead to more persistent membrane contacts and, even with a nanodisc, a corresponding increase in the molecular correlation time, resulting in an extensive line broadening, especially of the backbone resonances (44). In our case interactions are very transient to the extent that with a liposome (of 100 nm in diameter), effects are due to proximity of a spin label, such as paramagnetic ion gadolinium (Gd3+), far outweigh the effects due to transient attachment and large correlation times. In such a context, PRE appears to be a sensitive tool for transient interactions (51–54).
Fig. 4A shows the superimposed 15N-1H HSQC-TROSY spectra of full-length K-Ras4B.G12V with PIP2-doped DOPC lipid vesicles without (red) and with (green) a Gd3+ spin-labeled lipid. A decrease in peak intensity indicates proximity to the spin-labeled lipid, taken as binding to the liposome. Fig. 4B is a plot of the peak intensity ratio versus protein sequence (only to residue 159, as the remainder has not been assigned. Many signals in the center of the spectrum, fully shown in Fig. S3, overlap and, thus, are not clearly attributable, with some likely belonging to the HVR.) The intensity of a good number of NMR signals from the amide groups in the G-domain are perturbed, indicating the involvement of these residues in binding that brings them close to the spin label. Specifically, the perturbations are mainly in the catalytic lobe and fewer in the allosteric lobe. In the catalytic lobe, the perturbations are mostly located at strands β1, β2, and β3, the C terminus of α3. In the allosteric lobe, the perturbations are mainly at the termini of β6 and the loop between β6 and helix α5, occasionally at helix α3. Table S1 lists the residues that are implicated by these data to interact with the PIP2(4,5)-doped liposome. Those residues were mapped to the 3D structure of the K-Ras4B G-domain, as shown in Fig. 4C. The distribution of these residues suggests that not all residues involved in interaction can be simultaneously satisfied by a single protein–lipid interface. This indicates a dynamic equilibrium between at least two orientations of K-Ras4B on the PIP2 membrane, employing different sites of K-Ras for membrane association.
Figure 4.

Full-length K-Ras4B (1–188) interaction with PIP2-doped DOPC liposomes studied by NMR spectroscopy. A, representative areas of superimposed 15N-1H HSQC-TROSY spectra of full-length K-Ras4B.G12V recorded at 800 MHz and 37 °C, in the presence of PIP2-doped DOPC lipid vesicles without (red) and with (green) Gd3+ spin-labeled lipid (the full range spectrum is shown in Fig. S3). B, peak intensity change as a function of protein sequence. The residues decreased >15% in peak intensity are shown as blue bars, the residues increased >15% in peak intensity are shown as red bars, and the residues changed within ± 15% are shown as green bars. Residue positions not assigned in the NMR spectrum are left blank, but the data are shown as UN1 to UN27 at the end of the sequence. The K-Ras4B switch regions as well as the secondary structure are indicated at the bottom. C, peak intensity changes are mapped to the 3D structure of K-Ras4B.G12V (PDB 4TQ9) and are displayed as two orientations of the protein. The color scheme is same as B, with unassigned residues shown in gray, and the switch regions shown in orange (the location of the C terminus is indicated by an asterisk).
We further investigated the interaction of HVR-truncated K-Ras with the PIP2-doped liposome. Fig. 5A shows the plots of the peak intensity ratio versus protein sequence. Several residues in the G-domain still experience diminished peak intensity, confirming the interaction between truncated K-Ras4B and the PIP2 liposome. The perturbed residues in truncated K-Ras include part of the β2- and β3-strands in the catalytic lobe. Similar to the full-length protein, peaks from residues located in β6 and in the turn between β6 and helix α5 experience the most significant intensity changes, as well as for two unassigned residues. A mapping of perturbed residues onto the 3D K-Ras structure is shown in Fig. 5B. However, the extent of intensity change is diminished compared with the full-length K-Ras, consistent with the other experiments above, which showed that binding is more transient for HVR-truncated K-Ras. Furthermore, the perturbed residues are much less spread out in the protein sequence, possibly because the HVR–lipid interaction also creates longer range effects in the full-length protein. Nevertheless, again the distribution of these residues in the structure is better accommodated by two or more orientations of K-Ras4B on membrane than by a single orientation.
Figure 5.

Truncated K-Ras. G12V (1–169) interaction with PIP2-doped DOPC liposomes studied by NMR spectroscopy. The legend to A and B, is the same as for Fig. 4, B and C, respectively.
K-Ras4B on PI(4,5)P2 membrane-interface and orientations studied by simulation
All-atom classical MD simulations were also used to examine the K-Ras4B regions that interact with PIP2. Because the re-orientation of the K-Ras4A GTPase at membranes is slow (42), simulations were started with two orientations of K-Ras4B (orientation states OS1 and OS2) previously predicted by more extensive simulations at a POPS containing bilayer (40). Then simulations of K-Ras4B interacting with a PIP2 containing membrane were performed for 380 ns, sufficient to allow clustering of PIP2 near the protein. The results show that the two orientations have remained stable over the course of each of the trajectories. It should be noted that for our previous K-Ras4A (42) and K-Ras4B (40) simulations with PIP2, primarily the OS1 state was found to be populated (similar to O3 in Li and Buck (42)). The OS2 started simulation may not be able to interconvert to OS1 on the timescale of the current simulations, also having been stabilized by PIP2 contacts; such transition was seen, however, by Gregory et al. (43) using a membrane model with increased fluidity.
Snapshots for the final configurations of K-Ras4B with respect to the membrane are shown in Fig. 6A. In both orientations, the G-domain as well as the HVR domain contact the membrane. But the two orientations involve different regions of the G-domain. In the OS1 orientation, the interaction mainly involves α-helices 3 and 4 in the globular domain, whereas the OS2 orientation primarily involves the β-sheet and α-helix 2. Fig. 6B shows the specific residues that frequently contact the membrane during the simulation. The HVR region in both cases involves a large number of contacts with the membrane, due to the insertion of the farnesyl group into the membrane as well as the favorable interaction between the multiple lysine groups (stretch 175KKKKKKSKTK184 of the HVR) and the membrane. In the G-domain, the membrane contacting residues are mostly positively charged residues. But several negatively charged residues also participate in interactions with the membrane, such as Glu-3, Glu-107, and Asp-132 (Table S2). Although the positively charged residues directly interact with PIP2 phosphate groups in membrane, the interaction between some of the negatively charged residues and the PIP2 phosphate was revealed to be bridged by the Na+ ions, as shown in Li et al. (42) and in Fig. 6C, with representative interactions between Arg/Lys/Glu residues and PIP2 lipid. Overall, the electrostatic interactions likely play a major role in K-Ras interaction with the PIP2 in the membrane. Moreover, residues showing high contact frequency with the membrane in simulations are consistent with the NMR experimental results, not only the exact same residues but also close-by residues in the experiments (also considering gaps in the experimental data because not all assignments could be transferred to our spectra). Examples are Lys-5 in β1 (Lys-5 in experiments), Arg-41 in β2 (Ser-39 in experiments), Arg-97 in α3 (His-95 in experiments), Lys-128 in α4 (Asp-126 in experiments), Ser-136, Tyr-137, and Gly-138 in α4 (Ile-139 in experiments).
Figure 6.

Simulation of K-Ras4B at a membrane composed of POPC and PIP2. A, snapshots of the orientation of K-Ras4B.G12V at the PIP2 containing membrane at the end of simulation; left, OS1, and right, OS2 started simulation. B, contact frequency of K-Ras4B residues interacting with the PIP2 membrane (residues within 3 Å of the membrane surface are considered). Top: OS1 orientation; bottom, OS2 orientation. C, representative interactions between Arg/Lys/Glu residues and PIP2 lipids in each of the final structures.
The majority of lipid-binding defective mutants show decreased lipid affinity
Following the indications of PIP2-binding sites at several regions of the G-domain, we investigated the binding of several mutants of K-Ras.G12V with PIP2-doped liposomes by MST, as detailed above. The rationale for choosing the sites was severalfold. The NMR and simulations data above suggest a number of surface clusters of residues that are involved in K-Ras4B–PIP2 interactions. Thus we chose K16E (next to Ser-17), D47K, D92N, D126N, as well as K165E/K167E (at the end of the G-domain). E49K, R135A, K147E, and R164Q fulfill the above criteria but are also cancer-associated mutations (missense, nonsense but also as silent mutations) (56) and appear in the COSMIC database (57)). K88E was included as a residue that is not involved in a cluster and thus unlikely to be involved in PIP2 binding. K104M was included because Lys-104 is a target of posttranslational acetylation. In all cases, except for K16E, binding affinity to liposomes was reduced at least 3-fold, in many cases 9-fold or more (Table 1).
Table 1.
Binding affinities of K-Ras4B mutants interaction with 5% PI(4,5)P2-doped DOPC lipid vesicles measured by MST
A DOPC liposome doped with fluorescently labeled and unlabeled PI(4,5)P2 was titrated with a serial dilution of K-Ras4B proteins.
| K-Ras.G12V (1–188) mutants binding with PI(4,5)P2-doped DOPC liposome | Binding affinity (Kd) |
|---|---|
| μm | |
| K16E | 30.0 ± 1.7 |
| D47K | >1180 |
| E49K | 246 ± 44.5 |
| K88E | 118.5 ± 5.6 |
| D92N | 146.8 ± 22.3 |
| K104M | 262 ± 7.1 |
| D126N | 119 ± 16.3 |
| R135A | 141.25 ± 39.8 |
| R164Q | 129.9 ± 37.3 |
| K147E | 239.2 ± 50.2 |
| K165E and K167E | Too weak to be detected |
The effect of mutations on biological function: excluding an effect on intrinsic and regulated GAP and GEF activity of K-Ras4B by use of in vitro assays
We first examined the hydrolysis and exchange activity of K-Ras4B, including both intrinsic activity and stimulated activity by regulatory proteins p120Ras as a GTPase-activating protein (GAP), and SOS as a guanine exchange factor (GEF). If the mutants are greatly altered in their hydrolysis and exchange activity, it is clear that the mutations already have effects solely on the K-Ras protein, even though they probably also affect the lipid binding of the GTPase core region.
