

Summary
Metabolic diseases are often characterized by circadian misalignment in different tissues, yet how altered coordination and communication among tissue clocks relates to specific pathogenic mechanisms remains largely unknown. Applying an integrated systems biology approach, we performed 24-h metabolomics profiling of 8 mouse tissues simultaneously. We present a temporal and spatial atlas of circadian metabolism in the context of systemic energy balance, and under chronic nutrient stress (high fat diet, HFD). Comparative analysis reveals how the repertoires of tissue metabolism are linked and gated to specific temporal windows, and how this highly specialized communication and coherence among tissue clocks is rewired by nutrient challenge. Overall, we illustrate how dynamic metabolic relationships can be reconstructed across time and space, and how integration of circadian metabolomics data from multiple tissues can improve our understanding of health and disease.
Graphical Abstract
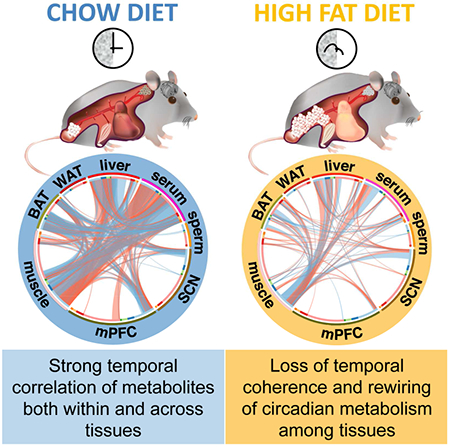
ETOC
Circadian metabolite profiles of 8 different tissues under standard and high fat diet uncovers highly specialized communication and coherence among tissue clocks and how this is rewired by nutrient challenge.
Introduction
Circadian clocks allow cells and tissues to compartmentalize metabolic pathways within precise temporal and spatial windows (Tu and McKnight, 2006). The importance of clock function in determining health is supported by animal studies and a wealth of epidemiological evidence (Lowrey and Takahashi, 2011). Specific metabolic pathways are key regulators of circadian clocks (Rey et al., 2016; Zwighaft et al., 2015) and oscillating metabolites can synchronize clocks, modulate transcription, and regulate chromatin accessibility (Aviram et al., 2016; Nakahata et al., 2009; O’Neill et al., 2008; O’Neill et al., 2011; Peek et al., 2013; Ramsey et al., 2009). Clocks across tissues are aligned under conditions of energy balance, while disruption of tissue communication is thought to increase risk of metabolic diseases (Roenneberg et al., 2012; Roenneberg and Merrow, 2016). Metabolism and circadian clocks are thus inextricably linked (Asher and Sassone-Corsi, 2015).
Metabolites reside at this important interface between circadian clocks and metabolism (Dyar and Eckel-Mahan, 2017) and are used as surrogate diagnostic and predictive biomarkers to define health or pathology (DeBerardinis and Thompson, 2012). Blood metabolite profiling is common but intrinsically limited, and few studies have combined metabolomics data from more than one tissue (Lu et al., 2017; Sugimoto et al., 2012). Providing essential temporal perspective, several groups have begun mapping metabolite dynamics over 24-h (Aviram et al., 2016; Chen et al., 2016; Dallmann et al., 2012; Davies et al., 2014; Dyar et al., 2014; Eckel-Mahan et al., 2012; Fustin et al., 2012; Giskeodegard et al., 2015; Kim et al., 2014; Krishnaiah et al., 2017; Martinez-Lozano Sinues et al., 2014; Masri et al., 2014). Integration of 24-h metabolomics data from multiple tissues is thus poised to push metabolite profiling beyond simple diagnostics and biomarker detection (Brown, 2016; Goodacre, 2007) and towards actionable knowledge for preventative medicine and better predictions of how metabolite profiles can be controlled to affect a desired metabolic outcome (German et al., 2005; Gooley and Chua, 2014).
Here we present global 24-h metabolite profiles of 8 different tissues under conditions of energy balance (standard chow) or chronic nutrient stress (HFD). We reveal how metabolic pathways are gated to specific temporal and spatial windows, and how novel tissue-specific and inter-organ 24-hr dynamics emerge under HFD. Our resource highlights temporal links between muscle insulin resistance, hepatic steatosis, atherosclerosis, and reduced energy expenditure.
Results
Temporal and Tissue-Specific Metabolite Profiling
We performed 24-h metabolomics profiling on 8 different murine tissues: suprachiasmatic nucleus (SCN), medial prefrontal cortex (mPFC), gastrocnemius skeletal muscle, interscapular brown adipose tissue (BAT), epididymal white adipose tissue (WAT), liver, serum, and cauda epididymal sperm (Figure 1A). Tissues were collected every 4-h across the light/dark cycle from a single cohort of C57BL/6J mice after 10 weeks of ad libitum access to standard chow or HFD. Mice fed HFD had rhythmic energy intake indistinguishable from chow-fed mice (Eckel-Mahan et al., 2013). Hundreds of metabolites covering a range of metabolite classes were detected in each tissue by mass spectrometry (LC/MS & GC/MS) (Figures 1B and 1C). All data are available on CircadiOmics (http://circadiomics.igb.uci.edu/) (Patel et al., 2012), including interactive networks to explore the data.
Figure 1. Global metabolite profiling of mouse tissues over 24-hours reveals common and tissue-specific metabolic signatures on chow and HFD.
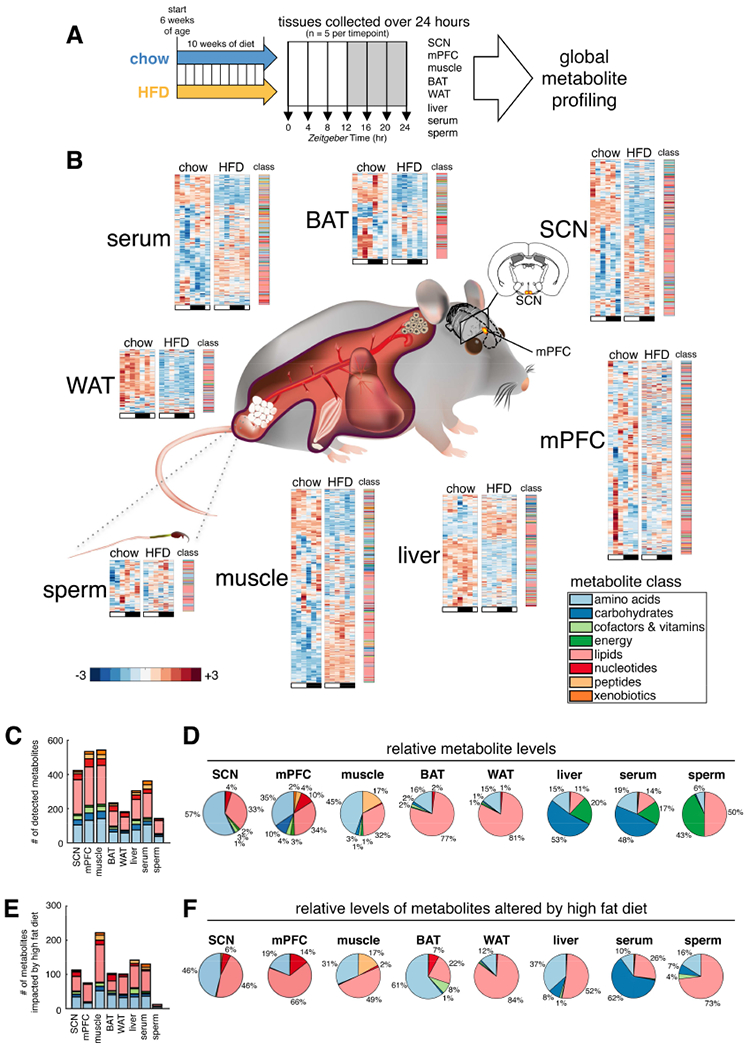
(A) Experimental design. Suprachiasmatic nucleus (SCN), medial prefrontal cortex (mPFC), gastrocnemius skeletal muscle, interscapular brown adipose tissue (BAT), epididymal white adipose tissue (WAT), liver, serum, and cauda epididymal sperm collected every four hours across the light/dark cycle from a single cohort of C57BL/6J mice. 6 time points were profiled for SCN, mPFC, muscle and sperm; a 7th replicate time point, ZT24, was additionally profiled for serum, liver, BAT and WAT.
(B) Tissue-specific metabolite heatmaps. Rows reflect normalized (z-score) metabolite abundance across the light/dark cycle (white bar=ZT0, 4, 8 & 24; black bar=ZT12, 16 & 20). Metabolite class is indicated at the right of each heatmap.
(C) Counts and class of detected metabolites for each tissue.
(D) Tissue metabolite class composition according to relative metabolite masses (sum of standardized abundances).
(E) Counts and class of metabolites significantly impacted by diet (diet effect p<0.05, linear regression model).
(F) HFD-altered metabolite class composition for each tissue. (relative metabolite masses affected by HFD).
Tissue-specific differences in metabolite number, temporal oscillations, and impact of HFD were apparent (Figure 1B). While class distribution was similar across tissues (Figure 1C), relative metabolite abundance reflected tissue composition (Figure 1D): WAT and BAT revealed mostly lipids; brain and muscle showed amino acids and lipids; liver and serum mostly carbohydrates and comparable amounts of amino acids, lipids, and energy metabolites; sperm contained mostly lipids and energy metabolites.
The impact of HFD varied among tissues (Figures 1C,E). While ~540 total metabolites were detected in both mPFC and muscle, >3-fold more metabolites were altered by HFD in muscle than in mPFC (232 significantly impacted in muscle versus 75 in mPFC). Overall, WAT, liver, BAT, muscle and serum were most impacted by HFD, with 40%-60% of detected metabolites showing significant alterations (Table 1). Brain and sperm metabolites appeared more resistant to diet-induced metabolite alterations, with SCN, mPFC and sperm showing alterations in only 27%, 14%, and 8% of metabolites, respectively.
Table 1.
Percent metabolites impacted by HFD (diet effect p<0.05, linear regression model)
| tissue | % |
|---|---|
| WAT | 63 |
| liver | 55 |
| BAT | 52 |
| muscle | 43 |
| serum | 41 |
| SCN | 27 |
| mPFC | 14 |
| sperm | 8 |
Subpathway enrichment analysis of significantly altered metabolites (Table S1) identified specific lipid classes altered by HFD, including long chain fatty acids, polyunsaturated fatty acids (PUFA), diacylglycerols, phospholipids, sphingolipids, glycerolipids, and lysolipids. Metabolites involved in amino acid metabolism, particularly the urea cycle metabolites arginine and proline, but also lysine, branched chain amino acids (BCAA) leucine, isoleucine and valine, and dipeptides were also highly impacted.
Altered abundance of metabolites suggested tissue pathology (Figure 1F). For example, carbohydrates comprised 53% of liver metabolites on chow, and made up only 8% altered by HFD, while lipids comprised only 11% on chow, yet 52% altered by HFD (Figure 1D), suggesting hepatic steatosis. Skeletal muscles on HFD exhibited a large accumulation of lipids known to negatively impact insulin sensitivity.
