

Abstract
Understanding the genetic and evolutionary impacts of stocking on wild fish populations has long been of interest as negative consequences such as reduced fitness and loss of genetic diversity are commonly reported outcomes. In an attempt to sustain a fishery, managers implemented nearly five decades of extensive stocking of over a million Muskellunge (Esox masquinongy), a native species in the Lower St. Lawrence River (Québec, Canada). We investigated the effect of this stocking on population genetic structure and allelic diversity in the St. Lawrence River in addition to tributaries and several stocked inland lakes. Using genotype by sequencing, we genotyped 643 individuals representing 22 locations and combined this information with forward simulations to investigate the genetic consequences of long‐term stocking. Individuals native to the St. Lawrence watershed were genetically differentiated from stocking sources and tributaries, and inland lakes were naturally differentiated from the main river. Empirical data and simulations within the St. Lawrence River revealed weak stocking effects on admixture patterns. Our data suggest that the genetic structure associated with stocked fish was diluted into its relatively large effective population size. This interpretation is also consistent with a hypothesis that selection against introgression was in operation and relatively efficient within the large St. Lawrence River system. In contrast, smaller populations from adjacent tributaries and lakes displayed greater stocking‐related admixture that resulted in comparatively higher heterozygosity than the St. Lawrence. Finally, individuals from inland lakes that were established by stocking maintained a close affinity with their source populations. This study illustrated a benefit of combining extensive genomic data with forward simulations for improved inference regarding population‐level genetic effects of long‐term stocking, and its relevance for fishery management decision making.
Keywords: admixture, Esox masquinongy, forward simulations, gene flow, genomic, stocking
1. INTRODUCTION
Many native species are undergoing steep population declines due to rapid and globally changing environments and overexploitation (Allendorf, 2017). In response, supplementation programs continue to be extensively applied to enhance natural populations to sustain exploitation and more recently for biodiversity conservation (Laikre, Schwartz, Waples, Ryman, & GeM Working Group, 2010). Fish populations, in particular, are subjected to intense commercial and recreational exploitation (Dunham, 2011). This leads to species transfers, sometimes over large distances, to supplement genetically and ecologically divergent populations. As a consequence, understanding how supplementation practices from nonindigenous sources may impact the evolutionary potential of wild populations remains a major concern (Araki, Cooper, & Blouin, 2007, 2009; Laikre & Ryman, 1996; Ryman & Laikre, 1991; Waples, Hindar, Karlsson, & Hard, 2016).
From a genetic standpoint, the potential negative impacts of stocking include the following: reduction in genetic diversity and effective population size due to the Ryman–Laikre effect (i.e., use of small numbers of individuals for breeding programs, Laikre & Ryman, 1996; Ryman & Laikre, 1991), genetic homogenization of wild populations (Araki & Schmid, 2010; Eldridge, Myers, & Naish, 2009; Eldridge & Naish, 2007; Lamaze, Sauvage, Marie, Garant, & Bernatchez, 2012; Perrier, Guyomard, Bagliniere, Nikolic, & Evanno, 2013), and ultimately the loss of locally adapted traits (Araki, Cooper, & Blouin, 2009; Ford, 2002; Lynch & O'Hely, 2001). Loss of local adaptation may arise from selection of traits associated with captive environments or because a divergent stocking source may exhibit reduced fitness in the local environment (Fraser et al., 2018). With admixture (impacting genome‐wide structure) and eventual introgression (where allelic variants are transferred from one differentiated population to another), differences in selective values can lead to outbreeding depression through disruption of co‐adapted gene complexes or from the breakup of epistatic interactions (Lynch, 1991; Tallmon, Luikart, & Waples, 2004). Such results have been documented in several occasions (e.g., Allendorf, Leary, Spruell, & Wenburg, 2001; Edmands, 2007; Le Cam, Perrier, Besnard, Bernatchez, & Evanno, 2015). These have raised concern regarding the consequences of stocking on long‐term maintenance and recovery of wild populations (Létourneau et al., 2018).
Conversely, positive effects of supplementation have also been reported. Several threatened populations recently isolated due to habitat fragmentation often display low genetic divergence and have not accumulated genetic incompatibilities. These small populations, however, are exposed to genetic drift whereby mildly deleterious mutations are not efficiently removed by selection, as would be the case in larger populations (Whitlock & Bürger, 2004). These mildly deleterious mutations may increase in frequency and rise to fixation (Glémin, 2003; Lynch et al., 1999; Wang, Hill, Charlesworth, & Charlesworth, 1999). This random drift load, however, becomes important when the selection coefficient is >50% of the effective population size (Whitlock, 2000) and can ultimately lead to extinctions of very small populations (Lynch, Conery, & Bürger, 1995). Similarly, inbreeding depression is of concern in these small populations (Bataillon & Kirkpatrick, 2000; Wang et al., 1999). In the long term, this may pose major threat to the adaptability of these populations to changing environment (Carlson, Cunningham, & Westley, 2014; Ralls et al., 2018).
Population supplementation, when done appropriately (see Miller et al., 2017), can have beneficial effects (Frankham, 2015; Frankham et al., 2011; Ralls et al., 2018). For instance, Frankham (2015) showed a genetic rescue effect after outcrossing that provided a 148% median fitness increase in wild populations exposed to stressful environments. Such effects have been shown to persist through several generations (Frankham, 2015; Frankham, 2016; Ralls et al., 2018), and, when applied sensibly, the probability of outbreeding depression is generally low (Frankham et al., 2011).
While admixture and introgression have been extensively documented in salmonid species (Finnegan & Stevens, 2008; Hansen, Fraser, Meier, & Mensberg, 2009; Létourneau et al., 2018; Perrier, Baglinière, & Evanno, 2013; Perrier, Guyomard et al., 2013), less emphasis has been placed on other taxonomic groups. Here, we examine genetic implications of long‐term stocking in Muskellunge Esox masquinongy, a representative of the Esocidae, the sister family of all salmonids (Rondeau et al., 2014). Muskellunge are distributed within particular temperate rivers and lakes from midwestern to eastern North America (Crossman, 1978), including the Great Lakes where it often occurs in sympatry with its congener, Northern Pike Esox lucius. In the St. Lawrence River, Muskellunge reaches sexual maturity between 5 and 7 years for males and 6 and 8 years for females (Farrell et al., 2007) and can grow up to more than 1.5 m and 20 kg that makes it a highly prized species for recreational trophy fishing. Given its typically low population density (~<1.0 fish/ha; Cloutier, 1987; Simonson, 2010) that limits sampling programs, more information is needed regarding its biology, genetic diversity, and population dynamics (Crane et al., 2015; Kapuscinski, Sloss, & Farrell, 2013). In many places, native Muskellunge have undergone pronounced population declines due to various factors including overharvesting, habitat degradation, and pollution (Brodeur, Hatin, & Bacon, 2013; Crossman, 1978; Farrell et al., 2007; Mongeau & Massé, 1976; Whillans, 1979). The decline has been more recently reinforced by viral hemorrhagic septicemia and round goby introduction (Farrell, Getchell, Kapuscinski, & LaPan, 2017).
In response to Muskellunge management challenges, widespread and sometimes intensive and long‐term stocking is used to rehabilitate native populations and supplement existing ones. Stocking was also performed as species introductions to support new recreational fisheries and as a predator to control invasive fish species (Crane et al., 2015; Jennings et al., 2010; Kapuscinski, Belonger, Fajfer, & Lychwick, 2007; Wingate, 1986). Studies regarding Muskellunge genetic divergence and stocking effects primarily used microsatellite markers and mainly focused on the Laurentian Great Lakes and Upper St. Lawrence River (Kapuscinski et al., 2013; Miller, Mero, & Younk, 2009, 2012; Turnquist et al., 2017; Wilson, Liskauskas, & Wozney, 2016). These studies revealed pronounced genetic differentiation among weakly connected populations at small spatial scales. They also demonstrated a generally reduced genetic diversity putatively associated with recent bottlenecks and/or strong genetic drift in populations of small effective size and restricted dispersal due to spawning site fidelity (Crossman, 1990; Farrell et al., 2007; Jennings, Hatzenbeler, & Kampa, 2011; Margenau, 1994; Miller, Kallemeyn, & Senanan, 2001). The impact of stocking has been documented in several populations where modest levels of admixture and a partial homogenization of population genetic structure have been reported (Scribner et al., 2015). In contrast, no study has documented native Muskellunge population structure further east, in the Lower St. Lawrence River basin of Québec (Canada), downstream of the Great Lakes. Moreover, no population genomic studies using thousands of markers distributed throughout the genome have been performed on this species to date.
Similar to populations throughout its distribution range, Lower St. Lawrence Muskellunge experienced a prolonged decline attributed to habitat loss (Robitaille & Cotton, 1992) and commercial overfishing that occurred until 1936 (Crossman, 1986; Dymond, 1939). Therefore, from 1951 to 1998 the Lower St. Lawrence was supplemented by over 1 million young Muskellunge (Brodeur et al., 2013; De La Fontaine, unpublished; Dumont, 1991; Mongeau, Leclerc, & Brisebois, 1980; Vézina, 1977; Vincent & Legendre, 1974). In particular, during 1951–1965, fish from both southwestern New York State (Chautauqua Lake, Ohio River basin) and Ontario's Kawartha Lakes (represented in our study by Pigeon Lake) were transferred to Lachine hatcheries in Québec to support a massive stocking program in the St. Lawrence River (De La Fontaine, unpublished; Dumont, 1991). Muskellunge from the Ohio basin are thought to rise from a distinct regional lineage (Koppelman & Philipp, 1986), but phylogeographic and systematic relationships of these two stocking sources are incompletely understood (Miller et al., 2017). Fish from those sources were also stocked in numerous lakes including Lake Joseph and Lake Tremblant, which themselves became widely used as a brood source from 1965 for hatcheries until the end of stocking in 1998 (De La Fontaine, unpublished; Dumont, 1991; Vézina, 1977; Vincent & Legendre, 1974). Besides stocking in those waterbodies, Muskellunge were also introduced in several lakes and rivers (details on stocking history and translocations are provided in Supporting Information Figure S1).
An improved understanding of the effects of admixture following stocking can also be advanced by application of individual‐based forward simulations (Hoban, 2014; Hoban, Bertorelle, & Gaggiotti, 2012). Simulated outcomes of population divergence from a variety of demographic scenarios (e.g., in terms of population size and differential mortality) may be used to identify the one that best explains empirically documented patterns (Guillaume & Rougemont, 2006; Hernandez, 2008; Lawrie, 2017; Messer, 2013). These methods, however, are rarely applied to investigate stocking effects on the genetic composition of wild fish (but see Perrier, Guyomard et al., 2013). It is also possible to generate increasingly complex and biologically realistic simulations for comparison with empirical data. Such simulations can aid in understanding supplementation efficacy and how it may generate admixture, while also helping to understand the limits of simple clustering tools (e.g., Alexander, Novembre, & Lange, 2009; Pritchard, Stephens, & Donnelly, 2000) in detection of genome‐wide effects of admixture on local ancestry.
Here, we combined empirical population genomics using a genotype‐by‐sequencing approach with forward simulations to document the extent and patterns of genetic admixture that resulted from past stocking events in the St. Lawrence River watershed. With this dataset and simulation tools, we (a) explored the effect of nearly five decades of stocking on wild populations and documented fine‐scale population genetic diversity and structure of the Muskellunge in the St. Lawrence River watershed, (b) compared expected patterns of admixture from simplified demographic simulations with empirical observations, and (c) used this information for management recommendations pertaining to stocking practices.
2. METHODS
2.1. Sampling
A total of 662 Muskellunge were sampled from 22 sites within the St. Lawrence River drainage, mainly from Québec (Canada) with a median sample size of 24 individuals per site (Figure 1, Table 1). This sampling represents the Upper and Lower St. Lawrence River, its main tributaries, and inland lake populations. Two sampling sites (Chautauqua Lake, CHQ, and Pigeon Lake, PIG) represented the original stocking sources used from 1951 to 1965 and two more recent secondary sources (Lake Joseph, JOS, 1965–1986, and Lake Tremblant, TRE, 1986–1997; both initially stocked with the CHQ and PIG source) were included in the baseline. Of all sampling locations, to the best of our knowledge, only Traverse Lake (TRA) had no record of past stocking and is presumed to be native. Most samples (fin clips preserved in 95% ethanol) were obtained from catch‐and‐release captures of professional fishing guides and anglers between 2010 and 2015. Samples were also collected by the Ministère des Forêts, de la Faune et des Parcs du Québec (MFFP), the Ontario Ministry of Natural Resources and Forestry (MNRF), and the New York Department of Environmental Conservation. A graphical representation of the major stocking operations performed in Québec is presented in Supporting Information Figure S1. The Upper St. Lawrence River is represented by pooled samples taken at a network of spawning locations within the Thousand Islands (TIN) where long‐term Muskellunge population monitoring occurs (Farrell et al., 2017; Kapuscinski et al., 2013).
Figure 1.
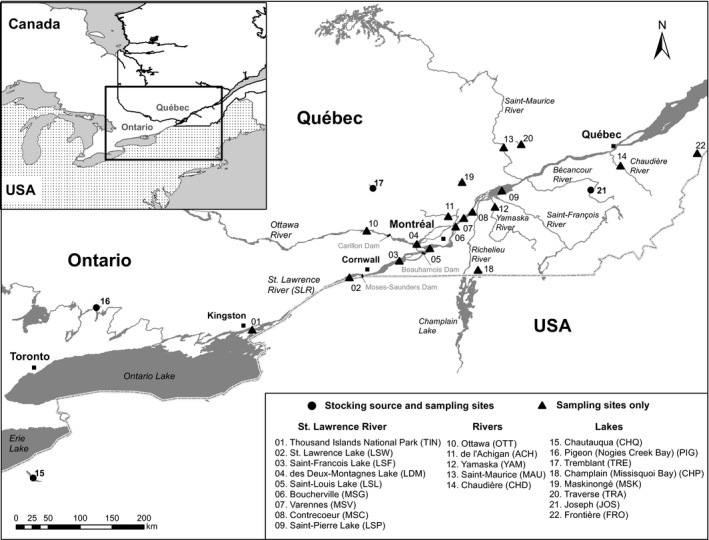
Map showing sampling locations. Two major physical barriers are present on the St. Lawrence system: Moses‐Saunders and Beauharnois‐Les Cèdres hydropower dams located at the upstream and downstream end of Lake Saint‐Françis, respectively
Table 1.
