

Abstract
IgE plays a key role in allergies by binding to allergens and then sensitizing mast cells through the Fc receptor, resulting in the secretion of proinflammatory mediators. Therefore, IgE is a major target for managing allergies. Previous studies have reported that oligomannose on IgE can be a potential target to inhibit allergic responses. However, enzymes that can modulate IgE activity are not yet known. Here, we found that the commercial receptor-destroying enzyme (RDE) (II) from Vibrio cholerae culture fluid specifically modulates IgE, but not IgG, and prevents the initiation of anaphylaxis. RDE (II)–treated IgE cannot access its binding site on bone marrow–derived mast cells, resulting in reduced release of histamine and cytokines. We also noted that RDE (II)–treated IgE could not induce passive cutaneous anaphylaxis in mouse ears. Taken together, we concluded that RDE (II) modulates the IgE structure and renders it unable to mediate allergic responses. To reveal the mechanism by which RDE (II) interferes with IgE activity, we performed lectin microarray analysis to unravel the relationship between IgE modulation and glycosylation. We observed that RDE (II) treatment significantly reduced the binding of IgE to Lycopersicon esculentum lectin, which recognizes poly-N-acetylglucosamine and poly-N-acetyllactosamine. These results suggest that RDE (II) specifically modulates branched glycans on IgE, thereby interfering with its ability to induce allergic responses. Our findings may provide a basis for the development of drugs to inhibit IgE activity in allergies.
Keywords: immunoglobulin E (IgE), allergy, Vibrio cholerae, mast cell, inflammation, anaphylaxis, autoimmune disorder, glycans, immune response, oligomannose
Introduction
Immunoglobulin ϵ (IgE) plays a key role in type I allergy (e.g. asthma and anaphylaxis). IgE was discovered about 50 years ago by Ishizaka and Ishizaka (1, 2) as a novel immunoglobulin able to induce allergic reactions in the skin. The study found that intracutaneous injection with only 1–2 ng/ml IgE could induce an erythema-wheal reaction in healthy subjects (2). Mast cells and blood basophils in the tissue were found to express a high affinity to the IgE receptor, FcϵRI2 (3, 4). By subsequent exposure to the allergen, IgE-binding mast cells released proinflammatory mediators, including histamine and cytokines, which cause an allergic response (5). Yamaguchi et al. (5) then demonstrated that IgE enhances the expression level of FcϵRI in mast cells and permits mast cells to increase production of proinflammatory mediators by antigen challenge. Taken together, IgE is considered one of the major targets for therapy against allergies. Omalizumab, which binds to the Fc region of IgE and inhibits binding to FcϵRI, has been previously found to be a successful therapy against certain allergies (3, 6, 7). However, omalizumab cannot displace IgE bound to FcϵRI, which leads to a delay of several weeks or months before the onset of any clinical benefits (7). As such, another approach to IgE is necessary to develop a therapy against allergy.
Glycosylation of immunoglobulin is considered to be important for its structure and function (8). Minor modifications of glycans on IgG (e.g. fucose depletion (9)) can have a significant impact on receptor binding and the effector functions (8). In contrast, IgE is the most heavily glycosylated antibody (10, 11). Human IgE has seven predicted N-glycosylation sites, whereas murine IgE has eight or nine (8, 10). Björklund et al. (12) reported that N-linked glycan is important for binding to FcϵRI. They indicated that Flavobacterium meningosepticum peptide:N-glycosidase F (PNGase F), which removes almost all N-linked oligosaccharides, reduces the binding level of IgE to FcϵRI in ELISA. Moreover, Shade et al. (10) reported that oligomannose on Asn-394 in human IgE and Asn-384 in murine IgE is important for the structural integrity of the immunoglobulin. Modifications at these sites by endoglycosidase F1 (Endo F1), which cleaves within the chitobiose core of high-mannose and some hybrid oligosaccharides from N-linked glycoproteins, abrogate IgE binding to FcϵRI. Therefore, the modification of oligomannose prevents the initiation of anaphylaxis by mast cells. In their study, they suggested that IgE oligomannose may be a potential therapeutic target. Wu et al. (11) also determined high-mannose glycans on the same site in IgE obtained from a patient with a novel hyper-IgE syndrome. However, PNGase F and Endo F1 cannot specifically modulate IgE because most sugar proteins have N-linked oligosaccharides. To target the glycan on IgE specifically for the therapy, more research is required to identify a specific enzyme that is able to modulate the glycan structure in IgE.
Previously, we coincidentally found that commercial receptor-destroying enzyme (RDE) (II) from Vibrio cholerae culture fluid (13) reduced the binding level of IgE to influenza virus antigen, hemagglutinin (HA) (Fig. 1A) (14). RDE (II) contains a neuraminidase (NA) (sialidase) and is usually used for preventing nonspecific binding of influenza HA to sialic acid in neutralizing assays. We genetically switched the constant region of anti-HA IgG to anti-HA IgA, IgM, IgD, and IgE and established all classes of anti-HA–coding plasmids. All variable regions were the same, and the binding affinity to influenza HA was considered as equal. However, RDE (II)–treated IgE could not neutralize virus infection despite there being sufficient activity of RDE (II)–treated IgG, IgA, and IgM (14). In this study, we showed that RDE (II) altered the structure of IgE and abrogated IgE binding to FcϵRI on mast cells, which is directly related to the mast cell degranulation, thereby preventing anaphylaxis. Furthermore, we identified that RDE (II)–treated IgE had reduced capacity of binding to Lycopersicon esculentum (LEL), which recognizes poly N-acetylglucosamine (GlcNAc) and poly-N-acetyllactosamine (LacNAc) (15–18), by lectin microarray analysis using 96 lectins (19, 20). Taken together, RDE (II) was found to have the specific characteristics necessary to modulate the glycan on IgE, when compared with IgG.
Figure 1.

RDE (II) inactivates anti-HA IgE but not anti-HA IgG. A, HEK293T cells were transfected with pCADEST1–anti-HA IgG or anti-HA IgE and anti-HA κ. One week later, the supernatants were collected and treated with RDE (II) for 6 h. Then, the antigen-binding level of anti-HA IgE and anti-HA IgG was analyzed by competitive ELISA. B, expression level of the RDE (II)–treated antibodies in the supernatant was analyzed by quantitative ELISA coated with anti-mouse Ig. C, supernatants that were treated with RDE (II) were blotted under nonreducing conditions. They were analyzed with HRP-conjugated light chain BP. Data are representative of at least two independent experiments and indicate the mean ± S.D. ***, p < 0.001 (Student's t test).
Results
RDE (II) reduces the binding activity of anti-HA IgE to the antigen and the antibodies against the constant region, but not anti-HA IgG
We previously generated the plasmid vector coding the antibody gene of anti-HA IgG and anti-HA IgE (14). Surprisingly, even the variable regions were conserved, wherein anti-HA IgE was not able to neutralize the influenza virus in vitro (14). For the neutralizing assay, the specimens were treated with RDE (II) (13), followed by incubation with influenza virus in the presence of trypsin, which cleaves the HA of the influenza virus (21). We also reconfirmed that the antigen-binding activity of anti-HA IgE treated with RDE (II) was reduced to the background level, although anti-HA IgG was almost not affected (Fig. 1A) (14). Moreover, we were almost unable to detect RDE (II)–treated IgE by antibodies against ϵ chain, despite RDE (II)–treated anti-HA IgG being detected at almost the same level as the untreated antibody (Fig. 1B). In our previous report (14), we could not detect RDE (II)–treated anti-HA IgE using antibodies against ϵ chain by Western blotting. However, the light chain of anti-HA IgG and anti-HA IgE was expressed from the same plasmid vector. Therefore, we measured the binding activity of light chain-binding protein (BP) (Fig. 1C). We detected bands of untreated and RDE (II)–treated IgE at over 250 kDa, which was much larger than predicted (around 200 kDa) (Fig. 1C) (3). Taken together, these results suggest that RDE (II) affects not only the antigen-binding region of anti-HA IgE but also the constant region, except for the light chain. Because anti-HA IgG was not affected by RDE (II), it is also suggested that the reactivity of RDE (II) is specific for anti-HA IgE.
