

Abstract
Large-scale sequencing efforts in amyotrophic lateral sclerosis (ALS) have implicated novel genes using gene-based collapsing methods. However, pathogenic mutations may be concentrated in specific genic regions. To address this, we developed two collapsing strategies: One focuses rare variation collapsing on homology-based protein domains as the unit for collapsing, and the other is a gene-level approach that, unlike standard methods, leverages existing evidence of purifying selection against missense variation on said domains. The application of these two collapsing methods to 3093 ALS cases and 8186 controls of European ancestry, and also 3239 cases and 11,808 controls of diversified populations, pinpoints risk regions of ALS genes, including SOD1, NEK1, TARDBP, and FUS. While not clearly implicating novel ALS genes, the new analyses not only pinpoint risk regions in known genes but also highlight candidate genes as well.
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease characterized by progressive motor neuron loss leading to paralysis and death, most often from respiratory failure. Roughly 60%–70% of familial and 10% of sporadic cases have an identifiable mutation in a known causal ALS gene, the majority of which are repeat expansions in C9orf72 and point mutations in SOD1 (Renton et al. 2014). Recent efforts in gene discovery, largely driven by advances in sequencing and identification of rare variants, have implicated and confirmed several new genes in ALS pathogenesis including TBK1, NEK1, ANXA11, and CCNF (Bannwarth et al. 2014; Johnson et al. 2014; Smith et al. 2014, 2017; Cirulli et al. 2015; Kenna et al. 2016; Williams et al. 2016; Mackenzie et al. 2017; Nicolas et al. 2018). Despite this progress, the majority of sporadic cases still remain to be resolved genetically.
The now established paradigm for case-control analyses of exome or genome sequencing data of complex diseases and traits involves a gene-based collapsing framework in which all qualifying variants in a gene are treated as equivalent. Genes are associated with the trait when they exhibit a significant excess of qualifying variants occurring anywhere in the gene. This approach has implicated disease genes in a growing number of other complex conditions beyond ALS, including idiopathic pulmonary fibrosis (IPF), myocardial infarction (MI), and Alzheimer's disease (Cruchaga et al. 2014; Do et al. 2015; Petrovski et al. 2017).
While clearly effective, the power of this approach is limited by the inclusion of benign variants that reduce statistical power. However, for genes where pathogenic mutations are localized to specific regions, such as functional domains, power can be increased by using these regions as the unit for the collapsing analysis. In ALS-associated genes, there are several examples of genes that show regionally localized pathogenic variation. For example, in TARDBP, highly penetrant ALS variants are concentrated in a glycine-rich domain near the C terminus (Pesiridis et al. 2009). Furthermore, the gene FUS, which has a similar structure as TARDBP, has pathogenic mutations clustering in two regions: exons 13–15 (encoding an Arg-Gly-Gly-rich domain and the nuclear localization signal) and exons 3, 5–6 (encoding Gln-Gly-Ser-Tyr-rich and Gly-rich domains) (Mackenzie et al. 2010).
Recognizing that undiscovered ALS-associated genes might similarly have specific domains where pathogenic variants cluster, we now apply two complementary regional approaches to gene collapsing analyses to identify localized signals of rare variation in a data set of 3093 ALS cases of European ancestry (2663 exomes and 430 whole genomes) compared with 8186 controls of matched ancestry (7612 control exomes and 574 whole genomes). We further apply these analyses to a set of samples of diversified ancestry origins, consisting of 3239 cases and 11,808 controls. We compare the regional approaches to the standard gene collapsing analysis and highlight the importance of a regional view specifically for ALS genetics.
Results
Collapsing analyses using homology-defined protein domains
The standard approach to gene discovery focuses on the burden of rare variants across an entire gene by comparing the frequency of qualifying variants in cases and controls (illustrated in Fig. 1A). The qualifying variants can be defined by various criteria such as function and allele frequency.
Figure 1.
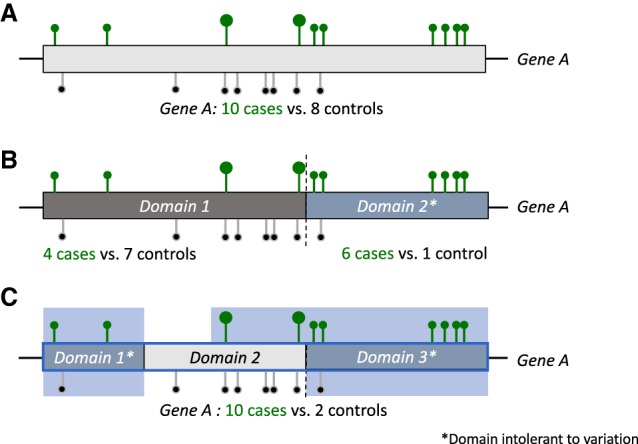
Gene and regional collapsing. (A) A standard gene-based approach for collapsing analysis of nonsynonymous and canonical splice rare variants in cases (green) and controls (black) on example Gene A. (B) A domain-unit–based regional approach in which only the domains that are intolerant to functional variation are considered as units for collapsing. (C) Intolerance-informed gene collapsing: a regional approach to gene collapsing in which the unit for collapsing is the entire gene, yet missense variants only qualify for the analysis if they reside in domains that are intolerant to variation (domain 1 and 3). Loss-of-function variants (big circles) continue to qualify regardless of whether they reside in a tolerant or intolerant domain of the gene. Bright blue background marks qualifying region.
In this study, we describe two additional approaches to rare variant collapsing: (1) a regional approach, in which the unit for collapsing is not the gene but rather the functional domains within the gene (Fig. 1B); and (2) a gene-based approach, in which the definition of qualifying variants is informed by regional intolerance to missense variation (Fig. 1C).
We first utilized the standard gene collapsing approach (Fig. 1A) to identify the burden of rare variants in a set of 3093 ALS cases and 8186 controls of European ancestry. The demographic features of our cohort reflect known epidemiological features of ALS, including male predominance and the distributions of age at onset and survival (Supplemental Table S1). Qualifying variants were defined as nonsynonymous coding or canonical splice variants that have a minor allele frequency (MAF) ≤0.1% in cases and controls (internal MAF) and also a ≤0.1% MAF imposed for each population represented in the ExAC Browser (Lek et al. 2016). High quality control (QC) metrics were further imposed on the variants (see Methods).