The measured intrinsic and stimulated hydrolysis rates are shown in Fig. 7A. The K-Ras4B.G12V constitutively active form was used as negative control. The activity of the other mutants were calculated relative to the WT (the stimulated hydrolysis activity of WT with p120Ras GAP was scaled to 100). Among the 11 mutations, the K16E mutation substantially decreases (essentially disrupted) the p120 GAP-stimulated GTP hydrolysis rate compared with the WT, whereas apart from the K104M and K165E/K167E mutations, all others increase the p120 GAP-stimulated GTP hydrolysis rate compared with the WT. The intrinsic and stimulated nucleotide exchange rates are shown in Fig. 7B. The K-Ras4B.S17N mutant is a dominant-negative form in vitro and was used as negative control. Among the 11 mutations, the K16E and K147E mutations substantially decrease the SOS GEF-stimulated exchange rate compared with that seen with the WT GTPase. The other mutations show little/no significant changes.
Figure 7.

GAP and GEF activity of designed K-Ras4B mutants. Assays were carried out for the intrinsic and p120Ras GAP-aided GTP hydrolysis activity and for the intrinsic and SOS GEF-stimulated nucleotide exchange activity. A, representative time courses of intrinsic and p120 GAP-stimulated GTP hydrolysis activities are shown for K-Ras WT and G12V. B, representative time courses of intrinsic and SOS GEF-stimulated exchange activities are shown for K-Ras WT and S17N. C, histograms of relative intrinsic and p120 GAP-stimulated hydrolysis activities of K-Ras constructs, with intrinsic hydrolysis activities shown in red, and stimulated hydrolysis rates shown in blue (WT K-Ras was scaled to 100%). D, histograms of relative intrinsic and SOS GEF-stimulated exchange activities of K-Ras constructs, with intrinsic exchange activities shown in red, and stimulated exchange rates shown in green (WT scaled to 100%). Note: the intrinsic rates in C and D and their uncertainties are plotted as multiplied by a factor of 3 to increase visibility.
Effect of mutations on K-Ras4B transforming activity
Next we wished to study the role of lipid binding in the biological activities of the mutants of oncogenic K-Ras4B.G12V in intact cells. We employed the established ability of this mutant protein to induce the loss of contact inhibition in confluent cultured fibroblasts (58), an accepted surrogate for oncogene-induced cell transformation (59). The cell transformation results of K-Ras4B mutants are shown in Fig. 8. Mutations of the following residues caused a significant impairment in the focus formation activity of K-Ras4B.G12V: Lys-16, Asp-47, Asp-92, Lys-104, Asp-126, and Lys-147. Among these, the effect of mutations K16E, D47E, and K147E is not attributable to disruption of lipid binding alone, because these mutations altered the GAP or GEF activity too (see Fig. 7). By contrast, we have shown that mutations D92N, D126N, and K104M have essentially unaltered nucleotide hydrolysis and exchange activities. The impairment in the focus formation, thus, lends direct support for the involvement of these residues in lipid binding and that the lipid binding is essential for the protein's biological activity.
Figure 8.
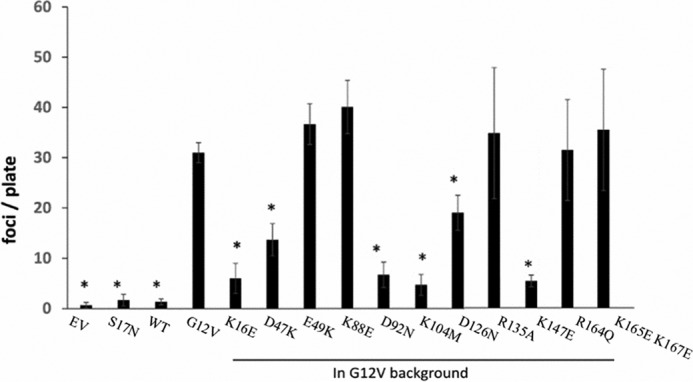
Transformation activity of designed K-Ras4B mutants as measured in foci formation of NIH3T3 cells. y axis of the histograms is the foci count per plate (asterisk indicates the transformation activity of mutants that are statistically significantly from G12V based on apaired t test, i.e. p < 0.05). EV indicates untransformed cells. K-Ras4B.G12V and K-Ras4B.S17N were included as positive control and negative control, respectively. The foci formation activity of the designed K-Ras4B mutants were all based on the oncogenic G12V background.
Effect of mutations on intracellular localization of K-Ras4B
We further examined how mutations of lipid-binding residues affect K-Ras4B's intracellular localization in cultured cells. Fig. 9 shows results from microscopy experiments, where transfected K-Ras4B alleles were visualized with anti-K-Ras immunofluorescence and the actin cytoskeleton highlighted with fluorescently-tagged phalloidin. As anticipated and reported earlier (60), we observed the localization of the constitutively active K-Ras (G12V) allele at the cells' periphery, along with cortical actin that marks the plasma membrane. However, different mutations of lipid-binding residues have different impact on K-Ras localization. The K104M mutation completely disrupted plasma membrane localization, indicating that this residue indeed is critical in lipid binding or for another process involving intracellular localization of K-Ras4B. This provided a direct explanation for the inactivity of this construct in the transformation assays (Fig. 8). On the contrary, the D92N and D126N alleles did not significantly affect the localization, and remained localized at the cells' periphery. But meanwhile transformation assays show that these two constructs substantially diminished the foci activity of NIH3T3 fibroblasts. The possible explanation is that these two residues indeed are involved in K-Ras–PIP2 membrane binding and tune the orientation of K-Ras at the membrane. Changes of K-Ras4B orientation on the membrane rendered K-Ras4B unable to bind certain downstream regulators, subsequently diminishing the transformation activity.
Figure 9.
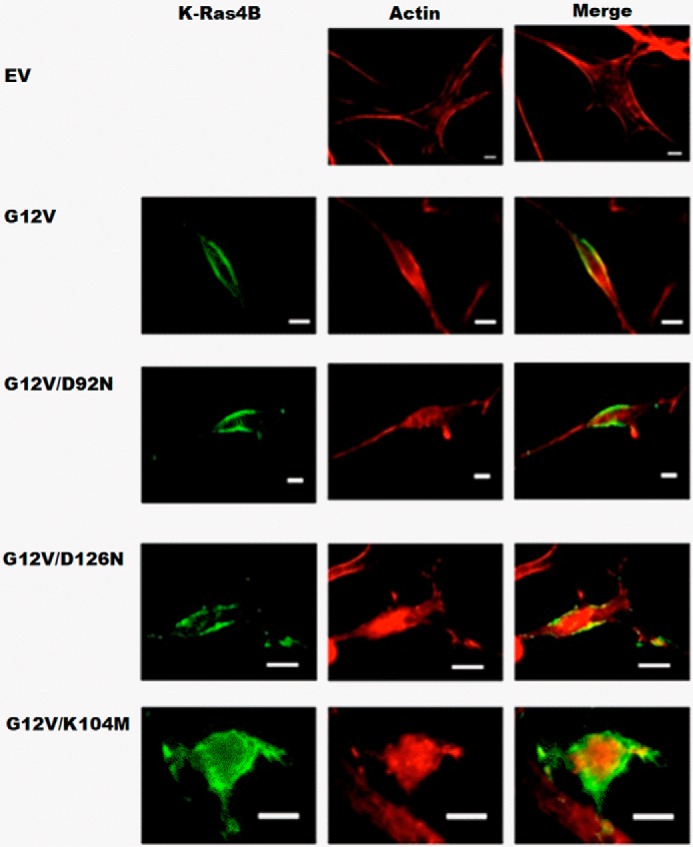
Intracellular localization of designed K-Ras4B mutants in cultured NIH3T3 cells using fluorescence microscopy. Transfected K-Ras4B alleles were visualized with anti-K-Ras immunofluorescence and the actin cytoskeleton highlighted with fluorescently-tagged phalloidin. Images are shown for Ev, K-Ras4B.G12V, and designed K-Ras mutants G12V/D92N, G12V/D126N, G12V/K104M. Ev, indicates untransformed cells, and K-Ras4B.G12V was included as positive control.
Discussion
Our experimental data from a lipid-strip assay, solution NMR spectroscopy, microscale thermophoresis, and also all atom molecular dynamics simulations clearly show that K-Ras4B directly interacts with PIP2. Binding is more extensive and appears stronger when several protein sites are involved simultaneously, consistent with the concept of binding avidity (multivalence) (61, 62). In fact, most mutations at the suggested binding sites substantially (3–6–fold) diminish affinity, but do not outright abolish it (Fig. 3, Table 1). We confirm that the interaction of K-Ras4B with PIP2 involves both the G-domain and the HVR region, as shown by experiments where the HVR was truncated. Furthermore, our NMR data and also previous simulations with K-Ras4A and PIP2, suggest that the G-domain samples multiple orientations relative to the PIP2 membrane (42). Such dynamic protein configurational states at the membrane are likely to be important for cell signaling kinetics (see below).
