

Abstract
Prostate cancer is a complex, heterogeneous disease that mainly affects the older male population with a high-mortality rate. The mechanisms underlying prostate cancer progression are still incompletely understood. Beta-adrenergic signaling has been shown to regulate multiple cellular processes as a mediator of chronic stress. Recently, beta-adrenergic signaling has been reported to affect the development of aggressive prostate cancer by regulating neuroendocrine differentiation, angiogenesis, and metastasis. Here, we briefly summarize and discuss recent advances in these areas and their implications in prostate cancer therapeutics. We aim to provide a better understanding of the contribution of beta-adrenergic signaling to the progression of aggressive prostate cancer.
Keywords: angiogenesis, beta-adrenergic signaling pathway, metastasis, neuroendocrine differentiation, prostate cancer
INTRODUCTION
Prostate adenocarcinoma (PAC) is the most prevalently diagnosed malignancy in men in the western world. Progression to castration-resistant prostate cancer (CRPC) is the major therapeutic challenge.1 Androgen signal is required for regeneration, function, and development of normal prostate and prostatic adenocarcinoma.2 Targeting androgen synthesis or blocking androgen receptor (AR) is a widely accepted hormonal treatment that is initially effective. However, most tumors acquire resistance to hormonal therapies and subsequently develop CRPC.3 An aggressive subset of CRPC, neuroendocrine prostate cancer (NEPC), expresses low or absent prostate-specific AR signaling, and elevated levels of stem cell and neuroendocrine markers.4 It is still unclear whether the AR-negative prostatic cancer cells arise directly from AR-positive adenocarcinomas or from native AR-low neuroendocrine cells. However, the majority of evidence in the literature supports the hypothesis that NEPC cells evolve from preexisting PAC cells through neuroendocrine differentiation (NED).1,5,6,7
Beta-adrenergic signaling has been implicated in regulating apoptosis, angiogenesis, NED, migration, and metastasis of prostate cancer cells. Epidemiologic reports have shown that repressing beta-adrenergic receptors (ADRBs) by specific beta-adrenergic antagonists (beta-blockers) is associated with reduced prostate cancer mortality.8,9 The dominant pathway downstream of ADRBs is the cyclic adenosine monophosphate-protein kinase A-cAMP response element-binding protein (cAMP-PKA-CREB) cascade.10 However, downstream targets of CREB activation in NED and in neoangiogenesis are largely unknown. Recently, we elucidated new mechanisms of angiogenesis and NED-mediated by beta-adrenergic signaling in prostate cancer progression.11,12,13 Several other reports have also revealed the effects of beta-adrenergic signaling in prostate cancer.14,15 In this minireview, we aim to provide a summary in the role of beta-adrenergic signaling pathway and related factors in prostate cancer progression, especially the effects on NED and angiogenesis.
PROSTATE CANCER PROGRESSION
The prostate is a multilobular exocrine gland, which together with the other major male accessory glands, the seminal vesicles, produces most of the seminal fluid volume.16 Normal development and function of the prostate are dependent on androgens such as testosterone, dehydroepiandrosterone, and dihydrotestosterone.1,17 Prostate tissue contains three epithelial cell types, including basal, luminal, and neuroendocrine cells.
A clear understanding of the detailed mechanisms of prostate cancer progression remains the goal of active research. Proliferation and survival of healthy prostate and early PAC are critically dependent on androgen stimulation.18 The activated phosphoinositide 3-kinase (PI3K)-Akt signal pathway is required to sustain PAC against apoptosis.19 Moreover, several tumor-suppressing genes are depressed, lost, or mutated in early PAC, such as phosphatase and tensin homolog (PTEN).20 Early PAC can be diagnosed by serum prostate-specific antigen (PSA) measurement, nuclear magnetic resonance (NMR), biopsy, and investigation of combinations of multiple tumor markers by tissue staining. Treatment for patients with PAC includes surgical removal of prostate, radiation, chemotherapy, and androgen deprivation therapy (ADT).21
Nearly all patients gain symptomatic relief and respond to hormonal therapy. However, blocking androgen synthesis or inhibiting AR function can rarely eradicate all PAC cells, some of which become resistant to ADT after months to years of treatment with a CRPC phenotype. As cancer progresses, different castration resistant mechanisms emerge, including restored or elevated AR signaling; other nuclear receptors, such as glucocorticoid receptor, bypassing AR; and complete AR independence.1,22 In restored or elevated AR signaling, high-affinity AR pathway antagonists, including enzalutamide and abiraterone, retain a transient ability to repress CRPC.23
In about 25% of patients with late-stage prostate cancer, a significant proportion of the cancer cells display small cell carcinoma (SCC) phenotype. Cells with this phenotype express neuroendocrine or stem cell markers such as synaptophysin and chromogranin A.24,25 Tumors with this phenotype are called NEPC or CRPC-NE.26 Due to high rates of metastasis and resistance to most therapies, the NEPC subset is invariably fatal, with most patients dying within 1–3 years of diagnosis.27 The incidence of this aggressive variant of CRPC with neuroendocrine phenotype was previously underestimated.28 To develop a therapeutic solution for CRPC/NEPC, a better understanding of the molecular events underlying NEPC progression is urgently needed.