Tissue-specific Impact of HFD on 24-h Metabolism
Comparing circadian metabolites (i.e. oscillating with a 24-h period) revealed major differences in how 24-h metabolism is organized among different tissues. Serum, BAT, liver, skeletal muscle, and mPFC are all highly dynamic tissues (Figure 2), with 20%-50% of metabolites showing 24-h oscillation regardless of diet. While lipids comprised the largest class detected in each tissue (Figure 1C), the numbers of cycling lipids varied widely among tissues on both diets (Figure 2). For example, we detected more cycling lipids in BAT, mPFC and serum on chow compared to SCN, muscle, WAT and liver. Furthermore, BAT, mPFC and serum all lost lipid oscillation on HFD, while SCN surprisingly gained ~100 de novo oscillating lipids. We also noted a marked temporal gating of 24-h cycling lipids to the light phase in mPFC, BAT, and serum on chow, with peaks slightly different in each tissue. Lipids peaked around ZT6 in mPFC, ZT6-ZT8 in serum, and ZT8-ZT10 in BAT.
Figure 2. Comparative analysis of circadian metabolites highlights heterogeneity of tissue circadian function.
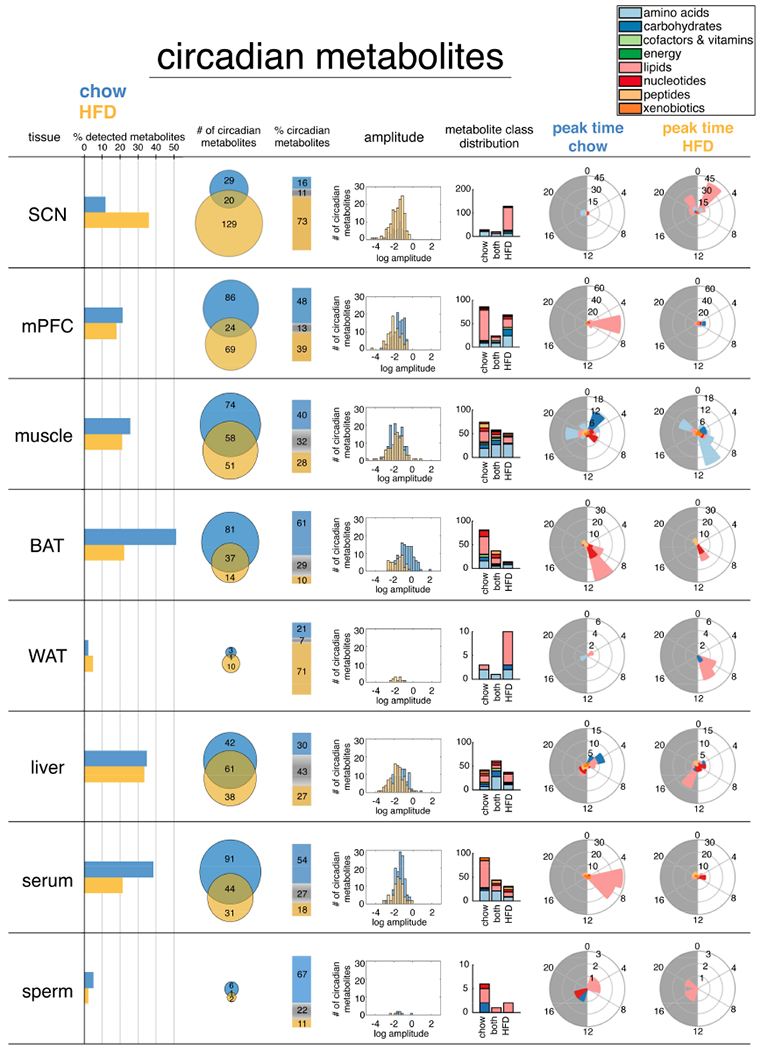
24-hr oscillating metabolites (p<0.05, JTK_CYCLE) for each tissue are plotted from left to right according to 1) relative % of detected metabolites, 2) absolute numbers 3) % distribution, 4) amplitude of oscillation, 5) class distribution, and 6&7) phase distribution.
Skeletal muscle showed a more diverse and temporally compartmentalized distribution of 24-h cycling metabolite peaks on chow. Carbohydrates peaked around ZT2, whereas nucleotides peaked around ZT8, and amino acids around ZT18. Amino acid peaks were drastically altered on HFD, shifting to around ZT8-ZT10 with a secondary peak at ZT20, highlighting a massive reorganization of muscle amino acid metabolism on HFD. Disease-associated metabolite enrichment (Xia and Wishart, 2016) of these revealed an association with “degradation of skeletal muscle”, highlighting a potential pathologic underpinning. Indeed, short-term HFD in mice accelerates skeletal muscle atrophy of the soleus upon denervation (Roseno et al., 2015), while 16-weeks of chronic HFD in rats causes gastrocnemius atrophy and upregulation of ubiquitin ligase Murf1 (Sishi et al., 2011).
Cauda epidydimal sperm (Figures S1A and S1B) displayed relatively few oscillating metabolites (Figure 2). However, glycolytic intermediates 2- and 3-phosphoglycerate oscillated only on chow (Figure S1C), while more than 40% of all lysolipids were significantly reduced during the dark phase on HFD (Figure S1D).
Circadian metabolite subpathway enrichment revealed common and tissue-specific metabolite classes (Table S2). Independent of diet, serum and muscle maintained robust 24-h oscillation of BCAA metabolites, while on HFD, both serum and muscle gained additional circadian enrichment of metabolites involved in glycine, serine and threonine metabolism. Muscle further gained 24-h oscillation of metabolites related to alanine and aspartate metabolism on HFD, while BAT gained oscillation of guanine-containing purine nucleotides.
Metabolite Correlations Reveal Tissue-specific Temporal Gating of Metabolic Pathways
Coherent temporal gating of metabolic pathways is essential for maintenance of tissue homeostasis (Noguchi et al., 2013). We hypothesized that metabolites sharing positive temporal correlation may have common origins or functions, and belong to related metabolic networks, whereas metabolites with negative correlation may reflect temporal gating of incompatible pathways. Furthermore, temporal correlation of metabolites can serve as a general index for synchronization of individual cells within a tissue, since dynamic pathways should be highly correlated among synchronized cells. We found extensive positive and negative temporal correlations among metabolites in all tissues (Figure 3A,B), and interestingly, many tissues lost metabolite correlations on HFD. Serum and BAT lost 98% and 74%, respectively, whereas liver and muscle lost only 34% and 39%, respectively. This suggests maintenance and/or reorganization of metabolic pathways in liver and muscle. On the other hand, the severe loss of intra-tissue metabolite correlations in serum has major implications for how misalignment among tissues may be exacerbated by nutritional challenge.
Figure 3. Tissue-specific metabolite correlations illustrate how temporal coherence and normal gating of metabolic pathways is maintained or altered by HFD.
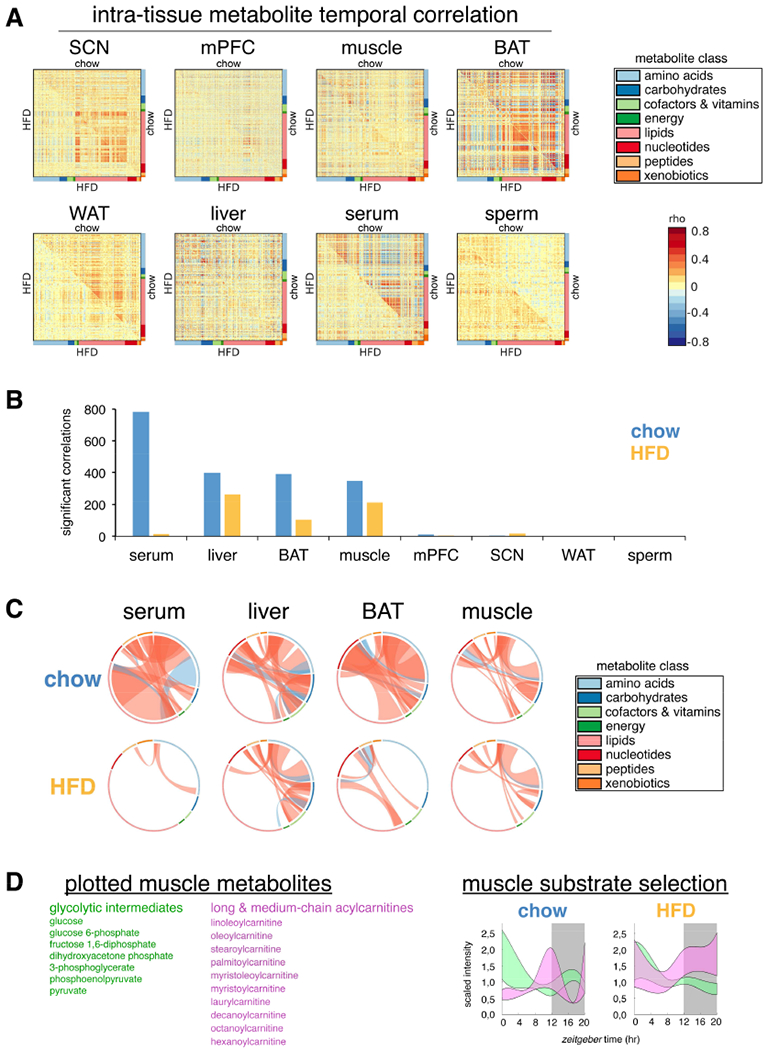
(A) Correlation heatmaps for metabolites in each tissue. Correlation coefficient rho is shown as red (positive) or blue (negative) as indicated.
(B) Number of significant metabolite correlations according to tissue and diet.
(C) Graphical visualization of significant metabolite correlations. Each ribbon indicates a significant metabolite correlation (positive=red, negative=blue) between or within each metabolite class. Ribbon thickness refers to number of significantly correlated metabolites. Metabolites were ordered according to metabolite class as indicated in colored bar around the circumference.
(D) “Fuzzy plots” show the range (colored area) between minimum and maximum abundance for members of each metabolic pathway. Cubic-splines interpolation estimated continuous abundance.
We next visualized significant positive and negative temporal correlations according to metabolite class (Figure 3C). Serum lipids showed the greatest degree of temporal correlation with other metabolites, especially amino acids. Importantly, these were severely reduced on HFD, with serum amino acids and peptides gaining correlations. Major positive correlations among BAT nucleotides, lipids and amino acids were lost on HFD. While muscle and liver similarly lost temporal correlation among metabolite classes, they also maintained substantial metabolite correlations on HFD, particularly among amino acids and carbohydrates.
Subpathway enrichment analysis of significantly correlated metabolites revealed maintenance of carbohydrate correlations on HFD, with muscle glycolytic intermediates and liver sugar metabolites retaining temporal correlation (Table S3), in agreement with enrichment analysis (Table S2). However, serum lost correlation among essential fatty acids on HFD, in addition to several medium chain fatty acids, and lysolipids, while muscle lost correlation among acylcarnitines, as well as TCA cycle metabolites. Furthermore, both serum and muscle gained correlations among BCAA and other amino acids on HFD, suggesting a coordinated response.
Using temporal associations, one can reconstruct metabolic pathways over 24-h. For example, muscle glycolytic intermediates and acylcarnitines respectively reflect cytosolic or mitochondrial ATP production. Both metabolite classes showed dynamic oscillation over 24-h (Figure 3D). On chow, peaks were confined to distinct temporal windows, reflecting delineation of diurnal fuel preference. Glycolytic intermediates declined across the light phase, whereas acylcarnitines increased towards the end. Acylcarnitines were lowest in the middle of the dark phase when glycolytic intermediates were increased. However, on HFD oscillation of glycolytic intermediates was blunted during the dark phase, while acylcarnitines remained at peak levels. These coordinated metabolite alterations imply impaired metabolic flexibility and may contribute to insulin resistance.