RDE (II) changes the structure of IgE but not IgG, followed by the reduction of the binding activity to anti-ϵ chain
To confirm whether RDE (II) affected the IgE antibody, purified mouse monoclonal IgE (anti-TNP IgE) and IgG (anti-T-2 mycotoxin IgG) were treated with RDE (II) and detected by Western blotting. Although RDE (II)–treated IgG was detected as several smaller bands, a single band over 150 kDa, which was the intact size of IgG antibodies (Fig. 2A, lanes 5–8), was detected and identified as the untreated IgG (Fig. 2A, lanes 1–4). In contrast, in the absence of RDE (II), the band of the purified IgE was mainly detected as over 250 kDa, as seen in Fig. 1C (Fig. 2A, lanes 9–12). We also detected a much smaller-sized band of RDE (II)–treated IgE, around 150 kDa (Fig. 2A, lanes 13–16). Moreover, we obtained the same results by CBB staining (Fig. 2B) and light chain BP (Fig. 2C). To analyze which heavy chain (ϵ chain) or light chain RDE (II) has a greater effect, we analyzed RDE (II)–treated IgE under reducing conditions. Using antibodies against the ϵ chain, we obtained a smaller band (around 50 kDa) than that of the untreated (over 75 kDa), depending on the serial dilution (Fig. 2D). Using light chain BP, the band size obtained was the same even after treating with RDE (II) (Fig. 2E). These results indicate that RDE (II) affects the ϵ chain and not the light chain, which is similar to that demonstrated in Fig. 1C. To confirm the influence of RDE (II) on the constant region, we quantified the binding activity of the antibodies against the constant region in the presence and absence of RDE (II). In the plate coated with anti-mouse immunoglobulins (Igs), RDE (II)–treated IgE levels decreased depending on the concentration of RDE (II) (Fig. 2F), although RDE (II)–treated IgG was stably detected at almost the same level as that of the untreated (Fig. 2G). Moreover, a 34-fold dilution was the titer of RDE (II) for IgE in the reduction of binding levels to coated anti-mouse Igs (Fig. 2F) and that of the band from over 75 to 50 kDa (Fig. 2, D and F). Taken together, these results suggest that the reduction of the size by RDE (II) correlates with the binding activity to antibodies against ϵ chain.
Figure 2.

RDE (II) changes the structure of IgE and reduces its binding activity to anti-ϵ chain but not that of IgG. A, serially diluted purified IgG (clone 15H6) and IgE (clone C38-2) (187.5 ng, ∼3-fold dilutions) were treated with RDE (II) at 37 °C overnight (12–20 h). They were blotted under nonreducing conditions with HRP-conjugated anti-mouse IgG and IgE. Ab, antibody. B, purified IgG and IgE (140 ng) were treated with RDE (II). They were analyzed with CBB staining. C, purified IgE and IgG were treated with RDE (II). They were blotted by SDS-PAGE under nonreducing conditions with HRP-conjugated light chain–binding protein (BP). D, purified IgE treated with diluted RDE (II) (3-fold dilutions, indicated number means multiplier (= n)) was blotted under reducing conditions with HRP-conjugated anti-mouse IgE. E, purified IgG and IgE were treated with RDE (II). They were blotted under reducing conditions with HRP-conjugated light chain BP. F and G, purified IgE (F) or IgG (G) were treated with diluted RDE (II) as indicated overnight (12–20 h). The levels of IgE (F) and IgG (G) were measured by quantitative ELISA. Data are representative of two independent experiments and indicate the mean ± S.D.
Next, we confirmed whether RDE (II) could reduce the binding activity of serum IgE to anti-ϵ chain ex vivo. We previously succeeded in inducing the expression of anti-HA IgE in the mouse serum by hydrodynamic injection with the plasmid vector encoding the antibody gene (14). We obtained serum from mice in which anti-HA IgE and anti-HA IgG antibody coding gene had been transferred, followed by incubation with RDE (II). The expression level of RDE (II)–treated anti-HA IgE was not detected in the serum at all, although 2400 ng/ml (= 103.4) of the untreated specimen was detected (Fig. 3A). On the contrary, RDE (II)–treated anti-HA IgG in serum was detected at the same level as that in the absence of RDE (II) (Fig. 3B). We also confirmed that the entire level of IgE in the mouse serum was reduced by treatment with RDE (II) (Fig. 3C). In contrast, the entire IgG level was the same as the level both of RDE (II)–treated and -untreated specimens (Fig. 3D). These results indicate that RDE (II) specifically reduces the binding of IgE to anti-ϵ antibodies but does not affect the level of IgG in serum. Therefore, this suggests that RDE (II) influences the construction of the ϵ chain and not the γ chain.
Figure 3.
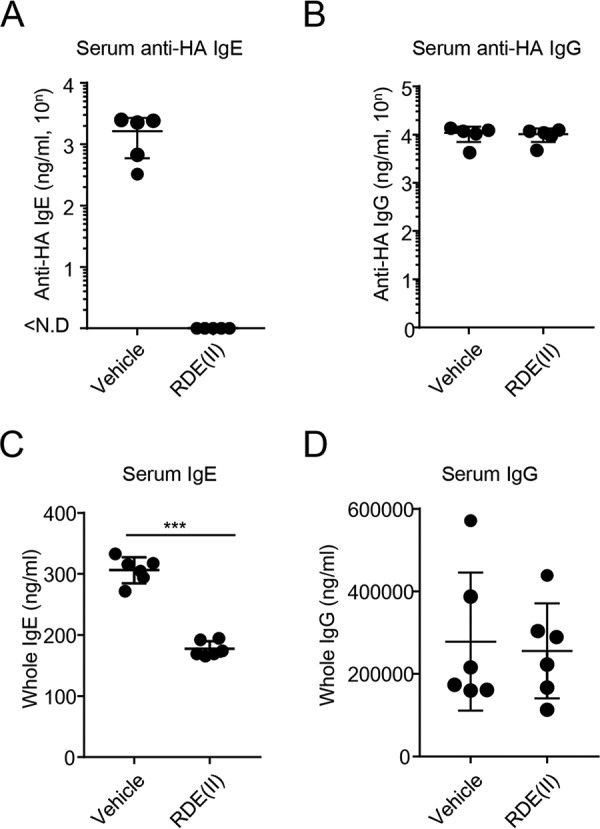
RDE (II) inactivates serum IgE antibodies but not IgG. A and B, BALB/c were injected with pCADEST1–anti-HA IgG or anti-HA IgE and pCADEST1–anti-HA κ by hydrodynamic injection. One day later, the serum was obtained and treated with RDE (II) overnight ex vivo. Then, the specimens were incubated at 56 °C for 30 min. Anti-HA IgE and IgG levels were analyzed by quantitative ELISA. C and D, serum, which was obtained from naïve mice, was treated with RDE (II) overnight (12–20 h). Total IgE (C) and IgG (D) levels were analyzed by quantitative ELISA. Data are representative of two independent experiments and indicate the mean ± S.D. ***, p < 0.001 (Student's t test).
RDE (II)–treated IgE could not induce anaphylaxis in bone marrow–derived mast cells (BMMCs)
RDE (II) treatment affects the construction of IgE. Therefore, we investigated whether RDE (II) alters IgE-mediated mast cell activation. To evaluate the stimulation and release of cytokines and histamines from BMMCs with RDE (II)–treated IgE, BMMCs were incubated with RDE (II)–treated anti-dinitrophenol (DNP) IgE (22). Furthermore, BMMCs were activated by cross-linking IgE with DNP–human serum albumin (HSA). The supernatants were obtained, and the induction levels of TNFα, IL-6, and histamine were measured, which are the classical activation markers of mast cells. Although untreated IgE-sensitized BMMCs released large amounts of TNFα, IL-6, and histamine, all of the levels were significantly reduced in RDE (II)–treated IgE-sensitized BMMCs (Fig. 4, A–C). To confirm the binding of RDE (II)–treated IgE to BMMCs, we sensitized the cells with untreated or RDE (II)–treated anti-DNP IgE. As shown in Fig. 4D, although untreated IgE could bind to BMMCs, RDE (II)–treated IgE could barely bind to BMMCs (Fig. 4D). However, the binding level of control IgG to bone marrow–derived macrophages (BMMs) was barely affected by RDE (II). We also confirmed that RDE (II) was not toxic for BMMCs, because propidium iodide–positive cells were barely detected in BMMCs with RDE (II)–treated IgE (Fig. S1). These data indicate that RDE (II)–treated IgE can no longer bind to BMMCs. Therefore, BMMCs are unable to release cytokines and histamine even in presence of the antigen.
Figure 4.