OVERVIEW OF BETA-ADRENERGIC SIGNALING PATHWAYS IN PROSTATE CANCER
Beta-adrenergic signaling can mediate acute and chronic stress responses induced by the sympathetic nervous system (SNS).10 The SNS regulates multiple cellular processes contributing to tumorigenesis and the tumor microenvironment via ADRB-mediated activation of PKA and exchange protein activated by adenylyl cyclase (EPAC).29,30,31 As cell surface sensors of the SNS, ADRBs in tumors and their surrounding stromal cells could be activated by stress hormones, catecholamines including norepinephrine (from sympathetic nerve fibers) and epinephrine (from circulating blood).32 It has been recently reported that SNS is essential for both normal prostate development and prostate tumor formation in murine models.15,33
In the human prostate, ADRB2 is enriched in prostatic luminal cells, while ADRB2 and ADRB3 are expressed in prostatic stromal cells.34,35 Interestingly, heterogeneous ADRB2 expression levels were found during prostate cancer development. High and low levels of ADRB2 protein were observed in metastatic prostatic cancer cells. Meanwhile, decreased ADRB2 mRNA levels were detected after androgen deprivation therapy.30,36,37 A study by Yu et al.36,38 found that ADRB2 transcription is repressed by enhancer of zeste homolog 2 (EZH2) and ERG, both upregulated in prostate tumorigenesis. In addition, ADRB2 is also a direct target of AR.39
As shown in Figure 1, once triggered by catecholamines, activated ADRBs raise the level of intracellular cAMP with Gs alpha guanine nucleotide binding protein.40 Then, ADRBs regulate several cell processes, mainly through the cAMP mediators PKA and EPAC.31,41 Growing evidence suggests that the influence of the SNS on prostate and prostatic cancer is predominantly mediated by PKA.10,14 Activated PKA is capable of further activating tumor-related factors, such as cAMP responsive element binding protein (CREB), and inhibits antiproliferative factors including Ras homolog gene family, member A (RhoA) and Rho-associated coiled-coil-containing protein kinase (ROCK).42 Recently, the cAMP/PKA pathway was shown to phosphorylate and inhibit the BCL-2-associated death promoter (BAD), inducing PAC resistance to apoptosis.43 The PI3K-AKT is another downstream pathway of PKA. PI3K-AKT can upregulate vascular endothelial growth factor (VEGF) in a hypoxia-inducible factor-1-alpha (HIF-1 alpha)-dependent manner, promoting angiogenesis.44 As described below, histone deacetylase 2 (HDAC2) and G-protein receptor kinase 3 (GRK3) can also be induced by CREB, which promotes NED and angiogenesis.11,12,13 EPAC and PKA may act independently, converge synergistically, or oppose each other in regulating a specific cellular function.31 EPAC signaling accounts for many cAMP-induced effects on cell morphology, motility, and secretion dynamics. However, treatment with an EPAC-specific analog did not induce any obvious phenotypes on LNCaP.45 On the other hand, continuous presence of catecholamines may lead to desensitization of receptor ADRBs through arrestin-mediated receptor protein internalization and receptor mRNA downregulation.46 Importantly, these cAMP/PKA signaling cascades can also be activated by radiation and hormonal therapy, which are known to induce NED.47,48
Figure 1.
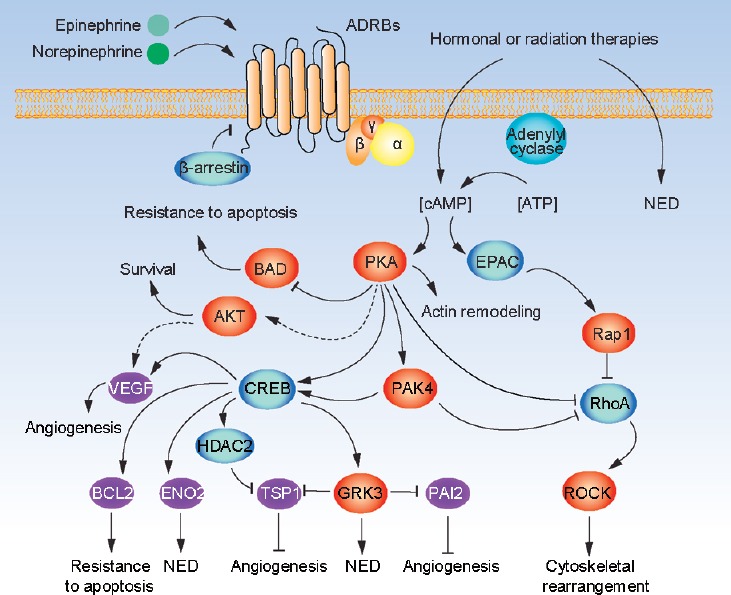
Beta-adrenergic signaling pathway in prostatic cancer. When beta-adrenergic receptors on prostate cancer cells are activated by epinephrine or norepinephrine, cellular cAMP level is increased. cAMP level in prostate cancer cells is also elevated by cancer treatments, such as radiation and hormonal therapies. cAMP activates PKA and EPAC. PKA can either directly stimulate or act through PAK4 to increase CREB activity. Activated PKA/CREB axis contributes to several biological processes relevant for cancer progression, such as (1) promoting angiogenesis through induction of VEGF, and repression of TSP1 and PAI2 through inducing HDAC2 and GRK3; (2) promoting NED, at least in part through direct induction of ENO2 as well as GRK3 that in turn elevates neuroendocrine markers in prostate cancer cells; (3) inhibiting apoptosis through CREB-induction of BCL2, and PKA-mediated phosphorylation of BAD (inhibiting) and AKT (activating); (4) together with EPAC, regulating cytoskeletal rearrangement and cellular migration through regulating ROCK activity via RhoA and PAK4. cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; CREB: cAMP response element-binding; VEGF: vascular endothelial growth factor; NED: neuroendocrine differentiation; TSP1: thrombospondin 1; PAI2: plasminogen activator inhibitor-2; HDAC2: histone deacetylase 2; GRK3: G-protein receptor kinase 3; BCL-2: B-cell lymphoma-2; BAD: BCL-2-associated death promoter.
BETA-ADRENERGIC REGULATION OF PROSTATE NEUROENDOCRINE DIFFERENTIATION
In the normal prostate, mature neuroendocrine cells are devoid of proliferative activity and interspersed among the epithelial basal and luminal cells. They maintain homeostasis of the surrounding epithelial cell populations by secreting neuropeptides and cell growth factors.49 During prostate cancer development, neuroendocrine cells continue producing cell-proliferation related factors to affect the surrounding PAC cells.50
De novo neuroendocrine prostate tumors are rarely diagnosed.51 Without treatment-induced transdifferentiation, rare AR-absent neuroendocrine prostate cells are found to mix with PAC cells de novo.24,25,52 On ADT, the cAMP/PKA pathway is activated.42 There are also significant increases in the numbers and the sizes of cell clusters that are positive for neuroendocrine markers, which are called NEPC cells.