Comparing genetic variation across 18,653 protein-coding genes found a genome-wide and study-wide significant (P < 6.7 × 10−7) case-enrichment only for SOD1 (P = 1.23 × 10−18) (Fig. 2A), with qualifying variants identified in 43 cases (1.39%) and only six controls (0.07%; OR = 19.2). TARDBP showed the second strongest enrichment (OR = 3.6, P = 1.02 × 10−4), but with 23 cases (0.74%) and 17 controls (0.21%) it did not achieve genome-wide significance. FUS harbored qualifying variants in 20 cases and 37 controls (OR = 1.43, P = 0.23) (Fig. 2A).
Figure 2.

Q-Q plots of gene- and domain-level collapsing. (A) The results for a standard gene-level collapsing of 3093 cases and 8186 controls; 18,065 covered genes passed QC with more than one case or control carrier for this test. The genes with the top associations and FUS gene are labeled. The genomic inflation factor, lambda (λ), is 1.10. (B) The results for the domain-based collapsing of 3093 cases and 8186 controls; 70,603 covered domains passed QC with more than one case or control carrier for this test. The genes with the top associations are labeled and genome-wide significant genes are in bold. λ = 1.046.
A gene-based analysis evaluating only rare loss-of-function (LoF) variants was also performed, identifying a genome-wide and study-wide significant case-enrichment of NEK1 variants (OR = 7.35, P = 1.85 × 10−10), with 33 cases (1.07%) compared to 12 controls (0.15%) (Supplemental Fig. S1).
As a negative control, we included a model for rare synonymous variants and did not observe any genes with significant enrichment. The genomic inflation factor, lambda (λ) for this model was 1.03 (Supplemental Fig. S2).
We hypothesized that genes with clustered mutations that had weak enrichments using this standard gene-based collapsing approach, such as TARDBP and FUS, could be identified by a collapsing method that uses functional gene regions (i.e., domains) as the unit for collapsing (Fig. 1B). For this analysis, we utilized a list of 89,522 gene domains covering the human coding sequence, as described previously (Gussow et al. 2016). In short, the coding sequence of each gene was aligned to a set of conserved protein domains based on the Conserved Domain Database (CDD) (Marchler-Bauer et al. 2013). The final domain coordinates for each gene were defined as the regions within the gene that aligned to the CDD and the unaligned regions between each CDD alignment. These domains were then used as the unit for collapsing compared with a standard gene-based collapsing approach (Fig. 1A,B; Supplemental Table S2).
This domain-based analysis was performed using the same cohort and coding model as the standard approach (European ancestry, nonsynonymous and canonical splice variants, internal and population MAF ≤0.1%). As hypothesized, the top three case-enriched domains reside in ALS genes: SOD1, TARDBP, and FUS (Fig. 2B). For SOD1, one domain that uniquely maps to a conserved domain (Cu-Zn superoxide dismutase) spans the majority of SOD1’s coding sequence and contains most of the variation found in 1.29% of cases and 0.07% of controls (OR = 17.9; P = 4.1 × 10−17) (Fig. 2B).
The glycine-rich TARDBP domain, where known mutations cluster, is now identified with genome-wide and study-wide significance (OR = 7; P = 5.84 × 10−7) (Fig. 2B). Of note, this glycine-rich domain covers exon 6 of TARDBP and was not mapped to a conserved domain from the CDD.
The same trend was observed for FUS, which shows the third strongest enrichment in this analysis (OR = 8.6; P = 3.6 × 10−5) (Fig. 2B). Specifically, qualifying variants were identified in 13 cases (0.42%) and four controls (0.05%) in the previously reported Arg-Gly rich domain covering exons 13–15, which also did not map to a conserved CDD domain (Pesiridis et al. 2009). Although not at genome-wide or study-wide significance, this represents a substantial improvement over the gene-based collapsing approach (OR = 1.43, uncorrected P = 0.23).
The fourth most case-enriched domain was a conserved armadillo repeat domain spanning exons 12–14 of PKP4 (plakophilin 4, also known as p0071). Qualifying variants occurred in 0.61% of ALS cases and 0.13% of controls (OR = 4.6, P = 4.1 × 10−5). While not genome-wide significant, PKP4 is a relevant candidate gene that has been previously linked to various ALS-related pathways (see Discussion).
Gene-based collapsing analyses informed by regional intolerance to missense variation
As we have demonstrated, domain-based collapsing effectively identifies genes where pathogenic variants are localized to single specific regions (e.g., TARDBP and FUS) and highlights suggestive candidates for further study (PKP4). However, to identify haploinsufficient genes where truncating variants and sufficiently damaging missense mutations could both contribute to risk of disease, the difficulty lies in determining which missense variants should qualify in the analysis. To address this challenge, we implemented a collapsing approach that leverages regional patterns of intolerance to missense variation (sub-RVIS) (Gussow et al. 2016; Traynelis et al. 2017) as a way to prioritize missense variants most likely to result in disease. In this ‘intolerance-informed’ approach, rare missense alleles were counted as qualifying if they resided in gene regions that are intolerant to missense variation, whereas LoF variants were counted as qualifying regardless of location within the gene (Fig. 1C).
As a measure of intolerance of gene regions, we applied a complementary approach to sub-RVIS (Gussow et al. 2016) for when there is limited resolution in the sequence region of interest. This approach uses the observed to expected missense ratio in a domain (OE-ratio), which is equivalent to a domain-based missense tolerance ratio (MTR) (Traynelis et al. 2017). In short, the expected rate leverages the underlying sequence context in the domain, and the observed rate is based on the rate of nonsynonymous variants identified in the subregion of interest based on the ExAC Browser, release 0.3 (see Methods; Lek et al. 2016).
We focus our intolerance-informed gene collapsing approach on domains that have intolerance below the median exome-wide OE-ratio, thus subselecting variants in genic regions that have greater evidence of purifying selection acting against nonsynonymous variation. As mentioned earlier, for each gene, variants in these intolerant regions are considered along with LoF variants independent of their location within the gene.