Genetic diversity parameters
| Sampling location | Code | Type | n | H o | H s | P | Pi | N e [95% CI] |
|---|---|---|---|---|---|---|---|---|
| Upper Saint Lawrence | ||||||||
| Thousand Islands Network | TIN | Stocked | 18 | 0.154 | 0.160 | 0.181 | 0.0033 | 362 [150–inf ] |
| St. Lawrence Lake | LSW | Stocked | 15 | 0.169 | 0.174 | 0.177 | 0.0034 | 669 [493–823] |
| Lower Saint Lawrence and inland lake | ||||||||
| Saint‐Louis Lake | LSL | Stocked | 55 | 0.099 | 0.101 | 0.305 | 0.0030 | 669 [493–823] |
| Saint‐Françis Lake | LSF | Stocked | 61 | 0.102 | 0.104 | 0.286 | 0.0030 | 669 [493–823] |
| Saint‐Pierre Lake | LSP | Stocked | 57 | 0.101 | 0.102 | 0.298 | 0.0030 | 669 [493–823] |
| Contrecoeur (St. Lawrence River) | MSC | Stocked | 16 | 0.163 | 0.168 | 0.176 | 0.0034 | 669 [493–823] |
| Boucherville (St. Lawrence River) | MSG | Stocked | 19 | 0.144 | 0.150 | 0.209 | 0.0033 | 669 [493–823] |
| Varennes (St. Lawrence River) | MSV | Stocked | 16 | 0.161 | 0.165 | 0.189 | 0.0034 | 669 [493–823] |
| Tributaries | ||||||||
| des Deux‐Montagnes Lake | LDM | Stocked | 56 | 0.110 | 0.114 | 0.287 | 0.0030 | 146 [80–298] |
| De l'Achigan River | ACH | Stocked | 25 | 0.149 | 0.154 | 0.170 | 0.0029 | 17 [11–178] |
| Chaudière River | CHD | Introduced | 15 | 0.196 | 0.198 | 0.150 | 0.0032 | 375 [84–inf] |
| Maskinongé Lake | MSK | Stocked | 25 | 0.131 | 0.136 | 0.236 | 0.0032 | 52 [24–145] |
| Ottawa River | OTT | Stocked | 7 | 0.255 | 0.277 | 0.110 | 0.0035 | 45 [17–inf] |
| Yamaska River | YAM | Stocked | 16 | 0.180 | 0.183 | 0.137 | 0.0030 | 62 [29–129] |
| Stocking sources | ||||||||
| Chautauqua Lake | CHQ | Source | 41 | 0.129 | 0.129 | 0.210 | 0.0028 | 137 [95–175] |
| Pigeon Lake | PIG | Source | 27 | 0.088 | 0.086 | 0.152 | 0.0019 | 79 [36–178] |
| Joseph Lake | JOS | Source | 30 | 0.139 | 0.142 | 0.190 | 0.0029 | 137 [95–175] |
| Tremblant Lake | TRE | Source | 29 | 0.152 | 0.157 | 0.160 | 0.0029 | 137 [95–175] |
| Stocked lake | ||||||||
| Champlain Lake | CHP | Stocked | 23 | 0.165 | 0.174 | 0.159 | 0.0031 | 14 [6–31] |
| Frontière Lake | FRO | Introduced | 31 | 0.145 | 0.148 | 0.170 | 0.0029 | 137 [95–175] |
| Unstocked lake | ||||||||
| Traverse Lake | TRA | Wild | 35 | 0.070 | 0.066 | 0.154 | 0.0016 | NA [NA] |
| Saint‐Maurice River | MAU | Wild | 26 | 0.172 | 0.174 | 0.161 | 0.0031 | 45 [25–77] |
Code = acronyms for each sampling site, n = number of individuals genotyped, H o = observed heterozygosity, H s = gene diversity, P = proportion of polymorphic loci. Pi = nucleotide diversity (Tajima, 1989), N e = effective population size with 95% CI obtained from jackknifing over individuals. Effective population size was computed by merging the major group within the St. Lawrence together. Similarly, all populations from the same stocking group (JOS, TRE, and FRO) were merged in order to increase the sample size. Effective population size could not be estimated for Traverse Lake when implementing a correction for linkage and resulted in an estimated of N e = 3 [95% CI = 2–4] without correction.
2.2. Molecular methods and SNP calling
A salt‐extraction protocol adapted from Aljanabi and Martinez (1997) was used to extract genomic DNA. Sample quality and concentration were checked on 1% agarose gels and a NanoDrop 2000 spectrophotometer (Thermo Scientific). Concentration of DNA was normalized to 20 ng/µl. Libraries were constructed following a double‐digest RAD (restriction‐site‐associated DNA sequencing; Andrews, Good, Miller, Luikart, & Hohenlohe, 2016) protocol modified from Mascher, Wu, Amand, Stein, and Poland (2013). Genomic DNA was digested with two restriction enzymes (PstI and MspI) by incubating at 37°C for 2 hr followed by enzyme inactivation by incubation at 65°C for 20 min. Sequencing adaptors and a unique individual barcode were ligated to each sample using a ligation master mix including T4 ligase. The ligation reaction was completed at 22°C for 2 hr followed by 65°C for 20 min for enzyme inactivation. Samples were multiplexed (n = 48 individuals) to insure fish from each sampling location were sequenced on a minimum of six different multiplexes to avoid pool effects. Libraries were size‐selected using a BluePippin prep (Sage Science), amplified by PCR, and sequenced for 96 individuals on a Ion Proton P1v2 chip that generated approximately 80 million reads per chip. Based on the number of reads for each individually barcoded sample, the prepared DNA was re‐pooled into a new library where the representation of samples with low reads counts was increased. This new library was sequenced on two additional Ion Proton chips to normalize the number of reads per sample.
2.3. Bioinformatics
Barcodes were removed, and reads were trimmed to 80 pb using cutadapt (Martin, 2011) allowing for an error rate of 0.2 and demultiplexed using the “process_radtags” module of Stacks v1.44 (Catchen, Hohenlohe, Bassham, Amores, & Cresko, 2013). They were aligned to the Northern Pike (Esox lucius) reference genome Eluc_V3 (Rondeau et al., 2014) using bwa‐mem (Li, 2013) with default parameters. Here, 19 randomly distributed individuals with <2.5 million reads were removed, and 643 of 662 Muskellunge were retained for subsequent analysis. Then, aligned reads were processed with Stacks v.1.44 for SNPs calling and genotyping. The “pstacks” module was used with a minimum depth of 3, and up to three mismatches were allowed in catalog assembly. We then ran the “populations” module to produce a vcf file that was filtered with a custom python script. To control for paralogs and HWE disequilibrium, SNPs were retained with a read depth higher than 5, and presence in 70% of each individual at each sampling location and had heterozygosity <0.60. We removed any SNPs that lacked presence in at least 70% of the dataset (max‐missing 0.70 in vcftools) that did not eliminate a variant. The resulting vcf file comprised 16,266 SNPs spread over 11,458 loci and represented the least stringent dataset used to estimate basic diversity parameters. This vcf file was subsampled to meet model assumptions (in particular the use of unlinked SNPs) underlying the population genetic analysis applied. A haplotype file was exported from the “populations” module in Stacks providing information on each RAD locus instead of single SNPs. These loci were then used to estimate a range of statistics described below and to perform FineRADstructure analysis.
2.4. Genetic diversity analysis
Hierfstat (Goudet, 2005) was used to compute patterns of observed heterozygosity and gene diversity after excluding monomorphic markers from each sampling location. We then computed the proportion of polymorphic loci in each sampling location using custom R scripts. Contemporary effective population size (N e) was estimated using the bias‐corrected version based on linkage disequilibrium and assuming random mating (Waples, 2006; Waples & Do, 2008) and implemented in NeEstimator (Do et al., 2014). We kept one SNP per locus and filtered the dataset for physical LD using plink (Chang et al., 2015). Windows of 50 SNPs were shifted by five SNPs each iteration and the routine removed any SNP with a variation inflation factor >2. Nonpolymorphic loci were removed in each population as well as singletons. Effective population size (N e) was computed after merging sampling locations into major genetic clusters and applying equations from Table 2 of Waples (2006). All burrow estimates for pairs of SNPs located on the same chromosome were removed prior to N e estimation. Finally, we estimated nucleotide diversity (Tajima, 1989) and differentiation statistics (D xy, D a, F st) for each RAD locus using mscalc (Ross‐Ibarra, Tenaillon, & Gaut, 2009; Ross‐Ibarra et al., 2008; Roux et al., 2011).
Table 2.
Simulation results for the 20 ms showing the lowest RMSE
| Mean admixture q‐value | Pairwise F ST | Polym | RMSE | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Admixture with SRC (grp1) | Admixture with LDM (grp2) | Admixture with SLR (grp3) | SLR versus LDM | SLR versus SRC | LDM versus SRC | (1% MAF) | |||||||||
| LDM | SLR | SRC | LDM | SLR | SRC | LDM | SLR | SRC | |||||||
| Model | Empirical value | 0.101 | 0.033 | 0.978 | 0.881 | 0.102 | 0.020 | 0.018 | 0.865 | 0.002 | 0.036 | 0.23 | 0.178 | 3,619 | |
| m23_N600_D50 | 0.070 | 0.037 | 1.000 | 0.916 | 0.077 | 0.000 | 0.015 | 0.887 | 0.000 | 0.012 | 0.206 | 0.293 | 3,619 | 0.05 | |
| m21_N600_D200 | 0.112 | 0.068 | 1.000 | 0.863 | 0.066 | 0.000 | 0.026 | 0.866 | 0.000 | 0.012 | 0.197 | 0.293 | 3,621 | 0.57 | |
| m23_N600_D400 | 0.063 | 0.034 | 0.989 | 0.904 | 0.096 | 0.011 | 0.032 | 0.870 | 0.000 | 0.012 | 0.209 | 0.297 | 3,616 | 0.94 | |
| m23_N600_D300 | 0.057 | 0.035 | 1.000 | 0.929 | 0.088 | 0.000 | 0.013 | 0.877 | 0.000 | 0.013 | 0.206 | 0.296 | 3,615 | 1.16 | |
| m21_N600_D300 | 0.116 | 0.073 | 1.000 | 0.875 | 0.070 | 0.000 | 0.010 | 0.858 | 0.000 | 0.012 | 0.193 | 0.286 | 3,615 | 1.21 | |
| m21_N600_D400 | 0.106 | 0.082 | 1.000 | 0.866 | 0.067 | 0.000 | 0.028 | 0.852 | 0.000 | 0.012 | 0.192 | 0.285 | 3,624 | 1.38 | |
| m09_N600_D0 | 0.116 | 0.074 | 1.000 | 0.867 | 0.098 | 0.000 | 0.017 | 0.828 | 0.000 | 0.011 | 0.194 | 0.285 | 3,625 | 1.60 | |
| m22_N600_D200 | 0.069 | 0.051 | 1.000 | 0.918 | 0.036 | 0.000 | 0.013 | 0.914 | 0.000 | 0.013 | 0.202 | 0.292 | 3,629 | 2.87 | |
| m22_N600_D400 | 0.076 | 0.053 | 1.000 | 0.910 | 0.049 | 0.000 | 0.012 | 0.898 | 0.000 | 0.012 | 0.203 | 0.295 | 3,630 | 2.92 | |
| m20_N600_D300 | 0.057 | 0.036 | 0.949 | 0.797 | 0.140 | 0.051 | 0.146 | 0.824 | 0.000 | 0.005 | 0.212 | 0.286 | 3,604 | 4.17 | |
| m10_N600_D0 | 0.087 | 0.048 | 0.968 | 0.844 | 0.118 | 0.031 | 0.068 | 0.833 | 0.001 | 0.013 | 0.200 | 0.294 | 3,638 | 5.20 | |
| m21_N600_D50 | 0.137 | 0.070 | 1.000 | 0.853 | 0.106 | 0.000 | 0.011 | 0.824 | 0.000 | 0.012 | 0.192 | 0.286 | 3,641 | 5.97 | |
| m11_N600_D0 | 0.058 | 0.041 | 0.989 | 0.905 | 0.033 | 0.011 | 0.037 | 0.926 | 0.000 | 0.013 | 0.206 | 0.297 | 3,597 | 6.17 | |
| m02_N600_D0 | 0.000 | 0.000 | 0.866 | 0.712 | 0.067 | 0.134 | 0.288 | 0.933 | 0.000 | 0.007 | 0.227 | 0.297 | 3,596 | 6.32 | |
| m22_N600_D300 | 0.073 | 0.047 | 1.000 | 0.911 | 0.064 | 0.000 | 0.016 | 0.888 | 0.000 | 0.013 | 0.203 | 0.297 | 3,596 | 6.38 | |
| m04_N600_D0 | 0.027 | 0.015 | 0.947 | 0.802 | 0.075 | 0.053 | 0.170 | 0.909 | 0.000 | 0.005 | 0.216 | 0.288 | 3,594 | 7.01 | |
| m17_N600_D0 | 0.034 | 0.020 | 0.946 | 0.762 | 0.112 | 0.054 | 0.204 | 0.868 | 0.000 | 0.006 | 0.218 | 0.291 | 3,592 | 7.50 | |
| m22_N600_D100 | 0.078 | 0.046 | 1.000 | 0.875 | 0.032 | 0.000 | 0.047 | 0.921 | 0.000 | 0.012 | 0.203 | 0.294 | 3,647 | 7.79 | |
| m23_N600_D200 | 0.066 | 0.038 | 1.000 | 0.923 | 0.054 | 0.000 | 0.011 | 0.908 | 0.000 | 0.014 | 0.208 | 0.298 | 3,650 | 8.59 | |
| m22_N600_D50 | 0.075 | 0.050 | 1.000 | 0.913 | 0.052 | 0.000 | 0.011 | 0.898 | 0.000 | 0.013 | 0.201 | 0.290 | 3,654 | 9.70 | |
SRC = source population used for stocking. Given the shared patterns of ancestry between the populations used for stocking (i.e., Chautauqua, Joseph, and Tremblant lakes), individuals were merged together and modeled as a single unit. Given that no sign of ancestry from Pigeon Lake was found in the St. Lawrence (SLR) or Deux‐Montagnes L. (LDM), this source was not included here. Based on our empirical results, the migration rate (m) was set 1.5 times higher in LDM than in SLR in which we included LSL, LSF, LSP, MSC, MSG, and MSV. Admixture and F ST values in bold are those observed in our empirical data. The group 1 (grp1), group 2 (grp2), and group 3 (grp3) represent admixture membership probability of individuals from LDM, SLR, and SRC, respectively, for K = 3. Polym = number of polymorphic sites with a MAF equal to 1% or more. RMSE = root‐mean‐squared error. The lower the RMSE, the closer a given model is to the empirical observation. Here, the 20 best models in terms of RMSE are displayed. Further details can be found in Supporting Information Table S3.