Unlike normal NE cells in the benign prostate, NEPC cells are proliferative, metastatic, and express prostate epithelial-specific markers.52,53 Comparisons of the molecular profiles of CRPC with and without NE phenotypes reveal similar genetic alteration profiles in these two subtypes.26 Loss or mutation of retinoblastoma (RB), PTEN, and tumor protein P53, as well as amplification or activation of N-MYC and aurora kinase A (AURKA), occur predominantly in NEPC.54 Moreover, prostate cancer-specific TMPRSS2-ERG (transmembrane protease serine 2 and ETS related gene fusion) gene fusion is detectable in nearly half of NEPC cases. A similar percentage is observed for PAC. This result supports the hypothesis that NEPC cells are derived from PAC cells.55
As a tumor suppressor, P53 mutations inactivate the Interleukin 8-CXC chemokine receptor 2-P53 (IL-8-CXCR2-P53) cascade, which may lead to NEPC development.56 RB loss-mediated RB-E2F1-MAD2 pathway inactivation may enhance apoptosis resistance of NEPC cells.57 The N-MYC and AURKA regulate the entry in mitosis, thereby affecting the PAC cell cycle in NED.58 Overexpressed N-MYC cooperates with EZH2 in NEPC,59 while EZH2 has been reported to affect AR, PI3K-AKT pathway, EMT, and NED.60,61,62 Growing evidence reveals many other functional genes are dysregulated in NED, including macrophage migration inhibitory factor (MIF), serine/threonine-protein kinase 1 (PLK1), focal adhesion kinase (FAK), SIAHE3 ubiquitin protein ligase 2 (Siah2), zinc finger protein Snail, forkhead box protein A2 (FoxA2), yes-associated protein 1 (YAP), and proto-oncogene tyrosine-protein kinase Src.14,54,56 Several triggers have been shown to promote NED in both AR-dependent and AR-independent cells in vitro. Among these triggers are cAMP/PKA signal activator, forskolin, IL-6, epidermal growth factor (EGF), and AR antagonists. However, removal of these factors could lead to reversion of cells to an epithelial carcinoma cell phenotype.42,63
Notably, cAMP alone can induce NED in several common prostate cancer lines including LNCaP, PC-3, and PC-3M.14 Overexpressing activated PKA can also drive NED of LNCaP cells. Conversely, inhibiting PKA-induced CREB phosphorylation at serine 133 will inhibit NED.64,65 As a result, the ADRB2-cAMP-PKA cascade is recognized as the key pathway in NED. However, only chronic PKA stimulation over prolonged periods of time may induce NED. Short-term induction with low-level PKA may activate AR and increase PAC cell proliferation in vitro.42,66,67 Neurite outgrowth is evidenced in cAMP-induced NED of LNCaP cells. As a target of the cAMP-PKA pathway, RhoA inactivation is reported to induce neurite outgrowth through ROCK-mediated cytoskeletal rearrangement.14,68
CREB phosphorylation is induced by PKA, PKB, P21-activated kinase 4 (PAK4, which is also directly induced by PKA), p90 ribosomal S6 kinase (P90RSK), and mitogen-activated protein kinase (MAPK).69 CREB promotes the expression of Cyclin A1, Cyclin D1, VEGF, type IV collagenase matrix metalloproteinase-2 (MMP-2), cell adhesion molecule MUC18/MCAM, BCL2, and enolase 2 (ENO2).69,70 Its activation plays a critical role in NED of prostate cancer cells. However, the downstream pathways and targets of CREB in NEPC cells are still largely unclear. Through unbiased shRNA and cDNA screens, we previously discovered that beta-adrenergic receptor kinase 2 (ADRBK2 or GRK3) promotes PAC progression. Furthermore, overexpressing GRK3 induces NE markers in PAC LNCaP cells, while silencing GRK3, represses NED, and proliferation of NEPC cells.12
In the investigation of GRK3 regulation in prostate cancer progression, we found GRK3 expression is higher in NEPC than in PAC. Furthermore, GRK3 expression positively correlates with CREB expression and activation in human prostate cancer.13 We further demonstrated that GRK3 is a direct target of CREB and is induced by PKA-CREB signaling activation. This represents a paradigm shift in the understanding of CREB-GRK3 signaling. GRK3 induction by CREB activation was previously observed in retinoblastoma and neuroblastoma cells. In this context, GRK3 induction by CREB inactivation was considered to be negative feedback regulation for GRK3 to desensitize ADRBs. This was considered as a mechanism to control CREB signaling upon activation of ADRBs.70 We showed that silencing GRK3 inhibits the NED induction by CREB activation. This result suggests that GRK3 is a critical mediator downstream of cAMP-PKA-CREB in inducing neuroendocrine phenotypes of PAC cells.13 We further found that GRK3 expression is positively correlated with CREB expression in 1000 human cancer cell lines (CCLE dataset).71 Therefore, it can be speculated that the CREB/GRK3 axis may be active in wide range of cancer cells and biological contexts.
BETA-ADRENERGIC SIGNALING IN PROSTATIC CANCER ANGIOGENESIS
In normal physiological conditions in adults, angiogenesis is turned on only transiently. However, for tumors to grow beyond 1–2 mm in diameter, an increase of blood supply and recruitment of new blood vessels should occur to provide oxygen and nutrients as well as to remove metabolite waste.72 There are several proteins known to induce and promote angiogenesis, including VEGF, IL-6, fibroblast growth factor-2 (FGF-2), transforming growth factor-beta (TGF-beta), and MMPs. On the other hand, thrombospondin 1 (TSP1), plasminogen activator inhibitor-2 (PAI2), and endostatin are shown to repress angiogenesis.73,74
In the human normal prostate, neuroendocrine cells are the primary source of VEGF. Elevated neuroendocrine marker expression has been linked to increased angiogenesis in prostate cancer.75,76 Furthermore, many cytokines involved in NED, such as IL-6, are recognized to regulate angiogenesis. This result indicates the existence of an interplay between NED signaling and angiogenesis. The induction of VEGF and IL-6 by chronic stress is associated with increased angiogenesis in ovarian carcinoma.77 Enhanced angiogenesis is another mechanism through which beta-adrenergic signaling promotes cancer progression. Epinephrine and norepinephrine-induced ADRB2 activation lead to VEGF induction in prostate cancer cell lines through the cAMP-PKA-PI3K-AKT-HIF-1 alpha pathway.14,44 Conversely, inhibition of ADRB2 by a beta blocker lead to a reduction of rat prostate blood vessel volume.78 Recently, Zahalka et al.15 found that the endothelial ADRB2 related pathway is critical for activation of an angiogenic switch between low-grade preneoplastic stage (low-PIN) and high-PIN. Loss of ADRB represses PAC angiogenesis by altering oxidative phosphorylation induced by cytochrome c oxidase assembly factor 6 (COA6).