Because intolerant coding regions are expected to have a lower rate of common variation, we included samples from diversified ancestries when applying intolerance-informed gene collapsing. The diversified population approach increased the total number of samples by 3768, to 3239 cases and 11,808 controls, thereby increasing the power of the analysis. For this approach, we applied similar rules for qualifying variants, including low population frequency (MAF ≤ 0.1% imposed for each population represented in ExAC), an internal MAF ≤ 0.04% (decreased from 0.1% due to a larger control cohort), coding annotation (nonsynonymous and splice variants), and high QC metrics, with the additional criteria of residing in the lower 50th percentile of OE-ratio domains.
The genomic inflation factor (λ) of the diversified populations intolerance-informed analysis was 1.14, slightly higher than the European-only cohort used for the standard gene-level analyses (λ = 1.1) (Fig. 2A). Yet, this inflation is much lower than for the standard gene-based analysis using a diversified population (λ = 1.25) (Supplemental Fig. S3), demonstrating the advantage of an intolerance-informed approach for reducing the genomic inflation due to variation in tolerant regions.
In this analysis, SOD1 achieved a slightly better enrichment than in either gene-based or domain-based analyses (OR = 20.31; P = 4.13 × 10−22) (Fig. 3A). TARDBP also had genome-wide and study-wide significant enrichment (OR = 4.95; P = 8.77 × 10−8) (Fig. 3A), which is due to the added power of a diversified versus European-only analysis (Fig. 3B).
Figure 3.

Intolerance-informed gene-level collapsing with unified/diversified ancestry samples. (A) A Q-Q plot presenting the results of the gene-based intolerance-informed collapsing of 3239 cases and 11,808 controls from diversified ancestries. Missense variants are aggregated only if they reside in an intolerant domain that is lower than the 50th percentile OE-ratio score, while loss-of-function variants are aggregated independent of location; 17,795 genes passed QC with more than one case or control carrier for this test. The genes with the top associations are labeled. λ = 1.14. (B) A Q-Q plot of a gene-based intolerance-informed collapsing of 3093 cases and 8186 controls of European ancestry; 18,135 genes passed QC with more than one case or control carrier for this test. The genes with the top associations are labeled and genome-wide significant genes are in bold. λ = 1.073.
LGALSL (galectin like, previously known as lectin, galactoside-binding-like) was the third gene to have a strong enrichment of qualifying variants in cases (OR = 14.63; P = 2.29 × 10−6) (Fig. 3A) that was not study-wide significant given the models tested. The enrichment of this gene originates from one specific domain that harbors variants for 12 cases (0.37%) and three controls (0.025%) with the addition of an African-American and a Latino case over the European-only analysis. The target domain harboring all LGALSL case-variants is a region comprising 378 bp that is mapped to a conserved protein domain intolerant to variation. Notably, LGALSL LoF variants were only identified in cases and absent from nearly 12,000 controls. To assess the rate of LoF variants in a larger control population, we looked at the ExAC cohort and found three LoF alleles in 60,033 individuals (Lek et al. 2016).
We also examined the effects of population and gender as possible covariates by performing the analysis using the Firth logistic regression and using gender and first five genotype principal components as covariates. We find that the inflation factor is reduced from λ = 1.14 to λ = 1.04, and the P-value for LGALSL is strengthened to 5.84 × 10−7 (Supplemental Fig. S4).
The case-enrichment of LGALSL was also replicated using an additional data set constructed by Cirulli et al. (2015), in which the top 51 genes identified in their best performing model (including LGALSL) were sequenced in a replication cohort. We subjected this data set of 830 cases and 1858 controls that were available at Columbia University to the same intolerance-informed analysis as the original cohort. We identify two cases (0.24%) and one control (0.054%) with qualifying variants in the LGALSL domain and an odds-ratio of 4.5. While not significant due to the small sample size, this analysis shows the same direction of case-enrichment in LGALSL. We further used the Cochran-Mantel-Haenszel (CMH) test to combine the original and replication cohorts, providing a final, combined P = 1.54 × 10−7 that is genome- and study-wide significant and a common odds-ratio of 11.35 for LGALSL.
We also performed a domain-level analysis to compare gene and domain results over the diversified population cohort. This analysis achieved similar signals for the top three genes: SOD1 (P = 2.56 × 10−20), TARDBP (P = 4.32 × 10−11), and LGALSL (P = 2.29 × 10−6) (Supplemental Fig. S5). While this analysis achieves similar results as the intolerance-informed gene-level analysis, there is an advantage to using the intolerance-informed analysis since it requires correcting for the lower number of genes (18,653) than the much higher number of genic domains (89,522).
Genome-wide associations with age-at-onset
We next examined whether qualifying variants in known ALS genes, or candidate genes identified by our novel approaches, influence age at symptom onset (AAO). We therefore examined the average AAO of cases in all genes that have at least three carriers (11,541 genes) against the average AAO of the rest of the cases (Supplemental Table S3).
We found that SOD1 variant carriers tended to be younger than the rest of the cohort (52.2 vs. 57.1 yr, P = 0.059; Mann-Whitney U test, ranked at position 705/11,541). Also, subjects harboring qualifying variants in ANXA11 showed delayed onset (63.8 yr, P = 0.037; Mann-Whitney U test, ranked at position 417/11,541), which is consistent with prior studies (Smith et al. 2017). No other known ALS genes showed significant influence on AAO.
Subjects harboring LGALSL qualifying variants showed a mean AAO that is 13 yr younger than the rest of the cohort (43.8 yr vs. 57.1, P = 8.1 × 10−4; Mann–Whitney U test). The AAO information was available for 11/12 variant carriers and 2767/3239 noncarriers, and LGALSL was ranked as the fifth highest gene with regard to significance of AAO difference.
The early onset in cases carrying LGALSL variants was further validated by a random sampling approach in which LGALSL carriers’ average AAO was significantly lower than 9983/10,000 randomly sampled sets of 11 cases (P = 0.0017) (Methods). We further examined the recruitment sites of these 11 LGALSL carriers that might explain the earlier age at onset. We find that they originate from five different recruitement sites, and the AAO per site did not reveal any confounding effects (Supplemental Table S4).
