

Abstract
Excitatory amino acid transporters (EAATs) buffer and remove synaptically released l-glutamate and maintain its concentrations below neurotoxic levels. EAATs also mediate a thermodynamically uncoupled substrate-gated anion conductance that may modulate cell excitability. Here, we demonstrate that modification of a cysteine substituted within a C-terminal domain of EAAT1 abolishes transport in both the forward and reverse directions without affecting activation of the anion conductance. EC50s for l-glutamate and sodium are significantly lower after modification, consistent with kinetic models of the transport cycle that link anion channel gating to an early step in substrate translocation. Also, decreasing the pH from 7.5 to 6.5 decreases the EC50 for l-glutamate to activate the anion conductance, without affecting the EC50 for the entire transport cycle. These findings demonstrate for the first time a structural separation of transport and the uncoupled anion flux. Moreover, they shed light on some controversial aspects of the EAAT transport cycle, including the kinetics of proton binding and anion conductance activation.
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system that contributes not only to fast synaptic neurotransmission, but also to complex physiological processes such as learning and memory (1). However, excessive levels of extracellular glutamate are excitotoxic and lead to neuronal death. High affinity sodium-dependent excitatory amino acid (EAA) transporters serve to buffer and remove extracellular glutamate and to maintain its concentrations below excitotoxic levels (2–4).
Inward transport mediated by this carrier family is coupled to the cotransport of two or three sodium ions (5–8) and a proton (7, 8) and the countertransport of a potassium ion (5, 8), resulting in a net influx of positive charge per EAA molecule transported. The carriers also appear to mediate a substrate-gated anion conductance, normally carried by chloride ions, that is not linked stoichiometrically to substrate translocation. Functional characterization of the cloned EAA transporters (EAATs 1–5) in Xenopus oocytes has shown that the relative proportion of the substrate activated steady-state current generated by the anion conductance varies with each subtype (9–11). In the case of the cerebellar carrier, EAAT4, and the retinal carrier, EAAT5, this chloride conductance represents over 95% of the observed steady-state current (9, 11). For EAAT1, the glial carrier examined in this report, the anion current represents less than one third of the total transporter current (10). One proposed role for the conductance is to dampen cell excitability, thus preventing a decrease in the transporter turnover rate that occurs with depolarization. In salamander cone cells, which express EAAT5, the chloride conductance may act as a feedback sensor to limit depolarization and thus additional glutamate release (12), whereas in retinal bipolar cells it appears to mediate part of the light response (13).
Although the molecular basis of the anion conductance has not yet been formally identified, it is thought to be intrinsic to the carrier because it exhibits the same ionic dependence and pharmacology as the transport activity and because it has been observed with all glutamate transporter subtypes examined thus far (9–11). Studies using patch clamp analyses to measure rapid transport kinetics have linked gating of the anion conductance to early steps in the transport cycle (14–18). Association of anion channel gating events with the translocation limb of the cycle is further supported by the observation that transporters locked in an exchange mode still mediate the anion conductance (19–23). However, because substrates activate the anion channel activity at membrane potentials (>70 mV) and temperatures (<4°C) that greatly restrict transport (14), translocation may not be required.
Relatively little is known about the structural features of the carrier that underlie its transport and anion channel activities. There is general agreement that the N-terminal half of the carrier forms six transmembrane domains, although the C-terminal half remains unresolved (4, 24). We recently proposed a model for the topology of the C-terminal half of EAAT1, in which two membrane-spanning domains (domains 7 and 11) flank a central region that contains at least one re-entrant loop domain (domain 8; ref. 25). Using similar approaches, other groups modeled this region of the GLT-1 (26, 27) and GltT (28) carriers as having two oppositely oriented re-entrant loops (domains 7 and 9 in EAAT1) and two alpha helices (domains 8 and 11 in EAAT1). Site-directed mutagenesis and substituted cysteine accessibility studies have identified a number of residues within highly conserved domains that appear to interact with substrates (23, 27–29), inhibitors (29, 30), and cotransported ions (21, 22, 29, 30).