Angiogenesis is a complicated process that depends on achieving a balance between activators and inhibitors of angiogenesis. The ADRB2/PKA/CREB pathway has been shown to induce pro-angiogenic factor VEGF. The anti-angiogenic proteins repressed by beta-adrenergic signaling had not been identified. Results from the study have recently shown two pathways, CREB-HDAC2 and CREB-GRK3, regulating angiogenesis at the downstream of cAMP-PKA-CREB cascade (Figure 1). GRK3 is a direct target of CREB.13 GRK3, in turn, represses two antiangiogenic factors TSP1 and PAI2, which accelerates angiogenesis of PAC.12 We also found angiogenesis was induced by chronic stress through CREB-mediated induction of HDAC2, which in turn epigenetically inhibits TSP1 in different prostate cancer models (PC-3 and LNCaP).11 This result is in contrast with work by Hassan et al.,43 which showed no increase of angiogenesis after stress induction in mouse xenograft of C4-2 cell line. The apparent conflict may be explained by the fact that the C4-2 cell line is already highly vascularized.
TSP1 signaling, through receptors CD36 or CD47, inhibits the PKA/CREB pathway, inducing platelet activation, inhibiting T cell activation, and mediates killing of breast cancer cells.79 With our finding that CREB represses TSP1 expression, an antagonism can be postulated between PKA/CREB and TSP1 signaling in certain biological processes. We confirmed that HDAC2 expression is induced by CREB as a direct target. Further, the HDAC2 expression is necessary for prostatic cancer angiogenesis induced by beta-adrenergic signaling.11 In the current paradigm, activated CREB recruits CREB binding protein (CBP), a histone acetyltransferase (HAT), to activate transcription of its target genes.80 Increased HDAC activity presumably counteracts CBP and decreases CREB-dependent transcription. Thus, it is counterintuitive that CREB elevates HDAC2 expression. The study results support a new model that HDAC2 is a critical mediator for CREB in enhancing angiogenesis and TSP1 repression.
ADRB2 SIGNALING PATHWAY ON PROSTATE CANCER METASTASIS
Metastasis is a process with multiple sequential steps. In this process, the cancer cell has to overcome restrictions from its primary tumor site, degrade extracellular matrix, and invade and settle in distant tissues. During metastasis, beta-adrenergic signaling participates in prostate cancer cell invasion and survival.14,81
Norepinephrine drives PC-3 metastasis both in vivo and in vitro. It is inhibited by ADRB1 blocker atenolol and ADRB2 blocker ICI118551, indicating that ADRBs are essential for norepinephrine's impact on PAC metastasis.37,82 In a study by Magnon et al.,33 sympathetic nerve ablation inhibited PC-3 metastasis in mice. They also observed a significant reduction of tumor volume in the primary site and metastasis of PC-3 xenografts in an ADRB2/ADRB3 double knockout mouse model. The data mentioned above suggest ADRB signaling is a key pathway for PAC metastasis. However, as mentioned in the section of prostate cancer progression, expression of ADRB2 in metastatic prostatic cancer cells is heterogeneous. Yu et al.36 found ADRB2 is downregulated in metastatic prostate cancer, especially in the aggressive subtype, compared to clinically localized tumors. ADRB2 knock-down promoted EMT of an immortalized prostatic epithelial cell line (RWPE-1), increasing mesenchymal cell markers vimentin and N-cadherin, and decreasing adhesion molecules beta-catenin and integrin-beta 4. These data suggest that low expression of ADRB2 is associated with the mesenchymal-like phenotype. During colonization, disseminated mesenchymal tumor cells may have the potential to differentiate into epithelial cells with upregulation of ADRB expression at the metastatic site.
An adrenergic effect has also been described for the survival of PAC cells. Epinephrine protects C4-2 cells from apoptosis in mice and may promote PAC progression.83 Surgical stress delays prostate cancer apoptosis, which could be prevented by blocking ADRB2 with ICI118551 in mice.43,84 Several pathways are downstream of the ADRB2-cAMP-PKA pathway that mediate the stress-induced resistance to apoptosis. Phosphorylation of BAD induced by PKA renders BAD unable to bind BCL2, thereby repressing the apoptosis. As noted in the section of beta-adrenergic regulation of prostate neuroendocrine differentiation, PAK4 is also able to regulate BCL2 through activating CREB.70 Moreover, PKA has been reported to directly phosphorylate actin monomers,85 suggesting that PKA may regulate actin remodeling in PAC metastasis.
CLINICAL STUDIES WITH BETA-ADRENERGIC EFFECTS IN PROSTATE CANCER
While most beta-blockers could interfere ADRB1/2, ADRB2 is highly expressed in prostatic luminal cells and required for PAC progression,34,35 As we know, beta-blockers have been safely used as cardiovascular therapeutics.86 Since beta-adrenergic signaling modulates PAC progression via multiple pathways, ADRB antagonists may be effective therapeutics targeting PAC cells and their surrounding microenvironment in cancer patients.
Several epidemiologic reports have investigated the association of usage of beta-blockers with prostate cancer–specific survival rate and found that repressing ADRBs by beta-blockers may reduce prostate cancer mortality,8,9 Furthermore, usage of beta-blockers has been linked to a better prognosis in several other solid cancer types, such as breast, lung, pancreas, colon, stomach, and ovarian cancer.87,88,89,90 There is still no direct evidence showing anti-PAC effects of beta-blockers in clinical oncology.10,91 However, several clinical trials are directly investigating the anti-tumor effects of beta-blockers in prostate cancer (ClinicalTrial.gov identifiers: NCT01857817, NCT02944201, and NCT03152786).