Discussion
Here, we present a regional approach to rare variant collapsing analyses and demonstrate its utility in ALS. This approach has two distinct forms: (1) aggregating rare variants on genic subregions defined using conserved protein domain annotations; and (2) aggregating rare variants on a gene unit but using the pattern of purifying selection to identify the most damaging missense variants and combine them with loss-of-function mutations occurring anywhere in the gene. Both approaches show improved sensitivity for known ALS genes, finding SOD1, NEK1, and TARDBP as genome-wide significant. We also find FUS’s Arg-Gly-rich domain within the top three associations in our domain-based regional collapsing, jumping from an insignificant OR = 1.43 to a high OR = 8.6. These findings underscore the utility of applying a regional approach to ALS genetics, especially in light of similar Gly-rich domains’ importance in mediating pathologic RNA-protein complexes (Rogelj et al. 2011).
This approach has also implicated a potential new candidate ALS gene, LGALSL, encoding the galectin like protein GRP (galectin-related protein, also known as HSPC159). We identified a case-enriched intolerant galectin-binding domain (Fig. 4A). While the functions of LGALSL remain largely unknown, members of the galectin family, including LGALS1 and LGALS3, have been implicated in ALS disease processes and progression. Specifically, LGALS1 has been identified as a component of sporadic and familial ALS-related neurofilamentous lesions (Kato et al. 2001) and is associated with early axonal degeneration in the SOD1G93A ALS mouse model (Kobayakawa et al. 2015). Furthermore, homozygous deletion of Lgals3 reportedly led to accelerated disease progression and reduced lifespan in SOD1G93A mice (Lerman et al. 2012). We also performed an analysis of age at onset that is independent of the predisposition analysis, showing a significant association between carriers of qualifying variants in LGALSL and early age at onset. This independent analysis supports LGALSL as a candidate ALS gene that might be responsible for a form of ALS with a younger age at onset.
Figure 4.

Distribution of functional coding variants across LGALSL and PKP4. The distribution of LGALSL (A) and PKP4 (B) coding variants across domains (LGALSL transcript ENST00000238875 and PKP4 transcript ENST00000389757). The y-axis corresponds to the total number of variants identified at a specific location. The blue boxes highlight the (A) LGALSL carbohydrate-binding domain and (B) PKP4 armadillo repeat domain 2 (ARM2) found to be enriched for variants in cases (green) compared to controls (black). Each domain's OE-ratio percentile is marked above for both tolerant (bright blue) and intolerant (orange) domains.
Further study of additional LGALSL mutation carriers will be required to confirm this observed genotype-phenotype correlation.
Regional collapsing analyses also highlighted PKP4 as a new candidate gene, with a single armadillo repeat domain strongly enriched for qualifying variants in cases (Fig. 4B). Evidence supporting PKP4’s role in ALS-linked processes including microtubule transport and endosomal processing, in addition to its local translation in ALS-mutant FUS granules, all provide evidence in favor of PKP4 as a risk factor for ALS (Keil et al. 2013; Yasuda et al. 2013; Keil and Hatzfeld 2014; Becher et al. 2017).
This study incorporated both exome and whole genome samples from a large cohort of over 3000 cases and close to 12,000 controls. Yet, despite these large cohorts, the standard gene collapsing approach identified only SOD1 and NEK1 (loss-of-function-specific model) as achieving genome-wide significance and failed to uncover other known signals for ALS risk factors. We were able to capture these signals, along with candidate novel signals, using a regional approach that is informed by missense variation intolerance. That being said, while confirming TARDBP, and suggesting LGALSL (0.42% of cases) as a candidate gene, the regional approach was still underpowered with the current sample size to show genome-wide significance for FUS and PKP4 that might reflect true associations. For example, relying on the aggregate variant frequencies observed in our case and control cohorts, we find that roughly 10,300 of each group are necessary to reach 80% power to detect genome-wide significance of FUS using the domain-level approach. Alternatively, the standard gene-level analysis would require nearly 89,000 in each cohort. For PKP4, a domain-level approach would require roughly 10,300 cases and controls each, compared to around 12,100 for a gene-level approach. This suggests that even with our signal optimization approaches, larger sequencing studies are required in ALS.
While being able to pinpoint candidate genes, the approaches used in this study are still experimental and stand alongside, not in place of, current methodologies of gene discovery analyses. Furthermore, other methods can and should be used to identify functional genic domains that might harbor signals of case enrichment. While the mapping of coding regions to CDD domains is one approach to identify functional domains that has proven beneficial for these analyses, it is possible that case enrichment could be further strengthened by using different functional domains. In addition, the measurement of intolerance that incorporates information from over 60,000 samples still lacks information on many genic domains that, in time and with larger population sequencing efforts, will increase the confidence in the assumption that intolerant regions harbor damaging variation. That said, we are confident that the continued application of regional approaches to collapsing analyses in ALS and other rare disorders will enable the identification of novel, rare risk factors in patient populations. These were previously difficult to identify given the presence of benign variants in intolerant genic regions.
Methods
Subject sources
ALS samples analyzed by whole-exome or -genome sequencing came from the Genomic Translation for ALS Care (GTAC study), the Columbia University Precision Medicine Initiative for ALS, the New York Genome Consortium, and the ALS Sequencing Consortium (IRB-approved genetic studies from Columbia University Medical Center, including the Coriell NINDS repository), University of Massachusetts at Worchester, Stanford University (including samples from Emory University School of Medicine, the Johns Hopkins University School of Medicine, and the University of California, San Diego), Massachusetts General Hospital Neurogenetics DNA Diagnostic Lab Repository, Duke University, McGill University (including contributions from Saint-Luc and Notre-Dame Hospital of the Centre Hospitalier de l'Université de Montréal [CHUM], [University of Montreal]), Gui de Chauliac Hospital of the CHU de Montpellier (Montpellier University), Pitié Salpêtrière Hospital, Fleurimont Hospital of the Centre Hospitalier Universitaire de Sherbrooke (CHUS) (University of Sherbrooke), Enfant-Jésus Hospital of the Centre hospitalier affilié universitaire de Québec (CHA) (Laval University), Montreal General Hospital, Montreal Neurological Institute and Hospital of the McGill University Health Centre, and Washington University in St. Louis (including contributions from Houston Methodist Hospital, Virginia Mason Medical Center, University of Utah, and Cedars Sinai Medical Center).
All subjects provided written, informed consent for genetic studies that had been IRB-approved at each contributing center.
