

Abstract
The noninvasive, quantitative ability of nuclear magnetic resonance (NMR) spectroscopy to characterize small molecule metabolites has long been recognized as a major strength of its application in biology. Numerous techniques exist for characterizing metabolism in living, excised, or extracted tissue, with a particular focus on 1H-based methods due to the high sensitivity and natural abundance of protons. With the increasing use of high magnetic fields, the utility of in vivo 1H magnetic resonance spectroscopy (MRS) has markedly improved for measuring specific metabolite concentrations in biological tissues. Higher fields, coupled with recent developments in hyperpolarization, also enable techniques for complimenting 1H measurements with spectroscopy of other nuclei, such as 31P and 13C, , and for combining measurements of metabolite pools with metabolic flux measurements. We compare ex vivo and in vivo methods for studying metabolism in the brain using NMR and highlight insights gained through using higher magnetic fields, the advent of dissolution dynamic nuclear polarization, and combining in vivo MRS and ex vivo NMR approaches.
Keywords: Dynamic nuclear polarization, in vivo magnetic resonance spectroscopy, in vivo and ex vivo metabolomics, brain metabolism
Graphical Abstract
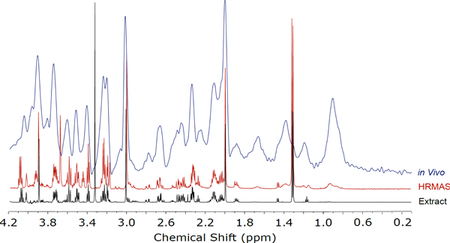
Comparison of 1H chemical shift spectra for the cortex region of a rat brain collected in vivo, after harvesting tissue and using HRMAS, and upon extracting small molecule metabolites from the tissue and reconstituting them in buffer for conventional NMR spectroscopy. In vivo data collected at 11.1 T (470 MHz); ex vivo data collected at 14.1 T (800 MHz).
Introduction
1H magnetic resonance spectroscopy (MRS)[1] has long been recognized as a means to monitor metabolism in the brain. Both its sensitivity and resolution improve with higher magnetic fields, and the FDA recently approved 7T systems, or ~300 MHz 1H, for clinical MRI studies in humans.[2] Most medical facilities currently utilize 1.5 or 3 T MRI scanners (64 and 128 MHz 1H, respectively). At these fields, metabolites with mM concentrations can readily be detected, and 7T scanners extend detection limits to below 0.1 mM.[3] Research facilities now support studies in humans at fields of up to 10.5 T with ongoing installations of magnets at 11.7 T providing exciting new platforms for continued development and exploitation of 1H MRS. Preclinical MRI systems, supporting MRS studies in rodent models, exist up to 21.1 T, or 900 MHz 1H.[4] Complimentary NMR experiments characterizing samples consisting of excised tissues or tissue-extracted metabolites can be performed using conventional NMR instruments with fields up to 23.5 T, or 1000 MHz 1H, with a further increase in sensitivity down into the μM range.
Here, we compare in vivo and ex vivo techniques at fields currently attainable for NMR and pre-clinical MRS and discuss metabolites which can readily be detected and are of particular interest for characterizing metabolism in healthy and diseased brains. By comparing ex vivo 1H NMR spectra of tissues and extracts at high fields to in vivo 1H MRS at a similar field (i.e. >11T), some interesting insights emerge that suggest, with adequate technology development, 1H MRS could become a highly sensitive tool for quantitation in real time of the majority of brain metabolites which exist at concentrations >1 mM in addition to enabling the study of several more minor metabolic species. The ability to distinguish and quantitate these metabolites is determined by their NMR characteristics, i.e. their chemical shifts, J-couplings, relaxation rates, and their overlap with other biomolecules. Govindaraju et al[5] provide a comprehensive tabulation of chemical shifts, J-couplings, and cerebral concentration ranges for common brain metabolites. Here we focus on how these parameters, coupled with tissue localization, manifest in comparing 1H spectra collected using a combination of in vivo and ex vivo approaches. For in vivo MRS and HRMAS NMR of intact tissues, tissue localization plays a large role in determining molecular diffusion rates as well as susceptibility to homogeneous and inhomogeneous broadening mechanisms. There are significant variations in achievable line shapes even within relatively homogenous tissue such as the brain. The achievable linewidth in vivo is also in part driven by the voxel size and shape. For the most useful metabolic information, the voxel size and placement is dictated by the metabolic considerations of the tissue, which can lead to a compromise on achievable line widths. Small molecules have T2 relaxation times on the order of seconds in extracted samples. In brain tissue, T2 relaxation times ranging from 90–140 msec have been measured in vivo at 9.4 T.[6] Under HRMAS conditions, relaxation rates for small molecules in brain tissue are on the order of a few hundred msec.
Other NMR-active nuclei, such as 31P and 13C, show potential for providing metabolic information complementary to 1H MRS. However, they have inherently lower sensitivity, with gyromagnetic ratios 2.5-fold and 4-fold lower than 1H, respectively. Nonetheless, with the development of higher magnetic fields, many recent studies have demonstrated their utility. The 31P isotope is naturally 100% abundant, so phosphorylated compounds at mM concentrations can be detected in a matter of minutes. [3b] Due to the 1.1% natural abundance of 13C, detection in vivo requires the use of 13C-enriched metabolic substrates. Many of the techniques developed in high resolution NMR for enhanced 13C detection have been adapted in vivo applications.[7] For these studies, subjects are typically infused with 13C-enriched substrate for one to two hours with spectral acquisition for 15–30” once steady state enrichment of downstream metabolites is achieved. Of particular interest to metabolic flux measurements is the concurrent development of dissolution dynamic nuclear polarization (dDNP) which has been demonstrated to enhance 13C signals >10,000 –fold,[8] ushering in an era of real-time measurements of metabolism using 13C-enriched substrates at conventional magnetic field strengths.
In this minireview, we demonstrate how high field 31P and dDNP-enhanced 13C MRS can be combined with 1H MRS and NMR to develop a more comprehensive picture of metabolism in the brain(Figure 1). We specifically focus on these techniques for collecting data as they can capture changes in brain metabolism on the order of a few minutes, a regime of particular interest for understanding metabolic shifts due to acute changes in the brain, including responses to drug administration or changes in glucose levels.
Figure 1.
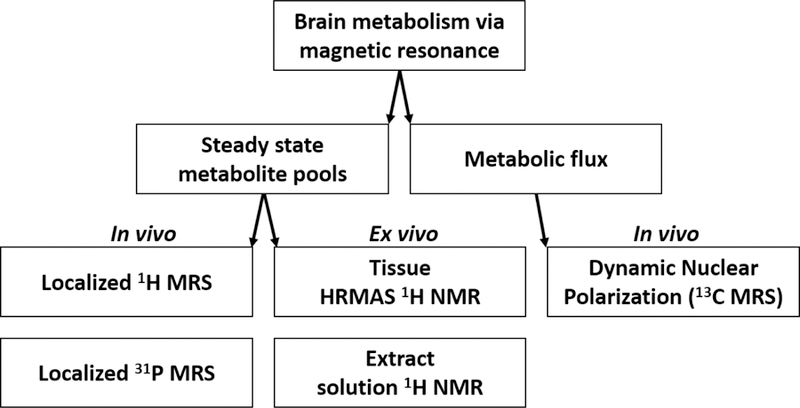
Flowchart of MR experiments discussed in this perspective.
1. Major brain metabolites
Neurotransmission is regulated via the flow of excitatory amino acids, particularly L-glutamate (Glu) and L-aspartate (Asp), and inhibitory amino acids, primarily γ-aminobutyric acid (GABA), glycine (Gly), and taurine (Taur). Normal brain concentrations are in the mM range readily enabling detection. However their chemical shifts overlap substantially with other brain metabolites in magnetic fields typical for clinical scanners making their quantitation difficult. J-based spectral editing schemes have been successfully developed to distinguish glutamate from its metabolic partner glutamine (Gln) and GABA at these lower fields.[9] At higher fields (9.4 T and above), increased chemical shift separation allows for some spectral deconvolution of the neurotransmitters, enabling quantitation of their relative concentrations.[10]
Small organic acids, including Acetate (Ac), Pyruvate (Pyr), Lactate (Lac), and Succinate (Succ), play critical roles in meeting the energy needs of the brain via consumption of glucose (Glc). A phosphocreatine (PCr) / creatine (Cr) balance maintains phosphate levels for ATP generation. Tight regulation of energy conversion ensures that physiologic concentrations of PCr/Cr are maintained at near constant levels. Due to their highly similar methyl proton chemical shifts, PCr and Cr are not easily resolved; their prominent resonances at ~3 ppm are typically highly invariant within brain structures making them an obvious choice, along with select others, for referencing changes in other metabolite concentrations.[11] The organic acids typically have well-resolved resonances, but for reasons discussed below, are often undetectable or unresolved from macromolecules in vivo. Using newly developed dissolution DNP capabilities, their interconversion can be measured in real time.[12]
Several abundant small molecule metabolites serve in regulatory capacities. N-Acetyl aspartate (NAA), and myo-Inositol (myo-Ins)) are easily detectable and play roles in osmoregulation, as precursors for neurotransmitters, and in energy storage. With distinct chemical shifts, levels of these metabolites can be estimated in vivo via spectral deconvolution even at clinical fields of 1.5 and 3T. Many studies documenting changes in their concentrations with age and disease have been documented.[13] NAA is commonly reported as a marker of neuronal health while myo-Ins is suggested to indicate glial status. At higher fields, choline (Chol) and phosphocholine (PC) can be resolved as reporters of changes in tissue health.