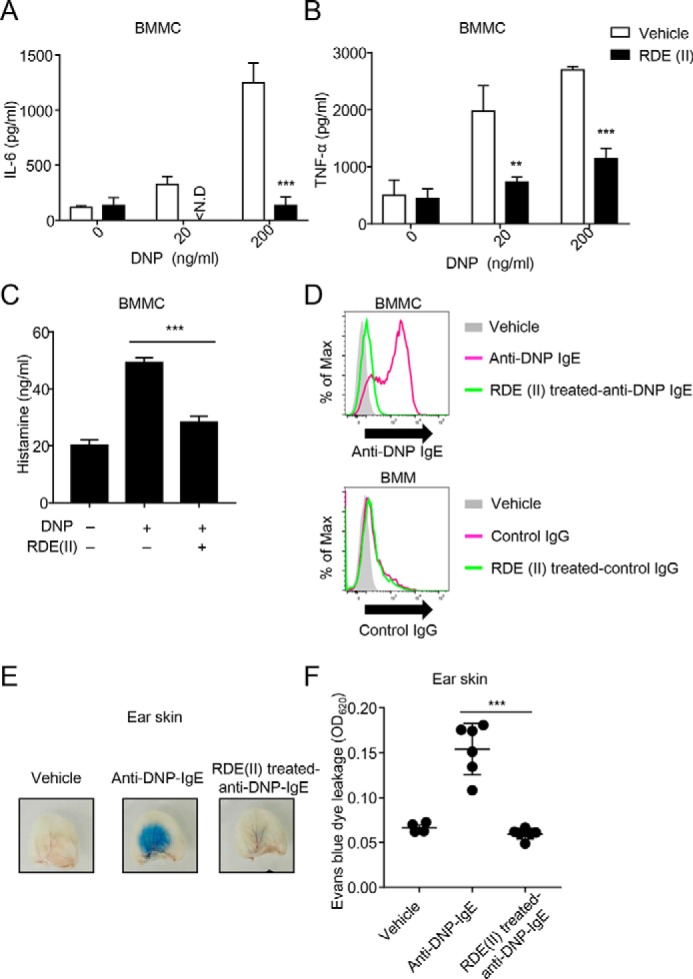
RDE (II)–treated IgE cannot induce anaphylaxis. A and B, effects of RDE (II)–treated IgE reduced the expression levels of IL-6 (A) or TNFα (B) from BMMCs. Two μg/ml anti-DNP IgE was treated with RDE (II) overnight (12–20 h), followed by incubation at 56 °C. Then, the antibodies sensed BMMC for 2 h, followed by incubation with HSA–DNP as indicated overnight (12–20 h). Then, the supernatant was collected and analyzed with quantitative ELISA for IL-6 (A) or TNFα (B). **, p < 0.01, and ***, p < 0.001 (Student's t test). N.D., not detected. C, 2 μg/ml anti-DNP IgE was treated with RDE (II) overnight (12–20 h). BMMCs were sensed overnight (12–20 h) and then incubated with 200 ng/ml HSA–DNP for 1 h. The supernatant was then obtained and analyzed with competitive ELISA for histamine. ***, p < 0.001 (one-way ANOVA). D, 20 μg/ml biotin-conjugated anti-DNP IgE or control IgG (MOPC-21) was treated with RDE (II) overnight (12–20 h). BMMCs or BMMs were incubated with the antibodies for 30 min, followed by incubation with APC-conjugated streptavidin. The binding level was analyzed by FACSCantoII. E, anti-DNP IgE was treated with RDE (II) overnight (12–20 h). The ears of mice were passively sensitized with 20 ng of RDE (II)–treated anti-DNP IgE. One day later (18–24 h), all mice were intravenously injected with 200 μg of DNP–HSA containing Evans blue dye. Thirty minutes later, the ears were obtained and observed in the transudation of Evans blue dye to determine vascular permeability. F, the absorbance of the extraction from the ear was measured at 620 nm. ***, p < 0.001 (one-way ANOVA). All data are representative of at least two independent experiments and indicate the mean ± S.D.
To evaluate whether RDE (II)–treated anti-DNP IgE has a similar effect in vivo, we examined IgE-mediated passive cutaneous anaphylaxis (PCA) in mouse models. Increased vascular permeability was observed in mice that were injected with untreated anti-DNP IgE (Fig. 4, E and F). On the contrary, vascular permeability was significantly decreased in mice with RDE (II)–treated anti-DNP IgE, as in the control (Fig. 4, E and F). These results correlated with those observed in the experimental data of BMMC.
Structure of IgE was affected by trypsin, but IgG was not, as with RDE (II)
RDE (II) significantly affects the structure and activity of IgE. Therefore, this suggests that the neutralizing titer of anti-HA IgE against influenza virus was reduced in vitro, compared with that of anti-HA IgG in our previous report (14). To cleave the HA of influenza virus to enable the virus to infect host cells, we incubated the cells in the presence of trypsin (21). We also confirmed the binding activity of the anti-ϵ chain to the trypsin-treated IgE. Although trypsin-treated IgG was detected at almost the same level as intact IgG (Fig. S2B), trypsin-treated IgE was significantly reduced by 2-fold (Fig. S2A). To evaluate whether trypsin affected the structure of IgE, we separated trypsin-treated IgE by Western blotting under nonreducing conditions. Interestingly, the main band of the IgE was reduced to around 150 kDa, as seen previously in RDE (II)–treated IgE (Fig. S2C, lanes 10–12). On the contrary, trypsin-treated IgG was detected at the same size as untreated IgG (Fig. S2C, lanes 4–6). Under reducing conditions, we also obtained a smaller band, around 50 kDa, than that of untreated IgE, depending on the serial dilution (Fig. S2D). These results suggest that trypsin also specifically affects the structure of IgE as RDE (II).
RDE (II) resists several protease inhibitors to modulate IgE
Both RDE (II) and trypsin significantly reduced the binding activity of IgE against anti-ϵ chain antibodies and affected the structure. It was considered that RDE (II) has protease activity like trypsin. To confirm our hypothesis, we used preincubated RDE (II) at 56 or 100 °C (56 °C-RDE (II) or 100 °C-RDE (II)), followed by treatment of IgE and IgG. 56 °C-RDE (II)–treated IgE indicated the same binding activity to anti-ϵ chain as intact RDE (II), which was reduced by 2-fold compared with the control (Fig. 5A). However, 100 °C-RDE (II)–treated IgE was not detected in the reduction of the binding activity to the anti-ϵ chain at the same level as the control (Fig. 5A). We also found that a band of ∼150 kDa was not detected in 100 °C-RDE (II)–treated IgE, and only a band over 250 kDa was detected (Fig. 5B). These results suggest that the activity of RDE (II) is caused by a function of an enzyme such as protease or glycosidase. To remove the possibility of low molecular weight compound-induced function, we dialyzed RDE (II) to exclude any molecule below 12–16 kDa. We confirmed that the band of IgE treated with dialyzed RDE (II) was also around 150 kDa, the same as that treated with intact RDE (II) (Fig. 5C). These results suggest that RDE (II) works as an enzyme for the modulation of IgE. Then, we analyzed the time course of the reduction by treatment with RDE (II). After incubation for 10 min, ladder bands between >250 and ∼150 kDa were detected (Fig. 5D, lane 2). The band over 250 kDa reduced and that around 150 kDa increased over time (Fig. 5D, lanes 3 and 4). After 360 min, only bands around 150 kDa were detected, with almost no bands over 250 kDa (Fig. 5D, lane 6). This result demonstrates that the band size of IgE is gradually reduced by RDE (II) and not abruptly reduced from over 250 kDa to around 150 kDa.
Figure 5.