Inter-tissue Clock Communication: Metabolite Coherence or Dissonance
Visualization of inter-tissue metabolite temporal correlations revealed metabolic coupling between tissues (Figure 4A). Importantly, serum emerged as a main source of metabolite correlations on chow, underscoring the role of circulating factors in temporal communication, as serum metabolites both direct and reflect tissue-specific metabolism. HFD was associated with a severe loss of temporal coherence (Figure 4B), specifically between serum and muscle, liver, and BAT (Figures 4C). Comparative inspection of cross-tissue correlations between serum, liver, muscle, and BAT further emphasized HFD-induced loss and reorganization of metabolite correlations (Figure 5). Normally abundant liver-serum and muscle-serum metabolite correlations were reduced ~60% on HFD (Figure S2A), while BAT-serum correlations were reduced 74%. Lipids and amino acids were the main metabolite classes correlated between serum and other tissues on chow, comprising ~60-70% of all significant inter-tissue metabolite correlations. Interestingly, BAT and serum nucleotides showed a relatively high degree of temporal correlation. Importantly, most cross-tissue metabolite correlations were lost on HFD (Figure S2B), with inter-tissue lipids losing ~80% in all tissues, and amino acids losing 35%-66%, depending on the tissue (Figure S2C). A majority of HFD inter-tissue metabolite correlations were not shared on chow. These de novo correlations were especially prevalent among muscle, serum, and liver amino acids, again highlighting coordinated metabolic responses linking these tissues.
Figure 4. Loss of inter-tissue metabolite correlations on HFD.
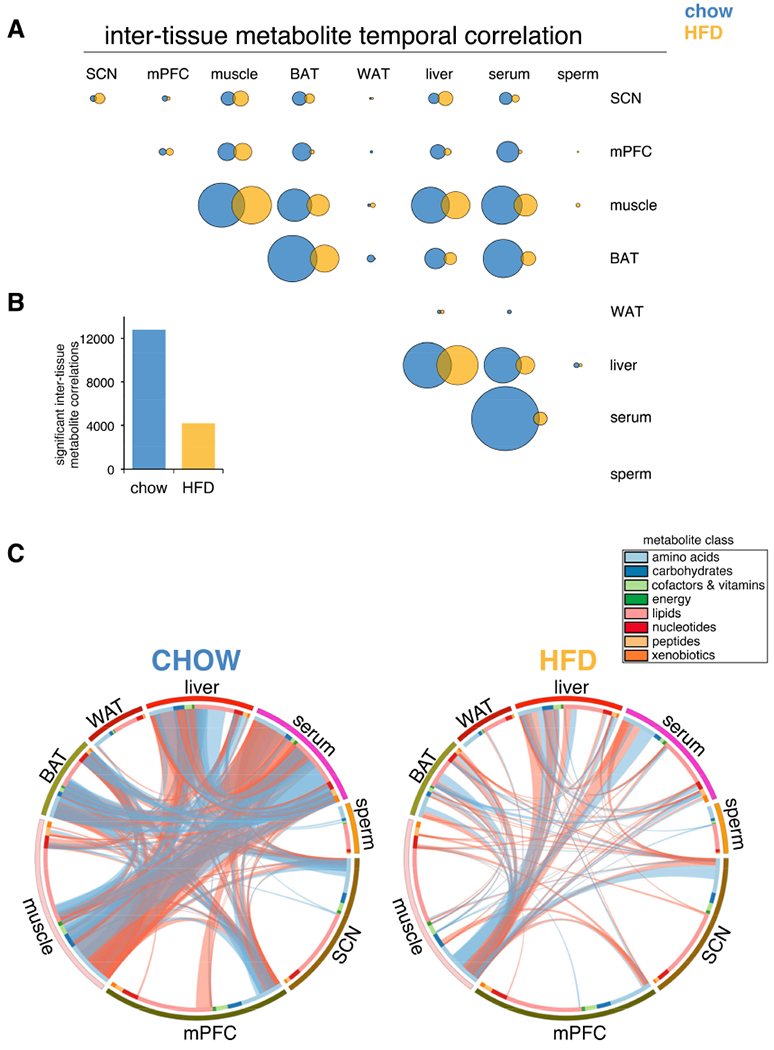
(A) Correlated metabolites between each tissue. Circle areas sized according to number of significant metabolite correlations.
(B) Significant inter-tissue metabolite correlations.
(C) Graphical summary of all inter-tissue metabolite temporal correlations. Each ribbon indicates a significant intertissue metabolite correlation (positive=red, negative=blue). Ribbon thickness refers to number of significantly correlated metabolites.
Figure 5. Serum, liver, muscle and BAT cross-tissue metabolite correlations severely altered by HFD.
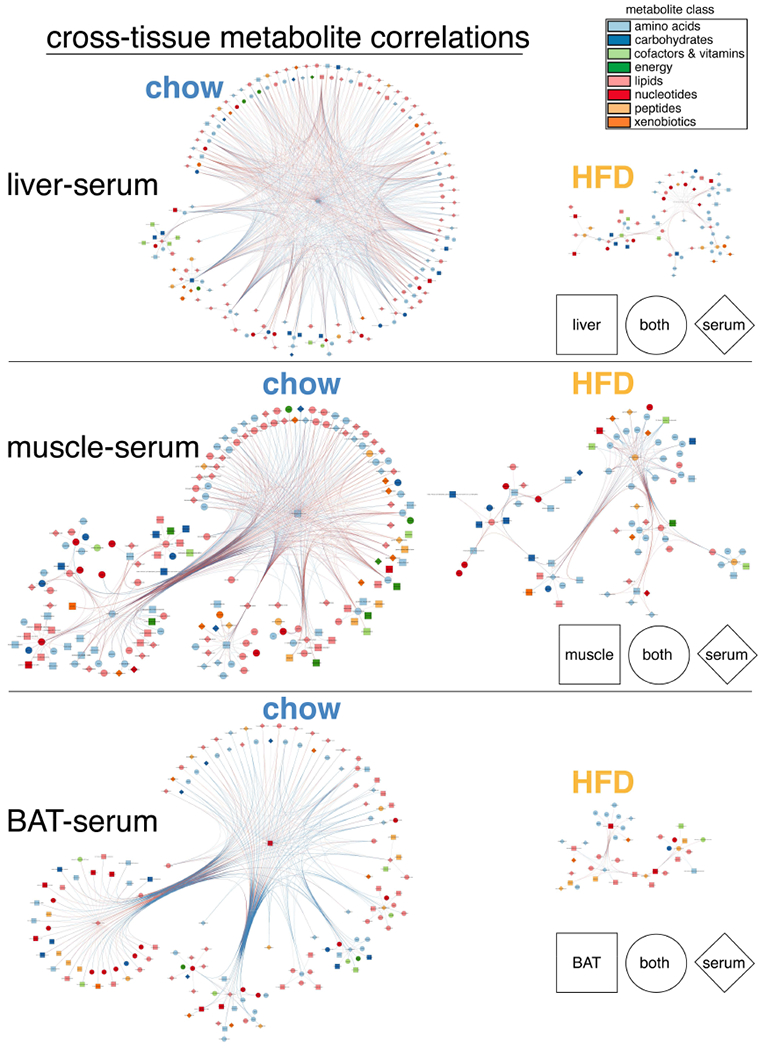
Networks of significantly correlated metabolites detected on chow or HFD. Each node refers to a single metabolite. Node shape indicates tissue or tissues showing correlation. Node color refers to metabolite class. Edges are drawn for each significant inter-tissue correlation, and edge colors refer to the sign of correlation coefficient (red=positive, blue=negative).
Increased Muscle Protein Turnover on HFD Linked to Increased Liver Gluco- and Glyceroneogenesis
Our data revealed significant HFD-specific metabolite correlations and de novo oscillations characteristic of increased muscle protein degradation, and linked to liver gluco- and glyceroneogenesis. We noted diurnal rhythms of glutamine and alanine in muscle and liver were gated to distinct temporal windows on chow, oscillating anti-phase to one another (Figure 6A). However, circulating alanine remained relatively stable across the circadian cycle in chow-fed mice, yet on HFD became highly oscillatory and increased during the light phase. This effect was even more pronounced in skeletal muscle on HFD, with alanine levels constitutively elevated across the light phase. There were no major HFD-induced changes in alanine levels in liver or WAT, whereas BAT showed slightly reduced levels.
Figure 6. Increased circadian oscillation of muscle and serum metabolites linked to increased liver precursors for gluco- and glyceroneogenesis.
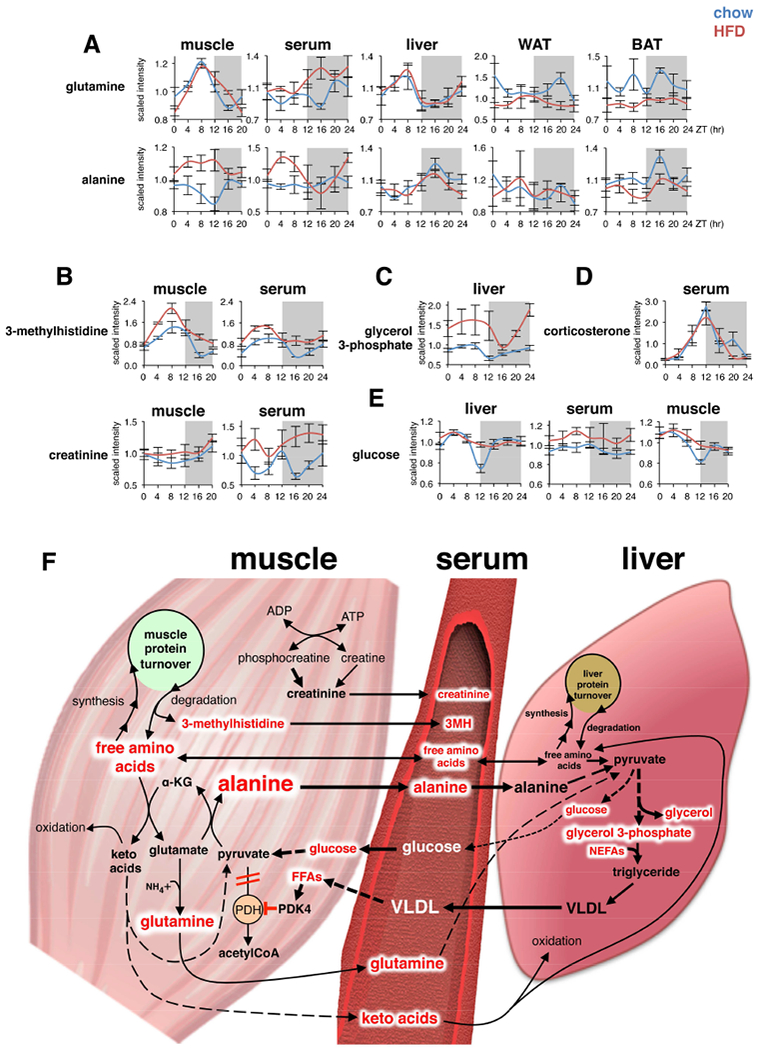
(A-E) Diurnal variations of selected metabolites in mouse tissues on chow or HFD (mean±SEM; n=5×group×time point).
(F) Simplified scheme showing interrelationships between HFD-induced metabolite alterations detected muscle, serum, and liver. Red text indicates metabolites significantly increased by HFD.