Involvement of the G-domain of K-Ras GTPases in binding to PS containing membranes has been previously reported in several papers. In a simulation study, Prakash et al. (40) reported the involvement of helices 3 and 4, and β-strands 1–3, as well as helix 2 on the opposite face of the catalytic domain in the K-Ras4B G12D interaction with POPS/POPC bilayers. More recently, the interaction of C terminally membrane-anchored K-Ras4B with PS was also studied experimentally by Ikura and colleagues (44, 63) using nanodiscs. This study revealed two, if not possibly more, orientation states, but also suggested a GTPase nucleotide and effector-bound state-specific shift between these states. However, as mentioned in the Introduction, allosteric residue/residue and residue/solvent networks (e.g. Ref. 64) have been described for Ras, which propagate changes that occur in the catalytic domain/switch regions to other regions in the protein. Thus, in principle it is possible that the interpretation of the NMR data by us (and others) in terms of an equilibrium between multiple orientations needs to be considered with caution. An argument against local or long-range conformational changes in our study is the lack of perturbation to the chemical shifts of the NMR resonances. Although in our study membrane–nanodisc/liposome interactions are very transient, this is not the case in other studies (40). Although it is possible that the allosteric change upon membrane binding is dynamic, as has been seen in some GTPases (65) and may contribute, similar to the spin labeling effect, to a broadening of resonances, we find the effects on NMR signal line widths are very local. The same argument about the transient nature of the membrane-bound state applies in in our study: the population of such states would not be large enough to detect any allosteric effects.
In general, our results for K-Ras4B at the PIP2 membrane are in good agreement with this experimental study of K-Ras4B at PS containing nanodiscs (40). Although orientation 1 (OS1) resembles the GTP state orientation, orientation 2 (OS2) resembles the GDP state orientation in their paper, there are also some differences, which likely arise because of the different nature of PIP2 compared with PS lipid. Specifically, PIP2 has a −3 charge compared with the −1 charge of PS and also has a larger headgroup. As we discussed in Li and Buck (42), the electrostatic interaction between K-Ras and PIP2 dominates the binding, and thus may account for the orientation change of K-Ras on PIP2-containing membranes (increased population of OS1-like states). However, it is worth noting that in the experimental study K-Ras was bound to the membrane via a thiol-reactive maleimide-functionalized lipid (40). By contrast, our K-Ras is not anchored to the membrane and thus our experiments report more on transient events, rather than tightly bound states or the transitions between them. Thus, the similarity between the results of these two studies indicate that the transient encounters occur with orientations, which are equivalent to those seen in the more persistently membrane-bound G-domain. This is consistent with the observation that there are no significant medium- or long-range contacts between the G-domain and the HVR in these simulations (40–42). Overall these findings are consistent with the emerging view that whereas the HVR plays a major role in lipid binding selectivity and in strengthening Ras-membrane interactions (33), the orientation states of the G-domain are closely related to the identity of the Ras GTPase, with the HVR playing a minor role (66).
The interactions of the G-domain with the membrane imply that they may help the GTPase to associate with membranes prior to C-terminal lipidation or in other settings, such as binding to the mitochondrial outer membrane, where lipidation is not needed (67); thus our in vitro biophysical study was carried out with the full-length but nonlipidated protein, or with nonlipidated HVR-truncated protein. Such interactions between the G-domain and PIP2 containing membranes could be especially relevant biologically in cases where the lipid group is not yet attached, or when it is occluded by other GTPase regulators, such as phosphodiesterase (PDEδ) (68), or other lipid binding/GTPase chaperone proteins (69). Although it is long established that lipidated HVRs helps Ras GTPases anchor to the membrane, decades of research into the development of compounds that interfere with HVR lipidation, processing, and subsequent binding to membranes have failed to provide effective therapeutic reagents (70, 71). Thus the binding interface between G-domain and membrane may provide a novel avenue to target K-Ras, as indeed has been very recently reported (63).
Electrostatic interactions likely play an important role in K-Ras4B binding with PIP2. As shown in the contact frequencies analysis of the simulation results, protein-charged residues have a high frequency of contact with PIP2 in the membrane. But different from our intuition, not only positive but also negative residues participated in binding with the anionic PIP2 membrane. Although the positively charged residues interact directly with the negatively charged PIP2 headgroup, negatively charged residues can be indirectly bridged with PIP2 by sodium ions. This is supported by NMR results also in that multiple residues identified by NMR in K-Ras4B binding with PIP2 are immediately adjacent to the corresponding charged residues listed in the contact frequency analysis of simulation. The role of negatively charged residues and bridging sodium ions was first suggested in our recent simulation study of K-Ras4A binding with PIP2 lipid (42). At the same time work by Hancock and colleagues (33) have also implicated a role of hydrogen bonding of Arg versus Lys side chains and even for hydrophobic interactions between the HVR and lipids. We do not know whether the interactions can also be cation specific, although preliminary experiments suggest a 3-fold weaker binding to PIP2 membranes in the presence of both 50 and 250 mm KCl, compared with NaCl. Such interactions need to be investigated further experimentally and computationally, due to their importance for K-Ras (e.g. a reduced Na+/K+ voltage gradient across the membrane has been shown to enhance interactions of K-Ras with PS containing membranes by Hancock and colleagues (72)) and other G-domain systems (G-protein gated channels (e.g. Ref. 73) and the A2A GPCR (74)). Calcium is known to bind to some small GTPases (75) but also causes a clustering of PIP2 (76, 77).
The interaction of K-Ras4B with a PIP2 containing membrane is dynamic rather than fixed, likely more so because, as noted in this study, K-Ras is not membrane anchored. Mentioned under “Results,” we were able to exploit the sensitivity of NMR to dynamics on a range of different timescales and record PRE-data upon binding to liposomes, doped with PIP2, and spin-labeled lipid. From our results, we propose there is an equilibrium of unbound protein and multiple orientations of K-Ras4B that are bound to the PIP2 membrane. This may be important for the diverse functions of K-Ras. The G-domain of Ras GTPases binds GDP/GTP and associates with effectors, GEFs and GAPs (8). The multiple orientations thus enable K-Ras to interact with certain regulatory and effector proteins at different contexts, whereas hindering the interaction with others. Indeed, a recent nuclear magnetic resonance (NMR) study of K-Ras proposed that orientation preference of K-Ras4B at the POPS bilayer membrane is nucleotide dependent (44). Specifically, the GTP state K-RAS4B binding with membrane occludes its interaction with effectors. On the contrary, GDP state K-Ras4B, as well as in the oncogenic G12D mutant, the G-domain interaction with membrane exposed its binding interface for effectors and regulatory proteins (44). Another simulation as well as experimental study showed that H-Ras sampled two major orientations on a DOPC bilayer, and that the population of the two orientations can be tuned by mutating residues in the G- versus the HVR domain (39). Simulation studies on K-Ras also suggested two major orientations of K-Ras at a DOPC bilayer (78), a l-α-dimyristoylphosphatidylcholine bilayer (79) and at POPC/POPS bilayer (40). So the multiple orientations of K-Ras4B on a PIP2 containing membrane may not be unique but are probably a common phenomenon in interactions of different GTPases with different lipids.
PIP2 binding to K-Ras4A (42) and here K-Ras4B, appears to stabilize an OS1-like state (denoted O3 in our previous paper), which has the effector and regulatory binding switch regions exposed to the cytoplasm and thus is expected to increase the signaling function of the GTPase, relative to other membrane-bound states that occlude these regions. How the membrane regulates the functions of K-Ras in a live cell environment, however, is rather complicated.
First, K-Ras can be posttranslationally modified by phosphorylation, nitrosylation, ubiquitination, or acetylation on certain residues, all of which may affect GTPase function and targeting. In the case of the mutation of Lys-104, this will affect a primary acetylation site. Yang et al. (26) showed that the K104Q mutation (an acetylation mimic) suppressed transformation activity (similarly to K104M in our case) but by contrast to our results did not affect localization to the plasma membrane. Another more recent study found K104Q compromised both GEF and GAP activity, but had no effect on cell transformation, possibly due to compensatory mechanisms in the cell (80). Our results are different in that K104M has normal GEF and GAP function, but is compromised in its localization and transformation ability. The former results may be explained by the methionine side chain being of similar geometry to lysine, in our case, whereas the latter may indeed also be due to a difference in residue side chain character, with glutamine still being able to hydrogen-bond and interact with PIP2, whereas a methionine may not be able.
Second, the concentration of PIP2 in membranes can vary, but is thought to be 2–3% overall (25). The lipid-strip assay presents pure lipid species at high concentrations and therefore is not very physiological. By contrast, our study with 5% PIP2-doped DOPC nanodiscs and liposomes is closer. It is becoming established that certain microdomains or compartments contain higher concentrations of PIP2 as well as other lipids (81, 82). In addition, membrane curvature and packing defects are likely to play a role, especially for protein-lipidated tail insertion or partial protein insertion into membranes. It is intriguing that PIP2 accumulates that on curved membrane segments (83) and may thus be helpful in stabilizing certain Ras orientation states in such compartments, such as the endoplasmic reticulum, where lipidation and processing of the Ras C-terminal HVR occurs (11). Although our liposomes have considerable curvature compared with the nanodiscs that are assumed to be flat, we notice a slightly higher binding affinity of PIP2 for the K-Ras G-domain in the former, this topic awaits further study.