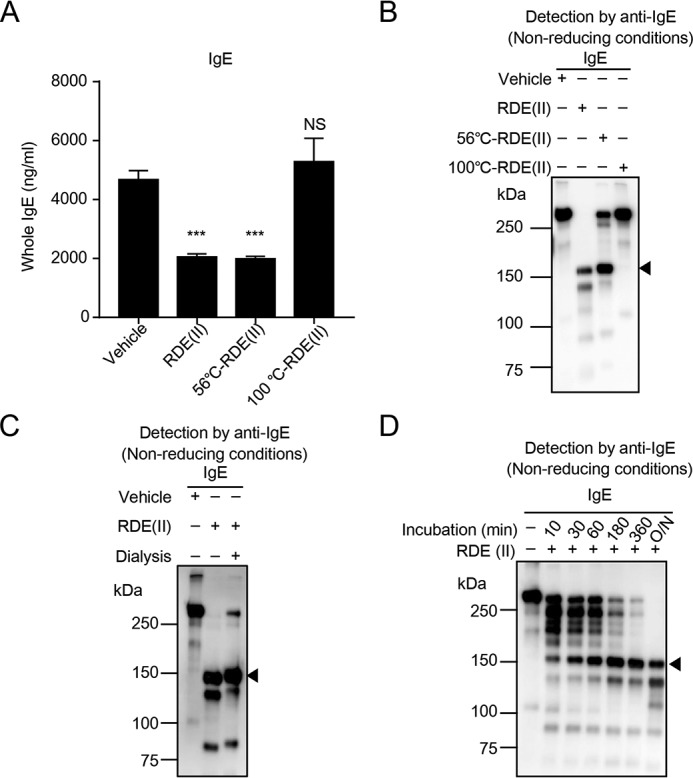
Characteristics of RDE (II) for modulating IgE. A and B, RDE (II) was preincubated at 56 or 100 °C for 30 min. Purified IgE was treated with the RDE (II) overnight (12–20 h). The level of IgE was measured by quantitative ELISA (A), and they were also blotted and analyzed with HRP-conjugated goat anti-mouse IgE (B). C, purified IgE was treated with dialyzed RDE (II) overnight (12–20 h). They were blotted by Western blotting under nonreducing conditions. D, purified IgE were treated with RDE (II) for the indicated time. They were blotted under nonreducing conditions. Data are representative of two independent experiments and indicate the mean ± S.D. ***, p < 0.001 (one-way ANOVA). NS, no significance.
RDE (II) was obtained from the culture fluid of V. cholerae, which contains some protease and sialidase (23). To evaluate the protease activity of RDE (II) for the modulation of IgE, we pretreated RDE (II) with paraoxon, diisopropyl fluorophosphate (DFP), as the serine–protease inhibitor (24) or bis-p-nitrophenyl phosphate (BNPP) as the carboxylesterase inhibitor (25). Paraoxon and BNPP could not inhibit the IgE modification function of both RDE (II) and trypsin (Fig. S3A). However, DFP could inhibit the modification function of trypsin (Fig. S3B, lanes 5 and 6) but not of RDE (II) (Fig. S3B, lanes 3 and 4). These results suggest that RDE (II) activity is not a result of carboxylesterase and serine–protease activity. Next, we evaluated the inhibitory effect against RDE (II) function with another protease inhibitor, cOmplete, which contains several inhibitors (e.g. α2-macroglobulin, see under “Experimental procedures”). cOmplete was also found to inhibit the modification of IgE by trypsin (Fig. S3D) but not RDE (II) (Fig. S3C), regardless of the concentration. In CBB staining, we could not detect any extra bands of IgE by incubation with cOmplete-treated RDE (II) (Fig. S3E). These results suggest that the function of RDE (II) is mainly not via protease.
Characteristic features of glycan alteration on RDE (II)–treated IgE
RDE from V. cholerae is a known NA (sialidase) (26). We examined the glycan structures of RDE (II)–treated IgE with lectin microarray using two types of IgE (Fig. 6, #1, anti-TNP IgE, and #2; anti-DNP IgE). To select a lectin probe, the mean-normalized data for untreated and RDE (II)–treated IgE were analyzed by Student's t test. The binding activity of RDE (II)–treated IgE for two lectins, LEL and Phaseolus vulgaris leucoagglutinin (PHA-L), was significantly reduced (p < 0.01) (Fig. 6A and Table S1). The binding of LEL was decreased by 3.5- or 7.1-fold after RDE (II) treatment (Fig. 6B) and that of PHA-L by 5.0- or 9.1-fold (Fig. 6C). This result demonstrates that RDE (II) influences LEL and PHA-L binding to the sugar chain of IgE. To confirm these results, we also carried out lectin-blotting assays. The signal intensity of LEL (Fig. 6D, lanes 1 and 2) and PHA-L (Fig. 6F, lanes 1 and 2) was significantly reduced in RDE (II)–treated IgE against each internal control (Fig. 6, E and G, lanes 1 and 2). Interestingly, the band of IgG was not detected with either LEL or PHA-L (Fig. 6, D and F, lanes 3 and 4), even though the band was detected with anti-IgG antibodies (Fig. 6, E and G, lanes 3 and 4). These results suggest that the glycan structures of IgE and IgG are different, which exercises an effect on RDE (II). Moreover, RDE (II)–treated IgE remained to be detected with several sialic acids (Fig. 6A and Table S1). Unlike treatment with RDE (II), we almost failed to detect a reduction in the band of IgE by treatment with NA (sialidase) (Fig. S4, lanes 3–9), similar to the situation with IgG (Fig. S4, lanes 12–18). These results suggest that sialidase is not primarily responsible for the modification of IgE by RDE (II).
Figure 6.

RDE (II) modulates the glycan on IgE. A, lectin microarray data of RDE (II)–treated IgE (#1 and #2) and -untreated (#1 and #2) were mean-normalized and then analyzed by Cluster 3.0. Clustering method was complete linkage. The heat map with clustering was acquired using Java TreeView. Yellow is positive, and blue is negative. B and C, indicated data (p < 0.01) represent the mean-normalized signal intensities of LEL (B) and PHA-L (C). The sugar residue or oligosaccharide structure in parentheses represents the binding preference. The glycan in parentheses represents carbohydrate specificity for each lectin. #1, anti-TNP IgE (C38-2); #2, anti-DNP IgE. D–G, relationship between the exercises of RDE (II) and the glycan structures. Purified IgE (C38-2) or IgG (15H6) was treated with the RDE (II) overnight (12–20 h). They were blotted under nonreducing conditions. The specimens were analyzed with biotin-conjugated LEL (D) or PHA-L (F), followed by incubation with HRP-conjugated streptavidin. E and G show internal controls for D and F, which are detected with HRP-conjugated anti-mouse IgE and IgG. Data in D to G are representative of two independent experiments.
The structure of IgE was also affected by trypsin. Therefore, we also analyzed the binding level of trypsin-treated IgE to LEL and PHA-L via lectin blotting. The signal intensity of trypsin-treated IgE with LEL (Fig. S5, left panel), and not PHA-L (Fig. S5, middle panel), was significantly reduced (Fig. S5, bar graph). These results suggest that at least poly-GlcNAc or poly-N-LacNAc, which was recognized by LEL, are important sugar chains in the structure of IgE.
RDE (II) is a potent enzyme and influences the structure of IgE
Glycan structures, which bind to LEL or PHA-L, are potentially important for the IgE structure. Previously, Björklund et al. (12) reported that N-linked glycan in IgE influences the receptor-binding structures. We confirmed that anti-HA IgE was not detected in the supernatant of HEK293T cells in the presence of tunicamycin, which inhibits N-glycosylation of glycoprotein (Fig. S6A, lanes 2 and 3). The expression of anti-HA IgG was detected even in the presence of tunicamycin (Fig. S6A, lanes 5 and 6). This result suggests that N-glycosylation influences the structure of IgE, compared with IgG. We then analyzed whether PNGase F reduces the band size of IgE. Under nondenaturing conditions, PNGase F slightly reduced the band size of IgE, unlike RDE (II)–treated IgE around 75 kDa (Fig. S6B, lanes 5 and 6). In contrast, under denaturing conditions, a reduction in the IgE band to ∼75 kDa was detected by treating with PNGase F (Fig. S6C, lanes 5 and 6). Shade et al. (10) also reported that oligomannose, an N-linked glycan, is indispensable for IgE binding to FcϵRI. However, we could not confirm that Endo H reduced the IgE band as RDE (II) treated IgE under both nondenaturing and denaturing conditions (Fig. S6, B and C, lanes 3 and 4). These results suggest that RDE (II) has a more potent effect against even the intact structure of IgE, compared with PNGase F and Endo H.
Discussion
In this study, we demonstrated that an enzyme from V. cholerae culture fluid (RDE (II)) can modulate only IgE and not IgG. The results suggest that LEL-binding sugar chains on IgE are important to retain the structure, allowing for binding to antigen and FcϵRI on mast cells. In a previous report, IgA and IgM were also found to not be affected by RDE (II) (14). Moreover, we were unable to detect the modulation of light chain by the RDE (II)-like ϵ chain using Western blotting (Figs. 1C and Fig. 2, C and E). This suggested that RDE (II) can modulate only the ϵ chain but not the light chain. Using light chain BP, we detected a band of RDE (II)–treated IgE around 150 kDa, which is the same size as the anti-ϵ chain (Fig. 2, A and C). This result indicates that RDE (II) did not separate IgE into an ϵ chain and a light chain. These results demonstrate that the enzymatic activity of RDE (II) is able to specifically inhibit the ϵ chain in the initiation of anaphylaxis.