Enhancing and repressing effects of beta-adrenergic activation on PAC development and progression have both been demonstrated.36,46,64,92 As discussed by Braadland et al.,14 cells expressing low levels of ADRB2 may represent a de-differentiated group with EMT phenotype and enhanced migration and invasion ability. On the other hand, cells expressing high levels of ADRB2 may represent differentiated group with anti-apoptosis, angiogenesis, and NED phenotypes. These paradoxical results may limit the use of beta-blockers for cancer treatments. Therefore, further research is needed to elucidate the dynamic effects of beta-blockers on cancer progression in mouse models and in patients.
PROSPECT AND FUTURE DIRECTION
Much remains to be elucidated regarding chronic stress, beta-adrenergic signaling, and PAC progression. The effects of low and high levels of ADRBs during PAC development remain controversial. Since NEPC tumor shares many genetic alterations with adenocarcinoma,26 it is believed that treatment-related NEPC cells are derived from PAC cells. This conclusion is also supported by lineage tracing using genetically engineered mouse models.7 Further in-depth investigations are required to determine whether ADRB signaling is a key contributor in NEPC progression after androgen deprivation.
In addition to PKA, EPAC is an important downstream mediator of ADRB-cAMP. It has been reported that EPAC induces TSP1 in endothelial cells, and using EPAC specific cAMP analog is able to regulate RhoA and AKT activity in prostatic epithelial cells.93,94,95 The synergy between EPAC-ras-related protein 1 (Rap1) and PKA-CREB in prostate cancer progression warrants further study.
We discovered that GRK3 promotes PAC progression.12 Detailed regulatory mechanisms of GRK3 induction of NED are a subject of active research. As GRK3 belongs to the GPCR kinase subfamily, suggesting that GRK3 may function through critical GPCR pathways. On the other hand, other GRKs are known to interact with or phosphorylate several non-GPCR targets.96,97 The role of GRK3 in NEPC progression can be better understood by further investigating these and other possible mechanisms.
Specific inhibitors targeting downstream key factors of beta-adrenergic pathway regulating proliferation, metastasis, angiogenesis, and NED may be suitable for PAC therapies. A few kinase inhibitors have been effectively and safely employed in current cancer therapeutics.98 As mentioned above, we have presented two candidate drug targets (GRK3 and HDAC2) downstream of ADRB-PKA-CREB. Unlike wild-type GRK3, the kinase dead mutant of GRK3 can no longer depress TSP1 and PAI2. This mutant also fails to induce NE marker expression in prostate cancer cells.12,13 These data suggest that specific GRK3 kinase inhibitors may be candidates for treating aggressive prostate cancer. HDAC inhibitors have been reported to repress angiogenesis and induce TSP1.99 We have revealed that HDAC2 is a major mediator involved in stress-induced angiogenesis. This result suggests specific inhibitors for HDACs, especially HDAC2 inhibitors, may be effective as adjuvant therapies to treat aggressive PAC and NEPC.
Angiogenesis and neurogenesis are closely linked. Proangiogenic factors VEGF and several neurosecretory peptides are known to promote angiogenesis in NEPC, which are highly vascularized.78,100 However, molecular mechanisms underlying the connection between NED and angiogenesis are largely unknown. In particular, much remains to be understood about the endogenous angiogenic inhibitors that are involved in angiogenesis regulation in NEPC.
Much research has focused on the effects of stress and beta-adrenergic signaling on the tumor cells. Notably, ADRBs on tumor and their stromal cells can be activated by adrenaline and noradrenaline from nerve fibers and blood. Recently, important work by Zahalka et al.15 showed that endothelial beta-adrenergic receptor signaling via adrenergic nerve-derived noradrenaline in the prostate stroma is critical for activation of an angiogenic switch, which thereby regulates PAC progression. Thus, more comprehensive analysis of the microenvironment surrounding prostate tumors is in urgent need to expand our understanding of NEPC progression and facilitate the development of new therapeutics and prognostic markers for aggressive prostate cancer.
AUTHOR CONTRIBUTIONS
YZ wrote the main manuscript and prepared the figure. WL edited and approved the final version of the manuscript. Both authors read and approved the final manuscript.
COMPETING INTERESTS
Both authors declare no competing interests.
ACKNOWLEDGMENTS
We are very grateful to the critical reading and editing of this paper by Dr. Georgina To’a Salazar. This work is supported by grants from the Cancer Prevention and Research Institute of Texas (CPRIT) and American Cancer Society (ACS) to W Li.