To further elucidate the structural elements underlying substrate transport and the anion conductance, we used a version of EAAT1 lacking cysteine residues (Cysless) to examine the impact of cysteine substitution and thiol modification on transporter-mediated currents in Xenopus oocytes. Here we report the characterization of a functional mutant of Cysless EAAT1, V449C, which is located within a previously unexplored hydrophobic domain near the C terminus of the carrier (domain 10) that is readily accessible from the extracellular aqueous milieu. When modified by sulfhydryl-specific methanethiosulfonate (MTS) reagents (31), V449C retains the ability to mediate the substrate-activated anion conductance, but no longer facilitates the accumulation of substrates. This study provides the first evidence of a structural alteration that separates activation of the anion conductance from the transport of substrates and offers a means to explore the structural and kinetic relationships underlying the two transporter-mediated activities.
Materials and Methods
Construction of Cysless and V449C.
Cysless (C186S, C252A, and C375G) and V449C (C186S, C252A, C375G, and V449C) mutants were created from the human EAAT1 cDNA by using a PCR-based mutagenesis strategy as described (29). Both mutants were sequenced by Dye Terminator Cycle Sequencing (ABI PRISM, Perkin–Elmer) and subcloned into pOTV for expression in Xenopus oocytes (32).
Expression in Oocytes.
Capped RNAs transcribed from linearized pOTV-EAAT1, Cysless, or V449C by using T7 RNA polymerase (mMessage mMachine, Ambion, Austin, TX) were microinjected into defolliculated stage IV or V oocytes (≈10 ng per oocyte). The oocytes were kept at 17°C for 2–8 days.
Electrophysiology.
Two-microelectrode voltage-clamp recordings were performed as described (11). Normal recording solution (ND96) contained (in mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 Hepes (pH 7.5) or 5 MES (pH 6.5). For sodium substitution experiments, choline chloride replaced sodium chloride. For the chloride substitution experiments, chloride salts were replaced with gluconate, bromide, or nitrate salts and KCl/agar bridges were used to avoid voltage offsets associated with buffer changes. High potassium, low sodium buffer contained (in mM): 58 NaNO3, 40 KNO3, 1.8 CaCl2, 1 MgCl2, and 5 Hepes (pH 7.5). To generate current–voltage (I–V) relationships, oocytes were voltage clamped at a holding potential of −60 mV, stepped to potentials from −80 to + 80 mV in 10-mV increments, and steady-state currents were measured. To avoid contaminating effects from transporter-independent, endogenous currents in oocytes, currents elicited by substrate are plotted as the difference between currents measured in the presence and absence of substrate.
Transport Assays.
Voltage-clamped uptake of radiolabeled L-glutamate or D-aspartate was performed by perfusing 200 nM L-[3H]glutamate or D-[3H]aspartate and 99.8 μM unlabeled L-glutamate onto oocytes clamped at −60 mV for 240 s, followed by a 120-s washout. Oocytes were solubilized in 1% SDS and counted in a scintillation counter.
Materials.
MTSET {[2-(Trimethylammonium)ethyl] methanethiosulfonate bromide} and MTSES [sodium (2-sulfonatoethyl)methanethiosulfonate] were obtained from Toronto Research Chemicals (Downsview, ON, Canada). L-[3H]glutamate and D-[3H]aspartate were from DuPont/NEN.
Results
To eliminate the possibility that the MTS derivatives could react with one or more endogenous cysteine residues in the EAAT1 transporter, we used a cysteineless version of the carrier (Cysless) as the template to create the V449C mutant. The Cysless carrier has been shown in both COS-7 cells and in Xenopus oocytes to have functional properties similar to EAAT1 (29). For brevity, the V449C mutant of Cysless EAAT1 will be referred to as V449C.
Sulfhydryl Modification of V449C Affects Substrate-Dependent Steady-State Currents.
