

Abstract
Summary
Prediction of transcription factor (TF) binding from epigenetics data and integrative analysis thereof are challenging. Here, we present TEPIC 2 a framework allowing for fast, accurate and versatile prediction, and analysis of TF binding from epigenetics data: it supports 30 species with binding motifs, computes TF gene and scores up to two orders of magnitude faster than before due to improved implementation, and offers easy-to-use machine learning pipelines for integrated analysis of TF binding predictions with gene expression data allowing the identification of important TFs.
Availability and implementation
TEPIC is implemented in C++, R, and Python. It is freely available at https://github.com/SchulzLab/TEPIC and can be used on Linux based systems.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Transcription Factors (TFs) are key players of transcriptional regulation. Prediction of TF binding is essential to gain a deeper understanding of their function. While experimental identification of TF binding is possible through laborious and expensive ChIP-seq assays, several computational approaches have been proposed to identify TF binding sites (TFBSs) (Jayaram et al., 2016). These predictions have been successfully augmented using epigenetics data (Cuellar-Partida et al., 2012; Pique-Regi et al., 2011; Sherwood et al., 2014). As delineated in Supplementary Section 1, TEPIC 2 builds upon and extends the functionality of existing TFBS prediction tools. Among other features, TEPIC 2 allows the direct aggregation of TFBS predictions on the gene level and uses these scores to gain novel insights on cell type specific functions of TFs via several machine learning analysis. This is a unique feature not supported by competitive TFBS prediction approaches (Supplementary Tables S1 and S2). Compared to its predecessor, TEPIC 2 has substantially lower runtime, contains an extended set of TF motifs, offers various means for downstream machine learning analyses as easy-to-use pipelines, and adds new functionalities to compute TF gene scores.
2 Features
The core functionalities of TEPIC 2 are to predict TFBS in user provided regions and to aggregate them to TF gene scores. The TF gene score computation has been modified to compute statistical features such as region length, region count, and the signal of an epigenetic assay within the considered regions. TEPIC 2 can compute a binary binding assessment, i.e. a TF binds or does not bind, based on p-values obtained using a set of background regions of similar characteristics as the input set. This feature complements the continuous TF affinity values of TRAP, which are not suitable for all downstream applications (Supplementary Section 4).
Additionally, the aforementioned TF gene scores can be used in several integrative analysis workflows (Supplementary Section 7, 8 and 9). INVOKE refers to a sparse linear regression model to reveal key TFs potentially regulating transcription. It highlighted several known tissue-specific regulators in liver hepatocytes and CD4+ T cells (Schmidt et al., 2017) and is also available as a web-server (Kehl et al., 2017). Besides, TEPIC 2 includes a sparse logistic regression classifier to infer TFs related to gene expression changes between samples (DYNAMITE). DYNAMITE has been successfully applied to discover regulators of CD4+ T cell differentiation (Durek et al., 2016). Recently, we combined TEPIC with DREM (Schulz et al., 2012) to uncover master regulatory TFs from paired time-series expression and epigenomics data (EPIC-DREM), which was used to analyze mesenchymal stem-cell differentiation of osteoblasts and adipocytes (Gerard et al., 2018).
Furthermore, we considerably extended the set of TF motifs readily available in TEPIC 2. Now, this resource contains 30 species-specific and six taxonomy-specific sets from JASPAR (Mathelier et al., 2016), as well as aggregated sets for humans, mice and vertebrates containing 561, 380 and 690 TF motifs (Supplementary Section 3). To streamline the training and interpretation of statistical models (Supplementary Fig. S1), we provide clustered versions of the merged TF motif files, representing families of binding motifs with high similarity (Pape et al., 2008).
3 Implementation
TEPIC 2 uses a parallelized C++ implementation of TRAP (Roider et al., 2007) that is considerably faster than the previous R implementation. Runtime was further reduced by using more efficient search algorithms and by enabling pre-filtered analyses of samples in minutes (Fig. 1a, Supplementary Table S5, Supplementary Fig. S2 and Supplementary Section 5). We evaluated the accuracy of TFBS predictions from TEPIC 2 using TF footprints called with HINT-BC (Gusmao et al., 2016) on ENCODE data (The ENCODE Project Consortium, 2012). In comparison to established tools for TFBS prediction using epigenomics data (Cuellar-Partida et al., 2012; Sherwood et al., 2014), TEPIC performs favorably in terms of area under the precision recall curve (AUPR) (Fig. 1b, Supplementary Fig. S3 and Supplementary Section 6). Details on samples used are provided in Supplementary Section 2. The machine learning pipelines included in TEPIC 2 are implemented in R. Both workflows deliver results that are easy to interpret, also for non-expert users, due to automated figure generation and extensive documentation. As input, the pipelines require standard file formats, e.g. bed files for candidate TFBS and tab delimited txt files containing gene expression data. TEPICs full functionality is brought to the user via start-to-finish pipelines, which are automatically installed with TEPIC 2.