REFERENCES
- 1.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Hsu CL, Chang C. Androgen receptor corepressors: an overview. Prostate. 2005;63:117–30. doi: 10.1002/pros.20170. [DOI] [PubMed] [Google Scholar]
- 3.Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res. 2014;20:2846–50. doi: 10.1158/1078-0432.CCR-13-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sargos P, Ferretti L, Gross-Goupil M, Orre M, Cornelis F, et al. Characterization of prostate neuroendocrine cancers and therapeutic management: a literature review. Prostate Cancer Prostatic Dis. 2014;17:220–6. doi: 10.1038/pcan.2014.17. [DOI] [PubMed] [Google Scholar]
- 5.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–8. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017;7:736–49. doi: 10.1158/2159-8290.CD-16-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grytli HH, Fagerland MW, Fossa SD, Tasken KA, Haheim LL. Use of beta-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate. 2013;73:250–60. doi: 10.1002/pros.22564. [DOI] [PubMed] [Google Scholar]
- 9.Grytli HH, Fagerland MW, Fossa SD, Tasken KA. Association between use of beta-blockers and prostate cancer-specific survival: a cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol. 2014;65:635–41. doi: 10.1016/j.eururo.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Cole SW, Sood AK. Molecular pathways: beta-adrenergic signaling in cancer. Clin Cancer Res. 2012;18:1201–6. doi: 10.1158/1078-0432.CCR-11-0641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hulsurkar M, Li Z, Zhang Y, Li X, Zheng D, et al. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene. 2017;36:1525–36. doi: 10.1038/onc.2016.319. [DOI] [PubMed] [Google Scholar]
- 12.Li W, Ai N, Wang S, Bhattacharya N, Vrbanac V, et al. GRK3 is essential for metastatic cells and promotes prostate tumor progression. Proc Natl Acad Sci U S A. 2014;111:1521–6. doi: 10.1073/pnas.1320638111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sang M, Hulsurkar M, Zhang X, Song H, Zheng D, et al. GRK3 is a direct target of CREB activation and regulates neuroendocrine differentiation of prostate cancer cells. Oncotarget. 2016;7:45171–85. doi: 10.18632/oncotarget.9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braadland PR, Ramberg H, Grytli HH, Tasken KA. Beta-adrenergic receptor signaling in prostate cancer. Front Oncol. 2014;4:375. doi: 10.3389/fonc.2014.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zahalka AH, Arnal-Estape A, Maryanovich M, Nakahara F, Cruz CD, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science. 2017;358:321–6. doi: 10.1126/science.aah5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walz J, Graefen M, Huland H. Basic principles of anatomy for optimal surgical treatment of prostate cancer. World J Urol. 2007;25:31–8. doi: 10.1007/s00345-007-0159-6. [DOI] [PubMed] [Google Scholar]
- 17.Lau KM, To KF. Importance of estrogenic signaling and its mediated receptors in prostate cancer. Int J Mol Sci. 2016;17:1434. doi: 10.3390/ijms17091434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Attard G, Cooper CS, de Bono JS. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell. 2009;16:458–62. doi: 10.1016/j.ccr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 19.Barrett CS, Millena AC, Khan SA. TGF-beta effects on prostate cancer cell migration and invasion require FosB. Prostate. 2017;77:72–81. doi: 10.1002/pros.23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wise HM, Hermida MA, Leslie NR. Prostate cancer, PI3K, PTEN and prognosis. Clin Sci (Lond) 2017;131:197–210. doi: 10.1042/CS20160026. [DOI] [PubMed] [Google Scholar]
- 21.Narizhneva NV, Tararova ND, Ryabokon P, Shyshynova I, Prokvolit A, et al. Small molecule screening reveals a transcription-independent pro-survival function of androgen receptor in castration-resistant prostate cancer. Cell Cycle. 2009;8:4155–67. doi: 10.4161/cc.8.24.10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–22. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beltran H, Tagawa ST, Park K, MacDonald T, Milowsky MI, et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30:e386–9. doi: 10.1200/JCO.2011.41.5166. [DOI] [PubMed] [Google Scholar]
- 24.Palmgren JS, Karavadia SS, Wakefield MR. Unusual and underappreciated: small cell carcinoma of the prostate. Semin Oncol. 2007;34:22–9. doi: 10.1053/j.seminoncol.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 25.Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol. 2014;38:756–67. doi: 10.1097/PAS.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Wang FW. Impact of age on clinical presentation, treatment, and cancer-specific survival of patients with small-cell carcinoma of the prostate. Clin Interv Aging. 2013;8:871–7. doi: 10.2147/CIA.S44772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang HT, Yao YH, Li BG, Tang Y, Chang JW, et al. Neuroendocrine prostate cancer (NEPC) progressing from conventional prostatic adenocarcinoma: factors associated with time to development of NEPC and survival from NEPC diagnosis – a systematic review and pooled analysis. J Clin Oncol. 2014;32:3383–90. doi: 10.1200/JCO.2013.54.3553. [DOI] [PubMed] [Google Scholar]
- 29.Baker JG, Hill SJ, Summers RJ. Evolution of beta-blockers: from anti-anginal drugs to ligand-directed signalling. Trends Pharmacol Sci. 2011;32:227–34. doi: 10.1016/j.tips.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramberg H, Eide T, Krobert KA, Levy FO, Dizeyi N, et al. Hormonal regulation of beta2-adrenergic receptor level in prostate cancer. Prostate. 2008;68:1133–42. doi: 10.1002/pros.20778. [DOI] [PubMed] [Google Scholar]
- 31.Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40:651–62. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15:563–72. doi: 10.1038/nrc3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 34.Goepel M, Wittmann A, Rubben H, Michel MC. Comparison of adrenoceptor subtype expression in porcine and human bladder and prostate. Urol Res. 1997;25:199–206. doi: 10.1007/BF00941983. [DOI] [PubMed] [Google Scholar]
- 35.Haynes JM. beta(2) and beta(3)-adrenoceptor inhibition of alpha(1)-adrenoceptor-stimulated Ca(2+) elevation in human cultured prostatic stromal cells. Eur J Pharmacol. 2007;570:18–26. doi: 10.1016/j.ejphar.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 36.Yu J, Cao Q, Mehra R, Laxman B, Yu J, et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell. 2007;12:419–31. doi: 10.1016/j.ccr.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 37.Palm D, Lang K, Niggemann B, Drell TL, Masur K, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer. 2006;118:2744–9. doi: 10.1002/ijc.21723. [DOI] [PubMed] [Google Scholar]