Substrate-elicited steady-state currents mediated by the EAA transporters reflect both electrogenic transport and a substrate-elicited anion conductance. To evaluate the effect of sulfhydryl modification on steady-state currents mediated by V449C, we measured the amplitude of the currents elicited by the application of 100 μM L-glutamate before and after a 4-min application of the sulfhydryl-specific reagents, MTSET and MTSES. As shown in Fig. 1, 1 mM MTSET reduced the amplitude of the steady-state currents mediated by the V449C transporter, whereas it had no effect on steady-state currents mediated by Cysless. The same findings were observed with application of 10 mM MTSES (data not shown). Next, we evaluated the effect of sulfhydryl modification on the voltage dependence of the steady-state currents. As shown in Fig. 2, the glutamate elicited I–V relationship generated by the unmodified V449C transporter is similar to that generated by Cysless and wild-type EAAT1 (the reversal potential, Erev = +30 mV). However, after a 2-min exposure to either 1 mM MTSET or 10 mM MTSES followed by a 2-min washout, the L-glutamate-elicited I–V curve in V449C is shifted to more negative potentials (Erev = −10mV) and the amplitude of the inward currents reduced. I–V curves generated by Cysless are not affected by exposure to the MTS derivatives.
Figure 1.

Effect of MTSET on Cysless and V449C transporters. (A) Application of 100 μM l-glutamate (open bar) generates a steady-state current in oocytes expressing the V449C carrier (Vm = −60 mV). After a 4-min application of 1 mM MTSET (shaded bar) and a 2-min washout, a second application of 100 μM l-glutamate (open bar) generates a steady-state current of smaller amplitude. (B) The same protocol carried out on the Cysless transporter results in steady-state currents of similar amplitude.
Figure 2.

Effect of MTS derivatives on the I–V relationships generated by the V449C, Cysless, and EAAT1 transporters. Voltage jumps were made both before (open squares) and after (filled squares) application of 1 mM MTSET (2 min) to oocytes expressing the V449C (A), Cysless (B), or EAAT1 (C) transporters. Control currents were subtracted from currents generated in the presence of 100 μM l-glutamate.
Characterization of Substrate Elicited Steady-State Currents After Modification.
Based on the shape and reversal potential of the substrate-activated I–V curves generated by the modified V449C carrier, we hypothesized that the observed currents reflect only the uncoupled anion conductance (i.e., no transport component). Consistent with this, the reversal potential of these currents was the same as the chloride equilibrium potential (ECl), which we determined by measuring the Erev of endogenous Ca2+-activated chloride channel currents in the same oocytes (data not shown). In addition, previous characterization of the EAATs has shown that the reversal potential of the steady-state current is substrate-dependent (9, 10) and that this dependence reflects the relative contribution of the two transporter-mediated functions (10). For EAAT1, as for the unmodified V449C carrier, the Erev of the steady-state currents evoked by substrates exhibits a rank order, D-aspartate < L-aspartate < L-glutamate (data not shown). After modification, however, the I–V curves elicited by all three substrates reverse at the same potential (Erev = −10 mV, data not shown), consistent with the idea that the currents are derived solely from the anion conductance.
It is well established that substrate binding and, thus, transport and substrate gating of the anion conductance depend on sodium ions (5–9, 19, 33). Therefore, we tested whether the current observed after modification of V449C is sodium-dependent by measuring L-glutamate-elicited I–V curves with choline substituted for sodium ions. As expected, no substrate-activated currents were observed either before or after modification under these conditions (Fig. 3).
Figure 3.

Sodium dependence of the steady-state currents generated by V449C. I–Vs generated by the application of 100 μM l-glutamate before (open symbols) and after (filled symbols) application of 1 mM MTSET and in the presence of extracellular sodium (squares) or choline (circles). Control currents were subtracted from currents generated in the presence of 100 μM l-glutamate.
To determine whether the substrate-activated current generated by the
modified V449C transporter is in fact carried exclusively by chloride
ions, we measured the reversal potential of the glutamate-elicited
currents as a function of the extracellular chloride concentration. As
shown in Fig. 4A, a 10-fold
decrease in chloride concentration outside resulted in a 58 ± 3
mV rightward shift in the reversal potential, consistent with the
Nernst equation prediction for a pure chloride conductance. A second
characteristic of the EAA transporter-mediated anion conductance is its
relative permeability to various anions (NO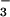 >
I− > Br− >
Cl− ≫ gluconate; refs. 10, 14, and 33). This
same permeability sequence (NO
>
I− > Br− >
Cl− ≫ gluconate; refs. 10, 14, and 33). This
same permeability sequence (NO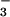 >
Br− > Cl− ≫ gluconate)
is observed with the V449C carrier before and after sulfhydryl
modification (Fig. 4B).