Third, membranes are a complex mixture of several lipids (e.g. Ref. 16) and local clustering of lipids (and the involvement of proteins in this process) is just beginning to be understood (62, 84). For example, it may be expected that the PS can, to some extent, mask, if not compete with protein–PIP2 interactions in physiological membranes. However, this could be protein specific as indicated by one recent study, where the apparent K-Ras4B–lipid interaction specificity could be switched by a lysine to arginine mutation or Ser-181 phosphorylation in the HVR region (33). K-Ras clustering seems to have the ability to sort certain lipids, depending on the character of the long acyl chain, noting that in most studies, such as ours, synthetic lipids are used. Thus, the cellular behavior may involve lipid species, also of PIP2 that have not been tested in vitro or in model system.
Fourth, several studies have provided evidence for the existence and biological relevance of Ras dimers, establishing a new mechanism for regulating Ras activity (85, 86). Research by the Hancock group and others (87–90) have shown that N-, K-, and H-Ras assemble into higher order oligomers and nanoclusters, and do so in an isoform-specific manner. However, recently, neither K-Ras dimerization, nor clustering could be shown on supported lipid bilayers with a range of lipid compositions, suggesting that additional proteins, e.g. the cytoskeleton as a scaffold, may be required (91). The situation in cancer cells is likely even more complicated, due to the different types of K-Ras mutations (recently by Ambrogio et al. (92)), and mutations/abnormal expressions of Ras-regulating proteins and signaling lipids too.
To resolve the mechanism of Ras regulation by membranes, further experimental and computational studies of Ras cancer mutants, of post-translationally modified forms of Ras, of Ras in complex with regulatory proteins, of dimers and oligomers of K-Ras, all at the membrane, will be needed. The issue of allostery within Ras has only been considered in a relatively few studies and whereas difficult to examine experimentally, networks showing either conformational and/or dynamic allostery can be analyzed computationally. In addition, most effector proteins bind to the Ras GTPases at the membrane. For example, our laboratory has recently published a simulation study of K-Ras-C Raf (CRD-RBD domain) complexes at a POPS membrane (93), which illustrated the complexity of such systems when the effector protein domains interact with the membrane as well.
Concluding Summary
The present data provide, to our knowledge, the first experimental study of Ras–membrane interaction using liposomes and NMR, and also provide a residue level structural report of the K-Ras G-domain binding to PIP2. Binding to PIP2 likely provides an additional mechanism to tune, if not regulate GTPase signaling activity in cells.
Experimental procedures
Protein constructs, expression, purification, and site-directed mutagenesis
The cDNA for human K-Ras4B (residues 1–188) was obtained from the cDNA Resource Center at Bloomsburg University and subcloned into pET28a using NdeI and BamHI restriction sites. HVR-truncated K-Ras4B (residues 1–169) and the mutants were made using the QuikChange Lightning site-directed mutagenesis kit (Agilent). A subgroup of residues that are identified in K-Ras–PIP2 membrane binding were selected for mutation, as detailed in the main text. Transformed Escherichia coli strain BL21(DE3) bacteria (Novagen) were grown at 37 °C in LB, and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for expression at 25 °C overnight. Bacterial pellets were resuspended in the following buffer: 20 mm Tris-HCl, 150 mm NaCl, 0.5 mm TCEP (tris(2-carboxyethyl)phosphine hydrochloride), 4.0 mm MgCl2, pH 7.5, with added protease inhibitors (phenylmethylsulfonyl fluoride, 1 mm; benzamidine, 10 mm; leupeptin, 42 μm; antipain, 3 μm). Following sonication, lysates were clarified by centrifugation and recombinant proteins were purified from the supernatant using nickel-nitrilotriacetic acid–agarose (Qiagen). K-Ras proteins were dialyzed against NMR buffer (20 mm Tris-HCl, 20 mm NaCl, 1 mm TCEP, and 4 mm MgCl2, pH 7.4). Purified proteins were >90% pure identified by SDS-PAGE. For NMR experiments, K-Ras was 15N uniformly labeled by growing bacteria in M9 medium containing 15NH4Cl as the sole nitrogen source.
Similar to other protocols, initially K-Ras.WT, G12V, and mutant proteins were loaded with nucleotides (GTP or its nonhydrolyzable analog, GMPPNP) by incubating 100 μm freshly purified protein with 0.5 mm nucleotide in 20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5 mm EDTA, 2 mm DTT at 30 °C for 10 min. The reaction was stopped by adding 50 mm MgCl2. A PD-10 Sephadex G-25 desalting column was used to remove the excess nucleotide, and to change the buffer to 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm TCEP, 4 mm MgCl2. To our surprise, this protocol did not work well and a TLC assay showed that K-Ras WT remained bound to GDP, whereas G12V was initially bound to GTP, but hydrolyzed to GDP over several hours at room temperature. Thus, to obtain fully loaded K-Ras, the more elaborate loading protocol with a phosphatase to hydrolyze GTP and GDP is needed (63). The catalytic domain of p120 RasGAP (a gift from Dr. R. Ahmadian) was subcloned into pET28 and expressed and purified as described previously (94). The SOS catalytic domain in a pProExHTb vector was gift from Dr. J. Kuriyan. This protein was expressed and purified as described (95). Proteins were placed at 4 °C on ice for immediate use or flash frozen with an additional 5% glycerol and stored at −80 °C.
Protein–lipid overlay assay
Nitrocellulose lipid strips (P6001 from Echelon Biosciences) were blocked with 3% fatty acid-free BSA in TBS (10 mm Tris, 150 mm NaCl, 1 mm TCEP, 4 mm MgCl2), pH 8.0, 0.1% Tween 20 (denoted TSB-T) for 1 h, and incubated for 1 h with 100 nm purified His-tagged K-Ras in the same TBS-T buffer, but adjusted to pH 7.0 at room temperature. The membranes were then washed three times in TBS-T buffer at pH 7.5 for 5 min each. After washing, membranes were incubated with monoclonal anti-His6 antibody (Pierce) at 1:1,000 dilution for 1 h at room temperature, followed by additional washing and incubation with goat anti-mouse IgG horseradish peroxidase-conjugated antibody (Santa Cruz) at 1:2,000. After final washing 6 times for 5 min each with TSB-T, pH 7.5, membrane-bound K-Ras was visualized by chemiluminescence (WesternBright ECL from ADVANSTA) for 1 min (96). The protein was detected for 1–20 min exposure using FluorChem E (Protein Simple) and the image was processed in Alphaview software (ProteinSimple) to reverse black/white).
Liposome preparation
All lipids and lipid headgroups were purchased from Avanti Polar Lipids Inc. For the full-length lipids, as a first step, the lipid-chloroform solutions were dried under nitrogen gas, and then hydrated in 20 mm Tris-HCl, pH 7.4, 20 mm NaCl, 2 mm TCEP, and 4 mm MgCl2. For MST experiments, fluorescent labeled lipids TopFluor® PI(3,5)P2, TopFluor® PI(4,5)P2, and TopFluor PI(3,4,5)P3 were mixed with DOPC at the stated ratios. Rehydrated lipids were extruded through polycarbonate membranes with 100 nm pore size (Avestin) using a mini-extruder and following the manufacturers' manual (Avanti Polar Lipids Inc.) to a final lipid concentration of 500 μm. The diameter of the prepared liposome was determined by dynamic light scattering to be 120 ± 20 nm.
MST
MST was measured with fluorescently labeled liposomes to a final concentration of 5 nm, titrated by a serial dilution of K-Ras (294–0.1436 μm). K-Ras was tested in NMR buffer (see below), supplemented with 2 mg/ml of BSA. Alternatively, the K-Ras protein was fluorescently labeled with NT-647 dye, using the Monolith Protein Labeling Kit RED-NHS (NanoTemper Technologies). Mixtures of the protein–liposome solutions were filled into hydrophobic glass capillaries (Nanotemper Technologies) and measured with a Nanotemper Monolith NT.115 system (75% light-emitting diode, 20% IR laser power). The protein–liposome dissociation constant (Kd) was obtained by fitting the binding curve with the quadratic solution for the formation of a protein–liposome complex (assuming 1:1 binding), calculated from the equation: [PT] = ½ × (([P0] + [T0] + Kd − (([P0] + [T0] + Kd)2 − 4 × [P0] × [T0])1/2). Where [P0] is the concentration of the total fluorescent K-Ras; [T0] is the total lipid concentration; [PT] is the concentration of the formed protein–lipid complex. The concentration of the complex ([PT]) is derived from the fraction of fluorescent molecules that formed the complex: X = [PT]/[P0], in which X directly corresponds to the signal obtained in the MST measurement (normalized fluorescence).
Preparation of nanodiscs and liposomes for NMR
The MSP1D1 protein was used to make nanodiscs (97). The pGBHPS-MSP vector encoding membrane scaffold protein (MSP) variant 1D1 was obtained from AddGene. MSP1D1 was expressed and purified according to established protocols (98, 99). Briefly, MSP1D1 was expressed in E. coli (BL21) grown in Terrific Broth, and 37 °C and when A600 reached 2.0, protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 1 h followed by a 2.5-h incubation at 28 °C. MSP1D1 was purified using nickel-nitrilotriacetic acid-agarose (Qiagen) resin, and His tag was cleaved with tobacco etch virus protease and further purified as described elsewhere (52, 100). The efficiency of cleavage was monitored using SDS-PAGE to be >90%. For the nanodisc preparation, PIs, PE-DTPA, and DOPC were mixed at a molar ratio of 10:5:85. The lipid solution was dried under nitrogen, followed by drying under high vacuum for at least 4 h to remove the residual organic solvent. The dry film was solubilized in 100 mm cholate in the NMR buffer (20 mm Tris-HCl, 20 mm NaCl, 2 mm TCEP, and 4 mm MgCl2 and 0.01% NaN3, pH 7.4) to a final phospholipid concentration of 36 mm. The solution was subjected to three freeze/thaw cycles, vortexed, and sonicated to clarity in an ultrasonic bath. MSPD1 was added to this lipid solution at a lipid:protein molar ratio at 80:1. The lipid–protein mixture was incubated for 1 h with gentle rotation at 20 °C. Immediately following the incubation, sodium cholate was removed from the MSPD1-lipid mixture by three sequential dialyzes against NMR buffer. After the dialysis step, nanodiscs were further purified by size exclusion chromatography in NMR buffer on Superdex 200 10/300 (GE Healthcare). Finally, nanodiscs were concentrated using Amicon centrifugal units of 10-kDa MWCO to the desired concentration of ∼250 μm NMR experiments. The nanodiscs were measured with dynamic light scattering and found to have an average diameter of 10 ± 1.8 nm.