Previous studies have reported that N-glycosylation of IgE is essential for its binding to FcϵRI and its function (10, 12). IgE has four constant domains (Cϵ1–4) and is heavily glycosylated (27). Shade et al. (10) reported that the N-linked oligomannose structure on Cϵ3 of IgE plays a crucial role to abrogate binding to FcϵRI. Therefore, they concluded that mutations of Cϵ3 or Endo F1 rendered IgE incapable of eliciting mast cell degranulation. However, we could not find this effect on PNGase F and Endo H by Western blotting (Fig. S6B). The longer treatment time of 72 h, as seen in Shade et al. (10) may reduce the band size of IgE. Moreover, although our result in Fig. S6C is consistent with previous reports (10, 12), IgE was only affected by PNGase F under denaturing conditions. On the contrary, RDE (II) was found to affect the structure of IgE after only a 10-min incubation under nondenaturing conditions (Fig. 5D). These results suggest that RDE (II) activity has a greater effect on the structure of IgE, when compared with PNGase F and Endo H.
The band size of mouse IgE from HEK293T cells was barely affected by RDE (II) (Fig. 1C), possibly due to the glycosylation in human cells (28, 29), although the binding level of anti-ϵ chain decreased considerably (Fig. 1B). However, RDE (II) decreased the band size of ϵ chain from mouse hybridoma (C38-2) to ∼50 kDa, which was smaller than the predicted size of 73 kDa (Fig. 2D). Moreover, any protease inhibitor could not inhibit the effect of RDE (II) (Fig. S3). Taken together, we considered that IgE was not directly digested by RDE (II) but instead primarily affected the sugar chains on ϵ chain. This result also suggests that not only one but several glycans on IgE are involved in modulation, because RDE (II) was found to gradually modulate the structure of IgE (Fig. 5D). Furthermore, our results (Figs. 1C and 2C) also suggested that some effects of RDE (II) are different between mouse and human IgE because of the difference in the glycan structure (10).
Through our lectin microarray using 96 lectins, we identified LEL and PHA-L (p < 0.01), which decreased the binding level to RDE (II)–treated IgE (Fig. 6). LEL recognizes N-GlcNAc β-1,4-linked N-GlcNAc oligomers up to four carbohydrate units ((GlcNAc)2–4) or Galβ1–4GlcNAcβ1–3 oligomers ((LacNAc)n) (15–18). PHA-L binds to tetra-antennary complex oligosaccharides (Gal(β1–4)GlcNAc(β1–6)Man). The current result suggests that RDE (II) modulates branched glycans on IgE, which play a crucial role in retaining the structure and function of IgE. Trypsin was observed to have a similar function to RDE (II) in the modulation of IgE (Fig. S2). Interestingly, trypsin-treated IgE was reduced to the binding level of only LEL and not PHA-L (Fig. S5). This result suggests that LEL-binding sugar chains are more important for the modulation of IgE than PHA-L. However, both lectins were unable to bind to IgG (Fig. 6, D and F). This suggests that the glycoform in the IgE structure is different from that in IgG. Taken together, these results suggest that may be possible for RDE (II) to specifically recognize the IgE-glycan and not IgG.
The binding to anti-ϵ antibody in ELISA and the change of the band size in Western blotting were correlated because the titer of RDE (II) was the same (Fig. 2, D and F). Therefore, this suggests that modulation of the structure by RDE (II) is important for the inhibition of IgE function. RDE (II)–treated IgE was also unable to bind to the antigen and was thereby incapable of neutralizing influenza virus infection (14). These results suggest that the enzyme in RDE (II) drastically changed both the Fc region and antigen-binding site of IgE and not IgG. It is possible that IgE is unable to bind not only to the FcϵRI receptor but also to the allergen as a result of RDE (II) treatment. Taken together, this indicates that RDE (II) may be able to inactivate the allergic function of IgE.
It is well known that RDE (II) has both protease and sialidase activity (23). However, unlike trypsin, we could not inhibit the function of RDE (II) via any protease inhibitor (Fig. S3). We also found that the main function of RDE (II) was not sialidase, because RDE (II)–treated IgE was still able to bind several sialic acids in the lectin array (Fig. 6A and Table S1), and the band size IgE was not reduced by sialidase (Fig. S4). V. cholerae has other glycosidase activities (30, 31). One is amidase, which is crucial for cell division and growth (30). Amidases release peptide side chains from the glycan strand, followed by the cleavage of septal peptidoglycan, which is composed of glycan chains with alternating β-1,4-linked N-GlcNAc and N-acetylmuramic acid (Mur-NAc) peptide residues (30). V. cholerae also has at least two chitinases, which synergistically hydrolyze chitin, (GlcNAc)n, into small oligomers (32, 33). It is possible that the amidase or chitinase in RDE (II) can modulate the structure of IgE; however, further studies are required to elucidate the relevance of RDE (II) in inhibiting the function of IgE.
One antibody drug, omalizumab, binds to the Fc fragment of IgE and is able to neutralize IgE by blocking the binding to FcϵRI (3, 6, 7). Therefore, the target of IgE is highly effective for therapy against allergies. However, omalizumab has a limitation, i.e. the delay in the onset of clinical benefits, which can take several weeks or months because omalizumab is unable to displace IgE bound to FcϵRI under the current treatment conditions (7). In this study, RDE (II) was found to decrease the binding level of not only the Fc region but also at the antigen-binding site (Fig. 1, A and B). Therefore, RDE (II) may be able to abrogate the function of even the IgE bound to FcϵRI by inhibiting the binding of the allergen. Moreover, we also found that RDE (II) had an effect not only in purified monoclonal IgE but also in IgE in the serum (Fig. 3). This suggests that RDE (II) is effective against polyclonal IgE, which have multiple kinds of glycan. In this study, RDE (II) showed specific characteristics for modulating the glycan on IgE, compared with IgG. Taken together, our results suggest that treatment with RDE (II) could be successful for therapy against allergy; however, further studies are needed to identify the specific enzyme in RDE (II), which modulates IgE.
Experimental procedures
Plasmid construction
In our previous report (14, 34), we generated the plasmids encoding the genes for the heavy chain (IgG and IgE) and the light chain (κ) of a neutralizing anti- hemagglutinin (HA) antibody (35). All constructions are based on the pCADEST1 vector (36).
Reagents
RDE (II) produced from V. cholerae Ogawa type 558 (Denka Seiken, Tokyo, Japan) was dissolved in 20 ml of 0.9% NaCl solution and stored at −20 °C until use. In some experiments, RDE (II) was dialyzed overnight against 100 volumes of PBS(−) using cellulose tubing (Viskase Co., Inc.) with a molecular mass cutoff of 12–16 kDa. One mg/ml acetylated trypsin (Sigma) was dissolved in sterilized distilled H2O. As demonstrated in Fig. S2, A and B, the reaction with trypsin was performed using minimum Eagle's medium (Nissui, Tokyo, Japan) containing 0.03% glutamine, 0.01 m HEPES, 0.2% BSA, and 0.075% NaHCO3 as the neutralizing assay, as mentioned in previous reports (14, 34). Neuraminidase (NA) from Arthrobacter ureafaciens (Nacalai Tesque, Tokyo, Japan), Endo H, and PNGase F (New England Biolabs Inc., Beverly, MA), purified mouse monoclonal anti-T-2 mycotoxin IgG (15H6) (Southern Biotech, Birmingham, AL), purified mouse monoclonal anti-TNP IgE (C38-2) (Pharmingen), and cOmplete (Roche Diagnostics, Mannheim, Germany), which is composed from several protease inhibitors (aprotinin, bestatin, calpain inhibitor, chymostatin, E-64, leupeptin, α2-macroglobulin, Pefabloc SC, pepstatin, phenylmethylsulfonyl fluoride, 1-chloro-3-tosylamido-7-amino-2-heptanone–HCl, and trypsin inhibitor), were purchased. BNPP (Sigma), which is a carboxylesterase inhibitor (25), paraoxon (Sigma) and DFP (Wako, Osaka, Japan), which are serine protease inhibitors, were also purchased.