The branched-chain amino acids Isoleucine (Ile), Leucine (Leu), and Valine (Val) are essential amino acids important for synthesis of neurotransmitters and proteins as well as energy homeostasis. In vivo they are difficult to distinguish from the macromolecular background, but they can be easily quantitated in brain extract samples. The nonessential amino acid Alanine (Ala) can be detected in vivo at higher fields, and has been reported as an indicator of ischemia.[14]
2. In vivo detection of metabolites
In vivo NMR spectroscopy is typically referred to as magnetic resonance spectroscopy or MRS.[15] It allows direct observation of in-vivo metabolite levels, and thus provides the most physiologically relevant data of any of the methods considered in this review. However, losses in resolution and sensitivity relative to ex vivo techniques limit the quantitation of specific metabolites. The significant broadening of resonances observed in vivo are due to the slower molecular motions of even small molecule metabolites in semi-solid tissues, local B0 variations driven by magnetic susceptibility differences between tissue types, and local paramagnetic broadening effects of metals and oxygen. While a range of potential nuclei can be observed in-vivo, typically 1H and 31P spectroscopy are most common due to sensitivity limitations for other nuclei. Less abundant spin-active nuclei, such as 13C, are only observed when isotopically enriched metabolic substrates are introduced, and real-time observation is only possibly with hyper-polarization techniques such as dissolution dynamic nuclear polarization (dDNP), discussed below.
MRS is commonly executed in a spatially localized manner to detect metabolism in specific tissues.[16] To select a particular region of interest, or voxel, gradient magnetic fields are applied with selective radiofrequency pulses. The size of the voxel selected for observation is determined by the strength of the gradients applied and the strength of the signals of interest (Figure 2). To fully localize a voxel, at least three rf pulses in conjunction with three orthogonal localization gradients are applied. The exact details depend on the specific sequence used. The width of each excitation slice, Δrs , the final voxel being the intersections of the three slices, depends on the bandwidth of the rf pulse (Δω) and the strength of the applied field gradient (Gslice), and the gyromagnetic ratio (γ).
Figure 2.

1H MRI gradient echo image of a rat brain showing the localized voxel identified for collecting 1H MRS data.
A shaped rf pulse is used to excite the specific bandwidth desired, with the frequency offset of the pulse set to select the position of the slice. Traditionally, basic pulse shapes, such as sinc pulses, have been used as a good compromise between rf pulse length and excitation profile. Most modern spectrometers will calculate more complex pulse shapes on the fly, which generally give improved slice excitation profiles without sacrificing pulse length and excitation bandwidth.[17] Minimal pulse lengths are desired to both shorten echo times and to maximize the excitation or refocusing bandwidth of the pulses. This helps minimize chemical shift offset error (CSO) introduced by all localization sequences. The CSO is a consequence of using signal frequency to spatially select a particular voxel in the sample.[18] Due to chemical shift, different metabolite peaks occur at slightly different frequencies. The difference in frequency is dependent on the B0 field strength of the magnet, and thus issues with CSO increase with magnetic field strength. The CSO essentially causes the signal from different parts of the NMR spectra to be localized to slightly different physical regions of the object. The spatial offset error, ΔrCS, is given by the ratio of the chemical shift difference between a particular metabolite resonance and the rf excitation frequency compared to the bandwidth of the rf pulse.
This error can lead to spectral artifacts, especially when unwanted signals, such as fat surrounding a tissue of interest, are ‘shifted’ into the observed volume. Ideally, using the highest bandwidth pulses possible will minimize this effect. Outer volume suppression (OVS) can also be used to suppress unwanted signals from surrounding tissues. There are numerous vendor-specific implementations of OVS, which are all based on spatial presaturation methods.[19] Trains of rf pulses are applied coincident with gradients to suppress signals from slices neighboring the voxel of interest. It should be noted that if OVS is used and the CSO is large there can be a significant decrease in signal with respect to chemical shift offset. This is due to the signal being obtained from an essentially decreasing voxel size as the difference in chemical shift from the offset frequency increases. As long as a consistent method and rf bandwidths are used between study subjects, this should not affect conclusions from within a given study. However, comparisons with different localization schemes or methods can be problematic.
2.1. 1H in vivo MRS
Due to the homogeneous nature of brain tissue, and the higher rates of molecular motion for small molecule metabolites, one can typically identify ~20–25 peaks at preclinical magnetic fields (Figure 3), which primarily arise from ~15–20 metabolites. The ultimate resolution achievable at high fields for standard 1H MRS methods is determined by B0 homogeneity, which is dependent on both the voxel placement[20] and shimming capabilities,[10b] and by the local dynamics of the metabolites, as demonstrated below. In Figure 3, the singlet 1H resonances have linewidths of 0.04 ppm. For well-resolved singlets, resolution as good as 0.02–0.025 ppm has been observed for mouse and rat MRS at 9.4 T.[6] However, many metabolite resonances have an underlying multiplet structure due to J-couplings, yielding lineshapes that are typically 0.1 ppm or greater. Due to their similar chemistries and the limited chemical shift range of 1H NMR, there is typically significant overlap between different metabolite resonances; macromolecules also contribute broad ‘background’ signals that overlap with the metabolite signals. Lipids can present a challenge to discriminating peaks in the 1 – 2 ppm range, due to the large signal from the alkyl chain groups present.
Figure 3.

1H and 31P MRS spectra within a rat brain at 11.1 T acquired on an AVIIIHD Bruker horizontal imaging system. 1H voxel is shown in Figure 2 while the 31P voxel encompasses almost then entire brain. Major brain metabolites are highlighted and are abbreviated as indicated in the text. Also shown are example spectral deconvolutions using a simple gaussian-lorentzian peak fitting algorithm for quantitation and the residual error between the experimental spectrum and the peak fits. No background correction has been performed, and a more refined fit could be achieved using the LC-model. The 1H spectrum is a STEAM sequence acquired at 470 MHz with 128 scans, an echo time of 5.6 ms and a recycle time of 2 s. The 1H MRS voxel size is 6.9 × 2.6 × 5.0 mm (~90 μl) and covers the cortex. The 31P spectra is also acquired on the same instrument using an ISIS sequence with 64 scans (8 complete ISIS scans) and a repetition time of 2 s. The 31P MRS voxel, 14.1 × 9.4 × 28.1 mm (~3.7 mL), approximately covers the entire brain.
Issues of signal normalization are also present. For comparative studies, it is essential to select a similar voxel position and volume between different subjects, but reproducibility can be limited by variations in animal position and size--signal variations due to coil tuning and positioning are much more significant than for ex-vivo NMR techniques. For this reason, resonance quantitation relies on normalization based on some aspect of the MRS spectra.[21] Common methods of normalization include acquisition of an unsuppressed water signal, normalization to a specific metabolite, or normalization to the ‘total’ signal. The first method of normalizing to the water signal from an unsuppressed scan theoretically corrects for differences in coil sensitivity, B1 field, and voxel size. The water content is also independent of metabolic changes that may affect other normalization techniques. Care must be taken to also correct for changes in receiver gain, which are often dynamically altered by modern hardware and can complicate signal normalization between scans. The second method is to normalize to a peak integral within the water suppressed spectra. A relatively high intensity peak such as creatine or NAA is chosen if it is known that the reference metabolite does not to vary between groups under investigation. Care must be taken in the analysis of data normalized in this method. All of the quoted metabolites are technically ratios, and if the chosen metabolite peak does vary significantly, either randomly or between groups, the real metabolic changes may be obscured. Finally, normalization can be done by using all of the identified peaks in the water-suppressed spectra. Normalizing to a total integral over the water-suppressed spectrum is not recommended due to the large overlaps with macromolecular peaks that can vary significantly due to changes in the voxel position and acquisition setup. Instead, normalization with this method should rely on the sum of the individual peaks areas (from either local integrals or peak deconvolution and fitting techniques). Either the sum of all the peaks can be used or a modified version of Probabilistic Quotient Normalization (PQN)[22] can be applied to calculate the most probable scaling factor. This technique requires 10 −15 peaks to produce adequate results, so all of the observable peaks in the 1H MRS spectra must be included in the analysis.
For 1H MRS, one of the first considerations for observing metabolites is suppression of the background signal from water, with an effective molarity of ~40 M which is thousands of times stronger than the metabolite concentrations. Water suppression is therefore a necessity for in vivo 1H MRS. Simple rf saturation of the water signal does not typically work well due to the breadth of the water signal. Instead, many water suppression techniques are based on CHESS (CHEmical Shift Selective saturation)[23] and use a train of frequency selective excitation pulses, coupled with spoiler gradients, to dephase and suppress the water signal. There are many modifications and improvements to the basic CHESS sequence, a full discussion of which is beyond the scope of this article. Many of these sequences are implemented on commercial instruments with some variation between equipment vendors. In a preclinical setting, VAPOR (VAriable Power rf pulses with Optimized Relaxation delays)[6] is the most commonly used water suppression approach. Additionally, many of the protons present in metabolites are in fast exchange with water protons at physiologic pH. For these reasons, 1H MRS spectra are typically only reported from 0 – 4.5 ppm, as resonances above 5 ppm are difficult to quantitate. In brain 1H MRS, metabolite signals below 4 ppm should be easily resolved with a flat baseline.