For the NMR experiments that detect binding via paramagnetic relaxation enhancement (PRE), nanodiscs and liposomes were prepared as above but were supplemented with 3.5% 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-diethylenetriaminepentaacetic acid (gadolinium salt; PE-DTPA (Gd3+)). The DOPC, PI(4,5)P2, PI(3,5)P2, PI(3,4,5)P3, as well as PE-DTPA (Gd3+) lipids were purchased from Avanti Polar Lipids. In NMR experiments, four kinds of samples were used: DOPC; DOPC + PE-DTPA (Gd3+) (96.5:3.5); DOPC + PIP2 (90:10); and DOPC + PIP2 + PE-DTPA (Gd3+) (86.5:10:3.5). Data from NMR experiments carried out with DOPC liposome and DOPC + PE-DTPA (Gd3+) were used as a control, validating that there is no significant interaction of K-Ras with nanodiscs or liposomes that consisted of DOPC lipid only (i.e. have no PIP2 component).
NMR spectroscopy
NMR spectra for K-Ras interaction with lipids using lipid headgroups and nanodiscs were recorded at 25 °C on a Bruker Avance II 800 MHz spectrometer equipped with a TXI cryoprobe. To improve the spectra quality, the NMR spectra for the K-Ras interaction with lipids using liposomes were recorded at 37 °C, yielding sharper lines and apparently, slightly more dispersed spectra. Although, the G12V mutant, the normally constitutively active protein, was used throughout, spectra at both temperatures closely correspond to the GDP form. All NMR measurements were carried out in NMR buffer (see above). For experiments using the lipid headgroup Ins(1,4,5)P3, the concentration of K-Ras protein was 150 μm, and the protein:IP3 ratio was to 1:3. In NMR experiments using nanodiscs as membrane mimetic, the concentration of K-Ras4B protein was 80 μm, the ratio of protein to nanodiscs was 1:1.2 (ratio of protein to total lipids is 1:96). In NMR experiments using liposomes, the concentration of K-Ras4B protein was 80 μm, the ratio of protein to the total lipid concentration of the liposomes was 1:100 (with 10 lipids as PIP2). The spectra were collected with 1024 × 180 (F1 × F2) points and 32, 80, and 96 scans and were processed with NMRPipe (101) and analyzed with Sparky software (Goddard TD and Kneller DG, SPARKY 3, University of California, San Francisco, CA).
Analysis of line broadening and chemical shift perturbation for NMR experiments
For PRE measurements, the peak intensities in the spectrum of K-Ras, in the presence of nanodiscs or liposomes containing PE-DTPA (Gd3+), were compared with those of a control sample prepared without spin label. The cross-peak intensities were measured using Sparky by Gaussian line fitting. Any small difference in protein concentration between samples was corrected by normalization of the calculated intensity ratios against the highest observed I*/Io (where I* is the peak intensities in the spectrum of K-Ras in the presence of nanodiscs or liposomes incorporating 3.5% PE-DTPA (Gd3+), and Io is that in the paramagnetic ion-free nanodiscs or liposomes). The weighted average chemical shift perturbation were calculated as: Δδavg = [(ΔδH)2 + (ΔδN/5)2/2]0.5. Ab initio NMR assignment of small GTPases are typically challenging projects (e.g. Cao et al. (102). Because the chemical shift perturbations of most resonances were small from the published NMR assignment for human K-Ras (residues 1–166) in the GDP-bound form at a physiological pH of 7.4 (46), assignments could be transferred with high confidence for the dispersed signals. Peaks that could not be assigned this way were in the crowded center of the spectrum and were followed as unassigned (UN) and are nevertheless, informative. All peak intensities were given uncertainties relative to spectral baseline noise and those uncertainties were propagated to the peak intensity change ratios plotted.
Molecular dynamics simulations
A membrane consisting of 284 POPC and 16 PIP2 lipid molecules was generated by the program/website CHARMM-GUI (103). The membrane was equilibrated for 100 ns at 310 K in solvent with counterions. Simulations were carried out with full-length K-Ras, which was adopted from a previous publication (40, 42) and was originally built by ligating the crystal structure of G12D K-Ras (PDB 4DSO, residues 1–173) to a K-Ras4B HVR and lipid anchor (tK, residues 174–185). This structure was chosen because it extends the C-terminal helix to residue 173 and for the results to be comparable to other simulations. The parameter for the farnesyl group at the C-terminal of the HVR region was produced by the CHARMM generalized force field (CGenFF) (104). Previously, Prakash and co-workers (40, 66) showed that K-Ras4B.G12D populates two major orientations relative to the membrane (OS1 and OS2). These were provided to us and adopted here with a D12V mutation, as the initial configurations. As before, the farnesyl group was preinserted into the membrane (42). The CHARMM36 force field including the CMAP correction was applied to the system (105, 106). The TIP3P model was used for water. The system was neutralized and provided a near-physiological ion concentration of 0.15 m NaCl. In the all-atom simulations, the electrostatic interaction was treated by the Particle-Mesh Ewald method. The van der Waals interaction was cut at 1.2 nm. The time step was set as 2 fs. Temperature was coupled by using Langevin thermostat at 310 K, whereas pressure was 1 bar controlled by the semi-isotropic Langevin scheme. All these systems ran for the first 30 ns using the NAMD/2.10 package (107), and then were transferred to the Anton supercomputer (108) for another 350 ns of simulation.
GTP hydrolysis assays
The kinetics of GTP hydrolysis were measured by monitoring the release of Pi using a fluorescently labeled, phosphate-binding protein sensor, as previously described (109). 2 μm K-Ras.GTP (loaded as described above) was incubated with 2 μm MDCC phospho-binding proteins (Thermo Scientific) until the baseline was stabilized (2–3 min). 75 nm p120-GAP was added to the reaction. Released Pi was monitored as changes in the fluorescence over time. The fluorescence was measured with excitation at 425 nm (5 nm bandwidth) and emission at 465 nm (7 nm bandwidth) in a Tecan Infinite M1000 microplate reader. All experiments were done in triplicates, and data shown represent averages and standard deviations. Reported values derive from the initial velocity of the hydrolysis reaction (fluorescence change over the first 5 min after mixing), and the relative activity (fold-difference between initial velocity with versus without p120-GAP, or mutants versus WT K-Ras).
Nucleotide exchange assays
Nucleotide exchange reaction was measured by monitoring the time-dependent fluorescence change of mant-GDP as it occupies the Ras nucleotide-binding pocket (110). 2 μm K-Ras. GTP was incubated with 1.25 μm mant-GDP (Jena Bioscience) in 20 mm HEPES, 50 mm NaCl, 4 mm MgCl2, 0.5 mm TCEP, pH 7.5, at 25 °C for 5 min for the baseline to stabilize. 200 nm SOS was added to the reaction and GEF activity was monitored by the fluorescence change of mant-GDP (excitation: 355 nm, emission 448 nm) over 30 min in a Tecan Infinite M1000 plate reader at 25 °C. Again the initial rates were used for assessment of the exchange kinetics. All experiments were done in triplicates. Data shown are averages and standard deviations. For each protein the initial rate and rate in the control were calculated (initial rate with versus without SOS, or mutants versus WT K-Ras).
Focus formation in cell assay
Approximately 250,000 NIH3T3 cells were seeded in triplicate in 35-mm dishes in Dulbecco's modified Eagle's medium containing 10% calf serum. The next day, the cells were transfected with 250 ng of the indicated K-Ras allele cDNA in the pCEFVL plasmid or together with 3 μg of empty pCEFVL vector using polyethylenimine (PEI; Polysciences Inc.) according to published protocol (55). Transfection mixture contained 3 μl of PEI, 1 μg of DNA/well in serum-free medium. After 24 h, each well was transferred into triplicate 10-cm dishes and grown in Dulbecco's modified Eagle's medium containing 2% calf serum for 2 weeks. The medium was replaced every 2 days. After washing with PBS, cells were fixed with 3.7% paraformaldehyde for 15 min, stained with crystal violet, and visible foci (>2 mm) were manually counted under a microscope. Values reported are averages of triplicate transfections and associated standard deviations.
Fluorescence microscopy
NIH3T3 cells were grown on collagen-coated coverslips, transfected with the indicated K-Ras constructed using PEI as described above, and cultured in serum-free medium for 17 h. After washing, cells were fixed in 3.7% paraformaldehyde, they were permeabilized with 0.2% Triton X-100, blocked with 2% BSA, and stained with anti-K-Ras antibody (clone 3B10–2F2, Abnova). Actin cytoskeleton was stained with Alexa Fluor 555-conjugated phalloidin. Slides were imaged on a Keyence BZ-X 700 fluorescence microscope.