2.5. Genetic differentiation and population admixture
Levels of genetic differentiation between sampling locations were computed using Weir and Cockerham's F st estimator θ (Weir & Cockerham, 1984) in vcftools v0.1.15, and resulting values were used to construct a heatmap and a dendrogram in R using custom script. Confidence intervals around F st were computed using Hierfstat with 1,000 bootstraps. Isolation by distance (IBD) was tested between pairwise genetic distances as F st/(1 − F st) (Rousset, 1997) and the pairwise distance following the river watershed network using a Mantel test in R with 10,000 permutations.
To better understand whether low genetic differentiation was due to high population connectivity, we also measured the level of migration within two sites at the extreme ends of the St. Lawrence mainstem sampling using an approach similar to Migrate (Beerli & Felsenstein, 2001). We performed coalescent simulations of two populations of independent size N 1 and N 2, connected by migration (M = 4N ref m), modeled independently, and therefore with possible asymmetry. We modified an ABC pipeline (described in Supplementary Materials: Appendix S1) and estimated the intensity of gene flow between the sites with conventional ABC procedures (Csilléry, Blum, Gaggiotti, & François, 2010).
Our simulation pipeline was also applied to examine whether strong genetic differentiation (as measured by F st) translated into a long divergence time. We estimated the divergence time between Chautauqua and Pigeon lakes, the two original sources of stocking, under the assumption that no gene flow existed between these contemporary isolated lakes. Under this hypothesis, the divergence history can be summarized by a model of strict isolation (SI) characterized by the effective population size of the two daughter populations (N1 and N2) that diverged from a common ancestral population of size Nanc at time Tsplit. Coalescent simulations were again used to generate genomic data and to compare these theoretical patterns to our empirical data using an ABC framework. Full details are provided in Supplementary Materials: Appendix S1. This analysis was replicated five times by comparing a randomly chosen site within the St. Lawrence to either Chautauqua or Pigeon Lake.
Next, ancestry and admixture proportions were inferred for each individual using Admixture (Alexander et al., 2009) with K‐values ranging from one to 25. Then the snmf function implemented in the LEA R package (Frichot & François, 2015) to estimate ancestry coefficients levels of K. Other DAPC model‐based clustering methods (Jombart, Devillard, & Balloux, 2010) produced highly congruent results to those obtained using Admixture or LEA. All admixture analyses were performed by maintaining a single SNP per locus. Keeping either a random SNP or the SNP with the highest minor allele frequency produced similar results (not shown), and we report only the results of analyses performed with SNPs showing the highest minor allelic frequency (MAF) at each locus. We then correlated the individuals “stocking source” ancestry coefficient to individual levels of observed heterozygosity (Spearman's rho) and tested it for significance with a t test.
Finally, we used a modification of the fineSTRUCTURE package (Lawson, Hellenthal, Myers, & Falush, 2012) implemented in FineRADstructure (Malinsky, Trucchi, Lawson, & Falush, 2018) to infer levels of population genetic structure and ancestry from haplotype data derived from RAD loci. Less than one percent of missing data were allowed. First, RADpainter was used to compute the co‐ancestry matrix. Then individuals were assigned to populations with fineSTRUCTURE using 100,000 MCMC iteration for burn‐in and the same number of sampled iteration with a thinning interval of 1,000.
2.6. Demographic simulations of historical stocking
Individual forward simulations were used to investigate the effect of stocking on admixture patterns and the contemporary genetic makeup of populations in the fluvial system of the Lower St. Lawrence River (a series of connected lakes with the mainstem). Tributaries were not simulated as data on their stocking intensity were not consistently available. The goal of the simulations was to estimate the effective migration rate from stocking sources to the St. Lawrence population necessary to explain observed admixture and benefit our understanding of stocking effects (i.e., levels of introgression). We focused on a restricted dataset to construct a simplified model of divergence made of three Muskellunge groupings along the Lower St. Lawrence fluvial system. The first group included fish from sites within the fluvial lakes and the mainstem separating lakes within the St. Lawrence River, namely lakes Saint‐Pierre (LSP), Saint‐Françis (LSF), and Saint‐Louis (LSL), as well as from three different sampling mainstem localities between Montréal and Sorel including Contrecoeur (MSC), Boucherville (MSG), and Varennes (MSV) sampling sites (n = 224 individuals, named SLR hereafter). A second population (lake des Deux‐Montagnes, LDM n = 56) was identified based on patterns of admixture and shared ancestry (see Section 3). Finally, a third population was the source of stocking populations and included as a single group the original site CHQ as well as JOS and TRE lakes (n = 100) that originated from stocking with CHQ (confirmed by admixture and FineRADstructure analysis, see Section 3). Finally, the other source of stocking, PIG from the Kawartha system was not considered since no contribution to the admixture patterns in the St. Lawrence River was detected (see Section 3).
We simulated a neutral 10,000 kb long chromosome assuming a uniform mutation rate µ of 1e‐8 per bp per generation and a recombination rate r of 1e‐8 per bp per generation using slim v2.6 (Haller & Messer, 2009). First, we simulated an ancestral ideal population of size N = 2,400 for 80,000 generations to reach equilibrium. The ancestral population was then split into three populations corresponding to the stocking source (SRC), the LDM, and the SLR with a reduced initial size of N = 800 (a range of different sizes were tested, see below). Given observed data, we allowed for a constant rate of migration between LDM and SLR with m SLR‐>LDM = 0.0005 and m LDM→SLR = 0.00025 (we explored parameters producing data similar to empirical observations). We implemented asymmetric dispersal to reflect the expected downstream biased dispersal (e.g., Paz‐Vinas, Loot, Stevens, & Blanchet, 2015), which was also observed in our data from LDM into SLR (see admixture results). Initially, no migration was allowed between the stocking sources (considered as a single unit based on patterns of shared ancestry) and either LDM or SLR. These populations kept diverging for 2,685 generations roughly corresponding to the postglacial divergence period (assuming a generation time of 6 years based on age at first spawning; Farrell et al., 2007). To reproduce initial stocking event about 15 generations ago, we introduced migration between stocking sources and LDM and SLR. Since more admixture was observed in LDM compared to SLR (see Section 3), migration into LDM was set 1.5 times higher than in SLR. We tested a range of migration rate from 1e‐6 to 0.1. After 82 685 generations, a set of individuals matching our empirical sample sizes were randomly sampled and exported into vcf format for computation of summary statistics corresponding to admixture levels, Weir & Cockerham F ST and the numbers of polymorphic site above the 1% MAF. We allowed for lower fitness of the stocking source population by implementing a mortality‐based filter (fitness callback in Slim v2, Haller & Messer, 2017). We explored a set of fitness filters where 50, 100, 200, and 400 randomly chosen stocked individuals died, implying a mortality rate of 6%, 12.5%, 25% and 50% for N = 800. This allowed testing outcomes if a small number of individuals reproduced, while additionally considering effects of a variety of mortality‐based filters such as lower local adaptation of migrant individuals. Finally, we tested the effect of varying demographic scenarios through population increase or decrease by multiplying the size of each descendant populations by 2 (N = 1,600), 0.75 (N = 600, closer to the estimated effective population size) and 0.5 (N = 400). The ancestral population size was multiplied accordingly. The number of individuals that died was kept constant, implying a varying mortality rate. Simulations were repeated 50 times to assess the variability of admixture inferences. We then computed the root‐mean‐squared error (RMSE) for each scenario and computed the distance between the distributions of summary statistics and the empirically observed one, allowing us to compare model fit to the data. Data could have been analyzed in an Approximate Bayesian Computation framework (Beaumont, Zhang, & Balding, 2002) to test realistic scenarios of migration and subsequent admixture. However, the forward simulations implemented were too slow to generate the required number of simulations (i.e., >500,000) for classical ABC analysis. All scripts used with Slim will be freely available Github at https://github.com/QuentinRougemont/fwd_sims
3. RESULTS
3.1. Genetic diversity
An average of 3.23 million reads per individual was sequenced. Genetic diversity indices measured on each rad loci revealed a median п value of 0.003. Median observed heterozygosity and gene diversity for polymorphic SNPs markers were 0.144 and 0.148, respectively (Table 1). Two sites stood out as displaying significantly lower observed heterozygosity compared with other sites, namely the populations from TRA (H o = 0.088, H s = 0.086, Wilcoxon‐test, W = 759 p < 0.001) and PIG (H o = 0.070, H s = 0.066, Wilcoxon‐test, W = 330, p < 0.001). Lower diversity for these populations was reflected by their п estimates of 0.0016 and 0.0019, respectively (Table 1). Lakes of the St. Lawrence system (des Deux‐Montagnes [LDM], Saint–Louis [LSL], Saint‐Françis [LSF], and Saint‐Pierre [LSP] Lakes) also displayed slightly lower diversity than median diversity values (median H o = 0.103, H s = 0.105) whereas the opposite was observed for sites located in the fluvial stretch of the St. Lawrence between LSL and LSP (MSG, MSC, and MSV; median H o = 0.156, H s = 0.161). The median proportion of polymorphic SNPs was 18% (SD = 6%), with the least polymorphic site being the Ottawa River (OTT; 11%) as expected given its small sample size (n = 7), whereas LSL was the most polymorphic location (31%). On average, there was more polymorphism within the St. Lawrence than in the tributaries or lakes (Table 1). There was 21 times more private polymorphisms into the St. Lawrence mainstem compared to those within tributaries. The St. Lawrence mainstem also contained 2.7 times more private polymorphism than all stocking sources merged together. Additionally, on average, there were 10 times more private polymorphisms in the stocking source when compared to tributaries and adjacent lakes. N e estimates returned low values ranging from 14 (CHP) to 2,308 (LSP) with four cases where the N e value could not be estimated, either because of paucity of polymorphic markers and/or limited sample size such as for OTT (result not shown). Pooling all Muskellunge according to their inferred genetic cluster (see below) resolved this problem and revealed for example a N e value of 669 (95% CI = 493–823) for the St. Lawrence group (Table 1). Without correction for physical linkage all N e values were downwardly biased, corroborating findings by Waples, Larson, and Wasples (2016).
3.2. Population differentiation
The global F ST estimate averaged over‐all pairwise comparisons, was 0.211 (ranging from 0.0006 between LSL and MSC to 0.709 between TRA and PIG). Five locations from the Lower St. Lawrence River (i.e., LSL vs. MSG, LSL vs. MSC, MSG vs. MSC and both MSC and MSG vs. MSV) had nonsignificant pairwise F ST values (Figure 2a; Supporting Information Table S1). When excluding all tributaries and inland lakes, F ST values averaged over all sites from the Thousand Islands upper river network of spawning sites (TIN) in the Upper St. Lawrence River downstream to LSP was 0.014 (Figure 2a). Here, the LDM site clustered separately from all sites located on the Upper and Lower St. Lawrence (Figure 2b). When removing the LDM site, the F ST among all other sampling locations dropped to 0.008, suggesting weak genetic differentiation throughout the mainstem of the St. Lawrence River. LSL, LSP, LSF, and LSW displayed significant but weak F ST (i.e., <0.009) between each other and between the three sites from the St. Lawrence section including MSC, MSG, and MSV. The uppermost St. Lawrence population (TIN) displayed higher differentiation when compared to all other sites with F ST ranging from 0.015 to 0.055.
Figure 2.