As stated above, voxel localization requires a minimum of three slice selective rf pulses. These can take the form of either refocusing or excitation pulses. There are many pulse sequences available for localization in combination with solvent suppression optimized for 1H MRS, and full reviews of many of the sequences can be found elsewhere.[15, 24] Here we will consider the most common types of sequences for 1H MRS. These can be broadly divided into sequences based on PRESS (Point RESolved Spectroscopy) and those based on STEAM (STimulated Echo Acquisition Mode). Each sequence comes with a mix of advantages and disadvantages, and no one sequence is optimal for all situations.
PRESS[25] is one of the simplest localization sequences. It has three rf pulses, a slice selective excitation followed by two slice-selective refocusing pulses. Typically, the echo time is relatively long, about 30 msec, and signal loss is primarily due to T2 relaxation. PRESS spectra exhibit significant CSO artefacts, particularly at higher field strengths. This is due to the bandwidth-limited refocusing pulses present in the sequence. The refocusing pulses also lead to a second issue of partial slice excitation. By definition, at the edges of the refocused slice, the proton spins will be excited rather than refocused. This leads to signal loss nearer the edges of the slices as well as significant unwanted excitation outside of the desired voxel. The unwanted excitation can be suppressed through spoiler gradients, phase cycling and OVS, but the signal loss can be harder to attenuate. Alternatives to traditional PRESS have been developed using adiabatic pulses, with LASER (Localization by Adiabatic Selective Refocusing)[26] and semi-LASER[27] now in wide spread use. LASER uses a hard excitation pulse followed by three sets of paired adiabatic refocusing pulses, while semi-LASER compromises with a slice-selective excitation followed by two pairs of adiabatic refocusing pulses. The adiabatic refocusing pulses are use in pairs to prevent phase dispersion which occurs across the slice. There are a number of advantages in using LASER based sequences when compared to traditional PRESS. The adiabatic pulses give excellent refocusing performance across the entire slice, which significantly decrease partial slice excitation issues. They also have a higher excitation bandwidth for a given peak power than traditional RF pulses, which helps to reduce the issues of CSO seen in traditional PRESS sequences. The use of adiabatic pulses has significant advantages when using surface transceive coils, which cannot otherwise produce good refocusing pulses due to their inhomogeneous B1 fields. Adiabatic refocusing pulses should always be used in transceive surface coils for this reason alone. There are however a number of drawbacks when compared to PRESS. The echo times for these sequences are long, typically at least double that seen in PRESS. This is less of a concern at lower fields, when T2 relaxation is typically slower, but can represent a more significant signal loss at higher fields. The total RF deposition of LASER is substantially higher than the PRESS sequence and can present issues related to the allowed specific absorption rate (SAR). Semi-LASER represents a compromise that lowers the amount of RF deposition without losing most of the advantages of LASER. While LASER is certainly to be favored with surface transceive coils, its advantages at higher fields and with volume coils, or volume transmit/ surface receive setups, is less clear. With modern MRS hardware and software, the use of on the fly Shinnar–Le Roux (SLR) calculated complex pulse shapes gives almost as good slice selection properties as the adiabatic hyperbolic secant pulses traditionally used in LASER without the need for paired refocusing pulses and increased echo time. PRESS with complex pulse shapes for optimal selectivity largely obviates the need for LASER and semi-LASER sequences in 1H MRS for preclinical studies where volume coil setups are readily available
STEAM[28] solves one of the main issues with PRESS, the bandwidth limited refocusing pulses, by replacing them with excitation pulses. This leads to the formation of a stimulated echo rather than a double spin echo as seen in PRESS. The stimulated echo allows a much shorter effective echo time, usually < 10 msec, and includes a mixing time, Tm, during which the spins undergo T1 evolution. Excitation pulses can be applied at a much higher bandwidth, reducing CSO issues. While there is signal loss at slice edges due to partial slice excitation effects, it is a less significant issue due to the nature of the stimulated echo. Signal loss during the excitation is from a mixture of T1 and T2 relaxation, and with T1 usually considerably larger then T2 in vivo this can help improve signal to noise for metabolites. However, stimulated echoes suffer one major drawback: ignoring relaxation effects, a stimulated echo only produces 50% of the SNR of a spin echo. At higher B0 fields, with significant CSO issues and shortened T2 for metabolites of interest, STEAM is the more favorable sequence choice despite this SNR penalty. At lower fields, where the overall signal intensity is lower, the CSO is more manageable and the longer T2 of metabolites present less of issue. Thus, PRESS based sequences are preferred. With surface transceive coils LASER or other adiabatic refocusing pulses, should be utilized.
Both sequences have numerous modifications for specific spectral editing purposes, such as DRYSTEAM[29] and MEGA-PRESS[9]. Spectral editing with the MEGA-PRESS sequence is often used to improve identification of GABA concentrations in vivo. GABA has a multiplet at ~3 ppm, which is very susceptible to J modulation. PRESS, and to a lesser extent STEAM due to its much shorter echo times, exhibit J-modulated attenuation of this signal in their resultant spectra due to the J-coupling not being effectively refocused during spin echo sequence. Due to the relatively broad peaks present in vivo, this effect is not immediately obvious in the peak shape, but it still leads to significant signal suppression of multiplets, particularly for GABA. At high fields, the use of STEAM with a short echo time can significantly reduce this effect. However, at lower fields where PRESS is typically used, this is less of a feasible option. A more general approach to combat J modulation is to potentially alter the PRESS sequence with the addition of a perfect echo, as used to good effect in ex vivo spectroscopy discussed below.
The chosen voxel size must strike a balance between SNR, spectral quality, and organ or tissue specificity. The available gradients and rf powers can also limit selectivity, although on most modern systems for preclinical MRS SNR tends to be the limiting factor. In principle SNR will decrease proportionally to the volume of the voxel, thus an isotropic 1-mm3 voxel will actually have only an 8th of the SNR of a 2-mm3 voxel. To obtain identical SNR, 64 times the number of scans would be required. This issue is further complicated by the need for voxel localized shimming. Most modern MRI scanners have multiple automated shimming and field mapping routines that can assist with obtaining an optimal shim. Best practice is to first obtain a basic FID shim on the whole sample, followed by automated B0 mapping focusing on the organ of interest, and finally narrowing down to the specific region of interest. Localized shimming using low order shims on the specific voxel is finally conducted in order to obtain the best line shape. For highest quality spectra, the voxel should not cross any interfaces, i.e. it should reside entirely within the brain. In practice, it is easier to shim on smaller isotropic voxels than larger non-isotropic shapes. In practice, a near isotropic voxel size of 1 – 2 mm3 works well for rodents and allows a specific brain region to be studied while still providing sufficient SNR.
Recent reviews of current practices in high field 1H MRS demonstrate the resolution that can be gained on moving from 3 T to 7 T, 14 T, and beyond. At 14.1 T, up to 19 brain metabolites can be measured and quantitated by spectral deconvolution.[10b] A commonly used deconvolution tool for quantitating 1H MRS spectra is LCModel which subtracts out background signals from macromolecules (obtained from a control spectrum) and models the spectral features of various brain metabolites at a given magnetic field based on their known chemical shifts and J-couplings.[30]
2.2. 31P in vivo MRS
31P is a 100% naturally abundant spin −½ nucleus, and thus in principle well suited to MRS experiments. A number of metabolites, such as ATP, NAD and Phosphocreatine, are at mM concentrations in tissue and can be easily detected. The main challenge with 31P MRS are the inherently very short T2’s of in vivo phosphate groups, which are typically < 20 msec and shorten with increasing field strength. This presents a challenge to even the shorter echo time MRS sequences, such as STEAM. For this reason, ISIS (Imaging Selected In vivo Spectroscopy)[31] has been commonly used for 31P MRS. ISIS is an interesting sequence, as it requires eight separate acquisition scans to be added together in a specific way to produce a fully localized signal. Each of the scans acquires an FID, rather than an echo, based upon inversion recovery sequences; the slice selective refocusing pulses precede a non-selective excitation pulse. The ISIS sequence is not motion tolerant, as it requires accurate summation of the eight scans. It is relatively slow, again due to the need to sum scans. A more recently proposed approach for use in animal models is the use of an OVS selected voxel. Essentially, six interleaved OVS bands can be used to suppress signal from everywhere in the sample except the voxel of interest. A simple non-selective excitation pulse is then used to acquire signal, which produces a pure FID. It is substantially faster than ISIS, and is very tolerant to poor B1 performance. This is important, as 31P experiments are typically performed using transceive surface coils which inherently have poor B1 homogeneity. Normalization of the signal intensities in a 31P MRS spectrum is typically achieved via total integration via a specific, invariant resonance, such as phosphocreatine. There are an insufficient number of resonances to use more complex normalization methods such as the PQN.