Antibody expression in vitro
HEK293T cells were maintained in Dulbecco's modified Eagle's medium (ThermoFisher Scientific, Waltham, MA) supplemented with 10% heat-inactivated fetal calf serum and penicillin–streptomycin–glutamine. HEK293T cells were co-transfected with the plasmid vectors using FuGENE HD transfection reagent (Promega, WI). After 1 week, the supernatants were obtained and treated with RDE (II) at 37 °C overnight (12–20 h), followed by incubation at 56 °C for 30 min as mentioned in our previous report (14). HEK293T cells were also transfected and incubated in the presence or absence of tunicamycin (Wako). Forty hours later, the supernatants were obtained.
Mice
BALB/c (SLC, Shizuoka, Japan) were maintained under specific pathogen-free conditions. Animal experiments were conducted in accordance with the approval of the Animal Research Committee of the Aichi Medical University.
ELISA
The antigen-binding level of anti-HA antibody treated with RDE (II) was determined as reported previously (14, 34, 37). Briefly, a 96-well plate was coated with HA protein purified from A/PR8 virus using affinity columns constructed by coupling of CNBr-activated Sepharose 4B beads (GE Healthcare UK Ltd., Buckinghamshire, UK) and anti-HA antibodies. The plate was incubated with TBS-containing bovine casein (Merck Millipore) for blocking. The plate was then incubated with serially diluted supernatants or serum. HA-specific antibodies were detected by using HRP-conjugated goat anti-mouse IgG (Southern Biotech) or IgE. Anti-HA IgG purified from a hybridoma (35) or anti-HA IgE obtained from the transfected-HEK293T cells were used as the standard (14, 34, 37). Finally, the expression level was detected using an ELISA POD substrate TMB kit (Nacalai Tesque). Absorbance was measured by Spectramax M5 (Molecular Devices). To detect whole IgG or IgE, a 96-well plate was coated with anti-mouse Igs (Southern Biotech).
Competitive ELISA
For competitive ELISA, we used biotin-conjugated anti-HA IgG antibodies purified from a hybridoma (35), along with HRP-conjugated streptavidin, as reported previously (14). The titer was determined by the inhibition curve based on the absorbance of standard anti-HA IgG antibody.
Antibody expression in vivo
We performed hydrodynamic injections (38, 39) using a previously described method (14). Briefly, 6–10-week-old BALB/c were injected in the tail vein with PBS-containing plasmid (e.g. 5 μg/1.6 ml), where the DNA volume was 8–12% of the body weight. The injection was performed over less than 5 s using a 27-gauge needle. One day later, the serum was obtained and treated with RDE (II) overnight at 37 °C, followed by the measurement of the expression level of anti-HA IgE and anti-HA IgG or whole IgE and IgG by quantitative ELISA.
Western blotting and CBB staining
Anti-HA IgE and anti-HA IgG in the supernatants or purified IgE (C38-2) and IgG (15H6) treated with RDE (II) were separated by SDS-PAGE (6 or 12%) under nonreducing or reducing conditions, followed by either transfer to a PVDF membrane (Immobilon-P, Merck Millipore) or stained with CBB (Bio-Rad). The membrane was blocked with Blocking One reagent (Nacalai Tesque) for 30 min, followed by incubation at room temperature with a mixture of HRP-conjugated goat anti-mouse IgG and IgE or light chain (kappa) BP (Santa Cruz Biotechnology). The specific bands were visualized using the enhanced chemiluminescence ECL substrate (GE Healthcare) on the ImageQuant LAS4000 system (GE Healthcare). All full-length blots were indicated in Fig. S7 and Fig. S8.
BMMC culture and stimulation
BMMCs were generated from BALB/c, as described previously (40, 41). Briefly, mouse bone marrow cells were harvested from the tibias and femurs and cultured for 4–6 weeks in RPMI 1640 medium (ThermoFisher Scientific) containing IL-3 obtained from IL-3–expressing CHO cells, 10% fetal calf serum, 1 mm pyruvic acid (Wako), 0.1 mm nonessential amino acids (Wako), 50 μm 2-mercaptoethanol, and penicillin–streptomycin–glutamine. Anti-DNP IgE, which was kindly provided by Dr. Fu-Tong Liu (University of California) (9), was treated with RDE (II) overnight (12–20 h), followed by incubation at 56 °C for 30 min. Then, BMMCs were incubated with the RDE (II)–treated anti-DNP IgE for 2 h or overnight (12–20 h), followed by incubation with HSA–DNP (Sigma) as indicated either overnight (12–20 h) or for 1 h. The supernatant was then collected and analyzed using a quantitative ELISA kit for IL-6 (ebioscience), TNFα, and histamine (Neogen Corp., Lexington, KY).
Flow cytometry to detect the binding of IgE to BMMCs
Twenty μg/ml biotin-conjugated anti-DNP IgE or mouse IgG1 isotype control antibody (MOPC-21; Biolegend, San Diego, CA) was treated with RDE (II) overnight (12–20 h). BMMCs or BMMs, which differentiated in the presence of L929 cell–conditioned medium as mentioned in a previous report (42), were incubated with the antibodies for 30 min at 4 °C, followed by incubation with APC-conjugated streptavidin (Biolegend). The binding level was analyzed by FACSCantoII (BD Biosciences).
IgE-mediated PCA
We performed mast cell-dependent PCA experiments, which is known as a model of in vivo type I allergy, as described previously (43–45). Briefly, 6–10-week-old BALB/c were primed with 20 ng/10 μl RDE (II)–treated anti-DNP IgE or were untreated in both ears under pentobarbital anesthesia (Somnopentyl, Kyoritsu, Tokyo, Japan). One day later, the mice were intravenously injected with 200 μg of HSA–DNP in 200 μl of PBS(−)-containing 0.5% Evans blue dye (Sigma) to visualize IgE-mediated anaphylaxis, in which vascular permeability increased. Thirty minutes later, an 8-mm ear punch was collected and minced in 50 μl of 1 n potassium hydroxide and incubated at 37 °C overnight. To extract the Evans blue dye, 260 μl of extraction buffer (0.4 n phosphoritic acid and 72% (v/v) acetone) was added to the specimen. The absorbance was determined at 620 nm by Spectramax M5.
Lectin array
The lectin microarray production and analysis were performed as described previously (19). Briefly, biotin-conjugated IgE (anti-TNP IgE (C38-2) and anti-DNP IgE) was concentrated with Dynabeads M280 streptavidin (ThermoFisher Scientific) and dissolved in the Matsunami spotting solution. After filtration, they were spotted on the Schott epoxy-coated glass slide using the MicroSys noncontact microarray printing robot. The Cy3-labeled streptavidin dissolved in the probing solution was applied to each chamber of the lectin microarray (80 μl/well) and incubated at 20 °C overnight. After washing the chambers with the probing solution, fluorescent images were immediately acquired using a Bio-Rex scan 200 evanescent field–activated fluorescence scanner (Rexxam Co. Ltd., Kagawa, Japan). Data were analyzed with the Array Pro analyzer version 4.5 (Media Cybernetics, Inc.). The net intensity value for each spot was determined by signal intensity minus background value. The lectin signals of triplicate spots were averaged and normalized to the highest signal intensity among 96 lectin conjugates immobilized on the array.
Lectin blotting
The binding activity of RDE (II)–treated IgE to the lectins was analyzed by lectin blotting as described previously (46). Briefly, purified IgG and IgE were separated by 6% SDS-PAGE, followed by transfer to a PVDF membrane and blocking. Then, the membrane was incubated with biotin-conjugated LEL (Vector Laboratories, Inc., Burlingame, CA) or PHA-L at room temperature for 2 h, followed by incubation with HRP-conjugated streptavidin (GE Healthcare) for 30 min.
Statistical analysis
All graphs were constructed using GraphPad Prism 7 (GraphPad Software). Data were analyzed using parametric one-way ANOVA or Student's t test, where p < 0.05 was considered statistically significant. Data are shown and compared as mean and ranges of more than five mice in different groups.