>
Br− > Cl− ≫ gluconate)
is observed with the V449C carrier before and after sulfhydryl
modification (Fig. 4B).
Figure 4.

Characterization of the anion conductance generated by the V449C carrier. (A) The reversal potential of the l-glutamate-elicited currents generated by the modified V449C transporter was measured at three concentrations of extracellular chloride (103.6, 55.6, and 7.6 mM). A 10-fold change in the extracellular chloride concentration resulted in a 58 ± 3 mV shift in the reversal potential. Each point is the average ± SEM from ten oocytes. (B) Extracellular anion substitutions. I–Vs elicited by 100 μM l-glutamate were measured with chloride (squares), bromide (triangles), or nitrate (circles) as the extracellular anion both before (closed symbols) and after (open symbols) modification by MTSET. Control currents were subtracted from currents generated in the presence of 100 μM l-glutamate.
Effect of Modification on V449C Transport Activity.
The absence of a substrate transport component in the current measured for the modified V449C transporter could be explained by one of two mechanisms: either the stoichiometry of substrate transport has become electroneutral, or transport itself has been abolished. To determine whether the V449C carrier mediates substrate transport following modification, we measured the uptake of 100 μM radiolabeled D-aspartate or L-glutamate under voltage clamp (Vm = −60 mV; Fig. 5). Before modification, V449C-expressing oocytes accumulated radiolabeled D-aspartate or L-glutamate 5–7-fold over background levels (uninjected oocytes). However, after a 4-min exposure to 1 mM MTSET, V449C-expressing oocytes accumulated the same amount of radiolabeled substrate as similarly treated uninjected oocytes. No change in the uptake of radiolabeled substrate was observed in Cysless expressing or uninjected oocytes after application of 1 mM MTSET. To demonstrate that transport does not occur even at high substrate concentrations, we also performed voltage-clamped uptake measurements with 1 mM radiolabeled L-glutamate. Similar to the results obtained with 100 μM L-glutamate and D-aspartate, the unmodified V449C carrier transported radiolabeled L-glutamate ≈5-fold more than uninjected oocytes, whereas the modified V449C was unable to accumulate radiolabeled L-glutamate over similarly treated uninjected oocytes (data not shown). Additional studies in transiently transfected COS-7 cells showed that modification of the V449C transporter with either MTSET or MTSES abolished transport activity over a range of L-glutamate concentrations from 1 μM to 1 mM (data not shown). In summary, our findings indicate that modification of the cysteine residue in V449C with MTSET abolishes the carrier's ability to facilitate substrate transport, but not to mediate the substrate-gated anion conductance.
Figure 5.

Radiolabeled uptake by the V449C and Cysless transporters held under voltage clamp. Oocytes were voltage clamped (Vm = −60mV) and 100 μM d-[3H]aspartate (shaded squares) or l-[3H]glutamate (open squares) was applied for 4 min followed by a 2-min washout. Each value is the average ± SEM for n ≥ 4.
Functional Properties of V449C Before and After Modification.
Based on kinetic models of the transport cycle, parameters reflecting activation of the anion conductance are likely to differ from those measured for the entire transport cycle (16, 17). To examine this, we measured and compared several kinetic properties of the carrier before and after modification, including apparent affinities for substrate and sodium ions, pH dependence, and the ability of potassium ions to stimulate efflux. After modification of the carrier with 1 mM MTSET or 10 mM MTSES, the EC50 for L-glutamate (Vm = −60mV) is ≈3-fold lower (EC50 = 35 ± 4 μM, n = 6, versus EC50MTSET = 12 ± 1 μM, n = 3, and EC50MTSES = 12 ± 2 μM, n = 6). Additionally, the EC50 of sodium ions to elicit L-glutamate (100 μM)-activated steady-state currents (Vm = −60mV) is also slightly lower (≈2-fold; EC50 = 40 ± 4 mM, Hill coefficient, nH = 1.8 ± 0.1, n = 9, versus EC50MTSET = 21 ± 5 mM, nH = 2.4 ± 0.3, n = 3, and EC50MTSES = 17 ± 4 mM, nH = 2.5 ± 0.3, n = 2). EC50 values for L-glutamate and sodium observed with Cysless were not changed by exposure to MTSET (data not shown). Thus, V449C exhibits slightly higher apparent affinities for substrates and for the cotransported ion sodium after modification with MTSET or MTSES.