31P MRS can be used to measure the pH of tissue by determining the chemical shift difference between inorganic phosphate and phosphocreatine.[32] Of greater interest in metabolic studies are magnetization transfer methods[33] that allow measurement of de novo ATP synthesis from inorganic phosphate (Pi) and the metabolic conversion of ATP to Phosphocreatine to be studied. The γ-ATP resonance is saturated with narrow bandwidth rf pulses. This causes an associated change in the Phosphocreatine signal, which is related to the conversation rate of ATP to PCr. The OVS selected voxel localization method is particularly suited to these techniques, as it does not require the summing of eight separate scans used by ISIS. As MRS progresses to higher magnetic fields, the possibility of resolving α-ATP, NADH, NAD+ becomes possible.[34] At 11.1 T, we note that these resonances are not clearly resolved but can be deconvoluted using published chemical shifts and J couplings (Figure 3)
3. Dissolution Dynamic Nuclear Polarization (dDNP) to monitor metabolic flux
Although 13C NMR has the possibility to provide key insights into metabolism in vivo, it is inherently insensitive due to the low gyromagnetic ratio (γ) of the carbon nuclei in addition to the 1.1% natural abundance of the magnetically active 13C isotope. dDNP is a methodology that overcomes the inherent SNR limitations of 13C and is rapidly enabling 13C MRS as a cost effective and clinically applicable alternative to PET modalities for tracking metabolic flux because of three major benefits: the ability to observe downstream metabolic products, a much faster tracer clearance time, and no exposure to radioactive isotopes of CT irradiation.[35] Carbon hyperpolarization is necessary for in vivo metabolic flux measurements via MRS due to the large amount of signal averaging that would otherwise be required to overcome the inherent insensitivity of the 13C isotope. dDNP enables carbon polarization enhancement which is more than four orders of magnitude compared to thermal equilibrium for in vivo 13C MRS.[8, 36] This is achieved by transferring polarization from high γ spins (unpaired electrons) to low γ nuclei (13C) in a metabolic substrate at low temperature (~1–2 K) via microwave irradiation. This polarization can be retained while the polarized sample is subsequently dissolved in heated buffer and injected into a subject. The polarized metabolite can then be observed in vivo as it is metabolized into well-characterized products. This technique enables the measurement of metabolic flux in real time due to drastically increasing the SNR for a specific polarized 13C nucleus. The significant gain in SNR by DNP is transient and decays as a function of nuclear T1 time. Therefore, isotopically enriched carbonyl carbon groups are often selected due to their longer relaxation times.[37] Because of the temporal constraint on DNP observation, which is especially apparent in vivo, biochemical kinetics can only be modeled through a limited number of enzymatic steps. This is both advantage and a disadvantage in monitoring the TCA cycle as the hyperpolarized substrate is only observable in a single turn while multi-turn isotopomer contributions can be ignored due to the relaxation timescale. Another limitation of DNP is that substrate hyperpolarization is on the order of hours while NMR observation is on the order of minutes so most hyperpolarization experiments are single shot experiments and are not under true steady state tracer conditions.
Numerous substrates have been the target of polarization studies because of their potential as biological tracers for in vivo applications and to study their inherent NMR properties, and optimal radical and 13C substrate concentrations have been well characterized and optimized for maximum polarization in the solid state for many compounds.[38] To date, the most studied substrate using dDNP and 13C MRS is the [1-13C] pyruvate as it has a relatively long carbon T1 relaxation time (a necessity for retaining polarization during the melting and 13C MRS process), it is naturally self-glassing,[39] and it plays a central role in metabolism. After dissolution, pyruvate and its metabolic substrates retain sufficient polarization for monitoring via 13C MRS for 2–5 minutes.
Multiple products are observed in a typical dDNP pyruvate experiment (Figure 4), and pyruvate-to-lactate conversion, in particular, can provide a semi-quantitative indicator of intracellular glycolytic production through lactate production. Lactate production is an especially important biomarker for cancer diagnosis though the “Warburg Effect”.[40] In the healthy brain, lactate in an important indicator of high glycolytic metabolism, furthermore, there has been significant evidence that lactate is exchanged between astrocytes and neuron though the “Lactate Shuttle”.[41] This shuttle is a dynamic intercellular coupling by which the astrocytes supplement neuronal oxidative function through the lactate intermediate. Although dDNP has not been shown to be able to discriminate cellular compartmentalization of the lactate pools, hyperpolarized lactate production could offer global insights into the lactate shuttle paradigm. This is especially the case when lactate production is compared to hyperpolarized bicarbonate pool, which is a product of oxidative pyruvate dehydrogenase (PDH) activity. Due to the transient nature of polarized 13C signals, rapid acquisition multiple one-dimensional 13C spectra is necessary for observation tracking of conversion and accurate kinetic modeling. Conversion of pyruvate to lactate can be modeled using simple first order kinetics, taking into account T1 relaxation of 13C polarization and the use of small tip angle rf “read” pulses (Figure 4). Although this basic model is appropriate for simple comparison of different metabolic states, models that are more complex have also been described.[42] These alternative kinetic models account for multiple physical compartments, intercellular/extracellular transport, and bidirectional kinetic exchange.
Figure 4.

13C MRS spectrum after injecting polarized [1-13C] pyruvate into a rat brain. The associated in-vivo brain kinetics monitored for conversion of polarized Pyr to Lac are also shown. The 13C spectra were localized using a small surface coil placed adjacent to the brain on the rat’s head. The repetition time was 1 s with a nominal 10° excitation pulse.
4. Quantitation of metabolites in tissues via high resolution magic angle sample spinning (HRMAS)
In-vivo 1H MRS techniques are non-invasive, and provide the most comprehensive, physiologically relevant data on metabolic activity in the brain of any MR method. However, significant line broadening is observed for in-vivo spectra, due in large part to magnetic susceptibility broadening as well as the reduced molecular motion which occurs in the semi-solid environment of the brain tissue.[43] Reduced molecular motion leads to only a partial averaging of dipolar couplings and chemical shift anisotropies.[44] Excised tissue, particularly brain tissue due to its semi-solid nature, is ideal for study via High Resolution Magic Angle Spinning (HRMAS) techniques.[45] HRMAS involves the use of a specialized NMR probe that can spin the tissue, packed into a rotor, at the magic angle of 54.7 ° relative to the main magnetic field, B0. Modern HRMAS probes represent a hybrid between traditional MAS probes developed for solid-state NMR applications and solution-state NMR probes, as they typically have a stator for tight control of the rotor spinning frequency and angle combined with a deuterium lock channel and a gradient coil that is orientated along the axis of rotation. They are designed to deliver radiofrequency fields typical of solution NMR pulse sequences, as small molecules have sufficient motion in tissues to reduces dipolar couplings and chemical shift anisotropies to on the order of a few kHz. Thus, when tissues are spun fast enough, the MAS rate is sufficient to remove the residual broadening of these interactions in addition to removing magnetic susceptibility broadening. The net effect is spectra in which small molecules exhibit sharp resonances while more immobile macromolecules contribute to broad peaks underlying these resonances (Figure 5). Typically, a sample volume of < 100 μL is used and spinning rates are lower than those used in traditional MAS methods, with rates of up to 5 kHz or even lower for particularly fragile samples.[46] For a well-shimmed brain tissue sample, line widths approaching those seen in a solution state sample can be achieved for small molecule metabolites, typically about 0.005 PPM. This allows for robust identification and quantitation of a significantly greater number of metabolites than in in-vivo MRS while still studying relatively intact tissue. An additional benefit is HRMAS probes are compatible with conventional NMR systems, and data can be collected at fields up to 23.5 T (1000 MHz 1H).
Figure 5.

Comparative 1H NMR spectra at 14.1 T from rat tissue via HRMAS and from tissue extract via conventional solution NMR. Spectra were acquired using a 600 MHz Bruker AVIIIHD NMR spectrometer. Major metabolites are highlighted and are abbreviated as indicated in the text. The HRMAS data was acquired on 20 mg of cortex tissue spinning at 5 kHz in a 4 mm rotor at 4 °C using a CPMG sequence with 128 scans, an effective echo time of 30 ms and a recycle time of 5 s. The extract NMR spectrum was acquired on extracted metabolites from 75 mg of tissue reconstituted in 500 μL of buffer using a 1D NOESY water presaturation sequence, with 32 scans and a recycle time of 5 s and a mixing time of 100 ms. Inset: HRMAS rotor and sample insert for tissue containment.
Sample preparation is important in obtaining the highest quality spectra from HRMAS. The first step of tissue harvest is most important for biological reproducibility and requires extensive training for proficiency. Because the brain is encased in the skull, isolation of the region of interest without damaging the tissue must be performed carefully, but also rapidly enough to prevent severe hypoxia. This procedure is typically performed using a guillotine and surgical scissors followed by careful dissection of the region of interest and rapid flash freezing of the tissue in liquid nitrogen for storage at −80 °C prior to collection of NMR data.[47] Accuracy in dissection is paramount, and use of a brain atlas and dissection matrix is recommended.[48]
HRMAS probes have a range of rotor sizes, but 4 mm rotors are a typical diameter and hold < 100 μL of sample. Tissues are typically packed into single use Kel-F rotor inserts (Figure 5 inset). These have a number of advantages, despite decreasing the sample volume, and thus in principle the SNR, to 50 μL or less. They allow a higher throughput of samples, as the rotors do not need extensive cleaning between samples, they reduce the chance of contamination between samples, and they enhance containment of tissues that are biohazardous. More importantly, they confine the tissue to the center of the rotor, which greatly improves the ease of shimming and helps eliminate some of the poor line shape issues sometimes observed with HRMAS. The inserts are loaded with a tissue plug such that no trapped air bubbles are present. It is possible to use a narrow gauge syringe to remove any air bubbles after the tissue is loaded into the insert. D2O is added to the sample at this point to provide a lock signal, but is minimized to prevent leaching of metabolites from the tissue to the surrounding D2O. Typically 5 – 10 % D2O by volume is sufficient. The insert is then sealed, again ensuring no are bubbles are present, and placed inside the MAS rotor.