Subject selection criteria
ALS subjects were diagnosed according to El Escorial revised criteria as suspected, possible, probable, or definite ALS by neuromuscular physicians at submitting centers. Subjects were considered sporadic if no first- or second-degree relatives had been diagnosed with ALS or died of an ALS-like syndrome. Because screening for known ALS gene mutations prior to sample submission was highly variable across the cohort, gene status was not considered a priori. Controls were selected from >45,000 whole-exome or -genome sequenced individuals housed in the IGM Data Repository. We excluded all individuals with a known diagnosis or family history of neurodegenerative disease, but not all had been specifically screened for ALS.
Sequencing
Sequencing of DNA was performed at Columbia University, the New York Genome Center, Duke University, McGill University, Stanford University, HudsonAlpha, and University of Massachusetts, Worcester. Whole-exome capture used Agilent All Exon kits (50MB, 65MB, and CRE), Nimblegen SeqCap EZ Exome Enrichment kits (V2.0, V3.0, VCRome, and MedExome), IDT Exome Enrichment panel, and Illumina TruSeq kits. Sequencing occurred on Illumina GAIIx, HiSeq 2000, or HiSeq 2500 sequencers according to standard protocols (Supplemental Tables S5, S6).
Illumina lane-level FASTQ files were aligned to the human reference genome (NCBI Build 37) using the Burrows–Wheeler Alignment Tool (BWA) (Li and Durbin 2009). Picard software (http://picard.sourceforge.net) removed duplicate reads and processed lane-level SAM files to create a sample-level BAM file. Genomes (n = 402) from the New York Genome Center were transferred as sample-level BAM files. We used GATK to recalibrate base quality scores, realign around indels, and call variants (McKenna et al. 2010).
While sequences were aligned to the genome reference build 37, we do not expect a change in the results by mapping to the newer build 38. This is because the collapsing utilized only the consensus coding sequence (CCDS) of human genes, which was already mature in build 37 and experienced only minor changes. Furthermore, none of the top genes presented in this work saw any changes (other than chromosomal base pair numbering) between build 37 (CCDS version r15) and that of build 38 (CCDS version r17).
Quality control
Several robust measures were taken to control for the effects that might arise from the different sequencing platforms, kits, and coverage that are the result of the various recruitment sites, sequencing centers, and technology changes through the years when the samples were collected and sequenced. These measures were taken in both sample- and variant-level filtering. The cohort of cases and controls underwent the following steps of construction and coverage harmonization that are explained below in detail: Following stringent QC and ancestry filters (if applied) and assessment of 10× coverage (Supplemental Tables S7, S8), a newly developed method was applied to account for the variability of coverage over the CCDS between samples originating from various sequencing kits and platforms. This method was used to remove samples that are considered coverage outliers. Following the removal of coverage outliers, a further genotype PCA analysis was used to remove remaining genotypic outliers. Following the cohort stratification and harmonization, tests were performed to remove variants that did not survive stringent QC metrics. The surviving high-quality variants were then subjected to a coverage binomial test to control for variant-level coverage differences between the cohorts (detailed below). Any site which did not pass a significance threshold was eliminated from consideration in the test of enrichment.
Cohort construction: sample quality and relatedness filters
The initial sample consisted of 4149 ALS cases and 15,107 controls. Samples reporting >8% contamination according to verifyBamID (Jun et al. 2012) were excluded. KING (Manichaikul et al. 2010) was used to ensure only unrelated (up to third-degree) individuals contributed to the analysis. For controls, where sample collection methods were not known, we excluded samples where X:Y coverage ratios did not match expected sex. For studies where sample collection and processing involved only ALS patients, mismatches were not exclusionary. Further, to be eligible, samples were further subjected to a CCDS 10-fold coverage principal components analysis (PCA) and an ancestry prediction filter (for European ancestry analysis).
Cohort construction: ancestry prediction
The ancestry classification model was trained using genotyped data from 5287 individuals of known ancestry and 12,840 well-genotyped and ancestry-informative markers that were limited to the human exome. The model was trained, tested, and validated on a set of individuals with ancestry as follows: non-Finnish European (N = 2911), Middle Eastern (N = 184), Hispanic (N = 368), East Asian (N = 539), South Asian (N = 529), and African (N = 756). Briefly, the sample × genotype matrix was scaled to have unit mean and standard deviation along each SNV and subjected to a principal component analysis. For training the classifier, the genotypes were projected onto the top six PCs and used as feature vectors. The classifier is a multi-layer perceptron with one hidden layer, a logistic activation function, L2 regularization term, alpha = 1 × 10−5, size of hidden layer = 6, and a L-BFGS solver. The classifier was implemented using the scikit-learn API in Python. A stratified 10-fold CV with 80:20 split of the training data was used to tune parameters using a grid search. Cross-validation performance on the cohort yielded precision/recall scores as follows: NFE: 0.99/1, AFR: 0.99/1, SAS: 0.99/1, EAS: 0.99/1, HIS: 0.93/0.97, ME: 0.93/0.77. Samples in this study were subjected to ancestry prediction using the model trained above by projecting their genotype vector to the training PCA model and running the classifier to obtain a given sample's ancestry probabilities for each of the trained populations.
For samples to qualify for a European ancestry analysis, they were required to have a European probability greater than 0.5 and an overall genotyping rate of 0.87 across the 12,840 well-genotyped and ancestry informative markers. Lower genotyping rates were considered as uninformative for ancestry prediction. In the case of low genotyping rate, we considered self-declared ethnicity of ‘White’ as qualifying for the European-based analysis.
Furthermore, once the final list was constructed, we applied an additional analysis to control for population stratification by using EIGENSTRAT (Price et al. 2006) to remove samples that were considered as genetic outliers. This ensured that the main cluster of samples was of European origin (see below).
Cohort construction: CCDS coverage PCA
To account for the variability of the coverage over the CCDS between cases and controls that originate from various sequencing centers, kits, and platforms, we developed a method to remove samples that are considered outliers due to coverage. This step was performed for samples that passed QC and ancestry prediction filters (if applied) and allowed for maximizing the coding region available for the analysis when harmonizing variant-level coverage between cases and controls.