Previous studies have shown that the transport of glutamate is thermodynamically coupled to the influx of protons, as well as sodium ions (8, 34, 35). In this model, protons bind to the carrier from the outside before activation of the anion conductance and are subsequently cotranslocated with the substrate. We examined the effect of pH on the EC50 of L-glutamate at V449C, both before and after modification by measuring substrate-elicited steady-state currents (Vm = −60 mV). Before modification, the L-glutamate EC50s were similar at pH 6.5 and pH 7.5 (EC50 = 34 ± 5 μM, n = 5; EC50 = 35 ± 5 μM, n = 6). However, after modification with MTSES (or MTSET; data not shown), the EC50 was significantly lower at pH 6.5 (EC50 = 3 ± 1 μM, n = 4) than at pH 7.5 (EC50 = 12 ± 2 μM, n = 7). Changing the pH did not significantly alter the maximum current amplitude either before or after modification (data not shown). These data are consistent with the idea that protons influence the activation of the anion conductance by modulating the apparent affinity of the substrate.
In both heterologous and native systems expressing EAATs, reversal of the transmembrane electrochemical gradients for sodium and potassium ions leads to an outward flux of intracellular glutamate (3) and the activation of the transporter-associated anion conductance (19, 21). To determine whether the modified carrier can catalyze reverse transport and/or activate the anion conductance when ionic gradients are altered to favor substrate efflux, we applied a high potassium (40 mM), low sodium (60 mM) buffer to oocytes expressing the V449C or Cysless carriers. In these experiments, we substituted nitrate for chloride to amplify the anion current, and determined the I–V relationship for the portion of the current that was sensitive to DL-threo-β-benzyloxyaspartate (DL-TBOA), before and after sulfhydryl modification (Fig. 6). DL-TBOA is a high-affinity, nontransported competitive inhibitor of both transport and the anion conductance mediated by EAAT1 (36), the Cysless EAAT1 (29), and V449C (data not shown). The use of DL-TBOA to resolve the transporter-mediated component of the current eliminates the nonspecific effects of changing potassium and sodium concentrations on oocytes. Before modification, the high potassium, low sodium buffer produced DL-TBOA-sensitive outward currents at positive potentials in oocytes expressing V449C or Cysless, reflecting both reverse transport and a transporter-associated anion current. After modification, however, application of 40 mM potassium/60 mM sodium to oocytes expressing V449C resulted in no DL-TBOA-sensitive outward current, similar to what was seen in uninjected oocytes. In contrast, DL-TBOA-sensitive currents generated by Cysless were not altered by exposure to MTSET. The lack of a DL-TBOA-sensitive outward current with V449C does not reflect a loss in affinity for DL-TBOA after modification, because DL-TBOA still inhibits the glutamate-elicited current under these conditions (data not shown). Thus, it appears that the modified V449C carrier is unable to undergo reverse transport or to activate the anion conductance when the electrochemical gradients driving transport are reversed.
Figure 6.

dl-TBOA-sensitive currents mediated by V449C and Cysless during reverse transport before and after modification. I–V curves elicited by 40 mM potassium nitrate (60 mM sodium nitrate) were measured from V449C- or Cysless-expressing or uninjected oocytes. I–Vs were subtracted from those measured in the presence of 100 μM dl-TBOA. Before modification, the I–V for V449C (A, open squares) was similar to Cysless (B, open squares). After application of 1 mM MTSET, the I–V for V449C (A, filled squares) was similar to uninjected oocytes (C).
Discussion
It has recently become apparent that many transporter families mediate uncoupled ion conductances in addition to transporting their designated substrate(s) (37–39). In the case of the EAATs, several biophysical studies have shown that the anion current is thermodynamically uncoupled from transport, although these two activities appear to be tightly associated within the transport cycle (14–18). Little is known, however, about the structural relationship between substrate transport and the ion channel activity. Here, we demonstrate for the first time that the two transporter-mediated activities are structurally separable. Modifying the side chain chemistry at one residue in the C-terminal half of the EAAT1 carrier abolishes substrate transport but not the substrate-gated anion flux.