Author contributions
S. Cao, D. M., and M. B. conceptualization; S. Cao, S. Chung, S. K., and Z.L. data curation; S. Cao, S. Chung, S. K., and Z. L. formal analysis; S. Cao, S. Chung, S. K., Z. L., D. M., and M. B. validation; S. Cao, S. Chung, S. K., Z. L., D. M., and M. B. visualization; S. Cao, S. Chung, S. K., Z. L., D. M., and M. B. methodology; S. Cao, S. Chung, S. K., Z. L., D. M., and M. B. writing-original draft; S. Cao, S. Chung, S. K., Z. L., D. M., and M. B. writing-review and editing; D. M. and M. B. supervision; D. M. and M. B. funding acquisition; D. M. and M. B. investigation; D. M. and M. B. project administration.
Supplementary Material
Acknowledgments
We thank Dr. J. Kuriyan (University of California, Berkley) for the generous gift of a bacterial expression vector of the human Sos GEF and Dr. R. Ahmadian (University of Düsseldorf) for p120 Ras GAP. We thank the Ohio Super Computer center for computational resources. Anton2 Computer time was provided by the Pittsburgh Supercomputing Center (PSC) through Grant R01GM116961 from the National Institutes of Health. A refrigerated centrifuge used for this project was in part funded by a donation in memory of Daniel Sheehan by the Sheehan Family Trust.
This work was supported by National Institutes of Health NIGMS Grant R01GM112491 (to the Buck lab). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S3.
- HVR
- hyper-variable region
- GEF
- GTPase exchange factor
- GAP
- GTPase-activating protein
- PIP(4,5)P2
- phosphatidylinositol (4,5)-phosphate
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- MST
- microscale thermophoresis
- HSQC
- heteronuclear single quantum coherence
- TROSY
- transverse relaxation-optimized spectroscopy
- PS
- phosphatidylserine
- TCEP
- tris(2-carboxyethyl)phosphine hydrochloride
- GMP-PNP
- guanyl-5′-yl imidodiphosphate
- PRE
- paramagnetic relaxation enhancement
- PDB
- Protein Data Bank
- PE
- phosphatidylethanolamine
- PA
- phosphatidic acid
- OS1
- orientation state 1
- OS2
- orientation state 2
- PEI
- polyethylenimine
- PI
- phosphatidylinositol
- DTPA
- diethylenetriamine pentaacetate
- LPA
- lipoprotein A
- LPC
- lipoprotein C.
References
- 1. Karnoub A. E., and Weinberg R. A. (2008) Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9, 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pylayeva-Gupta Y., Grabocka E., and Bar-Sagi D. (2011) RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer. 11, 761–774 10.1038/nrc3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malumbres M., and Barbacid M. (2003) RAS oncogenes: the first 30 years. Nat. Rev. Cancer. 3, 459–465 10.1038/nrc1097 [DOI] [PubMed] [Google Scholar]
- 4. Castellano E., and Santos E. (2011) Functional specificity of ras isoforms: so similar but so different. Genes Cancer 2, 216–231 10.1177/1947601911408081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cox A. D., and Der C. J. (2010) Ras history: the saga continues. Small GTPases 1, 2–27 10.4161/sgtp.1.1.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prior I. A., Lewis P. D., and Mattos C. (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 10.1158/0008-5472.CAN-11-2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., and Der C. J. (2014) Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 10.1038/nrd4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vetter I. R., and Wittinghofer A. (2001) The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 10.1126/science.1062023 [DOI] [PubMed] [Google Scholar]
- 9. Gorfe A. A. (2010) Mechanisms of allostery and membrane attachment in Ras GTPases: implications for anti-cancer drug discovery. Curr. Med. Chem. 17, 1–9 10.2174/092986710789957832 [DOI] [PubMed] [Google Scholar]
- 10. Nussinov R., Tsai C. J., and Mattos C. (2013) “Pathway drug cocktail”: targeting Ras signaling based on structural pathways. Trends Mol Med. 19, 695–704 10.1016/j.molmed.2013.07.009 [DOI] [PubMed] [Google Scholar]
- 11. Wright L. P., and Philips M. R. (2006) Thematic review series: Lipid posttranslational modifications: CAAX modification and membrane targeting of Ras. J. Lipid Res. 47, 883–891 10.1194/jlr.R600004-JLR200 [DOI] [PubMed] [Google Scholar]
- 12. Brunsveld L., Waldmann H., and Huster D. (2009) Membrane binding of lipidated Ras peptides and proteins: the structural point of view. Biochim. Biophys. Acta 1788, 273–288 10.1016/j.bbamem.2008.08.006 [DOI] [PubMed] [Google Scholar]
- 13. Chenette E. J., and Der C. J. (2011) Lipid modification of Ras superfamily GTPases: not just membrane glue enzymes. Enzymes 29, 59–95 10.1016/B978-0-12-381339-8.00005-6 [DOI] [Google Scholar]
- 14. Ahearn I. M., Haigis K., Bar-Sagi D., and Philips M. R. (2011) Regulating the regulator: post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 13, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Paolo G., and De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 10.1038/nature05185 [DOI] [PubMed] [Google Scholar]
- 16. Yang Y., Lee M., and Fairn G. D. (2018) Phospholipid subcellular localization and dynamics. J. Biol. Chem. 293, 6230–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wymann M. P., and Schneiter R. (2008) Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 9, 162–176 10.1038/nrm2335 [DOI] [PubMed] [Google Scholar]
- 18. Yamaguchi H., Yoshida S., Muroi E., Kawamura M., Kouchi Z., Nakamura Y., Sakai R., and Fukami K. (2010) Phosphatidylinositol 4,5-bisphosphate and PIP5-kinase Iα are required for invadopodia formation in human breast cancer cells. Cancer Sci. 101, 1632–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gagliardi P. A., di Blasio L., and Primo L. (2015) PDK1: a signaling hub for cell migration and tumor invasion. Biochim. Biophys. Acta 1856, 178–188 [DOI] [PubMed] [Google Scholar]
- 20. Lien E. C., Dibble C. C., and Toker A. (2017) PI3K signaling in cancer: beyond AKT. Curr. Opin. Cell Biol. 45, 62–71 10.1016/j.ceb.2017.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hedger G., Sansom M. S., and Koldsø H. (2015) The juxtamembrane regions of human receptor tyrosine kinases exhibit conserved interaction sites with anionic lipids. Sci. Rep. 5, 9198 10.1038/srep09198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kolay S., Basu U., and Raghu P. (2016) Control of diverse subcellular processes by a single multi-functional lipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]. Biochem. J. 473, 1681–1692 10.1042/BCJ20160069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chadli M., Rebaud S., Maniti O., Tillier B., Cortès S., and Girard-Egrot A. (2017) New tethered phospholipid bilayers integrating functional G-protein-coupled receptor membrane proteins. Langmuir 33, 10385–10401 10.1021/acs.langmuir.7b01636 [DOI] [PubMed] [Google Scholar]
- 24. McLaughlin S., and Murray D. (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611 10.1038/nature04398 [DOI] [PubMed] [Google Scholar]
- 25. McLaughlin S., Wang J., Gambhir A., and Murray D. (2002) PIP(2) and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175 10.1146/annurev.biophys.31.082901.134259 [DOI] [PubMed] [Google Scholar]
- 26. Yang M. H., Nickerson S., Kim E. T., Liot C., Laurent G., Spang R., Philips M. R., Shan Y., Shaw D. E., Bar-Sagi D., Haigis M. C., and Haigis K. M. (2012) Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. U.S.A. 109, 10843–10848 10.1073/pnas.1201487109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gresset A., Sondek J., and Harden T. K. (2012) The phospholipase C isozymes and their regulation. Subcell. Biochem. 58, 61–94 10.1007/978-94-007-3012-0_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Snyder J. T., Singer A. U., Wing M. R., Harden T. K., and Sondek J. (2003) The pleckstrin homology domain of phospholipase C-β2 as an effector site for Rac. J. Biol. Chem. 278, 21099–21104 10.1074/jbc.M301418200 [DOI] [PubMed] [Google Scholar]
- 29. El-Sibai M., and Backer J. M. (2007) Phospholipase Cγ negatively regulates Rac/Cdc42 activation in antigen-stimulated mast cells. Eur. J. Immunol. 37, 261–270 10.1002/eji.200635875 [DOI] [PubMed] [Google Scholar]
- 30. Walliser C., Retlich M., Harris R., Everett K. L., Josephs M. B., Vatter P., Esposito D., Driscoll P. C., Katan M., Gierschik P., and Bunney T. D. (2008) Rac regulates its effector phospholipase Cγ2 through interaction with a split pleckstrin homology domain. J. Biol. Chem. 283, 30351–30362 10.1074/jbc.M803316200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heo W. D., Inoue T., Park W. S., Kim M. L., Park B. O., Wandless T. J., and Meyer T. (2006) PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314, 1458–1461 10.1126/science.1134389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gulyás G., Radvánszki G., Matuska R., Balla A., Hunyady L., Balla T., and Várnai P. (2017) Plasma membrane phosphatidylinositol 4-phosphate and 4,5-bisphosphate determine the distribution and function of K-Ras4B but not H-Ras proteins. J. Biol. Chem. 292, 18862–18877 10.1074/jbc.M117.806679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou Y., Prakash P., Liang H., Cho K. J., Gorfe A. A., and Hancock J. F. (2017) Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 168, 239–251.e16 10.1016/j.cell.2016.11.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Magee A. I., Gutierrez L., McKay I. A., Marshall C. J., and Hall A. (1987) Dynamic fatty acylation of p21N-ras. EMBO J. 6, 3353–3357 10.1002/j.1460-2075.1987.tb02656.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker T. L., Zheng H., Walker J., Coloff J. L., and Buss J. E. (2003) Distinct rates of palmitate turnover on membrane bound cellular and oncogenic H-Ras. J. Biol. Chem. 278, 19292–19300 [DOI] [PubMed] [Google Scholar]