Increased alanine production and release by skeletal muscle normally occurs in response to increased protein catabolism during periods of starvation (Felig, 1973). The rate of muscle protein degradation is often clinically inferred by levels of 3-methylhistidine, a unique amino acid released by myofibrillar proteolysis and excreted in the urine (Elia et al., 1981). We detected 3-methylhistidine only in serum and skeletal muscle, with both tissues showing 24-h oscillation on chow and HFD (Figure 6B). However, peak 3-methylhistidine was significantly increased on HFD, suggesting increased muscle protein degradation. We also noted increased creatinine in serum on HFD, and a similar trend in muscle, indicating that muscle mass is maintained or even slightly increased despite protein breakdown, and suggests increased overall rates of muscle protein turnover on HFD.
The majority of alanine released from muscle is synthesized de novo by transamination of pyruvate, with nitrogen originating mostly from BCAA via glutamate (Odessey and Goldberg, 1979). Serum, muscle and liver BCAA showed 24-h oscillation on chow and HFD, with increased levels during the dark phase, and reduced levels during the light phase (Figure S3A). Serum and muscle from HFD mice also showed increased BCAA levels at various time points compared to chow mice, whereas BCAA levels in liver, WAT, and BAT were all significantly reduced on HFD. While most amino acids are transaminated in the liver, BCAA are transaminated mainly in skeletal muscle, resulting in formation of branched-chain keto acids (BCKA) α-ketoisocaproate, α-ketomethylvalerate, and α-ketoisovalerate. All three BCKAs were drastically reduced and lost 24-h oscillation in muscle on HFD (Figure S3B), yet serum BCKA levels were significantly increased, suggesting augmented export from muscle and/or reduced uptake from other tissues.
Another striking effect of HFD was the specific increase of glucogenic amino acids in muscle and serum, with reduction in liver, WAT and BAT (Figure S3A), possibly reflecting increased rates of gluco- and glyceroneogenesis in liver (Song et al., 2001) and reduced amino acid uptake and metabolism in WAT and BAT (Serra et al., 1994). Moreover, glycerol 3-phosphate, a major precursor for liver glyceride-glycerol and VLDL synthesis (Nye et al., 2008), was increased ~50% across the day/night cycle on HFD (Figure 6C), and oscillated in phase with muscle and serum alanine, indicating increased conversion of serum alanine (and other glucogenic amino acid substrates resulting from muscle protein degradation) to liver glyceride-glycerol (Nye et al., 2008).
Muscle protein degradation and liver gluco- and glyceroneogenesis are under tight hormonal control, mainly by opposing actions of circulating glucocorticoids and insulin (Hatting et al., 2018; Schiaffino et al., 2013; Shimizu et al., 2015). The temporally-linked rise and fall of muscle and serum 3-methylhistidine on chow largely reflected 24-h oscillation of endogenous serum corticosterone (Figure 6D) and insulin (Jouffe et al., 2016). Interestingly, serum corticosterone levels were not increased and oscillated normally on HFD. This is intriguing, considering associations between chronic HFD and insulin resistance of skeletal muscle and liver (Oakes et al., 1997) largely mirror metabolic complications associated with chronic glucocorticoid administration, including impaired insulin-dependent glucose uptake and metabolism by muscle, and reduced insulin-mediated suppression of hepatic glucose production leading to hyperglycemia (Dunford and Riddell, 2016). Our results indicate that chronic HFD may locally enhance sensitivity to the effects of endogenous glucocorticoids. Indeed, serum glucose remained constitutively elevated on HFD, while oscillation of glucose in liver and muscle in chow-fed mice was blunted and likewise elevated, but only at ZT12 (Fig. 6E), corresponding to the peak of endogenous glucocorticoids.
The proposed pathogenic relationships between muscle, serum, and liver metabolites increased on HFD are summarized in Figure 6F. Increased cycles of muscle protein turnover lead to increased production and release of alanine and other amino acids destined for uptake mostly by the liver. The importance of serum alanine is supported by arteriovenous differences in rat and human showing preferential hepatic uptake of alanine above all other amino acids (Pozefsky et al., 1969). Glutamine is the only amino acid released by muscle at higher rates than alanine, and it is destined mostly for kidneys and small intestine, which in turn release alanine (Snell and Duff, 1979). Excess amino acids not utilized for liver protein synthesis may be metabolized and converted to glucose and glyceride-glycerol, contributing to elevated glycemia, increased production of hepatic triacylglycerol (TAG) and secretion of VLDL. Hypertriglyceridemia and increased VLDL secretion are associated with metabolic diseases linked to poor diet, lack of exercise, and obesity, including insulin resistance and cardiovascular disease (Eckel et al., 2005). Increased muscle FFA may activate pyruvate dehydrogenase kinase 4 (PDK4), inhibiting pyruvate dehydrogenase, further promoting alanine production (Zhang et al., 2014). Our data suggest that muscle protein turnover, and particularly muscle alanine production and release, may play a previously underappreciated role in metabolic diseases.
De novo Oscillation of Purine Nucleotides in BAT on HFD Linked to Impaired UCP1 Activity
Tissue-specific changes in glycerol and non-esterified fatty acids (NEFA) reflect continuous turnover and transport of lipids among tissues in a state of so-called “dynamic equilibrium” (Guggenheim, 1991), in which total body fat content remains constant in the midst of high rates of degradation, inter-conversion and re-esterification of lipid stores within and across tissues over 24-h. Interestingly, HFD blunted oscillation of circulating NEFAs, increased levels of serum glycerol, and severely reduced NEFA levels and oscillation in WAT and BAT (Figure 7A), suggesting an impaired diurnal turnover of lipid stores in both fat depots. In contrast, liver glycerol and NEFAs were dramatically increased on HFD, similar to skeletal muscle, highlighting known pathological deposition of biologically active lipids associated with obesity and insulin resistance. The massive reduction in circadian BAT NEFAs is of interest, since they directly stimulate thermogenic proton leak via activation of uncoupling protein 1 (UCP1) (Nicholls and Locke, 1984).
Figure 7. Loss of circadian lipid oscillation in BAT linked to reduced purine catabolism, de novo oscillation of purine nucleotides, and impaired UCP1 activation.
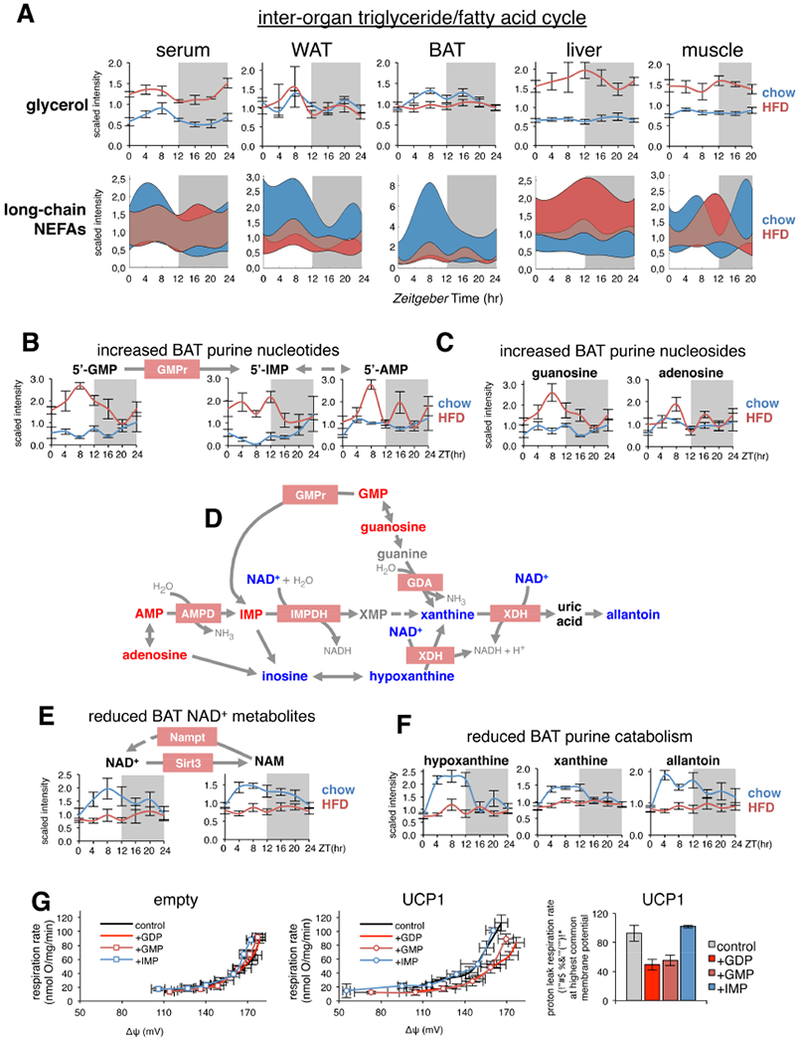
(A) Representative diurnal variations of substrates from the inter-organ triglyceride/fatty acid cycle in relevant tissues (mean±SEM; n=5×group×time point). “Fuzzy plots” of all detected medium and long-chain nonesterified fatty acids (NEFAs), indicate the range (colored area) between minimum and maximum abundance for all medium and long-chained NEFAs.
(B&C) Diurnal variations of BAT purine nucleotides and nucleosides (mean±SEM; n=5 × group × time point). GMP reductase (GMPr), converts GMP to IMP.
(D) Scheme showing how metabolites of BAT purine catabolism are interrelated and altered on HFD. Metabolites significantly increased by HFD are indicated by red text, while significantly reduced metabolites are blue. Black metabolites are unchanged, and grey metabolites were not measured. Enzymes are shown as pink boxes, and are all coded by circadian genes in BAT.
(E) Diurnal variations of NAD+ metabolites (mean±SEM; n=5×group×time point). Nampt and Sirt3 are coded by circadian genes in BAT.
(F) Diurnal variations of selected purine catabolism metabolites in BAT (mean±SEM; n=5×group×time point).
(G) Respiration driving proton leak measured in isolated mitochondria from control HEK293 cells (empty, left panel) or HEK293 cells ectopically expressing mouse UCP1 (middle panel) in the presence of selected purine nucleotides. Note that the upward shift in leak kinetics in the presence of UCP1 is prevented to a similar extent by GDP and GMP addition. Right panel shows data plotted at the highest common membrane potential.
In absence of activating fatty acids, extra-mitochondrial purine nucleotides restore energy conservation in BAT mitochondria by directly binding and inhibiting UCP1 (Shabalina et al., 2004). Guanine nucleotides show higher recoupling efficiency than adenine nucleotides due to their higher affinity for UCP1 and higher concentration in the mitochondrial intermembrane space (Jastroch et al., 2010). While we did not detect GDP, we noted that both 5’-GMP and its precursor guanosine gained de novo oscillation on HFD and showed a massive increase during the light phase, peaking at ZT8 (Figures 7B,C). BAT contains high levels of GMP reductase (GMPr), which catalyzes the reductive deamination of guanine nucleotides to inosine monophosphate (IMP), thus facilitating the activation of UCP1 by releasing endogenous inhibition by these nucleotides. IMP can be further converted to AMP, and dephosphorylated to form adenosine, an activator of brown adipocytes (Gnad et al., 2014). In agreement with increased 5’-GMP, IMP, AMP and adenosine were all increased in BAT from HFD mice, with a gradual reduction across the circadian cycle depending on their metabolic vicinity to GMP (Figures 7B,C).