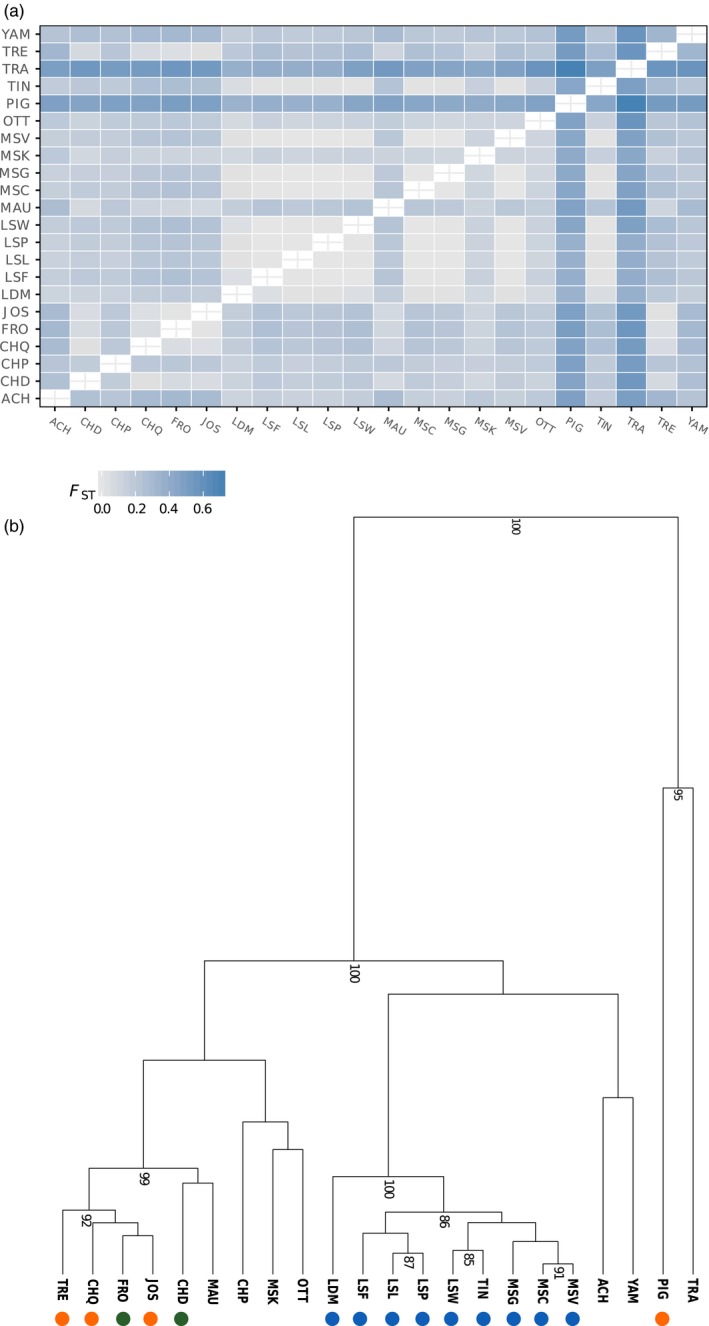
(a) Heatmap of pairwise F ST values between each sample site. (b) Clustering based on pairwise F ST values performed on each sample site. Only nodes with a bootstrap support higher than 80 are displayed. Orange dots = source of individuals used for stocking. Green dots = lake or river where Muskellunge were absent and have been introduced. Blue dots = site from the St. Lawrence River and des Deux‐Montagnes Lake
All tributaries were significantly differentiated from the St. Lawrence R. sites between LSL and LSP and displayed a much stronger level of differentiation than observed within the St. Lawrence R. itself, with an averaged F ST value between each tributary and all sites from the St. Lawrence R. of 0.20 for the Saint‐Maurice R. (MAU), 0.119 for OTT, 0.177 for the Chaudière R. (CHD), and 0.196 for the Yamaska R. (YAM). With population tree topology, the CHD and MAU populations, both with presumably historically low abundance that were subsequently stocked, clustered close to the stocking source, yet their distinctiveness was strongly supported (Figure 2b). Differentiation among Maskinongé L. (MSK), Ottawa R. (OTT) and Champlain Lake (CHP) was less clear as they appeared close to the source of stocking albeit the clustering was weakly supported in the tree (Figure 2b). There was a modest signal of IBD when all sites were included (mantel test r = 0.369 p = 0.0129) and considering only St. Lawrence R. sites (from TIN to LSP) a stronger IBD pattern was revealed (r = 0.799, p = 0.0046, Figure 2). Finally, pairwise D xy and net divergence (D a) did not revealed major difference between compared pairs of populations (Supporting Information Table S2).
3.3. Evidence for asymmetric gene flow
ABC estimates of migration rates and effective population size were performed using 1 million coalescent simulations. We chose the site LSP and LSP, located at the opposite ends on the Lower St. Lawrence and did not use the more upstream site (e.g., TIN) located upstream of dams artificially restricting upstream migration. Results indicated that posterior distribution of the migration rate was highly different from the prior distribution, resulting in narrow credible intervals (Supporting Information Figure S5). This migration was strongly asymmetric with a median value M down←up = 40 [95% credible intervals = 39–40] whereas migration from downstream to the upstream site was low with a median value M down←up = 0.067 [95% credible intervals = 0.018–0.140]. Effective population size was similar between downstream and upstream sites (Supporting Information Figure S5) This provided support for the hypothesis that low genetic differentiation was partly due to strong downstream directed dispersal. Relatively large effective population size may also contribute to this pattern (Supporting Information Figures S5 and S6).
3.4. Evidence for a relatively recent and shared history of divergence
Estimates of time since divergence were performed assuming that the divergence history could be summarized by a simplified model of SI (Supplementary results: Appendix S1). Posterior distribution of the divergence time between the two major sources of stocking were differentiated from the prior distribution, indicating that this parameter could confidently be interpreted. We found, that the median value was 0.023 coalescent units [95% CI = 0.010–0.045]. Assuming a mutation rate µ of 1e −8 bp per generation (see Supplementary results: Appendix S1 for treatment) and the 5 years generation time, this would translate to a divergence time of 23,000 years [10,000‐45,000]. Replicating this analysis using other sites (Supplementary results: Appendix S1, Supporting Information Figures S7–S17) resulted in lower estimates of divergence time (i.e., median range from 15,400 to 19,300 years, min = 6,500, max = 31,000).
3.5. Population structure and admixture
Analyses of admixture cross‐validation consistently produced low cross‐validation scores for K = 8 and K = 13 indicating the number of clusters likely lies between these values (Supporting Information Figure S3). Similar results were obtained from LEA cross‐entropy criterion with minimum values obtained around K = 12 to 13 while the DAPC provided the same results. For simplicity, only results from the admixture analysis for K = 8 and K = 13 are detailed below (see also Figure 3a). Further plots are presented in Supporting Information Figure S3 for additional K values (up to K = 19).
Figure 3.
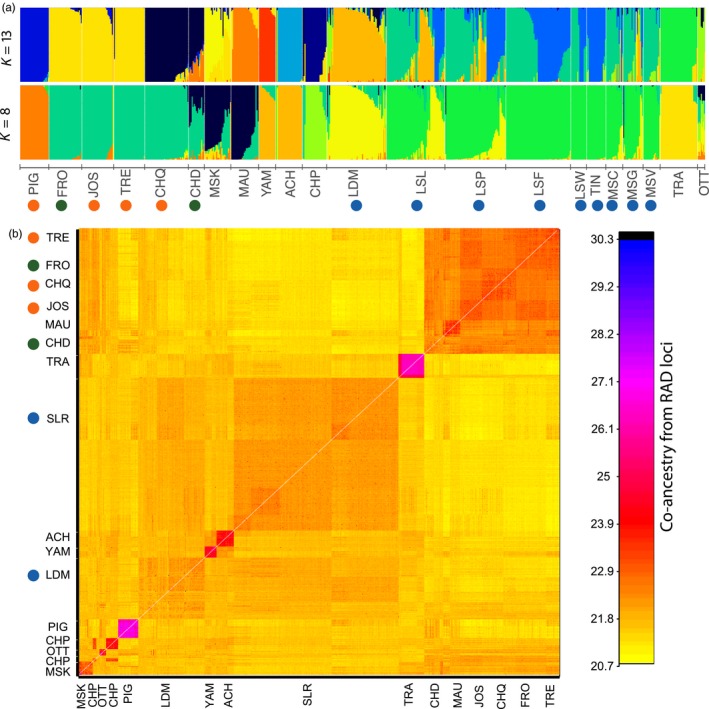
(a) Levels of admixture for K = 8 and K = 13. Each color represents a distinct cluster, and each bar represents an individual. See Figure 1 for the labels of each dot in the graph; (b) co‐ancestry matrix inferred by FineRADstructure. Each pixel represents individual co‐ancestry coefficient inferred based on short haplotype loci. The labels summarize the major groups according to names of sampling sites. The higher values of co‐ancestry coefficient sharing are depicted in darker colors, whereas lower values of co‐ancestry coefficient sharing appear in yellow colors
Considering K = 8 clustered together the major groups used for stocking (i.e., CHQ, JOS, TRE) together with the FRO introduced from the JOS population (mean q‐value for the four lakes = 0.975; Figure 3). Below, we considered that any individual from the St. Lawrence and lake who displayed a stocked ancestry coefficient above 0.10 to be admixed, as commonly done in the literature (e.g., Valiquette, Perrier, Thibault, & Bernatchez, 2014, Létourneau et al., 2018). We defined “stocked ancestry” as the group FRO‐JOS‐TRE‐CHQ and the group PIG.
Based on this criterion, we observed variable levels of admixture in all tributaries and lakes outside the mainstem of the St. Lawrence River. From MAU, 13 fish (50%) displayed a stocking source membership probability >0.10. Specifically, one individual was assigned to the “stocking” group with a q‐value >0.95 and remaining individuals displayed mixed membership probability suggesting backcrosses or presence of advanced generation hybrids (q‐values between 0.116 and 0.626). The remaining individuals (50%) from MAU displayed a q‐value >0.90 belonging to another group comprised of individuals from MAU, CHD and MSK. All 15 Muskellunge from CHD displayed mixed ancestry with membership probability of belonging to the “stocking” group ranging from 0.407 to 0.724 and individuals from MSK were admixed (q‐values ranging from 0.22 to 0.88). Among those, nine (36%) displayed q‐value of belonging to the “stocking group” ranging from 0.10 to 0.617 with no individual of apparent pure “stocking” origin (Figure 3a). Six (37.5%) and four individuals (57%), respectively, from YAM and OTT were admixed (q‐value > 0.10) with the stocking source (Figure 3a). None of these individuals was of pure stocking origin as they displayed q‐values ranging from 0.10 to 0.211 and from 0.10 to 0.365 for YAM and OTT, respectively. In OTT, all seven sampled Muskellunge were admixed and none was assigned to a particular cluster. The Achigan R. (ACH) and YAM populations clustered together (mean q‐value = 0.915) whereas TRA and CHP populations formed two separate clusters (q‐value = 0.96 and 0.81, respectively). Finally, the Champlain L. showed indicators of significant admixture from the stocking source for six individuals (30%). There were 2 F0 immigrants and 5 introgressed individuals with q‐values ranging from 0.10 to 0.525.
Sites from the St. Lawrence River from the uppermost (TIN) to the most downstream site (LSP) tended to form a single group with the exception of LDM that clearly clustered separately (q‐value = 0.84). In contrast with inland lakes and tributaries, weak evidence was detected for admixture with the stocking source population. Thus, 21 individuals (8%) from the St. Lawrence R. (from TIN to LSP) displayed introgression with the stocking source (q‐value ranging from 0.10 to 0.54) and no individual was of pure stocking origin. However, we observed more fish (n = 19, 33%) from LDM that were admixed with the stocking source (q‐value ranging from 0.10 to 0.50) but none were of pure stocking origin. While there was limited admixture with the stocking source, we found evidence for admixture among fish from different locations within the system. Here, 6% of individuals from LSL were classified as F0 immigrants (q‐value > 0.9) from the nearby LDM population and 42% were admixed with LDM (Figure 3a). It is noteworthy that considering the spatial location of individuals in LSL (all collected fish within the SLR were georeferenced by GPS), we observed a tendency for the most admixed individuals (78%) to be preferentially found in the northern part of LSL whereas 87% of pure individuals were located preferentially in the southern part of LSL. Finally, 17 Muskellunge (30%) from LSP displayed admixture with LDM whereas 11 individuals (22%) from the St. Lawrence R. including MSG, MSC, and MSV were admixed with LDM (q‐value>0.10).
Considering K = 13 revealed the same general pattern of differential admixture and weak admixture with the stocking source was observed at all sites within the mainstem of the St. Lawrence R., a somewhat more pronounced admixture in LDM and the highest admixture being observed in tributaries and inland lakes (except FRO where no Muskellunge occurred before stocking) (Figure 3a). However, K = 13 revealed more separation between some population clusters. Thus, Muskellunge from YAM and ACH tributaries were clearly assigned to two clusters corresponding to their local river. The original stocking source (CHQ) clustered independently (mean q‐value = 0.95) of derived individuals of JOS‐TRE stocking sources that grouped together (q‐value = 0.96). While FRO lake still clustered with JOS, fish from MAU clustered independently (q‐value = 0.87) but with one individual showing a F0 immigrant profile (q‐value > 0.95) from the JOS‐TRE stocking group and seven other individuals showing signs of introgression (0.10 < q‐value < 0.53) (Figure 4a,b). Five Muskellunge (20%) from MSK displayed a q‐value > 0.90, while 95% of remaining individuals showed introgression (0.13 < q‐value < 0.730) with the JOS‐TRE stocking source (Figure 3a, Figure 4). The CHP site tended to form a separate cluster (mean q‐value = 0.81) but with one F0 immigrant from CHQ stocking source and five individuals (21%) showing admixture (F1 like profile) from the JOS‐TRE group (Figure 3a). Individuals from the St. Lawrence River from TIN to LSP were now separated into two admixed groups. In particular, individuals from the uppermost TIN site were assigned to this cluster (mean q‐value = 0.87) while 31%, 26%, 12%, and 7% of individual, respectively, from LSF, LSW, LSL, and LSP were now assigned (q > 0.90) to this new group. In these same sites, respectively, 31%, 27%, 22%, and 37% of individuals, as well as 3.5% in LDM were admixed (Figure 3a, top panel). Finally, five individuals (9%) from LSL were assigned as putative F0 immigrants from LDM and 16 individuals (29%) displayed mixed membership probability. The same was observed in LSP with no F0 immigrants but 15 individuals (26%) admixed with LDM. Overall, Muskellunge from the St. Lawrence displayed lower stocking q‐value membership than the LDM or tributaries and inlands lakes (SLR range: 0.10 to 0.63, LDM range: 0.10 to 0.51, tributaries range: 0.10 to 0.99; see details and boxplots in Figure 4a). Similarly, the number of admixed individuals was lower in the St. Lawrence (8%) than in the LDM (31%) and tributaries and inland lakes (42%) (Figure 4b). Finally, there was a global significant and positive correlation between the level of stocking ancestry and the observed heterozygosity for K = 13 (rho spearman = 0.37, t = −15.578, df = 452.03, p‐value < 2.2e‐16) as well as for K = 8 (rho spearman = 0.26, t = 12.863, df = 513.38, p‐value < 2.2e‐16).
Figure 4.
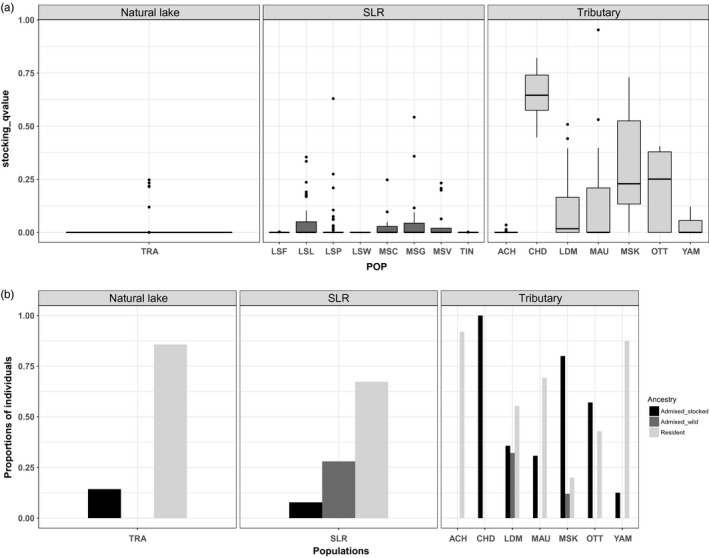
(a) Boxplot of “stocking source” ancestry coefficient (q‐values) in the single nonsupplemented population and in the different supplemented populations separated according to whether they occur in the St. Lawrence River itself (SLR) or its tributaries. (b) Proportion of individuals assigned to different classes of ancestry: individual assigned as “resident” if q‐value >0.9, individual assigned as of “admixed domestic ancestry” if q‐value of belonging to a “stocking” source cluster was above 0.1, individual assigned as of “wild ancestry” if q‐value of belonging to another foreign population was above 0.10.