Author contributions
T. Y., M. Inui, S. T., M. Itoh, I. I., A. N., H. Takagi, and S. A.-T. conceptualization; T. Y., K. I., J. K., and S. A.-T. resources; T. Y., N. S., and S. A.-T. data curation; T. Y., K. H., and S. A.-T. formal analysis; T. Y., S. T., and S. A.-T. funding acquisition; T. Y., T. I., N. S., H. Tateno, and S. A.-T. validation; T. Y., M. Inui, K. H., S. T., M. Itoh, I. I., A. N., H. Takagi, M. B., T. I., N. S., and H. Tateno investigation; T. Y., K. H., and H. Tateno visualization; T. Y., M. Inui, K. H., T. I., N. S., and H. Tateno methodology; T. Y. writing-original draft; T. Y. and S. A.-T. project administration; K. H. and H. Tateno software; S. A.-T. supervision; S. A.-T. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. Fu-Tong Liu (University of California) for supplying anti-DNP IgE. We also thank the Division of Laboratory Animal Research (Aichi Medical University) for providing the facilities for performing the animal experiments, and the Division of Advanced Research Promotion (Aichi Medical University) for technical instructions and assistance.
This work was supported by JSPS KAKENHI Grant Numbers 15K09491 (to S. A.-T.) and 16H05898 (to S. T.), Joint Research Project of the Institute of Medical Science grants, University of Tokyo Grant 2016-3008, Aichi Medical University for Unit Support Grant 3094117003, the TOYOAKI Scholarship Foundation, the Aikeikai Foundation, the Nitto Foundation, and Takeda Science Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S8 and Table S1.
- FcϵRI
- high-affinity fragment crystallizable ϵ receptor
- BMM
- bone marrow–derived macrophage
- BMMC
- bone marrow–derived mast cell
- BNPP
- bis-p-nitrophenyl phosphate
- (light chain) BP
- binding protein
- CBB
- Coomassie Brilliant Blue
- DFP
- diisopropyl fluorophosphate
- DNP
- dinitrophenol
- Endo F1
- endoglycosidase F1
- LacNAc
- N-acetyl-lactosamine
- RDE
- receptor-destroying enzyme
- HA
- hemagglutinin
- HSA
- human serum albumin
- HEK293T
- human embryonic kidney cells 293 that stably express the SV40 large T antigen
- LEL
- L. esculentum lectin
- NA
- neuraminidase (sialidase)
- PCA
- passive cutaneous anaphylaxis
- PHA-L
- P. vulgaris leucoagglutinin
- PNGase F
- peptide-N-glycosidase F
- TNP
- trinitrophenyl
- TNFα
- tumor necrosis factor-α
- HRP
- horseradish peroxidase
- PVDF
- polyvinylidene difluoride
- APC
- allophycocyanin
- ANOVA
- analysis of variance
- IL
- interleukin.
References
- 1. Ishizaka K., and Ishizaka T. (1967) Identification of γ-E-antibodies as a carrier of reaginic activity. J. Immunol. 99, 1187–1198 [PubMed] [Google Scholar]
- 2. Ishizaka K., and Ishizaka T. (2016) Identification of IgE. J. Allergy Clin. Immunol. 137, 1646–1650 10.1016/j.jaci.2015.12.1343 [DOI] [PubMed] [Google Scholar]
- 3. Balbino B., Conde E., Marichal T., Starkl P., and Reber L. L. (2018) Approaches to target IgE antibodies in allergic diseases. Pharmacol. Ther. 191, 50–64 10.1016/j.pharmthera.2018.05.015 [DOI] [PubMed] [Google Scholar]
- 4. Cookson W. (2004) The immunogenetics of asthma and eczema: a new focus on the epithelium. Nat. Rev. Immunol. 4, 978–988 10.1038/nri1500 [DOI] [PubMed] [Google Scholar]
- 5. Yamaguchi M., Lantz C. S., Oettgen H. C., Katona I. M., Fleming T., Miyajima I., Kinet J. P., and Galli S. J. (1997) IgE enhances mouse mast cell FcϵRI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE-dependent reactions. J. Exp. Med. 185, 663–672 10.1084/jem.185.4.663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Busse W., Corren J., Lanier B. Q., McAlary M., Fowler-Taylor A., Cioppa G. D., van As A., and Gupta N. (2001) Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J. Allergy Clin. Immunol. 108, 184–190 10.1067/mai.2001.117880 [DOI] [PubMed] [Google Scholar]
- 7. Oettgen H. C. (2016) Fifty years later: emerging functions of IgE antibodies in host defense, immune regulation, and allergic diseases. J. Allergy Clin. Immunol. 137, 1631–1645 10.1016/j.jaci.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shade K.-T., and Anthony R. (2013) Antibody glycosylation and inflammation. Antibodies 2, 392–414 10.3390/antib2030392 [DOI] [Google Scholar]
- 9. Natsume A., Niwa R., and Satoh M. (2009) Improving effector functions of antibodies for cancer treatment: enhancing ADCC and CDC. Drug Des. Devel. Ther. 3, 7–16 [PMC free article] [PubMed] [Google Scholar]
- 10. Shade K.-T., Platzer B., Washburn N., Mani V., Bartsch Y. C., Conroy M., Pagan J. D., Bosques C., Mempel T. R., Fiebiger E., and Anthony R. M. (2015) A single glycan on IgE is indispensable for initiation of anaphylaxis. J. Exp. Med. 212, 457–467 10.1084/jem.20142182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu G., Hitchen P. G., Panico M., North S. J., Barbouche M. R., Binet D., Morris H. R., Dell A., and Haslam S. M. (2016) Glycoproteomic studies of IgE from a novel hyper-IgE syndrome linked to PGM3 mutation. Glycoconj. J. 33, 447–456 10.1007/s10719-015-9638-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Björklund J. E., Karlsson T., and Magnusson C. G. (1999) N-Glycosylation influences epitope expression and receptor binding structures in human IgE. Mol. Immunol. 36, 213–221 10.1016/S0161-5890(99)00036-X [DOI] [PubMed] [Google Scholar]
- 13. Tyrrell D. A., and Horsfall F. L. Jr. (1952) A procedure which eliminates nonspecific inhibitor from human serum but does not affect specific antibodies against influenza viruses. J. Immunol. 69, 563–574 [PubMed] [Google Scholar]
- 14. Yamazaki T., Nagashima M., Ninomiya D., Ainai A., Fujimoto A., Ichimonji I., Takagi H., Morita N., Murotani K., Hasegawa H., Chiba J., and Akashi-Takamura S. (2018) Neutralizing antibodies induced by gene-based hydrodynamic injection have a therapeutic effect in lethal influenza infection. Front. Immunol. 9, 47 10.3389/fimmu.2018.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Immel F., Broussard C., Catherinet B., Plasseraud L., Alcaraz G., Bundeleva I., and Marin F. (2016) The shell of the invasive bivalve species Dreissena polymorpha: biochemical, elemental and textural investigations. PLoS ONE 11, e0154264 10.1371/journal.pone.0154264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferreira-Medeiros M., and Correa-Gillieron E. (2004) Recognition of N-acetylglucosamine (GlyNAc) and poly-N-acetyllactosamine residues in vessels of the rat pineal gland. Int. J. Morphol. 22, 285–290 10.4067/S0717-95022004000400008 [DOI] [Google Scholar]
- 17. Naidu R. A., Ingle C. J., Deom C. M., and Sherwood J. L. (2004) The two envelope membrane glycoproteins of tomato spotted wilt virus show differences in lectin-binding properties and sensitivities to glycosidases. Virology 319, 107–117 10.1016/j.virol.2003.10.012 [DOI] [PubMed] [Google Scholar]
- 18. Tateno H., Uchiyama N., Kuno A., Togayachi A., Sato T., Narimatsu H., and Hirabayashi J. (2007) A novel strategy for mammalian cell surface glycome profiling using lectin microarray. Glycobiology 17, 1138–1146 10.1093/glycob/cwm084 [DOI] [PubMed] [Google Scholar]
- 19. Tateno H., Toyota M., Saito S., Onuma Y., Ito Y., Hiemori K., Fukumura M., Matsushima A., Nakanishi M., Ohnuma K., Akutsu H., Umezawa A., Horimoto K., Hirabayashi J., and Asashima M. (2011) Glycome diagnosis of human induced pluripotent stem cells using lectin microarray. J. Biol. Chem. 286, 20345–20353 10.1074/jbc.M111.231274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tateno H., Kuno A., Itakura Y., and Hirabayashi J. (2010) A versatile technology for cellular glycomics using lectin microarray. Methods Enzymol. 478, 181–195 10.1016/S0076-6879(10)78008-3 [DOI] [PubMed] [Google Scholar]