After thiol modification, V449C fails to mediate the accumulation of radiolabeled D-aspartate or L-glutamate over background levels, indicating that substrate transport does not occur. Furthermore, modification of V449C does not appear to convert Cysless EAAT1 into a substrate exchanger, because transporters that act as exchangers still accumulate radiolabeled L-glutamate inside cells because of the high intracellular concentration of endogenous L-glutamate (≈10 mM; refs. 20 and 23). Finally, when the sodium and potassium concentrations are altered to favor reverse transport, neither reverse transport nor gating of the anion conductance occurs following modification of V449C. Together, these data suggest that the sulfhydryl-modified V449C has lost the ability to catalyze transport in either the forward or reverse direction.
Several results in this study indicate that following modification of V449C; the substrate-gated current reflects a pure anion conductance. First, we observed a shift in the substrate-activated I–V relationship after modification to more negative potentials (Erev = −10 mV) and a diminution of the inward current amplitudes. Second, the reversal potential is the same as the chloride equilibrium potential and changes as a function of extracellular chloride concentration in a manner consistent with the Nernst equation predictions for a pure chloride conductance. Additionally, the relative anion selectivity is the same as observed with EAAT1 and the unmodified V449C. Finally, it is thought that Erev reflects the relative contribution of the two components that comprise the current, electrogenic transport and the anion conductance, and that differences in the relative contributions underlies why Erev differs with the various EAA substrates (10). Consistent with the hypothesis that thiol modification of V449C selectively blocks electrogenic transport, we find that after modification Erev equals ECl for all three substrates.
Based on these findings, it appears that the modified V449C carrier is limited to states of the transport cycle that include the extracellular binding of substrate and cotransported ions and subsequent activation of the anion conductance, with no release of substrate internally. Although our data do not rule out the possibility that the modified carrier still undergoes substrate translocation and is impaired in the intracellular unbinding of substrate or cotransported ions, the simplest explanation is that modification prevents translocation of the fully bound transporter across the membrane. There are several ways in which the modification could block translocation. One way is if the modifying group prevents one or more conformational changes necessary for translocation in both the forward and reverse directions. In a previous study of EAAT1, it was found that lowering the temperature significantly decreased the amount of radiolabeled substrate transported and shifted the substrate-elicited I–V curves toward ECl; results similar to what we observe by modifying the V449C transporter (14). This temperature sensitivity of transport (Q10 ≈ 3), but not the anion conductance (Q10 ≈ 1.3), was suggested to be due to large conformational changes associated with translocation (14). Another possibility is if modification of the thiol group directly occludes the movement of substrate through the translocation pathway. This scenario, however, is less likely because the presence of a saturating concentration of substrate or inhibitor did not slow the rate of modification by the MTS derivatives (data not shown).
Our findings with the modified V449C carrier are also interesting in terms of transporter structure. For reasons mentioned previously, it is generally believed, although it has not been formally demonstrated, that the anion conductance is directly mediated by the carrier rather than by a separate channel that associates with the carrier. Recent studies of the multimeric structure of EAAT3 in oocytes suggest that the carrier forms a pentamer with what could be a central pore shared by all subunits (40). Thus, one could imagine that one or both functions of the carrier occur through this central pore rather than through the individual subunits. Because we have shown that the anion conductance occurs in the absence of substrate transport, our data imply that the pathway(s) for substrate and cotransported ions is to some degree structurally distinct from that for anions. Moreover, our findings suggest that it is possible to make a mutation in the carrier that eliminates the anion conductance without affecting the transport activity. Such a mutant would be a valuable tool for understanding the role of the anion conductance in synaptic transmission.
It is interesting that nontransported, competitive inhibitors of the EAATs, such as kainate (32) and DL-TBOA (36), do not by themselves elicit the anion conductance, and yet we have shown here that activation of the conductance does not require transport. This finding suggests that to activate the anion conductance, substrates go through additional steps beyond simply binding, and that nontransported inhibitors stabilize the transporter in a mode that prevents the entrance into an anion conducting substate.
