

Abstract
Summary
MicroRNAs (miRNAs) function as master regulators of gene expression. Recent studies demonstrate that miRNA isoforms (isomiRs) play a unique role in cancer development. Here, we present QuagmiR, the first cloud-based tool to analyze isomiRs from next generation sequencing data. Using a novel and flexible searching algorithm designed for the detection and annotation of heterogeneous isomiRs, it permits extensive customization of the query process and reference databases to meet the user 's diverse research needs.
Availability and implementation
QuagmiR is written in Python and can be obtained freely from GitHub (https://github.com/Gu-Lab-RBL-NCI/QuagmiR). QuagmiR can be run from the command line on local machines, as well as on high-performance servers. A web-accessible version of the tool has also been made available for use by academic researchers through the National Cancer Institute-funded Seven Bridges Cancer Genomics Cloud (https://cancergenomicscloud.org).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
MicroRNAs are a class of small non-coding RNAs (21–24 nucleotides long) with important inhibitory functions (Bartel, 2018). Since their initial discovery, thousands of miRNA genes have been identified in viral, plant, and animal genomes (Kozomara and Griffiths-Jones, 2014). It has been estimated that miRNAs regulate the expression for more than 60% of human transcripts (Friedman et al., 2009). Hence, it is not surprising that miRNA dysregulation contributes to many diseases including cancer (Lin and Gregory, 2015; Sayed and Abdellatif, 2011).
A single miRNA locus can generate multiple distinct isoforms (isomiRs) that differ in length and sequence composition (Morin et al., 2008; Neilsen et al., 2012). In the canonical miRNA biogenesis pathway, the ends of mature miRNAs are defined by the cleavage events of Drosha and Dicer (Ha and Kim, 2014). Imprecise cleavage by Drosha or Dicer generates isomiR sequences that match the parent gene but vary in length, a situation referred to as ‘templated’. IsomiRs also form via post-maturation editing of miRNA sequences. Exoribonucleases take away nucleotides from the 3′ end (trimming), producing shorter templated isomiRs. Terminal nucleotidyl transferases (TUTs) generate non-templated isomiRs by adding one or more bases (tailing). These post-maturation sequence modifications happen at the 3′ end rather than at the 5′ end of miRNAs. As a result, 3′ isomiRs are the most frequently observed types of isomiRs, both in terms of the number of miRNAs displaying these variations and their overall abundance.
In terms of the effect of isomiR generation on the miRNA function, 5′ isomiRs have a different seed sequence than the corresponding canonical miRNAs and therefore regulate a distinct set of target genes. Sequence modifications at the 3′ end do not change the seed sequence but are reported to play critical roles in regulating miRNA biogenesis and turnover (Fernandez-Valverde et al., 2010; Rüegger andGroßhans, 2012). Interestingly, the alteration in the isomiR profile rather than in the overall miRNA abundance correlates with cancer progression, which suggests a unique role of isomiRs in tumorigenesis (McCall et al., 2017; Telonis et al., 2015). These observations highlight the importance of having computational tools to analyze isomiR expression profiles from sequencing data to further investigate their functions.
Many challenges in the isomiR detection are due to their heterogeneous origin. Various combinations of 5′ and 3′, templated and non-templated, and trimmed and tailed isomiRs can exist at once, rendering the development of a single algorithm capable of mapping and efficiently processing all isomiRs nearly impossible (Ziemann et al., 2016). Furthermore, users with specific biological questions may focus on unique aspects of isomiR analysis. Current methods address these challenges by simply leaving certain types of isomiRs unmapped or requiring the user to perform additional post-run analyses, thereby hampering their applications. Here, we aim to provide the field with a highly customizable, cloud-based tool to comprehensively analyze isomiRs in a high-throughput and in an automated manner.
2 Algorithm
To process large-scale data with improved efficiency and accuracy, we developed QuagmiR, a novel algorithm for the isomiR analysis (Fig. 1). Each miRNA sequence is divided into three regions: 5′ part, 3′ part and a central region. The central region is unique to each miRNA and will be referred to as the ‘motif’. Many miRNA families such as miR-148a and miR-148b only differ from each other by a single nucleotide. Nonetheless, a set of 13mer motifs are able to distinguish close to 95% of miRNA sequences. With additional motifs of longer length, we uniquely identified every miRNA reported in miRBase (Kozomara and Griffiths-Jones, 2014) (Supplementary Fig. S1A). Reads matching a certain motif were considered as potential isomiRs for the corresponding miRNA.
Fig. 1.