Typically, samples are spun at rates up to 5 kHz and at temperatures of 2 – 4°C to minimize further degradation of the tissue. Samples must be individually shimmed to provide the best results. Automated shimming protocols based on the lock signal can be used as a starting point, but may be of limited use as the majority of the deuterium lock signal is from outside the tissue, rather than the sample regions containing the metabolites. Shimming on the water signal can also be misleading, but provides a useful starting point if no other method is available. Once the lower order shims have been adjusted based on the water signal, it is best to shim using a resonance from a relatively concentrated metabolite that produces a singlet peak in the tissue spectrum. Typically, either the Creatine (~3 ppm) or NAA (~2 ppm) peaks are used in brain tissue samples. A simple presaturation water suppression sequence can be used, and then the shims optimized on either of these peaks. The line width and symmetry of the line shape should be considered, but it is also important to minimize any tails or shoulders which occur on the peaks, as they can hamper later analysis of the metabolite composition in the tissue.
The main challenges in choosing an NMR sequence are twofold: The choice of water suppression technique and how to deal with broad macromolecule ‘background’ peaks. The water peak must be suppressed in order to obtain good spectra of the metabolites, but ideally the chosen technique will not significantly affect regions of the spectra containing signals from the metabolites. There are a number of review articles that deal with the many different suppression techniques available,[21] and most will produce adequate results in HRMAS of brain tissue samples. In practice, since the NMR signals from the metabolites of interest largely lie below 4.2 ppm, full suppression of the water peak is not required. Simple water presaturation-based sequences provide adequate results, although care must be taken in choosing an appropriate presaturation power to avoid signal suppression of metabolite peaks closer to the water peak, which will affect quantitation. More complex sequences, such as WATERGATE based options, can lead to differential diffusion weighting of the metabolite peaks.
Biological macromolecules, such as DNA, lipids, and proteins, present in the brain tissues are not as averaged by molecular motions compared to small molecule metabolites, and thus exhibit broad ‘background’ peaks in HRMAS spectra, which, if not attenuated, will limit quantitation of the metabolites. There are two main methods for handling these unwanted signals. The first is to acquire a largely unfiltered 1D 1H NMR spectra of the brain tissue using a 1D NOESY sequence.[47, 49] This gives an improved baseline over the standard pulse acquire sequence, but still includes large contributions from the macromolecules present. The unwanted broad peaks are then removed in the processing step, essentially by fitting broad peaks to them, either using something as simple as cubic spline regression or a more complicated peak fitting algorithm. The remaining small metabolite peaks can then be analyzed after subtraction of the macromolecule peaks. The main issue with this technique is the large variability which can occur between spectra in removing the macromolecule peaks while leaving behind ‘clean’ small metabolite spectra. The second method is to suppress the macromolecule signals during NMR acquisition. This is done using a T2 filter, most typically using a CPMG sequence.[50] The macromolecules have significantly shorter T2’s than the small metabolites, so applying a T2 filter of ~10 – 20 msec significantly suppresses the unwanted peaks. There is an SNR penalty introduced compared to the pulse acquire or 1D NOESY sequences, as well the introduction of T2 contrast. Depending on the analysis performed, this may not be an issue especially in comparing similar or identical tissue types. For improved quantitation, T2 correction can be optimized for various metabolites. This can be done by acquiring several T2 filtered spectra on the same sample with different echo times, although this does increase the total acquisition time per sample. A significant drawback of the CPMG sequence for analyzing mixtures of metabolites is the J modulation that occurs during the refocusing echoes,[51] which has a significant effect on subsequent data analyses. To reduce J modulation, the time between refocusing pulses can be shortened, but this can lead to sample heating due to the larger amount of radiofrequency energy deposition. In practice, an inter-echo time is chosen that experimentally gives the least obvious issues with J modulation for metabolites of interest. An alternative sequence, Periodic Refocusing of J Evolution by Coherence Transfer (PROJECT),[51b] initially proposed for use in solution NMR experiments (below), has successfully been demonstrated in low-rotational rate HRMAS experiments on delicate tissues.[46] It essentially introduces additional excitation pulses interspersed in the echo train and applied at a 90° phase offset relative to the original excitation pulse. The concept is based on ‘perfect’ echoes.[52] These additional pulses are intended to ‘refocus’ the J coupling evolution which underlies the original CPMG sequence. Here, we suggest its general applicability as a replacement to CPMG-based T2 filters for all HRMAS experiments. The specific implementation of the PROJECT sequence we use has an extended phase cycle that improves its robustness for slightly miscalibrated radiofrequency pulses. The PROJECT sequence provides identical T2 filtering, with no additional SNR penalty and a minimal increase in rf energy deposition compared to the traditional CPMG sequence.
With a properly prepared sample, HRMAS can produce spectra with a resolution approaching that seen in a solution state spectrum of a polar extract while retaining the physiological relevancy of intact, albeit deceased, tissue. While there are some unavoidable changes in metabolite pools which occur when the animal is sacrificed, such as the large production of lactate due to hypoxia, HRMAS of tissues still provides an excellent compromise between the loss of signal resolution seen in-vivo and the potential loss or conversion of metabolites when preparing highly processed polar extracts.
5. Quantitation of metabolites via high resolution NMR of tissue extracts
Of all the NMR technique we discuss, ex vivo extraction based analysis is the most quantitative, due primarily to the superior line shapes and higher resolution afforded by removal of macromolecules. Additionally, solution state NMR approaches enjoy the technical benefits of high, homogeneous magnetic fields, high throughput, and excellent reproducibility. NMR of extracted biological samples also has additional advantages over mass spectrometry-based small molecule profiling, as it is non-destructive and can identify positional labeling of isotopomers.[53] Furthermore, extract NMR can also be applied to almost any biological sample including blood, CSF, tissue, efferent tissue perforate, etc. Although classical solution state NMR remains the gold standard of NMR based small molecule profiling, there are significant limitations that should be considered. Compared to HRMAS and in vivo approaches, extract NMR is the most static measure of metabolism and the furthest removed from a natural biological state. Furthermore, as a part of the extraction process, there are additional risks of degradation, oxidation, or loss of volatile compounds and relative amounts of metabolites can strongly vary across different extraction protocols.
Broadly speaking, brain extract NMR has five basic steps: rapid tissue dissection (as described above for HRMAS of tissues), flash freezing of the tissue, pulverization of the tissue, extraction of polar and/or nonpolar metabolites, reconstitution of the metabolites in a defined solvent system, and NMR acquisition. Pulverization is typically accomplished by first grinding the frozen tissue in a mortar and pestle then homogenizing it using a hand held or bead homogenizer.[47] Here, we focus on a standard extraction of polar metabolites that is commonly used for isolating metabolites of interest to brain metabolism. Polar metabolites are extracted by suspending the pulverized tissue in a solution that contains equal volumes of chloroform, methanol, and water at 20 °C followed by centrifugation to remove the insoluble macromolecule fraction. To minimize oxidation of redox enzymes (NADPH, NADH), the solvents can be degassed with argon or helium.[54] The water-containing phase is then lyophilized to dryness and stored at −80 °C until NMR analysis is performed. Just prior to NMR acquisition, samples are reconstituted in degassed, buffered D2O (generally phosphate buffered saline) at neutral pH, with an internal NMR standard (TSP or DSS) added to a final concentration of 1–0.5 mM. NMR pulse sequences optimized for metabolomics analyses, discussed below, enable reproducible quantitation of metabolites while removing signals for residual macromolecules. If sample preparation protocols are consistently followed, metabolomics profiling by solution state NMR is a robust and reproducible technique that is quantitative and can complement HRMAS and in vivo spectral characterization due to well-resolved chemical shifts and J-couplings. Furthermore, because of the lyophilization step after extraction, water contamination is nominal, allowing for near-complete suppression of the water resonance and quantification of metabolomics resonances, such as glucose, very near to water. The dynamic range of solution NMR at high field enables quantitation of both major and minor metabolite species, with lower detection limits in the micromolar range.
The two gold standard pulse sequences for NMR based metabolomics are the 1D nuclear Overhauser enhancement spectroscopy (NOESY) sequence and the T2-weighted Carr−Purcell− Meiboom−Gill (CPMG), both with presaturation for water suppression.[47] Extraction of the cerebral tissue prior to NMR acquisition removes a majority of the non-polar compounds with short T2’s and reduces the need for a T2-filtered approach. Nonetheless, due to residual lipids remaining, often a T2-filtered approach should be considered to improve the quality of the NMR spectra. Although the CPMG sequence has been a well-regarded approach for decades for characterizing lipid-rich samples, such as brain tissue extracts, recent modifications developed to correct for homonuclear J-modulations during the refocusing echo train have improved this technique for quantifying mixtures of metabolites. As discussed for its use in HRMAS studies, the PROJECT sequence quenches J-modulation artifacts by inserting a 90° pulse at the center of a double spin echo loop of the CPMG sequence[51b] and has been rapidly adopted in metabolomics analyses.