Due to their important roles in metabolic regulation and signaling, intracellular concentrations of purine nucleotides are normally kept under tight control by continuous inter-conversion and catabolism (Bender, 2011). Degradation can follow alternate routes (Figure 7D), mostly relying on reduction of nicotinamide adenine dinucleotide (NAD+) to NADH, including conversion of IMP to xanthosine monophosphate (XMP) via IMP dehydrogenase (IMPDH), conversion of hypoxanthine to xanthine, and conversion of xanthine to uric acid via xanthine dehydrogenase (XDH). The XDH enzyme is also inter-convertible with xanthine oxidase (XO), which transfers electrons from xanthine to oxygen instead of NAD+, and produces superoxide anion and hydrogen peroxide rather than NADH (Nishino et al., 2008). Interestingly, similar to the liver (Eckel-Mahan et al., 2013), 24-h NAD+ oscillation was abolished in BAT on HFD, with NAD+ at constitutively low levels (Figure 7E), supporting a scenario of impaired BAT purine nucleotide catabolism on HFD.
NAD+ provides metabolic regulation in both mitochondrial and nuclear pools (Mori et al., 2005; Scher et al., 2007). Similarly, nicotinamide (NAM), produced from Sirt3 deacetylase activity, and precursor for NAD+ salvage via nicotinamide phosphoribosyltransferase (NAMPT) also lost oscillation on HFD (Figure 7E). Finally, 24-h oscillation of hypoxanthine, xanthine and allantoin were all severely blunted in BAT on HFD (Figure 7F), further suggesting reduced purine catabolism.
Sirt3 and Nampt are direct BMAL1 targets (Koike et al., 2012; Nakahata et al., 2009; Rey et al., 2011). Thus, HFD-induced attenuation of BAT clock function may directly impair Sirt3 and Nampt expression. Indeed, clock genes Bmal1 and Clock showed altered expression on HFD (Figure S4A). Also, Nampt and Sirt3 showed altered 24-h expression profiles (Figure S4B); however, Nampt showed modestly reduced expression during the light phase, while Sirt3 was significantly increased during the dark phase. Additional circadian genes in BAT, such as Gmpr (coding for GMP reductase), Impdh1 (coding for IMP dehydrogenase), and Ucp1, also showed drastic alterations in on HFD (Figure S4B and C). It is thus plausible that altered 24-h profiles of many BAT metabolites on HFD may reflect functional defects linked to local clock disruption/misalignment.
Accumulation of purine nucleotides in the absence of activating fatty acids results in UCP1 inhibition and reduced BAT-derived energy expenditure, but the significance of ~7-8-fold increased GMP levels in BAT on HFD (Figure 7B) is unclear due to its 50-fold lower binding affinity for UCP1 (Klingenberg and Huang, 1999). Endogenous levels of GMP and GDP in BAT are unknown, but 120-130uM GMP and 50uM GDP has been reported in mouse heart (McKee et al., 1999) and ~20-fold differences of GMP concentration in whole cell (~100uM) versus mitochondrial matrix (~4uM) of HeLa cells (Chen et al., 2016). To investigate the role of GMP and IMP per se in driving proton leak inhibition, we utilized isolated mitochondria from stable human embryonic kidney cells (HEK293) ectopically expressing mouse UCP1 (Hirschberg et al., 2011). Importantly, mouse UCP1 displays native behavior in this system (i.e. fatty acid and GDP sensitivity) without inducing artificial uncoupling. Addition of purines GDP, GMP or IMP to mitochondria from control HEK293 cells had no effect on respiration driving proton leak (Figure 7G). However, in presence of UCP1, there was an upwards shift in the proton leak kinetics (relationship between respiration rate and membrane potential) indicating increased uncoupling (Divakaruni and Brand, 2011) which was prevented to a similar extent after addition of GDP or GMP. At the highest common membrane potential, addition of GMP or GDP to UCP1-containing mitochondria caused a similar decrease in proton conductance (Figure 7G), while addition of IMP had no effect on respiration rate or membrane potential. This indicates the importance of a guanine moiety for UCP1 inhibition. Altogether, our data suggest that increased GMP in BAT on HFD is physiologically relevant, and may be sufficient to significantly impair proton leak, especially within the context of drastically reduced fatty acid levels on HFD (Figure 7A). This would severely reduce BAT contribution to global energy expenditure and promote expansion and accumulation of lipid stores.
Discussion
Homeostasis is maintained by coordinating physiological responses among organs and tissues (Roenneberg and Merrow, 2016). While circadian clocks are known to play various homeostatic roles (Eckel-Mahan and Sassone-Corsi, 2013), understanding how 24-h metabolism is coordinated among various tissue clocks has remained limited. Our study provides a comprehensive map of both temporal and spatial distribution of metabolites over 24-h, and reveals intra- and inter-tissue relationships under conditions of energy balance and imbalance. We uncover how metabolites are linked within and across various tissues over time, and how these connections are modified by nutrient stress. This uniquely systems-level view of how tissue metabolism is organized over time has the potential to reveal previously unappreciated metabolic relationships ripe for therapeutic exploitation.
For example, increased blood concentration of specific amino acids and their metabolites is associated with obesity and T2DM (Adams, 2011; Adibi, 1968; Felig et al., 1969). While BCAAs are predictive biomarkers for T2D risk (Batch et al., 2013; Newgard, 2012; Newgard et al., 2009; Wang et al., 2011), their origin is debatable (Herman et al., 2010; Herrero et al., 1997; Zhao et al., 2016). Our data support a scenario where increased circulating amino acids mostly reflect increased skeletal muscle protein turnover (Figures 6A&S3). Increased circulating levels of alanine and glutamine on HFD most likely originate from muscle proteolysis and metabolism of BCAA, with increased release of BCKA. Our atlas suggests further investigation of serum alanine as a biomarker for T2DM and cardiovascular disease risk is warranted, in keeping with findings on alanine aminotransferase (ALT) (Vozarova et al., 2002; Zhou et al., 2013) and recent indications in human T2DM patients (Isherwood et al., 2017). We thus propose a more general “risk network” of alanine-related metabolites and enzymatic biomarkers.
We also show how loss of BAT metabolite lipid oscillation in conjunction with de novo oscillation of purine nucleotides may be relevant for UCP1 function (Figure 7B). The pre-awakening hours (ZT8-ZT10) are particularly important for normal BAT physiology, and coincide with the peak of glucose uptake (van der Veen et al., 2012), and REV-ERBα expression, a critical regulator of circadian BAT function (Gerhart-Hines et al., 2013). Human BAT exhibits a similar “dawning” phenomenon, i.e. coupling glucose uptake rhythm to heat production before awakening (Lee et al., 2016), which also coincides with the peak of diurnal plasma catecholamine levels (Scheer et al., 2010). Interestingly, BAT circadian activation appears to be a clock-driven anticipatory process as it persists ex vivo, without external cues (Lee et al., 2016).
Loss of NAD+ oscillation during the light phase can explain impaired purine nucleotide degradation and may contribute to UCP1 inhibition by increasing GMP levels during this crucial time point. Notably, elevated levels of GMP, IMP and AMP are observed in erythrocytes from diabetic patients (Dudzinska and Hlynczak, 2004) and elevated blood purine nucleotides are common pathophysiological consequences of metabolic diseases (Park et al., 2015). Loss of NAD+ and NAM oscillations in BAT on HFD may be caused by circadian disruption of enzymes involved in their formation (Eckel-Mahan et al., 2013). Indeed, expression of core clock genes (Bmal1 and Clock) and BAT circadian genes regulating NAD+ formation (Sirt3 and Nampt) are altered by HFD (Figure S4A and B). Likewise, circadian expression of genes regulating purine nucleotide metabolism (Impdh1 and Gmpr) and thermogenesis (Ucp1) were severely impacted by HFD. Thus, BAT circadian function likely involves close coordination between transcriptional regulation and metabolites.
In summary, our atlas constitutes a rich starting point for hypothesis generation and validation by allowing for the temporal reconstruction of complex metabolic networks across tissues and under different feeding paradigms. Additional studies are needed to further elaborate subcellular metabolite localization and dynamics (Aguilar-Arnal et al., 2016; Aviram et al., 2016; Dyar and Eckel-Mahan, 2017; Mauvoisin et al., 2017), and to monitor inter-organ metabolite fluxes. While a wide range of intra- and inter-tissue metabolite correlations describing potentially novel predictive biomarkers and pathogenic networks remains to be explored, this resource can already serve as a basis for future studies integrating subcellular metabolomics data, clock gene mutants, additional feeding regimes and exercise protocols.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Paolo Sassone-Corsi (psc@uci.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethics Statement
All experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines of the University of California at Irvine in compliance with the Animal Welfare Act and other federal and state statutes and regulations relating to animal experiments.
Animals, diets and tissue collection
Six week old male C57BL6/J mice were purchased from JAX / Jackson Labs (Stock Number: 000664). Mice were were randomly assigned to experimental groups, maintained on a 12hr light/12hr dark cycle (ZT0 corresponds to lights on and ZT12 to lights off in the animal facility), and fed ad libitum for 10 weeks with either standard chow diet (Prolab RMH 2500) or high fat diet (HFD) composed of 60% Kcal from fat (Research Diets, D12492). Body weight was measured weekly. Animals were separated into individual cages 1 week before tissue collection. Five male mice for each time point/diet were used. Tissues were immediately collected after cervical dislocation and stored at −80°C until further processing/analysis. Serum was prepared from an abdominal/thoracic blood sample and stored at −80°C. Sperm was collected after swimming out from the caudal portion of the epididymis in non-capacitating media (MEM without BSA) for 10min at 37°C. Sperm cells were then washed with PBS twice at low centrifugation speed and the sperm pellet was rapidly frozen in liquid nitrogen and stored at −80°C.
Cell culture
Human embryonic kidney cells (HEK293) were cultured in Dulbecco’s Modified Eagle Medium (4500 mg/l glucose, +L-glutamine, – pyruvate) supplemented with 10% fetal calf serum (Biochrom), 50 μ g/ml gentamycin and 2.5 μ g/ml amphotericin B at 37 °C in a 5% CO2 humidified incubator as described (Jastroch et al., 2012). To passage, medium was removed and cells washed with PBS, trypsinised (0.2% trypsin), suspended in medium, centrifuged (500 g; 3min), resuspended in growth medium and seeded on new plates.
METHOD DETAILS
Global metabolite profiling
Non-targeted metabolite profiling, peak identification, and curation was performed by Metabolon (Durham, NC, USA) and by the Genome Analysis Center (GAC), Helmholtz Zentrum München (Neuherberg, Germany). Liver, serum, and sperm were processed and run by Metabolon on an HD3 system using described methods (Abbondante et al., 2016; Eckel-Mahan et al., 2012). Briefly, this analytical system combines a Linear Ion Trap MS/MS (LTQ XL, Thermo Scientific) coupled with UPLC (Acquity, Waters), and consists of 2 reverse phase (RP)/UPLC-MS/MS methods: 1) with positive ion mode electrospray ionization (ESI) optimized for acidic species, and 2) with negative ion mode ESI optimized for basic species. An additional GC/MS platform for volatile compounds was used in parallel. WAT and BAT samples were processed and run by the GAC on the same analytical system, with the exception of the GC/MS platform, and with curation again performed by Metabolon. Skeletal muscle and brain tissues (SCN & mPFC) were processed and run by Metabolon on their HD4 platform, which runs with High Resolution Accurate Mass (HRAM) MS/MS (QExactive, Thermo Scientific) also coupled with UPLC (Acquity, Waters). Overall, we processed and analyzed a total of 70 tissues each of liver, serum, BAT, and WAT (5 replicates × 2 groups × 7 time points, including additional time point ZT24), and 60 tissues each for SCN, mPFC, gastrocnemius skeletal muscle, and sperm (5 replicates × 2 groups × 6 time points). One biological replicate each from chow-fed SCN at ZT20 and from HFD-fed mPFC at ZT4 were lost during sample processing, leaving 4 remaining replicates each for these particular time points/diets. Two biological replicates from chow-fed mPFC at ZT4 were likewise lost, leaving 3 remaining replicates.