- 36. Rocks O., Peyker A., Kahms M., Verveer P. J., Koerner C., Lumbierres M., Kuhlmann J., Waldmann H., Wittinghofer A., and Bastiaens P. I. (2005) An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307, 1746–1752 10.1126/science.1105654 [DOI] [PubMed] [Google Scholar]
- 37. Prior I. A., Harding A., Yan J., Sluimer J., Parton R. G., and Hancock J. F. (2001) GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell. Biol. 3, 368–375 10.1038/35070050 [DOI] [PubMed] [Google Scholar]
- 38. Prior I. A., Muncke C., Parton R. G., and Hancock J. F. (2003) Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 160, 165–170 10.1083/jcb.200209091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gorfe A. A., Hanzal-Bayer M., Abankwa D., Hancock J. F., and McCammon J. A. (2007) Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J. Med. Chem. 50, 674–684 10.1021/jm061053f [DOI] [PubMed] [Google Scholar]
- 40. Prakash P., Zhou Y., Liang H., Hancock J. F., and Gorfe A. A. (2016) Oncogenic K-Ras binds to an anionic membrane in two distinct orientations: a molecular dynamics analysis. Biophys J. 110, 1125–1138 10.1016/j.bpj.2016.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Z., Cao S., Buck M. (2016) K-Ras at anionic membranes: orientation orientation … orientation: recent simulations and experiments. Biophys. J. 110, 1033–1035 10.1016/j.bpj.2016.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Z. L., and Buck M. (2017) Computational modeling reveals that signaling lipids modulate the orientation of K-Ras4A at the membrane reflecting protein topology. Structure 25, 679–689.e2 10.1016/j.str.2017.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gregory M. C., McLean M. A., and Sligar S. G. (2017) Interaction of KRas4b with anionic membranes: a special role for PIP2. Biochem. Biophys. Res. Commun. 487, 351–355 10.1016/j.bbrc.2017.04.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mazhab-Jafari M. T., Marshall C. B., Smith M. J., Gasmi-Seabrook G. M., Stathopulos P. B., Inagaki F., Kay L. E., Neel B. G., and Ikura M. (2015) Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. Proc. Natl. Acad. Sci. U.S.A. 112, 6625–6630 10.1073/pnas.1419895112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Banerjee A., Jang H., Nussinov R., and Gaponenko V. (2016) The disordered hypervariable region and the folded catalytic domain of oncogenic K-Ras4B partner in phospholipid binding. Curr. Opin. Struct. Biol. 36, 10–17 10.1016/j.sbi.2015.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vo U., Embrey K. J., Breeze A. L., and Golovanov A. P. (2013) 1H, 13C and 15N resonance assignment for the human K-Ras at physiological pH. Biomol. NMR Assign. 7, 215–219 [DOI] [PubMed] [Google Scholar]
- 47. Prosser R. S., Evanics F., Kitevski J. L., and Al-Abdul-Wahid M. S. (2006) Current applications of bicelles in NMR studies of membrane-associated amphiphiles and proteins. Biochemistry 45, 8453–8465 10.1021/bi060615u [DOI] [PubMed] [Google Scholar]
- 48. Glück J. M., Wittlich M., Feuerstein S., Hoffmann S., Willbold D., and Koenig B. W. (2009) Integral membrane proteins in nanodiscs can be studied by solution NMR spectroscopy. J. Am. Chem. Soc. 131, 12060–12061 10.1021/ja904897p [DOI] [PubMed] [Google Scholar]
- 49. Vinogradova O., and Qin J. (2012) NMR as a unique tool in assessment and complex determination of weak protein-protein interactions. Top. Curr. Chem. 326, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barrett P. J., Chen J., Cho M. K., Kim J. H., Lu Z., Mathew S., Peng D., Song Y., Van Horn W. D., Zhuang T., Sönnichsen F. D., and Sanders C. R. (2013) The quiet renaissance of protein nuclear magnetic resonance. Biochemistry 52, 1303–1320 10.1021/bi4000436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ubbink M., Worrall J. A., Canters G. W., Groenen E. J., and Huber M. (2002) Paramagnetic resonance of biological metal centers. Annu. Rev. Biophys. Biomol. Struct. 31, 393–422 10.1146/annurev.biophys.31.091701.171000 [DOI] [PubMed] [Google Scholar]
- 52. Mazhab-Jafari M. T., Marshall C. B., Stathopulos P. B., Kobashigawa Y., Stambolic V., Kay L. E., Inagaki F., and Ikura M. (2013) Membrane-dependent modulation of the mTOR activator Rheb: NMR observations of a GTPase tethered to a lipid-bilayer nanodisc. J. Am. Chem. Soc. 135, 3367–3370 10.1021/ja312508w [DOI] [PubMed] [Google Scholar]
- 53. Clore G. M. (2015) Practical aspects of paramagnetic relaxation enhancement in biological macromolecules. Methods Enzymol. 564, 485–497 10.1016/bs.mie.2015.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ceccon A., Tugarinov V., Bax A., and Clore G. M. (2016) Global dynamics and exchange kinetics of a protein on the surface of nanoparticles revealed by relaxation-based solution NMR spectroscopy. J. Am. Chem. Soc. 138, 5789–5792 10.1021/jacs.6b02654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Durocher Y., Perret S., and Kamen A. (2002) High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30, E9 10.1093/nar/30.2.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Šolman M., Ligabue A., Blaževitš O., Jaiswal A., Zhou Y., Liang H., Lectez B., Kopra K., Guzmán C., Härmä H., Hancock J. F., Aittokallio T., and Abankwa D. (2015) Specific cancer-associated mutations in the switch III region of Ras increase tumorigenicity by nanocluster augmentation. Elife 4, e08905 10.7554/eLife.08905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Forbes S. A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C. G., Ward S., Dawson E., Ponting L., Stefancsik R., Harsha B., Kok C. Y., Jia M., Jubb H., Sondka Z., Thompson S., De T., and Campbell P. J. (2017) COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 10.1093/nar/gkw1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin R., Cerione R. A., and Manor D. (1999) Specific contributions of the small GTPases Rho, Rac, and Cdc42 to Dbl transformation. J. Biol. Chem. 274, 23633–23641 [DOI] [PubMed] [Google Scholar]
- 59. Martin C. B., Mahon G. M., Klinger M. B., Kay R. J., Symons M., Der C. J., and Whitehead I. P. (2001) The thrombin receptor, PAR-1, causes transformation by activation of Rho-mediated signaling pathways. Oncogene 20, 1953–1963 [DOI] [PubMed] [Google Scholar]
- 60. van der Hoeven D., Cho K. J., Ma X., Chigurupati S., Parton R. G., and Hancock J. F. (2013) Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol. Cell. Biol. 33, 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barnard A., and Smith D. K. (2012) Self-assembled multivalency: dynamic ligand arrays for high-affinity binding. Angew. Chem. Int. Ed. Engl. 51, 6572–6581 10.1002/anie.201200076 [DOI] [PubMed] [Google Scholar]
- 62. Banjade S., and Rosen M. K. (2014) Phase transitions of multivalent proteins can promote clustering of membrane receptors. Elife 3, 10.7554/eLife.04123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fang Z., Marshall C. B., Nishikawa T., Gossert A. D., Jansen J. M., Jahnke W., and Ikura M. (2018) Inhibition of K-Ras4B by a unique mechanism of action: stabilizing membrane-dependent occlusion of the effector-binding site. Cell Chem. Biol. 25, 1327–1336.e4 [DOI] [PubMed] [Google Scholar]
- 64. Kearney B. M., Johnson C. W., Roberts D. M., Swartz P., and Mattos C. (2014) DRoP: a water analysis program identifies Ras-GTP-specific pathway of communication between membrane-interacting regions and the active site. J. Mol. Biol. 426, 611–629 10.1016/j.jmb.2013.10.036 [DOI] [PubMed] [Google Scholar]
- 65. Bouguet-Bonnet S., and Buck M. (2008) Compensatory and long-range changes in picosecond-nanosecond main-chain dynamics upon complex formation: 15N relaxation analysis of the free and bound states of the ubiquitin-like domain of human plexin-B1 and the small GTPase Rac1. J. Mol. Biol. 377, 1474–1487 10.1016/j.jmb.2008.01.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prakash P., and Gorfe A. A. (2017) Membrane orientation dynamics of lipid-modified small GTPases. Small GTPase 8, 129–138 10.1080/21541248.2016.1211067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sung P. J., Tsai F. D., Vais H., Court H., Yang J., Fehrenbacher N., Foskett J. K., and Philips M. R. (2013) Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc. Natl. Acad. Sci. U.S.A. 110, 20593–20598 10.1073/pnas.1306431110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Suladze S., Ismail S., and Winter R. (2014) Thermodynamic, dynamic and solvational properties of PDEδ binding to farnesylated cystein: a model study for uncovering the molecular mechanism of PDEδ interaction with prenylated proteins. J. Phys. Chem. B 118, 966–975 10.1021/jp411466r [DOI] [PubMed] [Google Scholar]
- 69. Zhang X., Cao S., Barila G., Edreira M. M., Hong K., Wankhede M., Naim N., Buck M., and Altschuler D. L. (2018) Cyclase-associated protein 1 (CAP1) is a prenyl-binding partner of Rap1 GTPase. J. Biol. Chem. 293, 7659–7673 10.1074/jbc.RA118.001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lobell R. B., Liu D., Buser C. A., Davide J. P., DePuy E., Hamilton K., Koblan K. S., Lee Y., Mosser S., Motzel S. L., Abbruzzese J. L., Fuchs C. S., Rowinsky E. K., Rubin E. H., Sharma S., et al. (2002) Preclinical and clinical pharmacodynamic assessment of L-778,123, a dual inhibitor of farnesyl:protein transferase and geranylgeranyl:protein transferase type-I. Mol Cancer Ther. 1, 747–758 [PubMed] [Google Scholar]