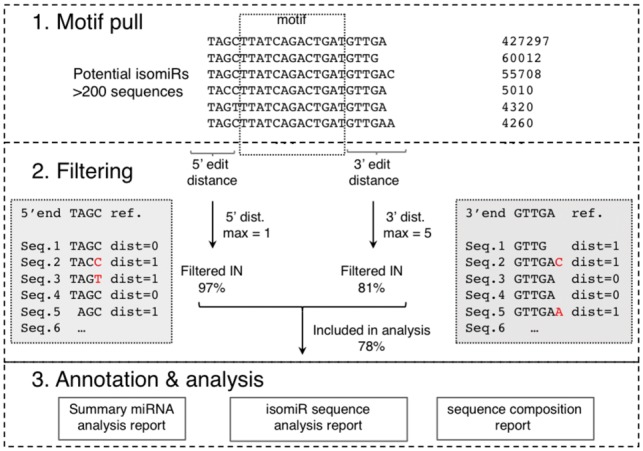
Flow diagram of the QuagmiR algorithm and outputs. Step 1. Collapse the FASTQ file and then pull reads that match the motif. Step 2. Filter reads based on the weighted Levenshtein distance on the substrings at the 5′ and 3′ ends of the read compared to the reference miRNA. Based on the maximum distance defined, reads are filtered out or in for further analysis. Step 3. Each read is annotated relative to the reference miRNA, and three separate reports are generated
The potential isomiR reads are further filtered according to the nucleotides that precede and follow the motif (5′ part and 3′ part, respectively). The pairwise sequence similarity between a read and the reference miRNA is calculated using the Levenshtein distances—the number of deletions, insertions, or substitutions required to transform one string into the other. The penalty for any change may be fine-tuned, although the default value is 1. The filtering parameters for the 5′ and 3′ regions may be set independently to capture the asymmetrical distribution of the sequence heterogeneity. By this approach, users can customize the mapping process to focus on particular types of isomiRs. For example, 3′ isomiRs can be specifically targeted by setting the 5′ distance to 0 and leaving the 3′ distance open to any value (set to −1).
Finally, isomiRs passing these filters are characterized by comparing them again to the canonical miRNA sequence. At the 5′ end, the variation of the start position, which is termed as the ‘fidelity index’, is calculated to infer the cleavage fidelity of Drosha (for 5p miRNAs) or Dicer (for 3p miRNAs) (Gu et al., 2012). At the 3′ end, based on the position(s) of non-alignment between the isomiR and the miRNA reference, the lengths of trimming (truncation) and tailing are determined.
Three files are outputted with every run of QuagmiR. The summary file describes for each miRNA: the raw counts, number of isomiRs, fidelity index of the 5′ end (Supplementary Fig. S1B), percentages of trimming and tailing (Supplementary Fig. S1C), and composition percentages on the non-templated tails. The isomiR sequence file contains all of the matched isomiR sequences with detailed information as listed above for each corresponding miRNA. The nucleotide composition file describes the coverage and nucleotide composition of all the isomiRs captured under each motif (Supplementary Fig. S1D). These reports can be obtained either for individual samples or cohorts of samples analyzed in batch and tabulated in an ‘R-friendly’ format. They are of special value for subsequent analysis aimed at elucidating the molecular mechanism behind isomiR biogenesis and regulation. In addition, a GFF3 file is generated for each run, allowing the user to compare the results generated by QuagmiR to those of previously established aligners documented in the miRTOP community (Pantano, 2016). Links to the further documentation regarding the QuagmiR installation and run parameters as well as other resources can be found in Supplementary Table S1.
3 Results design of QuagmiR on the cancer Genomics Cloud
QuagmiR can run both on local machines and high-performance computing platforms such as NIH-Biowulf. In addition, we have deployed QuagmiR on the Seven Bridges Cancer Genomics Cloud (CGC) (Lau et al., 2017) to promote broad availability. Development of the CGC was funded in part by the National Cancer Institute with the goal of enhancing the accessibility of cancer genomics data and facilitating reproducible and collaborative analysis in the cloud. Deployment on the CGC allows researchers to run QuagmiR via an intuitive graphical interface that permits simple changes to parameters and creation of integrated workflows with other analytical tools (Supplementary Fig. S2).
QuagmiR can be used to analyze both private datasets and the public datasets that are available to authorized researchers through the CGC, such as the >11 000 miRNA-seq samples from adult and pediatric cancers that are included in The Cancer Genome Atlas (TCGA) and Therapeutically Applicable Research to Generate Effective Treatments (TARGET) datasets. This enables researchers to perform big data analysis without downloading or storing sensitive data locally. Deployment of QuagmiR on the CGC also provides researchers with access to scalable and flexible computing resources on demand.
To analyze private datasets on the CGC using QuagmiR, researchers can upload sequencing files in the FASTQ format to the CGC and use them as inputs. The motifs and canonical sequences for all human miRNAs reported in miRBase v22 have been compiled and are provided as a default reference. Motif files corresponding to other organisms are also available, making it possible to perform QuagmiR analysis for miRNAs from all annotated genomes in miRBase v22 (250 in total). Alternatively, users can generate customized motif files via a script provided with QuagmiR.