We first randomly selected a set of 1000 CCDS genes for a coverage test. We next constructed a coverage matrix in which the rows are the samples used for the analysis and the columns are the number of bases covered at 10× in each of the 1000 random genes. Finally, we used the matrix in a principal-component analysis. Outliers were identified as being further than three standard deviations away from the center of the first four principal components (PCs).
In the Caucasian analysis, 3866 cases and 9426 controls passed initial QC and ancestry filters and were subjected to the coverage PCA filter. The coverage PCA maintained 3314 cases and 9214 controls.
In the diversified population analysis, 4075 cases and 14,494 controls passed initial QC and were subjected to the coverage PCA filter. The coverage PCA maintained 3468 cases and 13,957 controls.
Cohort construction: Eigenstrat PCA threshold adjustment
EIGENSTRAT (Price et al. 2006) PCA was used for removing genotypic outlier samples as a final cohort pruning step before running the collapsing analysis. The default threshold for removing outliers is six standard deviations from mean over the top 10 PCs. This process, including recalculation of the PCs, was repeated five times.
In the Caucasian analysis, 3208 cases and 8821 controls passed initial QC, ancestry, coverage PCA, and kinship filters and were subjected to the final EIGENSTRAT PCA filter. The EIGENSTRAT PCA maintained the final 3093 cases and 8186 controls used for the collapsing analysis, including 383 out of 420 whole genome cases that were mapped by the New York Genome Center (NYGC).
In the diversified analysis, 3353 cases and 13,373 controls passed initial QC, coverage PCA, and kinship filters and were subjected to the final EIGENSTRAT PCA filter. The default EIGENSTRAT PCA threshold removed all 420 NYGC whole genomes. This was the result of the addition to the Caucasian analysis of over 3000 exomes, which reduced the standard deviation and resulted in the exclusion of NYGC whole genomes in the third PC. As these samples were very high-quality and were included in the Caucasian only analysis, we adjusted the threshold of the third PC to seven standard deviations, thus maintaining 402 out of 420 NYGC whole genomes. In total, following the stratification phase, we maintained 3239 cases and 11,808 controls for the collapsing analysis.
Variant-level quality control
Quality thresholds were set based on previous studies (Cirulli et al. 2015; Epi4K consortium; Epilepsy Phenome/Genome Project 2017). Variants were required to have a quality score of at least 30, a quality by depth score of at least 2, genotype quality score of at least 20, read position rank sum of at least −3, mapping quality score of at least 40, mapping quality rank sum greater than −10, and a minimum coverage of at least 10. SNVs had a maximum Fisher's strand bias of 60, while indels had a maximum of 200. For heterozygous genotypes, the alternative allele ratio was required to be greater than or equal to 25%. Variants were excluded if they were marked by EVS as being failures (http://evs.gs.washington.edu/EVS/). Variants were annotated to Ensembl 73 using SnpEff (Cingolani et al. 2012).
Variant-level coverage harmonization between cases and controls
To ensure balanced sequencing coverage of evaluated sites between cases and controls, we imposed a statistical test of independence between the case/control status and coverage. For a given site, consider s the total number of cases, t the total number of controls, x the number of cases covered at 10×, and y the number of controls covered at 10×. We model the number of covered cases X as a binomial random variable
If case/control status and coverage status are independent, then
We test for this independence by performing a two-sided binomial test on the number of covered samples at given site, x.
In the collapsing analyses described below, a binomial test for coverage balance as described above was run as an additional qualifying criterion. Any site which resulted in a nominal significance threshold of 0.01 or lower was eliminated from further consideration.
Variant-level statistical analysis
Our primary model was designed to search for nonsynonymous coding or canonical splice variants that have less than 12 cases with a recurring variant in cases and controls (internal MAF) and also a ≤0.1% MAF imposed for each population represented in the ExAC Browser (Lek et al. 2016).
This model was tested in three forms: a standard gene-unit collapsing analysis; a domain-unit analysis; and an intolerance-informed gene collapsing analysis. A further gene-based analysis evaluating only rare loss of function variants was also performed. Two additional models examined other thresholds of population allele frequencies of ≤0.01% and an ultrarare 0% using the standard gene-based approach. Overall, the results of these analyses show SOD1 as the only genome-wide significant gene (Supplemental Tables S9, S10).
For each of the six models, we tested the list of 18,653 CCDS genes. For each gene, we counted the presence of at least one qualifying variant in the gene. A two-tailed Fisher's exact test (FET) was performed for each gene to compare the rate of cases carrying a qualifying variant compared to the rate in controls. For our study-wide significance threshold, after Bonferroni correction for the number of genes tested across the six nonsynonymous models, the study-wide multiplicity-adjusted significance threshold α = (0.05/[6 × 18653]) = 4.47 × 10−7. We did not correct for the synonymous (negative control) model.
OE-ratio intolerance for coding domains
The OE-ratio is calculated using the same approach as the missense tolerance ratio that is described by Traynelis et al. (2017). This approach uses the observed to expected missense ratio for the 89,522 domain coordinates that are described by Gussow et al. (2016).
For calculating a domain OE-ratio, the following requirements are applied: (1) adequate coverage—at least 50% of the bases within the domain must have at least a 10-fold coverage in the ExAC Browser, release 0.3 (Lek et al. 2016); and (2) at least five distinct variants (of any annotation) are required to perform a binomial exact test depletion of missense at uncorrected alpha of P < 0.05. There were 67,890 domains that passed the above requirements and were scored for their OE-ratio. The average size of the remaining unscored domains was usually very short (mean = 21 bp; median = 12), and they accounted for 0.77% of the protein-coding exome. Unscored domains were considered as below the intolerance ratio required for the intolerance-informed analysis (Fig. 3) to prevent loss of gene-level information. Once a domain lacking an OE-ratio is implicated in an analysis, its intolerance is examined using the average missense intolerance ratio (Traynelis et al. 2017) of the domain in question (http:// mtr-viewer.mdhs.unimelb.edu.au).
In the case of LGALSL, the last three codons of the coding transcript are a short independent domain that was not mapped to a conserved domain from the CDD. However, this small region is still considered part of the gal-binding domain by other databases (Finn et al. 2016). The MTR score for these three codons is below the 30th percentile of intolerance, marking this region at least as intolerant as the implicated galectin binding domain (OE-ratio percentile of 37).