Considering higher K values (from 14 to 16) resolved all rivers except the CHD (that was admixed with CHQ and LDM), only a value of K = 19 separated CHD individuals to their own cluster (median‐q > 0.90). The FRO and JOS lake remained clustered together (see Supporting Information Figure S1).
Results from co‐ancestry analyses at the haplotype level provided additional information about ancestry between individuals and revealed patterns consistent with admixture analyses at different K values and highlight the similarity observed at K = 8 and the finer differences for K = 13. In particular, two major blocks stood out corresponding to (a) the populations used for stocking and (b) the St. Lawrence R. Here, the CHQ, JOS, and TRE sources as well as the derived populations FRO and CHD appeared closely related (top panel in Figure 3b). All individuals from MAU, 24% from MSK and 9% from CHP shared ancestry with these source populations (Figure 3b). The second major block was made of individuals from the different sampling sites from the St. Lawrence River (TIN to LSP excluding tributaries) and displayed moderately shared co‐ancestry with LDM. In particular, 36% of LSL, 12% of LSP, and 16% of the MSC, MSG, and MSV group of individuals displayed closer co‐ancestry to LDM than to remaining individuals from SLR. Admixture analyses revealed one individual from JOS that displayed close ancestry to the St. Lawrence River fish. The seven individuals from OTT were closely related to a group of five individuals (9%) from LDM, indicating possible downstream dispersal from the Ottawa River to LDM or a common ancestral origin. All fish from ACH and YAM were well separated (K = 13 in admixture), but displayed shared ancestry, highlighting their close relationship (K = 8 in admixture). Finally, both PIG and TRA individuals formed well‐separated clusters of ancestry (Figure 3b).
3.6. Simulations of admixture
Results indicated that over all scenarios, N = 600 (closer to estimated N e in the SLR) systematically displayed lower RMSE (root mean squared errors) than those with higher N e (Supporting Information Table S4). For instance, only N = 600 resulted in a similar level of polymorphism (i.e., polymorphic SNPs at the 1% level) as to empirical data, while all other datasets were significantly different in number of polymorphic sites. Therefore, for simplicity, we present results for the N = 600 scenario (the 20 best model related to N = 600 are presented in Table 2); other scenarios for models with the lowest RMSE are presented in Supporting Information Table S3). The best scenarios in terms of RMSE were those where migration rate from the stocking source (SRC) to the St. Lawrence (LSW to LSP) ranged between m = 0.0025 and m = 0.005. Migration rate was systematically higher from SRC to LDM with values ranging from m = 0.00375 to m = 0.0075 (ms M20–M23, similar to M9–M11 but includes mortality filters; Table 2). None of the scenarios where migration rate was set higher (from m = 0.01 to m = 0.30) provided a good fit (Table 2, Supporting Information Table S3). In particular, M16 theoretically would reproduce most closely the expected level of migration (m ~ 0.5) due to the intensity of stocking, but poorly fitted the data (Supporting Information Table S3). Scenarios M1–M8, with lower migration rates (m < 0.001), did not properly reproduce observed data. While the level of genetic differentiation between SRC and SLR or between SRC and LDM were qualitatively close to that observed empirically in models M20–M23 (simulated F ST range ~0.192 to 0.298, Table 2), none of the scenarios were able to reproduce the observed divergence between SLR and LDM (mean F ST for the 20 best scenarios = 0.011 vs. 0.036 in our data set; Table 2). The difference between simulated and observed differentiation was significant (p < 0.005), an indication that some processes not considered in the model, such as a long isolation period or a smaller N e in LDM or that two populations originating from evolutionary divergent lineages, may have contributed to their divergence. Overall, scenarios with the mortality‐based filter produced the lowest RMSE, with the best scenario (RMSE = 0.05) where 50 individuals randomly died (Table 2). However, scenario M9 where no individual died also produced a low RMSE of 1.60. Several scenarios with mortality of 100–400 individuals presented intermediate RMSE values (Table 2), indicating that varying rates of mortality for N = 600 could explain empirical data. Increasing mortality rate to 500 individuals (i.e., 83%) resulted in RMSE of 1.73.
4. DISCUSSION
4.1. Genetic structure between distant lakes and isolation by distance within the St. Lawrence River
A common issue in population genetics is delineation of population structure confounded with isolation by distance (IBD), where genetic differentiation increases with geographic distance due to genetic drift under limited dispersal (Wright, 1943). Here, our data indicated both the occurrence of clusters of genetically differentiated groups (i.e., among distant and isolated water bodies) and significant isolation by distance within the St. Lawrence River, as reported in Muskellunge (Kapuscinski et al., 2013; Miller et al., 2017) and other species (Meirmans, 2012; Sexton et al., 2014). The behavior of the Muskellunge, especially its known spawning site fidelity (Farrell et al., 2007) and possible natal homing (Crossman, 1990; Jennings et al., 2011; Margenau, 1994; Miller et al., 2001) are factors that can contribute to both IBD and the establishment of population structure. Regarding the major clusters, a strict interpretation of F ST values and admixture results indicates that each lake and each tributary could be classified as a distinct population (Supporting Information Figure S3). However, finer analyses revealed shared co‐ancestry between many samples, especially between some stocked tributaries/lake with population stocking source. Moreover, coalescent analyses suggested a recent divergence of studied populations, and there is evidence for ample shared polymorphism between all populations. We therefore hypothesize that tributaries represent a bottlenecked subset of the larger St. Lawrence R. populations following their colonization by a small number of founders. Then, isolation of each individual group enhanced the effect of genetic drift (e.g., Turnquist et al., 2017), resulting in pronounced population‐level genetic differentiation. In the case of the Chaudière River, we further note that the connectivity with the St. Lawrence is already restricted by natural waterfall. In this particular case, the river display stronger footprint of introgression with individuals of stocking origin.”
The two clusters observed within the St. Lawrence R. are likely due to isolation by distance confounding the clustering methods (Bradburd, Coop, & Ralph, 2018; Meirmans, 2012). As expected, our coalescent analysis supported downstream biased dispersal, which likely explains the high connectivity within the St. Lawrence R. (Morrissey & de Kerckhove, 2009). Moreover, the presence of two dams on the St. Lawrence certainly contributes to restrict upstream dispersal and associated gene flow.
In tributaries, a variable proportion of fish from sites between mainstem LSL and LSP displayed shared ancestry with fish from the LDM tributary. Admixed individuals were preferentially located in the northern shore of LSL, whereas those on the southern shore where generally not admixed. Interestingly, this dichotomy between the northern and southern shores of LSL was also reported in the Northern Pike (Ouellet‐Cauchon, Mingelbier, Lecomte, & Bernatchez, 2014) and Yellow Perch Perca flavescens (Leclerc, Mailhot, Mingelbier, & Bernatchez, 2008). In both species, fish from the south shore of the LSL were more similar to those from elsewhere within the St. Lawrence R. than those from the north shore. LSL is characterized by contrasted water masses flowing from the St. Lawrence R. on the south shore and from the Ottawa River on the north shore (Hudon & Carignan, 2008; Leclerc et al., 2008). Therefore, it would be of interest to distinguish the respective contribution of ecological (e.g., temperature, pH, light extinction, etc.) and historical (e.g., glacial refugia) factors responsible for this repeated pattern of divergence observed between the north and south shores of LSL among different species.
4.2. Genetic impacts of stocking vary with supplementation history and population size
Correlation between stocking intensity and admixture have previously been documented in a range of species including mainly salmonids (Campos, Posada, & Morán, 2008; Finnegan & Stevens, 2008; Garcia‐Marin, Sanz, & Pla, 1999; Marie, Bernatchez, & Garant, 2010; Perrier, Baglinière et al., 2013; Perrier, Guyomard et al., 2013; Sønstebø, Borgstrøm, & Heun, 2008) Studies additionally revealed a tendency toward a decreasing proportion of admixture following stocking cessation (Hansen et al., 2009; Harbicht, Wilson, & Fraser, 2014; Létourneau et al., 2018; Perrier, Baglinière et al., 2013; Valiquette et al., 2014). Our results are congruent with these observations where PIG lake, initially used as a stocking source (from 1950 to 1965), left few traces of admixture in our populations. Significant “PIG” ancestry was detected in <2% of the individuals. Miller et al. (2009) documented a similar pattern with microsatellite DNA markers following introduction of three sources of Muskellunge including intrabasin transfer of progeny from Shoepack Lake Minnesota. The stocking resulted in admixture from each source and the deleterious effect of slow growth was detected in fish with Shoepack ancestry. Managers responded by altering stocking policies and the strain mixing has since declined over time via dilution (Miller et al., 2012). We hypothesize a similar dilution of individual ancestry through time has occurred with respect to the PIG stocking source although we are aware of no negative effects of its introduction.
Miller et al. (2012) compared genetic mixing following Muskellunge stocking of four strains released in supplemented and introduced populations that showed ancestry from each of the source populations, but also indicated that the quantity of stocking was unrelated to levels of admixture. Our results also indicated that the most extensively stocked populations (Lower SLR) displayed the lowest amount of stocking ancestry. This suggests that since the cessation of stocking in 1998, the majority of chromosomal blocks introgressed from the stocking source populations have been removed from the genome of local fish. In contrast, other supplemented tributaries and lakes (e.g., MSK, LDM, OTT) clearly displayed signs of shared ancestry with the stocking sources. Also, the MAU River, which has no recorded stocking to our knowledge, clustered with the stocking sources suggesting the possible occurrence of migrants from further upstream stocked waters in this watershed. Scribner et al. (2015) similarly demonstrated spread of source populations in Michigan watersheds following stocking as evidence of gene flow from stocked individuals.
The observation of low admixture in the St. Lawrence R. may be expected if the St. Lawrence exhibit a larger population size than currently estimated. Regarding N e , the estimated size might be downwardly biased due to the subdivided nature of the river and its tributaries and due to admixture (Waples & England, 2011). Moreover, coalescent estimates of N e (supplementary analyses) may be more relevant (Sjodin, 2005) but are different from estimates based on LD, highlighting classical difficulties in estimating and interpreting this parameter.
With these difficulties in mind, a first scenario to explain the lower admixture of the St. Lawrence R. involves a purely neutral admixture because of high connectivity (and putative high N e) in this system. Therefore, it is possible that the footprints of admixture between stocked fish (of apparently lower effective size) would quickly be diluted into the larger population of the St. Lawrence R. For instance, if stocked individuals are only able to hybridize with wild individuals, then the resulting F1 would mate with wild individuals. This would lead to a decline of the “stocked” genome as 1/2g generation (Phillips & Baird, 2015). This could quickly result in low levels of admixture that may not be detectable with global ancestry inference approaches. On the other hand, in the smaller stream populations with putative low effective population size, footprints of admixture are still apparent. In these small populations, stocking could potentially have some positive assets (Frankham, 2015). Given that all populations are likely postglacially derived, they are likely to be genetically compatible with small probability of outbreeding depression (Ralls et al., 2018). Therefore, stocking from slightly diverged populations may contribute to the maintenance of their genetic diversity and evolutionary potential (Carlson et al., 2014; Frankham, 2015).
A second demographic scenario fitting our empirical data involved differences in effective population size, but requires the action of selection. For instance, stocked individuals may display low survival, low reproductive success (hence resulting in little introgression), and/or selection could act against introgression, as supported by several of our models. Under this second scenario, the higher effective population size and pronounced connectivity of the Lower St. Lawrence R. may play a critical role in selection against introgression (Glémin, 2003). Alternatively, genetic drift may overcome selection against introgression in smaller, isolated populations (Frankham, Ballou, & Briscoe, 2010) leading to fixation of “foreign” alleles and higher footprints of admixture. Such variable outcomes of stocking on admixture have been documented in salmonids (Perrier, Guyomard et al., 2013). Low survival and/or reproductive success of stocked fish has been reported mostly in salmonid species characterized by putatively high rate of local adaptation (Araki, Cooper, & Blouin, 2007; Araki et al., 2009; Araki & Schmid, 2010; Christie, Ford, & Blouin, 2014; Thériault, Moyer, & Banks, 2010; Thériault, Moyer, Jackson, Blouin, & Banks, 2011). Similarly, in Muskellunge, low levels of admixture from the same Ohio strain used in our study, was observed following intensive stocking into West Virginia streams despite their high use (White, Kohli, & Converse, 2018). Our results do not necessarily invoke a mechanism of negative selection or lower fitness of individuals to explain observed pattern. Indeed some of our neutral simulations, with modest levels of migration and no mortality, also provided a good fit to the data.
Despite its ease of implementation, to the best of our knowledge, forward simulations have rarely been used to investigate the effect of stocking (but see Fernández‐Cebrián, Araguas, Sanz, & García‐Marín, 2014; Perrier, Guyomard et al., 2013). Here we assumed equal population size and used a small interval provided by LDNe estimates. However, estimates might be downwardly biased due to meta‐population structure, in which the true N e is higher than in a single randomly mating population. Ideally, future studies will combine whole genome analysis with forward simulations integrating both positive and deleterious mutations, to better understand their effect on patterns of local ancestry (Kim, Huber, & Lohmueller, 2018).
Levels of population genetic diversity were modest, consistent with previous studies on Muskellunge based on microsatellite data in other geographic areas (Miller et al., 2017; Miller et al., 2012; Turnquist et al., 2017; Wilson et al., 2016). Although the various biases associated to RADseq data may complicate the interpretation of genetic diversity estimates (Arnold, Corbett‐Detig, Hartl, & Bomblies, 2013; Cariou, Duret, & Charlat, 2016; Gautier et al., 2013), some general patterns emerged from our data.