- 38.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–54. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Massie CE, Adryan B, Barbosa-Morais NL, Lynch AG, Tran MG, et al. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007;8:871–8. doi: 10.1038/sj.embor.7401046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montminy M. Transcriptional regulation by cyclic AMP. Annu Rev Biochem. 1997;66:807–22. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–64. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merkle D, Hoffmann R. Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: cross-talk with the androgen receptor. Cell Signal. 2011;23:507–15. doi: 10.1016/j.cellsig.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 43.Hassan S, Karpova Y, Baiz D, Yancey D, Pullikuth A, et al. Behavioral stress accelerates prostate cancer development in mice. J Clin Invest. 2013;123:874–86. doi: 10.1172/JCI63324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park SY, Kang JH, Jeong KJ, Lee J, Han JW, et al. Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1alpha protein-dependent mechanism. Int J Cancer. 2011;128:2306–16. doi: 10.1002/ijc.25589. [DOI] [PubMed] [Google Scholar]
- 45.Jones SE, Palmer TM. Protein kinase A-mediated phosphorylation of RhoA on serine 188 triggers the rapid induction of a neuroendocrine-like phenotype in prostate cancer epithelial cells. Cell Signal. 2012;24:1504–14. doi: 10.1016/j.cellsig.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang P, He X, Tan J, Zhou X, Zou L. beta-arrestin2 mediates beta-2 adrenergic receptor signaling inducing prostate cancer cell progression. Oncol Rep. 2011;26:1471–7. doi: 10.3892/or.2011.1417. [DOI] [PubMed] [Google Scholar]
- 47.Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr Relat Cancer. 2007;14:531–47. doi: 10.1677/ERC-07-0061. [DOI] [PubMed] [Google Scholar]
- 48.Deng X, Liu H, Huang J, Cheng L, Keller ET, et al. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res. 2008;68:9663–70. doi: 10.1158/0008-5472.CAN-08-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terry S, Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front Oncol. 2014;4:60. doi: 10.3389/fonc.2014.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nouri M, Ratther E, Stylianou N, Nelson CC, Hollier BG, et al. Androgen-targeted therapy-induced epithelial mesenchymal plasticity and neuroendocrine transdifferentiation in prostate cancer: an opportunity for intervention. Front Oncol. 2014;4:370. doi: 10.3389/fonc.2014.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. 2010;39:707–12. doi: 10.1097/MPA.0b013e3181ec124e. [DOI] [PubMed] [Google Scholar]
- 52.Vashchenko N, Abrahamsson PA. Neuroendocrine differentiation in prostate cancer: implications for new treatment modalities. Eur Urol. 2005;47:147–55. doi: 10.1016/j.eururo.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Vlachostergios PJ, Papandreou CN. Targeting neuroendocrine prostate cancer: molecular and clinical perspectives. Front Oncol. 2015;5:6. doi: 10.3389/fonc.2015.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rickman DS, Beltran H, Demichelis F, Rubin MA. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat Med. 2017;23:1–10. doi: 10.1038/nm.4341. [DOI] [PubMed] [Google Scholar]
- 55.Beltran H. Update on the biology and management of neuroendocrine prostate cancer. Clin Adv Hematol Oncol. 2016;14:513–5. [PubMed] [Google Scholar]
- 56.Li Z, Chen CJ, Wang JK, Hsia E, Li W, et al. Neuroendocrine differentiation of prostate cancer. Asian J Androl. 2013;15:328–32. doi: 10.1038/aja.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20:890–903. doi: 10.1158/1078-0432.CCR-13-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825–41. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 59.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016;30:563–77. doi: 10.1016/j.ccell.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 61.Yang YA, Yu J. EZH2, an epigenetic driver of prostate cancer. Protein Cell. 2013;4:331–41. doi: 10.1007/s13238-013-2093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cindolo L, Cantile M, Vacherot F, Terry S, de la Taille A. Neuroendocrine differentiation in prostate cancer: from lab to bedside. Urol Int. 2007;79:287–96. doi: 10.1159/000109711. [DOI] [PubMed] [Google Scholar]
- 64.Deeble PD, Murphy DJ, Parsons SJ, Cox ME. Interleukin-6- and cyclic AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol Cell Biol. 2001;21:8471–82. doi: 10.1128/MCB.21.24.8471-8482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suarez CD, Deng X, Hu CD. Targeting CREB inhibits radiation-induced neuroendocrine differentiation and increases radiation-induced cell death in prostate cancer cells. Am J Cancer Res. 2014;4:850–61. [PMC free article] [PubMed] [Google Scholar]
- 66.Kasbohm EA, Guo R, Yowell CW, Bagchi G, Kelly P, et al. Androgen receptor activation by G(s) signaling in prostate cancer cells. J Biol Chem. 2005;280:11583–9. doi: 10.1074/jbc.M414423200. [DOI] [PubMed] [Google Scholar]
- 67.Gutierrez-Canas I, Juarranz MG, Collado B, Rodriguez-Henche N, Chiloeches A, et al. Vasoactive intestinal peptide induces neuroendocrine differentiation in the LNCaP prostate cancer cell line through PKA, ERK, and PI3K. Prostate. 2005;63:44–55. doi: 10.1002/pros.20173. [DOI] [PubMed] [Google Scholar]
- 68.Wells CM, Whale AD, Parsons M, Masters JR, Jones GE. PAK4: a pluripotent kinase that regulates prostate cancer cell adhesion. J Cell Sci. 2010;123:1663–73. doi: 10.1242/jcs.055707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao X, Li BX, Mitton B, Ikeda A, Sakamoto KM. Targeting CREB for cancer therapy: friend or foe. Curr Cancer Drug Targets. 2010;10:384–91. doi: 10.2174/156800910791208535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park MH, Lee HS, Lee CS, You ST, Kim DJ, et al. P21-activated kinase 4 promotes prostate cancer progression through CREB. Oncogene. 2013;32:2475–82. doi: 10.1038/onc.2012.255. [DOI] [PubMed] [Google Scholar]