In the case of COMMD1, the enrichment signal observed in Figure 3B is due to two very small domains at the beginning (11 aa) and the end (6 aa) of the gene, each harboring three cases and no controls. Since these domains are too small for scoring with the OE-ratio, they were considered as below the intolerance ratio required for the intolerance-informed analysis to prevent loss of gene-level information. When further testing the MTR score of these domains, the codons of the first short domain are indeed intolerant, but the other domain is tolerant to variation, which causes the frequency of cases with QVs in this gene to be 50% lower than observed in the original analysis.
LGALSL age-at-onset randomized testing scheme
The age-at-onset of LGALSL carriers was further validated using a random sampling approach. For this purpose, we sampled the AAO data 10,000 times, each time randomly selecting a group of 11 cases. The mean AAO of the original LGALSL cases (43.8) was lower than 9983 randomly selected AAO groups (p = 0.0017).
Data access
The aggregated genotypes of case and control cohorts used in this study are available for download as version 3.0 of the ALS database (http://www.alsdb.org).
Supplementary Material
Acknowledgments
We thank the following groups for contributing ALS samples, sequencing, or clinical data: the New York Genome Center ALS Consortium, including J. Kwan, D. Sareen, J.R. Broach, Z. Simmons, X. Arcila-Londono, E.B. Lee, V.M. Van Deerlin, E. Fraenkel, L.W. Ostrow, F. Baas, N. Zaitlen, J.D. Berry, A. Malaspina, P. Fratta, G.A. Cox, L.M. Thompson, S. Finkbeiner, E. Dardiotis, T.M. Miller, S. Chandran, S. Pal, E. Hornstein, D.J. MacGowan, T. Heiman-Patterson, M.G. Hammell, N.A. Patsopoulos, J. Dubnau, and A. Nath; the ALS Sequencing Consortium, including S.H. Appel, R.H. Baloh, R.S. Bedlack, W.K. Chung, S. Gibson, J.D. Glass, T.M. Miller, S.M. Pulst, J.M. Ravits, E. Simpson, and W.W. Xin; the Genomic Translation for ALS Care (GTAC) study, including L. Bruijn, S. Goutman, Z. Simmons, T.M. Miller, S. Chandran, S. Pal, G. Manousakis, S.H. Appel, E. Simpson, L. Wang, R.H. Baloh, R.S. Bedlack, D. Lacomis, D. Sareen, A. Sherman, and M. Penny.
The collection and sequencing of ALS cases for the New York Genome Center ALS Consortium was funded by the ALS Association and the Tow Foundation. Collection of samples, and sequencing for the GTAC study was funded by a partnership of the ALS Association and Biogen Idec. Sequencing of cases for the ALS Sequencing Consortium was funded by Biogen Idec.
We thank The Washington Heights–Inwood Columbia Aging Project (WHICAP) for the contribution of control samples. We also thank the WHICAP study participants and the WHICAP research and support staff for their contributions to this study: S. Kerns and H. Oster; K. Welsh-Bomer, C. Hulette, and J. Burke; F. McMahon and N. Akula; D. Valle, J. Hoover-Fong, and N. Sobriera; A. Poduri; S. Palmer; R. Buckley; and N. Calakos; The Murdock Study Community Registry and Biorepository Pro00011196; National Institute of Allergy and Infectious Diseases Center for HIV/AIDS Vaccine Immunology (CHAVI) (U19-AI067854); CHAVI Funding; R. Ottman; V. Shashi; E. Holtzman; S. Berkovic, I. Scheffer, and B. Grinton; The Epi4K Consortium and Epilepsy Phenome/Genome Project; C. Depondt, S. Sisodiya, G. Cavalleri, and N. Delanty; The ALS Sequencing Consortium (see above); The Washington University Neuromuscular Genetics Project; C. Woods, C. Village, K. Schmader, S. McDonald, M. Yanamadala, and H. White; G. Nestadt, J. Samuels, and Y. Wang; S. Schuman and E. Nading; D. Marchuk; D. Levy; E. Pras, D. Lancet, and Z. Farfel; Y. Jiang; T. Young and K. Whisenhunt; J. Milner; C. Moylan, A. Mae Diehl, and M. Abdelmalek; DUHS (Duke University Health System) Nonalcoholic Fatty Liver Disease Research Database and Specimen Repository; M. Winn and R. Gbadegesin; M. Hauser; S. Delaney; A. Need and J. McEvoy; A. Holden and E. Behr; M. Walker; M. Sum; Undiagnosed Diseases Network; National Institute on Aging (R01AG037212, P01AG007232).
The collection of control samples and data was funded in part by: Bryan ADRC NIA P30 AG028377; NIH RO1 HD048805; Gilead Sciences, Inc.; D. Murdock; National Institute of Allergy and Infectious Diseases Center for HIV/AIDS Vaccine Immunology (CHAVI) (U19-AI067854); Bill and Melinda Gates Foundation; NINDS Award# RC2NS070344; New York-Presbyterian Hospital; The Columbia University College of Physicians and Surgeons; The Columbia University Medical Center; NIH U54 NS078059; NIH P01 HD080642; The J. Willard and Alice S. Marriott Foundation; The Muscular Dystrophy Association; The Nicholas Nunno Foundation; The JDM Fund for Mitochondrial Research; The Arturo Estopinan TK2 Research Fund; UCB; Epi4K Gene Discovery in Epilepsy study (NINDS U01-NS077303) and The Epilepsy Genome/Phenome Project (EPGP - NINDS U01-NS053998); Biogen; The Ellison Medical Foundation New Scholar award AG-NS-0441-08; B57 SAIC-Fredrick Inc. M11-074; 1R01MH097971-01A1. This research was supported in part by funding from The Division of Intramural Research, NIAID, NIH; Funding from the Duke Chancellor's Discovery Program Research Fund 2014; an American Academy of Child and Adolescent Psychiatry (AACAP) Pilot Research Award; NIMH Grant RC2MH089915; Endocrine Fellows Foundation Grant; The NIH Clinical and Translational Science Award Program (UL1TR000040); NIH U01HG007672; The Washington Heights Inwood Columbia Aging Project; and The Stanley Institute for Cognitive Genomics at Cold Spring Harbor Laboratory. Data collection and sharing for the WHICAP project (used as controls in this analysis) was supported by The Washington Heights–Inwood Columbia Aging Project (WHICAP, PO1AG07232, R01AG037212, RF1AG054023) funded by the National Institute on Aging (NIA) and by The National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873. This manuscript has been reviewed by WHICAP investigators for scientific content and consistency of data interpretation with previous WHICAP Study publications. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
S.D. was funded by the Clinical and Translational Science Awards TL1 Training Award (5TL1TR001875-03); S.P. was funded by R.D. Wright Career Development Fellowship (National Health and Medical Research Council, 1126877); M.B.H. was funded by the Eleanor and Lou Gehrig ALS Center at Columbia University research fund, the ALS Association, Greater New York Chapter of the ALS Association, and Biogen Idec. Work in the Center for Genomics of Neurodegenerative Disease is supported by The ALS Association (grant number 15-LGCA-234) and The Tow Foundation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.243592.118.