Removal of macromolecules, spectral editing using T1 and/or T2 filters, superior water suppression, and high magnetic field homogeneity enable baseline resolution of many metabolite and ease of spectral deconvolution for more congested regions of the solution NMR spectra. Recent work on blood and urine samples has demonstrated the sensitivity of spectral assignments to buffer conditions, but with uniform buffer conditions or spectral determination of small changes in pH and ion concentrations, dozens of metabolites can be assigned and quantitated in an automated fashion.[55] As demonstrated in Figure 5, all the major brain metabolites are easily identifiable by inspection of a typical 1H NMR spectrum from a brain tissue polar extract. Spectral deconvolution allows quantitation and comparative analyses across samples. At fields of 14.1–18.8 T (600 – 800 MHz 1H), the majority of metabolite resonances are resolved into simple multiplets, the primary exceptions being Gln and, to a lesser extent, Glu amino acids due to overlap of the sidechain chemical shifts. This overlap necessitates consistent buffer pH across samples to prevent shifts in resonances affecting their ABX patterns.[56]
6. Brain metabolite standards for methods optimization and quality control
In order to optimize existing methods, to ensure reproducibility in quantitation over time and among instruments, and to develop new techniques, a high quality MRS phantom for brain metabolites is required. Ideally, it should replicate many of the features found in real samples, but with pre-determined properties, such as metabolite concentration, pH and buffering, salt concentrations, and molecular diffusion rates. This enables facile evaluation of newly developed sequences or analysis methods and instrument performance can be checked against a known sample. Traditionally, MRS sequences are qualified using a phantom based on “Braino”.[57] Braino is a solution developed by GE for testing of human MRI scanners that contains eight of the highest concentration, and hence most easily detected, metabolites in the brain (Table 1). Metabolite concentrations approximate to those seen in the brain are dissolved in buffer with a chemical shift reference, such as TSP, and gadolinium is added so that the metabolite relaxation times are near to those found in brain tissue. Braino is ideal for basic coil performance testing, but lacks many of the more complex features seen in real brain tissue and extract. As a simple solution, it also lacks the broader resonances seen in-vivo or in brain tissues via HRMAS. With few overlapping resonances, its potential for testing more advanced sequence, peak fitting, and deconvolution methods is also limited (Figure 6). Due to these limitations, we have developed a more complex mixture that we call “Braino 2.0” (Table 1). It increases the number of metabolites to 12, to more accurately reproduce the major spectral features seen brain extract spectra. The metabolite concentrations are derived from the metabolite ratios found via HRMAS NMR analysis of brain tissue from healthy Sprague Dawley rats (Figure 5), but are rounded to the nearest 0.5 mm in order to simplify the Brain 2.0 recipe. Both Braino and Braino 2.0 can be made with the acid or salt forms of the metabolites, as long as the final mixture is neutralized to physiological pH. The basic Braino 2.0 mixture is suitable for testing solution state sequences and spectral fitting routines. However, for testing of potential in-vivo sequences we add a thickening agent to the solution. By increasing the viscosity of the mixture, the molecular diffusion rates of the small metabolites are decreased so that their resonances more closely mimic those observed via in vivo 1H MRS. Sodium alginate is a particularly suitable thickening agent as the majority of its NMR resonances occur > 4 ppm, so they do not significantly overlap with the spectral regions used to quantify brain metabolites. It also does not require the resulting gel to be set or polymerized, and it is stable over a timescale of days to weeks. Over longer periods of time (months to years) the alginate chains can break down, reducing the viscosity of the gel. The concentration of alginate can be varied depending on the degree of broadening required and use of a thickening agent, rather than a relaxation agent, to increase spectral linewidths better mimics the line broadening mechanisms seen in relatively homogenous brain tissue. Thus, the “Braino 2.T” solution represents an ideal standardized imaging phantom for development and refinement of in vivo MRS sequences, while maintaining the reproducibility of metabolite concentrations seen in basic Braino phantoms.
Table 1:
Concentrations in Braino & Braino 2.0
| Braino (mM) | Braino 2.0 (mM) | |
|---|---|---|
| N-acetyl-L-Aspartic Acid | 12.5 | 6.5 |
| Choline Chloride | 3.0 | 0.5 |
| Creatine Monohydrate | 10.0 | 8.0 |
| Myo-Inositol | 7.5 | 2.0 |
| L-Glutamic Acid | 12.5 | 4.0 |
| Sodium Lactate | 5.0 | 3.0 |
| L-Phenylalanine | 1.0 | 2.5 |
| L-Alanine | 1.0 | 1.0 |
| γ-aminobutyric acid | - | 2.0 |
| Taurine | - | 3.5 |
| L-Aspartic Acid | - | 1.0 |
| Sodium Succinate | - | 0.5 |
| Trimethylsilylpropanoic acid (TSP) | 1.0 | 1.0 |
| Potassium Phosphate Monobasic | 50.0 | 50.0 |
| Gadodiamide (Omniscan™) | 0.5 | 0.5 |
| Sodium Azide | 1.0 | 1.0 |
Figure 6.

Comparison 1H NMR spectra of Braino, Braino 2.0, and Braino 2.T. See text and table for details on composition. Spectra were all acquired using a 5 mm solution NMR probe at 11.7 T using a Bruker 500 MHz AVIIIHD system. Automated gradient shimming was performed on all samples. All spectra were acquired at 25 °C with a water presaturation sequence. 32 scans were acquired with a recycle time of 3 s.
7. Comparison of results from magnetic resonances approaches at 11.1 – 14.1 T
A few studies have been published comparing in vivo data to HRMAS spectra[58] or HRMAS and extract spectra[59] for particular biological tissues. To our knowledge, this perspective is the first comparing all three types of 1H data in the same tissue type and conconcurrently evaluating information regarding metabolism achievable using 13C dissolution DNP data and 31P MRS data for similar timescales. By comparing the in vivo data from Figures 3 and 4 with the ex vivo data from Figure 5 and phantom data from Figure 6, several interesting insights emerge regarding how to quantitate particular metabolism pathways in the brain and future prospects for in vivo MRS at high fields. In Figure 7 we summarize the major metabolites which can be directly integrated or deconvoluted and quantified, rather than fit using model values for known metabolites, on progressing from real-time in vivo measurements, to longer scale in vivo experiments, to ex vivo tissue and ultimately tissue extracts.
Figure 7.

A simplified schematic map of neuron and astrocyte metabolism in the brain with metabolites color-coded for MR technique by which they can first be quantified using simple spectral deconvolution on progressing from in vivo to ex vivo methods. Lac (*) indicates the unusually high amounts seen ex vivo due to hypoxia.
The major neurotransmitters are all clearly visible via 1H MRS, but, due to their overlapping chemical shifts, Glu, Gln, and GABA remain difficult to quantify at 11.1 T. Asp is clearly distinguished even though it is present at a much lower concentration. Gly cannot be deconvoluted due to strong overlapping signals from myo-Ins. Taur can easily be tracked given its strong signal at ~3.4 ppm. All the neurotransmitter signals can easily be deconvoluted and quantified from either extract NMR or tissue HRMAS spectra. Their relative signal ratios are reasonably consistent between the two techniques. This suggests that ex vivo methods are accurate for measuring relative concentrations of neurotransmitters when tissue harvesting is a viable option. Of interest for future technique development is the fact that the neurotransmitter signals look very similar when comparing the spectrum of Braino 2.T to the 1H MRS spectrum. More intriguingly, these spectra suggest loss of resolution in vivo is primarily due to homogeneous broadening and that new methods which sufficiently overcome dipolar and J-coupling interactions in vivo may improve resolution for 1H MRS sufficiently to enable accurate quantitation of all the major neurotransmitters in vivo at fields of 7 – 11 T.
Several interesting phenomena emerge when comparing methods for quantifying organic acids. Ac, Pyr, and Succ not directly observable via 1H MRS; Lac heavily overlaps with macromolecules, limiting its accuracy of quantitation. Ac and Lac are clearly distinguished by HRMAS while Succ and Pyr are unobserved. Ac, Lac, and Succ are all clearly distinguished in NMR of tissue extract. Thus, the ability to monitor pyruvate conversion via dDNP stands out, as none of the other methods are able to quantify steady state levels of pyruvate. This is likely due to a lack of resolution for 1H MRS, but for ex vivo methods tissue hypoxia leads to almost complete conversion of pyruvate to lactate, even with efficient tissue harvesting methods, emphasizing the high energy needs within functioning brain tissue. Thus, any comparisons of pyruvate and lactate pools or conversion rates should rely on in vivo methods. In quantifying Ac, it is interesting to note the resonance is at just below 1.9 ppm in the extract NMR spectrum, but is at ~1.91 ppm in the HRMAS spectrum. This shift is due to differences in pH. Of more consequence to metabolite quantitation, the Ac peak is attenuated in the extract spectrum due to sublimation during the lyophilization step in sample preparation. In comparing HRMAS and extract NMR spectra, Succ displays the most unusual behavior—it is completely missing from the HRMAS spectrum while clearly visible in the extract NMR spectrum. This is likely due to Succ in the mitochondria being bound to or in close proximity to the metalloenzyme succinate dehydrogenase or other respiratory enzymes.