First, the population with the lowest level of genetic diversity (TRA) is strongly isolated, thus restricting opportunity for gene flow and implying small effective population size. Given the species life history traits, namely a long life span (15–30 years; Casselman, Robinson, & Crossman, 1999), a territorial and predatory behavior (Becker, 1983), small effective population sizes are expected (Romiguier et al., 2014).
Second, there was a significant and positive correlation between stocking ancestry and genetic diversity. Stocked populations from the Lower SLR and LDM, have been the most extensively stocked (>630,000 fish released) and displayed lower heterozygosity than remaining supplemented tributaries and inland lakes, or introduced populations from CHP, TRE, JOS, and FRO. Moreover, the most extensively stocked populations showed the lowest stocking ancestry. In Muskellunge, Scribner et al. (2015) found that stocked populations displayed higher genetic diversity when stocking was done using multiple strains. Similarly, Lake Trout (Salvelinus namaycush), Valiquette et al. (2014) and Ferchaud, Laporte, Perrier, and Bernatchez (2018) found that genetic diversity of stocked populations was higher than unstocked populations. Here, our results suggest that stocking could have some positive effects for populations found in the smaller tributaries and lakes. Some are currently disconnected from the St. Lawrence R. because of impassable dams. These populations are subject to drift due to their small effective population size that makes them prone to inbreeding depression (Sacherri et al., 1998; Wang et al., 1999). Stocking could contribute to reduce the probability of inbreeding depression by maintaining higher levels of genetic diversity (Frankham, 2015). Alternatively, it is unclear whether selection against introgression removed non‐native (e.g., stocked) alleles within the St. Lawrence or whether simple neutral admixture explains our results. For instance, Hansen et al. (2009) found that Danish Brown Trout (Salmo trutta) stocked for 60 years were subject to selection against non‐native alleles of stocking origin. Similarly, Muhlfeld et al. (2009) found a decline in fitness of Cutthroat Trout (Oncorhynchus clarkii) crossed with non‐native Rainbow Trout (O. mykiss) over generations. In our case, the time of divergence between stocked and population may not have been sufficient to generate differences in the load of deleterious mutations and to generate strong selection against introgression outcome. Therefore, it would be relevant for future studies to test whether selection against introgression was necessary and if it has been efficient in removal of non‐native alleles from stocked fish in the larger St. Lawrence as compared to smaller bottlenecked populations (Kim et al., 2018).
4.3. Conservation and management applications
Knowledge of the genetic structure of fish populations is essential for proper and sustainable stock management since it allows identification of groups of individuals that are genetically and often geographically unconnected, therefore implying at least some demographic independence. Our results indicate that Muskellunge samples studied here can be separated into the following groups: The first being the St. Lawrence watershed, characterized by an upstream‐to‐downstream pattern of dispersal and gene flow. The second group is des Deux‐Montagnes Lake with a shared co‐ancestry with the St. Lawrence R. population, especially fish from the north shore of Saint‐Louis L. The third group comprises tributaries draining into the St. Lawrence River. Each tributary has differentiated by genetic drift and display lower levels of polymorphism and lower effective population size possibly due to founder events. Therefore, ensuring the maintenance of connectivity of tributaries with the mainstem would contribute to maintenance of an overall large effective population size at the meta‐population scale, limiting the risks of inbreeding depression (Frankham, 2015). The fourth group is represented by lakes where Muskellunge has been introduced (Joseph L., Tremblant L. and Frontière L.). These populations exhibit high genetic similarity with the Chautauqua L. source of stocking. The fifth group includes stocked lakes where Muskellunge was initially present (Maskinongé and Champlain lakes) with each of these lakes representing a genetically distinct population. Finally, Traverse Lake represents the sixth group corresponding to a wild unstocked population with low genetic diversity. In isolated lacustrine and river systems originally unoccupied or with presumably low Muskellunge abundance, stocking efforts appear to have been successful in enhancing populations and contributed to support angling activity. This corroborates Dumont (1991) who reached similar conclusions following an analysis of data from the first half period of the stocking program in St. Louis L. located in the Lower St. Lawrence R.
In the smallest lakes, as well as in locations where a massive die‐off was observed (e.g., Thousand Islands; Farrell et al., 2017), supplementation may be necessary, and in such case, the most genetically diversified source should be favored.
In conclusion, our results show that Muskellunge populations are spatially structured into a set of different groups, whose differentiation was affected by the levels of stocking, natural connectivity, or geographic isolation. Our results highlight the necessity to go beyond the simple measurement of genetic differentiation and genetic structure to define subsequent management practice. We further suggest that fishery management and habitat protection measures should be applied globally in order to ensure high connectivity of the population and maintain their evolutionary potential at the highest. We also found that the genetic consequences of stocking on admixture and genetic diversity affected differentially Muskellunge from the St. Lawrence River, its tributaries, and inland lakes, whereby stocking apparently had little impact on the St. Lawrence R. and des Deux‐Montagnes L. populations but apparently led to more important admixture elsewhere. Disentangling the long‐term evolutionary consequences of this history of stocking would require further investigations from denser genome‐wide data.
CONFLICT OF INTEREST
None declared.
DATA AVAILABILITY
Raw sequence data are available at NCBI BioProject ID PRJNA512459. Pipeline for forward simulation is available on gitub at https://github.com/QuentinRougemont/fwd_sims.
Supporting information
ACKNOWLEDGEMENTS
We express our gratitude to Québec’s muskies anglers who collected most of the samples, especially Marc Thorpe, Mike Lazarus, and Mike Phillips. We thank Christopher Legard (New York State Department of Environmental Conservation) for collecting and sharing Chautauqua Lake samples. We thank Samuel Cartier for collecting Lake Champlain fish. Thanks to Chris Wilson (Aquatic Research and Monitoring Section Ontario Ministry of Natural Resources and Forestry) for sharing Pigeon Lake DNA samples and Christopher Wilson (Fish Culture Section, Ontario Ministry of Natural Resources and Forestry) for sharing hatcheries and stocking history between Ontario and Québec. Thanks to Chris Wilson (Aquatic Research and Monitoring Section Ontario Ministry of Natural Resources and Forestry) for sharing Pigeon Lake DNA samples. We also thank Nicolas Auclair, Florent Archambault, Rémi Bacon, Christian Beaudoin, Anabel Carrier, Chantal Côté, Julie Deschesnes, François Girard, Guillaume Lemieux, Louise Nadon, Yves Paradis, Geneviève Richard, and Éliane Valiquette for logistic, field, and laboratory assistance. Funding was provided by the Ministère des Forêts, de la Faune et des Parcs du Québec, and the Canadian Research Chair in Genomics and Conservation of Aquatic Resources.
Rougemont Q, Carrier A, Le Luyer J, et al. Combining population genomics and forward simulations to investigate stocking impacts: A case study of Muskellunge (Esox masquinongy) from the St. Lawrence River basin. Evol Appl. 2019;12:902–922. 10.1111/eva.12765
REFERENCES
- Alexander, D. H. , Novembre, J. , & Lange, K. (2009). Fast m‐based estimation of ancestry in unrelated individuals. Genome Research, 19, 1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljanabi, S. M. , & Martinez, I. (1997). Universal and rapid salt‐extraction of high quality genomic DNA for PCR‐based techniques. Nucleic Acids Research, 25(22), 4692–4693. 10.1093/nar/25.22.4692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf, F. W. (2017). Genetics and the conservation of natural populations: Allozymes to genomes. Molecular Ecology, 26(2), 420–430. 10.1111/mec.13948 [DOI] [PubMed] [Google Scholar]
- Allendorf, F. W. , Leary, R. F. , Spruell, P. , & Wenburg, J. K. (2001). The problems with hybrids: Setting conservation guidelines. Trends in Ecology & Evolution, 16(11), 613–622. 10.1016/S0169-5347(01)02290-X [DOI] [Google Scholar]
- Andrews, K. R. , Good, J. M. , Miller, M. R. , Luikart, G. , & Hohenlohe, P. A. (2016). Harnessing the power of RADseq for ecological and evolutionary genomics. Nature Reviews Genetics, 17(2), 81–92. 10.1038/nrg.2015.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, H. , Cooper, B. , & Blouin, M. S. (2007). Genetic effects of captive breeding cause a rapid, cumulative fitness decline in the wild. Science (New York, N.Y.), 318(5847), 100–103. 10.1126/science.1145621 [DOI] [PubMed] [Google Scholar]
- Araki, H. , Cooper, B. , & Blouin, M. S. (2009). Carry‐over effect of captive breeding reduces reproductive fitness of wild‐born descendants in the wild. Biology Letters, 5(5), 621–624. 10.1098/rsbl.2009.0315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, H. , & Schmid, C. (2010). Is hatchery stocking a help or harm?: Evidence, limitations and future directions in ecological and genetic surveys. Aquaculture, 308, S2–S11. 10.1016/j.aquaculture.2010.05.036 [DOI] [Google Scholar]
- Arnold, B. , Corbett‐Detig, R. B. , Hartl, D. , & Bomblies, K. (2013). RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Molecular Ecology, 22(11), 3179–3190. 10.1111/mec.12276 [DOI] [PubMed] [Google Scholar]
- Bataillon, T. , & Kirkpatrick, M. (2000). Inbreeding depression due to mildly deleterious mutations in finite populations: Size does matter. Genetics Research, 75(01), 75–81. 10.1017/S0016672399004048 [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A. , Zhang, W. , & Balding, D. J. (2002). Approximate Bayesian computation in population genetics. Genetics, 162(4), 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, G. C. (1983). Fishes of Wisconsin. Madison, WI: University of Wisconsin Press. [Google Scholar]
- Beerli, P. , & Felsenstein, J. (2001). Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proceedings of the National Academy of Sciences of the United States of America, 98(8), 4563–4568. 10.1073/pnas.081068098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradburd, G. S. , Coop, G. M. , & Ralph, P. L. (2018). Inferring continuous and discrete population genetic structure across space. Genetics, 210(1), 33–52. 10.1534/genetics.118.301333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur, P. , Hatin, D. , & Bacon, R. (2013). Suivi du maskinongé dans le Saint‐Laurent et le lac des Deux‐Montagnes. In: Atelier sur la faune aquatique 19–21 février 2013. Québec. Québec, Sainte‐Foy, 1‐2‐3 mars 1991.
- Campos, J. L. , Posada, D. , & Morán, P. (2008). Introgression and genetic structure in northern Spanish Atlantic salmon (Salmo salar L.) populations according to mtDNA data. Conservation Genetics, 9(1), 157–169. 10.1007/s10592-007-9318-y [DOI] [Google Scholar]
- Cariou, M. , Duret, L. , & Charlat, S. (2016). How and how much does RAD‐seq bias genetic diversity estimates? BMC Evolutionary Biology, 16, 240 10.1186/s12862-016-0791-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, S. M. , Cunningham, C. J. , & Westley, P. A. H. (2014). Evolutionary rescue in a changing world. Trends in Ecology & Evolution, 29(9), 521–530. 10.1016/j.tree.2014.06.005 [DOI] [PubMed] [Google Scholar]
- Casselman, J. M. , Robinson, C. J. , & Crossman, E. J. (1999). Growth and ultimate length of muskellunge from Ontario water bodies. North American Journal of Fisheries Management, 19(1), 271–290. [DOI] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013). Stacks: An analysis tool set for population genomics. Molecular Ecology, 22(11), 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C. C. , Chow, C. C. , Tellier, L. C. A. M. , Vattikuti, S. , Purcell, S. M. , & Lee, J. J. (2015). Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, M. R. , Ford, M. J. , & Blouin, M. S. (2014). On the reproductive success of early‐generation hatchery fish in the wild. Evolutionary Applications, 7(8), 883–896. 10.1111/eva.12183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier, L. (1987). Le maskinongé (Esox masquinongy). Dans: Problématique de la conservation et de la mise en valeur d’espèces de poissons d’eau douce au Québec Québec: Ministère du Loisir, de la Chasse et de la Pêche. [Google Scholar]
- Crane, D. P. , Miller, L. M. , Diana, J. S. , Casselman, J. M. , Farrell, J. M. , Kapuscinski, K. L. , & Nohner, J. K. (2015). Muskellunge and northern pike ecology and management: Important issues and research needs. Fisheries, 40(6), 258–267. 10.1080/03632415.2015.1038382 [DOI] [Google Scholar]
- Crossman, E. J. (1978). Taxonomy and distribution of North American esocids. American Fisheries Society Special Publication, 11, 13–26. [Google Scholar]
- Crossman, E. J. (1986). The noble muskellunge: A review In Gordon H. E. (Ed.), Managing muskies: A treatise on the biology and propagation of Muskellunge in North America (pp. 1–13). Bethesda, MD: American Fisheries Society. [Google Scholar]
- Crossman, E. J. (1990). Reproductive homing in muskellunge, Esox masquinongy . Canadian Journal of Fisheries and Aquatic Sciences, 47(9), 1803–1812. 10.1139/f90-205 [DOI] [Google Scholar]
- Csilléry, K. , Blum, M. G. B. , Gaggiotti, O. E. , & François, O. (2010). Approximate bayesian computation (ABC) in practice. Trends in Ecology & Evolution, 25(7), 410–418. 10.1016/j.tree.2010.04.001 [DOI] [PubMed] [Google Scholar]
- De La Fontaine . (unpublished)Muskellunge stocking in southern Quebec waters, not peer‐reviewed.