Fig. 1.
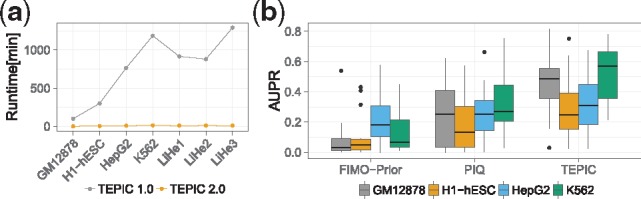
(a) Runtime comparison of TEPIC to TEPIC 2 using a subset of 458 human TFs. While the original implementation ran up to 1300 minutes to compute TFBS, TEPIC 2 is able to compute TF affinities for peaks in the vicinity of genes in at most 15 minutes. We used four cell line samples and three primary human hepatocyte samples (LiHe1–3) to conduct the runtime experiments. (b) We compared TEPIC TF affinities computed in footprints called with HINT-BC (Gusmao et al., 2016) in four different cell-lines in terms of AUPR against PIQ (Sherwood et al., 2014) and an extension of the widely used method Fimo, called Fimo-Prior (Cuellar-Partida et al., 2012). Notably, TF affinities computed with TEPIC outperform both PIQ and Fimo-Prior
4 Conclusion
TEPIC 2 is a fast and easy-to-use tool for TFBS prediction combined with integrative analysis capabilities for gene expression and epigenomic data. TFBS prediction and downstream machine learning pipelines for various analysis settings allow a deep, seamless exploration of epigenomic datasets supporting data driven hypothesis generation about the role of individual TFs in complex regulatory landscapes.
Supplementary Material
Acknowledgements
We thank Helge Roider who wrote the original C++ implementation of TRAP, as well as the DEEP and ENCODE consortia for processing and providing the data used in this project.
Funding
This work has been supported by the German Federal Ministry of Education and Research in Germany (BMBF) [01DP17005, 01KU1216A] and the Cluster of Excellence on Multimodal Computing and Interaction (DFG) [EXC248].
Conflict of Interest: none declared.
References
- Cuellar-Partida G. et al. (2012) Epigenetic priors for identifying active transcription factor binding sites. Bioinformatics, 28, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durek P. et al. (2016) Epigenomic profiling of human CD4+ T cells supports a linear differentiation model and highlights molecular regulators of memory development. Immunity, 45, 1148–1161. [DOI] [PubMed] [Google Scholar]
- Gerard D. et al. (2018) Temporal epigenomic profiling identifies AHR and GLIS1 as super-enhancer controlled regulators of mesenchymal multipotency. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusmao E. et al. (2016) Analysis of computational footprinting methods for DNase sequencing experiments. Nat. Methods, 13, 303–309. [DOI] [PubMed] [Google Scholar]
- Jayaram N. et al. (2016) Evaluating tools for transcription factor binding site prediction. BMC Bioinformatics. doi: 10.1186/s12859-016-1298-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehl T. et al. (2017) RegulatorTrail: a web service for the identification of key transcriptional regulators. Nucleic Acids Res., 45, W146–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A. et al. (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res., 44, D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape U.J. et al. (2008) Natural similarity measures between position frequency matrices with an application to clustering. Bioinformatics, 24, 350–357. [DOI] [PubMed] [Google Scholar]
- Pique-Regi R. et al. (2011) Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res., 21, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roider H.G. et al. (2007) Predicting trancription factor affinities to DNA from a biophysical model. Bioinformatics, 23, 134–141. [DOI] [PubMed] [Google Scholar]
- Schmidt F. et al. (2017) Combining transcription factor binding affinities with open-chromatin data for accurate gene expression prediction. Nucleic Acids Res., 45, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz M.H. et al. (2012) DREM 2.0: improved reconstruction of dynamic regulatory networks from time-series expression data. BMC Syst. Biol., 6, 104.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood R.I. et al. (2014) Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol., 32, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 741457–741474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.