For monitoring energy balance, 31P MRS at 11 T can clearly monitor ATP vs PCr levels. More intriguingly, while NADH and NAD+ are not fully resolved from the α-ATP peak, modeling of the NADH, NAD+ resonances using published studies on their field dependence[60] suggests they can be quantified. Similarly, recent work has shown redox potentials can be monitored in human brain at 7 T.[61] These results indicate that combining 31P MRS with 1H MRS and potentially 13C dDNP at 7 T is likely to give unprecedented insights into metabolism in the human brain and changes with development, aging, and disease. Similarly, while NAA and myo-Ins are distinguishable in 1H MRS at lower magnetic fields, at fields of 7T and above their measurements becomes more quantitative and thus can provide complimentary insights on osmoregulation and energy storage.
Finally, we note glucose and branched-chain amino acids (Ile, Leu, and Val) are only distinguishable in NMR of extracted tissue. While it is unlikely they will they will become quantifiable in 1H MRS at 7–14 T using current methods, we note that new techniques which overcome dipolar couplings and J modulation in vivo may enable their measurement using model-based fitting routines, such as LCmodel.
Neither in vivo MRS nor ex vivo NMR techniques can fully elucidate questions regarding small molecule brain metabolism. Here, we have discussed a range of ex vivo and in vivo MR techniques that provide complementary data. Instead of focusing on a single methodology, one can integrate data from all of these methods to produce a better cross section of information regarding brain metabolism, both in terms of steady-state metabolite pools and metabolic flux in order to better answer questions regarding brains metabolism. With future improvements to available hardware, pulse sequences and computer analysis techniques, ever more subtle changes in brain metabolism can be observed.
Acknowledgements
A portion of this work was performed in the McKnight Brain Institute at the National High Magnetic Field Laboratory’s AMRIS Facility, which is supported by National Science Foundation Cooperative Agreement No. DMR-1157490, the State of Florida, University of Florida Division of Sponsored Research. Instrumentation upgrades for the 11.1 T MRI/S system and the 600 MHz Avance IIIHD system were supported in part by NIH instrumentations awards S10RR025671 and S10RR031637, respectively. During the review of this work, the NMR community lost two wonderful colleagues in biological NMR spectroscopy, Michele Auger and Stefano Caldarelli. This perspective is dedicated to their memory.
REFERENCES
- [1].Tognarelli JM, Dawood M, Shariff MI, Grover VP, Crossey MM, Cox IJ, Taylor-Robinson SD, McPhail MJ, J Clin Exp Hepatol 2015, 5, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yarach U, In MH, Chatnuntawech I, Bilgic B, Godenschweger F, Mattern H, Sciarra A, Speck O, Magn Reson Med 2017, 78, 2250–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a. Tkác I, Oz G, Adriany G, Uğurbil K, Gruetter R, Magn Reson Med 2009, 62, 868–879; [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Henning A, Neuroimage 2018, 168, 181–198. [DOI] [PubMed] [Google Scholar]
- [4].a. Shemesh N, Rosenberg JT, Dumez JN, Muniz JA, Grant SC, Frydman L, Nature Com 2014, 5; [DOI] [PubMed] [Google Scholar]; b. Moser E, Laistler E, Schmitt F, Kontaxis G, Front. Physics 2017, 5, 15. [Google Scholar]
- [5].Govindaraju V, Young K, Maudsley AA, NMR Biomed 2000, 13, 129–153. [DOI] [PubMed] [Google Scholar]
- [6].Tkác I, Starcuk Z, Choi IY, Gruetter R, Magn Reson Med 1999, 41, 649–656. [DOI] [PubMed] [Google Scholar]
- [7].a. Rothman DL, De Feyter HM, de Graaf RA, Mason GF, Behar KL, NMR Biomed 2011, 24, 943–957; [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Sonnay S, Gruetter R, Duarte JMN, Front Neurosci-Switz 2017, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]; c. Valette J, Tiret B, Boumezbeur F, Anal Biochem Anal Biochem 2017, 529, 216–228. [DOI] [PubMed] [Google Scholar]
- [8].Ardenkjaer-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, Servin R, Thaning M, Golman K, Proc Natl Acad Sci U S A 2003, 100, 10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mescher M, Merkle H, Kirsch J, Garwood M, Gruetter R, NMR Biomed 1998, 11, 266–272. [DOI] [PubMed] [Google Scholar]
- [10].a. Gruetter R, Weisdorf SA, Rajanayagan V, Terpstra M, Merkle H, Truwit CL, Garwood M, Nyberg SL, Ugurbil K, J. Magn. Reson 1998, 135, 260–264; [DOI] [PubMed] [Google Scholar]; b. Xin L, Tkáč I, Anal Biochem 2017, 529, 30–39. [DOI] [PubMed] [Google Scholar]
- [11].Minati L, Aquino D, Bruzzone MG, Erbetta A, J Med Phys 2010, 35, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Marjanska M, Iltis I, Shestov AA, Deelchand DK, Nelson C, Ugurbil K, Henry PG, J. Magn. Reson 2010, 206, 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a. Kantarci K, Jack CR, Xu YC, Campeau NG, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Kokmen E, Tangalos EG, Petersen RC, Neurology 2000, 55, 210–217; [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Winsberg ME, Sachs N, Tate DL, Adalsteinsson E, Spielman D, Ketter TA, Biol. Psychiatry 2000, 47, 475–481; [DOI] [PubMed] [Google Scholar]; c. Chang L, Lee PL, Yiannoutsos CT, Ernst T, Marra CM, Richards T, Kolson D, Schifitto G, Jarvik JG, Miller EN, Lenkinski R, Gonzalez G, Navia BA, Consortium HM, Neuroimage 2004, 23, 1336–1347. [DOI] [PubMed] [Google Scholar]
- [14].Shimada N, Graf R, Rosner G, Heiss WD, J. Neurochem 1993, 60, 66–71. [DOI] [PubMed] [Google Scholar]
- [15].van der Graaf M, Eur Biophys J 2010, 39, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brown TR, Kincaid BM, Ugurbil K, Proc Natl Acad Sci U S A 1982, 79, 3523–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a. Pauly J, Le Roux P, Nishimura D, Macovski A, IEEE Med Imag 1991, 10, 53–65; [DOI] [PubMed] [Google Scholar]; b. Balchandani P, Pauly J, Spielman D, Magn Reson Med 2010, 64, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a. McKinnon G, Boesiger P, Magn. Reson. Med. Biol 1990, 4, 101–111; [Google Scholar]; b. Slotboom J, Mehlkopf AF, Bovee WMMJ, J. Magn. Reson. A 1994, 38–50. [Google Scholar]
- [19].Felmlee JP, Ehman RL, Radiology 1987, 164, 559–564. [DOI] [PubMed] [Google Scholar]
- [20].a. Haacke EM, Xu YB, Cheng YCN, Reichenbach JR, Magn Reson Med 2004, 52, 612–618; [DOI] [PubMed] [Google Scholar]; b. Wharton S, Bowtell R, Neuroimage 2010, 53, 515–525. [DOI] [PubMed] [Google Scholar]
- [21].Giraudeau P, Tea I, Remaud GS, Akoka S, J Pharm Biomed Analysis 2014, 93, 3–16. [DOI] [PubMed] [Google Scholar]
- [22].Dieterle F, Ross A, Schlotterbeck G, Senn H, Anal Chem 2006, 78, 4281–4290. [DOI] [PubMed] [Google Scholar]
- [23].Haase A, Frahm J, Hänicke W, Matthaei D, Phys Med Biol 1985, 30, 341–344. [DOI] [PubMed] [Google Scholar]
- [24].Zhu H, Barker PB, Methods Mol Biol 2011, 711, 203–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bottomley PA, Annals New York Acad Sci 1987, 508, 333–348. [DOI] [PubMed] [Google Scholar]
- [26].a. Slotboom J, Mehlkopf AF, Bovee W, J. Magn. Reson 1991, 95, 396–404; [Google Scholar]; b. Garwood M, DelaBarre L, J. Magn. Reson 2001, 153, 155–177. [DOI] [PubMed] [Google Scholar]
- [27].Scheenen TW, Klomp DW, Wijnen JP, Heerschap A, Magn Reson Med 2008, 59, 1–6. [DOI] [PubMed] [Google Scholar]
- [28].Frahm J, Bruhn H, Gyngell ML, Merboldt KD, Hänicke W, Sauter R, Magn Reson Med 1989, 9, 79–93. [DOI] [PubMed] [Google Scholar]