- Do, C. , Waples, R. S. , Peel, D. , Macbeth, G. M. , Tillett, B. J. , & Ovenden, J. R. (2014). NeEstimator v2: Re‐implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Molecular Ecology Resources, 14(1), 209–214. 10.1111/1755-0998.12157 [DOI] [PubMed] [Google Scholar]
- Dumont, P. (1991). Les ensemencements de maskinongé, de truite brune et de truite arc-en-ciel dans les eaux de la plaine de Montréal (pp. 30–42). Sainte-Foy: Conseil de l’aquiculture et des pêches du Québec, CAPQ. [Google Scholar]
- Dunham, R. A. (2011). Aquaculture and fisheries biotechnology (2nd ed.). Cambridge: CAB International; 10.1079/9781845936518.0000 [DOI] [Google Scholar]
- Dymond, J. R. (1939). The fishes of the Ottawa region. Royal Ontario Museum of Zoology; Retrieved from https://www.biodiversitylibrary.org/item/111705#page/7/mode/1up [Google Scholar]
- Edmands, S. (2007). Between a rock and a hard place: Evaluating the relative risks of inbreeding and outbreeding for conservation and management. Molecular Ecology, 16(3), 463–475. 10.1111/j.1365-294X.2006.03148.x [DOI] [PubMed] [Google Scholar]
- Eldridge, W. H. , Myers, J. M. , & Naish, K. A. (2009). Long‐term changes in the fine‐scale population structure of coho salmon populations (Oncorhynchus kisutch) subject to extensive supportive breeding. Heredity, 103(4), 299–309. 10.1038/hdy.2009.69 [DOI] [PubMed] [Google Scholar]
- Eldridge, W. H. , & Naish, K. A. (2007). Long‐term effects of translocation and release numbers on fine‐scale population structure among coho salmon (Oncorhynchus kisutch). Molecular Ecology, 16(12), 2407–2421. 10.1111/j.1365-294X.2007.03271.x [DOI] [PubMed] [Google Scholar]
- Farrell, J. M. , Getchell, G. , Kapuscinski, K. , & LaPan, S. R. (2017). Long‐term trends of St. Lawrence river muskellunge: Effects of viral hemorrhagic septicemia and round goby proliferation creates uncertainty for population sustainability. American Fisheries Society Symposium, 85.
- Farrell, J. M. , Klindt, R. M. , Casselman, J. M. , LaPan, S. R. , Werner, R. G. , & Schiavone, A. (2007). Development, implementation, and evaluation of an international muskellunge management strategy for the upper St Lawrence River. Environmental Biology of Fishes, 79(1–2), 111–123. 10.1007/s10641-006-9091-7 [DOI] [Google Scholar]
- Ferchaud, A. L. , Laporte, M. , Perrier, C. , & Bernatchez, L. (2018). Supplementation impacts on the population genetic structure of an exploited fish: Do thousands of SNPs and dozens of microsatellites tell us the same story? Evolutionary Applications, 11, 1153–1165. [Google Scholar]
- Fernández‐Cebrián, R. , Araguas, R. M. , Sanz, N. , & García‐Marín, J. L. (2014). Genetic risks of supplementing trout populations with native stocks: A simulation case study from current Pyrenean populations. Canadian Journal of Fisheries and Aquatic Sciences, 71(8), 1243–1255. 10.1139/cjfas-2013-0362 [DOI] [Google Scholar]
- Finnegan, A. K. , & Stevens, J. R. (2008). Assessing the long‐term genetic impact of historical stocking events on contemporary populations of Atlantic salmon, Salmo salar . Fisheries Management and Ecology, 15(4), 315–326. 10.1111/j.1365-2400.2008.00616.x [DOI] [Google Scholar]
- Ford, M. J. (2002). Selection in captivity during supportive breeding may reduce fitness in the wild. Conservation Biology, 16(3), 815–825. 10.1046/j.1523-1739.2002.00257.x [DOI] [Google Scholar]
- Frankham, R. (2015). Genetic rescue of small inbred populations: Meta‐analysis reveals large and consistent benefits of gene flow. Molecular Ecology, 24(11), 2610–2618. 10.1111/mec.13139 [DOI] [PubMed] [Google Scholar]
- Frankham, R. , Ballou, J. D. , & Briscoe, D. A. (2010). Introduction to conservation genetics. Cambridge: Cambridge University Press; 10.1017/CBO9780511809002 [DOI] [Google Scholar]
- Frankham, R. , Ballou, J. D. , Eldridge, M. D. B. , Lacy, R. C. , Ralls, K. , Dudash, M. R. , & Fenster, C. B. (2011). Predicting the probability of outbreeding depression. Conservation Biology, 25(3), 465–475. 10.1111/j.1523-1739.2011.01662.x [DOI] [PubMed] [Google Scholar]
- Frankham, R. (2016). Genetic rescue benefits persist to at least the F3 generation, based on a meta‐analysis. Biol. Conserv., 195, 33–36. [Google Scholar]
- Fraser, D. J. , Walker, L. , Yates, M. C. , Marin, K. , Wood, J. L. A. , Bernos, T. A. , & Zastavniouk, C. (2018). Population correlates of rapid captive‐induced maladaptation in a wild fish. Evolutionary Applications, 1–13. 10.1111/eva.12649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frichot, E. , & François, O. (2015). LEA: An R package for landscape and ecological association studies. Methods in Ecology and Evolution, 6(8), 925–929. 10.1111/2041-210X.12382 [DOI] [Google Scholar]
- Garcia‐Marin, J. l. , Sanz, N. , & Pla, C. (1999). Erosion of the native genetic resources of brown trout in Spain. Ecology of Freshwater Fish, 8(3), 151–158. 10.1111/j.1600-0633.1999.tb00066.x [DOI] [Google Scholar]
- Gautier, M. , Gharbi, K. , Cezard, T. , Foucaud, J. , Kerdelhué, C. , Pudlo, P. , … Estoup, A. (2013). The effect of RAD allele dropout on the estimation of genetic variation within and between populations. Molecular Ecology, 22(11), 3165–3178. 10.1111/mec.12089 [DOI] [PubMed] [Google Scholar]
- Glémin, S. (2003). How are deleterious mutations purged? Drift versus nonrandom mating. Evolution, 57(12), 2678–2687. 10.1111/j.0014-3820.2003.tb01512.x [DOI] [PubMed] [Google Scholar]
- Goudet, J. (2005). hierfstat, a package for r to compute and test hierarchical F‐statistics. Molecular Ecology Notes, 5(1), 184–186. 10.1111/j.1471-8286.2004.00828.x [DOI] [Google Scholar]
- Guillaume, F. , & Rougemont, J. (2006). Nemo: An evolutionary and population genetics programming framework. Bioinformatics (Oxford, England), 22(20), 2556–2557. 10.1093/bioinformatics/btl415 [DOI] [PubMed] [Google Scholar]
- Haller, B. C. , & Messer, P. W. (2017). SLiM 2: Flexible, interactive forward genetic simulations. Molecular Biology and Evolution, 34(1), 230–240. 10.1093/molbev/msw211 [DOI] [PubMed] [Google Scholar]
- Hansen, M. M. , Fraser, D. J. , Meier, K. , & Mensberg, K.‐L.‐D. (2009). Sixty years of anthropogenic pressure: A spatio‐temporal genetic analysis of brown trout populations subject to stocking and population declines. Molecular Ecology, 18(12), 2549–2562. 10.1111/j.1365-294X.2009.04198.x [DOI] [PubMed] [Google Scholar]
- Harbicht, A. , Wilson, C. C. , & Fraser, D. J. (2014). Does human‐induced hybridization have long‐term genetic effects? Empirical testing with domesticated, wild and hybridized fish populations. Evolutionary Applications, 7(10), 1180–1191. 10.1111/eva.12199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, R. D. (2008). A flexible forward simulator for populations subject to selection and demography. Bioinformatics (Oxford, England), 24(23), 2786–2787. 10.1093/bioinformatics/btn522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban, S. (2014). An overview of the utility of population simulation software in molecular ecology. Molecular Ecology, 23(10), 2383–2401. 10.1111/mec.12741 [DOI] [PubMed] [Google Scholar]
- Hoban, S. , Bertorelle, G. , & Gaggiotti, O. E. (2012). Computer simulations: Tools for population and evolutionary genetics. Nature Reviews. Genetics, 13(2), 110–122. 10.1038/nrg3130 [DOI] [PubMed] [Google Scholar]
- Hudon, C. , & Carignan, R. (2008). Cumulative impacts of hydrology and human activities on water quality in the St. Lawrence River (Lake Saint‐Pierre, Quebec, Canada). Canadian Journal of Fisheries and Aquatic Sciences, 65, 1165–1180. 10.1139/F08-069 [DOI] [Google Scholar]
- Jennings, M. J. , Hatzenbeler, G. R. , & Kampa, J. M. (2011). Spring capture site fidelity of adult muskellunge in inland lakes. North American Journal of Fisheries Management, 31(3), 461–467. 10.1080/02755947.2011.590118 [DOI] [Google Scholar]
- Jennings, M. J. , Sloss, B. L. , Hatzenbeler, G. R. , Kampa, J. M. , Simonson, T. D. , Avelallemant, S. P. , … Underwood, B. D. (2010). Implementation of genetic conservation practices in a muskellunge propagation and stocking program. Fisheries, 35(8), 388–395. 10.1577/1548-8446-35.8.388 [DOI] [Google Scholar]
- Jombart, T. , Devillard, S. , & Balloux, F. (2010). Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genetics, 11, 94 10.1186/1471-2156-11-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapuscinski, K. L. , Belonger, B. J. , Fajfer, S. , & Lychwick, T. J. (2007). Population dynamics of muskellunge in Wisconsin waters of Green Bay, Lake Michigan, 1989–2005. Environmental Biology of Fishes, 79(1–2), 27–36. 10.1007/s10641-006-9132-2 [DOI] [Google Scholar]
- Kapuscinski, K. L. , Sloss, B. L. , & Farrell, J. M. (2013). Genetic population structure of muskellunge in the great lakes. Transactions of the American Fisheries Society, 142(4), 1075–1089. 10.1080/00028487.2013.799515 [DOI] [Google Scholar]
- Kim, B. Y. , Huber, C. D. , & Lohmueller, K. E. (2018). Deleterious variation shapes the genomic landscape of introgression. PLOS Genetics, 14(10), e1007741 10.1371/journal.pgen.1007741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppelman, J. B. , & Philipp, D. P. (1986). Genetic applications in muskellunge management. In Hall G. E. (Ed.), Managing muskies. American Fisheries Society, Special Publication 15, American Fisheries Society, Bethesda, Maryland, pp. 111–121.
- Laikre, L. , & Ryman, N. (1996). Effects on intraspecific biodiversity from harvesting and enhancing natural populations. Ambio, 25(8), 504–509. [Google Scholar]
- Laikre, L. , Schwartz, M. K. , Waples, R. S. , & Ryman, N. , & GeM Working Group (2010). Compromising genetic diversity in the wild: Unmonitored large‐scale release of plants and animals. Trends in Ecology & Evolution, 25(9), 520–529. 10.1016/j.tree.2010.06.013 [DOI] [PubMed] [Google Scholar]
- Lamaze, F. C. , Sauvage, C. , Marie, A. , Garant, D. , & Bernatchez, L. (2012). Dynamics of introgressive hybridization assessed by SNP population genomics of coding genes in stocked brook charr (Salvelinus fontinalis). Molecular Ecology, 21(12), 2877–2895. 10.1111/j.1365-294X.2012.05579.x [DOI] [PubMed] [Google Scholar]
- Lawrie, D. S. (2017). Accelerating Wright‐Fisher forward simulations on the graphics processing unit. G3: Genes. Genomes, Genetics, 7, 3229–3236. 10.1534/g3.117.300103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, D. J. , Hellenthal, G. , Myers, S. , & Falush, D. (2012). Inference of population structure using dense haplotype data. PLoS Genetics, 8(1), e1002453 10.1371/journal.pgen.1002453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Cam, S. , Perrier, C. , Besnard, A.‐L. , Bernatchez, L. , & Evanno, G. (2015). Genetic and phenotypic changes in an Atlantic salmon population supplemented with non‐local individuals: A longitudinal study over 21 years. Proceedings of the Royal Society B: Biological Sciences, 282(1802), 20142765 10.1098/rspb.2014.2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc, E. , Mailhot, Y. , Mingelbier, M. , & Bernatchez, L. (2008). The landscape genetics of yellow perch (Perca flavescens) in a large fluvial ecosystem. Molecular Ecology, 17(7), 1702–1717. 10.1111/j.1365-294X.2008.03710.x [DOI] [PubMed] [Google Scholar]
- Létourneau, J. , Ferchaud, A.‐L. , Luyer, J. L. , Laporte, M. , Garant, D. , & Bernatchez, L. (2018). Predicting the genetic impact of stocking in Brook Charr (Salvelinus fontinalis) by combining RAD sequencing and ming of explanatory variables. Evolutionary Applications, 11(5), 577–592. 10.1111/eva.12566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v1 [q-bio.GN]
- Lynch, M. (1991). The genetic interpretation of inbreeding depression and outbreeding depression. Evolution, 45(3), 622–629. 10.2307/2409915 [DOI] [PubMed] [Google Scholar]
- Lynch, M. , Blanchard, J. , Houle, D. , Kibota, T. , Schultz, S. , Vassilieva, L. , & Willis, J. (1999). Perspective: Spontaneous deleterious mutation. Evolution, 53(3), 645–663. 10.2307/2640707 [DOI] [PubMed] [Google Scholar]
- Lynch, M. , Conery, J. , & Bürger, R. (1995). Mutational meltdowns in sexual populations. Evolution, 49(6), 1067–1080. 10.1111/j.1558-5646.1995.tb04434.x [DOI] [PubMed] [Google Scholar]
- Lynch, M. , & O’Hely, M. (2001). Captive breeding and the genetic fitness of natural populations. Conservation Genetics, 2(4), 363–378. 10.1023/A:1012550620717 [DOI] [Google Scholar]
- Malinsky, M. , Trucchi, E. , Lawson, D. J. , & Falush, D. (2018). RADpainter and fineRADstructure: Population inference from RADseq data. Molecular Biology and Evolution, 35(5), 1284–1290. 10.1093/molbev/msy023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margenau, T. L. (1994). Evidence of homing of a displaced muskellunge, Esox masquinongy . Journal of Freshwater Ecology, 9(3), 253–256. 10.1080/02705060.1994.9664892 [DOI] [Google Scholar]
- Marie, A. D. , Bernatchez, L. , & Garant, D. (2010). Loss of genetic integrity correlates with stocking intensity in Brook Charr (Salvelinus Fontinalis). Molecular Ecology, 19, 2025–2037. 10.1111/mec.2010.19.issue-10 [DOI] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.Journal, 17(1), 10–12. [Google Scholar]
- Mascher, M. , Wu, S. , Amand, P. S. , Stein, N. , & Poland, J. (2013). Application of genotyping‐by‐sequencing on semiconductor sequencing platforms: A comparison of genetic and reference‐based marker ordering in barley. PLoS ONE, 8, e76925 10.1371/journal.pone.0076925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirmans, P. G. (2012). The trouble with isolation by distance. Molecular Ecology, 21(12), 2839–2846. [DOI] [PubMed] [Google Scholar]
- Messer, P. W. (2013). SLiM: Simulating evolution with selection and linkage. Genetics, 194(4), 1037–1039. 10.1534/genetics.113.152181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, L. M. , Farrell, J. M. , Kapuscinski, K. L. , Scribner, K. , Sloss, B. L. , Turnquist, K. , & Wilson, C. C. (2017). A review of muskellunge population genetics: Implications for management and future research needs. American Fisheries Society Symposium, 85, 385–414. [Google Scholar]
- Miller, L. M. , Kallemeyn, L. , & Senanan, W. (2001). Spawning‐site and natal‐site fidelity by northern pike in a large lake: mark‐recapture and genetic evidence. Transactions of the American Fisheries Society, 130(2), 307–316. [DOI] [Google Scholar]
- Miller, L. M. , Mero, S. W. , & Younk, J. A. (2009). The Genetic legacy of stocking muskellunge in a northern Minnesota lake. Transactions of the American Fisheries Society, 138(3), 602–615. 10.1577/T08-114.1 [DOI] [Google Scholar]
- Miller, L. M. , Mero, S. W. , & Younk, J. A. (2012). The Impact of stocking on the current ancestry in twenty native and introduced muskellunge populations in Minnesota. Transactions of the American Fisheries Society, 141(5), 1411–1423. 10.1080/00028487.2012.697092 [DOI] [Google Scholar]
- Mongeau, J. R. , Leclerc, J. , & Brisebois, J. (1980). La répartition géographique des poissons, les ensemencements, la pêche sportive et commerciale, les frayères et la bathymétrie du fleuve Saint‐Laurent dans le bassin de Laprairie et les rapides de Lachine. Ministère du Loisir, de la Chasse et de la Pêche, Service de l’aménagement et de l’exploitation de la faune. Rapport technique No. 06–29. 145 p.