Metabolomics data processing and analysis
For further analysis, we used raw metabolomic intensity data (“origscale”, Table S4). Data processing closely followed procedures reported in (Shin et al., 2014). Run day correction was performed for each metabolite by setting the run day medians equal to 1. We removed metabolites with more than 50% missing values and transformed data to log10. Data points outside 4 times the standard deviation for each metabolite were considered as outliers and removed. Missing data were imputed by k-nearest-neighbor. (Missing Values: 5% BAT, 25% BrainPFC, 5% BrainSCN, 5% Liver, 17% Muscle, 6% Serum, 17% Sperm, 8% WAT).
Statistical analysis of metabolites
Heatmaps were generated using the median of 5 replicates for each time point. Hierarchical clustering was performed with euclidean distance and Ward’s minimum variance linkage algorithm. Metabolites were categorized according to Metabolon superpathways: Amino Acids, Carbohydrates, Cofactors & Vitamins, Energy, Lipids, Nucleotides, Peptides and Xenobiotics. To identify metabolites that show significant change over time and/or diet we used a linear regression model of the formula y ~ β0 + β1 * time + β2 * diet + β2 * diet * time + ε. Significance of each fixed effect term in the linear model was estimated using ANOVA. Enriched KEGG pathways were identified using hypergeometric distribution test. Subpathway enrichments were calculated using Fisher’s exact test. To identify 24-hr cycling metabolites (Table S5), we used the nonparametric test JTK_CYCLE (Hughes et al., 2010) using an adjusted p<0.05 as described in (Abbondante et al., 2016; Eckel-Mahan et al., 2012). Subpathway enrichment analyses were performed applying Fisher’s exact test. For data entered into CircadiOmics, the rhythmicity test, BIO_CYCLE is also used to define circadian oscillations, as described in (Agostinelli et al., 2016).
Robust Correlations
To identify significant temporal correlations among metabolites in each tissue, we applied a robust permutation test, performing 100 random permutations of the replicate samples and estimated correlation coefficient and significance (Pearson correlation). Metabolites with a correlation pvalue < 0.05 in 95% (99% within tissue circos plots) of all permutation tests were considered as significant. Cross-tissue correlation networks were generated by connecting metabolites that show significant positive and negative correlations between tissues (Bonferroni corrected). Calculations were done using MATLAB and Statistics Toolbox R2016b.
In vitro functional characterization of proton leak kinetics
HEK293 cells with stable expression of mouse UCP1 were generated as described (Jastroch et al., 2012). Briefly, mouse UCP1 ORF (NM009463) was cloned into pcDNA3.1. Subconfluent HEK293 cells were then transfected with either empty vector or mouse UCP1, and selected for stable transfection. Positive transfection was verified by Western blot using rabbit anti-UCP1 polyclonal antibody (Chemicon #3046). Several aliquots of a clonal cell line were stored in liquid nitrogen to control for passaging effects. For proton leak measurements, an aliquot of UCP1 expressing and wildtype cells was seeded on a 10 cm cell culture dish. Cells were transferred to four 10 cm dishes after 3 days, and on day 5 seeded on 2 500 cm2 dishes. Cell medium was replaced on day 7, and mitochondria were isolated on day 9. Proton leak kinetics were measured according to (Jastroch, 2012) by measuring mitochondrial membrane potential with a TPMP+ sensitive electrode simultaneously with oxygen consumption. In brief, 0.35 mg/ml of mitochondrial protein were incubated in buffer containing 0.3% defatted bovine serum albumin with either 500 uM inositol-monophosphate (IMP), or guanosine-monophosphate (GMP), or guanosine-diphosphate (GDP) for seven minutes before energization.
BAT qPCR
BAT mRNA was prepared from whole BAT tissue using Trizol (Invitrogen, 15596018). 1μg of RNA was used to synthesize cDNA using the Maxima H Minus cDNA Synthesis Master Mix (Thermo Scientific, M1662) and with an extension time of 60 min. at 50°C. qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, 1725270) using the following parameters:
95°C- 10 min.
95°C- 30 sec.
60°C- 1 min.
40 cycles of steps 2 and 3
95°C- 15 sec.
Amplification was performed using a QuantStudio 3 Real-Time PCR System. Analysis was performed using the 2ΔΔCT method and data were normalized relative to 18SrRNA expression. Primer sequences can be found in Table S6.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are expressed as means ± SEM unless stated otherwise. Statistical analysis was performed using a linear regression model or 2-way ANOVA. When ANOVA revealed significant genotype differences, further analysis was performed using Bonferroni’s multiple comparison test. Differences between groups were considered statistically significant for p < 0.05.
DATA AVAILABILITY
All raw 24-h metabolomics intensity data (“origscale”) has been deposited at (doi:10.17632/zndtmk8xc6.1), and can also be found in Table S4. Elaborated metabolomics data are available from CircadiOmics (http://circadiomics.igb.uci.edu).
Supplementary Material
Figure S1 (related to Fig.1): Spermatozoa collection and summary.
(A) Steps of spermatozoa collection. (B) MEFs, Mouse Embryonic Fibroblasts, 5, 25 and 40 thousand cells; Caput, spermatozoa harvested from caput epididymis, 0.1, 0.2 and 0.4 million cells; Cauda, spermatozoa harvested from cauda epididymis, 0.2, 0.5, 1 and 2 million cells. LAP2B, lamina-associated polypeptide 2β, present in somatic cells and immature germ cells; H3, Histone H3 and Actin were used as loading controls. (C-D) Main metabolite classes showing circadian function in mouse spermatozoa. (C) Graphs showing diurnal profiles of detected glycolytic intermediates. (D) Heat map of detected lysolipids showing ratio of HFD/chow for each timepoint. Green cells indicate metabolites reduced by HFD (dark green, p≤0.05; light green, 0.05≤p≤0.1; ANOVA). Pink cells indicate increased metabolites approaching significance (0.05≤p≤0.1).
Table S6 (related to STAR methods): qPCR Primer sequences
Figure S2 (related to Fig.5): Significant cross-tissue correlations between serum and liver, muscle, or BAT.
(A) Total number of correlated metabolites.
(B) Metabolite class distribution of correlated metabolites between serum and liver, muscle or BAT. (C) Metabolite class distribution of lost correlations between serum and liver, muscle and BAT.
Figure S3 (related to Fig.6): Diurnal variations of selected (A) amino acids and (B) branched-chain keto acids in mouse tissues under chow or HFD (mean±SEM; n=5 per group per time point).
Figure S4 (related to Fig.7): High fat diet alters 24-hour mRNA expression profiles of circadian genes in brown adipose tissue. Diurnal expression profiles of A) selected core circadian clock and B) selected circadian genes from brown adipose tissue (mean± SEM, expressed relative to 18S and normalized to chow ZT0; n=4 per group per time point; *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA with Bonferroni correction). C) Diet effect p-values determined by 2-way ANOVA.
Table S1 (related to Fig.1): Subpathway enrichment of metabolites altered by HFD (p=Fisher’s exact test)
Table S2 (related to Fig.2): Subpathway enrichment of circadian metabolites in each tissue (p=Fisher’s exact test)
Table S3 (related to Fig.3): Subpathway enrichment of correlated metabolites in each tissue (p=Fisher’s exact test)
Table S4 (related to STAR methods): Raw 24-h metabolomics intensity data (“origscale”)
Table S5 (related to STAR methods): JTK_CYCLE data
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-UCP1 polyclonal antibody | Chemicon | #3046 |
| Deposited Data | ||
| Mouse brown adipose, high fat diet | http://circadiomics.ics.uci.edu/ | MOUSE BROWN ADIPOSE DYAR 2018 HIGH-FAT |
| Mouse brown adipose, normal chow | http://circadiomics.ics.uci.edu/ | MOUSE BROWN ADIPOSE DYAR 2018 NORMAL-CHOW |
| Mouse liver, high fat diet | http://circadiomics.ics.uci.edu/ | MOUSE LIVER METABOLOME ECKEL-MAHAN 2013 HIGH-FAT |
| Mouse liver, normal chow | http://circadiomics.ics.uci.edu/ | MOUSE LIVER METABOLOME ECKEL-MAHAN 2013 NORMAL-CHOW |
| Mouse muscle, high fat diet | http://circadiomics.ics.uci.edu/ | MOUSE MUSCLE DYAR 2018 HIGH-FAT |
| Mouse muscle, normal chow | http://circadiomics.ics.uci.edu/ | MOUSE MUSCLE DYAR 2018 NORMAL-CHOW |
| Mouse prefrontal cortex, high fat diet | http://circadiomics.ics.uci.edu/ | MOUSE PREFRONTAL CORTEX NA 2018 HIGH-FAT |
| Mouse prefrontal cortex, normal chow | http://circadiomics.ics.uci.edu/ | MOUSE PREFRONTAL CORTEX NA 2018 NORMAL-CHOW |
| Mouse serum, high fat diet | http://circadiomics.ics.uci.edu/ | MOUSE SERUM ABBONDANTE 2016 HIGH-FAT |
| Mouse serum, normal chow | http://circadiomics.ics.uci.edu/ | MOUSE SERUM ABBONDANTE 2016 NORMAL-CHOW |
| Mouse sperm, high fat diet | http://circadiomics.ics.uci.edu/ | MOUSE SPERM DYAR 2018 HIGH-FAT |
| Mouse sperm, normal chow | http://circadiomics.ics.uci.edu/ | MOUSE SPERM DYAR 2018 NORMAL-CHOW |
| Mouse suprachiasmatic nucleus, high fat diet | http://circadiomics.ics.uci.edu/ | MOUSE SUPRACHIASMATIC NUCLEUS NA 2018 HIGH-FAT |
| Mouse suprachiasmatic nucleus, normal chow | http://circadiomics.ics.uci.edu/ | MOUSE SUPRACHIASMATIC NUCLEUS NA 2018 NORMAL-CHOW |
| Mouse white adipose, high fat diet | http://circadiomics.ics.uci.edu/ | MOUSE WHITE ADIPOSE DYAR 2018 HIGH-FAT |
| Mouse white adipose, normal chow | http://circadiomics.ics.uci.edu/ | MOUSE WHITE ADIPOSE DYAR 2018 NORMAL-CHOW |
| Raw “origscale” 24-h metabolomics intensity data from 8 different murine tissues under chow or high fat diet | doi:10.17632/zndtmk8xc6.1 | Table_S4.xlsx |
| Diets | ||
| Vivarium Chow- Prolab RMH 2500 | Lab Supply | 5P14 |
| High Fat Diet | Research Diets | D12492 |
| Mice | ||
| C57BL/6J | The Jackson Laboratory | 000664 |
| Plasmids | ||
| pcDNA3.1 | Invitrogen | V79020 |
| Mouse UCP1 ORF (NM009463) cloned into pcDNA3.1 | Martin Jastroch | n/a |
| Software | ||
| MATLAB and Statistics Toolbox R2016b | Mathworks | https://www.mathworks.com/products/new_products/release2016b.html |
| JTK_CYCLE | Hughes et al., 2010 | https://github.com/mfcovington/jtk-cycle |
| BIO_CYCLE | Agostinelli et al., 2016 | http://circadiomics.igb.uci.edu/static/downloadables/BioCycle.tar.gz |
| Cell lines | ||
| HEK293 | Sigma | 85120602 |
| Chemicals | ||
| TRIzol reagent | Invitrogen | 15596018 |
| SsoAdvanced Universal SYBR Green | Bio-Rad | Cat#4385618 |
| iScript cDNA reverse transcription kit | Bio-Rad | Cat#1708891 |
| DMEM, 4,500 mg/L glucose, + L-glutamine, -pyruvate | Gibco | 11965092 |
| FBS | Biochrom | S0115 |
| Gentamycin | Gibco | 15750060 |
| Amphotericin B | Sigma | A2942 |
| Superfect® Transfection Reagent | Qiagen | 301305 |
| Geneticin | Gibco | 10131035 |
| Fat-free BSA | Sigma | A3803 |
| GDP | Sigma | G7127 |
| GMP | Sigma | G8377 |
| IMP | Sigma | I4375 |
| Oligonucleotides | ||
| See Table S6 for qPCR primers | n/a | n/a |
Highlights.