Characterization of the V449C phenotype addresses two controversial aspects of the transport cycle: the step(s) where protons are involved and the step(s) where the anion conductance activated. Several groups have proposed a model in which a proton binds along with substrate from the outside before and during activation of the anion conductance, and is subsequently cotransported with the substrate (7, 34, 35). In an alternative model, a proton is transported into the cell during the countertransport of a potassium ion (18). Here we show that, in the absence of a complete transport cycle, protons increase the apparent affinity of L-glutamate to activate the anion conductance. Although it is possible that protons bind to an allosteric site, which alters V449C kinetics only after sulfhydryl modification, the most plausible explanation for these results is that protons modulate the extracellular binding of substrate before and during activation of the anion conductance. Most kinetic models of the transport cycle describe the existence of an anion-conducting state after the substrate and cotransported ions have bound to the extracellular side of the transporter and before their translocation (14–18). In an alternative model the anion conducting state has been proposed to follow the translocation of substrate and cotransported ions (19). Our finding that modification impairs substrate translocation but not the anion conductance supports the existence of an anion-conducting state before translocation. However, our data do not rule out activation of an additional anion conductance when substrates and transported ions are bound to the intracellular face of the carrier. Such an additional anion-conducting step would require a block in both the substrate and potassium translocation steps, because currents cannot be elicited in the modified carrier when the electrochemical gradients are reversed.
In this paper, we have demonstrated that two distinct substrate-activated functions of the glutamate transporter family can be separated by the modification of a single cysteine in EAAT1. The data provide further support for a model of the transport cycle in which protons modulate the affinity of substrates for gating of an anion conductance that is activated before substrate translocation. Additional studies of this mutant, including rapid kinetic analyses and fluorescent studies of conformational changes, will further increase our understanding of the functional and structural relationships that exist between these two carrier-mediated activities.
Acknowledgments
We thank A. Tzingounis, C. Jahr, and the members of the Amara laboratory for their helpful discussions, and K. Shimamoto for generously providing dl-TBOA. Also, we wish to acknowledge Y. Yang for her excellent technical assistance. This work was supported by the Howard Hughes Medical Institute, National Institutes of Health (NIH) grants (to S.G.A.), an National Institutes of Mental Health predoctoral fellowship (to R.P.S.), and the New Energy and Industrial Technology Development Organization (to Y.S.).
Abbreviations
- EAA
excitatory amino acid
- EAAT
EAA transporter
- MTS
methanethiosulfonate
- MTSET
[2-(trimethylammonium)ethyl] methanethiosulfonate bromide
- MTSES
sodium (2-sulfonatoethyl)methanethiosulfonate
- TBOA
threo-β-benzyloxyaspartate
- I–V
current–voltage
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Dingledine R, Borges K, Bowie D, Traynelis S F. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 2.Choi D W. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- 3.Nicholls D, Attwell D. Trends Pharmacol Sci. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- 4.Seal R P, Amara S G. Annu Rev Pharmacol Toxicol. 1999;39:431–456. doi: 10.1146/annurev.pharmtox.39.1.431. [DOI] [PubMed] [Google Scholar]
- 5.Kanner B I, Sharon I. Biochemistry. 1978;17:3949–3953. doi: 10.1021/bi00612a011. [DOI] [PubMed] [Google Scholar]
- 6.Stallcup W B, Bulloch K, Baetge E E. J Neurochem. 1979;32:57–65. doi: 10.1111/j.1471-4159.1979.tb04509.x. [DOI] [PubMed] [Google Scholar]
- 7.Erecinska M, Wantorsky D, Wilson D F. J Biol Chem. 1983;258:9069–9077. [PubMed] [Google Scholar]