- 71. Lobell R. B., Omer C. A., Abrams M. T., Bhimnathwala H. G., Brucker M. J., Buser C. A., Davide J. P., deSolms S. J., Dinsmore C. J., Ellis-Hutchings M. S., Kral A. M., Liu D., Lumma W. C., Machotka S. V., Rands E., et al. (2001) Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 61, 8758–8768 [PubMed] [Google Scholar]
- 72. Zhou Y., Wong C. O., Cho K. J., van der Hoeven D., Liang H., Thakur D. P., Luo J., Babic M., Zinsmaier K. E., Zhu M. X., Hu H., Venkatachalam K., and Hancock J. F. (2015) Signal transduction: membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 349, 873–876 10.1126/science.aaa5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Whorton M. R., and MacKinnon R. (2011) Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G-proteins, PIP2 and sodium. Cell 147, 199–208 10.1016/j.cell.2011.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ye L., Neale C., Sljoka A., Lyda B., Pichugin D., Tsuchimura N., Larda S. T., Pomès R., García A. E., Ernst O. P., Sunahara R. K., and Prosser R. S. (2018) Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 9, 1372 10.1038/s41467-018-03314-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Buhrman G., Holzapfel G., Fetics S., and Mattos C. (2010) Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. U.S.A. 107, 4931–4936 10.1073/pnas.0912226107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang Y. H., Collins A., Guo L., Smith-Dupont K. B., Gai F., Svitkina T., and Janmey P. A. (2012) Divalent cation-induced cluster formation by polyphosphoinositides in model membranes. J. Am. Chem. Soc. 134, 3387–3395 10.1021/ja208640t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Graber Z. T., Shi Z., and Baumgart T. (2017) Cations induce shape remodeling of negatively charged phospholipid membranes. Phys. Chem. Chem. Phys. 19, 15285–15295 10.1039/C7CP00718C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jang H., Muratcioglu S., Gursoy A., Keskin O., and Nussinov R. (2016) Membrane-associated Ras dimers are isoform-specific: K-Ras dimers differ from H-Ras dimers. Biochem. J. 473, 1719–1732 10.1042/BCJ20160031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Abankwa D., Gorfe A. A., Inder K., and Hancock J. F. (2010) Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. U.S.A. 107, 1130–1135 10.1073/pnas.0903907107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yin G., Kistler S., George S. D., Kuhlmann N., Garvey L., Huynh M., Bagni R. K., Lammers M., Der C. J., and Campbell S. L. (2017) A KRAS GTPase K104Q mutant retains downstream signaling by offsetting defects in regulation. J. Biol. Chem. 292, 4446–4456 10.1074/jbc.M116.762435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. van den Bogaart G., Meyenberg K., Risselada H. J., Amin H., Willig K. I., Hubrich B. E., Dier M., Hell S. W., Grubmüller H., Diederichsen U., and Jahn R. (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479, 552–555 10.1038/nature10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang J., and Richards D. A. (2012) Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol. Open 1, 857–862 10.1242/bio.20122071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koldsø H., Shorthouse D., Hélie J., and Sansom M. S. (2014) Lipid clustering correlates with membrane curvature as revealed by molecular simulations of complex lipid bilayers. PLoS Comput. Biol. 10, e1003911 10.1371/journal.pcbi.1003911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lu S. M., and Fairn G. D. (2018) Mesoscale organization of domains in the plasma membrane: beyond the lipid raft. Crit. Rev. Biochem. Mol. Biol. 53, 192–207 10.1080/10409238.2018.1436515 [DOI] [PubMed] [Google Scholar]
- 85. Inouye K., Mizutani S., Koide H., and Kaziro Y. (2000) Formation of the Ras dimer is essential for Raf-1 activation. J. Biol. Chem. 275, 3737–3740 10.1074/jbc.275.6.3737 [DOI] [PubMed] [Google Scholar]
- 86. Güldenhaupt J., Rudack T., Bachler P., Mann D., Triola G., Waldmann H., Kötting C., and Gerwert K. (2012) N-Ras forms dimers at POPC membranes. Biophys J. 103, 1585–1593 10.1016/j.bpj.2012.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Muratcioglu S., Chavan T. S., Freed B. C., Jang H., Khavrutskii L., Freed R. N., Dyba M. A., Stefanisko K., Tarasov S. G., Gursoy A., Keskin O., Tarasova N. I., Gaponenko V., and Nussinov R. (2015) GTP-Dependent K-Ras dimerization. Structure 23, 1325–1335 10.1016/j.str.2015.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nan X., Tamgüney T. M., Collisson E. A., Lin L. J., Pitt C., Galeas J., Lewis S., Gray J. W., McCormick F., and Chu S. (2015) Ras-GTP dimers activate the mitogen-activated protein kinase (MAPK) pathway. Proc. Natl. Acad. Sci. U.S.A. 112, 7996–8001 10.1073/pnas.1509123112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Plowman S. J., Muncke C., Parton R. G., and Hancock J. F. (2005) H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 102, 15500–15505 10.1073/pnas.0504114102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhou Y., and Hancock J. F. (2015) Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta 1853, 841–849 10.1016/j.bbamcr.2014.09.008 [DOI] [PubMed] [Google Scholar]
- 91. Chung J. K., Lee Y. K., Denson J. P., Gillette W. K., Alvarez S., Stephen A. G., and Groves J. T. (2018) K-Ras4B remains monomeric on membranes over a wide range of surface densities and lipid compositions. Biophys. J. 114, 137–145 10.1016/j.bpj.2017.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ambrogio C., Köhler J., Zhou Z. W., Wang H., Paranal R., Li J., Capelletti M., Caffarra C., Li S., Lv Q., Gondi S., Hunter J. C., Lu J., Chiarle R., Santamaría D., Westover K. D., and Jänne P. A. (2018) KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell 172, 857–868.e15 10.1016/j.cell.2017.12.020 [DOI] [PubMed] [Google Scholar]
- 93. Li Z. L., Prakash P., and Buck M. (2018) A “Tug of War” maintains a dynamic protein-membrane complex: molecular dynamics simulations of C-Raf RBD-CRD bound to K-Ras4B at an anionic membrane. ACS Cent. Sci. 4, 298–305 10.1021/acscentsci.7b00593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ahmadian M. R., Wiesmüller L., Lautwein A., Bischoff F. R., and Wittinghofer A. (1996) Structural differences in the minimal catalytic domains of the GTPase-activating proteins p120GAP and neurofibromin. J. Biol. Chem. 271, 16409–16415 [DOI] [PubMed] [Google Scholar]
- 95. Sondermann H., Soisson S. M., Boykevisch S., Yang S. S., Bar-Sagi D., and Kuriyan J. (2004) Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell 119, 393–405 10.1016/j.cell.2004.10.005 [DOI] [PubMed] [Google Scholar]
- 96. Dowler S., Kular G., and Alessi D, R. (2002) Protein lipid overlay assay. Sci. Signal. 129, pl6 [DOI] [PubMed] [Google Scholar]
- 97. Puthenveetil R., Nguyen K., and Vinogradova O. (2017) Nanodiscs and Solution NMR: preparation, application and challenges. Nanotechnol. Rev. 6, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kucharska I., Edrington T. C., Liang B., and Tamm L. K. (2015) Optimizing nanodiscs and bicelles for solution NMR studies of two b-barrel membrane proteins. J. Biomol. NMR 61, 261–274 10.1007/s10858-015-9905-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Denisov I. G., Grinkova Y. V., Lazarides A. A., and Sligar S. G. (2004) Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 126, 3477–3487 10.1021/ja0393574 [DOI] [PubMed] [Google Scholar]
- 100. Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., and Sligar S. G. (2009) Chapter 11: reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 10.1016/S0076-6879(09)64011-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 102. Cao S., Mao X., Liu D., and Buck M. (2013) Backbone assignment and secondary structure of Rnd1, an unusual Rho family small GTPase. Biomol. NMR Assign. 7, 121–128 10.1007/s12104-012-9391-0 [DOI] [PubMed] [Google Scholar]
- 103. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- 104. Vanommeslaeghe K., and MacKerell A. D. (2012) Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 52, 3144–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Huang J., and MacKerell A. D. (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 10.1002/jcc.23354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Buck M., Bouguet-Bonnet S., Pastor R. W., and MacKerell A. D. (2006) Importance of the CMAP correction to the CHARMM22 protein force field: dynamics of Hen lysozyme. Biophys. J. 90, L36–L38 10.1529/biophysj.105.078154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dror R. O., Jensen M. Ø., Borhani D. W., and Shaw D. E. (2010) Exploring atomic resolution physiology on a femtosecond to millisecond timescale using molecular dynamics simulations. J. Gen. Physiol. 135, 555–562 10.1085/jgp.200910373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sosa M. S., Lewin N. E., Choi S. H., Blumberg P. M., and Kazanietz M. G. (2009) Biochemical characterization of hyperactive β2-chimaerin mutants revealed an enhanced exposure of C1 and Rac-GAP domains. Biochemistry 48, 8171–8178 10.1021/bi9010623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Leonard D. A., Evans T., Hart M., Cerione R. A., and Manor D. (1994) Investigation of the GTP-binding/GTPase cycle of Cdc42Hs using fluorescence spectroscopy. Biochemistry 33, 12323–12328 10.1021/bi00206a040 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.