Comprehensive atlas of 24-hr metabolism reveals temporal cohesion among tissues
Nutrient stress disrupts circadian metabolites in a tissue-specific manner
Communication and inter-tissue metabolite correlations rewired by high fat diet
Multi-tissue metabolite correlations highlight a coordinated pathogenic response
Acknowledgments
This work was supported by NIH grant GM123558 and DARPA grant D17AP00002 (P.B.), DFG Emmy Noether NHU 275/1-1 (N.H.U.), the Alexander von Humboldt Foundation (M.H.T.), DK114037 (K.E.M.) and by INSERM (Institut National de la Sante et de la Recherche Medicale, France), KAUST (King Abdullah University of Science and Technology), National Institute of Health, and Novo Nordisk Foundation Challenge Grant NNF140C0011493 (P.S.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare no competing interests
References
- Abbondante S, Eckel-Mahan KL, Ceglia NJ, Baldi P, and Sassone-Corsi P (2016). Comparative Circadian Metabolomics Reveal Differential Effects of Nutritional Challenge in the Serum and Liver. J Biol Chem 291, 2812–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams SH (2011). Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Adv Nutr 2, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adibi SA (1968). Influence of dietary deprivations on plasma concentration of free amino acids of man. J Appl Physiol 25, 52–57. [DOI] [PubMed] [Google Scholar]
- Agostinelli F, Ceglia N, Shahbaba B, Sassone-Corsi P, and Baldi P (2016). What time is it? Deep learning approaches for circadian rhythms. Bioinformatics 32, i8–i17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Arnal L, Ranjit S, Stringari C, Orozco-Solis R, Gratton E, and Sassone-Corsi P (2016). Spatial dynamics of SIRT1 and the subnuclear distribution of NADH species. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, and Sassone-Corsi P (2015). Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell 161, 84–92. [DOI] [PubMed] [Google Scholar]
- Aviram R, Manella G, Kopelman N, Neufeld-Cohen A, Zwighaft Z, Elimelech M, Adamovich Y, Golik M, Wang C, Han X, et al. (2016). Lipidomics Analyses Reveal Temporal and Spatial Lipid Organization and Uncover Daily Oscillations in Intracellular Organelles. Mol Cell 62, 636–648. [DOI] [PubMed] [Google Scholar]
- Batch BC, Shah SH, Newgard CB, Turer CB, Haynes C, Bain JR, Muehlbauer M, Patel MJ, Stevens RD, Appel LJ, et al. (2013). Branched chain amino acids are novel biomarkers for discrimination of metabolic wellness. Metabolism 62, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender D (2011). Amino Acid Metabolism, Vol Wiley-Blackwell, Third Edition edn. [Google Scholar]
- Brown SA (2016). Circadian Metabolism: From Mechanisms to Metabolomics and Medicine. Trends Endocrinol Metab 27, 415–426. [DOI] [PubMed] [Google Scholar]
- Chen WW, Freinkman E, Wang T, Birsoy K, and Sabatini DM (2016). Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1337 e1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallmann R, Viola AU, Tarokh L, Cajochen C, and Brown SA (2012). The human circadian metabolome. Proc Natl Acad Sci U S A 109, 2625–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SK, Ang JE, Revell VL, Holmes B, Mann A, Robertson FP, Cui N, Middleton B, Ackermann K, Kayser M, et al. (2014). Effect of sleep deprivation on the human metabolome. Proc Natl Acad Sci U S A 111, 10761–10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Thompson CB (2012). Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, and Brand MD (2011). The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 26, 192–205. [DOI] [PubMed] [Google Scholar]
- Dudzinska W, and Hlynczak AJ (2004). Purine nucleotides and their metabolites in erythrocytes of streptozotocin diabetic rats. Diabetes Metab 30, 557–567. [DOI] [PubMed] [Google Scholar]
- Dunford EC, and Riddell MC (2016). The Metabolic Implications of Glucocorticoids in a High-Fat Diet Setting and the Counter-Effects of Exercise. Metabolites 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyar KA, Ciciliot S, Wright LE, Bienso RS, Tagliazucchi GM, Patel VR, Forcato M, Paz MI, Gudiksen A, Solagna F, et al. (2014). Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol Metab 3, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyar KA, and Eckel-Mahan KL (2017). Circadian Metabolomics in Time and Space. Front Neurosci 11, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel RH, Grundy SM, and Zimmet PZ (2005). The metabolic syndrome. Lancet 365, 1415–1428. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan K, and Sassone-Corsi P (2013). Metabolism and the circadian clock converge. Physiol Rev 93, 107–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, Dilag-Penilla SA, Dyar KA, Baldi P, and Sassone-Corsi P (2013). Reprogramming of the circadian clock by nutritional challenge. Cell 155, 1464–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, Mohney RP, Vignola KS, Baldi P, and Sassone-Corsi P (2012). Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci U S A 109, 5541–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia M, Carter A, Bacon S, Winearls CG, and Smith R (1981). Clinical usefulness of urinary 3-methylhistidine excretion in indicating muscle protein breakdown. Br Med J (Clin Res Ed) 282, 351–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P (1973). The glucose-alanine cycle. Metabolism 22, 179–207. [DOI] [PubMed] [Google Scholar]
- Felig P, Marliss E, and Cahill GF Jr. (1969). Plasma amino acid levels and insulin secretion in obesity. N Engl J Med 281, 811–816. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamada H, Komatsu R, Shimba S, and Okamura H (2012). Rhythmic nucleotide synthesis in the liver: temporal segregation of metabolites. Cell Rep 1, 341–349. [DOI] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Feng D, Emmett MJ, Everett LJ, Loro E, Briggs ER, Bugge A, Hou C, Ferrara C, Seale P, et al. (2013). The nuclear receptor Rev-erbalpha controls circadian thermogenic plasticity. Nature 503, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German JB, Hammock BD, and Watkins SM (2005). Metabolomics: building on a century of biochemistry to guide human health. Metabolomics 1, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giskeodegard GF, Davies SK, Revell VL, Keun H, and Skene DJ (2015). Diurnal rhythms in the human urine metabolome during sleep and total sleep deprivation. Sci Rep 5, 14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad T, Scheibler S, von Kugelgen I, Scheele C, Kilic A, Glode A, Hoffmann LS, Reverte-Salisa L, Horn P, Mutlu Sv et al. (2014). Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature 516, 395–399. [DOI] [PubMed] [Google Scholar]
- Goodacre R (2007). Metabolomics of a superorganism. J Nutr 137, 259S–266S. [DOI] [PubMed] [Google Scholar]
- Gooley JJ, and Chua EC (2014). Diurnal regulation of lipid metabolism and applications of circadian lipidomics. J Genet Genomics 41, 231–250. [DOI] [PubMed] [Google Scholar]
- Guggenheim KY (1991). Rudolf Schoenheimer and the concept of the dynamic state of body constituents. J Nutr 121, 1701–1704. [DOI] [PubMed] [Google Scholar]
- Hatting M, Tavares CDJ, Sharabi K, Rines AK, and Puigserver P (2018). Insulin regulation of gluconeogenesis. Ann N Y Acad Sci 1411, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, She P, Peroni OD, Lynch CJ, and Kahn BB (2010). Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem 285, 11348–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero MC, Remesar X, Blade C, and Arola L (1997). Muscle amino acid pattern in obese rats. Int J Obes Relat Metab Disord 21, 698–703. [DOI] [PubMed] [Google Scholar]
- Hirschberg V, Fromme T, and Klingenspor M (2011). Test systems to study the structure and function of uncoupling protein 1: a critical overview. Front Endocrinol (Lausanne) 2, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB, and Kornacker K (2010). JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J Biol Rhythms 25, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isherwood CM, Van der Veen DR, Johnston JD, and Skene DJ (2017). Twenty-four-hour rhythmicity of circulating metabolites: effect of body mass and type 2 diabetes. FASEB J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastroch M (2012). Expression of uncoupling proteins in a mammalian cell culture system (HEK293) and assessment of their protein function. Methods Mol Biol 810, 153–164. [DOI] [PubMed] [Google Scholar]
- Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, and Brand MD (2010). Mitochondrial proton and electron leaks. Essays Biochem 47, 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastroch M, Hirschberg V, and Klingenspor M (2012). Functional characterization of UCP1 in mammalian HEK293 cells excludes mitochondrial uncoupling artefacts and reveals no contribution to basal proton leak. Biochim Biophys Acta 1817, 1660–1670. [DOI] [PubMed] [Google Scholar]
- Jouffe C, Gobet C, Martin E, Metairon S, Morin-Rivron D, Masoodi M, and Gachon F (2016). Perturbed rhythmic activation of signaling pathways in mice deficient for Sterol Carrier Protein 2-dependent diurnal lipid transport and metabolism. Sci Rep 6, 24631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Mall C, Taylor SL, Hitchcock S, Zhang C, Wettersten HI, Jones AD, Chapman A, and Weiss RH (2014). Mealtime, temporal, and daily variability of the human urinary and plasma metabolomes in a tightly controlled environment. PLoS One 9, e86223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M, and Huang SG (1999). Structure and function of the uncoupling protein from brown adipose tissue. Biochim Biophys Acta 1415, 271–296. [DOI] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaiah SY, Wu G, Altman BJ, Growe J, Rhoades SD, Coldren F, Venkataraman A, Olarerin-George AO, Francey LJ, Mukherjee S, et al. (2017). Clock Regulation of Metabolites Reveals Coupling between Transcription and Metabolism. Cell Metab 25, 961–974 e964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Bova R, Schofield L, Bryant W, Dieckmann W, Slattery A, Govendir MA, Emmett L, and Greenfield JR (2016). Brown Adipose Tissue Exhibits a Glucose-Responsive Thermogenic Biorhythm in Humans. Cell Metab 23, 602–609. [DOI] [PubMed] [Google Scholar]
- Lowrey PL, and Takahashi JS (2011). Genetics of circadian rhythms in Mammalian model organisms. Adv Genet 74, 175–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Solmonson A, Lodi A, Nowinski SM, Sentandreu E, Riley CL, Mills EM, and Tiziani S (2017). The early metabolomic response of adipose tissue during acute cold exposure in mice. Sci Rep 7, 3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lozano Sinues P, Tarokh L, Li X, Kohler M, Brown SA, Zenobi R, and Dallmann R (2014). Circadian variation of the human metabolome captured by real-time breath analysis. PLoS One 9, e114422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C, Roqueta-Rivera M, Deng C, Osborne TF, Mostoslavsky R, et al. (2014). Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell 158, 659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvoisin D, Atger F, Dayon L, Nunez Galindo A, Wang J, Martin E, Da Silva L, Montoliu I, Collino S, Martin FP, et al. (2017). Circadian and Feeding Rhythms Orchestrate the Diurnal Liver Acetylome. Cell Rep 20, 1729–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee EE, Bentley AT, Smith RM Jr., and Ciaccio CE (1999). Origin of guanine nucleotides in isolated heart mitochondria. Biochem Biophys Res Commun 257, 466–472. [DOI] [PubMed] [Google Scholar]