4 Discussion
QuagmiR, similar to other methods (Patel et al., 2016; Ziemann et al., 2016), provides a robust computational pipeline for accurate mapping and expression analysis of miRNAs (Supplementary Fig. S3). However, QuagmiR excels at characterization and extensive analyses of 5′ and 3′ isomiRs. With its default settings, QuagmiR can detect more types of isomiRs than several other currently available tools (Supplementary Fig. S4). The additional strength of QuagmiR comes from its unique mapping algorithm, which provides users with the ability to customize the detection process. Furthermore, a novel aspect of QuagmiR is its unique treatment of isomiR sequence variations in the middle, 5′ part, and 3′ part, allowing it to efficiently distinguish bona fide isomiRs from false-positive reads. A potential caveat is that QuagmiR cannot detect isomiRs bearing sequence variations within the motif. To mitigate this issue, multiple miRNA motifs can be designed or the motifs can further be ‘relaxed’ by the inclusion of ambiguous nucleotides.
There are many potential applications of QuagmiR for small RNA analysis. For example, recent studies analyzing small RNA data with the miRDeep2 algorithm (Friedländer et al., 2008, 2012) have identified with high confidence hundreds or even thousands of low expression miRNAs (Londin et al., 2015). However, the vast majority of these miRNAs remain unannotated in reference genomes and databases. QuagmiR provides an excellent opportunity for researchers to work on characterizing isomiRs of these unannotated miRNAs. QuagmiR is also of interest for researchers working directly with small RNA-seq data from organisms where the reference genome is not available.
In addition, isomiR analysis of large-scale public datasets such as those available through the CGC could pave the way for a better understanding of the role of isomiRs in tumorigenesis. Lastly, from a methodological standpoint, QuagmiR customized motifs can provide a useful tool to analyze the processing of other small RNAs. For example, QuagmiR can be used to study shRNA maturation and help prevent misprocessings that diminish efficacy and increase off-target effects (Bofill-De Ros and Gu, 2016).
Funding
The Seven Bridges Cancer Genomics Cloud has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, Contract No. HHSN261201400008C and ID/IQ Agreement No. 17X146 under Contract No. HHSN261201500003I. X.B.D.R., K.C., S.C., M.J. and S.G. have been funded by the intramural research program of the National Cancer Institute, National Institutes of Health.
Conflict of Interest: NT, NS, SN, VV, EW and RM are employees and shareholders of Seven Bridges Genomics Inc.
Supplementary Material
References
- Bartel D.P. (2018) Metazoan microRNAs. Cell, 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barturen G. et al. (2014) sRNAbench: profiling of small RNAs and its sequence variants in single or multi-species high-throughput experiments. Methods in Next Generation Sequencing, 1, 21–31. [Google Scholar]
- Bofill-De Ros X., Gu S. (2016) Guidelines for the optimal design of miRNA-based shRNAs. Methods, 103, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A. et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emde A.-K. et al. (2010) MicroRazerS: rapid alignment of small RNA reads. Bioinformatics, 26, 123–124. [DOI] [PubMed] [Google Scholar]
- Fernandez-Valverde S.L. et al. (2010) Dynamic isomiR regulation in Drosophila development. RNA, 16, 1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer M.R. et al. (2008) Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol., 26, 407–415. [DOI] [PubMed] [Google Scholar]
- Friedländer M.R. et al. (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res., 40, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R.C. et al. (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res., 19, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S. et al. (2012) The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell, 151, 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M., Kim V.N. (2014) Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol., 15, 509–524. [DOI] [PubMed] [Google Scholar]
- Kozomara A., Griffiths-Jones S. (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res., 42, D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau J.W. et al. (2017) The cancer genomics cloud: collaborative, reproducible, and democratized-a new paradigm in large-scale computational research. Cancer Res., 77, e3–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Gregory R.I. (2015) MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer, 15, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londin E. et al. (2015) Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc. Natl. Acad. Sci. USA, 112, E1106–E1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall M.N. et al. (2017) Toward the human cellular microRNAome. Genome Res., 27, 1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin R.D. et al. (2008) Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res., 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilsen C.T. et al. (2012) IsomiRs–the overlooked repertoire in the dynamic microRNAome. Trends Genet., 28, 544–549. [DOI] [PubMed] [Google Scholar]
- Pantano L. et al. (2010) SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res., 38, e34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano L. et al. (2016) miRTOP: small RNA standard annotations, doi:10.5281/zenodo.45385. [Google Scholar]
- Patel P. et al. (2016) miTRATA: a web-based tool for microRNA Truncation and Tailing Analysis. Bioinformatics, 32, 450–452. [DOI] [PubMed] [Google Scholar]
- Rüegger S., Großhans H. (2012) MicroRNA turnover: when, how, and why. Trends Biochem. Sci., 37, 436–446. [DOI] [PubMed] [Google Scholar]
- Sayed D., Abdellatif M. (2011) MicroRNAs in development and disease. Physiol. Rev., 91, 827–887. [DOI] [PubMed] [Google Scholar]
- Telonis A.G. et al. (2015) Beyond the one-locus-one-miRNA paradigm: microRNA isoforms enable deeper insights into breast cancer heterogeneity. Nucleic Acids Res., 43, 9158–9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese D. et al. (2012) RazerS 3: faster, fully sensitive read mapping. Bioinformatics, 28, 2592–2599. [DOI] [PubMed] [Google Scholar]
- Ziemann M. et al. (2016) Evaluation of microRNA alignment techniques. RNA, 22, 1120–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.