References
- Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, Berg-Alonso L, Kageyama Y, Serre V, Moore DG, et al. 2014. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137: 2329–2345. 10.1093/brain/awu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher A, Eiseler T, Porzner M, Walther P, Keil R, Bobrovich S, Hatzfeld M, Seufferlein T. 2017. The armadillo protein p0071 controls KIF3 motor transport. J Cell Sci 130: 3374–3387. 10.1242/jcs.200170 [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6: 80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, et al. 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347: 1436–1441. 10.1126/science.aaa3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, et al. 2014. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature 505: 550–554. 10.1038/nature12825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R, Stitziel NO, Won HH, Jørgensen AB, Duga S, Angelica Merlini P, Kiezun A, Farrall M, Goel A, Zuk O, et al. 2015. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 518: 102–106. 10.1038/nature13917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K consortium; Epilepsy Phenome/Genome Project. 2017. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol 16: 135–143. 10.1016/S1474-4422(16)30359-3 [DOI] [PubMed] [Google Scholar]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44: D279–D285. 10.1093/nar/gkv1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussow AB, Petrovski S, Wang Q, Allen AS, Goldstein DB. 2016. The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biol 17: 9 10.1186/s13059-016-0869-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ, et al. 2014. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17: 664–666. 10.1038/nn.3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G, Flickinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, Kang HM. 2012. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am J Hum Genet 91: 839–848. 10.1016/j.ajhg.2012.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Kurita K, Seino T, Kadoya T, Horie H, Wada M, Kawanami T, Daimon M, Hirano A. 2001. Galectin-1 is a component of neurofilamentous lesions in sporadic and familial amyotrophic lateral sclerosis. Biochem Biophys Res Commun 282: 166–172. 10.1006/bbrc.2001.4556 [DOI] [PubMed] [Google Scholar]
- Keil R, Hatzfeld M. 2014. The armadillo protein p0071 is involved in Rab11-dependent recycling. J Cell Sci 127: 60–71. 10.1242/jcs.132266 [DOI] [PubMed] [Google Scholar]
- Keil R, Schulz J, Hatzfeld M. 2013. p0071/PKP4, a multifunctional protein coordinating cell adhesion with cytoskeletal organization. Biol Chem 394: 1005–1017. 10.1515/hsz-2013-0114 [DOI] [PubMed] [Google Scholar]
- Kenna KP, van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, van Rheenen W, van Eijk KR, Jones AR, Keagle P, et al. 2016. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet 48: 1037–1042. 10.1038/ng.3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayakawa Y, Sakumi K, Kajitani K, Kadoya T, Horie H, Kira J-I, Nakabeppu Y. 2015. Galectin-1 deficiency improves axonal swelling of motor neurones in SOD1G93A transgenic mice. Neuropathol Appl Neurobiol 41: 227–244. 10.1111/nan.12123 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman BJ, Hoffman EP, Sutherland ML, Bouri K, Hsu DK, Liu F-T, Rothstein JD, Knoblach SM. 2012. Deletion of galectin-3 exacerbates microglial activation and accelerates disease progression and demise in a SOD1G93A mouse model of amyotrophic lateral sclerosis. Brain Behav 2: 563–575. 10.1002/brb3.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, Neumann M. 2010. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9: 995–1007. 10.1016/S1474-4422(10)70195-2 [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, et al. 2017. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95: 808–816.e9. 10.1016/j.neuron.2017.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. 2010. Robust relationship inference in genome-wide association studies. Bioinformatics 26: 2867–2873. 10.1093/bioinformatics/btq559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, et al. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41: D348–D352. 10.1093/nar/gks1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle P, et al. 2018. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97: 1268–1283.e6. 10.1016/j.neuron.2018.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesiridis GS, Lee VM, Trojanowski JQ. 2009. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 18: R156–R162. 10.1093/hmg/ddp303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Todd JL, Durheim MT, Wang Q, Chien JW, Kelly FL, Frankel C, Mebane CM, Ren Z, Bridgers J, et al. 2017. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am J Respir Crit Care Med 196: 82–93. 10.1164/rccm.201610-2088OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909. 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- Renton AE, Chiò A, Traynor BJ. 2014. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17: 17–23. 10.1038/nn.3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogelj B, Godin KS, Shaw CE, Ule J. 2011. The functions of glycine-rich regions in TDP-43, FUS and related RNA-binding proteins. In RNA binding proteins (ed. Lorkovic Z), pp. 1–17. Springer, New York. [Google Scholar]
- Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, Scotter EL, Kost J, Keagle P, Miller JW, et al. 2014. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84: 324–331. 10.1016/j.neuron.2014.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BN, Topp SD, Fallini C, Shibata H, Chen HJ, Troakes C, King A, Ticozzi N, Kenna KP, Soragia-Gkazi A, et al. 2017. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci Transl Med 9: eaad9157 10.1126/scitranslmed.aad9157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis J, Silk M, Wang Q, Berkovic SF, Liu L, Ascher DB, Balding DJ, Petrovski S. 2017. Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome Res 27: 1715–1729. 10.1101/gr.226589.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, Zhang KY, Farrawell N, Vance C, Hu X, et al. 2016. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun 7: 11253 10.1038/ncomms11253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Zhang H, Loiselle D, Haystead T, Macara IG, Mili S. 2013. The RNA-binding protein Fus directs translation of localized mRNAs in APC-RNP granules. J Cell Biol 203: 737–746. 10.1083/jcb.201306058 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.