- Mori S, Kawai S, Shi F, Mikami B, and Murata K (2005). Molecular conversion of NAD kinase to NADH kinase through single amino acid residue substitution. J Biol Chem 280, 24104–24112. [DOI] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, and Sassone-Corsi P (2009). Circadian Control of the NAD+ Salvage Pathway by CLOCK-SIRT1. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB (2012). Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 15, 606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. (2009). A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9, 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, and Locke RM (1984). Thermogenic mechanisms in brown fat. Physiol Rev 64, 1–64. [DOI] [PubMed] [Google Scholar]
- Nishino T, Okamoto K, Eger BT, Pai EF, and Nishino T (2008). Mammalian xanthine oxidoreductase - mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J 275, 3278–3289. [DOI] [PubMed] [Google Scholar]
- Noguchi R, Kubota H, Yugi K, Toyoshima Y, Komori Y, Soga T, and Kuroda S (2013). The selective control of glycolysis, gluconeogenesis and glycogenesis by temporal insulin patterns. Mol Syst Biol 9, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye C, Kim J, Kalhan SC, and Hanson RW (2008). Reassessing triglyceride synthesis in adipose tissue. Trends Endocrinol Metab 19, 356–361. [DOI] [PubMed] [Google Scholar]
- O’Neill JS, Maywood ES, Chesham JE, Takahashi JS, and Hastings MH (2008). cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science 320, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill JS, van Ooijen G, Dixon LE, Troein C, Corellou F, Bouget FY, Reddy AB, and Millar AJ (2011). Circadian rhythms persist without transcription in a eukaryote. Nature 469, 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes ND, Bell KS, Furler SM, Camilleri S, Saha AK, Ruderman NB, Chisholm DJ, and Kraegen EW (1997). Diet-induced muscle insulin resistance in rats is ameliorated by acute dietary lipid withdrawal or a single bout of exercise: parallel relationship between insulin stimulation of glucose uptake and suppression of long-chain fatty acyl-CoA. Diabetes 46, 2022–2028. [DOI] [PubMed] [Google Scholar]
- Odessey R, and Goldberg AL (1979). Leucine degradation in cell-free extracts of skeletal muscle. Biochem J 178, 475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Sadanala KC, and Kim EK (2015). A Metabolomic Approach to Understanding the Metabolic Link between Obesity and Diabetes. Mol Cells 38, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VR, Eckel-Mahan K, Sassone-Corsi P, and Baldi P (2012). CircadiOmics: integrating circadian genomics, transcriptomics, proteomics and metabolomics. Nat Methods 9, 772–773. [DOI] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, et al. (2013). Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozefsky T, Felig P, Tobin JD, Soeldner JS, and Cahill GF Jr. (1969). Amino acid balance across tissues of the forearm in postabsorptive man. Effects of insulin at two dose levels. J Clin Invest 48, 2273–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, et al. (2009). Circadian Clock Feedback Cycle Through NAMPT-Mediated NAD+ Biosynthesis. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, and Naef F (2011). Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol 9, e1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey G, Valekunja UK, Feeney KA, Wulund L, Milev NB, Stangherlin A, Ansel-Bollepalli L, Velagapudi V, O’Neill JS, and Reddy AB (2016). The Pentose Phosphate Pathway Regulates the Circadian Clock. Cell Metab 24, 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roenneberg T, Allebrandt KV, Merrow M, and Vetter C (2012). Social jetlag and obesity. Curr Biol 22, 939–943. [DOI] [PubMed] [Google Scholar]
- Roenneberg T, and Merrow M (2016). The Circadian Clock and Human Health. Curr Biol 26, R432–443. [DOI] [PubMed] [Google Scholar]
- Roseno SL, Davis PR, Bollinger LM, Powell JJ, Witczak CA, and Brault JJ (2015). Short-term, high-fat diet accelerates disuse atrophy and protein degradation in a muscle-specific manner in mice. Nutr Metab (Lond) 12, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer FA, Hu K, Evoniuk H, Kelly EE, Malhotra A, Hilton MF, and Shea SA (2010). Impact of the human circadian system, exercise, and their interaction on cardiovascular function. Proc Natl Acad Sci U S A 107, 20541–20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher MB, Vaquero A, and Reinberg D (2007). SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev 21, 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, and Sandri M (2013). Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 280, 4294–4314. [DOI] [PubMed] [Google Scholar]
- Serra F, Gianotti M, Pons A, and Palou A (1994). Brown and white adipose tissue adaptive enzymatic changes on amino acid metabolism in persistent dietary-obese rats. Biochem Mol Biol Int 32, 1173–1178. [PubMed] [Google Scholar]
- Shabalina IG, Jacobsson A, Cannon B, and Nedergaard J (2004). Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem 279, 38236–38248. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Maruyama T, Yoshikawa N, Matsumiya R, Ma Y, Ito N, Tasaka Y, Kuribara-Souta A, Miyata K, Oike, Yv et al. (2015). A muscle-liver-fat signalling axis is essential for central control of adaptive adipose remodelling. Nat Commun 6, 6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SY, Fauman EB, Petersen AK, Krumsiek J, Santos R, Huang J, Arnold M, Erte I, Forgetta V, Yang TP, et al. (2014). An atlas of genetic influences on human blood metabolites. Nat Genet 46, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sishi B, Loos B, Ellis B, Smith W, du Toit EF, and Engelbrecht AM (2011). Diet-induced obesity alters signalling pathways and induces atrophy and apoptosis in skeletal muscle in a prediabetic rat model. Exp Physiol 96, 179–193. [DOI] [PubMed] [Google Scholar]
- Snell K, and Duff DA (1979). Muscle phosphoenolpyruvate carboxykinase activity and alanine release in progressively starved rats. Int J Biochem 10, 423–426. [DOI] [PubMed] [Google Scholar]
- Song S, Andrikopoulos S, Filippis C, Thorburn AW, Khan D, and Proietto J (2001). Mechanism of fat-induced hepatic gluconeogenesis: effect of metformin. Am J Physiol Endocrinol Metab 281, E275–282. [DOI] [PubMed] [Google Scholar]
- Sugimoto M, Ikeda S, Niigata K, Tomita M, Sato H, and Soga T (2012). MMMDB: Mouse Multiple Tissue Metabolome Database. Nucleic Acids Res 40, D809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, and McKnight SL (2006). Metabolic cycles as an underlying basis of biological oscillations. Nat Rev Mol Cell Biol 7, 696–701. [DOI] [PubMed] [Google Scholar]
- van der Veen DR, Shao J, Chapman S, Leevy WM, and Duffield GE (2012). A diurnal rhythm in glucose uptake in brown adipose tissue revealed by in vivo PET-FDG imaging. Obesity (Silver Spring) 20, 1527–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozarova B, Stefan N, Lindsay RS, Saremi A, Pratley RE, Bogardus C, and Tataranni PA (2002). High alanine aminotransferase is associated with decreased hepatic insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 51, 1889–1895. [DOI] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, et al. (2011). Metabolite profiles and the risk of developing diabetes. Nat Med 17, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, and Wishart DS (2016). Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr Protoc Bioinformatics 55, 14 10 11-14 10 91. [DOI] [PubMed] [Google Scholar]
- Zhang S, Hulver MW, McMillan RP, Cline MA, and Gilbert ER (2014). The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond) 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Han Q, Liu Y, Sun C, Gang X, and Wang G (2016). The Relationship between Branched-Chain Amino Acid Related Metabolomic Signature and Insulin Resistance: A Systematic Review. J Diabetes Res 2016, 2794591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Mo Y, Li H, Ran X, Yang W, Li Q, Peng Y, Li Y, Gao X, Luan X, et al. (2013). Alanine aminotransferase is associated with an adverse nocturnal blood glucose profile in individuals with normal glucose regulation. PLoS One 8, e56072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwighaft Z, Aviram R, Shalev M, Rousso-Noori L, Kraut-Cohen J, Golik M, Brandis A, Reinke H, Aharoni A, Kahana C, et al. (2015). Circadian Clock Control by Polyamine Levels through a Mechanism that Declines with Age. Cell Metab 22, 874–885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (related to Fig.1): Spermatozoa collection and summary.
(A) Steps of spermatozoa collection. (B) MEFs, Mouse Embryonic Fibroblasts, 5, 25 and 40 thousand cells; Caput, spermatozoa harvested from caput epididymis, 0.1, 0.2 and 0.4 million cells; Cauda, spermatozoa harvested from cauda epididymis, 0.2, 0.5, 1 and 2 million cells. LAP2B, lamina-associated polypeptide 2β, present in somatic cells and immature germ cells; H3, Histone H3 and Actin were used as loading controls. (C-D) Main metabolite classes showing circadian function in mouse spermatozoa. (C) Graphs showing diurnal profiles of detected glycolytic intermediates. (D) Heat map of detected lysolipids showing ratio of HFD/chow for each timepoint. Green cells indicate metabolites reduced by HFD (dark green, p≤0.05; light green, 0.05≤p≤0.1; ANOVA). Pink cells indicate increased metabolites approaching significance (0.05≤p≤0.1).
Table S6 (related to STAR methods): qPCR Primer sequences
Figure S2 (related to Fig.5): Significant cross-tissue correlations between serum and liver, muscle, or BAT.
(A) Total number of correlated metabolites.
(B) Metabolite class distribution of correlated metabolites between serum and liver, muscle or BAT. (C) Metabolite class distribution of lost correlations between serum and liver, muscle and BAT.
Figure S3 (related to Fig.6): Diurnal variations of selected (A) amino acids and (B) branched-chain keto acids in mouse tissues under chow or HFD (mean±SEM; n=5 per group per time point).
Figure S4 (related to Fig.7): High fat diet alters 24-hour mRNA expression profiles of circadian genes in brown adipose tissue. Diurnal expression profiles of A) selected core circadian clock and B) selected circadian genes from brown adipose tissue (mean± SEM, expressed relative to 18S and normalized to chow ZT0; n=4 per group per time point; *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA with Bonferroni correction). C) Diet effect p-values determined by 2-way ANOVA.
Table S1 (related to Fig.1): Subpathway enrichment of metabolites altered by HFD (p=Fisher’s exact test)
Table S2 (related to Fig.2): Subpathway enrichment of circadian metabolites in each tissue (p=Fisher’s exact test)
Table S3 (related to Fig.3): Subpathway enrichment of correlated metabolites in each tissue (p=Fisher’s exact test)
Table S4 (related to STAR methods): Raw 24-h metabolomics intensity data (“origscale”)
Table S5 (related to STAR methods): JTK_CYCLE data
Data Availability Statement
All raw 24-h metabolomics intensity data (“origscale”) has been deposited at (doi:10.17632/zndtmk8xc6.1), and can also be found in Table S4. Elaborated metabolomics data are available from CircadiOmics (http://circadiomics.igb.uci.edu).