- Mongeau, J. R. , & Massé, G. (1976). Les poissons de la région de Montréal, la pêche sportive et commerciale, les ensemencements, les frayères, la contamination par le mercure et les PCB. Ministère du Loisir de la Chasse et de la Pêche, Service de l’aménagement et de l’exploitation de la faune, Montréal, Québec. Rapport technique 06–13. xviii + 286p.
- Morrissey, M. B. , & de Kerckhove, D. T. (2009). The maintenance of genetic variation due to asymmetric gene flow in dendritic metapopulations. The American Naturalist, 174, 875–889. 10.1086/648311 [DOI] [PubMed] [Google Scholar]
- Muhlfeld, C. C. , Kalinowski, S. T. , McMahon, T. E. , Taper, M. L. , Painter, S. , Leary, R. F. , & Allendorf, F. W. (2009). Hybridization rapidly reduces fitness of a native trout in the wild. Biology Letters, 5, 328–331. 10.1098/rsbl.2009.0033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet‐Cauchon, G. , Mingelbier, M. , Lecomte, F. , & Bernatchez, L. (2014). Landscape variability explains spatial pattern of population structure of northern pike (Esox lucius) in a large fluvial system. Ecology and Evolution, 4(19), 3723–3735. 10.1002/ece3.1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz‐Vinas, I. , Loot, G. , Stevens, V. M. , & Blanchet, S. (2015). Evolutionary processes driving spatial patterns of intraspecific genetic diversity in river ecosystems. Molecular Ecology, 24(18), 4586–4604. 10.1111/mec.13345 [DOI] [PubMed] [Google Scholar]
- Perrier, C. , Baglinière, J.‐L. , & Evanno, G. (2013). Understanding admixture patterns in supplemented populations: A case study combining molecular analyses and temporally explicit simulations in Atlantic salmon. Evolutionary Applications, 6(2), 218–230. 10.1111/j.1752-4571.2012.00280.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier, C. , Guyomard, R. , Bagliniere, J.‐L. , Nikolic, N. , & Evanno, G. (2013). Changes in the genetic structure of Atlantic salmon populations over four decades reveal substantial impacts of stocking and potential resiliency. Ecology and Evolution, 3(7), 2334–2349. 10.1002/ece3.629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, B. L. , & Baird, S. J. E. (2015). Spatial sorting unlikely to promote maladaptive hybridization: Response to lowe, muhlfeld, and allendorf. Trends in Ecology & Evolution, 30(10), 564–565. 10.1016/j.tree.2015.08.005 [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralls, K. , Ballou, J. D. , Dudash, M. R. , Eldridge, M. D. B. , Fenster, C. B. , Lacy, R. C. , … Frankham, R. (2018). Call for a paradigm shift in the genetic management of fragmented populations. Conservation Letters, 11(2), e12412 10.1111/conl.12412 [DOI] [Google Scholar]
- Robitaille, J. A. , & Cotton, F. (1992). Bilan des connaissances sur le maskinongé (Esox masquinongy) et sur ses populations dans le Saint‐Laurent, Ministère du Loisir, de la Chasse et de la Pêche, Direction de la gestion des espèces et des habitats. Rapp. Tech. 1–55.
- Romiguier, J. , Gayral, P. , Ballenghien, M. , Bernard, A. , Cahais, V. , Chenuil, A. , … Galtier, N. (2014). Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515(7526), 261–263. 10.1038/nature13685 [DOI] [PubMed] [Google Scholar]
- Rondeau, E. B. , Minkley, D. R. , Leong, J. S. , Messmer, A. M. , Jantzen, J. R. , von Schalburg, K. R. , … Koop, B. F. (2014). The genome and linkage map of the northern pike (Esox lucius): Conserved synteny revealed between the salmonid sister group and the neoteleostei. PLoS ONE, 9(7), e102089 10.1371/journal.pone.0102089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross‐Ibarra, J. , Tenaillon, M. , & Gaut, B. S. (2009). Historical divergence and gene flow in the genus Zea . Genetics, 181(4), 1399–1413. 10.1534/genetics.108.097238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross‐Ibarra, J. , Wright, S. I. , Foxe, J. P. , Kawabe, A. , DeRose‐Wilson, L. , Gos, G. , … Gaut, B. S. (2008). Patterns of polymorphism and demographic history in natural populations of Arabidopsis lyrata . PLoS ONE, 3(6), e2411 10.1371/journal.pone.0002411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset, F. (1997). Genetic differentiation and estimation of gene flow from F‐statistics under isolation by distance. Genetics, 145(4), 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, C. , Castric, V. , Pauwels, M. , Wright, S. I. , Saumitou‐Laprade, P. , & Vekemans, X. (2011). Does speciation between Arabidopsis halleri and Arabidopsis lyrata coincide with major changes in a molecular target of adaptation? PLoS ONE, 6(11), e26872 10.1371/journal.pone.0026872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman, N. , & Laikre, L. (1991). Effects of supportive breeding on the genetically effective population size. Conservation Biology, 5(3), 325–329. 10.1111/j.1523-1739.1991.tb00144.x [DOI] [Google Scholar]
- Saccheri, I. , Kuussaari, M. , Kankare, M. , Vikman, P. , Fortelius, W. , & Hanski, I. (1998). Inbreeding and extinction in a butterfly metapopulation. Nature, 392(6675), 491–494. 10.1038/33136 [DOI] [Google Scholar]
- Scribner, K. , Howell, P. , Thomas, M. , Smith, K. , Hanchin, P. , Wolgamood, M. , & Whelan, G. (2015). Spatial genetic structure of suspected remnant and naturalized populations of muskellunge and evidence for introgression between stocked and native strains. Journal of Great Lakes Research, 41(4), 1131–1137. 10.1016/j.jglr.2015.08.011 [DOI] [Google Scholar]
- Sexton, J. P. , Hangartner, S. B. , & Hoffmann, A. A. (2014). Genetic isolation by environment or distance: Which pattern of gene flow is most common? Evolution, 68(1), 1–15. [DOI] [PubMed] [Google Scholar]
- Simonson, T. (2010). Muskellunge management update (ed. Resources WDoN), p. 40, Madison, WI.
- Sjodin, P. (2005). On the meaning and existence of an effective population size. Genetics, 169(2), 1061–1070. 10.1534/genetics.104.026799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sønstebø, J. H., Borgstrøm, R., & Heun, M. (2008). High genetic introgression in alpine brown trout (Salmo trutta L.) populations from Hardangervidda, Norway. Ecology of Freshwater Fish, 17(1), 174–183. 10.1111/j.1600-0633.2007.00271.x. [DOI] [Google Scholar]
- Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123(3), 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallmon, D. A. , Luikart, G. , & Waples, R. S. (2004). The alluring simplicity and complex reality of genetic rescue. Trends in Ecology & Evolution, 19(9), 489–496. 10.1016/j.tree.2004.07.003 [DOI] [PubMed] [Google Scholar]
- Thériault, V. , Moyer, G. R. , & Banks, M. A. (2010). Survival and life history characteristics among wild and hatchery coho salmon (Oncorhynchus kisutch) returns: How do unfed fry differ from smolt releases? Canadian Journal of Fisheries and Aquatic Sciences, 67(3), 486–497. 10.1139/F09-200 [DOI] [Google Scholar]
- Thériault, V. , Moyer, G. R. , Jackson, L. S. , Blouin, M. S. , & Banks, M. A. (2011). Reduced reproductive success of hatchery coho salmon in the wild: Insights into most likely mechanisms. Molecular Ecology, 20(9), 1860–1869. 10.1111/j.1365-294X.2011.05058.x [DOI] [PubMed] [Google Scholar]
- Turnquist, K. N. , Larson, W. A. , Farrell, J. M. , Hanchin, P. A. , Kapuscinski, K. L. , Miller, L. M. , … Sloss, B. L. (2017). Genetic structure of muskellunge in the Great Lakes region and the effects of supplementation on genetic integrity of wild populations. Journal of Great Lakes Research, 43(6), 1141–1152. 10.1016/j.jglr.2017.09.005 [DOI] [Google Scholar]
- Valiquette, E. , Perrier, C. , Thibault, I. , & Bernatchez, L. (2014). Loss of genetic integrity in wild lake trout populations following stocking: Insights from an exhaustive study of 72 lakes from Québec, Canada. Evolutionary Applications, 7(6), 625–644. 10.1111/eva.12160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vézina, R. (1977). Les introductions de maskinongé, Esoxmasquinongy, au Québec et leurs résultats, p. 129–135. In: Compte rendu du 10e atelier sur les poissons d'eau chaude. Ministère du Tourisme, de la Chasse et de la Pêche du Québec, Service de l'Aménagement de la Faune.
- Vincent, B. , & Legendre, V. (1974). Répartition géographique du maskinongé, Esox masquinongy, dans le district des Laurentides. Compilation 1972. Ministère du Tourisme, de la Chasse et de la Pêche du Québec, Service de l'Aménagement de la Faune et Service de la Recherche biologique.
- Wang, J. , Hill, W. G. , Charlesworth, D. , & Charlesworth, B. (1999). Dynamics of inbreeding depression due to deleterious mutations in small populations: Mutation parameters and inbreeding rate. Genetical Research, 74(2), 165–178. 10.1017/S0016672399003900 [DOI] [PubMed] [Google Scholar]
- Waples, R. S. (2006). A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conservation Genetics, 7, 167–184. 10.1007/s10592-005-9100-y [DOI] [Google Scholar]
- Waples, R. S. , & Do, C. (2008). ldne: A program for estimating effective population size from data on linkage disequilibrium. Molecular Ecology Resources, 8(4), 753–756. 10.1111/j.1755-0998.2007.02061.x [DOI] [PubMed] [Google Scholar]
- Waples, R. S. , & England, P. R. (2011). Estimating contemporary effective population size on the basis of linkage disequilibrium in the face of migration. Genetics, 189(2), 633–644. 10.1534/genetics.111.132233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. S. , Hindar, K. , Karlsson, S. , & Hard, J. J. (2016). Evaluating the Ryman‐Laikre effect for marine stock enhancement and aquaculture. Current Zoology, 62(6), 617–627. 10.1093/cz/zow060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. K. , Larson, W. A. , & Waples, R. S. (2016). Estimating contemporary effective population size in non‐model species using linkage disequilibrium across thousands of loci. Heredity, 117(4), 233–240 dx.doi.org/10.1038/hdy.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. , & Cockerham, C. (1984). Estimating F‐statistics for the analysis of population‐structure. Evolution, 38(6), 1358–1370. 10.2307/2408641 [DOI] [PubMed] [Google Scholar]
- Whillans, T. H. (1979). Historic transformations of fish communities in three great lakes bays. Journal of Great Lakes Research, 5(2), 195–215. 10.1016/S0380-1330(79)72146-0 [DOI] [Google Scholar]
- White, M. M. , Kohli, B. A. , & Converse, P. E. (2018). Historical and contemporary gene flow and genetic structure of Muskellunge in the Ohio River drainage. Transactions of the American Fisheries Society, 147, 1067–1077. 10.1002/tafs.10101 [DOI] [Google Scholar]
- Whitlock, M. C. (2000). Fixation of new alleles and the extinction of small populations: Drift load, beneficial alleles, and sexual selection. Evolution, 54(6), 1855–1861. 10.1111/j.0014-3820.2000.tb01232.x [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C. , & Bürger, R. (2004). Fixation of new mutations in small populations In Ferrière R., Dieckmann U., & Couvet D. (Eds.), Evolutionary conservation biology (pp. 155–170). Cambridge: Cambridge University Press, International Institute for Applied Systems Analysis. [Google Scholar]
- Wilson, C. C. , Liskauskas, A. P. , & Wozney, K. M. (2016). Pronounced genetic structure and site fidelity among native muskellunge populations in Lake Huron and Georgian Bay. Transactions of the American Fisheries Society, 145(6), 1290–1302. 10.1080/00028487.2016.1209556 [DOI] [Google Scholar]
- Wingate, P. J. (1986). Philosophy of muskellunge management. American Fisheries Society Special Publication, 15, 199–202. [Google Scholar]
- Wright, S. (1943). Isolation by distance. Genetics, 28(2), 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence data are available at NCBI BioProject ID PRJNA512459. Pipeline for forward simulation is available on gitub at https://github.com/QuentinRougemont/fwd_sims.