- 8.Zerangue N, Kavanaugh M P. Nature (London) 1998;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 9.Fairman W A, Vandenberg R J, Arriza J L, Kavanaugh M P, Amara S G. Nature (London) 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 10.Wadiche J I, Amara S G, Kavanaugh M P. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 11.Arriza J L, Eliasof S, Kavanaugh M P, Amara S G. Proc Natl Acad Sci USA. 1997;94:4155–4160. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Picaud S, Larsson H P, Wellis D P, Lecar H, Werblin F. Proc Natl Acad Sci USA. 1995;92:9417–9421. doi: 10.1073/pnas.92.20.9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grant G B, Dowling J E. J Neurosci. 1995;15:3852–3862. doi: 10.1523/JNEUROSCI.15-05-03852.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wadiche J I, Kavanaugh M P. J Neurosci. 1998;18:7650–7661. doi: 10.1523/JNEUROSCI.18-19-07650.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otis T S, Jahr C E. J Neurosci. 1998;18:7099–7110. doi: 10.1523/JNEUROSCI.18-18-07099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otis T S, Kavanaugh M P. J Neurosci. 2000;20:2749–2757. doi: 10.1523/JNEUROSCI.20-08-02749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grewer C, Watzke N, Wiessner M, Rauen T. Proc Natl Acad Sci USA. 2000;97:9706–9711. doi: 10.1073/pnas.160170397. . (First Published August 8, 2000; 10.1073/pnas.160170397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Auger C, Attwell D. Neuron. 2001;28:547–558. doi: 10.1016/s0896-6273(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 19.Billups B, Rossi D, Attwell D. J Neurosci. 1996;16:6722–6731. doi: 10.1523/JNEUROSCI.16-21-06722.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zerangue N, Kavanaugh M P. J Biol Chem. 1996;271:27991–27994. doi: 10.1074/jbc.271.45.27991. [DOI] [PubMed] [Google Scholar]
- 21.Kavanaugh M P, Bendahan A, Zerangue N, Zhang Y, Kanner B I. J Biol Chem. 1997;272:1703–1708. doi: 10.1074/jbc.272.3.1703. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Bendahan A, Zarbiv R, Kavanaugh M P, Kanner B I. Proc Natl Acad Sci USA. 1998;95:751–755. doi: 10.1073/pnas.95.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bendahan A, Armon A, Madani N, Kavanaugh M P, Kanner B I. J Biol Chem. 2000;275:37436–37442. doi: 10.1074/jbc.M006536200. [DOI] [PubMed] [Google Scholar]
- 24.Slotboom D J, Konings W N, Lolkema J S. Microbiol Mol Biol Rev. 1999;63:293–307. doi: 10.1128/mmbr.63.2.293-307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seal R P, Leighton B H, Amara S G. Neuron. 2000;25:695–706. doi: 10.1016/s0896-6273(00)81071-5. [DOI] [PubMed] [Google Scholar]
- 26.Grunewald M, Bendahan A, Kanner B I. Neuron. 1998;21:623–632. doi: 10.1016/s0896-6273(00)80572-3. [DOI] [PubMed] [Google Scholar]
- 27.Grunewald M, Kanner B I. J Biol Chem. 2000;275:9684–9689. doi: 10.1074/jbc.275.13.9684. [DOI] [PubMed] [Google Scholar]
- 28.Slotboom D J, Sobczak I, Konings W N, Lolkema J S. Proc Natl Acad Sci USA. 1999;96:14282–14287. doi: 10.1073/pnas.96.25.14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seal R P, Amara S G. Neuron. 1998;21:1487–1498. doi: 10.1016/s0896-6273(00)80666-2. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Kanner B I. Proc Natl Acad Sci USA. 1999;96:1710–1715. doi: 10.1073/pnas.96.4.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlin A, Akabas M H S. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 32.Arriza J L, Fairman W A, Wadiche J I, Murdoch G H, Kavanaugh M P, Amara S G. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eliasof S, Jahr C E. Proc Natl Acad Sci USA. 1996;93:4153–4158. doi: 10.1073/pnas.93.9.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zerangue N, Kavanaugh M P. J Physiol. 1996;493:419–423. doi: 10.1113/jphysiol.1996.sp021393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watzke N, Rauen T, Bamberg E, Grewer C. J Gen Physiol. 2000;116:609–622. doi: 10.1085/jgp.116.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- 37.Lester H A, Mager S, Quick M W, Corey J L. Annu Rev Pharmacol Toxicol. 1994;34:219–249. doi: 10.1146/annurev.pa.34.040194.001251. [DOI] [PubMed] [Google Scholar]
- 38.Sonders M S, Amara S G. Curr Opin Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- 39.DeFelice L J, Blakely R D. Biophys J. 1996;70:579–580. doi: 10.1016/S0006-3495(96)79604-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eskandari S, Kreman M, Kavanaugh M P, Wright E M, Zampighi G A. Proc Natl Acad Sci USA. 2000;97:8641–8646. doi: 10.1073/pnas.97.15.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]