- [29].Moonen CT, van Zijl PCM, J. Magn. Reson 1990, 88, 28–41. [Google Scholar]
- [30].a. Provencher SW, Magn Reson Med 1993, 30, 672–679; [DOI] [PubMed] [Google Scholar]; b. Provencher SW, NMR Biomed 2001, 14, 260–264. [DOI] [PubMed] [Google Scholar]
- [31].Ordidge RJ, Connelly A, Lohman JAB, J. Magn. Reson 1986, 66, 283–294. [Google Scholar]
- [32].Moon RB, Richards JH, J Biol Chem 1973, 248, 7276–7278. [PubMed] [Google Scholar]
- [33].a. Du F, Zhu XH, Qiao H, Zhang X, Chen W, Magn Reson Med 2007, 57, 103–114; [DOI] [PubMed] [Google Scholar]; b. Brindle KM, Blackledge MJ, Challiss RAJ, Radda GK, Biochem 1989, 28, 4887–4893. [DOI] [PubMed] [Google Scholar]
- [34].Zhu XH, Lu M, Chen W, J. Magn. Reson 2018, 292, 155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].a. Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, Ferrone M, van Criekinge M, Chang JW, Bok R, Park I, Reed G, Carvajal L, Small EJ, Munster P, Weinberg VK, Ardenkjaer-Larsen JH, Chen AP, Hurd RE, Odegardstuen LI, Robb FJ, Tropp J, Murray JA, Sci Transl Med 2013, 5, 198ra108; [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Kurhanewicz J, Vigneron DB, Brindle K, Chekmenev EY, Comment A, Cunningham CH, Deberardinis RJ, Green GG, Leach MO, Rajan SS, Rizi RR, Ross BD, Warren WS, Malloy CR, Neoplasia 2011, 13, 81–97; [DOI] [PMC free article] [PubMed] [Google Scholar]; c. Gallagher FA, Bohndiek SE, Kettunen MI, Lewis DY, Soloviev D, Brindle KM, J Nucl Med 2011, 52, 1333–1336. [DOI] [PubMed] [Google Scholar]
- [36].a. Nelson SJ, Vigneron D, Kurhanewicz J, Chen A, Bok R, Hurd R, Appl Magn Reson 2008, 34, 533–544; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yen YF, Nagasawa K, Nakada T, Magn Reson Med Sci 2011, 10, 211–217. [DOI] [PubMed] [Google Scholar]
- [37].Keshari KR, Wilson DM, Chem Soc Rev 2014, 43, 1627–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Keshari KR, Wilson DM, Chem Soc Rev 2014, 43, 1627–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lama B, Collins JHP, Downes D, Smith AN, Long JR, NMR Biomed 2016, 29, 226–231. [DOI] [PubMed] [Google Scholar]
- [40].a. Albers MJ, Bok R, Chen AP, Cunningham CH, Zierhut ML, Zhang VY, Kohler SJ, Tropp J, Hurd RE, Yen YF, Nelson SJ, Vigneron DB, Kurhanewicz J, Cancer Res 2008, 68, 8607–8615; [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Day SE, Kettunen MI, Gallagher FA, Hu DE, Lerche M, Wolber J, Golman K, Ardenkjaer-Larsen JH, Brindle KM, Nat Med 2007, 13, 1382–1387. [DOI] [PubMed] [Google Scholar]
- [41].Magistretti PJ, Allaman I, Nat Rev Neurosci 2018, 19, 235–249. [DOI] [PubMed] [Google Scholar]
- [42].a. Harrison C, Yang C, Jindal A, DeBerardinis RJ, Hooshyar MA, Merritt M, Dean Sherry A, Malloy CR, NMR Biomed 2012, 25, 1286–1294; [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Bankson JA, Walker CM, Ramirez MS, Stefan W, Fuentes D, Merritt ME, Lee J, Sandulache VC, Chen Y, Phan L, Chou PC, Rao A, Yeung SC, Lee MH, Schellingerhout D, Conrad CA, Malloy C, Sherry AD, Lai SY, Hazle JD, Cancer Res 2015, 75, 4708–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].a. Verkman AS, Trends Biochem Sci 2002, 27, 27–33; [DOI] [PubMed] [Google Scholar]; b. Lei Y, Han H, Yuan F, Javeed A, Zhao Y, Progress in Neurobiology 2017, 157, 230–246. [DOI] [PubMed] [Google Scholar]
- [44].Garroway AN, J. Magn. Reson. (1969) 1982, 49, 168–171. [Google Scholar]
- [45].a. Cheng LL, Lean CL, Bogdanova A, Wright SC Jr., Ackerman JL, Brady TJ, Garrido L, Magn Reson Med 1996, 36, 653–658; [DOI] [PubMed] [Google Scholar]; b. Maas WE, Laukien FH, Cory DG, J Amer Chem Soc 1996, 118, 13085–13086. [Google Scholar]
- [46].André M, Dumez J-N, Rezig L, Shintu L, Piotto M, Caldarelli S, Anal Chem 2014, 86, 10749–10754. [DOI] [PubMed] [Google Scholar]
- [47].Beckonert O, Keun HC, Ebbels TM, Bundy J, Holmes E, Lindon JC, Nicholson JK, Nature Protocols 2007, 2, 2692–2703. [DOI] [PubMed] [Google Scholar]
- [48].a. Bock NA, Kovacevic N, Lipina TV, Roder JC, Ackerman SL, Henkelman RM, J Neurosci 2006, 26, 4455–4459; [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Ma Y, Hof PR, Grant SC, Blackband SJ, Bennett R, Slatest L, McGuigan MD, Benveniste H, Neuroscience 2005, 135, 1203–1215. [DOI] [PubMed] [Google Scholar]
- [49].Mckay RT, Concept Magn Reson A 2011, 38A, 197–220. [Google Scholar]
- [50].Meiboom S, Gill D, Rev Sci Instr 1958, 29, 688–691. [Google Scholar]
- [51].a. Allerhand A, J Chem Phys 1966, 44, 1–9; [Google Scholar]; b. Aguilar JA, Nilsson M, Bodenhausen G, Morris GA, Chem Commun (Camb) 2012, 48, 811–813. [DOI] [PubMed] [Google Scholar]
- [52].Takegoshi K, Ogura K, Hikichi K, J. Magn. Reson 1989, 84, 611–615. [Google Scholar]
- [53].Markley JL, Brüschweiler R, Edison AS, Eghbalnia HR, Powers R, Raftery D, Wishart DS, Curr Opin Biotechnol 2017, 43, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gowda GAN, Abell L, Lee CF, Tian R, Raftery D, Anal Chem 2016, 88, 4817–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Takis PG, Schäfer H, Spraul M, Luchinat C, Nat Commun 2017, 8, 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Slomp G, App Spec Rev 1969, 2, 263–351. [Google Scholar]
- [57].Schirmer T, Auer DP, NMR Biomed 2000, 13, 28–36. [DOI] [PubMed] [Google Scholar]
- [58].a. Coquery N, Stupar V, Farion R, Maunoir-Regimbal S, Barbier EL, Remy C, Fauvelle F, Metabolomics 2015, 11, 1834–1847; [Google Scholar]; b. Croitor-Sava AR, Martinez-Bisbal MC, Laudadio T, Piquer J, Celda B, Heerschap A, Sima DM, Van Huffel S, Meas Sci Technol 2011, 22; [Google Scholar]; c. Dietz C, Ehret F, Palmas F, Vandergrift LA, Jiang YN, Schmitt V, Dufner V, Habbel P, Nowak J, Cheng LL, NMR Biomed 2017, 30; [DOI] [PMC free article] [PubMed] [Google Scholar]; d. Elkhaled A, Jalbert L, Constantin A, Yoshihara HAI, Phillips JJ, Molinaro AM, Chang SM, Nelson SJ, NMR Biomed 2014, 27, 578–593; [DOI] [PMC free article] [PubMed] [Google Scholar]; e. Jimenez-Xarrie E, Davila M, Gil-Perotin S, Jurado-Rodriguez A, Candiota AP, Delgado-Mederos R, Lope-Piedrafita S, Garcia-Verdugo JM, Arus C, Marti-Fabregas J, J Cerebr Blood F Met 2015, 35, 828–834; [DOI] [PMC free article] [PubMed] [Google Scholar]; f. Lee DW, Kim SY, Kim JH, Lee T, Yoo C, Nam YK, Jung JY, Shin HC, Kim HY, Kim DJ, Choe BY, Neurochem Int 2013, 62, 502–509; [DOI] [PubMed] [Google Scholar]; g. Opstad KS, Wright AJ, Bell BA, Griffiths JR, Howe FA, J Magn Reson Imaging 2010, 31, 289–297; [DOI] [PubMed] [Google Scholar]; h. Righi V, Tugnoli V, Mucci A, Bacci A, Bonora S, Schenetti L, Oncol Rep 2012, 28, 1461–1467; [DOI] [PubMed] [Google Scholar]; i. Selnaes KM, Gribbestad IS, Bertilsson H, Wright A, Angelsen A, Heerschap A, Tessem MB, NMR Biomed 2013, 26, 600–606; [DOI] [PubMed] [Google Scholar]; j. Swanson MG, Vigneron DB, Tabatabai ZL, Males RG, Schmitt L, Carroll PR, James JK, Hurd RE, Kurhanewicz J, Magn Reson Med 2003, 50, 944–954; [DOI] [PubMed] [Google Scholar]; k. Tugnoli V, Schenetti L, Mucci A, Parenti F, Cagnoli R, Righi V, Trinchero A, Nocetti L, Toraci C, Mavilla L, Trentini G, Zunarelli E, Tosi MR, Int J Mol Med 2006, 18, 859–869. [PubMed] [Google Scholar]
- [59].a. Decelle EA, Cheng LL, NMR Biomed 2014, 27, 90–99; [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Ratai EM, Pilkenton S, Lentz MR, Greco JB, Fuller RA, Kim JP, He J, Cheng LL, González RG, NMR Biomed 2005, 18, 242–251. [DOI] [PubMed] [Google Scholar]
- [60].Lu M, Zhu XH, Zhang Y, Chen W, Magn Reson Med 2014, 71, 1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W, Proc Natl Acad Sci U S A 2015, 112, 2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]