- 21. Klenk H. D., Rott R., Orlich M., and Blödorn J. (1975) Activation of influenza A viruses by trypsin treatment. Virology 68, 426–439 10.1016/0042-6822(75)90284-6 [DOI] [PubMed] [Google Scholar]
- 22. Liu F. T., Bohn J. W., Ferry E. L., Yamamoto H., Molinaro C. A., Sherman L. A., Klinman N. R., and Katz D. H. (1980) Monoclonal dinitrophenyl-specific murine IgE antibody: preparation, isolation, and characterization. J. Immunol. 124, 2728–2737 [PubMed] [Google Scholar]
- 23. Nakayama M., Ogawa Y., and Yamazi Y. (1986) Sialidase and protease activities of commercial RDE products used for the HI test of influenza viruses. Nihon Ika Daigaku Zasshi. 53, 534–536 10.1272/jnms1923.53.534 [DOI] [PubMed] [Google Scholar]
- 24. Hattori M., Isomura S., Yokoyama E., Ujita M., and Hara A. (2005) Extracellular trypsin-like proteases produced by Cordyceps militaris. J. Biosci. Bioeng. 100, 631–636 10.1263/jbb.100.631 [DOI] [PubMed] [Google Scholar]
- 25. Ohura K., Sakamoto H., Ninomiya S., and Imai T. (2010) Development of a novel system for estimating human intestinal absorption using Caco-2 cells in the absence of esterase activity. Drug Metab. Dispos. 38, 323–331 10.1124/dmd.109.029413 [DOI] [PubMed] [Google Scholar]
- 26. Ada G. L., French E. L., and Lind P. E. (1961) Purification and properties of neuraminidase from Vibrio cholerae. J. Gen. Microbiol. 24, 409–425 10.1099/00221287-24-3-409 [DOI] [PubMed] [Google Scholar]
- 27. Arnold J. N., Wormald M. R., Sim R. B., Rudd P. M., and Dwek R. A. (2007) The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 25, 21–50 10.1146/annurev.immunol.25.022106.141702 [DOI] [PubMed] [Google Scholar]
- 28. Inamura M., Itakura M., Okamoto H., Hoka S., Mizoguchi A., Fukazawa Y., Shigemoto R., Yamamori S., and Takahashi M. (2006) Differential localization and regulation of stargazin-like protein, γ-8 and stargazin in the plasma membrane of hippocampal and cortical neurons. Neurosci. Res. 55, 45–53 10.1016/j.neures.2006.01.004 [DOI] [PubMed] [Google Scholar]
- 29. Sharma R., van Damme E. J., Peumans W. J., Sarsfield P., and Schumacher U. (1996) Lectin binding reveals divergent carbohydrate expression in human and mouse Peyer's patches. Histochem. Cell Biol. 105, 459–465 10.1007/BF01457659 [DOI] [PubMed] [Google Scholar]
- 30. Möll A., Dörr T., Alvarez L., Chao M. C., Davis B. M., Cava F., and Waldor M. K. (2014) Cell separation in Vibrio cholerae is mediated by a single amidase whose action is modulated by two nonredundant activators. J. Bacteriol. 196, 3937–3948 10.1128/JB.02094-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park J. K., Wang L.-X., Patel H. V., and Roseman S. (2002) Molecular cloning and characterization of a unique β-glucosidase from Vibrio cholerae. J. Biol. Chem. 277, 29555–29560 10.1074/jbc.M202978200 [DOI] [PubMed] [Google Scholar]
- 32. Mondal M., Nag D., Koley H., Saha D. R., and Chatterjee N. S. (2014) The Vibrio cholerae extracellular chitinase ChiA2 is important for survival and pathogenesis in the host intestine. PLoS ONE 9, e103119 10.1371/journal.pone.0103119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meibom K. L., Li X. B., Nielsen A. T., Wu C.-Y., Roseman S., and Schoolnik G. K. (2004) The Vibrio cholerae chitin utilization program. Proc. Natl. Acad. Sci. U.S.A. 101, 2524–2529 10.1073/pnas.0308707101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamazaki T., Nagashima M., Ninomiya D., Arai Y., Teshima Y., Fujimoto A., Ainai A., Hasegawa H., and Chiba J. (2011) Passive immune-prophylaxis against influenza virus infection by the expression of neutralizing anti-hemagglutinin monoclonal antibodies from plasmids. Jpn. J. Infect. Dis. 64, 40–49 [PubMed] [Google Scholar]
- 35. Asanuma H., Matsumoto-Takasaki A., Suzuki Y., Tamura S., Sata T., Kusada Y., Matsushita M., and Fujita-Yamaguchi Y. (2008) Influenza PR8 HA-specific Fab fragments produced by phage display methods. Biochem. Biophys. Res. Commun. 366, 445–449 10.1016/j.bbrc.2007.11.135 [DOI] [PubMed] [Google Scholar]
- 36. Ainai A., Kawase T., Ida A., Maeda Y., Ohba H., Ikeda Y., Sato H., Takahashi M., and Chiba J. (2006) Renewal of EBV-hybridoma method: efficient generation of recombinant fully human neutralizing IgG antibodies specific for tetanus toxin by use of tetroma cells. Hum. Antibodies 15, 139–154 [PubMed] [Google Scholar]
- 37. Tamura S., Ito Y., Asanuma H., Hirabayashi Y., Suzuki Y., Nagamine T., Aizawa C., and Kurata T. (1992) Cross-protection against influenza virus infection afforded by trivalent inactivated vaccines inoculated intranasally with cholera toxin B subunit. J. Immunol. 149, 981–988 [PubMed] [Google Scholar]
- 38. Liu F., Song Y., and Liu D. (1999) Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6, 1258–1266 10.1038/sj.gt.3300947 [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi N., Nishikawa M., Hirata K., and Takakura Y. (2004) Hydrodynamics-based procedure involves transient hyperpermeability in the hepatic cellular membrane: implication of a nonspecific process in efficient intracellular gene delivery. J. Gene Med. 6, 584–592 10.1002/jgm.541 [DOI] [PubMed] [Google Scholar]
- 40. Yoshida K., Ito M., and Matsuoka I. (2017) Divergent regulatory roles of extracellular ATP in the degranulation response of mouse bone marrow–derived mast cells. Int. Immunopharmacol. 43, 99–107 10.1016/j.intimp.2016.12.014 [DOI] [PubMed] [Google Scholar]
- 41. Kitaura J., Song J., Tsai M., Asai K., Maeda-Yamamoto M., Mocsai A., Kawakami Y., Liu F. T., Lowell C. A., Barisas B. G., Galli S. J., and Kawakami T. (2003) Evidence that IgE molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the FcϵRI. Proc. Natl. Acad. Sci. U.S.A. 100, 12911–12916 10.1073/pnas.1735525100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maruzuru Y., Ichinohe T., Sato R., Miyake K., Okano T., Suzuki T., Koshiba T., Koyanagi N., Tsuda S., Watanabe M., Arii J., Kato A., and Kawaguchi Y. (2018) Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe 23, 254–265.e7 10.1016/j.chom.2017.12.014 [DOI] [PubMed] [Google Scholar]
- 43. Maeda-Yamamoto M., Inagaki N., Kitaura J., Chikumoto T., Kawahara H., Kawakami Y., Sano M., Miyase T., Tachibana H., Nagai H., and Kawakami T. (2004) O-Methylated catechins from tea leaves inhibit multiple protein kinases in mast cells. J. Immunol. 172, 4486–4492 10.4049/jimmunol.172.7.4486 [DOI] [PubMed] [Google Scholar]
- 44. Hata D., Kawakami Y., Inagaki N., Lantz C. S., Kitamura T., Khan W. N., Maeda-Yamamoto M., Miura T., Han W., Hartman S. E., Yao L., Nagai H., Goldfeld A. E., Alt F. W., Galli S. J., et al. (1998) Involvement of Bruton's tyrosine kinase in Fcϵ RI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 187, 1235–1247 10.1084/jem.187.8.1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Joo H. M., Kang S. J., Nam S. Y., Yang K. H., Kim C. S., Lee I. K., and Kim J. Y. (2015) The inhibitory effects of low-dose ionizing radiation in IgE-mediated allergic responses. PLoS ONE 10, e0136394 10.1371/journal.pone.0136394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao R., Qin W., Qin R., Han J., Li C., Wang Y., and Xu C. (2017) Lectin array and glycogene expression analyses of ovarian cancer cell line A2780 and its cisplatin-resistant derivate cell line A2780-cp. Clin. Proteomics 14, 20 10.1186/s12014-017-9155-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.