- 71.Cancer Cell Line Encyclopedia Consortium; Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature. 2015;528:84–7. doi: 10.1038/nature15736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Armaiz-Pena GN, Cole SW, Lutgendorf SK, Sood AK. Neuroendocrine influences on cancer progression. Brain Behav Immun. 2013;30(Suppl):S19–25. doi: 10.1016/j.bbi.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eisermann K, Fraizer G. The androgen receptor and VEGF: mechanisms of androgen-regulated angiogenesis in prostate cancer. Cancers (Basel) 2017;9 doi: 10.3390/cancers9040032. pii: E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Russo G, Mischi M, Scheepens W, De la Rosette JJ, Wijkstra H. Angiogenesis in prostate cancer: onset, progression and imaging. BJU Int. 2012;110:E794–808. doi: 10.1111/j.1464-410X.2012.11444.x. [DOI] [PubMed] [Google Scholar]
- 75.Borre M, Nerstrom B, Overgaard J. Association between immunohistochemical expression of vascular endothelial growth factor (VEGF), VEGF-expressing neuroendocrine-differentiated tumor cells, and outcome in prostate cancer patients subjected to watchful waiting. Clin Cancer Res. 2000;6:1882–90. [PubMed] [Google Scholar]
- 76.Heinrich E, Trojan L, Friedrich D, Voss M, Weiss C, et al. Neuroendocrine tumor cells in prostate cancer: evaluation of the neurosecretory products serotonin, bombesin, and gastrin-impact on angiogenesis and clinical follow-up. Prostate. 2011;71:1752–8. doi: 10.1002/pros.21392. [DOI] [PubMed] [Google Scholar]
- 77.Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–44. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]
- 78.Plecas B, Glavaski A, Solarovic T. Propranolol treatment affects ventral prostate blood vessels and serum testosterone concentrations in adult rats. Andrologia. 1997;29:109–14. doi: 10.1111/j.1439-0272.1997.tb00472.x. [DOI] [PubMed] [Google Scholar]
- 79.Manna PP, Frazier WA. CD47 mediates killing of breast tumor cells via Gi-dependent inhibition of protein kinase A. Cancer Res. 2004;64:1026–36. doi: 10.1158/0008-5472.can-03-1708. [DOI] [PubMed] [Google Scholar]
- 80.Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, et al. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–6. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 81.Armaiz-Pena GN, Lutgendorf SK, Cole SW, Sood AK. Neuroendocrine modulation of cancer progression. Brain Behav Immun. 2009;23:10–5. doi: 10.1016/j.bbi.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lang K, Drell TL, Lindecke A, Niggemann B, Kaltschmidt C, et al. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int J Cancer. 2004;112:231–8. doi: 10.1002/ijc.20410. [DOI] [PubMed] [Google Scholar]
- 83.Sastry KS, Karpova Y, Prokopovich S, Smith AJ, Essau B, et al. Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J Biol Chem. 2007;282:14094–100. doi: 10.1074/jbc.M611370200. [DOI] [PubMed] [Google Scholar]
- 84.Hassan S, Karpova Y, Flores A, D’Agostino R, Jr, Kulik G. Surgical stress delays prostate involution in mice. PLoS One. 2013;8:e78175. doi: 10.1371/journal.pone.0078175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Howe AK. Regulation of actin-based cell migration by cAMP/PKA. Biochim Biophys Acta. 2004;1692:159–74. doi: 10.1016/j.bbamcr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 86.Foody JM, Farrell MH, Krumholz HM. Beta-blocker therapy in heart failure: scientific review. JAMA. 2002;287:883–9. doi: 10.1001/jama.287.7.883. [DOI] [PubMed] [Google Scholar]
- 87.Schuller HM. Beta-adrenergic signaling, a novel target for cancer therapy? Oncotarget. 2010;1:466–9. doi: 10.18632/oncotarget.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population- based study. J Clin Oncol. 2011;29:2635–44. doi: 10.1200/JCO.2010.33.5422. [DOI] [PubMed] [Google Scholar]
- 89.Holmes S, Griffith EJ, Musto G, Minuk GY. Antihypertensive medications and survival in patients with cancer: a population-based retrospective cohort study. Cancer Epidemiol. 2013;37:881–5. doi: 10.1016/j.canep.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 90.Powe DG, Entschladen F. Targeted therapies: using beta-blockers to inhibit breast cancer progression. Nat Rev Clin Oncol. 2011;8:511–2. doi: 10.1038/nrclinonc.2011.123. [DOI] [PubMed] [Google Scholar]
- 91.Shah SM, Carey IM, Owen CG, Harris T, Dewilde S, et al. Does beta-adrenoceptor blocker therapy improve cancer survival? Findings from a population-based retrospective cohort study. Br J Clin Pharmacol. 2011;72:157–61. doi: 10.1111/j.1365-2125.2011.03980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cox ME, Deeble PD, Lakhani S, Parsons SJ. Acquisition of neuroendocrine characteristics by prostate tumor cells is reversible: implications for prostate cancer progression. Cancer Res. 1999;59:3821–30. [PubMed] [Google Scholar]
- 93.Doebele RC, Schulze-Hoepfner FT, Hong J, Chlenski A, Zeitlin BD, et al. A novel interplay between Epac/Rap1 and mitogen-activated protein kinase kinase 5/extracellular signal-regulated kinase 5 (MEK5/ERK5) regulates thrombospondin to control angiogenesis. Blood. 2009;114:4592–600. doi: 10.1182/blood-2009-04-217042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grandoch M, Rose A, ter Braak M, Jendrossek V, Rubben H, et al. Epac inhibits migration and proliferation of human prostate carcinoma cells. Br J Cancer. 2009;101:2038–42. doi: 10.1038/sj.bjc.6605439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Misra UK, Pizzo SV. Evidence for a pro-proliferative feedback loop in prostate cancer: the role of Epac1 and COX-2-dependent pathways. PLoS One. 2013;8:e63150. doi: 10.1371/journal.pone.0063150. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 96.Cant SH, Pitcher JA. G protein-coupled receptor kinase 2-mediated phosphorylation of ezrin is required for G protein-coupled receptor-dependent reorganization of the actin cytoskeleton. Mol Biol Cell. 2005;16:3088–99. doi: 10.1091/mbc.E04-10-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Penela P, Murga C, Ribas C, Lafarga V, Mayor F., Jr The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. 2010;160:821–32. doi: 10.1111/j.1476-5381.2010.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chessum N, Jones K, Pasqua E, Tucker M. Recent advances in cancer therapeutics. Prog Med Chem. 2015;54:1–63. doi: 10.1016/bs.pmch.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 99.Byler TK, Leocadio D, Shapiro O, Bratslavsky G, Stodgell CJ, et al. Valproic acid decreases urothelial cancer cell proliferation and induces thrombospondin-1 expression. BMC Urol. 2012;12:21. doi: 10.1186/1471-2490-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yazdani S, Kasajima A, Tamaki K, Nakamura Y, Fujishima F, et al. Angiogenesis and vascular maturation in neuroendocrine tumors. Hum Pathol. 2014;45:866–74. doi: 10.1016/j.humpath.2013.09.024. [DOI] [PubMed] [Google Scholar]